

Abstract
To maintain genome stability, organisms depend on faithful chromosome segregation, a process affected by diverse genetic pathways, some of which are not directly linked to mitosis. In this study, we set out to explore one such pathway represented by an undercharacterized gene, SNO1, identified previously in screens of the yeast knockout (YKO) library for mitotic fidelity genes. We found that the causative factor increasing mitotic error rate in the sno1Δ mutant is not loss of the Sno1 protein, but rather perturbation to the mRNA of the neighboring convergent gene, CTF13, encoding an essential component for forming the yeast kinetochore. This is caused by a combination of the Kanamycin resistance gene and the transcriptional terminator used in the YKO library affecting the CTF13 mRNA level and quality . We further provide a list of gene pairs potentially subjected to this artifact, which may be useful for accurate phenotypic interpretation of YKO mutants.
Keywords: SNO1, CTF13, chromosome instability, YKO collection, convergent gene pairs
Introduction
Cells must duplicate and equally partition their genomes with high fidelity. Diseases such as cancer are characterized by the disruption of processes involved in maintaining genome stability (Negrini et al. 2010). Chromosome instability (CIN), referring to increased rates of chromosome missegregation, is a hallmark of most solid tumors and plays a role in tumor evolution (Bakhoum et al. 2018; Potapova et al. 2013; Watkins et al. 2020). As such, it is of importance to understand the genetic pathways responsible for controlling accurate chromosome segregation.
Using budding yeast as a model eukaryotic cell to study CIN has advantages as this system is easily paired with whole genome screening. Considerable progress has been made toward identifying genes responsible for suppressing CIN using classic screening tools such as the chromosome transmission fidelity (CTF), A-like faker, and loss of heterozygosity assays (Novoa et al. 2018; Spencer et al. 1990;). At least 692 yeast genes were found to be involved in chromosome segregation in studies using these tools (Stirling et al. 2011; Yuen et al. 2007). One outstanding challenge is to characterize the roles of unanticipated and uncharacterized hits from these studies and learn how they may act to suppress CIN. The unexpected genes from these screens have the potential to shed light on previously unknown physiological processes affecting genome stability and lead research toward new potential targets or therapies for high CIN diseases.
One such gene that has consistently appeared in budding yeast screens related to altered CIN as well as genome instability is SNO1 (Novarina et al. 2020;Stirling et al. 2011; Yuen et al. 2007). Sno1 is annotated as a protein of uncharacterized function on the Saccharomyces Genome Database (SGD) with a predicted function in the pyridoxine biosynthesis pathway although its glutaminase activity has only been demonstrated in vitro (Dong et al. 2004; Padilla et al. 1998). We set out to determine what role the Sno1 protein might play in determining the fidelity of chromosome segregation. Here, we demonstrate that the protein is dispensable for mitotic function and its presence is not required for high-fidelity chromosome segregation, but rather a previously unexplored artifact introduced by the generation of the yeast knockout (YKO) deletion collection (Giaever et al. 2002) causes increased CIN in the SNO1 deletion background. Our data suggest that this occurs by altering the 3′ untranslated region (UTR) of mRNA from the neighboring convergent gene, CTF13, an essential protein involved in kinetochore formation. Of interest to the budding yeast research community, this YKO library artifact originates from the combination of the Kanamycin resistance gene with the associated TEF1 terminator sequence and has the potential to affect other convergent gene pairs in the genome-wide deletion library.
Materials and methods
Yeast strain genotypes can be found in Supplementary Table S1. Polymerase chain reaction (PCR) and real-time quantitative polymerase chain reaction (qPCR) primers can be found in Supplementary Tables S2 and S3, respectively. Relevant plasmids are listed in Supplementary Table S4.
Construction of mutant strains
In all cases, mutant strains were generated fresh into the qCTF background strain (RLY8492). PCR products for homologous recombination transformations were generated in one of two ways depending on availability of preexisting mutants. If a mutant already existed such as a YKO collection mutant, genomic DNA (gDNA) was extracted from relevant YKO collection mutants and PCR was used to amplify the KO Kanamycin resistance (KanMX originating from pFA6-KanMX4) cassette with 400 base pairs up and downstream the open reading frame (ORF) using 5′/3′ UTR check primers designed using Primers-4-Yeast software (Supplementary Table S2; Yofe and Schuldiner 2014). When generating a novel mutant, we amplified the KanMX gene directly from the pFA6a-KanMX6 plasmid (or the pFA6a-KanMX6-TADH1 plasmid when noted) using 40–80 base pairs homology to the genomic region to be targeted. pFA6-KanMX4 and pFA6a-KanMX6 have identical KanMX sequences including the promoter, Kanamycin resistance gene, and the terminator (Wach 1996). Henceforth, we refer to KanMX4/6 interchangeably as “KanMX.” Deletion mutants with URA3, NatMX, and HphMX markers were generated using the same method with specific details described in Supplementary Table S1. Transformants were generated using a standard lithium acetate homologous/PEG/single stranded carrier DNA protocol (Gietz and Schiestl 2007).
Yeast media
All qCTF strains were grown in SD-Leu (Sunrise Science Products, Cat #: 1707-500) liquid media or solid plates prior to performing qCTF assays to maintain selection of the mini-chromosome (MC). During the 24-h nonselective period of the qCTF assay, cells were grown in SD-Complete liquid media (Sunrise Science Products, Cat #: 1701-500). After transformant generation and selection for inserted selectable markers, all other (non-qCTF background) strains were grown in yeast-extract-peptone-dextrose (YPD) unless otherwise noted.
qCTF flow cytometry assay
The qCTF assay was performed as previously described (Zhu et al. 2015) with the following changes: cells were not sonicated and were analyzed on an Attune NxT Acoustic Focusing Cytometer with Autosampler (Thermo, Cat #: A24861). All transformations of flow cytometry and optical density data to CIN rate were performed as per the previously outlined mathematical formula with the assumption of equaling doubling time between MC+ and MC− cells of individual strains. Each boxplot for a qCTF assay is comprised of a range of 3–12 biological replicates for a single mutant. We sometimes include independently isolated transformants for strains (denoted as #1, #2, #3, etc.).
RNA extractions
A hot acid–phenol chloroform extraction method was used to isolate total RNA. Overnight cultures of yeast were refreshed in 12–50 mL of media to mid-log phase (OD600 ∼0.4–0.6). Then the cultures were harvested to pellets in 2 mL screw-cap tubes and frozen in liquid nitrogen. Cell pellets were thawed and resuspended in 300 μL of lysis buffer (8.4 mM EDTA, 60 mM NaOAc pH 5.5) with 20 μL of 20% SDS and heated at 65°C for 1–3 min; 325 μL of 65°C preheated acid phenol (VWR, Cat #: 0981) was added to samples and incubated while shaking for 15 min at 65°C. Samples were cooled for 5 min on ice and pelleted at 14,000 RPM for 10 min in a tabletop centrifuge at 4°C. The aqueous layer was added to 325 μL fresh room temperature acid phenol and vortexed for 30 s to 1 min followed by a second centrifugation under the same conditions. The aqueous layer was added to 325 μL chloroform (Sigma, Cat #: 288306-100ML) in a fresh 2 mL screw-cap tube. Samples were vortexed for 30 s at room temperature and centrifuged at maximum speed in a tabletop centrifuge for 2 min. The top aqueous layer was transferred into an RNAse-free tube and 3.0 M NaOAc pH 5.5 was added to bring the final concentration to 300 mM followed by addition of one volume of isopropanol to precipitate RNA at −80°C overnight. RNA was pelleted at 14,000 RPM in a tabletop centrifuge at 4°C for 30 min, washed in 70% ethanol, dried, and resuspended in 25–50 μL 10 mM Tris pH 8 on the same day the samples were to be used for cDNA generation.
qPCR
All total RNA samples were treated with RNAse-free DNAse I (NEB, Cat #: M0303S) for 10–15 min at 37°C followed by the addition of 5 mM EDTA and heat inactivation at 65°C for 10 min. cDNA for qPCR was generated using random hexamer primers and the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Cat #: 4368814). cDNA (corresponding to 1 pg to 100 ng of total RNA) and 130 nM each of forward and reverse primers were added to the 2X PerfeCTa® SYBR® Green FastMix®, ROX™ master mix (QuantaBio Cat #: 95073) following dispensing into 384-well plates that were subsequently loaded into a BioRad CFX384 Touch Real-Time PCR Detection System. Minus reverse transcriptase (RT) controls were routinely performed and samples were only included in analysis if this control produced at least a five-cycle difference in average Cq value when compared to the samples with RT. qPCR primer sequences as well as validated efficiencies can be found in Supplementary Table S3. Data were analyzed using the comparative CT method as previously described (Schmittgen and Livak 2008).
3′ rapid amplification of cDNA ends
3′ rapid amplification of cDNA ends (3′ RACE) was performed as previously described with the following adjustments (Scotto-Lavino et al. 2006). Five micrograms of total RNA was used as substrate for cDNA generation using SuperScript III Reverse Transcriptase (Invitrogen, Cat #: 18080044) with the 52 nucleotide “QT” primer (Scotto-Lavino et al. 2006). Q5 High-Fidelity Polymerase (NEB, Cat #: M0491S) was used for subsequent rounds of PCR amplification from the cDNA library using an annealing temperature of 64°C and an extension duration of 3 min at 72°C for 30 cycles. A second, nested PCR using the same cycling parameters was performed to decrease the nonspecific amplification from the first round of PCR using a 1:20 diluted template. All PCR reactions were assembled on ice and transferred directly to preheated thermocyclers at denaturing temperatures. 3′RACE primers can be found in Table S2. Following nested PCR, 3′ RACE PCR products were purified and concentrated (NEB, Cat #: T1030S) prior to sequencing. When noted, 3′ RACE products were gel extracted (NEB, Cat #: T1020S) from the highest signal PCR bands prior to sequencing. Primer MRG316 was used for Sanger sequencing all 3′ RACE PCR products (Supplementary Table S2).
Western blotting
Overnight cultures of cells were refreshed in YPD to log phase for 3.5–5 h at 30°C. Samples were then normalized to OD600 of 1.0 and 2.5 mL was pelleted, washed once with chilled H2O, and snap frozen in liquid nitrogen. Cell pellets were thawed and resuspended into 120 μL of 1x Bolt™ LDS sample buffer (Invitrogen, Cat #: B0008) supplemented with 40 mM DTT (Thermo Scientific, Cat #: R0861). Samples were boiled for 10 min and then vortexed at top speed with approximately 100 μL of 0.5 mm diameter glass beads (BioSpec Products, Cat #: 11079105) for 1 min and 30 s at 4°C. Lysates were boiled again for 10 min, spun down at maximum speed in a tabletop centrifuge for 1 min, and the supernatant was removed from the debris and glass beads into a fresh tube; 27 μL of this protein suspension was loaded for each sample into a 10-well 4–12% NuPAGE Bis-Tris gel (Invitrogen, Cat #: NW04120BOX) along with 10 μL Precision Plus Protein™ Dual Color Standard (Bio-Rad, Cat #: 1610374). SDS-PAGE gels were run according to manufacturer’s recommendation. The gel was next transferred to a PVDF membrane (Introgen, Cat #: IB24002) using the iBlot™ 2 transfer system. Membranes were blocked in 5% bovine serum albumin (BSA, Sigma, Cat #: A9647) in 1x TBS with 0.1% Tween-20 (1x TBST) for 1 h at room temperature followed by overnight incubation in primary antibody in blocking solution at 4°C. Pgk1 mouse monoclonal antibody (1:3000 dilution) and HA rabbit monoclonal antibody (1:1000 dilution) were purchased from Invitrogen (Cat #: 459250) and Cell Signaling (Cat #: 3724S), respectively. Following eight washes in 1x TBST, the membrane was then incubated in a 1:5000 dilution in 1x TBST of the species appropriate HRP-linked secondary antibody (Cell Signaling, Cat #: 7074S or 7076S) for 1 h at room temperature. After four 1x TBST washes, membranes were incubated in 2 mL of ECL reagent (Bio-Rad, Cat #: 170-5060) for 1–5 min following membrane exposure on a LI-COR Odyssey® Fc Imaging system. Quantification was performed using Image Studio Lite software.
Spot dilution assay
Log phase cultures of cells were normalized to an OD600 of 0.62 and 200 μL of each culture was pelleted, washed once with sterile H2O, resuspended in 200 μL sterile H2O, and transferred to a flat bottom 96-well plate; 20 μL of the most concentrated sample was then serially diluted into 180 μL of sterile H2O in a new well for a total of four 10-fold dilutions; 5 μL of each dilution was then dropped onto YPD or YPD plates supplemented with 200 mg/L Geneticin (G418; Thermo Scientific, Cat #: 11811023) and grown for 48 h at 30°C prior to image collection.
Statistical analysis
Statistical analysis for the qCTF assay was performed via a one-way ANOVA test on the mean fold-change for samples using the stat_compare_means() function from the ggpubR package (version 0.2.3) with default significance cutoffs (not significant [ns]: P > 0.05, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001). If the result of the one-way ANOVA test was significant, then a Tukey’s post hoc test was performed to identify which groups significantly differed from the relevant control strain using the tukey_hsd() function from the rstatix package (version 0.6.0) using the same significance cutoffs listed above. All significance cutoffs overlaid onto plots are generated from the Tukey’s test. Statistical analysis for qPCR and Western blot data was performed using the same statistical testing method. Unless otherwise noted, all qPCR experiments were performed using both biological and technical triplicates. Cq values for technical triplicates for each biological replicate were averaged and these Cq averages were used in the subsequent statistical analysis. Statistical analysis was only performed on qPCR data if there was a complete set of at least three biological replicates.
Code to extract convergent gene pairs from the SGD
A Python script was written to retrieve a list of all ORFs and chromosomal coordinates from the SGD for S288C reference strain genome. In the resulting table, coding strands for each ORF are denoted by a + or − for the Watson or Crick strand, respectively. To identify convergent gene pairs, the script iteratively compares the coding strand of pairs of genes. If both genes were either +/+ or −/−, meaning they are expressed from the same strand, the algorithm skips to compare the next two gene pairs. If the first gene in the pair is + and the second gene pair is −, then they are labelled convergent and this gene pair including its associated information were stored locally to a .txt file.
Data availability
All yeast strains and plasmids are available upon request. Source code for the convergent gene pair mining from the SGD is available online (https://github.com/mgordo34/Convergent_Gene_Script). Supplementary Table S1 contains the genotypes of budding yeast strains used in this study and the sources of any strains not generated in the present study as well as the primer sets and templates DNA sequences used for homologous recombination transformations and aliases for relevant strains. Supplementary Table S2 contains a list of primer names and sequences used for PCR, 3′ RACE, and DNA sequencing. Supplementary Table S3 contains qPCR primer sequence sets, the ORFs they anneal to, and their experimentally calculated efficiencies. Supplementary Table S4 contains relevant plasmids developed and used during the course of this study. Supplementary Table S5 contains one of the tables generated from the source code for mining convergent gene pairs from the SGD. This table lists nonoverlapping convergent gene pairs, the number of base pairs between each stop codon, the chromosomal coordinates of each gene, and the chromosome they reside on. The source code also produces a table of overlapping convergent gene pairs. Supplemental Material is available at figshare: https://doi.org/10.25387/g3.14195846.
Results
The Sno1 protein is dispensable for high-fidelity chromosome segregation
In a previous study, we developed a quantitative tool for measurements of CTF in yeast called the qCTF assay (Zhu et al. 2015). This tool improves upon the standard CTF assay (Spencer et al. 1990) for measuring the missegregation rate of a mini-chromosome (MC) by enabling high-throughput experimentation to generate single-cell quantitative data via flow cytometry, as opposed to the standard scoring of colored colonies in the sectoring assay. To construct the qCTF yeast background, a short linear MC made from the left arm of chrIII containing the MATα locus as well as a selectable marker was introduced into a MATa haploid yeast strain. The MATα locus on the MC represses the expression of green fluorescent protein (GFP) fused to the MATa-specific gene, MFA1, which is highly expressed in the MATa strain. Upon loss of the MC, GFP molecules irreversibly accumulate in the cell, which allows for the detection of the missegregation event using flow cytometry.
As SNO1 was implicated in previous screens for genes whose deletion affect CIN, we applied the qCTF assay to validate and quantify the effect of a SNO1 ORF deletion. We engineered fresh qCTF knockout (KO) strains through homologous recombination gene replacement that introduced a Kanamycin resistance (KanMX) gene in place of the target gene using the same PCR-based deletion strategy used to generate the YKO collection (Baudin et al. 1993; Wach et al. 1994). We chose to add the KanMX KO alleles directly to the qCTF background strain as opposed to engineering the qCTF components into the KO library due to the accumulation of secondary mutations that are known to occur within the KO library (Puddu et al. 2019; Teng et al. 2013). Because each of our mutants contained the KanMX gene, we additionally generated a control qCTF strain with a KanMX integrated in the TRP1 locus as a baseline to compare with our mutants. The resulting strains were used to measure a “CIN rate,” which is defined as the frequency of MC loss/cell divisions. Our control strain had a CIN rate of ∼0.00016, equivalent to 1.6 MC missegregations per every ten thousand divisions, which is comparable to our previous data as well as that of the standard CTF assay (Zhu et al. 2015). CIN rates are transformed into fold-changes, which are calculated by dividing the CIN rate of a given mutant by the CIN rate of an appropriate qCTF control strain.
Three independent transformants of a sno1Δ mutant showed a ∼40- to 53-fold increase in the CIN rate in the qCTF background (Figure 1A). To validate the functional relevance of the Sno1p in suppressing CIN, we ablated Sno1p function by incorporating a single nonsense point mutation within the SNO1 ORF at cysteine 84 to prevent full-length protein expression. This mutation was generated through negative selection to generate a “scar-less” genome edit harboring no selectable marker and was sequenced to confirm the presence of the nonsense mutation at the endogenous SNO1 ORF (Supplementary Figure S1A and S1B; Gray et al. 2005). This sno1-nonsense point mutant in the qCTF background did not elevate the CIN rate significantly compared to the wild-type control in two independent mutant isolates, suggesting that the high CIN phenotype in the sno1Δ mutant was not attributed to the loss of the Sno1p (Figure 1B). We also created a sno1Δ::URA3 strain, which did not have elevated CIN rates (Supplementary Figure S1A and S1C). Taken together, these data point to a lack of necessity for the Sno1p in maintaining proper chromosome segregation but rather suggest a potential artifact induced by the sno1Δ::KanMX mutant of the YKO collection.
Figure 1.

Sno1p is not necessary for high fidelity in chromosome segregation. (A) CIN rate measured using the qCTF assay for three independent isolates of sno1Δ mutants compared to a control strain (KanMX inserted into the TRP1 locus). Boxplot parameters for all qCTF figures are as follows: lower and upper hinges correspond to the first and third quartiles (the 25th and 75th percentiles). The hinge between the upper and lower hinge represents the median of the data. The upper whisker extends from the hinge to the largest value no further than 1.5 * IQR from the hinge (where IQR is the inter-quartile range, or distance between the first and third quartiles). The lower whisker extends from the hinge to the smallest value at most 1.5 * IQR of the hinge. Individual data points are overlaid on the boxplots. P-values are generated from a Tukey’s post hoc test. Default significance cut offs were used as follows: not significant (ns): p > 0.05, *: p ≤ 0.05, **: p ≤ 0.01, ***: p ≤ 0.001, ****: p ≤ 0.0001. (B) CIN rate measurements for two independent isolates of the sno1-nonsense (sno1-ns) mutant in which Cysteine 84 has been mutated to a stop codon. See Supplementary Figure S1B for Sanger sequencing of gDNA from sno1-ns strains to confirm the nonsense point mutation genotype. P-values are generated from a Tukey’s post hoc test. (C) Graphical representation of the SNO1 locus on chrXIII and corresponding KanMX insertions. The SNO1 ORF is displayed as a white arrow while KanMX insertions are depicted with colored arrows representing the size of DNA sequence replaced. In each case, the KanMX gene size is consistent, which is represented via blown up KanMX arrows in the sno1Δ and sno1Δ2 cartoons. All other KanMX sequences are represented in shorthand by the colored arrows. All plots are drawn to scale and aligned to the SNO1 ORF (grey dashed line). (D) CIN rate measurements for the SNO1 KanMX insertion mutants. P-values indicated as astericks centered at the bottom of each box plot are all pairwise comparisons to the control strain generated from a Tukey’s post hoc test. P-values indicated as astericks and brackets at the top of the graph are pairwise comparisons marking the only KanMX insertion mutants that are significantly different from the sno1Δ mutant.
When generating the YKO collection, the KanMX gene cassette from the plasmid pFA6a-KanMX encoding aminoglycoside phosphotransferase under the control of the TEF1 promoter and terminator from Ashbya gossypii was used to replace nonessential ORFs (Giaever et al. 2002; Wach et al. 1994). We therefore hypothesized that the placement of the KanMX gene within the SNO1 locus, rather than perturbation of the SNO1 ORF per se, may contribute to the CIN phenotype in this mutant. To test this, we generated a series of KanMX insertions at the SNO1 locus and measured the CIN rates of these mutants (Figure 1C). These KanMX insertion mutants either deleted portions of the SNO1 ORF or deleted a portion of the intergenic regions upstream of the SNO1 ORF. One of these mutants, sno1Δ6, had an entirely unperturbed SNO1 ORF with only 251 base pairs of upstream intergenic sequence containing the SNO1 promoter replaced with KanMX beginning 51 base pairs upstream of the ORF. All of these mutants significantly increased the CIN rate over 40-fold compared to the control strain, suggesting that the presence of the KanMX gene induces high CIN when inserted at various positions around the SNO1 locus (Figure 1D). While each of these mutants is significantly different from baseline level of CIN the control strain, only sno1Δ2 and sno1Δ6 have CIN rates that are significantly elevated compared with the standard YKO collection sno1Δ mutant.
The expression orientation of KanMX at the SNO1 locus differentially influences CIN
In an effort to study the SNO1 locus, we also assayed the CIN rate in a YKO collection mutant deleting the most proximal upstream nonessential gene to SNO1, SNZ1 (Figure 2A). The SNZ1 and SNO1 genes are arranged in a divergent orientation and share a common promoter sequence (Padilla et al. 1998). The snz1Δ CIN rate was not significantly increased compared to the wild-type control (Figure 2B), while a different KanMX insertion downstream of the SNZ1 ORF in an intergenic region increased CIN more than 70-fold (called snz1Δ2, Figure 2, A and C). When an ORF is replaced using the conventional YKO collection method, the KanMX gene is expressed from the same coding strand of DNA as the deleted ORF. For example, the coding strand for SNO1 is the Crick strand and in the YKO collection sno1Δ mutant, the KanMX gene is also expressed from the Crick strand. SNZ1 is expressed from the opposite, Watson, strand of SNO1, which means the KanMX gene in the snz1Δ mutant is also expressed from the opposite strand compared to SNO1, highlighting a potential difference that may be responsible for the increased CIN observed in the sno1Δ mutant.
Figure 2.

KanMX induces CIN when present at the SNO1 locus oriented in the same direction as the SNO1 coding strand. (A) Graphical representation of chrXIII showing the divergent SNZ1 (yellow) and SNO1 (blue) gene pair. KanMX deletions are depicted in grey. As noted in Figure 1C, the size of the KanMX is consistent whereas the grey arrow represents the amount of DNA sequence replaced. Right and left pointing arrow orientation represents gene expression from the Watson and Crick strand, respectively. (B) CIN rate measurements for two independent isolates of the snz1Δ strain using the qCTF assay. P-values are generated from a Tukey’s post hoc test. (C) CIN rate measurements for the snz1Δ2 strain using the qCTF assay. P-values are generated from a Tukey’s post hoc test. (D) CIN rate measurements for the a control strain, sno1Δ-Watson (sno1ΔW) strain, and the conventional sno1Δ-Crick (sno1ΔC) strain. P-values are generated from a Tukey’s post hoc test. (E) CIN rate measurements for the snz1Δ-Crick (snz1ΔC) strains compared to conventional snz1Δ-Watson (snz1ΔW) YKO strain. P-values are generated from a Tukey’s post hoc test.
To determine whether the KanMX expression orientation affects the CIN rate in an SNO1 deletion mutant, we generated two complementary sno1Δ mutants that express KanMX from either the Crick strand (as in the YKO collection) or the Watson strand (Figure 2A, “sno1Δ-Watson”). Only the SNO1 deletion expressing KanMX from the Crick orientation causes a significant increase in the CIN rate compared to the control for this experiment (Figure 2D). Similarly, switching the snz1Δ allele such that the KanMX gene is encoded in the Crick orientation raised the CIN rate significantly higher than the snz1Δ mutant from the YKO collection (Figure 2E). We further tested the effect of altering the KanMX expression orientation in the sno1Δ6 mutant, which has an unperturbed SNO1 ORF coding sequence, and found that the majority of the elevated CIN was significantly reduced. However, the CIN rate was still elevated ∼14- to 19-fold compared to the control levels (Supplementary Figure S2A). The remainder of elevated CIN in the sno1Δ6-Watson mutant points to an additional source of CIN independent of KanMX orientation within the sno1Δ6 background, consistent with the significantly different CIN rate between the sno1Δ and sno1Δ6 reported earlier (Figure 1D).
Finally, the effect of KanMX gene orientation on CIN does not affect all genes that are known CIN mutants, as when we reverse the KanMX gene direction in a different CIN mutant, csm3Δ, there was no significant difference in the CIN rate from the conventional KO allele (Supplementary Figure S2B). Together, these findings suggest a localized cis effect of the KanMX gene cassette on the CIN rate at the SNO1 locus with coding expression from the Crick strand, rather than a direct effect due to the absence of the Sno1p. This cis effect is present when KanMX is inserted at distances up to 1.3 kilobase pairs upstream of the SNO1 ORF as in the snz1Δ2 mutant, which was the farthest distance we tested (Figure 2, A and C).
sno1Δ affects the abundance of mRNA from the downstream gene CTF13
Given that Sno1p is not responsible for preventing CIN, we next determined whether there may be neighboring elements that are disrupted in the sno1Δ mutant and lead to increased CIN. Aside from SNZ1, the next closest ORF to SNO1 is the downstream gene, CTF13. CTF13 is an essential yeast gene encoding a component of the CBF3 inner kinetochore complex that binds to the CDEIII centromere element (Biggins 2013). We noted that SNO1 is arranged in a “tail-to-tail” (also referred to as convergent) orientation with CTF13, such that the 3′ UTRs of both genes are overlapping with only 23 base pairs of intergenic sequence separating the two genes’ stop codons (Figure 3A). Given its close proximity to the SNO1 gene, CTF13 mRNA may be altered in the YKO sno1Δ mutant. Because CTF13 is annotated as both a haploinsufficient and an essential gene in the SGD, a reduced abundance of CTF13 mRNA could explain the heightened CIN in a sno1Δ mutant from the YKO collection. To test this, we performed reverse transcription quantitative PCR (qPCR) to estimate the relative abundance CTF13 mRNA in the sno1Δ strain. When compared with the wild-type control, the sno1Δ strains showed significant reduction in CTF13 mRNA levels (Figure 3B). This trend was consistent for a second independent qPCR primer set probing the CTF13 ORF (Supplementary Figure S3A). Furthermore, we found this trend of reduced CTF13 mRNA to be consistent in the sno1Δ6 mutant, which also had increased CIN rates due to the KanMX insertion whereas the sno1Δ::URA3 mutant had wild-type abundance of CTF13 mRNA (Supplementary Figure S3B and S3C). These findings suggest that the KanMX sequence, inserted at varying positions in the SNO1 locus, perturbs the abundance CTF13 mRNA independent of an unaltered copy of the SNO1 ORF.
Figure 3.
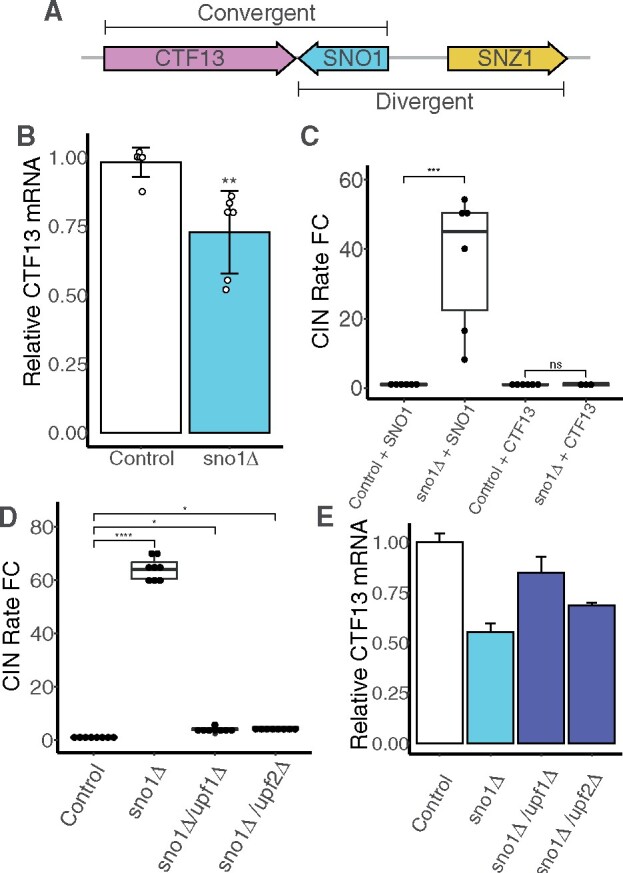
CTF13 mRNA abundance is reduced in the sno1Δ mutant. (A) Graphical representation of the locus on ChrXIII containing CTF13 (pink), SNO1 (blue), and SNZ1 (yellow) drawn to scale. SNO1 and CTF13 are convergent gene pairs. SNO1 and SNZ1 are divergent gene pairs sharing a common promoter sequence. (B) qPCR analysis of CTF13 mRNA levels expressed as fold changes performed on control and sno1Δ strains using a CTF13 ORF primer set (set #3, see Supplementary Table S3). Six independent replicates were performed each including technical triplicates. Each values is normalized to the level of ACT1 mRNA. Technical triplicates are averaged and pooled together and standard deviation is calculated for Cq means of the independent strains. P-values are generated from a Tukey’s post hoc test. (C) CIN rate measurements for control strain and sno1Δ strain with either an additional copy of SNO1 or CTF13 integrated into an ectopic position on chrIV. P-values are generated from a Tukey’s post hoc test. (D) CIN rate measurements for sno1Δ and NMDΔ double mutants (sno1Δ/upf1Δ and sno1Δ/upf2Δ). P-values are generated from a Tukey’s post hoc test. (E) qPCR analysis of CTF13 mRNA extracted from strains listed in Figure 3D using a CTF13 ORF primer set (set #3, see Supplementary Table S3). Only one independent isolate for each strain was used in this experiment and the standard deviation is measured from the technical triplicates. This result was validated using the same cDNA samples with CTF13 primer set #1 (data not shown).
If the CIN phenotype of the sno1Δ mutant was due to reduced CTF13 mRNA, increased dosage of the CTF13 gene should prevent high CIN phenotype observed in this background. To test this, we examined the effect of adding an extra copy of the CTF13 gene in the sno1Δ background into an intergenic location on chromosome IV (native CTF13 is located on chromosome XIII). Adding the second copy of the CTF13 ORF to sno1Δ completely suppressed the high CIN back to the wild-type level (Figure 3C). On the other hand, integrating a copy of the SNO1 ORF into the same location on chromosome IV in the sno1Δ background was not sufficient to rescue the high CIN phenotype (Figure 3C). This supports the idea that the causative factor increasing CIN in the sno1Δ mutant is reduced CTF13 mRNA.
Disrupting the nonsense-mediated decay pathway increases CTF13 mRNA level and reduces CIN in sno1Δ mutants
CTF13 mRNA is known to be affected by the nonsense-mediated decay (NMD) pathway such that inactivating NMD leads to increased CTF13 mRNA (Dahlseid et al. 1998). This is thought to occur bya mechanism independent of altering the half-life of the CTF13 mRNA but rather through affecting other transcripts involved in CTF13 gene expression. Indeed, NMD deletion (upf1Δ) led to a fivefold increase in the CTF13 mRNA when averaged across two independent CTF13 qPCR primer sets (Supplementary Figure S3D). We therefore reasoned that inactivating the NMD pathway may result in increased abundance of the CTF13 mRNA to a level sufficient to reduce the CIN phenotype in the sno1Δ YKO collection mutant. To this end, we constructed double mutants with sno1Δ combined with deletion of either UPF1 or UPF2, genes encoding two of the essential components of the NMD pathway. The resulting double mutants had markedly reduced CIN rates much closer to that of the control strain (Figure 3D). qPCR confirmed that the CTF13 mRNA abundance was indeed increased in sno1Δ upf1Δ and sno1Δ upf2Δ double mutants compared to the sno1Δ single mutant, although not back to the wild-type levels (Figure 3E).
We also used several KanMX insertion mutants to determine whether inactivated NMD could also suppress the CIN in these backgrounds. In our high-throughput qCTF analysis, sno1Δ2 and the sno1Δ6 were found to have significantly increased CIN even compared to the sno1Δ mutant (Figure 1D), but sno1Δ3 does not. This was surprising as sno1Δ3 deletes a portion of the SNO1 locus that encompasses both the sno1Δ2 and the sno1Δ6 deleted sequences plus an extra 97 base pair between the two. Our secondary analysis revealed that sno1Δ3 mutant also had significantly elevated CIN compared to the sno1Δ strain (Supplementary Figure S3E). NMD inactivation in either the sno1Δ2 or the sno1Δ6 mutant completely suppressed CIN back to the control levels, whereas the sno1Δ3 CIN rate was unaltered (Supplementary Figure S3E). NMD inactivation led to an increase in CTF13 mRNA compared to the single sno1Δ6 mutant (Supplementary Figure S3C), consistent with CIN suppression. This suggests that loss of the sequence uniquely deleted in the sno1Δ3 mutant has an additional effect on CTF13 gene expression unrelated to NMD (see Discussion).
CTF13 mRNA is abnormally truncated in the sno1Δ background
Several yeast terminators have been noted to function bidirectionally such that they can be read from either the sense or antisense strand (Heidmann et al. 1992; Uwimana et al. 2017). The bidirectionality is proposed to have evolved to keep transcriptional noise reduced in a genome that is highly compacted. The TEF1 terminator from the pFA6a-KanMX cassette used in the YKO library may be read bidirectionally such that the CTF13 transcript is prematurely terminated in the sno1Δ background. This hypothesis predicts two changes to the CTF13 mRNA in the sno1Δ background. First, the transcript would have a different length compared to the wild-type transcript. Second, the composition of the 3′ UTR would be altered. To address both of these predictions, we used 3′ RACE to map the sequence and length of the 3′ end of the CTF13 transcript in both the wild-type and sno1Δ background (Scotto-Lavino et al. 2006). CTF13 is known to have an unusually long 3′ UTR estimated to be greater than 2 kilobases (Dahlseid et al. 1998), which we were able to detect using the 3′ RACE method combined with nested PCR (Figure 4A, lane 2). When we assayed the length of the CTF13 3′ UTR in the sno1Δ mutant, a smaller 3′ UTR was observed as a faster migrating PCR product, supporting the notion that the KanMX gene cassette imparts an ectopic transcriptional termination sequence that truncates the CTF13 mRNA (Figure 4A, lane 3). Upon sequencing the 3′ UTR of the truncated CTF13 mRNA transcript in the sno1Δ mutant, we found no evidence of a polyA tail but rather we found heterogeneous mRNA ends, evidenced by unclear sequencing reads (Figure 4B, Full Sanger sequencing in S5). 3′ UTR sequence dictates mRNA stability as well as translational efficiency, providing a possible link to explain reduced CTF13 mRNA and even reduced Ctf13p.
Figure 4.

CTF13 mRNA length and 3′ end composition are altered in the sno1Δ mutant. (A) Nested reverse transcriptase (RT)-PCR products from a 3′ RACE assay using a CTF13 gene specific primer (MRG577) and QI primer (see Supplementary Table S2) run on a 1% agarose gel stained with ethidium bromide. Lanes 1 + 7: 5 μL 1 kb ladder (NEB # N3232L). Lane 2: Control strain, wild-type SNO1-CTF13 locus. Lane 3: sno1Δ::Kan, Lanes 4-6: 3 independent isolates of sno1Δ::Kan-TADH1. All strains were engineered into the qCTF background. (B) Sanger sequencing of gel extracted 3′ RACE PCR products for the sno1Δ strain trimmed to a 76 base pair window within the CTF13 3′ UTR. This includes the final base calls from this sequenced fragment. Purple Ns are reads that could not be accurately assigned using the sangerseqR package in R (Hill et al. 2014). Full Sanger sequencing data is available in Figure S5. (C) Graphical representation of the two plasmids used to generate sno1Δ mutants. Plasmid maps are displayed as linear fragments zoomed into the KanMX promoter, gene, and terminator segment. pFA6a-KanMX (top) contains the TEF1 promoter and terminator (PTEF1 and TTEF1, respectively) sequence from A. gossypii. pFA6a-KanMX-TADH1 (bottom, referred to as MGP026 in Supplementary Table S4) was cloned using the pFA6a-KanMX as a backbone and the TTEF1 was replaced with a 166 base pair ADH1 terminator (TADH1) sequence from S. cerevisiae. Maps drawn to scale. (D) Sanger sequencing of the 3′ RACE product for the CTF13 mRNA in the sno1Δ::Kan-TADH1 strain trimmed to a 78 base pair window within the CTF13 3′ UTR. The putative polyA tail signal is shown underlined in black. Full Sanger sequencing results available in Supplementary Figure S6. A black bar is used to indicate the putative polyA reads from this sequencing experiment. (E) qCTF assay CIN rate measurements from three independent isolates of the sno1Δ::Kan-TADH1 (TADH1 #1-3) strain and sno1Δ strain. P-values are generated from a Tukey’s post hoc test. (F) qPCR analysis of CTF13 mRNA levels performed on control, sno1Δ, and sno1Δ::Kan-TADH1 (TADH1) strains using a CTF13 ORF primer sets (set #3, see Supplementary Table S3). Three independent replicates were performed with technical triplicates. Each values is normalized to the level of ACT1 mRNA. Technical triplicates are averaged and pooled together and standard deviation is calculated for means of the independent strains. P-values are generated from a Tukey’s post hoc test. (G) qPCR analysis of CTF13 mRNA levels expressed as fold changes performed on control and sno1Δ::Kan-TADH1 (labeled TADH1 for shorthand) in the presence and absence of functional NMD using a CTF13 ORF primer set (set #3, see Supplementary Table S3). Three independent replicates were performed each including technical triplicates. Each values is normalized to the level of ACT1 mRNA. Technical triplicates are averaged and pooled together and standard deviation is calculated for means of the independent strains. Statistical analysis is generated from a Tukey’s post hoc test. (H) Western blot analysis for 3xHA-CTF13 protein levels in a SNO1 WT background or a sno1Δ::Kan-TADH1 background. Pgk1 was used as a loading control for quantification in Figure 4I. Exposure times were as follows: 5 min and 10 s for α-HA blot, 1 min and 2 s for α-Pgk1 blot. Uncropped membrane images including the molecular weight are provided in Figure S8E). (I) Quantification of the membrane shown in Figure 4H. Ratios between the HA and Pgkl signal were determined using Image Studio Lite. Statistical analysis was performed using a Tukey’s post hoc test and error bars represent the standard deviation.
Addition of homogenous and polyadenylated CTF13 transcripts suppresses CIN in the sno1Δ background
If the CTF13 mRNA is unstable in the sno1Δ background, we reasoned that generating a homogeneous pool of polyadenylated CTF13 transcripts should overcome the CIN phenotype in this mutant. As such, we replaced the TTEF1 from the common plasmid used to generate YKO collection mutants, pFA6a-KanMX, with a budding yeast ADH1 terminator (TADH1) and generated a novel sno1Δ mutant (henceforth called sno1Δ::Kan-TADH1, Figure 4C). We chose the TADH1 as it is known to create polyadenylated transcripts as well as function bidirectionally in budding yeast (Heidmann et al. 1992; Uwimana et al. 2017). Using the same PCR-based 3′ RACE technique, we confirmed that the CTF13 transcript was indeed truncated in the sno1Δ::Kan-TADH1 strain (Figure 4A, lanes 4–6) and that it contained a clear polyA signal at the 3′ end (Figure 4D, Full Sanger sequencing in Figure S6).
In this new sno1Δ::Kan-TADH1 mutant with polyadenylated CTF13 transcripts, the CIN rate was indistinguishable from the control strain (Figure 4E). This result implies that shortened length of the CTF13 transcript alone is insufficient to increased CIN. qPCR revealed that the CTF13 mRNA levels were significantly reduced in the sno1Δ::Kan-TADH1 strain such that the amount of CTF13 mRNA was not significantly different between the sno1Δ and sno1Δ::Kan-TADH1 strains (Figure 4F). However, when the sno1Δ::Kan-TADH1 strain was tested at a later time point, the cells had accumulated near wild-type levels of CTF13 mRNA (Figure 4G), suggesting that the initial observation of reduced CTF13 mRNA may be a transient state and the strain eventually accumulate more CTF13 transcripts. Notably, we never observed sno1Δ mutants with wild-type levels of CTF13 mRNA during the course of this study, further supporting the sustained instability of the CTF13 transcript in that background. Additionally, disruption of NMD in the sno1Δ::Kan-TADH1 strain led to an increase in the relative abundance of CTF13 mRNA (Figure 4G).
Given that the CTF13 3′ UTR appears to be efficiently polyadenylated with the bidirectional TADH1 sequence downstream of CTF13, we next asked whether a homogenous pool of polyadenylated CTF13 transcripts within a sno1Δ6 mutant would be sufficient to overcome the more penetrant CIN increase in this background. To this end, we inserted the 166 base pair TADH1 sequence between SNO1 and CTF13 using the “scar-less” method of negative selection (Supplementary Figure S4A, “CTF13-TADH1”). Sequencing a PCR product from genomic DNA and 3′ RACE were used confirm the presence of the terminator sequence and that the CTF13 transcript was both truncated and polyadenylated (Supplementary Figure S4B). The presence of the CTF13-TADH1 sequence was sufficient to replenish the CTF13 mRNA back to wild-type levels in the sno1Δ6 background as well as reduce the CIN rate back to wild-type levels (Supplementary Figure S4C and S4D). This result suggests that the all of the CIN in the sno1Δ6 mutant can be explained by CTF13 mRNA defects.
The combination of the KanMX and TEF1 terminator affects both the steady-state quantity and quality of CTF13 mRNA in sno1Δ mutants
The above results suggest that the TTEF1 sequence is necessary to cause elevated CIN when the KanMX gene is present in the Crick orientation at the SNO1 locus. To test whether TTEF1 sequence is sufficient to induce CIN, we generated SNO1 deletions using two other markers, pFA6a-NatMX and pFA6a-HphMX, which confer resistance to the drugs nourseothricin and hygromycin, respectively (Supplementary Figure S7A). Importantly, both of these KO cassettes use the same promoter as KanMX and TTEF1 immediately following the drug resistance gene as in the pFA6a-KanMX plasmid (Supplementary Figure S7A and S7B).
3′ RACE first confirmed that the CTF13 mRNA in each of these strains was truncated (Supplementary Figure S7C). We also confirmed that the CTF13 mRNA was truncated in the sno1Δ::URA3 strain, indicating that PURA3 also functions bidirectionally (Supplementary Figure S7C, lane 4). Surprisingly, neither the sno1Δ::Nat mutant nor the sno1Δ::Hph mutant had elevated CIN rates as was observed in the sno1Δ::Kan mutant (Supplemental Figure S7D), suggesting the TTEF1 sequence is insufficient to cause increased CIN. The sno1Δ::Nat mutant had wild-type levels of CTF13 mRNA (Supplementary Figure S7D), whereas the sno1Δ::Hyg mutant had reduced abundance of the CTF13 mRNA similar to what was observed in the sno1Δ::Kan mutant (Supplemental Figure S7E). We sequenced 3′ RACE cDNA ends to determine potential qualitative differences in CTF13 mRNA among these strains. All three biological replicates of the sno1Δ::Nat CTF13 mRNA had a consistent sequence at the end of the 3′ UTR (Supplemental Figure S7F) corresponding to TTEF1 sequence, whereas in the sno1Δ::Hyg mutant, two of three replicates matched the same CTF13 3′ UTR sequence of the sno1Δ::Nat mutant, while one replicate indicated a longer heterogenous CTF13 mRNA end (Supplementary Figure S7G and S7H). These data suggest that in the sno1Δ::Kan (and occasionally in the sno1Δ::Hyg strain), the CTF13 transcript extends into the drug resistance marker ORF. This seems to correlate with transcript instability, but the causal relationship is unclear. This observation highlights the complexity of mRNA perturbation by TTEF1 and suggests that some sequence features of KanMX may also be involved.
Presence of the TADH1 sequence is sufficient to increase Ctf13p levels in a sno1Δ mutant
To determine whether Ctf13p level may account for the different CIN rates between the sno1Δ YKO collection mutant and the sno1Δ::Kan-TADH1 mutant, we used negative selection to endogenously tag the CTF13 ORF with 3x hemagglutinin (3xHA; Supplementary Figure S8A). Importantly, we inserted the 3xHA tag at the N-terminus of CTF13 to avoid adding any extra DNA sequence that may influence the natural CTF13 3′ UTR. Following sequencing-based confirmation of the in-frame 3xHA-CTF13 tag at the endogenous ORF (Supplementary Figure S8A), we further generated sno1Δ and sno1Δ::Kan-TADH1 deletions in the 3xHA-CTF13 background. Of note, although colonies formed following the initial generation of the 3xHA-CTF13/sno1Δ double mutant, we were unable to isolate single colonies on fresh selective agar plates and as such could not accurately genotype the isolates (Supplementary Figure S8B and S8D).
The 3xHA-CTF13 tag itself seems to cause a very mild growth defect compared to an untagged CTF13 allele. As such it is possible that the 3xHA tag reduces the efficiency of this essential kinetochore protein and or results in increased Ctf13p degradation, which may explain the pronounced defect in combination with a sno1Δ (Supplementary Figure S8C). In characterizing this double mutant, we noticed that releasing selective pressure allowed the 3xHA-CTF13/sno1Δ strain to grow with only a mild growth defect (Supplementary Figure S8C). It is possible that the sno1Δ is lethal in the 3xHA-CTF13 background and, if selection is removed, the cell population adapts, perhaps by shutting down expression of KanMX via mutation or epigenetic regulation. Furthermore, if any aneuploids exist in the original transformed population (i.e., disome XIII that contains the SNO1-CTF13 locus), these strains can have continued growth even in the presence of G418 selection. Unfortunately, we were unable to confirm the synthetic lethality of the 3xHA-CTF13/sno1Δ strain using tetrad dissection analysis because the close proximity of SNO1 and CTF13 would likely limit crossing over during meiosis.
Although we could not confidently generate a pure 3xHA-CTF13/sno1Δ double mutant, we were able to generate multiple 3xHA-CTF13/sno1Δ::Kan-TADH1 double mutants, which grew qualitatively similarly to the 3xHA-CTF13 background strain and could grow well in the presence of selective pressure (Supplementary Figure S8C and S8D). Comparing the Ctf13p levels between wild-type and sno1Δ::Kan-TADH1 log phase cultures, Ctf13p levels were elevated in the sno1Δ::Kan-TADH1 in three repeated experiments (Figure 4, H and I and Supplementary Figure S8E). Overall, the data presented suggest that the elevated Ctf13p in the sno1Δ::Kan-TADH1 strain is sufficient to buffer against increased CIN.
Reduced mRNA within other convergent gene pairs in the YKO collection
Although we encountered the KanMX-induced perturbation of mRNA of a neighboring gene in the unique context of the SNO1 and CTF13 convergent gene pair, other convergent gene pairs could be affected similarly in the YKO collection. We computationally mined the SGD to extract convergent gene pairs genome wide. This list, which does not include overlapping ORFs, contains 1572 gene pairs encompassing 3144 ORFs and roughly 46.6% of the 6746 ORFs currently annotated in the SGD (Cherry et al. 2012; Engel et al. 2014; Supplementary Table S5). We further mapped the distribution of the distances between nonoverlapping convergent gene pairs genome-wide and found the median distance to be 236 base pairs (Figure 5A). Although there does not seem to be a minimum distance requirement for the KanMX artifact, there are only 17 convergent gene pairs that have even less intergenic region between stop codons than the SNO1-CTF13 gene pair.
Figure 5.

Genome-wide analysis of convergent gene pairs suggests KanMX convergent gene interference can be conserved. (A) Histogram depicting the distribution of intergene distance between all nonoverlapping convergent gene pairs in budding yeast using 100 bins. All distances that are greater than 2000 base pairs are plotted together in a single bin. Red dashed line depicts the median of the dataset at 236 base pairs. (B) Schematic of the SPC19/RAV2 and the CIN1/MNE1 gene pairs on their respective chromosomes. Distances are to scale. (C) qPCR analysis of mRNA levels of CTF13, CIN1, and SPC19 from sno1Δ, mne1Δ, and rav2Δ, respectively. Three, two, and four independent biological replicates were used for the CTF13 primer set (set #3), and MNE1, SPC19, respectively. Each value is normalized to the level of ACT1 mRNA. Technical triplicates are averaged and pooled together and standard deviation is calculated for means of the independent strains. P-values are generated from a Tukey’s post hoc test.
To ask whether other convergent gene pairs may be affected when one gene in the pair is deleted in the YKO, we measured RNA abundance in YKO mutants of two other convergent gene pairs: RAV2/SPC19 and MNE1/CIN1 (Figure 5B). The RAV2 and SPC19 gene pair is separated by 82 base pairs between their stop codons, while the MNE1 and CIN1 gene pair is separated by 30 base pairs. RNA was extracted from mne1Δ and rav2Δ strains freshly generated using pFA6a-KanMX in the BY4741 background and used primer sets in qPCR experiment for the neighboring convergent gene: CIN1 and SPC19, respectively. Both the CIN1 and SPC19 mRNA levels were reduced compared to the wild-type control when the neighboring genes mne1Δ or rav2Δ were deleted (Figure 5C). This amount of mRNA reduction is similar to what is observed for a freshly generated sno1Δ mutant in the same BY4741 background (Figure 5C). Importantly, this result suggests that the CTF13 mRNA perturbation is conserved among multiple budding yeast strain backgrounds in which SNO1 is deleted, as all other data were collected using the qCTF background. This neighboring gene reduction was significant in the case of the rav2Δ mutant, as is the case in the sno1Δ mutant. However, the CIN1 mRNA reduction in the mne1Δ strain was not significant, highlighting the complexity of this TTEF1 induced artifact. This result suggests that the cis effect observed for the SNO1-CTF13 gene pair likely occurs to other gene pairs in the YKO collection.
Discussion
We began our study by characterizing a potential role for SNO1 in faithful chromosome segregation, as the Sno1p is an uncharacterized protein that consistently appeared in screens using the YKO mutants to assay CIN and genome instability. From our initial experiments, it became clear that Sno1p is not required to suppress CIN, as a nonsense point mutation within the SNO1 ORF does not elevate the CIN rate. Furthermore, addition of a functional copy of the SNO1 ORF to a sno1Δ background does not decrease the high CIN rate. Evidence points to a specific effect of the KanMX deletion construct used in the YKO collection in elevating the CIN rate of the sno1Δ mutant, as three independent selectable markers (URA3, HphMX, and NatMX) also disrupting the SNO1 ORF did not increase CIN. By altering independent portions of the KanMX construct, we were able to pinpoint the increased CIN to a previously unappreciated artifact present within the YKO collection. The transcriptional terminator TTEF1 has an ectopic terminator sequence that appears to be read from the opposite strand such that the neighboring gene arranged in a convergent orientation to the deleted gene may have perturbed mRNA levels. The mRNA depletion artifact only occurs when the Kanamycin resistance gene is present upstream of the TTEF1, as this phenotype was undetectable or not dominant in the NatMX or HphMX context, respectively.
In the case of SNO1, the neighboring convergent gene CTF13 has truncated and reduced mRNA levels. This seems to underlie the increased CIN in a sno1Δ strain, as elevated CIN can be reduced by adding another copy of CTF13 in a separate genomic locus or by raising the CTF13 mRNA level via NMD inactivation. Furthermore, this effect appears to be specific to convergent gene pairs, as when we altered the KanMX orientation to be tandem with the CTF13 gene (as in the sno1Δ-Watson or sno1Δ6-Watson mutant), CIN was significantly reduced. This observation led us to focus on 3′ UTR composition and determine a difference in the quality of the 3′ ends of the CTF13 mRNA in the sno1Δ mutant, which is truncated and lacks a polyA tail, two factors likely affecting other aspects of the RNA regulation and translation. When CTF13 mRNA was polyadenylated as in the sno1Δ::Kan-TADH1 mutant, CIN was suppressed and CTF13 mRNA eventually reached wild-type levels, which correlated to elevated levels of the Ctf13p.
Given the CTF13 3′ UTR is naturally very long and likely encompasses the complete antisense SNO1 ORF sequence, it is tempting to speculate that the presence of the Sno1p may be irrelevant for chromosome segregation. Instead, the SNO1 ORF DNA sequence could act as a stabilizer or enhancer for the CTF13 mRNA. Recent studies have shed light on a novel mode of posttranscriptional control of convergent gene expression through the formation of RNA duplexes that sometimes contain the complete antisense ORF of one mRNA within the 3′ UTR of the convergent gene’s mRNA (Sinturel et al. 2015). However, this is not mechanistically relevant for our findings considering that several of the KanMX mutants we screened (i.e., sno1Δ6, snz1Δ2-Crick, and snz1Δ2) have completely unaltered SNO1 ORFs but still experience significantly elevated CIN. Furthermore, when a copy of the CTF13 ORF was added into an endogenous locus in a sno1Δ background, CIN was suppressed independent of the presence of a complete SNO1 ORF. These findings together argue against the necessity for the SNO1 ORF sequence in maintaining proper chromosome segregation.
Our data confirm previous findings that NMD plays a role in the downregulation of CTF13 mRNA level, as inactiving components of NMD increased the CTF13 mRNA level in wild-type, sno1Δ, sno1Δ::Kan-TADH1, and sno1Δ6 backgrounds. Given that these mRNAs have different CTF13 3′ UTRs as evidenced by 3′ RACE, it is unlikely that each of them would be a direct substrate for NMD. In fact, the natural CTF13 mRNA was previously proposed to be an indirect substrate of NMD, as the mRNA’s half-life was unchanged in the presence and absence of NMD (Dahlseid et al. 1998). It was suggested that transcripts encoding for proteins responsible for CTF13 transcription are depleted by NMD (Dahlseid et al. 1998). NMD inactivation results in differing levels of CTF13 mRNA in our mutant strains: the sno1Δupf1Δ double mutant has sub-wild-type CTF13 mRNA levels (although increased relative to the sno1Δ single mutant) but the sno1Δ::Kan-TADH1/upf1Δ double mutant has higher CTF13 mRNA than the wild-type strain. This difference may reflect that CTF13 transcription is increased equally in both strains, with extra transcripts retained efficiently in the sno1Δ::Kan-TADH1 strain due to the stable polyA tail, whereas extra transcripts are still subject to continuous instability in the sno1Δ strain.
NMD inactivation did not rescue CIN in sno1Δ3, which had an extra 97 base pair deletion compared to sno1Δ2 and sno1Δ6 combined. This 97 base pair region has predicted binding motifs for transcription factors encoded by ASH1, AZF1, CBF1, GCR1, PHO4, and PHO2 (De Boer and Hughes 2012; Monteiro et al. 2020). This sequence also contains the divergent promoter for SNO1 and SNZ1, and some of these transcription factors such as Pho4p are known to regulate the expression of this gene pair (Nishizawa et al. 2008). Of particular interest, GCR1 has a long intron that is known to be regulated by mRNA decay pathways including NMD (Kawashima et al. 2014). As such, GCR1 can encode for multiple RNA and protein isoforms that appear to be regulated by glucose availability (Hossain et al. 2016). This finding further highlights the complexity of gene regulation in proximity to CTF13.
One outstanding question is the identity of the sequences that may be read from the reverse direction when the KanMX ORF sequence is combined with the A. gossypii TTEF1 and what molecular players are involved in mRNA perturbation. A. gossypii possesses the smallest genome size of any free-living eukaryote at 9.7 megabases (Dietrich et al. 2004). The A. gossypii genome was found to be highly compacted containing only seven chromosomes with an average distance of 341 base pairs between ORFs. About 95% of A. gossypii genes have homologues in Saccharomyces cerevisiae. Compared to S. cerevisiae, the A. gossypii genome has increased gene density and this is believed to be achieved through shortening intergenic regions as opposed to shortening ORFs (Wendland and Walther 2011). The more streamlined and compact A. gossypii genome points for an even stronger need to prevent cryptic transcription from the closely spaced neighboring genes and as such it is reasonable to hypothesize that many A. gossypii terminators would function bidirectionally.
Although we are currently unsure of the necessary sequence elements within the KanMX cassette that contribute to the mRNA perturbation phenotype, a manual DNA sequence inspection found at least three Nab3p binding sites and one Nrd1p binding site within the TTEF1 sequence. Nrd1p and Nab3p function together with the Sen1p helicase to form an transcriptional termination complex that is utilized specifically for short transcripts such as snRNAs, snoRNAs, and cryptic unstable transcripts (Arndt and Reines 2015; Carroll et al. 2004; Steinmetz et al. 2001). Nrd1p and Nab3p bind the transcript as it emerges from the transcription bubble and act to recruit other factors such as the TRAMP complex and the nuclear exosome. Thus, it is possible that the exosome is targeted to the CTF13 transcript in the sno1Δ background. Indeed, conserved proteins with homology to budding yeast NRD1 and NAB3 are both annotated on the A. gossypii genome database (Gattiker et al. 2007), suggesting this pathway may also exist in A. gossypii. However, our results also suggest that the TTEF1 sequence was insufficient to cause CIN or reduced CTF13 mRNA quantity or quality for example when present in the sno1Δ::Nat mutant. An additional feature may be that the CTF13 mRNA in the original sno1Δ mutant contains portions of the distal end of the Kanamycin resistance gene ORF in the transcript.
Although the SNO1 YKO mutant was annotated as having elevated genome instability in each of three independent CIN screening tools (Yuen et al. 2007) as well as the qCTF assay as demonstrated in this study, another screen measuring gross chromosomal rearrangements found no difference in a SNO1-SNZ1 double KO mutant (Kanellis et al. 2007). Of note, this study deleted both genes together with a LEU2 auxotrophic selectable marker and the orientation of the expression of that marker is not reported. Nevertheless, we suspect that the reason this SNO1 KO mutant from this study does not have increased genomic instability is due to a difference in selectable markers (we do not detect the Nrd1p and Nab3p binding sites in the LEU2 terminator sequence), a difference in the expression orientation of this marker, or a combination of both scenarios.
The effect that we measured may be extrapolated to other gene pairs arranged in convergent orientations, as the two other example convergent gene pairs we tested had reduced mRNA levels of one gene when the other in the pair was deleted. There is at least one similar example in the literature with the convergent gene pair VPS36 and CDC73 on chrXII whose stop codons are separated by 203 base pairs of intergenic sequence. Deletion of all or portions of CDC73 with KanMX resulted in a subsequent decrease of VPS36 mRNA levels detected by qPCR (Rodrigues and Lydall 2018). This finding and our data point to an artifact that exists within the YKO collection effecting the interpretation of phenotypes of closely spaced convergent gene pairs. We have compiled a resource of all the genes arranged in a similar convergent/tail-to-tail orientation as a guide for future studies (Supplementary Table S5). Researchers studying YKO mutants for genes in this list should validate phenotypic observations through alternative methods of gene perturbation. To this end, the plasmid generated in this study (pFA6a-KanMX-TADH1) is particularly useful for the community as it contains the universal primer sites, F1 and R1, present on commonly used pFA6a based plasmids that can be used for PCR-based gene replacement.
Of further interest to the yeast research community, our finding raises the possibility of the construction of new partially defective budding yeast mutants in which the KanMX construct is inserted directly after the stop codon in a convergent orientation with specific genes of interest. The concept for these mutants is similar to the Decreased Abundance by mRNA Perturbation (DAmP) mutants (Breslow et al. 2008; Schuldiner et al. 2005), which place the KanMX construct in a tandem “tail to head” orientation with the gene of interest. In the case of the DAmP mutants, the mRNA perturbation is assumed to be due to the extension of the native 3′ UTR, which is known to perturb mRNA stability (Muhlrad and Parker 1999). The hypothetical convergent KanMX mRNA perturbation would result in an ectopic termination sequence coupled with 3′ UTR heterogeneity based off our current findings.
Finally, other researchers have suggested a concept known as the “neighboring gene effect” within the YKO collection and have created systematic ways to identify genes that may have incorrect annotations due to this problem (Ben-Shitrit et al. 2012). However, this resource does not mention a specific issue introduced by the combination of KanMX with the TEF1 terminator sequence, which can affect the mRNA of neighboring genes within convergent pairs and thus likely contributes to this neighboring gene effect. Without a full understanding of the underlying mechanisms, our findings offer a cautionary tale to the yeast research community.
Acknowledgments
The authors thank Karole D’Orazio, Boris Zinshteyn, and Rachel Green for advice on RNA biology and methods and Jeffrey Corden for helpful discussion related to mRNA degradation processes.
Funding
This work was supported partially by the grant R35 GM118172 from the National Institute of Health (NIH) to R.L. M.R.G was supported by a training grant, T32 GM007445, from the NIH to the Biochemistry, Cellular, and Molecular Biology (BCMB) graduate program at Johns Hopkins Medical Institute.
Conflicts of interest
None declared.
Literature cited
- Arndt K M, Reines D. 2015. Termination of Transcription of Short Noncoding RNAs by RNA Polymerase II. Annu Rev Biochem. 84:381–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhoum SF, Ngo B, Laughney AM, Cavallo J-A, Murphy CJ, et al. 2018. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature. 553:467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudin A, Ozier-Kalogeropoulos O, Denouel A, Lacroute F, Cullin C.. 1993. A simple and efficient method for direct gene deletion in Saccharomyces cerevisiae. Nucl Acids Res. 21:3329–3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shitrit T, Yosef N, Shemesh K, Sharan R, Ruppin E, et al. 2012. Systematic identification of gene annotation errors in the widely used yeast mutation collections. Nat Methods. 9:373–378. [DOI] [PubMed] [Google Scholar]
- Biggins S. 2013. The composition, functions, and regulation of the budding yeast kinetochore. Genetics. 194:817–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Boer CG, Hughes TR.. 2012. YeTFaSCo: a database of evaluated yeast transcription factor sequence specificities. Nucl Acids Res. 40:D169–D179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslow DK, Cameron DM, Collins SR, Schuldiner M, Stewart-Ornstein J, et al. 2008. A comprehensive strategy enabling high-resolution functional analysis of the yeast genome. Nat Methods. 5:711–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll KL, Pradhan DA, Granek JA, Clarke ND, Corden JL.. 2004. Identification of cis elements directing termination of yeast nonpolyadenylated snoRNA transcripts. Mol Cell Biol. 24:6241–6252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry JM, Hong EL, Amundsen C, Balakrishnan R, Binkley G, et al. 2012. Saccharomyces genome database: the genomics resource of budding yeast. Nucl Acids Res. 40:D700–D705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlseid JN, Puziss J, Shirley RL, Atkin AL, Hieter P, et al. 1998. Accumulation of mRNA coding for the ctf13p kinetochore subunit of Saccharomyces cerevisiae depends on the same factors that promote rapid decay of nonsense mRNAs. Genetics. 150:1019–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich FS, Voegeli S, Brachat S, Lerch A, Gates K, et al. 2004. The Ashbya gossypii genome as a tool for mapping the ancient Saccharomyces cerevisiae genome. Science. 304:304–307. [DOI] [PubMed] [Google Scholar]
- Dong YX, Sueda S, Nikawa JI, Kondo H.. 2004. Characterization of the products of the genes SNO1 and SNZ1 involved in pyridoxine synthesis in Saccharomyces cerevisiae. Eur J Biochem. 271:745–752. [DOI] [PubMed] [Google Scholar]
- Engel SR, Dietrich FS, Fisk DG, Binkley G, Balakrishnan R, et al. 2014. The Reference genome sequence of Saccharomyces cerevisiae: then and now. G3 (Bethesda). 4:389–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattiker A, Rischatsch R, Demougin P, Voegeli S, Dietrich FS, et al. 2007. Ashbya genome Database 3.0: a cross-species genome and transcriptome browser for yeast biologists. BMC Genomics. 8:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaever G, Chu AM, Ni L, Connelly C, Riles L, et al. 2002. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 418:387–391. [DOI] [PubMed] [Google Scholar]
- Gietz RD, Schiestl RH.. 2007. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc. 2:31–34. [DOI] [PubMed] [Google Scholar]
- Gray M, Piccirillo S, Honigberg SM.. 2005. Two-step method for constructing unmarked insertions, deletions and allele substitutions in the yeast genome. FEMS Microbiol Lett. 248:31–36. [DOI] [PubMed] [Google Scholar]
- Heidmann S, Obermaier B, Vogel K, Domdey H. 1992. Identification of pre-mRNA polyadenylation sites in Saccharomyces cerevisiae. Mol Cell Biol. 12:4215–4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JT, Demarest BL, Bisgrove BW, Su Y, Smith M, et al. 2014. Poly peak parser: method and software for identification of unknown indels using sanger sequencing of polymerase chain reaction products. Dev Dyn. 243:1632–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossain MA, Claggett JM, Edwards SR, Shi A, Pennebaker SL, et al. 2016. Posttranscriptional regulation of Gcr1 Expression and activity is crucial for metabolic adjustment in response to glucose availability. Mol Cell. 62:346–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanellis P, Gagliardi M, Banath JP, Szilard RK, Nakada S, et al. 2007. A screen for suppressors of gross chromosomal rearrangements identifies a conserved role for PLP in preventing DNA lesions. PLoS Genet. 3:e134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima T, Douglass S, Gabunilas J, Pellegrini M, Chanfreau GF.. 2014. Widespread use of non-productive alternative splice sites in Saccharomyces cerevisiae (H. D. Madhani, Ed). PLoS Genet. 10:e1004249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro PT, Oliveira J, Pais P, Antunes M, Palma M, et al. 2020. YEASTRACT+: a portal for cross-species comparative genomics of transcription regulation in yeasts. Nucl Acids Res. 48:D642–D649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhlrad D, Parker R.. 1999. Aberrant mRNAs with extended 3’ UTRs are substrates for rapid degradation by mRNA surveillance. RNA. 5:1299–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negrini S, Gorgoulis VG, Halazonetis TD.. 2010. Genomic instability an evolving hallmark of cancer. Nat Rev Mol Cell Biol. 11:220–228. [DOI] [PubMed] [Google Scholar]
- Nishizawa M, Komai T, Morohashi N, Shimizu M, Toh-e A.. 2008. Transcriptional repression by the Pho4 transcription factor controls the timing of SNZ1 expression. Eukaryot Cell. 7:949–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novarina D, Desai R, Vaisica JA, Ou J, Bellaoui M, et al. 2020. A genome-wide screen for genes affecting spontaneous direct-repeat recombination in saccharomyces cerevisiae. G3 Genes, Genomes, Genet. 10:1858–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novoa CA, Ang JS, Stirling PC.. 2018. The A-like faker assay for measuring yeast chromosome III stability, In: Methods Mol Biol. 1672:1-9: [DOI] [PubMed] [Google Scholar]
- Padilla PA, Fuge EK, Crawford ME, Errett A, Werner-Washburne M.. 1998. The highly conserved, coregulated SNO and SNZ gene families in Saccharomyces cerevisiae respond to nutrient limitation. J Bacteriol. 180:5718–5726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potapova TA, Zhu J, Li R.. 2013. Aneuploidy and chromosomal instability: a vicious cycle driving cellular evolution and cancer genome chaos. Cancer Metastasis Rev. 32:377–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puddu F, Herzog M, Selivanova A, Wang S, Zhu J, et al. 2019. Genome architecture and stability in the Saccharomyces cerevisiae knockout collection. Nature. 573:416–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues J, Lydall D.. 2018. Cis and trans interactions between genes encoding PAF1 complex and ESCRT machinery components in yeast. Curr Genet. 64:1105–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ.. 2008. Analyzing real-time PCR data by the comparative CT method. Nat Protoc. 3:1101–1108. [DOI] [PubMed] [Google Scholar]
- Schuldiner M, Collins SR, Thompson NJ, Denic V, Bhamidipati A, et al. 2005. Exploration of the function and organization of the yeast early secretory pathway through an epistatic miniarray profile. Cell. 123:507–519. [DOI] [PubMed] [Google Scholar]
- Scotto-Lavino E, Du G, Frohman MA.. 2006. 3′ End cDNA amplification using classic RACE. Nat Protoc. 1:2742–2745. [DOI] [PubMed] [Google Scholar]
- Sinturel F, Navickas A, Wery M, Descrimes M, Morillon A, et al. 2015. Cytoplasmic control of sense-antisense mRNA pairs. Cell Rep. 12:1853–1864. [DOI] [PubMed] [Google Scholar]
- Spencer F, Gerring SL, Connelly C, Hieter P.. 1990. Mitotic chromosome transmission fidelity mutants in Saccharomyces cerevisiae. Genetics. 124:237–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmetz EJ, Conrad NK, Brow DA, Corden JL.. 2001. RNA-binding protein Nrd1 directs poly(A)-independent 3′-end formation of RNA polymerase II transcripts. Nature. 413:327–331. [DOI] [PubMed] [Google Scholar]
- Stirling PC, Bloom MS, Solanki-Patil T, Smith S, Sipahimalani P, et al. 2011. The complete spectrum of yeast chromosome instability genes identifies candidate CIN cancer genes and functional roles for ASTRA complex components (M. Snyder, Ed.). PLoS Genet. 7:e1002057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng X, Dayhoff-Brannigan M, Cheng W-C, Gilbert CE, Sing CN, et al. 2013. Genome-wide consequences of deleting any single gene. Mol Cell. 52:485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uwimana N, Collin P, Jeronimo C, Haibe-Kains B, Robert F.. 2017. Bidirectional terminators in Saccharomyces cerevisiae prevent cryptic transcription from invading neighboring genes. Nucl Acids Res. 45:6417–6426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wach A. 1996. PCR-Synthesis of marker cassettes with long flanking homology regions for gene disruptions in S. cerevisiae. Yeast, 12, 259–265. [DOI] [PubMed] [Google Scholar]
- Wach A, Brachat A, Pöhlmann R, Philippsen P.. 1994. New heterologous modules for classical or PCR‐based gene disruptions in Saccharomyces cerevisiae. Yeast. 10:1793–1808. [DOI] [PubMed] [Google Scholar]
- Watkins TBK, Lim EL, Petkovic M, Elizalde S, Birkbak NJ, et al. 2020. Pervasive chromosomal instability and karyotype order in tumour evolution. Nature. 126–132. 587: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendland J, Walther A.. 2011. Genome evolution in the eremothecium clade of the saccharomyces complex revealed by comparative genomics. G3 Genes. G3 (Bethesda). 1:539–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yofe I, Schuldiner M.. 2014. Primers-4-Yeast: a comprehensive web tool for planning primers for Saccharomyces cerevisiae. Yeast. 31:77–80. [DOI] [PubMed] [Google Scholar]
- Yuen KWY, Warren CD, Chen O, Kwok T, Hieter P, et al. 2007. Systematic genome instability screens in yeast and their potential relevance to cancer. Proc Natl Acad Sci U S A. 104:3925–3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Heinecke D, Mulla WA, Bradford WD, Rubinstein B, et al. 2015. Single-cell based quantitative assay of chromosome transmission fidelity. G3 (Bethesda). 5:1043–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All yeast strains and plasmids are available upon request. Source code for the convergent gene pair mining from the SGD is available online (https://github.com/mgordo34/Convergent_Gene_Script). Supplementary Table S1 contains the genotypes of budding yeast strains used in this study and the sources of any strains not generated in the present study as well as the primer sets and templates DNA sequences used for homologous recombination transformations and aliases for relevant strains. Supplementary Table S2 contains a list of primer names and sequences used for PCR, 3′ RACE, and DNA sequencing. Supplementary Table S3 contains qPCR primer sequence sets, the ORFs they anneal to, and their experimentally calculated efficiencies. Supplementary Table S4 contains relevant plasmids developed and used during the course of this study. Supplementary Table S5 contains one of the tables generated from the source code for mining convergent gene pairs from the SGD. This table lists nonoverlapping convergent gene pairs, the number of base pairs between each stop codon, the chromosomal coordinates of each gene, and the chromosome they reside on. The source code also produces a table of overlapping convergent gene pairs. Supplemental Material is available at figshare: https://doi.org/10.25387/g3.14195846.