

This study demonstrates that the Pseudoalteromonas strain HM-SA03, isolated from the venomous blue-ringed octopus, Hapalochalaena sp., is a biosynthetically talented organism, capable of producing alterochromides and potentially six other specialized metabolites. We identified a pseudoalterobactin biosynthesis gene cluster and proposed a pathway for the production of the associated siderophore.
KEYWORDS: Pseudoalteromonas, Alteromonas, nonribosomal peptide, polyketide, siderophore, genome, antiSMASH, bacteriocin, lanthipeptide
ABSTRACT
Pseudoalteromonas species produce a diverse range of biologically active compounds, including those biosynthesized by nonribosomal peptide synthetases (NRPSs) and polyketide synthases (PKSs). Here, we report the biochemical and genomic analysis of Pseudoalteromonas sp. strain HM-SA03, isolated from the blue-ringed octopus, Hapalochlaena sp. Genome mining for secondary metabolite pathways revealed seven putative NRPS/PKS biosynthesis gene clusters, including those for the biosynthesis of alterochromides, pseudoalterobactins, alteramides, and four novel compounds. Among these was a novel siderophore biosynthesis gene cluster with unprecedented architecture (NRPS-PKS-NRPS-PKS-NRPS-PKS-NRPS). Alterochromide production in HM-SA03 was also confirmed by liquid chromatography-mass spectrometry. An investigation of the biosynthetic potential of 42 publicly available Pseudoalteromonas genomes indicated that some of these gene clusters are distributed throughout the genus. Through the phylogenetic analysis, a particular subset of strains formed a clade with extraordinary biosynthetic potential, with an average density of 10 biosynthesis gene clusters per genome. In contrast, the majority of Pseudoalteromonas strains outside this clade contained an average of three clusters encoding complex biosynthesis. These results highlight the underexplored potential of Pseudoalteromonas as a source of new natural products.
IMPORTANCE This study demonstrates that the Pseudoalteromonas strain HM-SA03, isolated from the venomous blue-ringed octopus, Hapalochalaena sp., is a biosynthetically talented organism, capable of producing alterochromides and potentially six other specialized metabolites. We identified a pseudoalterobactin biosynthesis gene cluster and proposed a pathway for the production of the associated siderophore. A novel siderophore biosynthesis gene cluster with unprecedented architecture was also identified in the HM-SA03 genome. Finally, we demonstrated that HM-SA03 belongs to a phylogenetic clade of strains with extraordinary biosynthetic potential. While our results do not support a role of HM-SA03 in Hapalochalaena sp. venom (tetrodotoxin) production, they emphasize the untapped potential of Pseudoalteromonas as a source of novel natural products.
INTRODUCTION
The genus Pseudoalteromonas, formerly classified as Alteromonas (1), encompasses a group of Gram-negative Gammaproteobacteria commonly found in seawater and marine sediments and in epiphytic associations with seaweed, sponges, and ascidians. Pigmented species, in particular, produce a wide variety of bioactive compounds (specialized metabolites) of ecological and pharmaceutical significance, including numerous broad-spectrum antibiotics and potent siderophores (2).
The vast majority (∼46%) of antimicrobial compounds isolated from Pseudoalteromonas thus far are alkaloids (3); however, recent genome mining studies suggest that this genus may also be a rich source of nonribosomal peptides (NRPs), polyketides (PKs), and hybrid compounds (4–6). NRPs and PKs are a structurally diverse group of specialized metabolites synthesized nonribosomally by large multimodular enzyme complexes known as nonribosomal peptide synthetases (NRPSs) and polyketide synthases (PKSs) (7). They are produced by numerous species of bacteria and filamentous fungi and have a wide range of bioactivities, including antibiotic, immunosuppressant, anticancer, and anticholesteremic activities.
The building blocks of NRPs (amino acids) and PKs (acyl-CoAs) are incorporated in an assembly line fashion by NRPSs and PKSs, respectively. These enzyme complexes are minimally comprised of an initiation domain, an extension domain, and a termination domain but may include various other tailoring domains (e.g., methyltransferase, epimerase, reductase, etc.) (7). NRPSs and PKSs are usually encoded within large operon-like gene clusters alongside additional stand-alone tailoring and transport enzymes.
Surprisingly, very few NRPs/PKs from Pseudoalteromonas have been matched to their corresponding biosynthesis gene clusters (BGCs). The best-characterized examples are the alterochromides (4, 8), thiomarinols (9–11), indolmycin (12), and pentabromopseudilin (13), which display broad-spectrum antibiotic activity, and pseudochelin (14), which has siderophoric properties. Further genomic investigation of Pseudoalteromonas is likely to uncover additional NRPS/PKS biosynthesis pathways, which could provide valuable insight into the success of this genus in marine econiches and facilitate the discovery and development of novel bioactive compounds.
The reduction in cost of genome sequencing coupled with faster and more powerful bioinformatic methods has expedited the unearthing of novel natural products and their BGCs in numerous other microbial genera (15). In general, more BGCs exist in an organism than the number of known compounds reported from that organism. This is highlighted by the reports of the Salinispora tropica and Salinispora arenicola genomes (16, 17), which revealed 49 natural product biosynthesis clusters and assisted in the structure elucidation of the polyene macrolactam salinilactam A (16). Such genomic approaches have also been used to great success in the actinobacterium Actinosynnema mirum, where genome-guided approaches facilitated the discovery of an unusual siderophore and the first reported BGC from that species (18).
In the present study, we use similar genome-based approaches to facilitate the discovery of new specialized metabolite BGCs in a novel strain, Pseudoalteromonas sp. HM-SA03, isolated from the venomous (tetrodotoxin-producing) blue-ringed octopus, Hapalochalaena sp. Extracts of HM-SA03 were previously shown to inhibit the growth of Staphylococcus aureus, and molecular screening revealed that the strain possesses multiple NRPS and PKS genes, which could potentially be involved in tetrodotoxin biosynthesis (19). Given that Pseudoalteromonas spp. are known to produce bioactive natural products and that relatively few NRPS or PKS biosynthesis pathways have been discovered in this genus, we sequenced the HM-SA03 genome with the aim of comprehensively assessing its potential for specialized metabolite production. Interrogation of the HM-SA03 genome revealed numerous complex BGCs encoding NRPSs and PKSs and enabled prediction of their corresponding natural products, including pseudoalterobactins, alterochromides, and several novel compounds. A phylogenomic analysis of 42 publicly available Pseudoalteromonas genomes additionally revealed that HM-SA03 belongs to a subclade of Pseudoalteromonas species with remarkable biosynthetic potential.
RESULTS AND DISCUSSION
HM-SA03 genome annotation and complex biosynthesis pathway mining.
The HM-SA03 genome assembly produced using SOAPdenovo, using a k-mer value of 71, resulted in a 5,248,267-bp assembly consisting of 119 scaffolds and 494 unscaffolded contigs with an N50 value of 106,644 bp and a maximum contig length of 182,387 bp. The GC content of the genome was estimated to be 43.1%. By comparison, the next best assembly was performed with Velvet using a k-mer value of 63, producing a genome size of 5,218,927 bp consisting of 91 scaffolds and 298 unscaffolded contigs with an N50 value of 101,219 bp and maximum contig length of 165,931 bp. The SOAPdenovo assembly was chosen for further analyses because it resulted in more and longer scaffolds than those generated using Velvet.
Gene detection and annotation were performed via the Rapid Annotation using Subsystem Technology (RAST) Server, resulting in the prediction of 4,735 protein-encoding genes and 90 RNAs. Through a combination of software-assisted (antiSMASH, 2metDB) and manual annotation, a total of nine BGCs were identified, namely, two bacteriocin clusters, one NRPS cluster, four NRPS-PKS hybrid clusters, one aryl polyene/NRPS, and one lanthipeptide/NRPS cluster. Some of the NRPS/PKS genes from HM-SA03 and their associated BGCs share significant homology to those from published Pseudoalteromonas genomes. However, structure prediction and biosynthetic pathway analyses have not been performed on these gene clusters, which are thus still considered “orphans.”
Because HM-SA03 was isolated from the venomous blue-ringed octopus, we hypothesized that it could be a primary producer of tetrodotoxin (19). We therefore carefully scrutinized the nine BGCs for amidinotransferases and NRPSs incorporating arginine, two plausible mechanisms for the biosynthesis of the tetrodotoxin guanidinium moiety (20). However, these genes were not detected. Our results concur with the lack of tetrodotoxin production in HM-SA03 cultures (19). However, we cannot discount the possibility that the genes for tetrodotoxin biosynthesis are unusual and therefore beyond the detection and analysis capabilities of antiSMASH and 2metDB.
Characterized biosynthesis gene clusters in the Pseudoalteromonas HM-SA03 genome.
Alterochromides. Mining of the HM-SA03 genome revealed an ∼30 kb (14-open reading frame [ORF]) gene cluster encoding fatty acid synthases, NRPSs and several tailoring and transport enzymes (Fig. 1; see Table S1 in the supplemental material). The gene cluster had an identical composition and arrangement to the alterochromide (alt) gene cluster of Pseudoalteromonas piscicida JCM 20779 (4) and an overall inferred amino acid sequence similarity of >97% (Table S1). It was thus concluded that the newly identified HM-SA03 gene cluster encoded an alterochromide biosynthesis pathway.
FIG 1.

Alterochromide (alt) gene cluster from HM-SA03, ∼30 kb. For MIBiG, BLASTp, and CD-Search results, see Table S1.
Amino acid substrate specificity predictions, based on analysis of the adenylation domain substrate-binding pockets of the three encoded NRPSs (AltK, AltL, and AltM) indicated that they were most likely to incorporate threonine, valine, two asparagines, and a leucine moiety. The amino acid composition of their predicted product showed similarities to the peptide-derived component of alterochromides from the sponge isolate Pseudoalteromonas maricaloris KMM 636T (8).
Mass spectrometry evidence confirmed the production of alterochromides A and B in culture extracts of HM-SA03 (Fig. 2), and although no brominated alterochromides were detected, this suggests that the alterochromide gene cluster (alt) in HM-SA03 is indeed functional. While the cluster does encode a putative halogenase, AltN, the absence of brominated alterochromides in this study is likely due to the lack of bromide supplementation in the fermentation medium. The biosynthetic pathway mirrors that reported by Moore and coworkers (4) in which the assembly is colinear with respect to gene architecture.
FIG 2.

Positive mode electrospray ionization (ESI) mass spectra of HM-SA03 crude extract. Sodiated parent ions indicate the production of des-brominated alterochromides A and B by Pseudoalteromonas HM-SA03. Similar fragmentation patterns are observed between alterochromide A and alterochromide B, indicating a common structure.
The assembly of the nonribosomal peptide portion of alterochromide, encoded by altK, altL, and altM, appears to be straightforward; however, the lipoinitiation and NRPS loading steps are less obvious (Fig. 3). The lipid precursor is putatively biosynthesized by AltA to AltJ from tyrosine, which is deaminated to provide coumaric acid that then undergoes chain extension and partial reduction to form the fatty acyl starter unit for the NRPS pathway. We propose that the predicted centrally located type II thioesterase, AltJ, serves to police the chain length of the fatty acid-derived starter units prior to uploading onto the NRPS complex (21). AltJ may also have relaxed substrate specificity, thus accounting for various-length starters observed in the alterochromides.
FIG 3.

Biosynthetic pathway for the production of alterochromide A in HM-SA03. Tyrosine is converted to coumaric acid by tyrosine ammonia lyase (TAL) and undergoes malonyl extension and is reduced to form the starter unit (R) for further NRPS-mediated biosynthesis.
The variability of the lipid chain length in alterochromides could also be due to the promiscuity of the initiation C domain. The relaxed specificity of a starter C domain toward fatty acyl-CoA substrates has also been observed in the biosynthesis of the lipopeptide antibiotic, calcium-dependent antibiotic (CDA) (22).
Pseudoalterobactin. A putative gene cluster for the biosynthesis of pseudoalterobactin was identified in HM-SA03 (Fig. 4, Table S2). A biosynthetic gene cluster for either pseudoalterobactin (23) or the structurally similar alterobactin (24) has never been identified, let alone characterized. The proposed biosynthetic gene cluster for pseudoalterobactin (pab) spans 53 kb and contains seven genes encoding NRPSs and one encoding a type I PKS. Two cassettes putatively encoding chorismate and 2-isopropylmalate biosynthesis flank the NRPS/PKS genes. Three genes, pabQOM, likely encoding the biosynthesis of the 2,3-dihydroxybenzoate (DHB) starter unit are also present in the gene cluster. Numerous siderophore and iron receptor, regulation, and transport proteins, PabDEKLR, are also encoded within the proposed gene cluster. The presence of an MbtH domain-containing protein reinforces the classification of this genomic locus as an NRPS-dependent siderophore BGC. The MbtH domain-containing protein, PabA, which is required in many NRPS-dependent siderophore biosynthesis pathways (25) is thought to be essential for the correct biosynthesis of siderophores in vivo.
FIG 4.
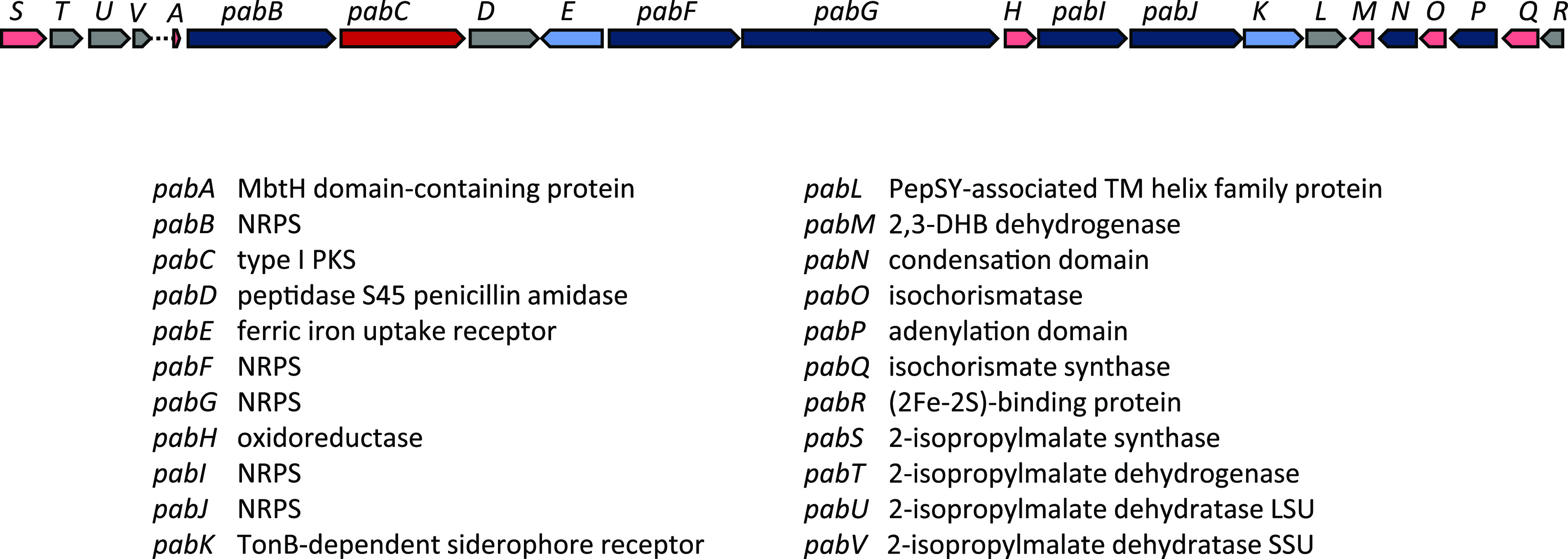
Pseudoalterobactin (pab) gene cluster from HM-SA03, ∼53 kb. For MIBiG, BLASTp, and CD-Search results, see Table S2.
The starter unit DHB is presumably activated by a CoA-ligase domain located at the N terminus of the NRPS protein PabB and subsequently condenses with l-lysine before undergoing PKS and NRPS catalyzed chain extensions encoded by pabBCFGIJ. Finally, the terminal PabJ thioesterase catalyzes the cyclization and release of the peptide chain from the complex to yield the final pseudoalterobactin product (Fig. 5). A consensus for the substrate specificity of the second adenylation domain of PabG was unable to be achieved and is likely to result in broad substrate specificity.
FIG 5.

Proposed biosynthetic pathway of pseudoalterobactin in HM-SA03. PabI is proposed to function iteratively to incorporate two hydroxyaspartic acid residues into the compound. A lack of hydroxylation enzymes and a presence of 2-isopropylmalate metabolism enzymes indicate the intact incorporation of hydroxyaspartic acid, rather than a downstream (postincorporation) hydroxylation.
Intriguingly, the activation of the DHB starter unit seems to be encoded by a redundant set of proteins, PabP, PabO, and PabN, whose genes are adjacent to the DHB biosynthesis genes, downstream and in the reverse orientation to the NRPS and PKS genes (Fig. 4). PabP is an adenylation domain-containing protein with substrate specificity for DHB. PabO encodes isochorismatase (2,3-dihydro-2,3-dihydroxybenzoate synthetase) and also contains a thiolation domain. This domain may be involved in the tethering of DHB to the NRPS. PabN encodes a condensation domain with homology to starter-type domains. These starter condensation domains have substrate specificity for unusual starter units, including benzoates and fatty acids. It is unknown at this stage whether one or both of these alternative pathways for DHB incorporation are functional.
Another unusual feature of this gene cluster is the proposed iteration of PabI, which is responsible for the activation and tethering of aspartic acid onto the NRPS. Unlike most NRPS modules, PabI does not contain a functional condensation domain. The PabI condensation domain is believed to be inactive, due to a mutation in the second histidine of the conserved HHxxxDG motif, which is critical to the correct function of the catalytic domain. However, both PabF and PabG have terminal condensation domains, which are proposed to replace the inactive condensation functionality of PabI (Fig. 5). The adenylation domains preceding the terminal condensation domains are both selective for amino acids with carbonyl-containing side chains. Such an iterated pathway adheres exactly with the backbone structure of pseudoalterobactin.
The hydroxylation of the PabI-activated aspartate is proposed to be catalyzed by PabH, a SyrP homologue. SyrP, has been shown to be responsible for the α-ketoglutarate-dependent hydroxylation of aspartate in syringomycin biosynthesis (26). Additionally, a set of four genes located upstream from NRPS genes, pabSTUV, are responsible for the metabolism of 3-isopropylmalate, which is structurally similar to hydroxyaspartic acid, with an isopropyl group substituting for an amine. These enzymes may act upon the two hydroxy-aspartic acid residues to give rise to hitherto unknown analogues.
Though some reported pseudoalterobactins are sulfated at the para position of the aromatic ring, there is no obvious enzyme encoded by the pab gene cluster in HM-SA03 to catalyze this sulfur transfer tailoring reaction. A proposed cysteine desulfurase, located 10 kb downstream from the last NRPS gene, may provide sulfur to the pseudoalterobactins, though the distance from the NRPS may render this unfeasible. Alternatively, an enzyme acting in trans and, therefore, not clustered with the NRPS/PKS genes, may be involved in the sulfonation of pseudoalterobactins.
Alteramide. One of the smallest BGCs in HM-SA03, alm, encodes a hybrid NRPS-PKS (Fig. 6, Table S3). Genome mining identified a gene encoding an NRPS module with ornithine adenylation specificity and a hybrid iterative type I PKS module. Manual annotation of the genes flanking the hybrid NRPS-PKS revealed two FAD-dependent oxidoreductases (phytoene dehydrogenase superfamily) and a hydroxylase (sterol desaturase/sphingolipid hydroxylase, fatty acid hydroxylase superfamily). The antiSMASH results indicated that these genes, including the hybrid NRPS-PKS were homologous to those involved in the biosynthesis of polycyclic tetramate macrolactams (27–29).
FIG 6.

Alteramide (alm) gene cluster from HM-SA03, ∼17 kb. For MIBiG, BLASTp and CD-Search results, see Table S3.
Intriguingly, the PKS modules of the hybrid NRPS-PKS involved in the biosynthesis of these macrolactams were proposed to be a hybrid iterative type I PKS. These PKSs differ from their modular counterparts because they assemble polyketide chains via a cyclical process, similar to type II PKS systems. Iterative type I PKSs are common in fungal polyketide biosynthesis but relatively uncommon in bacteria. However, a study by Clardy and coworkers (27) identified these gene clusters in multiple bacterial genomes, including the frontalamide-producing Streptomyces. The architecture of the alm gene cluster in HM-SA03 is similar to other gene clusters for frontalamide-like compounds. All of these clusters consist of a hybrid NRPS-PKS gene encoding malonyl and ornithine amino acid specificity, at least one phytoene dehydrogenase located downstream from the NRPS-PKS, and a hydroxylase. A key difference is the location of the hydroxylase enzyme, which is usually found upstream of the hybrid NRPS-PKS but is downstream from the hybrid NRPS-PKS in the alm cluster. Additionally, an alcohol dehydrogenase and cytochrome P450 monooxygenase, which are present in many gene clusters for frontalamide-like compounds, were absent from the alm gene cluster.
A two-stage, iterative, polyketide biosynthesis pathway for frontalamide derived from a fungal iterative type I PKS (ftl) has been proposed based on a combination of gene cluster analysis and biosynthetic precedents (27). Due to the similarity of the ftl and alm gene clusters, a similar pathway is proposed for alteramide A assembly in HM-SA03 (Fig. 7). Successive rounds of PKS biosynthesis produce partially reduced polyketides that are condensed onto the two amino groups of ornithine. The identification of an iterative type I PKS module in HM-SA03 represents only one of a few iterative type I PKS identified in gammaproteobacteria. The peptide-polyketide chain is then cyclized internally and released. Further cyclizations between the polyketide chains produce the 5-membered rings of alteramide.
FIG 7.
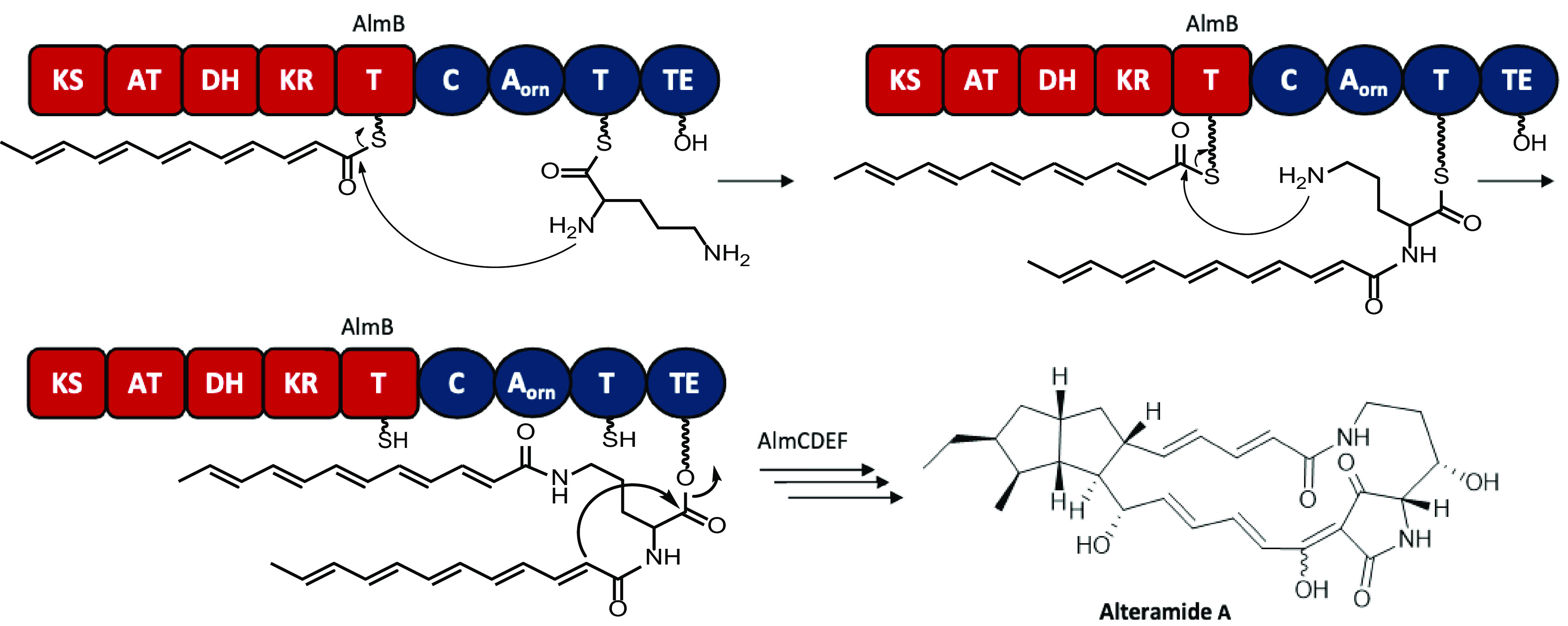
Proposed biosynthesis pathway of alteramide A in HM-SA03. Successive rounds of PKS biosynthesis produce partially reduced polyketides that are condensed onto the two amino groups of ornithine. The peptide-polyketide chain is cyclized internally and released. Further cyclizations between the polyketide chains produce the 5-membered rings of alteramide A. Stereochemistry of alteramide A is from reference 56.
Orphan and unknown biosynthesis pathways.
An unusual hybrid NRPS-PKS. A second siderophore biosynthesis pathway, encoded by the newly named sid gene cluster (Fig. 8, Table S4) is proposed to produce a compound with no structural homology to any known siderophores. Unusually, this gene cluster consists of alternating NRPS and PKS modules. Such genetic architecture in hybrid NRPS-PKS biosynthesis is unprecedented.
FIG 8.

Hybrid PKS-NRPS siderophore (sid) gene cluster from HM-SA03, ∼44.9 kb. For MIBiG, BLASTp, and CD-Search results, see Table S4.
The predicted siderophore is derived from a salicylic acid starter unit whose formation is catalyzed by an isochorismate synthase, SidH, which converts chorismate to isochorismate, and SidR, which hydrolyzes the isochorismate side chain (Fig. 9). We propose that salicylic acid is activated by SidI, a putative NRPS. Though the substrate specificity of the first adenylation domain of SidI was not predictable, its proximity to salicylate biosynthesis genes suggests a role in the adenylation of salicylic acid. Similarly, in Pseudomonas aeruginosa PAO1, phcBA encodes salicylate biosynthesis and is clustered with genes involved in the initial steps of pyochelin biosynthesis (30).
FIG 9.

Biosynthesis pathway for a putative siderophore encoded within a hybrid NRPS-PKS gene cluster in HM-SA03. The architecture of this gene cluster is unusual due to the alternation between NRPS and PKS modules.
sidJ-P encode alternating PKS and NRPS genes, which are predicted to successively incorporate cysteine, acetate, threonine, acetate, cysteine, acetate, and threonine. Numerous examples of mixed NRPS-PKS biosynthesis gene clusters are known; however, the occurrence of six alternating NRPS and PKS proteins, which are each encoded by discrete genes, is unique. It is also plausible that the assembly of the sid product is noncolinear with respect to the gene architecture in this case.
The cysteine-incorporating SidI and SidM NRPS modules also possess cyclization (Cy) domains which facilitate the formation of the thiazolines. SidM also possesses an oxidase that likely oxidizes the thiazoline to its corresponding thiazole. The PKS SidL contains an O-methyltransferase, which methylates threonine. While rare, O-methylated threonine residues have been reported in several other natural products, including depsipeptides (31, 32). The mature hybrid peptide-polyketide chain is released from the NRP via a thioesterase domain. The peptide-polyketide chain can be released as a linear acid or cyclized to form a heterocyclic peptide. In the case of the sid cluster product, the predicted linear peptide is shown (Fig. 9), as the cyclized product cannot be predicted with confidence.
Numerous examples of phenolate-containing siderophores with thiazoles exist, including yersiniabactin (33) and pyochelin (30). Assuming collinearity, an assembled compound as presented here is likely to be a novel scaffold.
Other gene clusters. The remaining three NRPS/PKS biosynthetic gene clusters identified in the HM-SA03 genome appear to encode chemical structures that have no similarity to previously identified compounds (Fig. S1). All three of these gene clusters encode NRPS biosynthesis, putatively, a heptapeptide NRP, a hybrid NRP-PK, and a hybrid lanthipeptide-NRP. MIBiG, BLASTp, and CD-Search results for the individual genes comprising these BGCs are appended in Tables S5, S6, and S7; putative linear peptides are appended in Table S8. The presence of numerous amino acid residues with potential iron-coordinating groups in all three structures suggests their possible roles as siderophores. However, the lack of siderophore and iron regulatory genes offers no support to these predictions. Furthermore, many of these gene clusters contain adenylation domains with unknown substrate specificities. This ambiguity in adenylation domain substrate prediction arises due to difficulties differentiating similar amino acid side chains (e.g., aspartate and asparagine) or if the adenylation domain utilizes an unusual substrate that has no precedents in other NRPS biosynthesis pathways, such as a nonproteinogenic amino acid. These ambiguous amino acid specificities challenge chemical structure predictions in these gene clusters. Lanthipeptide-NRP hybrid gene clusters were previously reported within Actinobacteria; however, their unique biosynthesis is yet to be elucidated. It is currently suggested that there might be cross talk between ribosomally synthesized lanthipeptides and NRPSs to form hybrid products (34). This is based on a similar process observed with pheganomycin biosynthesis, Streptomyces cerratus, where the linking of the two precursors is catalyzed by the peptide ligase Pgm1 (35). Despite these observations, there are limited precedents within the literature to aid the elucidation of the aforementioned cluster in HM-SA03 and whether it produces a hybrid product.
Pseudoalteromonas HM-SA03 is a member of a biosynthetically potent clade.
Several gene clusters identified in HM-SA03 were homologous to those found in other Pseudoalteromonas strains. Large numbers of biosynthetic pathways have been reported from actinobacteria, myxobacteria, and cyanobacteria; however, the biosynthetic potential of gammaproteobacteria, including the genus Pseudoalteromonas, has been largely overlooked. Therefore, mining and comparison of biosynthetic gene clusters from 42 Pseudoalteromonas genomes archived in GenBank was performed.
Genome sizes range from 3.4 to 6.2 Mbp, and our survey suggests that genome size is positively correlated with the number of specialized metabolite gene clusters (Fig. 10). Such correlation between genome size and biosynthetic potential has been documented for other biosynthetically potent taxa, including actinobacteria (36) and cyanobacteria (37).
FIG 10.

Correlation between genome size and number of NRPS/PKS gene clusters in Pseudoalteromonas species. Genomes represented by red circles are members of a highly biosynthetically potent (HBP) phylogenetic clade.
Based on a phylogenetic reconstruction of 16S rRNA genes, a highly biosynthetically potent (HBP) clade was identified. Nineteen sequenced strains, each containing an average of 9.84 specialized metabolite gene clusters grouped into this clade (Fig. 11). In contrast, members outside the HBP clade had an average of 2.78 BGCs. Three Pseudoalteromonas members of the HBP clade, P. spongiae UST010723-006, P. piratica OCN003, and P. spongiae SAO4-4, lacked genes encoding NRPS/PKS pathways. Although the 16S rRNA gene phylogeny of these species supports their position within the HBP clade, P. spongiae UST010723-006 has numerous morphological differences compared to its nearest relatives, including an absence of flagella and motility (38).
FIG 11.
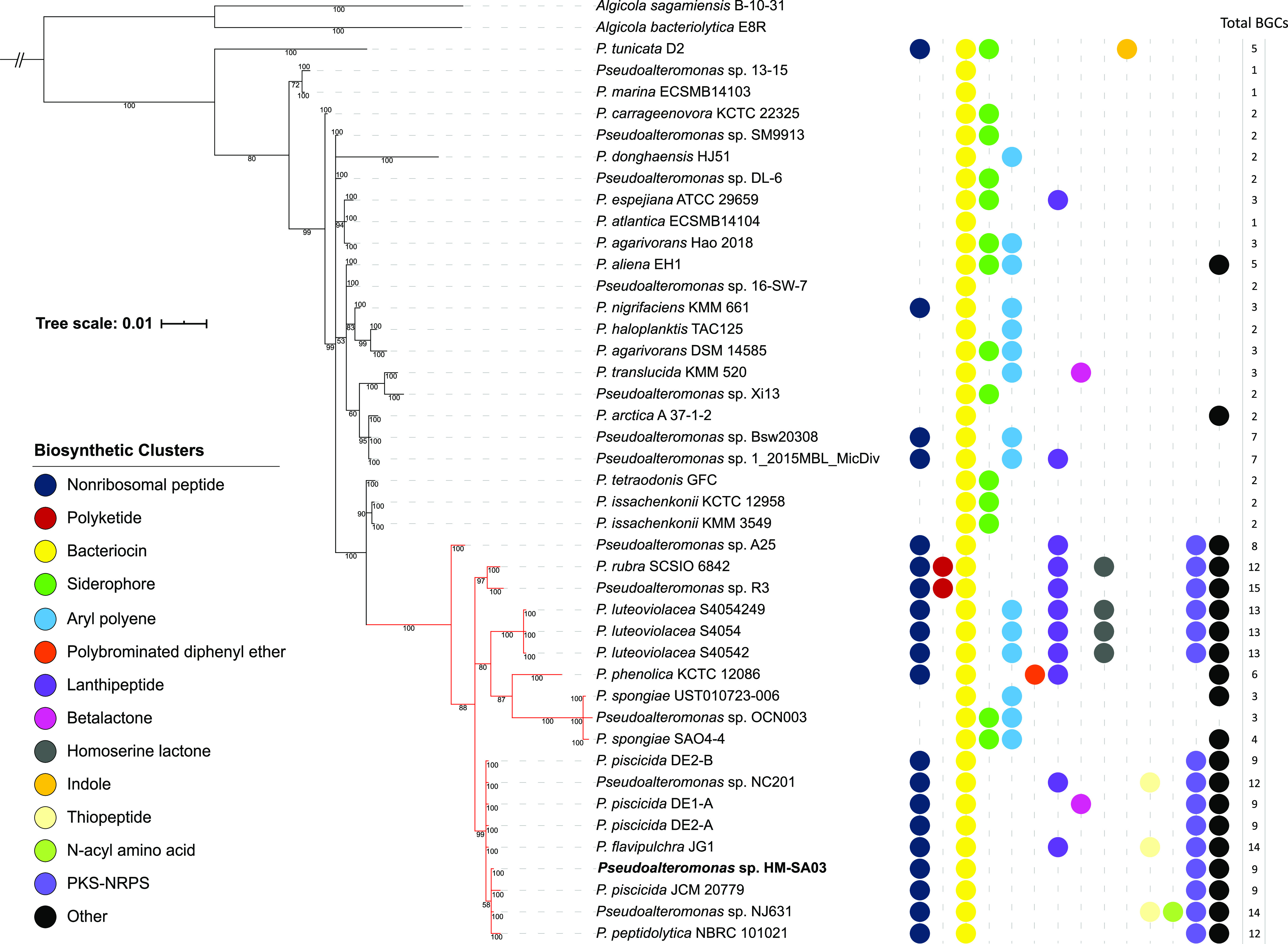
Phylogenetic reconstruction of Pseudoalteromonas 16S rRNA genes and relative distribution of biosynthesis gene clusters in this genus. Algicola sequences were used as artificial outgroups. The Pseudomonas sp. HM-SA03 sequence is bolded. The highly biosynthetically potent (HBP) clade has red branches. Colored circles indicate the presence of putative BGCs in the corresponding genome as predicted by antiSMASH. Scale represents nucleotide substitutions per base pair. Bootstrap values at nodes are given as percentages.
Therefore, this may be an example where the observed phenotypic morphology is contrary to the 16S rRNA phylogeny. No known natural products have been isolated from these strains; however, experiments have shown that P. spongiae UST010723-006 biofilms have the ability to promote the attachment of sponge larvae (39), while P. piratica OCN003 has been shown to be a coral pathogen causing Montipora white syndrome (40).
Scrutiny of the genetic architecture of NRPS/PKS gene clusters from members of the HBP clade revealed that many were conserved, particularly within the inner clade (Fig. 11). Out of a total of 36 unique biosynthetic pathways, 18 were observed to occur in more than one strain within this inner clade. In particular, four pathways (alterochromides, alteramide, and two unknown) were present in a majority of sequenced members of this clade (Fig. 12). Interestingly, the HBP clade coincides exactly with pigmentation in Pseudoalteromonas. It has long been known that pigmentation in this genus is an indicator of the production of bioactive compounds. Therefore, it is likely that these biosynthetic pathways may be responsible, in part, for the pigmentation observed.
FIG 12.
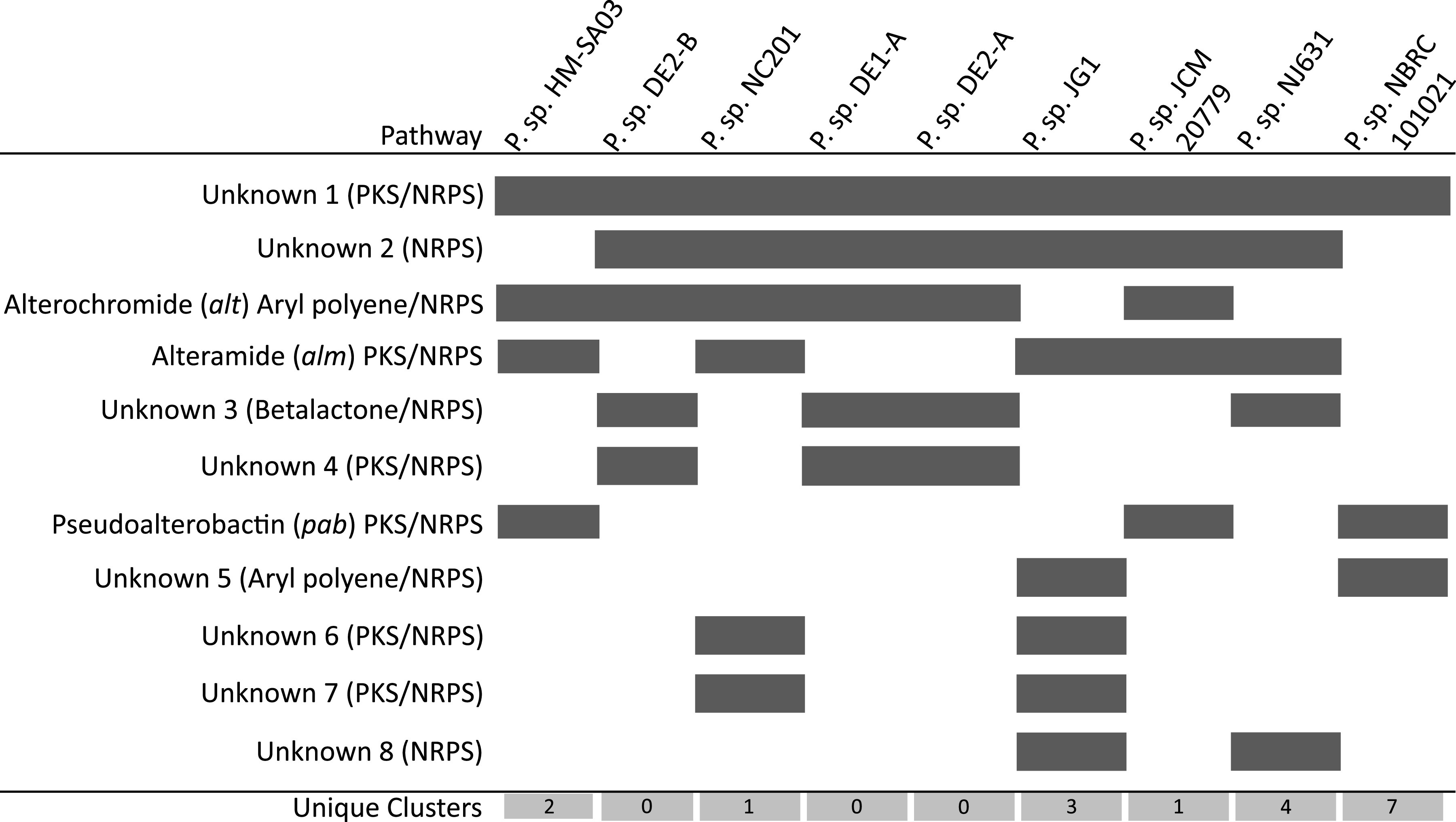
Conserved NRPS/PKS biosynthetic pathways in inner HBP clade Pseudoalteromonas genome sequences.
A highly biosynthetically potent clade of Pseudoalteromonas, as identified in this study, is supported by examples in the literature, including P. luteoviolacea, which is known to produce thiomarinols, xenorhabdins, violacein, and various other secondary metabolites (3). Furthermore, a recent study (5) investigated the biosynthetic potential of Gram-negative bacteria, including Pseudoalteromonas spp. The genomes of pigmented species, P. luteoviolacea, P. piscicida, and P. rubra, which are related to members of the HBP clade identified in this study, were observed to have between four and eight NRPS/PKS gene clusters. Conversely, NRPS/PKS gene clusters were not detected in P. agarivorans and P. ruthenica, which are unrelated to members of the HBP clade. Additionally, an analysis of the pangenome of various Pseudoalteromonas species from Antarctic regions found that they not only lacked pigmentation, but also had far fewer BGCs in their genomes compared to pigmented strains, particularly those in the HBP clade (41).
The identification of members from the proposed HBP clade assists in screening of Pseudoalteromonas strains with a higher potential for NRPS/PKS pathways, as well as a means of dereplicating strains for further chemical and bioassay investigations. Very few of the Pseudoalteromonas species in this clade have been investigated for bioactive natural products, and a majority of the BGCs are orphans. Therefore, members of this clade represent a new source for the discovery of novel bioactive small molecules.
Conclusions.
The results of this study highlight the significant biosynthetic potential of the genus Pseudoalteromonas for the production of specialized metabolites. The identification of seven BGCs associated with the production of PKS and NRPS products in the blue-ringed octopus isolate, HM-SA03, renders it a part of a group of sequenced Pseudoalteromonas strains with rich biosynthetic potential. Bioinformatics-assisted structure prediction of the products encoded by these gene clusters putatively characterizes the biosynthesis of alterochromide (NRP)-, alteramide (NRP-PK, alkaloid)-, and pseudoalterobactin (NRP-PK, siderophore)-like compounds. Furthermore, this study identified four gene clusters with no known homology to characterized BGCs, and their products could also therefore be novel. Unfortunately, no tetrodotoxin BGC was identified in the HM-SA03 genome, suggesting that this compound is produced by another symbiotic microorganism or by the blue-ringed octopus itself. Nonetheless, a highly biosynthetically potent clade of Pseudoalteromonas has been identified by this research. Members of this clade contain up to 10 NRPS/PKS per genome and represent an excellent phylogenetic target for the isolation of bioactive compounds.
MATERIALS AND METHODS
Sample preparation and genome sequencing.
Pseudoalteromonas sp. HM-SA03 (19) was grown in 0.5% peptone in filtered seawater at 23°C for 24 h. The cell culture was centrifuged at 4,200 × g, and a subset of the biomass was used for DNA extraction as previously described (42).
Genome sequencing and comparative analyses were performed at the Ramaciotti Centre for Genomics. Genomic DNA was sequenced using the Illumina HiSeq system following the manufacturer’s standard protocol. The sample was prepared using the Illumina paired-end sample preparation kit, and the library was purified using a QIAquick PCR purification kit (Qiagen). The sample was run at 8 pM of paired-end 102-bp chemistry. The run was performed using the genome analyzer Sequencing Control Software (SCS) v2.6 (Illumina).
HM-SA03 genome assembly.
The SolexaQA package (43) was used to trim reads to the longest contiguous read segment above a 0.05 P value. Quality-trimmed reads shorter than 50 bp were discarded. De novo genome assembly was performed with SOAPdenovo (44) using k-mer values between 21 and 91. These k-mer values represent the minimum read overlap during the assembly of contiguous DNA sequences (contigs). Contigs shorter than 200 bp were discarded from the final assembly. The final genome assembly was submitted to the NCBI database under accession number PRJNA400113.
Gene prediction and annotation.
The HM-SA03 draft genome was submitted to Integrated Microbial Genomes (IMG) for gene prediction and annotation (45). Additionally, secondary metabolite biosynthesis clusters were identified using a combination of 2metDB (46) and antiSMASH v5.1.2 (47, 48). Both software packages used profile hidden Markov models (pHMMs) of known biosynthesis gene domains to identify secondary metabolite genes and their domain architecture in query sequences. Substrates for PKS ketosynthase, NRPS adenylation (A), and CoA ligase domains were also predicted using these programs. All secondary metabolite gene clusters retrieved were manually checked, and further confirmation of domain architecture was performed using NCBI Conserved Domain Database (CDD) search (CD-Search) (49, 50).
Phylogenetic tree reconstruction.
A phylogenetic study of 16S rRNA gene sequences from 42 Pseudoalteromonas strains, including HM-SA03, was performed in order to investigate their evolution and subsequently map their biosynthetic potential (based on antiSMASH results). Species were selected based on genome completeness, and 16S rRNA nucleotide sequences were obtained from within genome sequences, where possible. For species where the complete 16S rRNA gene was not annotated in the genome database, the GenBank nucleotide sequence was used. A total of 42 Pseudoalteromonas sequences and two outgroup (Algicola spp.) sequences were aligned using ClustalW2 (51). Phylogenetic trees were constructed using MrBayes v3.2.6 (52) with a GTR+I+G substitution model, as recommended by jModelTest v2.1.3 (53). Two parallel chains were run for 1.25 million total generations, with a sample frequency of 250, until the trees converged (standard deviation of split frequencies, <0.01).
Genus-wide comparison of Pseudoalteromonas biosynthesis gene clusters.
A total of 42 Pseudoalteromonas genomes were analyzed for specialized metabolite BGCs using IMG Atlas of Biosynthetic Gene Clusters (ABC) (54). Those with BGCs were further analyzed using antiSMASH v5.1.2, to determine their domain architecture and predict the products of these pathways. Each antiSMASH result was manually assessed to determine if the pathway encoded a known compound, and all predicted clusters were then organized into sequence similarity networks using default settings in BiG-SCAPE (55) to interrogate pathway conservation across all Pseudoalteromonas genomes in this study. To avoid overestimation of BGCs, results describing single or orphan modules or domains, which may be a result of fragmented genome assemblies, were not included in the final analysis.
Small molecule extraction of Pseudoalteromonas HM-SA03 cultures.
HM-SA03 medium supernatant was extracted by adsorption onto 20 g/liter Amberlite XAD-7HP resin (Merck) for 1 h. The resin was filtered and washed with 10 ml MilliQ water to remove interfering medium components. Adsorbed compounds were eluted twice with 10 ml methanol, and the combined washes were evaporated to dryness under reduced pressure. An uninoculated culture was extracted using the same methodology and used as a control, for comparative purposes, during downstream analyses.
Analysis of Pseudoalteromonas HM-SA03 organic extracts by liquid chromatography-mass spectrometry (LC-MS).
Organic extracts of Pseudoalteromonas HM-SA03 cultures were analyzed using a Thermo Fisher Scientific Quantum Access coupled with a Thermo Fisher Scientific Accela pump and an HTC PAL autosampler. Separation was achieved using a BEH C18 2.1 mm by 50 mm 1.9-μm UHPLC column (Waters) using a gradient of 0.1% formic acid in MilliQ water against acetonitrile at 400 μl/min. The gradient system was 0 to 5 min 0% B, linearly ramped to 100% B at 25 min, held for 1 min, and returned to 0% B for 3 min. Column eluate was ionized using a positive mode electrospray source.
Data availability.
The final HM-SA03 genome assembly is available from the NCBI database under accession PRJNA400113.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Ramaciotti Centre for Genomics for performing the genome sequencing and the Bioanalytical Mass Spectrometry Facility at the Mark Wainwright Analytical Centre for performing the mass spectrometry experiments. This research includes computations using the Linux computational cluster Katana supported by the Faculty of Science, UNSW Australia. We thank S. E. Ongley for proofreading the manuscript.
This project and a fellowship to B.A.N. were funded by the Australian Research Council.
We declare no conflicts of interest.
Footnotes
This is contribution 30 from the Brett Neilan group.
Supplemental material is available online only.
REFERENCES
- 1.Gauthier G, Gauthier M, Christen R. 1995. Phylogenetic analysis of the genera Alteromonas, Shewanella, and Moritella using genes coding for small-subunit rRNA sequences and division of the genus Alteromonas into two genera, Alteromonas (emended) and Pseudoalteromonas gen. nov., and proposal of twelve new species combinations. Int J Syst Bacteriol 45:755–761. doi: 10.1099/00207713-45-4-755. [DOI] [PubMed] [Google Scholar]
- 2.Bowman JP. 2007. Bioactive compound synthetic capacity and ecological significance of marine bacterial genus Pseudoalteromonas. Mar Drugs 5:220–241. doi: 10.3390/md504220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Offret C, Desriac F, Le Chevalier P, Mounier J, Jegou C, Fleury Y. 2016. Spotlight on antimicrobial metabolites from the marine bacteria Pseudoalteromonas: chemodiversity and ecological significance. Marine Drugs 14:129. doi: 10.3390/md14070129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ross AC, Gulland LE, Dorrestein PC, Moore BS. 2015. Targeted capture and heterologous expression of the Pseudoalteromonas alterochromide gene cluster in Escherichia coli represents a promising natural product exploratory platform. ACS Synth Biol 4:414–420. doi: 10.1021/sb500280q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Machado H, Sonnenschein EC, Melchiorsen J, Gram L. 2015. Genome mining reveals unlocked bioactive potential of marine Gram-negative bacteria. BMC Genomics 16:158. doi: 10.1186/s12864-015-1365-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Graca AP, Viana F, Bondoso J, Correia MI, Gomes L, Humanes M, Reis A, Xavier JR, Gaspar H, Lage OM. 2015. The antimicrobial activity of heterotrophic bacteria isolated from the marine sponge Erylus deficiens (Astrophorida, Geodiidae). Front Microbiol 6:389. doi: 10.3389/fmicb.2015.00389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weissman KJ. 2015. The structural biology of biosynthetic megaenzymes. Nat Chem Biol 11:660–670. doi: 10.1038/nchembio.1883. [DOI] [PubMed] [Google Scholar]
- 8.Speitling M, Smetanina OF, Kuznetsova TA, Laatsch H. 2007. Bromoalterochromides A and A′, unprecedented chromopeptides from a marine Pseudoalteromonas maricaloris strain KMM 636T. J Antibiot (Tokyo) 60:36–42. doi: 10.1038/ja.2007.5. [DOI] [PubMed] [Google Scholar]
- 9.Shiozawa H, Shimada A, Takahashi S. 1997. Thiomarinols D, E, F and G, new hybrid antimicrobial antibiotics produced by a marine bacterium; isolation, structure, and antimicrobial activity. J Antibiot (Tokyo) 50:449–452. doi: 10.7164/antibiotics.50.449. [DOI] [PubMed] [Google Scholar]
- 10.Fukuda D, Haines AS, Song Z, Murphy AC, Hothersall J, Stephens ER, Gurney R, Cox RJ, Crosby J, Willis CL, Simpson TJ, Thomas CM. 2011. A natural plasmid uniquely encodes two biosynthetic pathways creating a potent anti-MRSA antibiotic. PLoS One 6:e18031. doi: 10.1371/journal.pone.0018031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qin Z, Huang S, Yu Y, Deng H. 2013. Dithiolopyrrolone natural products: isolation, synthesis and biosynthesis. Mar Drugs 11:3970–3997. doi: 10.3390/md11103970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Du YL, Alkhalaf LM, Ryan KS. 2015. In vitro reconstitution of indolmycin biosynthesis reveals the molecular basis of oxazolinone assembly. Proc Natl Acad Sci U S A 112:2717–2722. doi: 10.1073/pnas.1419964112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Agarwal V, El Gamal AA, Yamanaka K, Poth D, Kersten RD, Schorn M, Allen EE, Moore BS. 2014. Biosynthesis of polybrominated aromatic organic compounds by marine bacteria. Nat Chem Biol 10:640–647. doi: 10.1038/nchembio.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Korp J, Winand L, Sester A, Nett M. 2018. Engineering pseudochelin production in Myxococcus xanthus. Appl Environ Microbiol 84:e01789-18. doi: 10.1128/AEM.01789-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fischbach MA, Walsh CT. 2006. Assembly-line enzymology for polyketide and nonribosomal peptide antibiotics: logic, machinery, and mechanisms. Chem Rev 106:3468–3496. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]
- 16.Udwary DW, Zeigler L, Asolkar RN, Singan V, Lapidus A, Fenical W, Jensen PR, Moore BS. 2007. Genome sequencing reveals complex secondary metabolome in the marine actinomycete Salinispora tropica. Proc Natl Acad Sci U S A 104:10376–10381. doi: 10.1073/pnas.0700962104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Penn K, Jenkins C, Nett M, Udwary DW, Gontang EA, McGlinchey RP, Foster B, Lapidus A, Podell S, Allen EE, Moore BS, Jensen PR. 2009. Genomic islands link secondary metabolism to functional adaptation in marine Actinobacteria. ISME J 3:1193–1203. doi: 10.1038/ismej.2009.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giessen TW, Franke KB, Knappe TA, Kraas FI, Bosello M, Xie X, Linne U, Marahiel MA. 2012. Isolation, structure elucidation, and biosynthesis of an unusual hydroxamic acid ester-containing siderophore from Actinosynnema mirum. J Nat Prod 75:905–914. doi: 10.1021/np300046k. [DOI] [PubMed] [Google Scholar]
- 19.Chau R, Kalaitzis JA, Wood SA, Neilan BA. 2013. Diversity and biosynthetic potential of culturable microbes associated with toxic marine animals. Mar Drugs 11:2695–2712. doi: 10.3390/md11082695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chau R, Kalaitzis JA, Neilan BA. 2011. On the origins and biosynthesis of tetrodotoxin. Aquat Toxicol 104:61–72. doi: 10.1016/j.aquatox.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 21.Yeh E, Kohli RM, Bruner SD, Walsh CT. 2004. Type II thioesterase restores activity of a NRPS module stalled with an aminoacyl-S-enzyme that cannot be elongated. Chembiochem 5:1290–1293. doi: 10.1002/cbic.200400077. [DOI] [PubMed] [Google Scholar]
- 22.Kraas FI, Giessen TW, Marahiel MA. 2012. Exploring the mechanism of lipid transfer during biosynthesis of the acidic lipopeptide antibiotic CDA. FEBS Lett 586:283–288. doi: 10.1016/j.febslet.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 23.Kanoh K, Kamino K, Leleo G, Adachi K, Shizuri Y. 2004. Pseudoalterobactin A and B, new siderophores excreted by marine bacterium Pseudoalteromonas sp. KP20-4. J Antibiotics 35:871–875. doi: 10.7164/antibiotics.56.871. [DOI] [PubMed] [Google Scholar]
- 24.Deng J, Hamada Y, Shioiri T. 1995. Total synthesis of alterobactin A, a super siderophore from an open-ocean bacterium. J Am Chem Soc 117:7824–7825. doi: 10.1021/ja00134a035. [DOI] [Google Scholar]
- 25.Lautru S, Oves-Costales D, Pernodet J-L, Challis GL. 2007. MbtH-like protein-mediated cross-talk between non-ribosomal peptide antibiotic and siderophore biosynthetic pathways in Streptomyces coelicolor M145. Microbiology (Reading) 153:1405–1412. doi: 10.1099/mic.0.2006/003145-0. [DOI] [PubMed] [Google Scholar]
- 26.Singh GM, Fortin PD, Koglin A, Walsh CT. 2008. β-hydroxylation of the aspartyl residue in the phytotoxin syringomycin E: characterization of two candidate hydroxylases AspH and SyrP in Pseudomonas syringae. Biochemistry 47:11310–11320. doi: 10.1021/bi801322z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blodgett JA, Oh D-C, Cao S, Currie CR, Kolter R, Clardy J. 2010. Common biosynthetic origins for polycyclic tetramate macrolactams from phylogenetically diverse bacteria. Proc Natl Acad Sci U S A 107:11692–11697. doi: 10.1073/pnas.1001513107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu F, Zaleta-Rivera K, Zhu X, Huffman J, Millet JC, Harris SD, Yuen G, Li X-C, Du L. 2007. Structure and biosynthesis of heat-stable antifungal factor (HSAF), a broad-spectrum antimycotic with a novel mode of action. Antimicrob Agents Chemother 51:64–72. doi: 10.1128/AAC.00931-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shigemori H, Bae MA, Yazawa K, Sasaki T, Kobayashi J. 1992. Alteramide A, a new tetracyclic alkaloid from a bacterium Alteromonas sp. associated with the marine sponge Halichondria okadai. J Org Chem 57:4317–4320. doi: 10.1021/jo00041a053. [DOI] [Google Scholar]
- 30.Serino L, Reimmann C, Visca P, Beyeler M, Chiesa VD, Haas D. 1997. Biosynthesis of pyochelin and dihydroaeruginoic acid requires the iron-regulated pchDCBA operon in Pseudomonas aeruginosa. J Bacteriol 179:248–257. doi: 10.1128/jb.179.1.248-257.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taniguchi M, Suzumura K, Nagai K, Kawasaki T, Takasaki J, Sekiguchi M, Moritani Y, Saito T, Hayashi K, Fujita S, Tsukamoto S, Suzuki K. 2004. YM-254890 analogues, novel cyclic depsipeptides with Galpha(q/11) inhibitory activity from Chromobacterium sp. QS3666. Bioorg Med Chem 12:3125–3133. doi: 10.1016/j.bmc.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 32.Reher R, Kuschak M, Heycke N, Annala S, Kehraus S, Dai HF, Muller CE, Kostenis E, Konig GM, Crusemann M. 2018. Applying molecular networking for the detection of natural sources and analogues of the selective Gq protein inhibitor FR900359. J Nat Prod 81:1628–1635. doi: 10.1021/acs.jnatprod.8b00222. [DOI] [PubMed] [Google Scholar]
- 33.Pfeifer BA, Wang CCC, Walsh CT, Khosla C. 2003. Biosynthesis of yersiniabactin, a complex polyketide-nonribosomal peptide, using Escherichia coli as a heterologous host. Appl Environ Microbiol 69:6698–6702. doi: 10.1128/aem.69.11.6698-6702.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Q, Doroghazi JR, Zhao X, Walker MC, van der Donk WA. 2015. Expanded natural product diversity revealed by analysis of lanthipeptide-like gene clusters in actinobacteria. Appl Environ Microbiol 81:4339–4350. doi: 10.1128/AEM.00635-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Noike M, Matsui T, Ooya K, Sasaki I, Ohtaki S, Hamano Y, Maruyama C, Ishikawa J, Satoh Y, Ito H, Morita H, Dairi T. 2015. A peptide ligase and the ribosome cooperate to synthesize the peptide pheganomycin. Nat Chem Biol 11:71–76. doi: 10.1038/nchembio.1697. [DOI] [PubMed] [Google Scholar]
- 36.Wang H, Fewer DP, Holm L, Rouhiainen L, Sivonen K. 2014. Atlas of nonribosomal peptide and polyketide biosynthetic pathways reveals common occurrence of nonmodular enzymes. Proc Natl Acad Sci U S A 111:9259–9264. doi: 10.1073/pnas.1401734111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kalaitzis JA, Lauro FM, Neilan BA. 2009. Mining cyanobacterial genomes for genes encoding complex biosynthetic pathways. Nat Prod Rep 26:1447–1465. doi: 10.1039/b817074f. [DOI] [PubMed] [Google Scholar]
- 38.Lau SCK, Tsoi MMY, Li X, Dobretsov S, Plakhotnikova Y, Wong P-K, Qian P-Y. 2005. Pseudoalteromonas spongiae sp. nov., a novel member of the γ-Proteobacteria isolated from the sponge Mycale adhaerens in Hong Kong waters. Int J Syst Evol Microbiol 55:1593–1596. doi: 10.1099/ijs.0.63638-0. [DOI] [PubMed] [Google Scholar]
- 39.Huang Y-L, Dobretsov S, Xiong H, Qian P-Y. 2007. Effect of biofilm formation by Pseudoalteromonas spongiae on induction of larval settlement of the polychaete Hydroides elegans. Appl Environ Microbiol 73:6284–6288. doi: 10.1128/AEM.00578-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Beurmann S, Ushijima B, Videau P, Svoboda CM, Smith AM, Rivers OS, Aeby GS, Callahan SM. 2017. Pseudoalteromonas piratica strain OCN003 is a coral pathogen that causes a switch from chronic to acute Montipora white syndrome in Montipora capitata. PLoS One 12:e0188319. doi: 10.1371/journal.pone.0188319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bosi E, Fondi M, Orlandini V, Perrin E, Maida I, de Pascale D, Tutino ML, Parrilli E, Lo Giudice A, Filloux A, Fani R. 2017. The pangenome of (Antarctic) Pseudoalteromonas bacteria: evolutionary and functional insights. BMC Genomics 18:93. doi: 10.1186/s12864-016-3382-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wilson K. 2001. Preparation of genomic DNA from bacteria, Curr Protoc Mol Biol Chapter 2:Unit 2.4. doi: 10.1002/0471142727.mb0204s56. [DOI] [PubMed] [Google Scholar]
- 43.Cox MP, Peterson DA, Biggs PJ. 2010. SolexaQA: at-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinformatics 11:485. doi: 10.1186/1471-2105-11-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, He G, Chen Y, Pan Q, Liu Y, Tang J, Wu G, Zhang H, Shi Y, Liu Y, Yu C, Wang B, Lu Y, Han C, Cheung DW, Yiu S-M, Peng S, Xiaoqian Z, Liu G, Liao X, Li Y, Yang H, Wang J, Lam T-W, Wang J. 2012. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience 1:18. doi: 10.1186/2047-217X-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Markowitz VM, Chen I-MA, Palaniappan K, Chu K, Szeto E, Grechkin Y, Ratner A, Jacob B, Huang J, Williams P, Huntemann M, Anderson I, Mavromatis K, Ivanova NN, Kyrpides NC. 2012. IMG: the integrated microbial genomes database and comparative analysis system. Nucleic Acids Res 40:D115–D122. doi: 10.1093/nar/gkr1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bachmann BO, Ravel J. 2009. Methods for in silico prediction of microbial polyketide and nonribosomal peptide biosynthetic pathways from DNA sequence data. Methods Enzymol 458:181–217. doi: 10.1016/S0076-6879(09)04808-3. [DOI] [PubMed] [Google Scholar]
- 47.Medema MH, Blin K, Cimermancic P, de Jager V, Zakrzewski P, Fischbach MA, Weber T, Takano E, Breitling R. 2011. antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res 39:W339–W346. doi: 10.1093/nar/gkr466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blin K, Shaw S, Steinke K, Villebro R, Ziemert N, Lee SY, Medema MH, Weber T. 2019. antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res 47:W81–W87. doi: 10.1093/nar/gkz310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Lu F, Marchler GH, Mullokandov M, Omelchenko MV, Robertson CL, Song JS, Thanki N, Yamashita RA, Zhang D, Zhang N, Zheng C, Bryant SH. 2011. CDD: a conserved domain database for the functional annotation of proteins. Nucleic Acids Res 39:D225–D229. doi: 10.1093/nar/gkq1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lu S, Wang J, Chitsaz F, Derbyshire MK, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Marchler GH, Song JS, Thanki N, Yamashita RA, Yang M, Zhang D, Zheng C, Lanczycki CJ, Marchler-Bauer A. 2020. CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res 48:D265–D268. doi: 10.1093/nar/gkz991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 52.Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61:539–542. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Posada D. 2008. jModelTest: phylogenetic model averaging. Mol Biol Evol 25:1253–1256. doi: 10.1093/molbev/msn083. [DOI] [PubMed] [Google Scholar]
- 54.Palaniappan K, Chen IA, Chu K, Ratner A, Seshadri R, Kyrpides NC, Ivanova NN, Mouncey NJ. 2020. IMG-ABC v.5.0: an update to the IMG/Atlas of Biosynthetic Gene Clusters Knowledgebase. Nucleic Acids Res 48:D422–D430. doi: 10.1093/nar/gkz932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Navarro-Muñoz JC, Selem-Mojica N, Mullowney MW, Kautsar SA, Tryon JH, Parkinson EI, De Los Santos ELC, Yeong M, Cruz-Morales P, Abubucker S, Roeters A, Lokhorst W, Fernandez-Guerra A, Cappelini LTD, Goering AW, Thomson RJ, Metcalf WW, Kelleher NL, Barona-Gomez F, Medema MH. 2020. A computational framework to explore large-scale biosynthetic diversity. Nat Chem Biol 16:60–68. doi: 10.1038/s41589-019-0400-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moree WJ, McConnell OJ, Nguyen DD, Sanchez LM, Yang YL, Zhao X, Liu WT, Boudreau PD, Srinivasan J, Atencio L, Ballesteros J, Gavilan RG, Torres-Mendoza D, Guzman HM, Gerwick WH, Gutierrez M, Dorrestein PC. 2014. Microbiota of healthy corals are active against fungi in a light-dependent manner. ACS Chem Biol 9:2300–2308. doi: 10.1021/cb500432j. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The final HM-SA03 genome assembly is available from the NCBI database under accession PRJNA400113.