

This work describes the development of a lactobacillus CRISPR-based editing system for genome manipulations in three Lactobacillus species belonging to the lactic acid bacteria (LAB), which are commonly known for their long history of use in food fermentations and as indigenous members of healthy microbiotas and for their emerging roles in human and animal commercial health-promoting applications. We exploited the established CRISPR-SpyCas9 nickase for flexible and precise genome editing applications in Lactobacillus acidophilus and further demonstrated the efficacy of this universal system in two distantly related Lactobacillus species.
KEYWORDS: Lactobacillus acidophilus, CRISPR, Cas9, nickase, genome editing, probiotic, lactic acid bacteria, Lactobacillus
ABSTRACT
Diverse Lactobacillus strains are widely used as probiotic cultures in the dairy and dietary supplement industries, and specific strains, such as Lactobacillus acidophilus NCFM, have been engineered for the development of biotherapeutics. To expand the Lactobacillus manipulation toolbox with enhanced efficiency and ease, we present here a CRISPR (clustered regularly interspaced palindromic repeats)-SpyCas9D10A nickase (Cas9N)-based system for programmable engineering of L. acidophilus NCFM, a model probiotic bacterium. Successful single-plasmid delivery system was achieved with the engineered pLbCas9N vector harboring cas9N under the regulation of a Lactobacillus promoter and a cloning region for a customized single guide RNA (sgRNA) and editing template. The functionality of the pLbCas9N system was validated in NCFM with targeted chromosomal deletions ranging between 300 bp and 1.9 kb at various loci (rafE, lacS, and ltaS), yielding 35 to 100% mutant recovery rates. Genome analysis of the mutants confirmed precision and specificity of the pLbCas9N system. To showcase the versatility of this system, we also inserted an mCherry fluorescent-protein gene downstream of the pgm gene to create a polycistronic transcript. The pLbCas9N system was further deployed in other species to generate a concurrent single-base substitution and gene deletion in Lactobacillus gasseri ATCC 33323 and an in-frame gene deletion in Lactobacillus paracasei Lpc-37, highlighting the portability of the system in phylogenetically distant Lactobacillus species, where its targeting activity was not interfered with by endogenous CRISPR-Cas systems. Collectively, these editing outcomes illustrate the robustness and versatility of the pLbCas9N system for genome manipulations in diverse lactobacilli and open new avenues for the engineering of health-promoting lactic acid bacteria.
IMPORTANCE This work describes the development of a lactobacillus CRISPR-based editing system for genome manipulations in three Lactobacillus species belonging to the lactic acid bacteria (LAB), which are commonly known for their long history of use in food fermentations and as indigenous members of healthy microbiotas and for their emerging roles in human and animal commercial health-promoting applications. We exploited the established CRISPR-SpyCas9 nickase for flexible and precise genome editing applications in Lactobacillus acidophilus and further demonstrated the efficacy of this universal system in two distantly related Lactobacillus species. This versatile Cas9-based system facilitates genome engineering compared to conventional gene replacement systems and represents a valuable gene editing modality in species that do not possess native CRISPR-Cas systems. Overall, this portable tool contributes to expanding the genome editing toolbox of LAB for studying their health-promoting mechanisms and engineering of these beneficial microbes as next-generation vaccines and designer probiotics.
INTRODUCTION
Lactobacillus acidophilus is an indigenous member of the human gastrointestinal microbiota and is one of the most widely formulated probiotic strains in fermented dairy products, functional foods, and dietary supplements (1). A member of the phylum Firmicutes, L. acidophilus is a monophyletic Gram-positive lactic acid bacterial species (2) that is a facultative anaerobe, non-spore-forming, and homofermentative. The model strain L. acidophilus NCFM is a human isolate which has been broadly commercialized since the 1970s (3). The health-promoting attributes of NCFM have been well documented, such as the reduction of cold- and flu-like symptom incidence and duration in children and the modulation of intestinal visceral pain receptors and immune cell functions (4–6). The intrinsic ability of L. acidophilus NCFM to survive gut passage, in combination with its amenability to industrial production and genetic manipulations, has sparked interest in developing this strain into designer probiotics with enhanced functionality, and it constitutes an ideal chassis for oral delivery of mucosal vaccines and biotherapeutics (7, 8). Efforts under way to further elucidate the molecular mechanisms involved in the health-promoting effects of L. acidophilus also hinge on efficient molecular tools that enable genetic manipulation to decipher gene functions responsible for health-promoting attributes.
The first-generation genome manipulation system in L. acidophilus was based on single-crossover homologous recombination driven by conditional plasmid replication to generate targeted gene knockouts (9, 10). This system relied on the concurrent use of a broad-host-range nonreplicative pWV01-derived vector (Ori+ RepA−) carrying a gene fragment homologous to the target deletion and a temperature-sensitive helper plasmid, pTRK669, which provides repA in trans for conditional replication of the pORI-based plasmids. A markerless gene replacement system with upp-based counterselection was subsequently developed which has drastically increased the efficiency for generating single and multiple chromosomal deletions and gene insertions by providing direct selection for double-crossover recombinants (11, 12). Nonetheless, despite the convenience of efficient counterselection, the gene replacement procedures typically take a minimum of 2 weeks after plasmid transformation for single- and double-crossover event selections. In addition, the upp-based system requires prior establishment of an isogenic upp-null mutant as a background host to confer resistance against the 5-fluorouracil counterselective agent.
The discovery and characterization of prokaryotic CRISPR-Cas (clustered regularly interspaced short palindromic repeats–CRISPR-associated protein)-adaptive immune systems and the subsequent repurposing of Cas effectors for genome editing have revolutionized biology and genetics in the past decade (13–22). CRISPR-Cas systems confer adaptive immunity in prokaryotes against phages and foreign genetic elements, as well as other biological functions beyond immunity (23, 24). The sequence-specific targeting of nucleic acids via RNA-guided CRISPR-associated (Cas) effector proteins presents unique opportunities for repurposing these molecular machines into programmable genome editing tools (15). In particular, the popular Cas9 endonuclease from the Streptococcus pyogenes (SpyCas9) class 2 type II CRISPR-Cas system can be codelivered with a target-specific chimeric single guide RNA (sgRNA) to drive precise double-stranded-DNA cleavage (15). The portability of the Cas9 single effector protein provides convenience for heterologous expression in a wide range of hosts along with a customized homing “spacer” sequence in the sgRNA to guide Cas9 cleavage to the complementary protospacer adjacent to Cas9-specific sequence recognition, termed the protospacer-adjacent motif (PAM), in the host chromosome (17, 25–27). Precise deletions, insertions, or point mutations can be readily achieved by codelivering a DNA template that serves as a repair template to guide and budge the host DNA repair pathways (18, 26–29). Implementation of CRISPR-Cas editing in prokaryotes has gradually gained more traction, with CRISPR-based editing tool kits recently having been developed for Escherichia coli (27, 30–32), Bacillus (33–35), Lactococcus lactis (36), Clostridium (37, 38), Corynebacterium (39), and actinomycetes (40–42) (reviewed in reference 43). Nickase variants of SpyCas9 with inactivated RuvC (Cas9D10A) or HNH (Cas9H840A) nuclease domain were developed for genome editing, which results in single-stranded incision at the target DNA, thus greatly improving editing efficiencies in bacteria that lack efficient pathways to repair double-stranded-DNA breaks (15–17, 32, 34, 35, 38).
The potential of CRISPR-SpyCas9-based genome editing was recently reported for several Lactobacillus species. In Lactobacillus reuteri, the use of CRISPR-Cas9 coupled with recombinase T (RecT)-mediated single-stranded DNA (ssDNA) oligonucleotide recombineering provided high-efficiency selection of edited cells for oligonucleotide-mediated chromosomal deletions up to a 1-kb region (44). A similar approach using Cas9-based RecT-assisted ssDNA recombineering was compared with plasmid-borne recombineering template to perform point mutations in three different Lactobacillus plantarum strains (45). The comparison revealed variability in editing efficiencies based on strains and methods of repair template delivery. More recently, Zhou et al. (46) employed Cas9/RecT-assisted ssDNA recombineering in combination with host-derived DNA adenine methylase to improve the efficiency of ssDNA recombineering-mediated point mutations in L. plantarum. Gene deletion was further approached with Cas9/RecT-assisted double-stranded DNA (dsDNA) recombineering, in conjunction with the coexpression of host prophage-derived exonuclease analog and a putative host-nuclease inhibitor to improve homologous recombination of the dsDNA template and host chromosome. To enhance editing efficiency, the dsDNA template was protected from cytoplasmic exonuclease degradation by the incorporation of phosphorothioate bonds (46). To overcome the toxicity of Cas9-induced double-stranded breaks, Song et al. (47) achieved efficient gene deletion and insertion in Lactobacillus casei using a Cas9D10A nickase variant in combination with target-specific sgRNA and plasmid-borne repair templates. In strains of L. plantarum and Lactobacillus brevis with inefficient homology-directed repair (HDR), even with nonlethal Cas9 nickase-induced nicks, Huang et al. (48) combined the host-derived prophage recombinases RecE and RecT with the native Cas9 to achieve efficient gene deletions and gene replacement.
As a genetically and functionally diverse group of widespread species, the genus Lactobacillus is enriched with CRISPR-Cas immune systems (49). For species that possess functional CRISPR-Cas systems (for example, Lactobacillus crispatus), the endogenous type I-E CRISPR-Cas system can be hijacked for genome editing with plasmid delivery of customized gRNA and repair templates that coopt and redirect the immune system toward self and drive efficient genome editing (50). On the other hand, species and strains lacking functional CRISPR-Cas systems, such as L. acidophilus, need the deployment of plasmid-encoded portable effectors for genome editing (49, 51). CRISPR-based genome editing in this species and others lacking intact CRISPR-Cas systems will require heterologous CRISPR and Cas expression systems. Here, we report the development of a Lactobacillus SpyCas9-based genome editing tool kit for programmable editing in L. acidophilus, which can also be widely applicable to other health-promoting and industry-relevant Lactobacillus species. Due to the general lack of robust DNA repair mechanisms, such as nonhomologous-end-joining (NHEJ) DNA repair pathway in Lactobacillus, we exploited the established Cas9D10A nickase variant (Cas9N) for targeted gene deletion and insertion in L. acidophilus to circumvent double-strand-break-induced lethality. The portability of the resulting pLbCas9N system was further demonstrated in two phylogenetically distant species, Lactobacillus gasseri and Lactobacillus paracasei. The high editing efficiency of the pLbCas9N system in L. paracasei indicates that the heterologous CRISPR-Cas9N targeting activity was not interfered with by the host’s endogenous type I-E CRISPR-Cas system, highlighting applications of the pLbCas9N system beyond species devoid of native CRISPR-Cas systems.
RESULTS AND DISCUSSION
Establishment of a CRISPR-SpyCas9D10A nickase (Cas9N) system in L. acidophilus NCFM.
The Cas9N-sgRNA genome editing system for L. acidophilus is composed of a Gram-positive-E. coli shuttle vector system harboring cas9N under the regulation of the P6 Lactobacillus promoter, sgRNA driven by the endogenous promoter of elongation factor-Tu gene (tuf), an editing template consisting of 1-kb homologous arms flanking the editing site, and a chloramphenicol (Cm) selection marker. Preliminary strategies for developing a Cas9N genome editing system entailed either delivering the Cas9N and target-specific sgRNA in tandem with the repair template in a single-plasmid system or delivering the latter two components on separate plasmids in a two-plasmid delivery system in order to minimize the size of vector constructs. For both single- and two-plasmid-system approaches, delivery of Cas9N based on the Gram-positive-E. coli shuttle plasmid pGK12 (52) was unsuccessful due to instability of the cas9N expression cassette in various E. coli cloning hosts tested, as well as in L. acidophilus. Subsequently, successful construction of the Cas9N expression plasmid was achieved in a Gram-positive broad-host-range pNZ-based plasmid (53), pTRK687 (54). Expression of Cas9N is driven by the P6 promoter, an established high-expression promoter in lactobacilli (55), with restriction sites located downstream of the P6-cas9N cassette for cloning of sgRNA and homologous editing templates. The resulting 7.1-kb Cas9N vector, designated pLbCas9N (pTRK1203) (Table 1), is stable in selecting E. coli cloning hosts and was transformable into L. acidophilus NCFM with relatively high efficiency at ≥2 × 103 CFU/μg of vector DNA. To confirm the stability of the cas9N insert in NCFM, total DNA was extracted from two selected pLbCas9N transformants for PCR amplification of cas9N. Sequencing of the PCR amplicons confirmed intact cas9N in both transformants, verifying the fidelity of pLbCas9N replication in L. acidophilus.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Genotype or characteristic | Source or reference |
|---|---|---|
| Strains | ||
| L. acidophilus | ||
| NCK56 | NCFM strain, human intestinal isolate | 79 |
| NCK2773 | NCFM carrying a 300-bp in-frame deletion within rafE | This study |
| NCK2774 | NCFM carrying a 1,086-bp in-frame deletion within lacS | This study |
| NCK2676 | NCFM carrying a 1,919-bp deletion within ltaS | This study |
| NCK2777 | NCFM with 731-bp mCherry gene translational cassette inserted downstream of pgm | This study |
| L. gasseri | ||
| NCK334 | ATCC 33323 strain, human isolate, type strain | ATCC |
| NCK2775 | ATCC 33323 carrying a premature stop codon and a 562-bp deletion within 2crr (LGAS_0710) | This study |
| L. paracasei | ||
| NCK2639 | Lpc-37 (ATCC SD5275) Florafit strain, isolated from a dairy source | DuPont |
| NCK2776 | Lpc-37 carrying a 1,299-bp in-frame deletion within glgA (LPC37PB_RS04825) | This study |
| E. coli | ||
| MC1061 | Cloning host, Strr | 80 |
| Plasmids | ||
| pTRK687 | 3.018 kb; pNZ-based shuttle vector derivative, contains P6 promoter, Cmr | 54 |
| pTRK1203 | pLbCas9N; 7.149 kb; pTRK687 with SpyCas9D10A gene cloned downstream of P6 promoter | This study |
| pTRK1248 | 7.566 kb; pTRK1203 containing NCK56 Ptuf-sgRNA for rafE targeting | This study |
| pTRK1204 | 9.568 kb; pTRK1203 containing NCK56 Ptuf-sgRNA-editing template for ΔrafE | This study |
| pTRK1249 | 7.566 kb; pTRK1203 containing NCK56 Ptuf-sgRNA for lacS targeting | This study |
| pTRK1205 | 9.591 kb; pTRK1203 containing NCK56 Ptuf-sgRNA-editing template for ΔlacS | This study |
| pTRK1254 | 9.604 kb; pTRK1203 containing NCK56 Ptuf-sgRNA-editing template for ΔltaS | This study |
| pTRK1255 | 11.374 kb; pTRK1203 containing NCK56 Ptuf-sgRNA-editing template for mCherry gene insertion | This study |
| pTRK1256 | 9.235 kb; pTRK1203 containing NCK334 Ptuf-sgRNA-editing template for Δ2crr | This study |
| pTRK1257 | 9.586 kb; pTRK1203 containing NCK2639 Ptuf-sgRNA-editing template for ΔglgA | This study |
Gene deletions in L. acidophilus using the pLbCas9N system.
To evaluate the functionality of the pLbCas9N system in L. acidophilus NCFM, three targeted chromosomal deletions of various sizes were performed within the (i) raffinose ABC transporter substrate-binding protein gene rafE (deletion of 300 bp), (ii) lactose permease gene lacS (deletion of 1,086 bp), and (iii) phosphoglycerol transferase gene ltaS (deletion of 1,919 bp) of the lipoteichoic acid biosynthesis pathway (Fig. 1). Each editing construct consisted of the NCFM tuf promoter (Ptuf) upstream of a target-specific sgRNA along with the editing template, the latter composed of 1-kb homologous regions flanking the targeted site as a repair template (Fig. 1A). The repair templates were customized such that a portion of the PAM was removed in the repair template to circumvent subsequent Cas9N cleavage once editing took place in cells with the mutated allele (Fig. 1B). In the case of rafE and lacS deletions, several bases downstream of the PAM were also removed to create in-frame deletions. In order to ensure that adequate homologous regions adjacent to the Cas9N incision site was provided for the host’s HDR pathway, the repair templates also included a 30- to 40-bp short homologous region downstream of the PAM site prior to the targeted deletion regions. The selected gRNAs for both rafE and lacS loci targeted the noncoding strand, whereas the gRNA for ltaS was designed to target the coding strand (Fig. 1B), with the single nick by Cas9D10A occurring at the corresponding targeted strands.
FIG 1.

Heterologous expression of Cas9N and sgRNA via the pLbCas9N system for CRISPR-guided gene deletions in L. acidophilus NCFM. (A) Schematic overview of constructing a 300-bp in-frame deletion within the rafE gene. Target-specific sgRNA and an editing template consisting of 1-kb DNA fragments homologous to the regions flanking the target deletion were cloned into the pLbCas9N vector. Expression of Cas9N and sgRNA encoded in the generated plasmid, pLbCas9N_rafE (pTRK1204), resulted in a single nick cleavage at the targeted site within rafE. Homology-directed repair (HDR) of the DNA nick by the host, assisted by the editing template in pLbCas9N_rafE, leads to precise in-frame deletion at the target site in the rafE mutant. (B and C) Gene deletions of various sizes and in different genomic loci using the pLbCas9N system and transformation efficiencies of the rafE and lacS editing and control plasmid variants. Cells were transformed with pLbCas9N plasmids (pTRK1203 to -1205 and pTRK1248 and -1249) (Table 1) with and without target-specific sgRNA and editing templates (ET) for HDR. Cas9N-targeted single-nick cleavage did not significantly affect transformation efficiencies (sgRNA+, ET−), indicating that Cas9N expression was atoxic and cells were capable of DNA repair to overcome Cas9N cleavage with the provision of homologous editing templates. Sanger sequencing of the PCR amplicons verified the precise deletion genotype profiles, along with the targeted PAM removal (and several bases downstream of PAM to create in-frame deletions for rafE and lacS). A 30- to 40-bp short homologous region downstream of the PAM prior to the targeted deletion regions was also included in the editing template, to ensure adequate homologous regions adjacent to the incision site for host’s HDR pathway. Protospacers/guide spacer sequences and PAM are highlighted in blue and red, respectively. PCR screening of editing plasmid transformants (using the primer pairs shown on the left for respective deletion targets) revealed deletion alleles present in 35% (rafE, transformants 6, 7, 10, 13, 14, and 17) to 100% (lacS and ltaS, with 17 of 33 and 18 of 41 screened transformants shown, respectively) of the screened transformants. Left and right arrows represent oligonucleotide primers used for screening of deletion genotypes (Table 2). (D) Phenotypic analysis of both ΔrafE (NCK2773) and ΔlacS (NCK2774) mutants confirmed the genotypes and impaired ability of the mutants to grow on raffinose and lactose, respectively, as the sole carbon source. (E) Scanning electron microscopy imaging (magnification, ×2,500) showing that inactivation of the lipoteichoic acid biosynthetic pathway resulted in an elongated cell morphotype of the ΔltaS mutant (NCK2676), which is consistent with published observations (56). Bar, 10 μm.
Each editing construct was cloned into pLbCas9N downstream of cas9N, resulting in pTRK1204 (rafE) (Fig. 1A), pTRK1205 (lacS), and pTRK1254 (ltaS) CRISPR-editing plasmids (Table 1). Transformation efficiencies for the editing plasmids targeting rafE, lacS, and ltaS were 1.3 log (Fig. 1C), 1.9 log (Fig. 1C), and 1.1 log lower than that for the pLbCas9N control vector. For both transformation experiments targeting rafE and lacS, cells were transformed with pLbCas9N plasmids with and without target-specific sgRNA and editing templates for HDR (Fig. 1C). Cas9N-targeted single-nick cleavage did not significantly affect transformation efficiencies (Fig. 1C, sgRNA+ ET− [pTRK1248 and pTRK1249]), indicating that Cas9N expression was atoxic. Cells were capable of DNA repair to overcome Cas9N cleavage with the provision of homologous editing templates (Fig. 1C, sgRNA+ ET+ [pTRK1204 and pTRK1205]), although the lower transformation efficiencies observed with pLbCas9N editing plasmids containing both sgRNA and editing template could be attributed to the larger size of the editing plasmids (Table 1). Colony PCR screening of randomly selected transformants with chromosomal-specific primers flanking the deletion targets revealed the presence of deletion alleles in 35% (6/17), 100% (33/33), and 100% (41/41) of rafE, lacS, and ltaS loci, respectively (Fig. 1B). The observed high editing efficiencies for lacS and ltaS indicated effective expression of both cas9N and sgRNA and the absence of Cas9N toxicity. In addition, L. acidophilus was capable of repairing the single nick generated by Cas9N via HDR with the concurrent provision of a homologous repair template. The lower editing efficiency observed for rafE deletion likely resulted from suboptimal targeting of the selected gRNA sequence. PCR screening of the pTRK1204 transformants also showed that some of the rafE deletion mutants were present in a mixed-genotype population. Purification of ΔrafE mutants was achieved by streaking and isolating pure deletion mutants on MRS medium with Cm.
Sanger sequencing of the PCR amplicons generated from the targeted sites at rafE, lacS, and ltaS showed precise nucleotide deletions matching the provided editing templates (Fig. 1B). The purity of each deletion mutant was further verified by PCR using one of the primers that anneal to the deletion region, where the PCR amplicon was generated only from the parent control and not the deletion mutants. For curing of the editing plasmids after successful deletion, the mutants were subcultured in MRS broth without antibiotics and plated to obtain isolated colonies. Replica plating of selected colonies on both MRS and MRS supplemented with Cm showed that 70 to 100% of the mutant colonies were Cm sensitive, indicating loss of the editing plasmids after just one passage in MRS in the absence of antibiotic selective pressure. Plasmid curing was further confirmed by the absence of PCR amplicons using primers specific for the pLbCas9N backbone (Table 2). The efficient rate of plasmid curing enables successive rounds of editing to generate multiple mutations in the same strain. Phenotypic analyses of the mutants confirmed the respective deletion genotypes, whereby impaired growth of both ΔrafE and ΔlacS mutants was observed when raffinose and lactose were provided as the sole carbon sources, respectively (Fig. 1D). Morphological analysis by scanning electron microscopy (SEM) of the ΔltaS mutant revealed a slightly elongated cell morphotype compared to wild-type cells (Fig. 1E), which is consistent with previous observations in L. acidophilus mutant defective in the lipoteichoic acid biosynthetic pathway (56).
TABLE 2.
Oligonucleotide primers used in this study
| Use and oligonucleotide | Sequence (5′→3′)a |
|---|---|
| Amplification of cas9N | |
| spycas9n-F | GTA ATA CTG CAG AAA GAG GAG AAA GGA TCT ATG GAT |
| spycas9n-R | TTA GTA CTG CAG TTA GTC ACC TCC TAG CTG AC |
| Screening of ΔrafE deletion | |
| lac_rafE-F | ATA TGT CAA AAT GTT TAT AAG GC |
| lac_rafE-R | TTC CAT AAT TTG CTT AGT TGT C |
| Screening of ΔlacS deletion | |
| lac_lacS-F | CCA AAG GAA TGC AGA GAT CG |
| lac_lacS-R | TGC AGG AGC ATC ATA AGT TGG |
| Screening of ΔltaS deletion | |
| lac_ltaS-F | GAT TCA GGA TAA TCT TCT TCT GG |
| lac_ltaS-R | AAG TAA ATG TGT CTT ACT CAA TTC C |
| Amplification of fragments for pTRK1255 construction (mCherry gene insertion) | |
| lac_pgmCy-1F | AGG AGG AAC TAT ATC CGG ATG TCG AGA TCT AGA TAG TTT TGC TAG TGA TTT GG |
| lac_pgmCy-1R | ACT AAT TTA ATA TAG GAG ATA TTT CAT GGT TTC AAA G |
| lac_pgmCy-2F | GAA ATA TCT CCT ATA TTA AAT TAG TCG TCC AAC TTT TC |
| lac_pgmCy-2R | AGA AGG TTT TTA TAT TAC AGC TCC AGA TCT CAT CAT CTT TAT AGG TTG AAG CG |
| Screening of mCherry gene insertion | |
| lac_mCherry-F | AAT TAT CCC TGT AGC CCA CG |
| lac_mCherry-R | GCT GAT TTC TTT ACA CTA GCA GG |
| Screening of Δ2crr deletion | |
| lga_2crr-F | CAT TAT ATT TCA AGT CAT TCT TCT GC |
| lga_2crr-R | GGC TCA CTA GGT TTG TAC TAC |
| Screening of ΔglgA deletion | |
| lpc_glgA-F | TCA CGG ACC ACA TTC GTA GC |
| lpc_glgA-R | TGT TGT CAC CTC ATG CTT TGC |
| pLbCas9N-specific primers (to verify curing of editing plasmid in mutants) | |
| P6-F | ATT TCT TCA CAA ATA ATT CAC GCT T |
| NC-R | AAT CGC TTT AGC ATC TAC TCC |
Restriction enzyme sites are underlined.
pLbCas9N-guided chromosomal insertion in L. acidophilus.
To further demonstrate the versatility of the pLbCas9N system, a 711-bp gene encoding an NCFM codon-optimized mCherry fluorescent protein was targeted for chromosomal insertion immediately downstream of the pgm gene to create a polycistronic transcript (Fig. 2A). The assembled plasmid construct harbors an editing template consisting of an mCherry translational cassette (a native NCFM ribosomal binding site upstream of the start codon of the mCherry gene) flanked by 1.5-kb homologous arms each corresponding to the upstream and downstream regions of the insertion site. The selected protospacer and PAM were located on the minus strand, downstream of the transcriptional terminator of pgm. The final 11.4-kb plasmid construct, pTRK1255 (Table 1; Fig. 2A), was electroporated into NCFM, with a transformation efficiency 2 log lower than that of the pLbCas9N control vector. Initial screening of the pTRK1255 transformants did not yield any mCherry gene integrant.
FIG 2.

Chromosomal insertion of the mCherry gene in L. acidophilus using the pLbCas9N system. (A) The mCherry-encoding gene was targeted for chromosomal integration downstream of pgm for coexpression driven by the native pgm promoter. The mCherry translational cassette along with 1.5-kb homologous arms and a sgRNA targeting the minus strand downstream of the transcriptional terminator (dark gray rectangles) were cloned into the pLbCas9N vector to generate the pLbCas9N_mCherry editing plasmid (pTRK1255). Left and right arrows represent primers used for screening of mCherry gene integrants (Table 2). (B) PCR screening of pLbCas9N_mCherry gene transformants following subculturing steps revealed that 5/33 colonies contained mixed genotypes with the mCherry gene integrated at the targeted site (3.9-kb amplicons) in a subpopulation of the cells. (C) PCR screening of isolated colonies from transformant 9 (B) showing that the majority of the colonies are pure NCFM::mCherry gene integrants. (D) Sanger sequencing of the 3.9-kb PCR amplicon showed precise insertion of the mCherry gene cassette between pgm and the native transcriptional terminator (term). Asterisks indicate stop codons; the protospacer and PAM are highlighted in blue and red, respectively. Alignment with the wild-type (WT) sequence demonstrates the insertion junction and the elimination of PAM in the mCherry gene integrant. (E) Phase-contrast and fluorescence microscope examinations of the NCK2777 mCherry-expressing integrant compared to wild-type NCFM cells (magnification, ×40). mCherry fluorescence was detected in NCK2777 cells, confirming coexpression of the mCherry gene with pgm driven by the pgm promoter.
To trigger continuous Cas9N editing within the cell population and enrichment of the integrant population, the electroporated cell suspension after overnight recovery was subcultured with 0.1% inoculum for three passages in fresh MRS broth supplemented with Cm, followed by diluting and plating of the third-passage culture onto MRS medium with Cm selection. As a result of three subculturing passages, 15% (5/33) of the colonies screened contained integrant populations mixed with the wild-type genotype (Fig. 2B). One of these colonies was streaked on MRS plates with Cm to obtain isolated colonies. PCR screening of random colonies showed that 65% (11/17) were pure populations of mCherry gene integrants (Fig. 2C). PCR amplification and sequencing at the insertion site verified precise integration of the mCherry gene cassette downstream of pgm, prior to the transcriptional terminator (Fig. 2D). One of the integrants, designated NCK2777, was selected for the detection of mCherry fluorescent signals. Expression of mCherry proteins was confirmed by the observed fluorescent phenotype of NCK2777 under epifluorescence microscopy (Fig. 2E). Due to the general inherently low efficiencies of chromosomal gene insertion, the additional steps for enrichment of Cas9N targeting and recovery of positive integrants demonstrated the efficacy of the pLbCas9N system for precise gene insertion in L. acidophilus.
Whole-genome sequencing of L. acidophilus mutants confirmed target specificity of pLbCas9N system.
To verify the target specificity of the pLbCas9N editing plasmids, the three deletion mutants from L. acidophilus NCFM after plasmid curing were subjected to whole-genome sequencing and sequence comparison to the wild-type NCFM genome. Mapping of the Illumina short reads from ΔrafE, ΔlacS, and ΔltaS mutants against the NCFM reference genome showed the absence of sequence coverage at the targeted deletion regions in all the mutants (Fig. 3). Comparison of the assembled genomes (generated from hybrid assembly of sequences from Illumina short reads and Oxford Nanopore long reads) further confirmed the specificity of the system in achieving precise editing outcomes at all three genomic loci, with no off-targeting effects (Fig. 3). Interestingly, an insertion of a transposase gene occurred within the lysA gene in both the ΔrafE and ΔlacS mutants (Fig. 4A, B, and D). A closer examination of the Nanopore sequence data of the parent strain in the corresponding lysA region also revealed a small subpopulation of the cells with transposase-interrupted lysA, indicating preexisting, naturally occurring population polymorphism at this region (Fig. 4C). PCR analysis of additional ΔrafE and ΔlacS clones did not reveal transposase insertion within lysA (Fig. 4D). Overall, these results provide evidence that genome targeting by the pLbCas9N editing plasmids did not trigger random transposase duplications in L. acidophilus.
FIG 3.

Whole-genome sequencing of L. acidophilus deletion mutants confirmed the target specificity of the pLbCas9N-based editing plasmids for constructing deletions within (A) rafE, (B) lacS, and (C) ltaS, respectively, as depicted in Fig. 1. Mapping of the Illumina sequencing reads of the mutants to the parent genome (purple regions) and Mauve alignments of the whole genomes of parent and mutants (green regions) confirmed that the genome content variation (outlined by the yellow box) in each mutant is confined to the targeted deletion regions (represented by regions with no read coverage in the mutants). Bottom panels represent zoom-in visualization of the respective deletion regions within the yellow box, showing precise removal of the deletion targets.
FIG 4.

Detection of a spontaneous transposase gene disruption in lysA gene in a subpopulation of the L. acidophilus ΔrafE mutant. Whole-genome alignment (A) and mapping of Oxford Nanopore sequencing reads (B) of ΔrafE mutant (NCK2773) against the parent genome revealed lysA gene disruption by a transposase coding sequence in the mutant, a locus independent of the CRISPR-targeting rafE region. (C) Mapping of Oxford Nanopore sequencing reads from the parent at the corresponding region also revealed that a subset of the reads did not map contiguously to the full-length wild-type lysA gene, indicating spontaneous population polymorphism in this region. (D) PCR amplification of the lysA region in parent and all pLbCas9N-generated mutants showed that transposase disruption within lysA occurred in two of the six mutant isolates (non-target specific), confirming that the observed transposase disruption is due to clonal expansion (from population polymorphism) and not to targeting by Cas9N. Asterisks indicate mutant clones (NCK2773, NCK2774, and NCK2676) (Table 1) previously selected for genome sequencing and analysis.
Expansion of pLbCas9N-mediated genome editing in Lactobacillus paracasei and Lactobacillus gasseri.
The pLbCas9N system was constructed with a pNZ-based rolling circle high-copy-number Gram-positive shuttle vector backbone that replicates in streptococci, lactococci, and lactobacilli. In addition, the constitutive P6 promoter, an L. acidophilus native promoter for an ArsC family transcriptional regulator designed for cas9N expression, has also been commonly used for heterologous gene expression in other lactobacilli. To demonstrate the versatility of the pLbCas9N system, targeted gene deletions were performed in L. gasseri and L. paracasei, two species belonging to phylogenetic clades distinct from L. acidophilus. L. gasseri strain ATCC 33323, a neotype strain of human origin and commensal of the oral, intestinal, fecal, and vaginal microbiotas of juveniles and adults (57, 58), does not have a native CRISPR-Cas system. L. paracasei strain Lpc-37 is a commercial probiotic strain originally isolated from a dairy source which possesses an endogenous type I-E CRISPR-Cas system. The pLbCas9N system was used in L. gasseri ATCC 33323 to simultaneously generate a single base substitution to introduce a premature stop codon followed by a 562-bp deletion within the gene coding for a response regulator (2-CRR; lgas0710) of a two-component regulatory system (Fig. 5A). In L. paracasei Lpc-37, the pLbCas9N system was designed to target an in-frame deletion of 1,299 bp within the glgA gene, encoding a glycogen synthase of the putative glycogen biosynthesis pathway (Fig. 5B). Expression of sgRNAs was driven by the native Ptuf promoters identified from the corresponding strains. As described previously, the customized Ptuf-sgRNA cassette along with the 2-kb repair template (1-kb homologous regions flanking the targeted site) was cloned downstream of the P6-cas9N expression cassette for each genome editing plasmid, generating pLbCas9N_2crr (pTRK1256) and pLbCas9N_glgA (pTRK1257) (Table 1).
FIG 5.
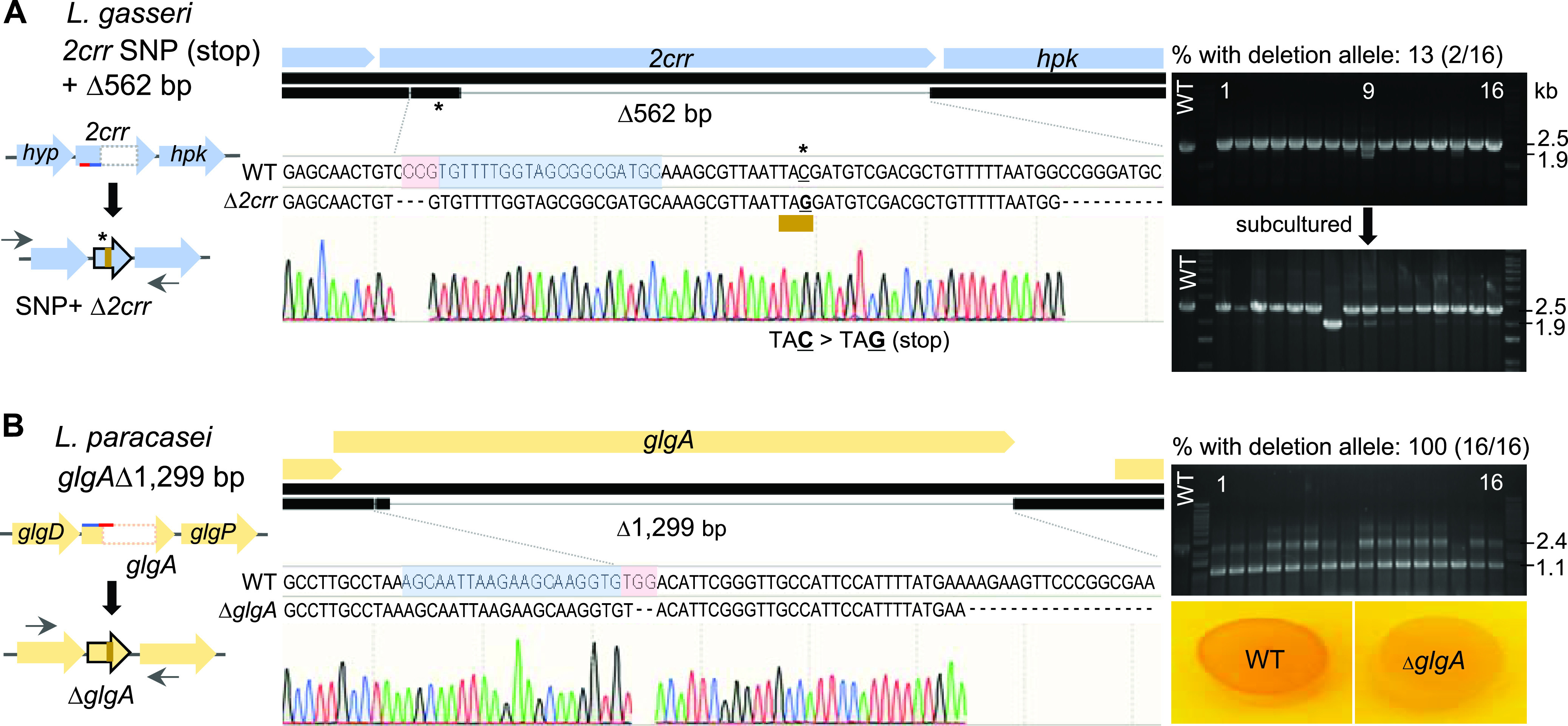
Portability of the pLbCas9N system in L. gasseri and L. paracasei. (A) In L. gasseri ATCC 33323, the editing template cloned into pLbCas9N was designed to create a premature stop codon by introducing a base change (C to G; indicated with an asterisk) at the 5′ region of the 2crr gene, followed by a 562-bp deletion within the same gene. The single base change and deletion in the selected mutant (NCK2775) were confirmed by Sanger sequencing. Initial PCR screening of 16 pLbCas9N_2crr (pTRK1256) transformants showed that two transformants (no. 9 and 14) contained subpopulations of cells carrying 2crr deletion at the targeted site (1.9-kb amplicon). Subsequent PCR screening of isolated colonies from transformant 9 after subculturing in MRS with Cm yielded one pure 2crr mutant (NCK2775) and additional colonies containing subpopulations with the 2crr deletion. (B) For L. paracasei Lpc-37, the pLbCas9N system was used to construct an in-frame deletion within the glgA gene. All 16 selected transformants contained the ΔglgA allele (1.1-kb amplicon), with the majority in mixed genotype populations containing unedited cells that can be further subcultured and purified to obtain pure mutant population. A glycogen staining assay confirmed the absence of glycogen biosynthesis in the ΔglgA mutant (NCK2776), as indicated by the lack of iodine staining of the mutant cells. Oligonucleotide primers used for screening deletion mutants are indicated with left and right arrows (Table 2). Blue- and red-highlighted bases represent protospacer sequences and PAM, respectively.
Similar to the transformation efficiencies previously observed with the editing plasmids in L. acidophilus, the transformation of pTRK1256 and pTRK1257 into L. gasseri ATCC 33323 and L. paracasei Lpc-37 resulted in 1.3-log and 1.1-log reductions of transformation efficiencies in the respective strains, compared to transformation of the pLbCas9N vector control. PCR-based screening of the transformants showed the presence of the deletion alleles in 13% (2/16) and 100% (16/16) of the ATCC 33323 and Lpc-37 transformants, respectively (Fig. 5A and B). In both L. gasseri and L. paracasei, transformants containing mixed wild-type and mutated genotypes were easily purified by culturing in MRS supplemented with Cm to enrich for the mutated genotype and subsequent purification of the mutant population. Sequencing of PCR amplicons generated from 2crr loci in selected ATCC 33323 mutants demonstrated precise base changes that resulted in a premature stop codon at the 5′ end of 2crr (TAC to TAG), along with downstream deletion of a 562-bp region within 2crr (Fig. 5A). Similarly, removal of a 1,299-bp region within the glgA gene of Lpc-37 was confirmed by sequencing of the glgA PCR amplicon from the mutants (Fig. 5B). No apparent phenotypic change was observed in the Δ2crr mutant (NCK2775) compared to the parent ATCC 33323, although the ortholog of 2CRR_Lgas0710 in L. acidophilus NCFM is essential for bile tolerance in vitro (59). As expected, an iodine staining assay for intracellular glycogen detection in L. paracasei showed a deficiency of glycogen accumulation in the ΔglgA mutant compared to the parent strain (Fig. 5B). This result indicated that glgA inactivation disrupted the glycogen biosynthetic pathway in the mutant and consequently its inability to synthesize intracellular glycogen. Overall, the editing efficacy of the pLbCas9N as demonstrated in L. gasseri and L. paracasei is promising for the exploitation of the system in other lactobacilli.
Conclusion.
In this study, we developed a Cas9N-based genome editing system for programmable genome engineering in L. acidophilus and demonstrated the portability of the system in two other Lactobacillus species, namely, L. gasseri and L. paracasei. Overall, the observed editing efficiencies (up to 100%) and diverse editing outcomes highlighted the robustness and versatility of the pLbCas9N system, including (i) flexible site-specific manipulation at different chromosomal loci, (ii) a varying deletion size range, and (iii) a customizable repair template design for multiple concurrent mutations. Recovery of deletion mutants can be achieved within 1 week posttransformation, thus significantly accelerating precise gene deletion throughput for functional studies of probiotic mechanisms compared to conventional double-crossover recombination. In some instances where edited cells were present together with unedited cells, a subsequent purification step was applied to the mixed-genotype culture to recover pure mutant populations. The instability of pLbCas9N editing plasmids in the absence of antibiotic selective pressure enabled efficient curing of the plasmid after editing and generation of iterative genome manipulations in the same host. Aside from the broad-host-range replicon and the modular design of the pLbCas9N system, the portability of the system is also conferred by its compatibility in hosts with intact or degenerate CRISPR-Cas systems (e.g., L. acidophilus and L. paracasei).
The GRAS (generally regarded as safe) status of most lactobacillus species has led to the recent emergence of various Lactobacillus strains as attractive candidates for engineered biotherapeutics. Hence, the pLbCas9N system represents a valuable tool for precise chromosomal manipulation of genes and pathways of interests and for exploiting highly expressed chromosomal regions (e.g., downstream of the constitutive highly expressed pgm gene) for gene insertion to achieve high and stable coexpression of target genes without relying on conventional heterologous plasmid expression. The tool and approaches presented in this work will serve as additional modalities for the timely development of L. acidophilus, and potentially other health-promoting Lactobacillus species, as next-generation mucosal vaccine and biotherapeutic delivery vehicles.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The strains and plasmids used in this study are listed in Table 1. E. coli transformants were selected and propagated at 30°C in Luria-Bertani (LB) medium (Difco Laboratories, Detroit, MI) supplemented with 15 μg/ml of Cm (Sigma, St. Louis, MO). Lactobacillus strains were propagated in MRS broth (Difco) statically under ambient atmospheric conditions or on MRS agar (1.5% [wt/vol]; Difco) under anaerobic conditions at 37°C. Lactobacillus plasmid transformants were selected in the presence of 5 μg/ml of Cm for both L. acidophilus and L. paracasei and 7.5 μg/ml for L. gasseri.
DNA manipulations and transformation.
Genomic DNA was isolated using a Quick-DNA fungal/bacterial kit (Zymo Research, Irvine, CA) according to the manufacturer’s protocol. Plasmid DNA from E. coli was purified using a QIAprep spin miniprep kit (Qiagen Inc., Valencia, CA). Restriction enzymes and a Quick dephosphorylation kit (New England Biolabs [NEB], Ipswich, MA) were used per the manufacturer’s instructions. DNA ligation was performed using Instant Sticky-end Ligase master mix (NEB). DNA fragments for plasmid construction were assembled using NEBuilder HiFi DNA assembly master mix (NEB) based on the manufacturer’s recommendations. Oligonucleotide primers for PCR and Sanger sequencing were synthesized by Integrated DNA Technologies (Coralville, IA). Gene fragments and sgRNA sequences were synthesized by Genewiz (South Plainfield, NJ). For cloning and Sanger sequencing purposes, PCR amplicons were generated using Q5 Hot Start high-fidelity 2× master mix (NEB) according to the supplier’s instructions. Routine PCR amplifications for screening of transformants were performed by using standard protocols and Choice-Taq Blue DNA polymerase (Thomas Scientific, Swedesboro, NJ). PCR amplicons were gel-purified using Monarch DNA gel extraction kits (NEB) followed by Monarch PCR and DNA cleanup kits (NEB), or with E-Gel CloneWell II agarose gels (Thermo Fisher Scientific), followed by further purification of the recovered products using Monarch PCR and DNA cleanup kits. Sanger sequencing to verify sequence integrity of PCR amplicons and plasmid constructs was performed by Genewiz.
E. coli chemically competent cells were prepared and transformed based on procedures previously described by Hanahan (60). Transformed cells were recovered with SOC medium (Corning Mediatech, Manassas, VA) at 30°C with aeration for 3 h prior to plating on selective medium and incubation at 30°C. L. acidophilus, L. gasseri, and L. paracasei cells were prepared for electroporation as described previously (11). Prepared cell aliquots (200 μl) were electroporated with 1 to 2 μg of each pLbCas9N plasmid construct, and transformed cells were recovered in 1.8 ml of MRS medium overnight at 37°C, followed by dilution and plating on selective medium.
Construction of a lactobacillus SpyCas9D10A-expressing vector for targeted genome editing.
For construction of a lactobacillus Cas9 nickase-expressing vector, the SpyCas9D10A sequence along with its ribosomal binding site was amplified from pCas9(D10A) (Table 2), a gift from Xiao Wang (Addgene plasmid 74495; RRID, Addgene_74495) (32), with PstI restriction sites added on both ends of the SpyCas9D10A amplicons. The purified 4.1-kb amplicon was digested with PstI and ligated with similarly digested and dephosphorylated pTRK687 vector. The ligation mix was transformed into E. coli cloning hosts, and transformants were selected on LB plates containing Cm. The resulting 7.1-kb Cas9 nickase expressing vector, pLbCas9N or pTRK1203, was maintained in E. coli MC1061 cloning host, and the stability of the cas9N insert was verified by restriction digest analysis and Sanger sequencing.
Design and construction of pLbCas9N-based editing plasmids.
To design sgRNA for targeted gene deletions and gene insertion, SpyCas9 target sites at each of the targeted editing locus were scanned and predicted using the CRISPy-web server (61). For each SpyCas9 target candidate, the seed region of the guide sequence was manually scanned along the genome using Geneious to verify the absence of potential off-target matches. The 19- to 20-nucleotide guide region along with the gRNA scaffold and termination signal was designed according to a protocol by Mali (https://media.addgene.org/cms/files/hCRISPR_gRNA_Synthesis.pdf). The predicted promoter sequence for the housekeeping tuf gene of each respective Lactobacillus strain (NCFM, ATCC 33323, or Lpc-37), along with the target-specific sgRNA, and an editing/repair template consisting of 1-kb homologous arms flanking the target site were designed as a single gene block with added BglII restriction sites on both ends and synthesized by Genewiz. Each gene block was digested with BglII and ligated into similarly digested and dephosphorylated pLbCas9N vector.
For chromosomal insertion of the mCherry-encoding gene between the stop codon of pgm and its transcriptional terminator in L. acidophilus, the mCherry amino acid sequence was obtained from pRSET-B mCherry (62) and the corresponding 711-bp gene sequence, codon optimized for L. acidophilus NCFM, was generated using the web-based JCat (Java Codon Adaptation Tool) (63). The Ptuf-sgRNA, a partial sequence of the repair template (homologous to the 1.5-kb flanking region downstream of the insertion site), and the codon-optimized mCherry gene with ribosomal-binding region of pgm added at the 5′ end were synthesized as a gene block and amplified with the primer pair lac_pgmCy-1F/lac_pgmCy-1R (Table 2), while the remaining portion of the repair template (homologous to the 1.5-kb flanking region upstream of insertion site) was amplified from the chromosome with the primer pair lac_pgmCy-2F/lac_pgmCy-2R. Both fragments were subsequently assembled into BglII-digested pLbCas9N using NEBuilder HiFi DNA assembly master mix. Ligation and assembly mixes were transformed into E. coli MC1061 cells, and transformants were recovered on LB supplemented with Cm at 30°C incubation. The sequence integrity of all plasmid inserts was verified by restriction digest analysis and Sanger sequencing prior to transformation into Lactobacillus hosts.
Isolation of edited mutants and plasmid curing.
Transformants of pLbCas9N editing plasmids were screened for targeted deletions or insertion by colony PCR with chromosomal-specific primers flanking the regions homologous to the editing templates (Table 2). Pure population of edited cells were purified by streaking on MRS plates with Cm and PCR analysis. The absence of wild-type cells was confirmed by PCR with one of the primers that annealed specifically to the deleted region and thus produced amplicons only in the presence of cells retaining wild-type genotypes. For plasmid curing, pure mutant strains were propagated in MRS broth without Cm, and overnight cultures (ca. 10 generations) were plated to obtain isolated colonies. Selected colonies were replica plated on MRS and MRS with Cm plates. Sensitivity to Cm exhibited by colonies indicated loss of the pLbCas9N editing plasmids, which was further confirmed by establishing the absence of PCR amplicons using primers specific to the pLbCas9N backbone (Table 2) (expected amplicon size of 960 bp).
Carbohydrate growth experiments and intracellular glycogen assays.
For carbohydrate growth experiments, L. acidophilus NCFM and its mutant derivatives NCK2773 (ΔrafE) and NCK2774 (ΔlacS) were grown in semidefined medium (64) (SDM) with glucose substituted for raffinose or lactose. Stationary-phase cultures grown in MRS broth (16 h of growth) were inoculated at 1% (vol/vol) into 96-well microplate wells (Corning Costar, Corning, NY) in triplicate, each containing 200 μl of SDM supplemented with 1% of the carbohydrate. Microplates were sealed with clear Thermalseal film (ISC Bioexpress, Kaysville, UT), incubated at 37°C in a Fluostar Optima microplate reader (BMG Labtech, Cary, NC), and optical density of the cells was monitored at 600 nm for 48 h. Qualitative detection of intracellular glycogen in L. paracasei Lpc-37 and NCK2776 (ΔglgA) mutant strains by iodine-staining method was performed as described previously (65).
SEM.
L. acidophilus NCFM and NCK2676 (ΔltaS) strains were grown in MRS medium to an optical density at 600 nm (OD600) of 0.6 to 0.8 (mid-log phase) and pelleted by centrifugation at 3,166 × g at room temperature. Cell pellets were resuspended in a fresh 1:1 (vol/vol) fixative mixture containing 3% glutaraldehyde in 0.1 M cacodylate (pH 5.5) and stored at 4°C until processed. Samples were processed for SEM by the Center for Electron Microscopy (CEM) at North Carolina State University and viewed with a JEOL JEM-5900LV SEM at 15 kV. Images were acquired digitally using a JEOL digital scan generator at a resolution of 1,280 by 960 pixels.
Fluorescence microscopy.
L. acidophilus NCFM and NCK2777 NCFM::mCherry gene integrant strains were grown in MRS broth at 37°C under ambient atmosphere conditions for 16 h. Aliquots (8 μl) of cultures were placed on microscope glass slides, and mCherry fluorescence was visualized at a magnification of ×40 under a Nikon Eclipse E600 microscope equipped with an AT-TRITC/Cy3/TagRFP/Alexa Fluor 546 filter set (excitation at 540 nm, emission at 605 nm).
Whole-genome sequencing of L. acidophilus mutants.
Overnight cultures grown in MRS medium (30 ml) were harvested, and frozen cell pellets were submitted to the High-Throughput Sequencing and Genotyping Unit of the Roy J. Carver Biotechnology Center at the University of Illinois for genomic extraction, whole-genome sequencing, and genome assembly. Genomic DNA was extracted using the MasterPure Gram Positive DNA purification kit (Lucigen, Middleton, WI). For Illumina MiSeq paired-end sequencing, shotgun genomic libraries were prepared with the Hyper Library construction kit from Kapa Biosystems (Roche). The library pool was quantitated by qPCR and sequenced on one ISeq flow cell for 151 cycles from each end of the fragments, which yielded paired-end read lengths of 250 nucleotides (nt). Fastq files were generated and demultiplexed with the bcl2fastq v2.20 conversion software (Illumina), and adapters were trimmed from the 3′ ends of the reads. For Oxford Nanopore sequencing, the genome DNA samples were converted into individual barcoded libraries with the NBD104 (barcoding) and LSK109 (library) kits from Oxford Nanopore (Oxford, UK).
The libraries were pooled and sequenced on two SpotON R10.1 RevD FLO-MIN106 flow cells for 48 h, using a GridIONx5 sequencer. Base calling was performed with Guppy (ver. 3.2.6), and demultiplexing and adapter removal were performed with Porechop (ver. 0.2.3) (66). For hybrid assembly of the genome sequences, Illumina MiSeq and Oxford Nanopore long reads were checked for quality prior to and after trimming using FastQC v0.11.8 (67). MiSeq reads were trimmed using Trimmomatic v0.38 (68) with parameters set to ILLUMINACLIP:TruSeq3-PE-2.fa:2:15:10 LEADING:28 TRAILING:28 MINLEN:30, retaining reads longer than 30 bp. Long reads were adapter trimmed with Porechop v0.2.3 (66) and length filtered to a minimum of 1 kb with seqtk v1.3 (69). Unicycler v0.4.8 (70) assembled the trimmed MiSeq and uncorrected Oxford Nanopore reads in a hybrid assembly using the default “normal” mode. Within Unicycler, the MiSeq reads were assembled with SPAdes v3.11.1 (71), and the resulting long-anchor contigs were assembled together with the Oxford Nanopore reads by an optimized version of miniasm (72) and Racon v0.5.0 (73). Pilon v1.22 (74) was used within Unicycler to iteratively polish the assembly with the MiSeq reads. The circularized genome assemblies were annotated using Prokka v1.14.6 (75) with parameters set for L. acidophilus. Assemblies were evaluated for completeness using BUSCO v3.0.1 (76) with the appropriate bacterial lineage.
To compare the genome sequences of the NCFM mutants (ΔrafE, ΔlacS, and ΔltaS) with the parent strain, sequencing reads from Illumina MiSeq (average, 2.3 million paired-end reads/sample) and Oxford Nanopore (average, 2 billion bases/sample) were mapped to NCFM reference genome (NC_006814) using Bowtie2 mapper (77) within Geneious software v11.1.15 with default settings. Assembled genomes of the NCFM mutants were aligned with the reference genome using Mauve plugin in Geneious, with the progressiveMauve alignment algorithm (78).
Data availability.
Genome sequence data for the L. acidophilus NCFM ΔrafE (NCK2773), ΔlacS (NCK2774), and ΔltaS (NCK2676) mutants have been deposited in the NCBI Sequence Read Archive (SRA) database under BioProject ID PRJNA681755 (BioSample accession numbers SAMN16967693 to SAMN16967695).
ACKNOWLEDGMENTS
This work was supported in part by the North Carolina Agricultural Foundation and DuPont Nutrition & Health USA, Inc.
We thank Valerie Lapham at the NCSU College of Agricultural and Life Sciences Center for Electron Microscopy for assistance in SEM imaging. We also express our gratitude to Alvaro Hernandez and Chris Wright at the University of Illinois at Urbana-Champaign (UIUC) Roy Carver Biotechnology Center for genome sequencing services, and Christopher Fields and Kimberly Walden at the UIUC Roy Carver Biotechnology Center and High-Performance Biological Computing for bioinformatics service and expertise in genome assembly.
REFERENCES
- 1.Morovic W, Hibberd AA, Zabel B, Barrangou R, Stahl B. 2016. Genotyping by PCR and high-throughput sequencing of commercial probiotic products reveals composition biases. Front Microbiol 7:1747. doi: 10.3389/fmicb.2016.01747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bull MJ, Jolley KA, Bray JE, Aerts M, Vandamme P, Maiden MC, Marchesi JR, Mahenthiralingam E. 2014. The domestication of the probiotic bacterium Lactobacillus acidophilus. Sci Rep 4:7202. doi: 10.1038/srep07202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gilliland SE, Speck ML, Morgan CG. 1975. Detection of Lactobacillus acidophilus in feces of humans, pigs, and chickens. Appl Microbiol 30:541–545. doi: 10.1128/AM.30.4.541-545.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leyer GJ, Li S, Mubasher ME, Reifer C, Ouwehand AC. 2009. Probiotic effects on cold and influenza-like symptom incidence and duration in children. Pediatrics 124:e172–e179. doi: 10.1542/peds.2008-2666. [DOI] [PubMed] [Google Scholar]
- 5.Rousseaux C, Thuru X, Gelot A, Barnich N, Neut C, Dubuquoy L, Dubuquoy C, Merour E, Geboes K, Chamaillard M, Ouwehand A, Leyer G, Carcano D, Colombel JF, Ardid D, Desreumaux P. 2007. Lactobacillus acidophilus modulates intestinal pain and induces opioid and cannabinoid receptors. Nat Med 13:35–37. doi: 10.1038/nm1521. [DOI] [PubMed] [Google Scholar]
- 6.Konstantinov SR, Smidt H, de Vos WM, Bruijns SC, Singh SK, Valence F, Molle D, Lortal S, Altermann E, Klaenhammer TR, van Kooyk Y. 2008. S layer protein A of Lactobacillus acidophilus NCFM regulates immature dendritic cell and T cell functions. Proc Natl Acad Sci U S A 105:19474–19479. doi: 10.1073/pnas.0810305105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mohamadzadeh M, Duong T, Sandwick SJ, Hoover T, Klaenhammer TR. 2009. Dendritic cell targeting of Bacillus anthracis protective antigen expressed by Lactobacillus acidophilus protects mice from lethal challenge. Proc Natl Acad Sci U S A 106:4331–4336. doi: 10.1073/pnas.0900029106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kajikawa A, Zhang L, Long J, Nordone S, Stoeker L, LaVoy A, Bumgardner S, Klaenhammer T, Dean G. 2012. Construction and immunological evaluation of dual cell surface display of HIV-1 Gag and Salmonella enterica serovar Typhimurium FliC in Lactobacillus acidophilus for vaccine delivery. Clin Vaccine Immunol 19:1374–1381. doi: 10.1128/CVI.00049-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leenhouts K, Buist G, Bolhuis A, ten Berge A, Kiel J, Mierau I, Dabrowska M, Venema G, Kok J. 1996. A general system for generating unlabelled gene replacements in bacterial chromosomes. Mol Gen Genet 253:217–224. doi: 10.1007/s004380050315. [DOI] [PubMed] [Google Scholar]
- 10.Russell WM, Klaenhammer TR. 2001. Efficient system for directed integration into the Lactobacillus acidophilus and Lactobacillus gasseri chromosomes via homologous recombination. Appl Environ Microbiol 67:4361–4364. doi: 10.1128/aem.67.9.4361-4364.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goh YJ, Azcarate-Peril MA, O'Flaherty S, Durmaz E, Valence F, Jardin J, Lortal S, Klaenhammer TR. 2009. Development and application of a upp-based counterselective gene replacement system for the study of the S-layer protein SlpX of Lactobacillus acidophilus NCFM. Appl Environ Microbiol 75:3093–3105. doi: 10.1128/AEM.02502-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Douglas GL, Goh YJ, Klaenhammer TR. 2011. Integrative food grade expression system for lactic acid bacteria. Methods Mol Biol 765:373–387. doi: 10.1007/978-1-61779-197-0_22. [DOI] [PubMed] [Google Scholar]
- 13.Ishino Y, Shinagawa H, Makino K, Amemura M, Nakata A. 1987. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol 169:5429–5433. doi: 10.1128/jb.169.12.5429-5433.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mojica FJ, Diez-Villasenor C, Soria E, Juez G. 2000. Biological significance of a family of regularly spaced repeats in the genomes of Archaea, Bacteria and mitochondria. Mol Microbiol 36:244–246. doi: 10.1046/j.1365-2958.2000.01838.x. [DOI] [PubMed] [Google Scholar]
- 15.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. 2012. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gasiunas G, Barrangou R, Horvath P, Siksnys V. 2012. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci U S A 109:E2579–E2586. doi: 10.1073/pnas.1208507109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. 2013. RNA-guided human genome engineering via Cas9. Science 339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilkinson R, Wiedenheft B. 2014. A CRISPR method for genome engineering. F1000Prime Rep 6:3. doi: 10.12703/P6-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barrangou R, Horvath P. 2017. A decade of discovery: CRISPR functions and applications. Nat Microbiol 2:17092. doi: 10.1038/nmicrobiol.2017.92. [DOI] [PubMed] [Google Scholar]
- 21.Knott GJ, Doudna JA. 2018. CRISPR-Cas guides the future of genetic engineering. Science 361:866–869. doi: 10.1126/science.aat5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramachandran G, Bikard D. 2019. Editing the microbiome the CRISPR way. Philos Trans R Soc Lond B Biol Sci 374:20180103. doi: 10.1098/rstb.2018.0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. 2007. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 24.Westra ER, Buckling A, Fineran PC. 2014. CRISPR-Cas systems: beyond adaptive immunity. Nat Rev Microbiol 12:317–326. doi: 10.1038/nrmicro3241. [DOI] [PubMed] [Google Scholar]
- 25.Horvath P, Romero DA, Coute-Monvoisin AC, Richards M, Deveau H, Moineau S, Boyaval P, Fremaux C, Barrangou R. 2008. Diversity, activity, and evolution of CRISPR loci in Streptococcus thermophilus. J Bacteriol 190:1401–1412. doi: 10.1128/JB.01415-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J. 2013. RNA-programmed genome editing in human cells. Elife 2:e00471. doi: 10.7554/eLife.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. 2013. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol 31:233–239. doi: 10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Selle K, Barrangou R. 2015. Harnessing CRISPR-Cas systems for bacterial genome editing. Trends Microbiol 23:225–232. doi: 10.1016/j.tim.2015.01.008. [DOI] [PubMed] [Google Scholar]
- 29.Cui L, Bikard D. 2016. Consequences of Cas9 cleavage in the chromosome of Escherichia coli. Nucleic Acids Res 44:4243–4251. doi: 10.1093/nar/gkw223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang Y, Chen B, Duan C, Sun B, Yang J, Yang S. 2015. Multigene editing in the Escherichia coli genome via the CRISPR-Cas9 system. Appl Environ Microbiol 81:2506–2514. doi: 10.1128/AEM.04023-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reisch CR, Prather KL. 2015. The no-SCAR (Scarless Cas9 Assisted Recombineering) system for genome editing in Escherichia coli. Sci Rep 5:15096. doi: 10.1038/srep15096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Standage-Beier K, Zhang Q, Wang X. 2015. Targeted large-scale deletion of bacterial genomes using CRISPR-nickases. ACS Synth Biol 4:1217–1225. doi: 10.1021/acssynbio.5b00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Westbrook AW, Moo-Young M, Chou CP. 2016. Development of a CRISPR-Cas9 tool kit for comprehensive engineering of Bacillus subtilis. Appl Environ Microbiol 82:4876–4895. doi: 10.1128/AEM.01159-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li K, Cai D, Wang Z, He Z, Chen S. 2018. Development of an efficient genome editing tool in Bacillus licheniformis using CRISPR-Cas9 nickase. Appl Environ Microbiol 84:e02608-17. doi: 10.1128/AEM.02608-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu D, Huang C, Guo J, Zhang P, Chen T, Wang Z, Zhao X. 2019. Development and characterization of a CRISPR/Cas9n-based multiplex genome editing system for Bacillus subtilis. Biotechnol Biofuels 12:197. doi: 10.1186/s13068-019-1537-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guo T, Xin Y, Zhang Y, Gu X, Kong J. 2019. A rapid and versatile tool for genomic engineering in Lactococcus lactis. Microb Cell Fact 18:22. doi: 10.1186/s12934-019-1075-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y, Zhang ZT, Seo SO, Choi K, Lu T, Jin YS, Blaschek HP. 2015. Markerless chromosomal gene deletion in Clostridium beijerinckii using CRISPR/Cas9 system. J Biotechnol 200:1–5. doi: 10.1016/j.jbiotec.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 38.Xu T, Li Y, Shi Z, Hemme CL, Li Y, Zhu Y, Van Nostrand JD, He Z, Zhou J. 2015. Efficient genome editing in Clostridium cellulolyticum via CRISPR-Cas9 nickase. Appl Environ Microbiol 81:4423–4431. doi: 10.1128/AEM.00873-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu J, Wang Y, Lu Y, Zheng P, Sun J, Ma Y. 2017. Development of a CRISPR/Cas9 genome editing toolbox for Corynebacterium glutamicum. Microb Cell Fact 16:205. doi: 10.1186/s12934-017-0815-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cobb RE, Wang Y, Zhao H. 2015. High-efficiency multiplex genome editing of Streptomyces species using an engineered CRISPR/Cas system. ACS Synth Biol 4:723–728. doi: 10.1021/sb500351f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang H, Zheng G, Jiang W, Hu H, Lu Y. 2015. One-step high-efficiency CRISPR/Cas9-mediated genome editing in Streptomyces. Acta Biochim Biophys Sin (Shanghai) 47:231–243. doi: 10.1093/abbs/gmv007. [DOI] [PubMed] [Google Scholar]
- 42.Tong Y, Charusanti P, Zhang L, Weber T, Lee SY. 2015. CRISPR-Cas9 based engineering of actinomycetal genomes. ACS Synth Biol 4:1020–1029. doi: 10.1021/acssynbio.5b00038. [DOI] [PubMed] [Google Scholar]
- 43.Mougiakos I, Bosma EF, de Vos WM, van Kranenburg R, van der Oost J. 2016. Next generation prokaryotic engineering: the CRISPR-Cas toolkit. Trends Biotechnol 34:575–587. doi: 10.1016/j.tibtech.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 44.Oh JH, van Pijkeren JP. 2014. CRISPR-Cas9-assisted recombineering in Lactobacillus reuteri. Nucleic Acids Res 42:e131. doi: 10.1093/nar/gku623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leenay RT, Vento JM, Shah M, Martino ME, Leulier F, Beisel CL. 2018. Genome editing with CRISPR-Cas9 in Lactobacillus plantarum revealed that editing outcomes can vary across strains and between methods. Biotechnol J 14:e1700583. doi: 10.1002/biot.201700583. [DOI] [PubMed] [Google Scholar]
- 46.Zhou D, Jiang Z, Pang Q, Zhu Y, Wang Q, Qi Q. 2019. CRISPR/Cas9-assisted seamless genome editing in Lactobacillus plantarum and its application in N-acetylglucosamine production. Appl Environ Microbiol 85:e01367-19. doi: 10.1128/AEM.01367-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Song X, Huang H, Xiong Z, Ai L, Yang S. 2017. CRISPR-Cas9(D10A) nickase-assisted genome editing in Lactobacillus casei. Appl Environ Microbiol 83:e01259-17. doi: 10.1128/AEM.01259-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang H, Song X, Yang S. 2019. Development of a RecE/T-assisted CRISPR-Cas9 toolbox for Lactobacillus. Biotechnol J 14:e1800690. doi: 10.1002/biot.201800690. [DOI] [PubMed] [Google Scholar]
- 49.Crawley AB, Henriksen ED, Stout E, Brandt K, Barrangou R. 2018. Characterizing the activity of abundant, diverse and active CRISPR-Cas systems in lactobacilli. Sci Rep 8:11544. doi: 10.1038/s41598-018-29746-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hidalgo-Cantabrana C, Goh YJ, Pan M, Sanozky-Dawes R, Barrangou R. 2019. Genome editing using the endogenous type I CRISPR-Cas system in Lactobacillus crispatus. Proc Natl Acad Sci U S A 116:15774–15783. doi: 10.1073/pnas.1905421116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Altermann E, Russell WM, Azcarate-Peril MA, Barrangou R, Buck BL, McAuliffe O, Souther N, Dobson A, Duong T, Callanan M, Lick S, Hamrick A, Cano R, Klaenhammer TR. 2005. Complete genome sequence of the probiotic lactic acid bacterium Lactobacillus acidophilus NCFM. Proc Natl Acad Sci U S A 102:3906–3912. doi: 10.1073/pnas.0409188102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kok J, van der Vossen JM, Venema G. 1984. Construction of plasmid cloning vectors for lactic streptococci which also replicate in Bacillus subtilis and Escherichia coli. Appl Environ Microbiol 48:726–731. doi: 10.1128/AEM.48.4.726-731.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.De Vos WM. 1987. Gene cloning and expression in lactic streptococci. FEMS Microbiol Lett 46:281–295. doi: 10.1016/0378-1097(87)90113-3. [DOI] [Google Scholar]
- 54.Sturino JM, Klaenhammer TR. 2002. Expression of antisense RNA targeted against Streptococcus thermophilus bacteriophages. Appl Environ Microbiol 68:588–596. doi: 10.1128/aem.68.2.588-596.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Djordjevic G, Bojovic B, Miladinov N, Topisirovic L. 1997. Cloning and molecular analysis of promoter-like sequences isolated from the chromosomal DNA of Lactobacillus acidophilus ATCC 4356. Can J Microbiol 43:61–69. doi: 10.1139/m97-009. [DOI] [PubMed] [Google Scholar]
- 56.Selle K, Goh YJ, Johnson BR, O'Flaherty S, Andersen JM, Barrangou R, Klaenhammer TR. 2017. Deletion of lipoteichoic acid synthase impacts expression of genes encoding cell surface proteins in Lactobacillus acidophilus. Front Microbiol 8:553. doi: 10.3389/fmicb.2017.00553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lauer E, Kandler O. 1980. Lactobacillus gasseri sp. nov., a new species of the subgenus Thermobacterium. Zentralbl Bakteriol Mikrobiol Hyg Abt 1 Orig C 1:75–78. [Google Scholar]
- 58.Azcarate-Peril MA, Altermann E, Goh YJ, Tallon R, Sanozky-Dawes RB, Pfeiler EA, O'Flaherty S, Buck BL, Dobson A, Duong T, Miller MJ, Barrangou R, Klaenhammer TR. 2008. Analysis of the genome sequence of Lactobacillus gasseri ATCC 33323 reveals the molecular basis of an autochthonous intestinal organism. Appl Environ Microbiol 74:4610–4625. doi: 10.1128/AEM.00054-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pfeiler EA, Azcarate-Peril MA, Klaenhammer TR. 2007. Characterization of a novel bile-inducible operon encoding a two-component regulatory system in Lactobacillus acidophilus. J Bacteriol 189:4624–4634. doi: 10.1128/JB.00337-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hanahan D. 1985. Techniques for transformation of E. coli, p 109–135. In Glover DM (ed), DNA cloning: a practical approach, vol 1. IRL Press Ltd., Oxford, England. [Google Scholar]
- 61.Blin K, Pedersen LE, Weber T, Lee SY. 2016. CRISPy-web: an online resource to design sgRNAs for CRISPR applications. Synth Syst Biotechnol 1:118–121. doi: 10.1016/j.synbio.2016.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sarabipour S, King C, Hristova K. 2014. Uninduced high-yield bacterial expression of fluorescent proteins. Anal Biochem 449:155–157. doi: 10.1016/j.ab.2013.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grote A, Hiller K, Scheer M, Munch R, Nortemann B, Hempel DC, Jahn D. 2005. JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res 33:W526–W531. doi: 10.1093/nar/gki376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kimmel SA, Roberts RF. 1998. Development of a growth medium suitable for exopolysaccharide production by Lactobacillus delbrueckii ssp. bulgaricus RR. Int J Food Microbiol 40:87–92. doi: 10.1016/s0168-1605(98)00023-3. [DOI] [PubMed] [Google Scholar]
- 65.Goh YJ, Klaenhammer TR. 2013. A functional glycogen biosynthesis pathway in Lactobacillus acidophilus: expression and analysis of the glg operon. Mol Microbiol 89:1187–1200. doi: 10.1111/mmi.12338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wick RR. 2017. Porechop. https://github.com/rrwick/Porechop.
- 67.Andrews S. 2010. FastQC: a quality control tool for high throughput sequence data http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- 68.Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li H. 2018. seqtk. https://github.com/lh3/seqtk.
- 70.Wick RR, Judd LM, Gorrie CL, Holt KE. 2017. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol 13:e1005595. doi: 10.1371/journal.pcbi.1005595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li H. 2016. Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics 32:2103–2110. doi: 10.1093/bioinformatics/btw152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vaser R, Sovic I, Nagarajan N, Sikic M. 2017. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res 27:737–746. doi: 10.1101/gr.214270.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, Earl AM. 2014. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Seemann T. 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 76.Waterhouse RM, Seppey M, Simao FA, Manni M, Ioannidis P, Klioutchnikov G, Kriventseva EV, Zdobnov EM. 2018. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol Biol Evol 35:543–548. doi: 10.1093/molbev/msx319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Darling AC, Mau B, Blattner FR, Perna NT. 2004. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res 14:1394–1403. doi: 10.1101/gr.2289704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Barefoot SF, Klaenhammer TR. 1983. Detection and activity of lactacin B, a bacteriocin produced by Lactobacillus acidophilus. Appl Environ Microbiol 45:1808–1815. doi: 10.1128/AEM.45.6.1808-1815.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Casadaban MJ, Cohen SN. 1980. Analysis of gene control signals by DNA fusion and cloning in Escherichia coli. J Mol Biol 138:179–207. doi: 10.1016/0022-2836(80)90283-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Genome sequence data for the L. acidophilus NCFM ΔrafE (NCK2773), ΔlacS (NCK2774), and ΔltaS (NCK2676) mutants have been deposited in the NCBI Sequence Read Archive (SRA) database under BioProject ID PRJNA681755 (BioSample accession numbers SAMN16967693 to SAMN16967695).