

Integrin α5β1 structures in resting and active states with ligand fibronectin reveal key interactions for integrin opening.
Abstract
Integrin α5β1 is a major fibronectin receptor critical for cell migration. Upon complex formation, fibronectin and α5β1 undergo conformational changes. While this is key for cell-tissue connections, its mechanism is unknown. Here, we report cryo–electron microscopy structures of native human α5β1 with fibronectin to 3.1-angstrom resolution, and in its resting state to 4.6-angstrom resolution. The α5β1-fibronectin complex revealed simultaneous interactions at the arginine-glycine-aspartate loop, the synergy site, and a newly identified binding site proximal to adjacent to metal ion–dependent adhesion site, inducing the translocation of helix α1 to secure integrin opening. Resting α5β1 adopts an incompletely bent conformation, challenging the model of integrin sharp bending inhibiting ligand binding. Our biochemical and structural analyses showed that affinity of α5β1 for fibronectin is increased with manganese ions (Mn2+) while adopting the half-bent conformation, indicating that ligand-binding affinity does not depend on conformation, and α5β1 opening is induced by ligand-binding.
INTRODUCTION
Integrin α5β1 is the primary fibronectin (FN) receptor (1), responsible for cell migration and adhesion. Integrin α5β1-FN interactions are of particular interest as both proteins are ubiquitously expressed in various cell and tissue types to maintain the communication between cells and the extracellular matrix (ECM). Furthermore, α5β1 represents an integrin prototype that is fundamental to cellular processes, yet the conformational change through its molecular interaction with FN, a major component of the ECM, is not well understood.
Serving as a bidirectional link between the cell and its environment, integrin α5β1 functions as a control hub for the intracellular focal adhesion machinery. Focal adhesion consists of hundreds of signaling and mechanosensing factors, transmitting extracellular signals to downstream players, including the actin cytoskeleton (2–4). The resulting remodeling and changes in the dynamic behavior of the actin cytoskeleton direct cell shape formation and cell motility, hence, dictating cell fate (2). Integrin α5β1 connects to the FN ligand, a major component of the ECM, through its extracellular domains and to the actin cytoskeleton through its cytoplasmic tail domains in combination with accessory factors (5). Failure of integrin function and corresponding impairment of the focal adhesion machinery is related to numerous diseases (6), including aggressive cancer formation due to uncoordinated migration. Integrin α5β1 particularly contributes to cancer metastasis, reflected by its up-regulation in invasive cancer cells (7, 8).
FN, the binding partner of integrin α5β1, is a fibrous dimer acting as a core component of the meshwork formation of the ECM. FN is a large protein that comprises repetitive modules of type I, II, and III domains, which show a “beads on a string” morphology (9, 10). FN is recognized by the large N-terminal extracellular domain of integrin α5β1, termed ectodomain, via the common integrin ligand motif “arginine-glycine-aspartate” (RGD) found in the FN fragment III containing domain 10 (FN10). During cell movement, as bound integrin is exposed to mechanical force, it grips FN through its so-called synergy site with the sequence DRVPHSRN in FN9 (11, 12) and anchors cells with an unknown mechanism. Thus, how FN engages integrin α5β1 and activates cells remains enigmatic.
Despite being a major class of integrin, α5β1 has been proposed not to follow the canonical integrin open-close paradigm, which has been derived mostly from studies on αIIbβ3 and αvβ3 integrins (13–18). In this classical β3-based model, integrins exhibit an acutely bent conformation on resting cells in which the heads of the ectodomains point toward the plasma membrane and the legs and transmembrane domains of α and β subunits are in close contact with each other (5, 13, 17–20). In this “unprimed” state, integrins show low affinity for their ligands with their heads occluding access to the ligand-binding site by facing toward the plasma membrane (5, 15). In the presence of Mn2+, which mimics intracellular signals in vitro, classical integrins become “primed” and show an increased affinity for their ligands (5, 17, 21). The upright conformation releases the ligand-binding integrin heads from the steric hindrance by the membrane and therefore renders them more potent to capture ligands. The binding of the RGD peptide to the groove at the tip of the integrin heads (14, 16, 22) translates conformational changes from the metal ion–dependent adhesion site (MIDAS), leading to the allosteric swing-out motion of the β hybrid domain (15) and further to the opening of the legs. However, this mode of action does not apply to integrin α5β1. Truncated integrin α5β1 ectodomains do not undergo the large conformational change in the presence of the RGD peptide alone (21). Moreover, negative-stain electron microscopy (EM) observations hinted that the α5β1 ectodomain may not bend completely in its resting state (23), which challenges the view that bending occludes the ligand-binding site. These observations raised the question whether integrin α5β1 uses a unique mechanism with only weak coupling between its global conformational change and its ligand-binding affinity. Considering its critical biological roles in cell attachment and metastatic detachment, it is imperative to uncover how the activation of α5β1 differs from the canonical model and how FN mediates the opening of α5β1.
Here, we report the cryo-EM structures of native human integrin α5β1 purified from human placenta (i) in its open conformation in complex with FN and the stabilizing antibody TS2/16 and (ii) in its resting state employing an incomplete bent conformation. The complex of integrin α5β1 with FN7-10 shows three simultaneous interactions critical for the stabilization of integrin opening, namely, (i) at the synergy site of FN9, (ii) at the RGD loop of FN10, and (iii) at the adjacent to MIDAS (ADMIDAS) of the integrin β1 βI domain. The dual binding of FN9 and FN10 oriented FN10 to stabilize the large opening of α5β1. In particular, key interactions centered at R1445 and Y1446 of FN and D137 of ADMIDAS in β1 βI domain provide an additional anchoring point that triggers the opening of α5β1, which was previously unknown. Through this interaction, D137 of the β1 βI domain moves toward FN, resulting in a translocation of helix α1, which then allosterically induces the known conformational changes (5, 13–15, 17, 21) for integrin opening. In the resting state, integrin α5β1 uses a unique, incompletely bent conformation. The incomplete bending does not sterically hinder the access of FN to the integrin head. Side-by-side structural and biochemical comparison indicated that integrin α5β1 changes its affinity for FN while still in its half-bent conformation. The integrin opening is observed when FN is bound rather than FN binding occurring as a consequence of integrin opening. Our study, thus, reveals a mechanism of integrin opening by FN in which FN structural domain acts as a scaffold to facilitate and the opening of integrin.
RESULTS
Ligand-bound native integrin α5β1 displays an extended conformation
To gain mechanistic insights into the regulation of integrin α5β1 on a molecular level, full-length native integrin α5β1 (Fig. 1 and fig. S1A) was purified from human placenta (24) and embedded into membrane-scaffolding nanodiscs (NDs) (fig. S1, B and C) (25). To understand integrin opening and ligand engagement, we determined the structure of integrin α5β1 in complex with its ligand FN7-10 in the presence of MnCl2 (Fig. 1, A and C, and figs. S2 and S3) and the stabilizing antibody fragment TS2/16 Fv-clasp (26) by cryo-EM at an overall resolution of 3.5 Å, with the head resolved up to 3.1 Å (figs. S2 and S3). Although the C-terminal portion including the flexible lower legs, the transmembrane helices, and the cytoplasmic tails was not visible from the cryo-EM map, the corresponding negative-staining analysis showed the full integrin complex embedded into the NDs (fig. S1C). The overall arrangement was similar to that observed in negative-stain EM images of the recombinant ectodomain fragment of unliganded α5β1 (23). These results assured a successful reconstitution of native integrin into the membrane scaffold.
Fig. 1. Cryo-EM analysis of FN-bound integrin α5β1 reveals an open conformation.
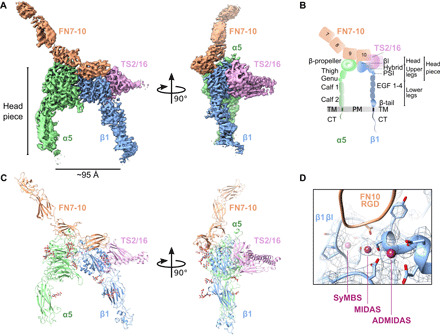
(A) Two views of the FN7-10 (orange)–bound α5β1 (green, α subunit; blue, β subunit) cryo-EM structure, stabilized by TS2/16 (pink). The headpiece reveals an open conformation. (B) Schematic of integrin α5β1 domain architecture and nomenclature of different parts (headpiece, head, upper, and lower legs), including FN7-10 and TS2/16. TM, transmembrane; PM, plasma membrane; CT, cytoplasmic tail. (C) Molecular model based on the cryo-EM density, showing the same two views as (A). (D) Close-up and overlay of cryo-EM density with the modeled synergistic metal ion–binding site (SyMBS or LIMBS), MIDAS, and ADMIDAS within the βI domain.
The structure revealed an arrangement of the integrin, where the globular head domains of α and β subunits were in close contact with each other (Fig. 1, A and C). FN formed a dual contact with FN9 contacting the α5 head and FN10 contacting both the α5 and the β1 head domains. The α5β1 domains were in a wide open, extended conformation, with the upper legs about 95 Å apart from each other (Fig. 1, A and C). The opening of the headpiece largely agreed with the arrangement seen in the crystal structures of truncated headpieces of other integrin isoform αIIbβ3 in complex with ligand-mimetic compounds (16, 27). The integrin βI domain contains three functional metal ion pockets exhibiting clear densities, namely, MIDAS, ADMIDAS, and the synergistic metal ion–binding site [SyMBS, also called LIMBS (ligand-associated metal-binding site)] (Fig. 1D). Consistent with previous assignments (14), we assigned the densities as Mn2+ ions, which were used in excess during sample preparation. The structures of the integrin heads, the bound FN9-10, and TS2/16 were well resolved, reaching to a maximal resolution of 3.1 Å; however, the leg portions and FN7-8 resolved at 4 to 7 Å (fig. S2C), indicating higher flexibility of the lower legs, the downstream domains, and the parts of FN distant from the integrin heads.
FN9-10 locks the integrin α5β1 opening by a gear-like mechanism
The α5β1–FN7-10 complex revealed the involvement of the RGD motif (residues 1493 to 1495) in FN10 and the synergy site (residues 1373 to 1380, DRVPHSRN) in FN9, both facing toward the integrin heads simultaneously (Fig. 2, A to D). The RGD loop was previously shown to be flexible in an isolated FN10 domain by nuclear magnetic resonance (28). In our structure, however, in complex with the headpiece of α5β1, the loop was fixed in the pocket between α and β integrin heads (Fig. 2, A to C), resembling structures of single RGD peptides bound to the integrin headpiece (fig. S4) (29). In addition, we observed that the succeeding P1497 in FN10 made a prominent van der Waals contact with the backbone of helix α1, adjacent to S134, and packed against Y133 in the β1 βI domain. S134 was also part of the MIDAS metal coordination sphere (Fig. 2B). Engagement of MIDAS was reported to induce the translocation of the α1 and α7 helices in the βI domain, triggering a piston-like movement, which then leads to the swing-out motion of the hybrid domain and the separation of the α and β legs (5, 13–15, 17, 21).
Fig. 2. Binding of integrin α5β1 to FN7-10 occurs through an extensive interface, including RGD, FN synergy site, and juxta-ADMIDAS area.
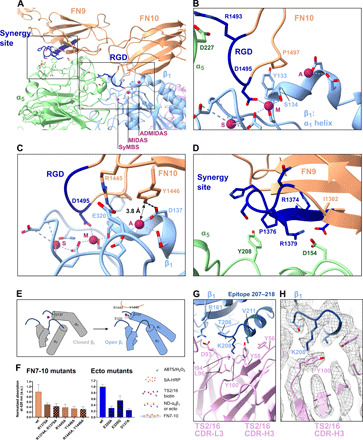
(A) Overview of the α5β1-FN interface. (B) The RGD loop of FN10 inserted into the pocket between α5 and β1 subunits. It binds to Q221, D227(α5), and the MIDAS cation in β1. Newly found contacts are made between P1497(FN10)-Y133(β1) and P1497(FN10)-S134(β1). A, ADMIDAS; M, MIDAS; S, SyMBS (LIMBS). (C) Key interactions in the ADMIDAS region: salt bridge R1445(FN10)-E320(β1); hydrogen bond Y1446(FN10)-D137(β1). (D) The synergy site of FN9 contacts α5: salt bridge R1379(FN9)-D154(α5); interaction of P1376(FN9)-Y208(α5). R1374 is not visibly engaged. (E) Arrangement of secondary structure elements around ADMIDAS and FN. (F) Solid-phase binding assays show the affinity of FN7-10 to integrin ectodomain mutants. Left: Binding of ND-α5β1 to FN7-10 mutants in the presence of Mn2+ (normalized to FN7-10 wild type). Middle: Binding of recombinant, unclasped α5β1 ectodomain mutants to FN7-10 in the presence of Mn2+ (normalized to wild-type ectodomain). Bars represent means + SD of three measurements. Right: Assay strategy. a.u., arbitrary units; wt, wild type. (G) Hydrophobic interactions between the antibody TS2/16 and β1 centered around K208(β1). (H) Close-up of the cation-π interaction between K208(β1) and Y100(TS2/16 CDR-H3) overlaid with cryo-EM density (gray).
However, to date, none of the tested RGD motif–containing peptides alone were sufficient to induce the large opening of integrin α5β1 (29, 30). That indicates that our structure contains key to optimally induce and stabilize the opening of integrin. A closer look at the interaction surfaces between FN and integrin allowed us to identify these interactions (Fig. 2C). Adjacent to the RGD loop, we found that R1445 in FN10 formed a salt bridge with E320 in integrin β1 and that Y1446 in FN10 was in hydrogen bonding distance to D137 in integrin β1. D137 is also part of ADMIDAS, which coordinates the metal ion. The hydroxyl oxygen of Y1446 was located close to Mn2+ (at a distance of 3.8 Å). The close approach of R1445 and Y1446 toward the ADMIDAS coordinating residues (juxta-ADMIDAS) would move helix α1 and its upstream portion closer to FN (Fig. 2E), facilitating a large tilt of the β head. Together, the observed arrangement of FN10 R1445 and Y1446 at ADMIDAS and P1497 at MIDAS is part of the critical pivoting point for the induction of the large conformational change of integrin in coordination with the concomitant insertion of the RGD motif into the interaction pocket at the integrin head in the groove between α5 and β1.
The third contacting point was at the interface between the FN9 synergy site and the α head (Fig. 2, A and D). This interaction was facilitated by a salt bridge (R1379-D154) and a proline-aromatic interaction between P1376 in FN9 and Y208 in α5 (Fig. 2D). In particular, the R1379-D154 interaction agrees with previous biochemical identifications of D154 as a key residue for the interaction with the synergy site (30) and of R1379 in FN9 as a residue involved in the force-induced reinforcement of the integrin-FN binding, called catch-bond formation (31, 32). Our structure explains the molecular basis of the markedly reduced cell adhesion activity of the triple mutant R1374A/P1376A/R1379A (33), although in this conformation, we saw no apparent interaction of FN R1374 with the integrin surface.
To validate the importance of the interactions derived from our structure analysis, we engineered point mutants of FN7-10 juxta-ADMIDAS R1445A, Y1446A, and R1445A/Y1446A as well as of the synergy site R1379A and R1374A/R1379A. We tested the binding of α5β1 to the FN7-10 mutants by a solid-phase equilibrium binding assay (Fig. 2F, left). For this enzyme-linked immunosorbent assay (ELISA assay)–like assay, wells were coated with FN7-10, and the binding of α5β1 to FN7-10 was measured. Compared to FN7-10 wild type, a reduction of binding was observed with all engineered mutants. This is also in excellent agreement with a previous cell biological report that an R1445A mutant displayed reduced cellular adhesion (33). We further tested binding of recombinant α5β1 ectodomain mutants on FN7-10, including α5β1 (E320A), α5β1 (E320Q), and α5β1 (D137A) (Fig. 2F, middle). Similar to FN7-10 mutants, all integrin mutants also displayed decreased binding to FN7-10 in comparison to the wild-type ectodomain.
Overall, the FN molecule covered the top of the α- and β-integrin heads (Fig. 2A). The structure of FN9-10 was rigid reaching to a local resolution of 3.1 Å, while the upstream F7-F8 was less well resolved (local resolution of 4 to 7 Å), indicating its flexibility (fig. S2C). The arrangement of FN9-10 showed an interdomain tilt angle between FN9 and FN10 of 28°, which is more tilted than 12° observed in the crystal structure of FN7-10 [Protein Data Bank (PDB) ID: 1fnf (12)] (root mean square deviation (RMSD) of 1.59 Å for the FN9-10 portion). This tilt accommodated the docking of the RGD motif into the groove between the integrin heads. We also note that our integrin-bound FN7-9 and the FN7-9 alone in the crystal structure had a similar topological arrangement with an RMSD of 1.04 Å, while FN10 was tilted away. This explains a previous observation that fixing the interdomain tilt angle of FN9-10 to 28° increases the affinity for integrin (34). Our results suggest that the bending of the FN9-10 connection (35) functions as a critical factor to scaffold the open conformation of integrin.
TS2/16 recognizes integrin α5β1 without interfering with key residues for conformational change
TS2/16 is a stimulatory antibody that recognizes the head domain of the β1 subunit [reviewed in (36)]. It is reported to reduce the off-rate of integrin-ligand interactions, thus promoting the integrin-ligand association. In our cryo-EM three-dimensional (3D) reconstruction, TS2/16 contacted the side of the βI domain with a buried surface area of 114.9 Å2 (Figs. 1A, pink, and 2G) and centered around the reported epitope of residues 207 to 218 in the β1 head (37). Residue K208 of the βI domain made a prominent contact by reaching into a hydrophobic pocket on the surface of TS2/16 and formed a cation-π interaction with Y100a of TS2/16 complementarity-determining region H3 (CDR-H3) (Fig. 2H). In addition, D93 of TS2/16 CDR-L3 formed a salt bridge to R161 in the βI domain. The recognition surface in β1 was distant from the sites where the movements occur during the large conformational change, which explains why TS2/16 also recognizes the closed conformation of integrin α5β1 (38). In this regard, we also note that the opening of integrin by FN7-10 occurred less frequently (fig. S2D) in the absence of TS2/16 and showing that TS2/16 is not essential for the opening of integrin α5β1 but it stabilizes the opening conformation.
GlycanN275 of α5 reinforces the FN engagement to integrin α5β1
Integrin was purified from a natural source, allowing us to evaluate the physiological relevance of posttranslational modifications, particularly glycosylation (Fig. 3). The identified N-glycosylation sites (Fig. 3A) validated the previous structural visualizations of recombinantly prepared samples (29, 30) and agreed well with previous biophysical analyses [α, (39); β, (40)]. Of the 14 and 12 predicted potential N-glycosylation sites in α and β subunits, 6 and 4 were found in our cryo-EM density map, respectively (Fig. 3A). The glycan attached to N275 (in the α5 head) reached out to the interface between FN9 and FN10 (Fig. 3, B and C). The glycan was surrounded by a positively charged surface and sandwiched between residues R220 and Y226 of integrin α5, which contacted a mannose and an N-acetylglucosamine moiety, respectively. S223 (α5) further restricted the flexibility of the glycan chain (Fig. 3B). To evaluate the relevance of N275 glycan for the binding to FN, we introduced a point mutation N275A into recombinant α5β1 ectodomains (41) and tested the binding of α5(N275A)β1 to FN7-10 by the solid-phase equilibrium binding assay (Fig. 3E). Compared to the wild-type ectodomains, N275A showed a reduced binding to FN by ~60%, reinforcing the importance of glycanN275 for FN binding. There is also an N-glycan found at the corresponding position on integrin αv (N266 of the αv head) (Fig. 3D) (42, 43); however, the previously reported structure of αvβ3-FN10 showed a different FN contact surface (42). We interpret that the αvβ3-FN10 structure lacks FN9, and therefore, the recognition of the glycan by FN was likely not fully engaged. Our structure, thus, offers a direct structural explanation why glycosylation is essential for integrin’s interaction with FN and also demonstrates the relevance of the glycosylation in cells and in vivo (44, 45).
Fig. 3. N-glycosylation of integrin α5β1 bridges interface to FN.
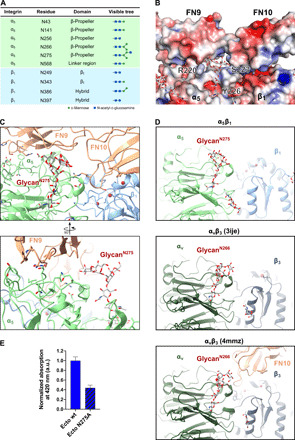
(A) Table listing the N-glycosylation sites and the types of glycan trees as determined in our structure. (B) Electrostatic potential map of the FN-integrin interface and the N275-glycan. (C) Two views of the N275-glycan on α5, which is part of the extensive α5β1-FN interface. The N275-glycan contacts with FN9. (D) Comparison of glycan position between integrin α5β1 and integrin αvβ3 [PDB ID: 3ije (43)] and αvβ3 in complex with FN10 [PDB ID: 4mmz (42)]. The N275-glycan (α5β1) is a conserved glycosylation site among FN-recognizing integrins such as αvβ3. (E) Solid-phase binding assay to measure the binding affinity of integrin α5(N275A)β1 ectodomain mutant in comparison to wild-type ectodomain. Both wild-type and N275A ectodomain mutant were unclasped and binding to FN7-10 wild type evaluated in the presence of Mn2+. Bars represent means + SD of three measurements. Unspecific binding of integrin to bovine serum albumin (BSA)–coated control wells was subtracted from the total absorbance values. Normalized binding is expressed as the ratio of mutant absorbance relative to wild-type absorbance (ratio = 1 represents wild-type binding).
Integrin α5β1 in its resting state shows incomplete bending
To understand the transition of the change of the integrin α5β1 conformation, we obtained the cryo-EM structure of integrin α5β1 in its resting conformation (Fig. 4 and figs. S5 to S7) in the presence of Ca2+ and Mg2+, but without Mn2+. Unlike the known resting structures of β3-containing integrins such as αvβ3 or αIIbβ3 (13, 20) showing an acutely bent conformation, α5β1 exhibited only a half-bent conformation (Fig. 4, A to F) with an angle of ~90° at the knee between the lower legs and the headpiece. Compared with the ligand-bound open conformation of integrin α5β1, the lower legs of both α and β subunits in resting integrin were less flexible, enough to be clearly visible. That is likely due to the parallel closure of the α and β legs (Fig. 4G), while the contact points of the legs are lost in the open conformation as a result of the opening of the upper legs. That shows that ligand binding to the head domains and the subsequent swing-out of the hybrid domain destabilizes the leg-leg interaction (Fig. 5A).
Fig. 4. Resting integrin α5β1 reveals an incompletely bent conformation.
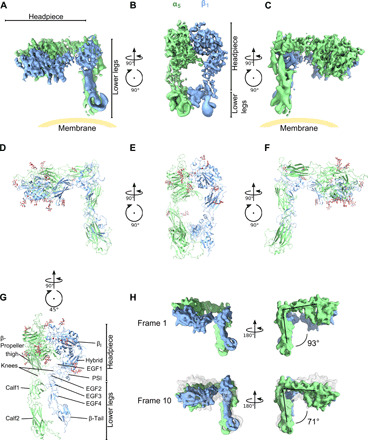
(A to C) Three views of the resting integrin α5β1 cryo-EM structure. The position of the membrane is guided in yellow. Note that precise direction of the membrane cannot be determined because of the lack of membrane density from the structure. (D to F) The structure of the headpieces and modeled structure using homology models of PDB ID: 3fcs (13) for the lower legs. The headpiece reveals a closed conformation, with an orthogonal angle between the headpiece and legs. (G) The lower legs of integrin α5β1 are parallelly contacting each other, between EGF1/2 (β1) and calf1 (α5). (H) Multibody analysis revealed flexibility between the headpiece and lower legs, with an angle varying from 71° to 93° between the first analyzed state (“frame 1”) and the last state (“frame 10”). See fig. S7 for all the frames.
Fig. 5. Resting integrin α5β1 is disadvantageous for FN binding.
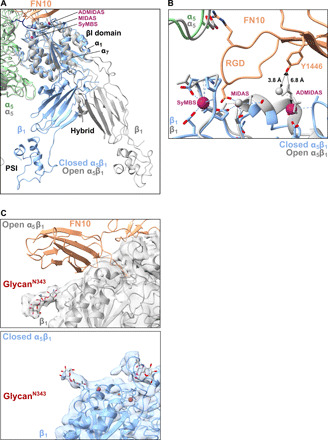
(A) An overlay of the βI, hybrid, and PSI domains of resting (blue) and FN-bound integrin (gray) shows the movement of the α1 and α7 helices in the βI domain and the subsequent swing-out motion of the hybrid domain. (B) ADMIDAS is farther from FN10 (Y1446) in the resting βI domain (blue) than in the open βI domain (gray). (C) Comparison of the position of glycanN343 on β1 with (top) and without (bottom) ligand binding. The glycanN343 hinders the FN binding in the resting structure.
The upper part of the ectodomain was the most stable portion of the whole structure, with a resolution reaching to 4.6 Å in the core of the integrin head (figs. S5, B and C, and S6), partially resolving the side chains (fig. S5D). The integrin heads were tilted in the closed conformation, making the three FN binding sites uncoordinated. In particular, a superposition of both structures showed that the ADMIDAS area was moved away from the possible reach of FN10 Y1446 (Fig. 5B, 6.8 Å in distance) so that ADMIDAS would not be readily available for FN binding. Furthermore, we found that the N-glycan at N343 of the βI domain presented a steric hindrance to the docking of FN10 when the head was closed (Fig. 5C), while it moved away from the FN binding site when integrin α5β1 used an open conformation (Fig. 5C). That motion suggests that N343 has a critical inhibitive function in ligand binding.
In contrast, the resolution of the lower legs was insufficient for model building. Therefore, rigid-body molecular fitting of individual domains was performed using available α5β1 structures and homology models of calf1 and calf2 for the α subunit and plexin-semaphorin-integrin (PSI), epidermal growth factor 1 (EGF1) to EGF4 and β-tail domains for the β subunit. The main contacting surface between the lower legs of α5 and β1 was the interface between the α-thigh/calf1 domain and β-EGF2/3 (Fig. 4G), which were aligned in parallel similarly to the available structures of acutely bent integrins (13, 20). Subsequent multibody analysis, implemented in the processing software RELION3 (46, 47), of the lower legs and the headpiece revealed fluctuation at the knee with variation of the angle between the upper and lower legs ranging between 71° and 93° (Fig. 4H, fig. S7, and movie S1), suggesting that the integrin is not rigidly fixed in this resting conformation but bears a certain degree of flexibility. It should be noted, however, that our analysis did not indicate subpopulation of molecules in the acutely bent conformation observed for the β3 integrins. Unlike the acutely bent conformation, no interaction between the hybrid domain and the lower legs of the β subunit was evident in our structure. Nevertheless, the interaction of the hinge-like interface at the knees, between calf1 and EGF2/3, was robust enough to hold the half-bent conformation.
Mn2+ is necessary for integrin α5β1 ligand binding but does not induce a large conformational change
To closely follow the key steps leading from the resting to the fully open conformation of integrin α5β1, we investigated the conformations of integrin in the presence of buffer containing various concentrations of Mn2+. Cryo-EM analysis showed that integrin α5β1 used the half-bent resting conformation even at the extreme concentration of 100 mM Mn2+ (Fig. 6A). Together with previous reports showing that the integrin α5β1 headpiece does not change its conformation when it binds to RGD peptides (29, 30), it highlights that neither the addition of Mn2+ nor that of RGD-containing peptide is enough to induce a large conformational change of the β subunit. That is in stark contrast to αvβ3, for which Mn2+ was sufficient to induce full opening (23).
Fig. 6. FN-binding of NDs containing α5β1 is enhanced by Mn2+.
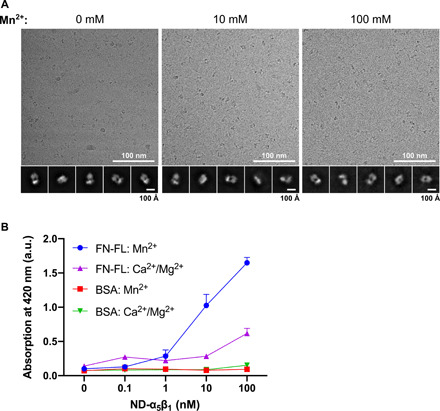
(A) Cryo-EM image of α5β1 with various concentrations of Mn2+. 2D class averages are shown, revealing no integrin opening. (B) Solid-phase binding assays to measure integrin binding to full-length FN (FN-FL) in the presence of activating (Mn2+) or resting (Ca2+/Mg2+) buffer conditions and varying integrin α5β1 concentration. Three individual experiments were conducted, and all conditions were measured and averaged (points represent means + SD). Control measurements of α5β1 on BSA are shown.
These findings led us to hypothesize that the large conformational change of integrin α5β1 is only stabilized as a result of FN binding rather than being the cause of FN binding. To test this notion, we performed solid-phase equilibrium binding assays (Fig. 6B). The binding of α5β1 to FN was measured in the presence or absence of Mn2+. When Mn2+ was added, we observed a marked increase in the binding of α5β1 to FN compared with that under control conditions without Mn2+, indicating that the affinity of integrin α5β1 for FN was increased. These results demonstrate that the affinity of integrin α5β1 for FN was increased with integrin still mostly using a half-bent conformation.
Furthermore, to systematically survey the global conformations of integrin α5β1, we performed reconstitution assays using size exclusion chromatography (SEC), followed by surveillance of the conformations using negative-stain EM (Fig. 7). While vitrified integrin samples for cryo-EM are instrumental in structural analyses of robustly folded populations, negative-stain EM allows the visualization of flexible entities that do not align well because of the low signal-to-noise ratio of cryo-EM images and would, therefore, be discarded during image analysis. First, the effect of Mn2+ on ligand binding was examined. Integrin α5β1 was mixed with FN7-10 and TS2/16 in the presence and absence of Mn2+ and subjected to analytical SEC (Fig. 7, A and B). The elution profiles revealed no observable shift in integrin α5β1 with or without Mn2+, indicating no large structural change, which was further confirmed by negative-stain observations {Fig. 7, A [green line/middle SDS–polyacrylamide gel electrophoresis (PAGE)], and C}. Those results were in agreement with our cryo-EM observations (Fig. 6A) showing the half-bent conformation. On the other hand, α5β1 coeluted with FN7-10 in the presence of Mn2+, indicating their interaction (Fig. 7A, right side, blue lines, peak marked with asterisk), whereas no binding took place without Mn2+ (Fig. 7A, left side). Subsequent negative-stain EM observation and conformational assessment revealed an increase in the proportion of molecules in the open conformation (41%), compared with that under the same condition without Mn2+ (0%) or without FN7-10 but with Mn2+ (7%) (Fig. 7C), although the FN density was only partially visible because of dissociation during the negative-stain preparation. Together, our results demonstrated that the large extension and opening of integrin are not coupled to the affinity of integrin for FN; instead, it is stabilized as a result of FN binding.
Fig. 7. Integrin α5β1 exists in an equilibrium of conformational states that can be altered by a combination of ligands FN7-10.
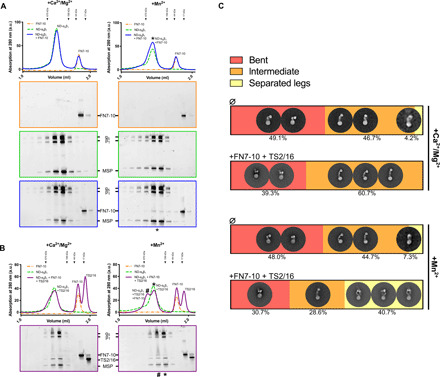
(A) FN7-10 binding to α5β1 was tested by SEC under resting (Ca2+/Mg2+) and activating (Mn2+) buffer conditions. In the presence of Mn2+, FN7-10 binding is detected by SDS-PAGE. The peak containing ND-α5β1 and FN7-10 is marked with asterisk. MSP, membrane scaffolding protein used to form NDs. The positions of molecular weight standards for gel filtration are marked with arrowheads on the chromatograms. (B) The effect of the antibody TS2/16 on α5β1 binding to FN7-10 was tested in the presence or absence of MnCl2. When Mn2+, FN7-10 and TS2/16 are mixed together with ND-α5β1, a peak shift can be observed (#). The positions of molecular weight standards for gel filtration are marked with arrowheads on the chromatograms. (C) Summary of the negative staining quantification of ND-α5β1 alone or mixed with FN7-10 and TS2/16 under resting (Ca2+/Mg2+) and activating (Mn2+) buffer conditions. Particles were sorted into three states: “bent,” “intermediate,” and “separated legs.” For each condition, ~20,000 particles were initially picked, and those displaying a clear signal (i.e., ~5000 to 8000) were selected for the final quantification. Examples of 2D classes are shown.
DISCUSSION
Integrin is the master adhesion receptor, controlling numerous signaling pathways and determining cell fates, such as migration and polarization (2). There is a strong incentive to understand the molecular mechanism of the activation of integrins because of their biological and medical relevance. In this study, we successfully purified human integrin α5β1 from a natural source (human placenta) and reconstituted the complex with FN in a membrane scaffold to facilitate a comprehensive biochemical and structural analysis. Our strategy has two important advantages: (i) Our results were obtained with native proteins in a biologically relevant context, and (ii) we were able to perform a direct and straightforward analysis without adjusting the conditions or impairing complex integrity.
Our study provides the first structure of the open, elongated conformation of integrin α5β1 in complex with FN. We found that three binding sites of the FN ligand at FN9 and FN10 were simultaneously engaged (Fig. 2), which explains the conundrum why the binding of RGD peptides was not sufficient for integrin opening in previous studies of α5β1 (29, 30). In particular, we identified that R1445 and Y1446 in FN10 approach close to the ADMIDAS region, which caused the translocation of ADMIDAS and downstream helix α1, leading to the stabilization of the open conformation of the β1 βI domain (Figs. 1 and 5A). That movement further allosterically triggers the swing-out motion to extend and open the α5β1 legs (15).
Mechanisms of integrin opening
The toolbox prepared for this study, including physiologically relevant full-length integrin together with our cryo-EM pipeline, allowed us to investigate the molecular response during integrin α5β1 ligand engagement. Our analysis revealed that the behavior of α5β1 is unexpectedly distinct from that of the well-studied β3-containing integrins, which show a tightly coupled conformational change upon addition of either ligand-mimetic peptides or specific ions. For the FN receptor α5β1, the extended, open conformation was observed only when both Mn2+ and FN were present (Fig. 7C). That means that FN binding does not require prior integrin opening, but instead, the open conformation of α5β1 becomes dominant as a result of the FN binding. This model of ligand-driven conformational changes of integrin is somewhat reminiscent of an alternative model described for β3 integrins (48, 49). We further showed that binding of Mn2+ is key to facilitate the interaction between FN and α5β1 without altering the conformational dynamics. We surmise that Mn2+ coordination at ADMIDAS might play a critical role in increasing the affinity for FN. Once the juxta-ADMIDAS FN binding site is primed, FN may engage with all three binding sites on the integrin surface. The binding fosters the translocation of ADMIDAS, which then, together with the synergy site, facilitates the RGD-driven allosteric conformational change leading to the downstream opening of the legs (Fig. 8). On the other hand, without Mn2+, integrin is not competent to stably capture FN, as the interaction is transient and short-lived without Mn2+ in vitro (Fig. 7, A and B).
Fig. 8. Model of integrin α5β1 opening.
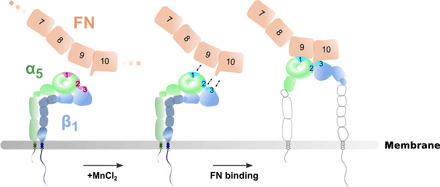
Integrin α5β1 exists in an equilibrium of conformational states, fluctuating between bending and extension. FN binding secures the extended and open conformation of integrin α5β1 through three binding sites (marked as 1, 2, and 3) at the integrin heads. Mn2+ primes binding sites, SyMBS (LIMBS), ADMIDAS, and MIDAS and brings integrin to a ready state in which it is ready to bind to FN. Full engagement of FN takes place, resulting in the integrin α5β1 opening. Note that lower leg opening was not visible in our integrin α5β1 structure and, therefore, the opened lower leg is based on previous biochemical observation (15).
Comparing protein ligand–attached structures of different integrin isotypes offers insights into the ligand-induced opening of integrins. Besides our α5β1-FN complex, two truncated integrins bound to their protein ligands have been reported, namely, αVβ6-proTGF-β1 and αVβ8-proTGF-β1 (50, 51). Each integrin structure revealed its own distinct mode of ligand recognition and opening of the headpiece. For αVβ6-proTGF-β1, an additional helix is formed immediately after the proTGF-β1 RGD motif. The helix then forms hydrophobic interactions with integrin, which appears to be important to move the specificity-determining loop 1 (SDL1), the loop upstream of the helix α1 (50). Furthermore, proTGF-β1 recognizes integrin β6 SDL2 as a secondary interaction surface (50). In contrast, the SDL2 (residues 176 to 195) of integrin β1 did not exhibit any interaction with FN (fig. S8). Integrin αVβ8 does not open its legs even after ligand binding, and notably, its ADMIDAS does not contain metal ion (51). While various mechanisms of ligand-integrin interactions exist, these observations implicate the relevance of ADMIDAS for integrin opening.
Implication of integrin-ligand coupling in cellular function
Our conformational analysis together with previous studies (23, 38) showed that α5β1 is only half-bent in its resting state and the closure of α5β1 is not tightly fixed but is rather a dynamic shift within the equilibrium of different states, showing 51% of extended-leg conformation in the presence of resting buffer without Mn2+ (Fig. 7C). Even within the half-bent structure, our multibody analysis hints at flexibility between the legs and the heads (Fig. 4H and fig. S7). That concept, termed molecular breathing, has been previously discussed with regard to integrins (13, 17). The half-bent conformation of α5β1 is still physically accessible to FN, but we speculate that a breathing motion toward extension might increase the probability of docking of the three interaction sites to FN. Last, when FN binds, integrin α5β1 is globally extended with its legs open, which is facilitated by the molecular constraint of the beads-on-string arrangement of FN9-10.
It is intriguing that the ligand recognition mechanisms of integrin isoforms appear to be fine-tuned for their specific ligands, presumably, depending on how robust an interaction has to be for their biological functions. The αVβ8 integrin is mostly expressed in immune cells, and it is plausible that the closed-extended conformation is advantageous for searching and binding to its ligands and subsequent cellular activation (51, 52). αIIbβ3 is enriched on the surface of platelets, and there is a clear distinction between its closed-inaccessible and open-extended conformations, because the aggregation of platelets has to be tightly regulated to avoid false activation and detrimental effects on blood circulation. In contrast, α5β1 is ubiquitously expressed throughout various tissues and is generally involved in cellular responses that are slower than those of the immune system. Therefore, the loose closure of α5β1 might reflect that functional aspect and, thus, provides a less acute response to extracellular cues. The differences among integrin isoforms may provide important regulatory features and may be related to the different functions of the isoforms within specific cell types.
Integrin-mediated intracellular focal adhesion
We demonstrated a stepwise mechanism of a large conformational change in α5β1 from a half-bent closed to an extended open complex (summarized in Fig. 8). However, in a cellular context, it is unknown how ECM components activate integrin through the recognition of integrin heads (outside-in) or how intracellular factors such as the integrin activators talin and kindlin activate integrin through the binding to the integrin tail (inside-out). It will be particularly important to elucidate which cellular factors trigger integrin α5β1 to assume the primed state observed in vitro by the presence of Mn2+ in which integrin can readily associate with FN. Moreover, once integrin α5β1 is engaged with FN, force is applied through the intracellular focal adhesion machinery bound to the integrin tail (3). It has been proposed that the FN synergy site is involved in the reinforcement of the interaction with integrin under applied force (32). Although a partial synergy site mutation (R1374A/R1379A) did not affect the viability of mice, it caused a bleeding disorder and accompanying cellular biological experiments indicated that the site is not essential for the formation of focal adhesion but is critical for focal adhesion maturation (53). Those findings support the hypothesis that the key function of the FN synergy site–integrin interaction is to provide a site for catch-bond formation when force is applied (31–33). In that context, we note that FN R1374, which has been reported as a key residue for the force-transduction function (32, 33), was not engaged in the interaction with integrin in our structure. That suggests the possibility that the interaction surface might alter to engage FN R1374 in the presence of applied force. In this study, we observed a comprehensive picture of the molecular interaction of FN and integrin α5β1 in the absence of force, which can be used as a basis for further analyses.
FN acts as a dimer in its full-length context and polymerizes into a fibrous form when integrated in the ECM network. The polymerization is induced by interaction with integrin α5β1 (1). In turn, we speculate that FN within the ECM might also act as a platform to induce integrin clustering, which contributes to stabilize the large intracellular multilayered focal adhesion machinery consisting of hundreds of proteins (4). Furthermore, the phenomenon of molecular breathing has been observed for the intracellular focal adhesion proteins talin (54) and vinculin (55) as well, which, together with integrin, may play an important role in the dynamic behaviors of focal adhesions. Molecular breathing might be advantageous in the cellular context of focal adhesion formation that requires associated molecules to promptly respond to diverse stimuli. It also allows a degree of flexibility in the recruitment and disassociation of various factors from focal adhesions, thus enabling fast assembly and disassembly during cell migration.
MATERIALS AND METHODS
Integrin α5β1 purification
Human endogenous integrin α5β1 was purified from placenta by adapting a previously published protocol (24). Deidentified human placentas were obtained from Ludwig Maximilian University (LMU) Klinikum Munich, Klinik und Poliklinik für Frauenheilkunde und Geburtshilfe, Campus Großhadern. Amnion and umbilical cords were first removed from the human placentas. The remaining placenta tissue was then cut into small pieces, mixed with tris-buffered saline (TBS) [50 mM tris and 150 mM NaCl (pH 7.5)], 1 mM MgCl2, 1 mM CaCl2, and protease inhibitor cocktail (Roche cOmplete) and homogenized. Debris was removed by centrifugation at 12,000g for 15 min at 4°C (Beckman centrifuge, J14 rotor). The supernatant was then sonicated (Vs70T, BANDELIN) and centrifuged again at 10,000g for 10 min at 4°C (Beckman Avanti J-26 centrifuge, JA-14 rotor). Next, the membrane fraction was pelleted by ultracentrifugation (150,000g, 4°C; Beckman, Ti 45 rotor), and the pellet was washed five times in 50 ml of 50 mM tris (pH 7.5), 1 M NaCl, 1 mM MgCl2, 1 mM CaCl2, and protease inhibitor cocktail. The membrane fraction was then solubilized by incubation with detergent [1% Triton X-100 in TBS (pH 7.5), 1 mM MgCl2, 1 mM CaCl2, and protease inhibitor cocktail] overnight at 4°C. After ultracentrifugation (150,000g, 4°C; Beckman, Ti70 rotor), the supernatant was incubated with 2 ml of KH72 monoclonal antibody–coupled Sepharose beads overnight at 4°C. The beads with bound protein were then washed five times with 10 ml of TBS (pH 7.5), 1 mM MgCl2, 1 mM CaCl2, and 0.1% Triton X-100 in a gravity flow column. Protein was eluted with 20 ml of 20 mM Pipes (pH 7.0), 150 mM NaCl, 1 mM CaCl2, 3 M MgCl2, and 0.1% Triton X-100. Three molars of MgCl2 is effective for elution (56).
After determination that protein was present by SDS-PAGE, fractions containing protein were pooled and dialyzed overnight at 4°C against TBS (pH 7.5), 1 mM MgCl2, 1 mM CaCl2, and 0.1% Triton X-100. The dialyzed samples were concentrated by ultrafiltration (Amicon) at a maximum speed of 2,500g in 5-min steps. The protein was then flash-frozen and stored at −80°C.
Clasped integrin α5β1 ectodomains preparation and purification
Recombinant integrin α5β1 ectodomains and its variants were prepared according to the previous reports (21, 23). Briefly, human embryonic kidney 293F cells (Life Technologies) were cotransfected with plasmids coding for the ectodomains of α5 (residues 1 to 950) and β1 (residues 1 to 691), followed by a Tobacco etch virus (TEV) protease recognition site, ACID or BASE-Cys peptide and a 6×His tag. After 2 days of expression, the culture supernatant was collected, and the protein was purified the same way as the native integrin α5β1 using KH72 antibody–coupled Sepharose beads, followed by gel filtration (Superdex 200, GE Healthcare) in TBS buffer. The peak fractions were concentrated, flash-frozen, and stored at −80°C.
Preparation of KH72 monoclonal antibody
Hybridoma cells expressing the KH72 monoclonal antibody were a gift from K. Miyake (University of Tokyo). Cells were cultured in RPMI 1640 medium (Gibco; with 25 mM Hepes and l-glutamine) supplemented with 10% fetal bovine serum, kanamycin (0.1 mg/ml), 2 mM l-glutamine, and 10 μM β-mercaptoethanol. The cells were grown at 37°C with 5% CO2 in T125 flasks and were split at confluency every 2 to 3 days by gently shaking the cells off and transferring ~30% of the volume to a new flask with fresh medium. Supernatant medium containing the expressed KH72 monoclonal antibody was collected, frozen, and stored at −20°C.
For purification of KH72 from the collected hybridoma medium, 1 liter of culture supernatant was thawed in a water bath at 37°C. Dead cells were removed by centrifugation at 4000g for 10 min at 4°C (Beckman, JLA-8.1000 rotor). The supernatant was filtered using a 0.22-μm-pore polyethersulfone (PES) membrane filter (Corning). The pH was then adjusted with NaOH to pH 8.2, and the sample was loaded onto 2 ml of Protein A Agarose (Pierce, Thermo Fisher Scientific) in a gravity column. The resin was then washed five times with 20 ml of 20 mM tris (pH 8.8) and 150 mM NaCl. Bound protein was eluted with low-pH immunoglobulin G elution buffer (Pierce, Thermo Fisher Scientific) and collected in 1-ml fractions, which were immediately neutralized by addition of tris (pH 8.8) to a final concentration of 150 mM. Fractions containing purified KH72, as determined by SDS-PAGE, were pooled and concentrated using an Amicon ultrafiltration device to a concentration of ~1 mg/ml. Coupling to N-hydroxysuccinimide (NHS)-Sepharose (GE Healthcare) was performed in a coupling buffer containing 200 mM NaHCO3 and 500 mM NaCl (pH 8.3). KH72 was dialyzed against a coupling buffer for 4 hours at room temperature using 7000 molecular weight cutoff (MWCO) Slide-A-Lyzer dialysis tubes with one exchange of the buffer. Then, ~1 mg of KH72 was added to 250 μl of activated NHS-Sepharose and incubated overnight at 4°C. The next day, 150 mM tris (pH 8.8) was added to stop the reaction, and the KH72-bound Sepharose was stored in a solution of 50 mM tris and 150 mM NaCl (pH 7.5) containing 0.05% NaN3.
Preparation of membrane scaffold protein
Membrane scaffold protein (MSP) was purified using a previously published protocol (25) with the following details. MSPE3D1 or MSPΔH5 (57) with an N-terminal 6×His tag was expressed in BL21 gold DE3 cells grown in Terrific broth medium containing kanamycin (50 μg/ml). The cells were grown until the optical density at 600 nm (OD600) reached 2 at 37°C. Then, expression was induced by incubation with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) overnight at 18°C. The cells were then harvested and lysed by sonication in phosphate-buffered saline (Sigma-Aldrich), 1% Triton X-100, and phenylmethylsulfonyl fluoride. After removal of debris by centrifugation at 20,000g for 30 min at 4°C (Beckman centrifuge, JA 25.50 rotor), the supernatant was filtered with a 0.22-μm-pore PES membrane filter (Corning), incubated on an Ni–nitrilotriacetic acid column (5 ml; Protino, Macherey-Nagel), and washed with 50 ml of each of the following buffers: first with 40 mM tris (pH 8), 300 mM NaCl, and 1% Triton X-100; then with 40 mM tris (pH 8), 300 mM NaCl, and 50 mM sodium cholate; and, last, with 40 mM tris (pH 8), 300 mM NaCl, and 5 mM imidazole. Protein was then eluted with 40 mM tris (pH 8), 300 mM NaCl, and 500 mM imidazole. Fractions containing protein, as determined by SDS-PAGE, were pooled and concentrated using an Amicon ultrafiltration device (10,000 MWCO), and the buffer was exchanged through SEC to 20 mM tris (pH 7.4), 100 mM NaCl, and 0.5 mM EDTA using a Superdex 200 column on an ÄKTA pure (GE Healthcare) machine. The protein peak was then pooled, concentrated using an Amicon ultrafiltration device (10,000 MWCO) to a concentration of ~4 mg/ml, flash-frozen, and stored at −80°C.
Purification of FN7-10
FN7-10 constructs were purified as described previously (12) with the following adaptations. FN7-10 was expressed in Escherichia coli BL21 gold (DE3) in ZY autoinduction medium containing ampicillin (100 μg/ml). Cells were grown until the OD600 reached ~1.5 to 2 at 37°C and then overnight at 18°C. Cells were pelleted and resuspended in 50 mM tris (pH 7.5), 200 mM NaCl, 1 mM EDTA, 1% Triton X-100, and protease inhibitor cocktail (Roche cOmplete) and stirred for 30 min at room temperature. Cells were then lysed by sonication (VS 70 T, BANDELIN), and the lysate was cleared by centrifugation at 76,000g for 30 min at 4°C (Beckman centrifuge, JA 25.50). The supernatant was subjected to precipitation by 40% (NH4)2SO4. After addition of solid (NH4)2SO4, the solution was stirred for 30 min at 4°C and pelleted by centrifugation at 76,000g for 30 min at 4°C (Beckman centrifuge, JA 25.50). The pellet was resuspended in 20 mM tris (pH 7.5) and 50 mM NaCl, filtered using a 0.22-μm-pore PES membrane filter (Corning), and loaded onto a HiTrap Q column (5-ml column volume; Cytiva). Bound protein was washed with 10 column volumes of 20 mM tris (pH 7.5) and 50 mM NaCl and then eluted in a gradient from 50 to 500 mM NaCl. The peak containing FN7-10 (as determined by SDS-PAGE) was pooled, concentrated using an Amicon ultrafiltration device (10,000 MWCO), and subjected to SEC using a Superdex 200 column on an ÄKTA pure (GE Healthcare) machine in 50 mM tris (pH 7.5) and 150 mM NaCl. The protein peak was pooled, concentrated using an Amicon ultrafiltration device (10,000 MWCO) to a concentration of 10 to 20 mg/ml, flash-frozen, and stored at −80°C.
TS2/16 Fv-clasp purification
TS2/16 Fv-clasp protein was purified as described previously (26). VH-SARAH and VL-SARAH fragments were individually expressed in BL21 gold DE3 cells grown in LB medium. The cells were grown until the OD600 reached ~0.6 at 37°C. Expression was then induced by incubation with 0.4 mM IPTG for 4 hours at 37°C. The TS2/16 Fv-clasp protein was expressed in inclusion bodies. After the cells were lysed by sonication, the inclusion bodies were isolated by centrifugation and solubilized in 50 mM tris, 150 mM NaCl, 6 M guanidine-HCl, and 375 μM β-mercaptoethanol (pH 8). VH-SARAH and VL-SARAH were then mixed at a 1:1 molar ratio in a total volume of 12 ml. To induce protein refolding, 50 mM tris, 4 M urea, 0.4 M l-arginine, 375 μM oxidized glutathione (GSSG), and 150 mM NaCl (pH 8) were added, bringing the total volume up to 300 ml (25× dilution), and the mixture was incubated for 4 hours at 4°C. Then, 300 ml of 50 mM tris, 0.4 M l-arginine, 375 μM GSSG, and 150 mM NaCl (pH 8) was added (2× dilution) and incubated for 16 hours at 4°C. After precipitation by 80% (NH4)2SO4, the protein was dissolved and dialyzed in TBS. The protein was then subjected to SEC on a HiLoad 16/600 Superdex 200 prep grade column (GE Healthcare) in 20 mM tris and 150 mM NaCl (pH 8). The peak containing TS2/16 Fv-clasp, as determined by SDS-PAGE, was then pooled, concentrated, diluted in 20 mM tris and 50 mM NaCl (pH 7.5), and further subjected to MonoQ chromatography. Elution was carried out in a linear gradient from 100 to 500 mM NaCl. The major eluting peak contained fully folded TS2/16 Fv-clasp and was pooled, concentrated using an Amicon ultrafiltration device (10,000 MWCO), flash-frozen, and stored at −80°C.
Assembly of integrin α5β1 into NDs
NDs containing integrin α5β1 were assembled according to a previously published protocol (25) with the following adaptations. Isoform MSPE3D1 was used for all experiments, except that we used MSPΔH5 to test the effects of different Mn2+ concentrations on the integrin conformation in cryo-EM (Fig. 6A). Lipid stock (from Folch fraction I, bovine brain, Sigma-Aldrich) was stored in chloroform:methanol:H2O (20:9:1) at a concentration of 25 mg/ml at −20°C. Before use, the required amount of lipid was dried in a stream of nitrogen and further vacuum-dried for 2 hours. The resulting lipid film was solubilized in 20 mM Hepes (pH 7), 150 mM NaCl, and 1% n-dodecyl–D-maltopyranoside on a thermoshaker for 1 hour at 37°C, resulting in a final concentration of 25 mg/ml. MSPE3D1 or MSPΔH5, α5β1, and lipids were mixed in a ratio of ~29:1:3460 for MSPE3D1 or ~ 45:1:110 for MSPΔH5 and then incubated for 1 hour at room temperature. SM-2 Bio-Beads (Bio-Rad) were added to the mix to remove detergent overnight at 4°C. The mixture was then subjected to SEC (Superose 6i GL10/300 on an ÄKTA pure, GE Healthcare) in TBS (pH 7.5), 1 mM MgCl2, and 1 mM CaCl2. The peak containing ND with inserted α5β1 (ND-α5β1; as determined by SDS-PAGE and negative-stain EM) was then pooled, concentrated (Amicon; 50,000 MWCO) at a maximum speed of 4,000g until an absorption at 280 nm of ~1.2 to 1.6 mg/ml (~5 μM) was reached, flash-frozen, and stored at −80°C.
Solid-phase integrin-FN binding assay
Solid-phase binding assays were performed on the basis of a previously published protocol (30) with some adaptations. Ninety-six–well microtiter plates were coated with 50 μl per well of either full-length FN (0.01 mg/ml; Sigma-Aldrich) in TBS (pH 7.5) or FN7-10 in TBS and its variant or 5% (w/v) bovine serum albumin (BSA) (Sigma-Aldrich) in TBS (pH 7.5) and incubated overnight at 4°C. The wells were then blocked with 5% BSA (200 μl per well) in TBS (pH 7.5) for 1 hour at room temperature. Then, ND-α5β1 or the recombinant α5β1 ectodomains were added in various concentrations in either TBS (pH 7.5), 1 mM MgCl2, and 1 mM CaCl2 or TBS (pH 7.5) and 1 mM MnCl2. Unclasping of ectodomains was achieved by overnight incubation with TEV protease at room temperature, at a ratio of 1:30 (w/w) and confirmed via SDS-PAGE. For Fig. 6B, the concentration of ND-α5β1 was varied in the range between 0.1 and 100 nM; for all other assays, a fixed concentration of 10 nM for both ND-α5β1 and ectodomains was used. The mixture (total volume, 50 μl per well) was then incubated for 3 hours at room temperature. The wells were then washed three times with 200 μl per well of the respective buffers (containing either MgCl2/CaCl2 or MnCl2), and antibody (50 μl per well) (TS2/16-biotin, 0.005 mg/ml) in the respective buffers containing additional 0.1% BSA was added. After incubation for 2 hours at room temperature, the wells were washed three times with 200 μl per well of the respective buffers, and streptavidin–horseradish peroxidase (SA-HRP; 1.25 μg/ml; Life Technologies) in the respective buffers containing additional 0.1% BSA was added at 50 μl per well. After incubation for 1 hour at room temperature, the wells were washed four times with 200 μl per well of the respective buffers, and 2,2’-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS)/H2O2 solution (100 μl per well; Sigma-Aldrich; ready to use) was added. Absorption at 420 nm was measured after the addition of ABTS/H2O2 using a plate reader (Tecan, Infinite M1000 PRO). Graphs were generated and assessed using GraphPad Prism software.
Analytical SEC
To assess the binding between ND-α5β1 and FN7-10 in the presence or absence of TS2/16 Fv-clasp, the reaction components were mixed, incubated, and injected into a Superose 6i column (S6i, 3.2/300 on ÄKTAmicro system, GE Healthcare), either in TBS (pH 7.5), 1 mM MgCl2, and 1 mM CaCl2 or in TBS (pH 7.5) and 1 mM MnCl2. When the effect of manganese was tested, ND-α5β1 was first incubated with MnCl2 at a final concentration of 10 mM for 30 min at 4°C.
For the ND-α5β1/FN7-10 runs, 0.08 nmol of each component were mixed, incubated for 30 min at 4°C, and loaded onto the S6i column. For the TS2/16 Fv-clasp runs, ND-α5β1 and FN7-10 were first mixed and incubated for 30 min at 4°C. Then, TS2/16 Fv-clasp was added to a final ND-α5β1:FN7-10:TS2/16 Fv-clasp molar ratio of 1:2:2, and the mixture was incubated for 10 min at 4°C. If a component was left out for a control run, then it was replaced with a corresponding amount of buffer.
Electron microscopy
Negative-stain EM
For negative-stain EM, 5 μl of sample (protein concentration ranging from ~4 to 12 μg/ml) was incubated on glow-discharged carbon-coated copper grids (homemade carbon layer) for 1 min at room temperature and then washed twice in 100-μl drops of H2O. Excess H2O was removed by touching the rim of the grids with blotting paper. The grids were then stained with 5 μl of 2% uranyl acetate solution for 1 min at room temperature, and excess uranyl acetate solution was removed by touching the rim of the grids with blotting paper. Datasets were collected using a Titan Halo (FEI) equipped with a Falcon 2 detector in nanoprobe mode at an operating voltage of 300 kV. Micrographs were recorded with EPU software (Thermo Fisher Scientific) at ×58,000 magnification (1.847 Å per pixel) with varying defoci from −1.5 to −2.5 μm in 0.5-μm steps at an exposure of 4 s with 10 frames per micrograph and a total dose of 105 e/Å2. Recorded, gain-normalized frames were aligned, motion-corrected, and dose-weighted using MotionCor2 software (58). Defocus values were assessed using either Gctf (59) or CTFFIND4 software (60). Using RELION3 (46), ~300 particles were manually picked and boxed out with a box size of 240 pixels. 2D alignment and classification were then performed. The resulting five initial 2D class averages were used as templates for automated picking with Gautomatch software (www2.mrc-lmb.cam.ac.uk/research/locally-developed-software/zhang-software/). For all quantifications, 20,000 particles (with a box size of 340 pixels) were aligned and 2D classified in RELION3. After the first round of 2D classification, ~10,000 particles with clear signals were selected for the second round, which was used for quantification of the particles into the bent, intermediate, and extended conformational states (related to Fig. 7C). For the representative 2D classes displayed in fig. S1C, the whole dataset of resting ND-α5β1 (81,249 particles in total) were boxed out (with a box size of 240 pixels) and submitted to alignment and 2D classification in RELION3.
Cryo-EM sample preparation
FN-bound and resting α5β1 were used at concentrations of 0.15 and 0.3 mg/ml, respectively. For the samples testing the effect of different Mn2+ concentrations (Fig. 6A), a concentration of 0.2 mg/ml was used for ND-α5β1 (MSPΔH5). Three microliters of ND-α5β1 supplemented with 0, 10, or 100 mM MnCl2 (final concentrations) was incubated for 1 hour at 4°C. Then, the sample was placed on a glow-discharged (GloQube; 30 s, 20 mA) R 1.2/1.3 Cu 200-mesh grid (QUANTIFOIL), blotted for 3.5 s, and plunge-frozen immediately in liquid ethane-propane using a Vitrobot Mark IV (FEI) at 4°C and 100% humidity.
Cryo-EM data collection for screening and initial reconstruction
The assessment of the large conformational change by cryo-EM, as well as the initial screening and reconstruction necessary for the final structural analysis, was performed as follows. A screening dataset for FN-bound α5β1 was collected using EPU software (Thermo Fisher Scientific) and a Talos Arctica (Thermo Fisher Scientific) equipped with a Falcon 3 direct detector in nanoprobe mode with the following parameters: operating voltage of 200 kV, ×92,000 magnification (1.61 Å per pixel), varying defoci from −2 to −3 μm, 0.5-μm steps, exposure of 3 s, 40 frames per micrograph, and total dose of 67 e/Å2. The recorded, gain-normalized frames were aligned, motion-corrected, and dose-weighted using MotionCor2. Defocus values were assessed by Gctf. Using RELION3, 754 particles were manually picked and boxed out (with a box size of 300 pixels). Then 2D alignment and classification were performed. Of four generated 2D averaged classes, the three classes with distinct patterns were used as a template for automated picking with Gautomatch. Of 105,351 particles that were initially picked, four of 2D classes containing 10,949 particles (with a box size of 340 pixels) were selected. Those four classes were also used as a template for automated picking with Gautomatch of the dataset used for the high-resolution structural analysis of FN-bound α5β1 described below. The 10,949 particles of the four classes were further used to reconstruct an initial model using the initial model option in RELION3. After a 3D classification run using the initial model as a reference, of eight generated classes, the reconstruction with the most prominent features (16,765 particles) was selected as an initial reference for 3D reconstruction of the dataset of FN-bound integrin α5β1 for high-resolution analysis, as described below.
For resting integrin α5β1, 2600 micrographs were recorded using SerialEM software (61) and a K2 Summit direct detector with a quantum energy filter (20- and 40-eV slit width) (Gatan) with the following parameters: counting mode, ×130,000 magnification (1.06 Å per pixel), varying defoci from −1.5 to −3.5 μm, 0.2-μm steps, and total dose of 51 e/Å2 (6-s exposure, 30 frames, and frame dose of 1.7 e/Å2). The micrograph frames were gain-normalized, motion-corrected, aligned, and dose-weighted using MotionCor2. Defocus and image resolution values were assessed using Gctf. Then, 502 particles were manually picked and boxed out with a box size of 220 pixels, and then 2D alignment and classification were performed using RELION3. Six selected 2D class averages were used as a template for automated picking with Gautomatch. A total of 103,534 automatically picked particles were boxed out with a box size of 300 pixels, binned to 150 pixels, and used to generate an initial 3D map using the ab initio reconstruction option in cryoSPARC (62). After the model was imported from cryoSPARC into RELION3 and further sorting by 3D classification, 23,332 remaining particles were refined. The refined 3D map was used as a reference for the main dataset of resting integrin α5β1 used for high-resolution analysis as described below.
For testing the effect of various Mn2+ concentrations on integrin α5β1 conformation, three datasets were obtained using a Talos Arctica and a Glacios (Thermo Fisher Scientific). The datasets for α5β1 at final concentrations of 10 and 100 mM Mn2+ were collected using EPU software and a Talos Arctica with a Falcon 3 direct detector with the following parameters: nanoprobe mode, operating voltage of 200 kV, ×92,000 magnification (1.61 Å per pixel), varying defoci from −2 to −3 μm, 0.5-μm steps, exposure of 3 s, 40 frames per micrograph, and total dose of 67 e/Å2. The recorded, gain-normalized frames were aligned, motion corrected, and dose-weighted using MotionCor2. Defocus values were assessed using CTFFIND4. A total of 965 and 88 micrographs were acquired for 10 and 100 mM Mn2+, respectively. Automated picking with Gautomatch was performed, and 202,156 and 18,822 particles were picked for 10 and 100 mM Mn2+, respectively. Those particles were boxed out with a box size of 200 pixels; 2D alignment and classification were performed using RELION3. Representative images and classes are displayed in Fig. 6A. The dataset for the control sample with 0 mM MnCl2 was collected using SerialEM software and a Glacios electron microscope equipped with a K2 Summit direct detector with the following parameters: nanoprobe mode, operating voltage of 200 kV, ×22,000 magnification (1.885 Å per pixel), varying defoci from −1.5 to −3 μm, 0.5-μm steps, exposure of 15 s, 50 frames per micrograph, and total dose of 58.5 e/Å2. The recorded frames were gain-normalized, aligned, motion-corrected, and dose-weighted using MotionCor2 software. Defocus values were assessed using CTFFIND4 software. A total of 1935 micrographs were acquired with automated picking by Gautomatch. The particles were boxed out with a box size of 174 pixels. Then, 2D alignment and classification were performed in RELION3. Representative images and classes are displayed in Fig. 6A. After particle alignment and 2D classification, only densities for the ectodomains of α5β1 were visible under both conditions. Despite extensive efforts to optimize the cryo-EM grid preparation, we were not able to visualize the ND density. However, SEC, corresponding negative-staining, peptide sequencing, and mass spectrometry showed full coverage of the sequence. Therefore, we concluded that the lack of membrane density was due to the flexible movement of the leg portions and also the specific characteristics of the cryo-EM sample preparation, such as the interaction of the membrane scaffolding with the air-water interface. That interaction in particular is known to cause local instability on embedded proteins during cryo-EM sample preparation, which might cause the integrin to compromise its rigidity at the lower leg portion. Therefore, the remaining transmembrane helices and cytoplasmic domains would be too flexible to appear as shown in the 2D and 3D classes, a notion supported by the progressive decrease in local resolution at the lower leg and the further downstream portion of the 3D reconstruction.
Cryo-EM data collection for high-resolution structural analysis
Datasets for high-resolution image analyses of FN-bound (Fig. 1) and resting (Fig. 4) integrin α5β1 were collected using SerialEM software and a Titan Krios (Thermo Fisher Scientific) equipped with a K3 Summit direct detector and a quantum energy filter (20-eV slit width) (Gatan) in nanoprobe mode at an operating voltage of 300 kV. For the resting integrin α5β1 dataset (Fig. 4), 2818 micrographs were recorded at ×105,000 magnification (0.85 Å per pixel) in counting mode with varying defoci from −0.5 to −3.2 μm in 0.3-μm steps at a total dose of either 65.8 e/Å2 (2-s exposure, 20 frames, and frame dose of 3.13 e/Å2) or 82.3 e/Å2 (2.5-s exposure, 50 frames, and frame dose of 1.65 e/Å2). The frames of the combined micrographs were gain-normalized, motion-corrected, aligned, and dose-weighted using MotionCor2. Defocus and image resolution values were assessed using either Gctf or CTFFIND4. A total of 1424 particles were manually picked and boxed out with a box size of 300 pixels. Then, 2D alignment and classification were performed using RELION3. Six of the 2D class averages with distinct features were used as a template for automated picking with Gautomatch. A total of 496,356 automatically picked particles were boxed out with a box size of 300 pixels, downscaled to 150 pixels, aligned, and 2D-classified in RELION3. After initial sorting, 255,514 particles remained and were further sorted into eight classes by 3D classification. The reference for the 3D classification was the reconstruction obtained from the screening dataset, which was then low pass–filtered to 30 Å. The 98,750 particles with the most prominent features were selected from the reconstruction for further processing. Those particles were rescaled to the original box size of 300 and further refined in the RELION3 processing pipeline, resulting in the final density map of the whole molecule with 6.2-Å total resolution, as determined by the gold-standard Fourier shell correlation criterion (FSC = 0.143) in the RELION3 postprocessing option. Using Chimera (63) and the implemented Segger plugin (64, 65), the map of the whole molecule was divided into the headpiece and the lower legs (see fig. S6) and then low pass–filtered to 15 Å. The envelopes were then used to generate masks, which were subsequently used for the multibody refinement option in RELION3 (47). The multibody refinement displayed movement between the two parts. Next, partial signal subtraction of either the headpiece or the lower leg signal was performed within RELION3, taking their different orientations as determined by multibody refinement into account. With focused 3D refinement after subtraction, the headpiece resolution was improved to 4.6 Å, as determined by the FSC = 0.143 criterion using the RELION3 postprocessing option. The resolution of the lower leg part was more flexible and determined to be 8 Å according to the local resolution analysis.
For the FN7-10–bound and TS2/16 Fv-clasp–bound α5β1 dataset (Fig. 1), 9431 micrographs were recorded at ×64,000 magnification (1.38 Å per pixel) in counting mode with varying defoci from −0.8 to −2.6 μm in 0.3-μm steps at a total dose of 59.7 e/Å2 (7.39-s exposure, 37 frames, and frame dose of 1.61 e/Å2). Initial image processing steps were automatically performed on a graphics processing unit workstation running the modular Focus software (66). Those steps included gain normalization, motion correction, and dose weighting by MotionCor2; contrast transfer function estimation by Gctf; and particle picking by Gautomatch, using a template obtained from the screening dataset of the same sample (see above). A total of 4,218,951 particles were boxed out with a box size of 360 pixels, initially downscaled to 90 pixels, aligned, and 2D classified in RELION3. After initial sorting, 1,492,856 particles remained and were further sorted by 3D classification using a 30-Å low pass–filtered map obtained from a previous dataset created on the same sample grid (see the paragraph below for details on how 3D reference was initially obtained). From the six initially generated 3D classes, the two with the clearest signal were selected, which resulted in 680,610 particles. Those particles were rescaled to their original size (360 pixels) and further refined in the RELION3 processing pipeline, resulting in the final density map of the whole molecule with 3.5-Å total resolution, as determined by the FSC = 0.143 criterion in the RELION3 postprocessing option. Using Chimera (63) and the implemented Segger plugin (64, 65), the map was divided into two parts: the stable core, comprising the β-propeller, βI and hybrid domains, and the FN 9-10 and TS2/16 Fv-clasp; and the flexible, protruding densities, combining FN7-8 and the remaining integrin leg densities (thigh domain and PSI) (see Fig. 1 and fig. S3). Both maps were imported into RELION3, low pass–filtered to 15 Å, and used to generate masks. Within RELION3, the signal of the flexible, protruding parts was partially subtracted, and the remaining density was subjected to focused 3D refinement, using the whole molecule as a reference and applying the mask of the “core.” That procedure improved the resolution of the core part to 3.1 Å, as determined by the FSC = 0.143 criterion in the RELION3 postprocessing option.
Model building
To prepare an initial model of the FN-bound and TS2/16-bound integrin α5β1, the available crystal structures 4wk0 [α5β1 headpiece (29)], 3vi3 [α5β1 headpiece with upper legs (30)], 5xcx [TS2/16 Fv-clasp (26)], and 1fnf [FN7-10 (12)] were fit as rigid bodies into the EM density maps using Chimera (63)). The model was further refined by manual building and real-space refinement in Coot (67), using maps of the core density of integrin (integrin α5β1, FN9-10, and TS2/16 Fv-clasp) at a resolution of 3.1 Å and that of the whole molecule at a resolution of 3.5 Å as references. B factors were then added, and the structure was validated using PHENIX (68). The final structure fitted well to the cryo-EM density, with a correlation coefficient around the model (CCmask) of 0.75 (table S1). Note that the B factors of the periphery of the model (FN7 and integrin legs) are high because of the flexibilities of the corresponding portions.
For the resting integrin α5β1, the crystal structures 4wjk (29) and 3vi3 (30) showing the headpiece and headpiece with upper legs, respectively, were used for initial fitting in Chimera. The missing parts of the lower legs (calf1 and calf2, PSI, EGF1 to EGF4, and β-tail domains) were prepared by homology modeling based on the crystal structure 3fcs [ectodomain of integrin αIIbβ3 (13)] using MODELLER (69). The initial model was manually adjusted, and domains were linked using Coot. B factors were added, and the structure was validated using PHENIX. The final structure fit to the cryo-EM density, with a correlation coefficient around the model (CCmask) of 0.66 (table S1). Note that the B factors of the periphery of the model (integrin lower legs) are high because of the flexibilities of the corresponding portions.
Acknowledgments
We thank C. Hübener (LMU Klinikum Munich, Klinik und Poliklinik für Frauenheilkunde und Geburtshilfe, Campus Großhadern) for providing deidentified human placentas. We are grateful to R. Izawa for work in the early stages of establishing the purification protocol of integrin α5β1 from human placenta. We thank S. Schkölziger for technical support. We acknowledge E. Conti and the Max Planck Institute of Biochemistry cryo-EM (D. Bollschweiler and T. Schäfer) and biochemistry core facilities for support and infrastructure. We also thank members of the Mizuno and Takagi laboratories for discussions. Funding: N.M. acknowledges the Max Planck Society, Boehringer Ingelheim Foundation Plus 3 Program, and the European Research Council (ERC-CoG, 724209) and German Research Foundation, DFG SFB863, for funding. N.M. is a recipient of the EMBO Young Investigator award. N.M. is further supported by the Intramural Research Program of the National Heart Lung and Blood Institute and by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the NIH. This work was performed under the International Collaborative Research Program of the Institute for Protein Research, Osaka University (ICR-18-05). Author contributions: S.S., J.T., C.B., and N.M. conceived, supervised, and designed the project. K.M. initiated the integrin purification from human placenta, and S.S. and R.V.N. performed cloning and protein purification. S.S. with the help of D.D. performed cryo-EM data collection and image processing. S.S. performed protein functional assays. Molecular modeling was performed by C.B. S.S., C.B., and N.M. wrote the manuscript with the help of all the authors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. 3D structure has been deposited to the Protein and Electron Microscopy Data Bank (PDB ID: 7NWL, EMD-12634 for integrin a5b1 in complex with FN7-10 and TS2/16; and PDB ID: 7NXD, EMDB-12637 for integrin a5b1 in half-bent conformation).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/19/eabe9716/DC1
REFERENCES AND NOTES
- 1.Sechler J. L., Corbett S. A., Schwarzbauer J. E., Modulatory roles for integrin activation and the synergy site of fibronectin during matrix assembly. Mol. Biol. Cell 8, 2563–2573 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hynes R. O., Integrins: Bidirectional, allosteric signaling machines. Cell 110, 673–687 (2002). [DOI] [PubMed] [Google Scholar]
- 3.Kechagia J. Z., Ivaska J., Roca-Cusachs P., Integrins as biomechanical sensors of the microenvironment. Nat. Rev. Mol. Cell Biol. 20, 457–473 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Zaidel-Bar R., Itzkovitz S., Ma’ayan A., Iyengar R., Geiger B., Functional atlas of the integrin adhesome. Nat. Cell Biol. 9, 858–867 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Campbell I. D., Humphries M. J., Integrin structure, activation, and interactions. Cold Spring Harb. Perspect. Biol. 3, a004994 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Winograd-Katz S. E., Fässler R., Geiger B., Legate K. R., The integrin adhesome: From genes and proteins to human disease. Nat. Rev. Mol. Cell Biol. 15, 273–288 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Schaffner F., Ray A. M., Dontenwill M., Integrin α5β1, the fibronectin receptor, as a pertinent therapeutic target in solid tumors. Cancer 5, 27–47 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang R., Rofstad E. K., Integrins as therapeutic targets in the organ-specific metastasis of human malignant melanoma. J. Exp. Clin. Cancer Res. 37, 92 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Engel J., Odermatt E., Engel A., Madri J. A., Furthmayr H., Rohde H., Timpl R., Shapes, domain organizations and flexibility of laminin and fibronectin, two multifunctional proteins of the extracellular matrix. J. Mol. Biol. 150, 97–120 (1981). [DOI] [PubMed] [Google Scholar]
- 10.Erickson H. P., Carrell N., McDonagh J., Fibronectin molecule visualized in electron microscopy: A long, thin, flexible strand. J. Cell Biol. 91, 673–678 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wierzbicka-Patynowski I., Schwarzbauer J. E., The ins and outs of fibronectin matrix assembly. J. Cell Sci. 116, 3269–3276 (2003). [DOI] [PubMed] [Google Scholar]
- 12.Leahy D. J., Aukhil I., Erickson H. P., 2.0 A crystal structure of a four-domain segment of human fibronectin encompassing the RGD loop and synergy region. Cell 84, 155–164 (1996). [DOI] [PubMed] [Google Scholar]
- 13.Zhu J., Luo B. H., Xiao T., Zhang C., Nishida N., Springer T. A., Structure of a complete integrin ectodomain in a physiologic resting state and activation and deactivation by applied forces. Mol. Cell 32, 849–861 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu J., Zhu J., Springer T. A., Complete integrin headpiece opening in eight steps. J. Cell Biol. 201, 1053–1068 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luo B.-H., Carman C. V., Springer T. A., Structural basis of integrin regulation and signaling. Annu. Rev. Immunol. 25, 619–647 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xiao T., Takagi J., Coller B. S., Wang J.-H., Springer T. A., Structural basis for allostery in integrins and binding to fibrinogen-mimetic therapeutics. Nature 432, 59–67 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takagi J., Petre B. M., Walz T., Springer T. A., Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell 110, 599–511 (2002). [DOI] [PubMed] [Google Scholar]
- 18.Eng E. T., Smagghe B. J., Walz T., Springer T. A., Intact αIIbβ3 integrin is extended after activation as measured by solution x-ray scattering and electron microscopy. J. Biol. Chem. 286, 35218–35226 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xie C., Zhu J., Chen X., Mi L., Nishida N., Springer T. A., Structure of an integrin with an αI domain, complement receptor type 4. EMBO J. 29, 666–679 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xiong J. P., Stehle T., Diefenbach B., Zhang R., Dunker R., Scott D. L., Joachimiak A., Goodman S. L., Arnaout M. A., Crystal structure of the extracellular segment of integrin αVβ3. Science 294, 339–345 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takagi J., Strokovich K., Springer T. A., Walz T., Structure of integrin α5β1 in complex with fibronectin. EMBO J. 22, 4607–4615 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xiong J.-P., Stehle T., Zhang R., Joachimiak A., Frech M., Goodman S. L., Arnaout M. A., Crystal structure of the extracellular segment of integrin αVβ3 in complex with an Arg-Gly-Asp ligand. Science 296, 151–155 (2002). [DOI] [PubMed] [Google Scholar]
- 23.Miyazaki N., Iwasaki K., Takagi J., A systematic survey of conformational states in β1 and β4 integrins using negative-stain electron microscopy. J. Cell Sci. 131, jcs21675 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Pytela R., Pierschbacher M. D., Argraves S., Suzuki S., Ruoslahti E., [27] Arginine-glycine-aspartic acid adhesion receptors. Methods Enzymol. 144, 475–489 (1987). [DOI] [PubMed] [Google Scholar]
- 25.Denisov I. G., Grinkova Y. V., Lazarides A. A., Sligar S. G., Directed self-assembly of monodisperse phospholipid bilayer nanodiscs with controlled size. J. Am. Chem. Soc. 126, 3477–3487 (2004). [DOI] [PubMed] [Google Scholar]
- 26.Arimori T., Kitago Y., Umitsu M., Fujii Y., Asaki R., Tamura-Kawakami K., Takagi J., Fv-clasp: An artificially designed small antibody fragment with improved production compatibility, stability, and crystallizability. Structure 25, 1611–1622.e4 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Springer T. A., Zhu J., Xiao T., Structural basis for distinctive recognition of fibrinogen γC peptide by the platelet integrin αIIbβ3. J. Cell Biol. 182, 791–800 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Main A. L., Harvey T. S., Baron M., Boyd J., Campbell I. D., The three-dimensional structure of the tenth type III module of fibronectin: An insight into RGD-mediated interactions. Cell 71, 671–678 (1992). [DOI] [PubMed] [Google Scholar]
- 29.Xia W., Springer T. A., Metal ion and ligand binding of integrin α5β1. Proc. Natl. Acad. Sci. U.S.A. 111, 17863–17868 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nagae M., Re S., Mihara E., Nogi T., Sugita Y., Takagi J., Crystal structure of α5β1 integrin ectodomain: Atomic details of the fibronectin receptor. J. Cell Biol. 197, 131–140 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kong F., García A. J., Mould A. P., Humphries M. J., Zhu C., Demonstration of catch bonds between an integrin and its ligand. J. Cell Biol. 185, 1275–1284 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Friedland J. C., Lee M. H., Boettiger D., Mechanically activated integrin switch controls α5β1 function. Science 323, 642–644 (2009). [DOI] [PubMed] [Google Scholar]
- 33.Redick S. D., Settles D. L., Briscoe G., Erickson H. P., Defining fibronectin’s cell adhesion synergy site by site-directed mutagenesis. J. Cell Biol. 149, 521–527 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Altroff H., Schlinkert R., van der Walle C. F., Bernini A., Campbell I. D., Werner J. M., Mardon H. J., Interdomain tilt angle determines integrin-dependent function of the ninth and tenth FIII domains of human fibronectin. J. Biol. Chem. 279, 55995–56003 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Copié V., Tomita Y., Akiyama S. K., Aota S., Yamada K. M., Venable R. M., Pastor R. W., Krueger S., Torchia D. A., Solution structure and dynamics of linked cell attachment modules of mouse fibronectin containing the RGD and synergy regions: Comparison with the human fibronectin crystal structure. J. Mol. Biol. 277, 663–682 (1998). [DOI] [PubMed] [Google Scholar]
- 36.Byron A., Humphries J. D., Askari J. A., Craig S. E., Mould A. P., Humphries M. J., Anti-integrin monoclonal antibodies. J. Cell Sci. 122, 4009–4011 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takada Y., Puzon W., Identification of a regulatory region of integrin beta 1 subunit using activating and inhibiting antibodies. J. Biol. Chem. 268, 17597–17601 (1993). [PubMed] [Google Scholar]
- 38.Su Y., Xia W., Li J., Walz T., Humphries M. J., Vestweber D., Cabañas C., Lu C., Springer T. A., Relating conformation to function in integrin α5β1. Proc. Natl. Acad. Sci. U.S.A. 113, E3872–E3881 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Isaji T., Sato Y., Zhao Y., Miyoshi E., Wada Y., Taniguchi N., Gu J., N-glycosylation of the β-propeller domain of the integrin α5 subunit is essential for α5β1 heterodimerization, expression on the cell surface, and its biological function. J. Biol. Chem. 281, 33258–33267 (2006). [DOI] [PubMed] [Google Scholar]
- 40.Isaji T., Sato Y., Fukuda T., Gu J., N-glycosylation of the I-like domain of beta1 integrin is essential for beta1 integrin expression and biological function: Identification of the minimal N-glycosylation requirement for alpha5beta1. J. Biol. Chem. 284, 12207–12216 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takagi J., Erickson H. P., Springer T. A., C-terminal opening mimics “inside-out” activation of integrin α5β1. Nat. Struct. Biol. 8, 412–416 (2001). [DOI] [PubMed] [Google Scholar]
- 42.Van Agthoven J. F., Xiong J.-P., Alonso J. L., Rui X., Adair B. D., Goodman S. L., Arnaout M. A., Structural basis for pure antagonism of integrin αVβ3 by a high-affinity form of fibronectin. Nat. Struct. Mol. Biol. 21, 383–388 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xiong J.-P., Mahalingham B., Alonso J. L., Borrelli L. A., Rui X., Anand S., Hyman B. T., Rysiok T., Müller-Pompalla D., Goodman S. L., Arnaout M. A., Crystal structure of the complete integrin αVβ3 ectodomain plus an α/β transmembrane fragment. J. Cell Biol. 186, 589–600 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zheng M., Fang H., Hakomori S., Functional role of N-glycosylation in alpha 5 beta 1 integrin receptor. De-N-glycosylation induces dissociation or altered association of alpha 5 and beta 1 subunits and concomitant loss of fibronectin binding activity. J. Biol. Chem. 269, 12325–12331 (1994). [PubMed] [Google Scholar]
- 45.Gu J., Isaji T., Sato Y., Kariya Y., Fukuda T., Importance of N-glycosylation on α5β1 integrin for its biological functions. Biol. Pharm. Bull. 32, 780–785 (2009). [DOI] [PubMed] [Google Scholar]
- 46.Zivanov J., Nakane T., Forsberg B. O., Kimanius D., Hagen W. J., Lindahl E., Scheres S. H., New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nakane T., Kimanius D., Lindahl E., Scheres S. H. W., Characterisation of molecular motions in cryo-EM single-particle data by multi-body refinement in RELION. eLife 7, 1485 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xiong J.-P., Stehle T., Goodman S. L., Arnaout M. A., New insights into the structural basis of integrin activation. Blood 102, 1155–1159 (2003). [DOI] [PubMed] [Google Scholar]
- 49.Arnaout M. A., Goodman S. L., Xiong J.-P., Structure and mechanics of integrin-based cell adhesion. Curr. Opin. Cell Biol. 19, 495–507 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dong X., Zhao B., Iacob R. E., Zhu J., Koksal A. C., Lu C., Engen J. R., Springer T. A., Force interacts with macromolecular structure in activation of TGF-β. Nature 542, 55–59 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Campbell M. G., Cormier A., Ito S., Seed R. I., Bondesson A. J., Lou J., Marks J. D., Baron J. L., Cheng Y., Nishimura S. L., Cryo-EM reveals integrin-mediated TGF-β activation without release from latent TGF-β. Cell 180, 490–501.e16 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cormier A., Campbell M. G., Ito S., Wu S., Lou J., Marks J., Baron J. L., Nishimura S. L., Cheng Y., Cryo-EM structure of the αvβ8 integrin reveals a mechanism for stabilizing integrin extension. Nat. Struct. Mol. Biol. 25, 698–704 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Benito-Jardón M., Klapproth S., Gimeno-LLuch I., Petzold T., Bharadwaj M., Müller D. J., Zuchtriegel G., Reichel C. A., Costell M., The fibronectin synergy site re-enforces cell adhesion and mediates a crosstalk between integrin classes. eLife 6, 562 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dedden D., Schumacher S., Kelley C. F., Zacharias M., Biertümpfel C., Fässler R., Mizuno N., The architecture of talin1 reveals an autoinhibition mechanism. Cell 179, 120–131.e13 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kelley C. F., Litschel T., Schumacher S., Dedden D., Schwille P., Mizuno N., Phosphoinositides regulate force-independent interactions between talin, vinculin, and actin. eLife 9, e56110 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fujii Y., Kaneko M., Neyazaki M., Nogi T., Kato Y., Takagi J., PA tag: A versatile protein tagging system using a super high affinity antibody against a dodecapeptide derived from human podoplanin. Protein Expr. Purif. 95, 240–247 (2014). [DOI] [PubMed] [Google Scholar]
- 57.Hagn F., Nasr M. L., Wagner G., Assembly of phospholipid nanodiscs of controlled size for structural studies of membrane proteins by NMR. Nat. Protoc. 13, 79–98 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zheng S. Q., Palovcak E., Armache J. P., Verba K. A., Cheng Y., Agard D. A., MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang K., Gctf: Real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rohou A., Grigorieff N., CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mastronarde D. N., Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005). [DOI] [PubMed] [Google Scholar]
- 62.Punjani A., Rubinstein J. L., Fleet D. J., Brubaker M. A., cryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017). [DOI] [PubMed] [Google Scholar]
- 63.Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E., UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 64.Pintilie G. D., Zhang J., Goddard T. D., Chiu W., Gossard D. C., Quantitative analysis of cryo-EM density map segmentation by watershed and scale-space filtering, and fitting of structures by alignment to regions. J. Struct. Biol. 170, 427–438 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pintilie G., Zhang J., Chiu W., Gossard D., Identifying components in 3D density maps of protein nanomachines by multi-scale segmentation. IEEE NIH Life Sci. Syst. Appl. Workshop. 2009, 44–47 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Biyani N., Righetto R. D., McLeod R., Caujolle-Bert D., Castano-Diez D., Goldie K. N., Stahlberg H., Focus: The interface between data collection and data processing in cryo-EM. J. Struct. Biol. 198, 124–133 (2017). [DOI] [PubMed] [Google Scholar]
- 67.Emsley P., Lohkamp B., Scott W. G., Cowtan K., Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Afonine P. V., Poon B. K., Read R. J., Sobolev O. V., Terwilliger T. C., Urzhumtsev A., Adams P. D., Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D Struct. Biol. 74, 531–544 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Webb B., Sali A., Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinformatics 47, 5.6.1–5.6.37 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/19/eabe9716/DC1