

Abstract
The CADM family of proteins consists of four neuronal specific adhesion molecules (CADM1, CADM2, CADM3 and CADM4) that mediate the direct contact and interaction between axons and glia. In the peripheral nerve, axon-Schwann cell interaction is essential for the structural organization of myelinated fibres and is primarily mediated by the binding of CADM3, expressed in axons, to CADM4, expressed by myelinating Schwann cells. We have identified—by whole exome sequencing—three unrelated families, including one de novo patient, with axonal Charcot-Marie-Tooth disease (CMT2) sharing the same private variant in CADM3, Tyr172Cys. This variant is absent in 230 000 control chromosomes from gnomAD and predicted to be pathogenic. Most CADM3 patients share a similar phenotype consisting of autosomal dominant CMT2 with marked upper limb involvement. High resolution mass spectrometry analysis detected a newly created disulphide bond in the mutant CADM3 potentially modifying the native protein conformation. Our data support a retention of the mutant protein in the endoplasmic reticulum and reduced cell surface expression in vitro. Stochastic optical reconstruction microscopy imaging revealed decreased co-localization of the mutant with CADM4 at intercellular contact sites. Mice carrying the corresponding human mutation (Cadm3Y170C) showed reduced expression of the mutant protein in axons. Cadm3Y170C mice showed normal nerve conduction and myelin morphology, but exhibited abnormal axonal organization, including abnormal distribution of Kv1.2 channels and Caspr along myelinated axons. Our findings indicate the involvement of abnormal axon-glia interaction as a disease-causing mechanism in CMT patients with CADM3 mutations.
Keywords: Charcot-Marie-Tooth disease, CMT, CADM3, cell-adhesion molecule
Rebelo et al. identify a unique pathogenic variant in the gene encoding the neuronal adhesion molecule CADM3 in three unrelated families with axonal Charcot-Marie-Tooth disease (CMT2) with upper limb involvement. Extensive functional studies and a new mouse model reveal the contribution of the mutation to CMT2 pathogenesis.
Introduction
In the peripheral nervous system, axon-glia interactions are essential for myelination and organization of the different axonal domains required for efficient saltatory conduction.1 Distinct sets of cell-cell adhesion molecules are known to mediate the interaction between axons and Schwann cells in different nerve domains. Those molecules are well characterized in the paranodal and juxtaparanodal regions. In the paranodal region the axonal proteins Caspr and contactin interact with neurofascin 155 (NF155) in glia, and in the juxtaparanodes Caspr2 binds to Tag1.1 In the internodes, the axon-glia interaction is mediated by more recently characterized cell adhesion molecules, the CADM family of proteins, also known as nectin-like (Necl) and synaptic cell adhesion (SynCAM) molecules.2,3 CADM proteins consist of four members that are differentially expressed in Schwann cells and axons; CADM1 (Necl2), CADM2 (Necl3), and CADM3 (Necl1) are found in axons, while CADM2 and CADM4 (Necl4) are present in myelinating Schwann cells. CADM proteins are composed of three extracellular immunoglobulin-like (Ig) domains where cell-cell adhesion takes place, a transmembrane domain and a short cytoplasmic region.4 Cell-cell contact is made through different combinations of homophilic and heterophilic interactions. However, the predominant interaction is mediated between CADM3 expressed in axons and CADM4 located on Schwann cells.2,3,5 Those interactions are essential for long-range axon-guidance, myelination and maintenance of the axonal architecture domain. Besides cell-cell adhesion, CADM proteins also play a role as cell signalling molecules. They can inhibit neuregulin-mediated activation of the PI3K/AKT pathway.6 Interestingly, ablation of CADM4 in mice results in myelin abnormalities and phenotype that resembles Charcot-Marie-Tooth disease (CMT).7 Mice lacking CADM4 exhibit impaired motor function, slower nerve conduction velocity and myelin abnormalities including neuropathy-like focal hypermyelination, and developed abnormal axon-glial contact and axonal organization.7 Severe myelin abnormalities, including very short internodes were also observed in transgenic mice expressing a dominant-negative mutant of CADM4 lacking its cytoplasmic domain (Cadm4dCT).7,8 Furthermore, CADM proteins collaborate with other axon-glia CAMs to regulate oligodendrocyte myelination in the CNS.9 In contrast, mice lacking the other Cadm family of genes, Cadm1, Cadm2 and Cadm3, showed no myelin or any other neurological abnormalities,7 suggesting genetic redundancy in mice. However, according to gnomAD, the four CADM genes in humans have high constraint for loss-of-function mutations, displaying extremely intolerant loss-of-function metrics (pLI ≥ 0.9 and observed/expected < 0.2), suggesting that humans are less tolerant to loss of CADM genes.
Here we report a pathogenic variant in CADM3 in three unrelated families sharing the same unusual CMT disease with predominant upper limb involvement and atypical pyramidal features. Our genetic findings and functional studies unveil a novel pathological mechanism in CMT involving axon-glial interactions.
Materials and methods
CMT patients and families
All study participants were ascertained by board-certified neurologists M.E.S. and M.M.R. All subjects’ consent was obtained according to the Declaration of Helsinki and it was approved by the ethical committee of the institution in which the work was performed.
Whole-exome and Sanger sequencing
Whole-exome sequencing was performed in the probands for the three CMT families. The SureSelect Human All Exon Kit (Agilent) was used for in-solution enrichment, and the HiSeq 2500 instrument (Illumina) was used to produce 100 bp paired-end sequence reads. The Burrows-Wheeler aligner (version: 0.7.10) and Freebayes (version 1.1.0) were used for sequence alignment and variant calling. Whole-exome data were uploaded into the GENESIS 2.0 software and analysed using strict filters for allele frequency, function and conservation. Mutations in known CMT-associated genes were absent in all families (Supplementary Table 1). Sanger sequencing confirmed the missense variant in CADM3 detected by whole-exome sequencing, and it also revealed de novo inheritance mode of segregation in one family and autosomal dominant segregation in another family.
Paternity test
Parental consents (paternal and maternal) were obtained according to the Declaration of Helsinki and were approved by the ethical committee of the institution in which the proband was evaluated. Fragment analysis was performed to verify paternity and maternity of Family 1 in order to confirm de novo mutation in the proband. Five different microsatellite markers were amplified by multiplex PCR using the Qiagen multiplex PCR kit: D6S1552, D6S1624, D61517, MOG-CA. PCR products were run on an ABI3730 automated sequencer (Applied Biosystems) using LIZ600 (orange) size standard. Results were analysed with GeneMapper (version 4.0).
Generation of knock-in mice
Knock-in mice containing the human CADM3 mutation Y172C were generated using the CRISPR/Cas9 system. SgRNA (crRNA and tracrRNA) and repair DNA oligonucleotides were ordered from IDT. All reagents were prepared according to the manufacturer’s instructions and were microinjected or electroporated with a cas9 purified protein, into fertilized oocytes of C57BL/6J mice. SgRNA sequence CCACAGAAACCCATAATCAC, repair DNA sequence TGAACTGTGACTTCATGGTGCCTCTCCTTCCTATCCTGGCCA TTCCCTATGCATGGCAGGGATCCCTCAGAAGCCTATTATTACTGGTTGTAAGTCATCATTGCGGGAAAAGGAGACAGCCACTCTAAATTGTCAGTCTTCTGGGAGCAA. The repair sequence included silent mutations in the area corresponding to the guide sequence including a restriction enzyme site. Mosaic mice were screened by primers that were specific for the edited allele (forward: CTCCACCCCACCTACACTAC; reverse: GTAATAATAGGCTTCTGAGG), and with primers flanking the edited area of which the products were further used for restriction fragment length polymorphism (RFLP) (forward: CTCCACCCCACCTACACTAC; reverse: TGTCTGCCATTCATTTCCACT, product was cut with BamHI). Positive mosaics confirmed by Sanger sequencing were crossed on wild-type C57BL/6J mice. All positive F1 mice were sequenced as well (as mosaic mice can contain more than one form of edited alleles). A single sequence-verified male and female F1 mice were crossed to establish a working colony, in parallel a single sequence-verified male was crossed on wild-type females to establish a family crossed continuously on a wild-type background. All mice were genotyped at 3 weeks after birth. Mice were monitored daily by staff and veterinary personnel for health and activity. Mice were given ad libitum access to water and standard mouse chow with 12-h light/dark cycles. Colonies were maintained in a specific pathogen-free (SPF) barrier facility, with quarterly testing of pathogens in sentinel animals housed in the same room. All animal procedures were approved by the Institutional Animal Care and Use Committee and performed in strict adherence to Weizmann Institute Animal Care and Use guidelines, following the NIH, European Commission, and Israeli guidelines. Cadm3 (Cadm3tm1.1Pele) and Cadm4 (Cadm4tm1.1Pele) mice were previously described.7 Mice carrying the Y170C mutation were designated Cadm4Y170C.Pele.
For reverse transcription (RT)-PCR analysis, total RNA was isolated from freshly dissected brains using TRI Reagent® (Sigma-Aldrich), cDNAs were obtained with SuperScriptTM II reverse transcriptase (Invitrogen) using oligo-dT. Specific PCR primer sets were designed according to mRNA sequences (NCBI). The following primer pairs were used for mRNA-based RT-PCR analysis: Cadm3 wild-type allele CCTATTTAATCCCGGCGACTGC and TAACCAGTGATTATGGGTTTCTG, mutant allele CCTATTTAATCCCGGCGACTGC and GTAATAATAGGCTTCTGAGGG; actin GTGGTGGTGAAGCTGTAGCCACGCT and GAGCACCCTGTGCTGCTCACCGAGG.
Generation of constructs
Plasmids encoding the open-reading frame (ORF) of CADM3 transcript1 (NM_021189.4) fused with either an HA tag or GFP tagged at the C-terminus end were obtained from Genecopoeia. Plasmid encoding the open-reading frame of CADM4 transcript 1 (NM_145296.1) fused with a myc-tag at the C-terminus was also obtained from Genecopoeia. Site-direct mutagenesis was performed to generate plasmids with the patients’ mutation corresponding to Tyr172Cys. Point mutation was introduced by PCR using the Q5 Site-Direct Mutagenesis kit (NEB) with the following primers: 5′-ATCACTGGTTGTAAATCTTCATTACG-3′ and 5′-GATGGGCTTCTGTGGAATTC-3′. Addition of N-terminus HA tag and the A519G mutation to rat Cadm33 (corresponding to rat and human Tyr170Cys and Tyr172Cys, respectively), were generated using the Site Directed Mutagenesis (SDM) TPCR. Primers (5′-GCGCCCGGCGGGGCCAATTACCCATACGATGTTCCAGATTACGCTCTTTCCCAGGACGGCTACTG-3′; 5′-TTCCCGCAACGATGACTTACAACCAGTGATTATGGGTTTC-3′) and (5-GCAGGCGAGGAGCAGGAGC-3′; 5′-GGGAGCAAACCTGCAGCC-3′) were used. The addition of C-terminus Myc tag was done by PCR using (5′-ACGACTCACTATAGGGAGACC-3′) and (5′-CTAGCGGATCCTACAGATCCTCTTCTGAGATGAGTTTTTGTTCGATGAAATATTCCTTCTTGTCATC-3′) primers. The ORF of all constructs was confirmed by DNA sequencing.
Endoplasmic reticulum stress analysis
RT4 Schwann cells stably expressing a dual luciferase reporter assay of XbpI splicing10 were used to monitor activation of the unfolded protein response (UPR). CADM3 constructs were transfected with Lipofectamine 3000 (Thermofisher) into RT4 Schwann cells. Firefly luciferase and nano-luciferase activity was assayed as described by Bai et al.10 Endoplasmic reticulum (ER) localization was analysed by confocal microscopy in cells co-stained with an HA antibody and the ER marker calnexin.
Co-immunoprecipitation assay and western blot
HEK293T cells, 48–72 h after transfection, were lysed in solubilization buffer (50 mM HEPES pH 7, 150 mM NaCl, 5 mM EGTA, 10% glycerol, 1% TritonTM X-100 and 1% Proteinase Inhibitors mix) (Calbiochem 539134). Lysates were centrifuged for 20 min at 4°C and the supernatants were kept at −20°C. Sample buffer ×5 (0.5 M Tris HCl pH 6.8, 50% glycerol, 5% SDS and 3.7% DTT) was added and the samples were boiled for 5 min and were loaded for separation on a 10–12% polyacrylamide gel. The gel was transferred to a polyvinylidene difluoride (PVDF) membrane for blotting. The blots were blocked in TBS-T (Tris-buffered saline with 0.1% Tween 20) containing 3% bovine serum albumin (BSA) for 1 h at room temperature and incubated for 1 h at room temperature and then for 1 h or for overnight at 4°C with different primary antibodies diluted in the blocking buffer. Following three washes in TBS-T, blots were incubated with horseradish-peroxidase-coupled secondary antibodies for 30 min at room temperature and were washed again with TBS-T.
Three-month-old mice SNs were dissected and immediately frozen in dry ice. Homogenization was performed in RIPA buffer (25 mM Tris-HCl pH 7.4, 1% NP40, 0.5% sodium deoxycholate, 0.1% SDS,150 mM NaCl) supplemented with 1% protease inhibitors cocktail (Calbiochem 539134). Homogenates were incubated on ice for 40 min, centrifuged at 21 000g for 30 min and supernatant was collected and kept at −80°C.
For the preparation of mice brain membranes, 3-month-old mice brains were homogenized in membrane homogenization buffer (20 mM HEPES pH 7.5, 0.32 M sucrose, 1 mM EGTA, 1.5 mM MgSO4) supplemented with 1% protease inhibitors cocktail (Calbiochem 539134). The homogenates were centrifuged at 1000g for 10 min at 4°C for removal of nuclei and heavy cell debris. Supernatants were collected and centrifuged at 20 000g for 1 h at 4°C. The pellets were resuspended in membrane extraction solubilization buffer (2% NP40, 2 mM MgCl2, 2 mM PMSF) supplemented with 1% protease inhibitors cocktail (Calbiochem 539134) and kept at −80°C. Protein concentration was determined using a BCA kit (Pierce). Sample buffer X5 (0.5 M Tris-HCl pH 6.8, 50% glycerol, 5% SDS, 3.7% DTT and bromophenol blue) was added and the samples were boiled for 5 min. Equal amounts of protein were loaded for separation on a 10% SDS-PAGE gel and transferred to PVDF membrane [semi-dry transfer using Trans-Blot Turbo system (25 V, 10 min)] (Bio-Rad). Membranes were then incubated with blocking solution (3% BSA in TBS-T) for 30 min at room temperature and then reacted for 1 h at room temperature or overnight at 4°C with the appropriate primary antibodies diluted in TBS-T. Membranes were then washed in TBS-T (3 × 5 min) and incubated with horseradish-peroxidase-coupled secondary antibody for 45 min at room temperature. Membranes were washed again and reacted with ECL (Thermo Scientific). Membranes were visualized using the ChemiDoc MP imaging system (Bio-Rad).
Extraction of membrane proteins
Transfected HEK293T cells were lysed with homogenization buffer (20 mM HEPES pH 7.5, 0.32 M sucrose, 1 mM EGTA, 1.5 mM MgSO4, 1% proteinase inhibitors mix) (Calbiochem 539134) using homogenizer. The lysates were centrifuged at 3000g for 10 min at 4°C to remove nuclei and cell debris. The supernatants were collected and centrifuged again for 60 min at 20 000g. The pellets were resuspended in solubilization buffer (2% IGEPAL, 2 mM MgCl2, 1% proteinase inhibitors mix), incubated for 30 min with alternate vortex and again centrifuged at 20 000g for 30 min. Samples were kept at −20°C and later analysed by immunoblot, as described above, using rabbit anti HA tag antibody.
Cell surface biotinylation
The surface biotinylation assay was carried out on transfected HEK293T cells at 48–72 h after transfection. Cells were washed three times with ice-cold PBS+Ca+Mg pH 8 and biotinylated with 1 mg/ml Sulfo-NHS-biotin (Thermo Scientific, 21217) as previously described.11 Proteins were immunoprecipitated by anti-Myc tag antibody and anti-mouse IgG agarose beads (Sigma, A6531) rotated overnight at 4°C. The beads were washed once with ice-cold solubilization buffer and twice with ice-cold TNTG (50 mM Tris, 150 mM NaCl, 0.1% TritonTM X-100, 10% glycerol) and eluted with the SDS-PAGE sample buffer following 5 min boiling. Samples were then subjected to western blot analysis, as described above. Membranes were blotted with horseradish-peroxidase-coupled streptavidin for biotinylated protein detection.
Binding assay
For Fc-fusion protein binding experiments, transfected COS7 cells were washed with fresh Dulbecco’s modified Eagle medium (DMEM) and incubated for 45 min at room temperature with conditioned media containing 0.5–1 μg/ml of the different Fc-fusion CADM proteins, preincubated with Cy3 anti human Fc antibody. The cells were washed twice with PBS to remove unbound proteins and were analysed by immunofluorescence, as previously described.
Electron microscopy
Wild-type and homozygous mice were sacrificed, and their sciatic nerves exposed and fixed by continuous dripping of fresh Karnovsky fixative for 20 min (fixative containing 4% paraformaldehyde, 0.1 M sodium cacodylate, and 2.5% glutaraldehyde, pH 7.4, in PBS). Sciatic nerves were then carefully dissected, placed in fresh fixative, and left to rotate overnight at room temperature. Samples were then transferred to 4°C until processed by an electron microscopy professional, as previously described.12 Semithin sections (1.5 μm) were stained with toluidine blue and analysed under a light microscope (Nikon Eclipse E800).
Electrophysiology
Sciatic nerve conduction velocity measurements were performed by M.E.S. (University of Iowa, IA USA) on adult wild-type (n = 5) and homozygous (CADM3Y172C/Y172C) mice (n = 3). Mice were anaesthetized with a mix of ketamine/xylazine (87.5 mg/kg ketamine, 12.5 mg/kg xylazine). Anaesthetized mice were placed on a heating pad (constant 37°C) to maintain constant body temperature throughout the electrophysiological study (Neuromax, XLTEK). Sciatic nerve motor nerve conduction velocity was recorded stimulating a distal (ankle) and a proximal (sciatic notch) site along the nerve with platinum monopolar needle electrodes. One pair of stimulating electrodes was positioned percutaneously at the sciatic notch. A second pair was inserted adjacent to the tibial nerve at the ankle. Compound muscle action potentials (CMAPs) were recorded from the intrinsic foot muscle using needle electrodes. CMAP amplitudes were measured from baseline to the peak of negative deflection.
Disulphide bond analysis by mass spectrometry
HEK293T cells were transfected with either Y172C-CADM3-HA or Y132C-CADM3-HA. Protein lysates were extracted and immunoprecipitated with magnetic beads containing HA antibodies. Immunoprecipitation eluate was submitted for analysis to MtoZ Biolabs. Briefly, protein samples were digested with trypsin and analysed on liquid chromatography with tandem mass spectrometry (LC-MS-MS), as previously described.13 Raw data were analysed by BiopharmFinder 4.0.
Transfection and immunocytochemistry
L-cells stably transfected with the CADM3 constructs—either wild-type, WT-CADM3-GFP, or mutant, Y172C-CADM3-GFP—were plated in glass-bottom 4-well chambers (Ibidi). Cells were co-transfected with CADM4-Myc using Lipofectamine® 3000 (ThermoFisher). Twenty-four hours after transfection cells were fixed with 4% paraformaldehyde (PFA) and permeabilized with cold methanol. Cells were incubated with primary antibodies, diluted in 2% BSA for 2 h. Cells were subsequently washed three times with PBS and incubated with the secondary antibodies. COS7 and HEK293T cells were grown in DMEM medium (Gibco), which contained 10% foetal bovine serum (FBS; Gibco) and 1% Penicillin/Streptomycin (Gibco). HEK293T transfection was preformed using the calcium phosphate method and COS7 transfection was carried out using Lipofectamine® 2000 reagent (Invitrogen). Transfected COS7 cells seeded on 13 mm glass cover slips (Thermo Scientific) were fixed in 4% PFA for 10 min and were blocked for 1 h in blocking solution (5% normal goat serum without 0.1% TritonTM X-100 in PBS) at room temperature. Fixed cells were then washed in PBS and incubated for 1 h at room temperature or overnight at 4°C with the relevant mixture of primary antibodies diluted in blocking solution. Coverslips were then washed in PBS and incubated with secondary antibodies diluted in blocking solution for 45 min at room temperature. Coverslips were washed again in PBS and then mounted with Fluoromount-GTM (SouthernBiotech).
Nerve immunofluorescence
Sciatic nerves were dissected and immersed in 4% PFA for 20 min at room temperature. Nerves were transferred to 20% sucrose (in PBS) and stored at 4°C till teasing. Nerves were desheathed and teased using fine forceps on SuperFrost PlusTM slides (Menzel-Glaser; Thermo Scientific), air dried overnight, and then kept frozen at −20°C until use. When staining for Kv1.2 or NrCAM, samples were postfixed for 2–5 min using cold methanol (−20°C) and washed with PBS. Samples were incubated for 1 h in blocking solution (5% normal goat serum and 0.1% TritonTM X-100 in PBS) at room temperature. If no post-fixation was performed, blocking solution contained 0.5% TritonTM X-100. Samples were incubated overnight at 4°C with the relevant mixture of primary antibodies diluted in blocking solution (5% normal goat serum and 0.1% TritonTM X-100, in PBS). Nerve samples were then washed in PBS and incubated with secondary antibodies for 45 min at room temperature (same blocking solution as for the primary antibodies). Samples were washed again in PBS, mounted as above, and observed using an Axioskop2 microscope equipped with an ApoTom imaging system (Carl Zeiss) fitted with a Hamamatsu ORCA-ER CCD camera. Images were acquired and processed using the Zen 2012 software (Carl Zeiss), Photoshop CC2019 software (Adobe) and PowerPoint 2019 software (Microsoft).
Antibodies
Primary antibodies that were used: monoclonal rabbit anti HA (Cell Signaling, C29F4), mouse anti-Myc (9E10), anti-GFP (Cell signaling), rabbit anti-extracellular domain Cadm3 (190), rabbit anti-extracellular domain of Cadm3 (polyclonal antibodies 876 and 8783), mouse anti-GAPDH (Millipore, MAB374), rabbit anti-Kv1.214 rabbit anti-Caspr (polyclonal antibodies 60-6215), and rat anti-NrCAM (RP12B316). Fluorophore-coupled secondary antibodies were purchased from Jackson Laboratories. Alexa Fluor 555 from ThermoFisher, and Janelia Fluor 646 from Novus Biologicals. Fc fusion CADM4 protein was used for the binding experiments.3
STORM imaging
Imaging experiments were carried out using a Nikon eclipse Ti2 microscope equipped with Nikon Instruments (NSTORM). For two colour dSTORM imaging, Janelia 646, and Janelia 549 secondary antibodies were used with MEA STORM imaging buffer and were imaged continuously with 10 000 frames collected per filter range at a frequency of 20 ms. The images were acquired using a 100×, 1.49 NA objective, and imaged onto a Hamamatsu C11440 ORCA-flash 4.0 camera. Stochastic optical reconstruction microscopy (STORM) analysis was carried out with Nikon Elements Analysis 5.02.01 for identification of molecules. Molecule list files were then exported from Nikon elements to be further analysed using software Clus-Doc software in MATLAB R2018b. Cluster analysis, specifically DBSCAN function, was carried out after manually selecting the region of interest.
Data availability
The data presented in this study are available from the corresponding author upon request.
Results
Identification of CADM3 as a disease-causing gene
Whole-exome sequencing was performed in three unrelated index patients diagnosed with unusual neuropathy with marked upper limb involvement (Table 1 and Supplementary Tables 2 and 3). Pathogenic variants in known CMT genes were absent in the three families. Sequencing data were processed and analysed by bioinformatics tools including BWA aligner, FreeBayers, GATK and GENESIS.17 The GENESIS platform currently harbours 9569 whole exomes/genomes from individuals with different phenotypes, including 1257 CMT affected individuals. CADM3 was identified by using a strategy of searching for very rare (‘private’) alleles shared across these CMT families. The required filters were non-synonymous variants, never observed in gnomAD [MAFgnomad = 0, highly conserved (GERP > 3)]18 and predicted to cause functional damage (combined annotation dependent depletion, CADD > 25).19 This approach yielded only one heterozygous variant: c.515A>G (NM_021189.4), p.Tyr172Cys (chr1:159163243, hg19), which was confirmed by Sanger sequencing (Fig. 1A). This CADM3 variant co-segregated with disease in all three families. The sporadic patient in Family 1 carried Tyr172Cys as a de novo variant; confirmed by paternity testing (Fig. 1A and Supplementary Fig. 1), which suggests an independent mutation event rather than a founder effect. Family 2 was sporadic, and DNA of the parents was not available. Family 3 showed dominant segregation of mutation in both affected individuals (mother and son) (Fig. 1A). This variant is highly conserved across species, predicted to be pathogenic by seven different prediction tools including DANN, DEOGEN2, EIGEN, FATHMM-MKL, M-CAP, MutationAssessor and MutationTaster. A variant is predicted to be ‘likely pathogenic’ (>90% certainty) based on ACMG21 standards (PS2, PM2 and PP3). Further, the Tyr172Cys allele was absent from our internal controls (8312 non-CMT WES/WGS chromosomes on the GENESIS platform). A Fisher’s exact test statistic formally supports a significance value of P < 0.01 (Supplementary Table 2).
Table 1.
Clinical features of CADM3 patients
| Individuals | Age at exam, years | CMTNSv2 | Distal weakness LL | Proximal weakness LL | Distal weakness UL | Proximal weakness UL | Vibration LL | Vibration UL | Pinprick LL | Pinprick UL | Central signs | Age of onset |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Family 1-II:1 | 30 | 18 | 3,5 | 5,5 | 0,0,0 | 5,5,5 | Abs toes (0/8) | Normal | Reduced ankle | Normal | None | 1 year |
| Family 2-II:1 | 7 | ND | 0,0 | 5,5 | 4,5,5 | 5,5,5 | Normal toes (8/8) | Normal | Normal | Normal | None | 3 years |
| Family 3-II:1 | 14 | ND | 1,4 | 5,5 | 1,2,3 | 5,5,5 | Knee (4/8) | Normal | Reduced ankle | Reduced to wrist, ulnar nerve distribution | Brisk reflexes | 3 years (slow runner) |
| Family 3-I:2 | 49 | ND | 3,4,3 | 5,5 | 4+,5,5 | 5,5,5 | Normal | Normal | Normal | Reduced right ulnar nerve, finger tips left | Brisk reflexes | 29 years (ulnar neuropathy) |
Motor weakness based on MRC scale (0–5). Lower limb distal weakness assessed by anterior tibialis and gastrocnemius; lower limb proximal weakness assessed by ilio psoas and quadriceps; upper limb distal weakness assessed by first dorsal interosseous, abductor pollicis brevis, and adductor digiti minimi, upper limb proximal weakness assessed by deltoids, biceps brachii, and triceps. Vibration based on Rydell tuning fork with ‘5’ on scale of ‘8’ being considered normal. Both motor and sensory evaluations were based on worst score observed of the two limbs. CMTNSv2 scores are separated into <10 (mild), 11–20 (moderate) or >20 (severe) impairment.20 Abs = absent; LL = lower limb; ND = not done; UL = upper limb.
Figure 1.

CADM3 mutation in CMT2 families. (A) Pedigree and Sanger sequencing traces of CMT2 families sharing the same heterozygous CADM3 variant Tyr172Cys. (B) Diagram showing CADM3 mutation at exon 5 (c.515A>G) and its corresponding position in the protein (Y172C) located in the extracellular domain between Ig1 and Ig2 loops. (C) Diagram showing native disulphide bonds represented by connected arrows (green) and new disulphide bond C172-C186 (red) generated by the mutation Y172C, which was detected by liquid chromatography with tandem mass spectrometry (LC-MS-MS).
Clinical description of individuals with CADM3 variants
Family 1
Patient II:1 was a 30-year-old female presenting with a pronounced neuropathy affecting upper and lower extremities. She was born 3 months prematurely weighing only 2 lb 9 oz and required intubation and care in the intensive care unit. She had a patent ductus arteriosus that required surgery. She did not walk independently until 18 months of age, required bracing to walk between the ages of 2 and 3 years and ankle foot orthotics (AFOs) to ambulate between the age of 5 and 12. She had multiple surgeries in both ankles after the age of 12 throughout her teenage years including tendon transfers and eventually bone fusions on the right ankle. She also had tendon transfers on the right forearm. As a child she had pronounced difficulties with balance. She required adaptations on her bicycle at 8 years of age because she was unable to use hand or foot brakes. At age 30 she utilizes AFOs to ambulate and has virtually no use of her left hand for fine motor movements. On neurological examination she has mild scoliosis, pronounced atrophy in both arms below her elbows and in both legs below her knees. Her left wrist strength was weak to extension and flexion (4−/5) while her right wrist extension was 4− but 5/5 for flexion. Left grip strength was 3/5 and 4+/5 on the right. Finger extensors were 2/5 on the left and 4−/5 on the right. Her intrinsic hand muscles, abductor pollicis brevis, first dorsal interosseous and abductor digiti minimi were all 0/5 on the left. Her abductor pollicis brevis was 3/5 on the right and the first dorsal interosseous and abductor digiti minimi were 1/5. In the lower extremities her anterior tibialis was 4/5 on the left and 4+/5 on the right. Her ankle extenders were 3+/5 bilaterally and inversion was full strength. She had no movement of either great toe. Touch sensation was reduced at both great toes but normal in her ankles and fingers. Vibratory sensation was absent at her toes, but normal at her ankles and fingers. Position sense was reduced in her fingers and left great toe. Deep tendon reflexes were absent at her ankles but present in her knees and upper extremities. There is no family history of these symptoms.
Family 2
Patient II:1 was a 7-year-old male who was first noted to have weakness at 3 years of age. Early developmental milestones were normal, including rolling by 5 months, sitting by 6 months, walking by 12 months, normal fine motor and language skills. He gradually developed complete foot drop and cannot walk without AFOs. He denies sensory symptoms. On neurological examination he has facial weakness, nasal speech, no voluntary movement in the lower extremities below his knees and severe foot deformity. He has wrist and finger contractures with moderate weakness in his first dorsal interosseous muscle of the hand. While he had weakness in wrist extension and flexion, his contractures prevented grading strength for these muscles. Vibratory sense was decreased in fingers bilaterally, position sense and cutaneous sensation were normal in all four extremities. Deep tendon reflexes were absent. Nerve conduction studies were mildly reduced in the ulnar and peroneal nerves with pronounced reduction of the CMAP amplitudes and were slow in the median nerve with a markedly reduced CMAP amplitude (Supplementary Tables 2 and 3). Sensory nerve action potential amplitudes were unobtainable. He also had nerve and muscle biopsy. A sural nerve biopsy showed a reduced number of large myelinated fibres and very few groups of small regenerating fibres (Fig. 2). Of interest, we observed a number of abnormally myelinated fibres with thick folded myelin and focal myelin swelling or hypermyelination. The muscle biopsy reported a neurogenic atrophy.
Figure 2.

Nerve biopsy from a CADM3 patient. (A) A cross-section of sural nerve from a patient of Family 2 showing a reduced number of large myelinated fibres and very few groups of small regenerating fibres. Of interest, we overserved a number of abnormally myelinated fibres with thick folded myelin. A teased fibre from the same biopsy (B) shows such a focal myelin swelling or hypermyelination, a histological feature of demyelinating peripheral neuropathies.
Family 3
Patient II:1 had normal birth and milestones. Around the age of 3 he was noted to have flat feet and between 3 and 4 years of age, became slow at running and began to turn in his feet. He was prescribed AFOs at the age of 5 and from the age of 8 was no longer able to run. He had multiple operations in his feet, including bilateral arthrodesis at the age of 11. He also complained of impaired dexterity of his hands from the first decade of life. There was no mention of sensory symptoms in the lower limbs, but he complained of sensory symptoms in the ulnar distribution bilaterally, which improved after ulnar nerve decompression, although muscle weakness did not. Examination at the age of 14 years showed distal atrophy and severe weakness of intrinsic hand muscles, ankle dorsiflexion and eversion, but preserved strength of ankle plantar flexion and of proximal muscle groups (Table 1). Reflexes were brisk throughout. Tone was normal and plantars were down-going. MRI of transversal T2 section of the brain and sagittal T2 section of the cervical spine showed normal appearance of the brain and cervical spine (Supplementary Fig. 2). Pinprick sensation was normal in the lower limbs but reduced in the ulnar nerve distribution bilaterally, being worse on the left where is also involved other nerve territories up to the distal arm. Vibration sensation was also reduced in the lower limbs up to the knee.
His mother, Case I:2 now aged 49, had no definite symptoms until the fourth decade of life, when she started complaining of cramping in her right hand and progressive sensory loss in the ulnar nerve territory. She underwent two right ulnar nerve decompression, which improved the sensory symptoms, although weakness in her right hand continued to progress. In the last decade she noticed similar symptoms in her left hand. When asked, she admits she always thought she had thin hands compared to the rest of her body and as a teenager she used to get occasional cramping in her legs. Her current examination showed distal atrophy and moderate weakness in her hands and reduced sensation distally in her upper limbs, worse on the right and in the ulnar nerve territory (Supplementary Table 3). Lower limb muscle bulk, strength and sensation were normal, but she had difficulty walking on her heels. Reflexes were pathologically brisk throughout. Muscle tone was normal and Babinski reflex was absent.
In conclusion, Cases II-1 and I-1 had a phenotype consistent with Silver syndrome, e.g. an upper limb predominant motor predominant sensory-motor neuropathy with brisk reflexes throughout. There was a prominent involvement of ulnar nerves, with partial improvement of sensory symptoms after ulnar nerve decompression. Nerve conduction studies were consistent in both cases with an axonal sensory-motor neuropathy with intermediate reduction of conduction velocities (Supplementary Tables 2 and 3).
Tyr172Cys mutation interferes with the native disulphide bond conformation
The mutation is located in the extracellular domain adjacent to the second Ig (Ig2) loop, essential for cell-cell adhesion (Fig. 1B). The Ig-like loops are formed by three pairs of cysteines connected by disulphide bonds at positions 84–144, 186–243, 288–333 (Fig. 1C). All six cysteines are conserved across the four different CADMs showing multiple sequence alignment (Supplementary Fig. 3). Interestingly, those are the only cysteines present in the entire CADM3 sequence, with the exception of the cysteines located at the membrane signal peptide that is supposed to be cleaved.22 Because the mutation creates a new cysteine, we hypothesized that this new cysteine forms a disulphide bond interfering with conformation of native disulphide bonds. To test this hypothesis, HEK293T cells were transfected with a plasmid encoding the HA-tagged human or the Y172C mutant CADM3 to pull-down the proteins using an HA antibody. The purified CADM3 proteins were then digested with trypsin and subjected to liquid chromatography with tandem mass spectrometry (LC-MS-MS). Analysis of the mass spectrometry data identified a non-native disulphide bond between the mutant site 172C and 186C (Fig. 1C and Supplementary Fig. 4). In addition, the native disulphide bond between 186C and 243C was also detected, suggesting either disulphide bond shuffling or co-occurrence of different folded molecules.
CADM3 mutation activates the unfolded protein response
To investigate whether the disulphide bond rearrangement could cause activation of the UPR due to protein misfolding, we used a dual luciferase reporter assay of XBP1 splicing using stably transfected RT4 Schwann cells.10 These cells encode XBP1, a key modulator of the UPR, fused to luciferase reporter (firefly and nano-luciferase) sequences, which are separated by an intronic sequence. XBP1 is fused out of frame with the luciferases, and both are pulled into frame only when XBP1 is spliced. During the process of UPR, IRE1α is activated and it splices XBP1, leading to the coding sequence of XBP1 to come into frame with the reporters, resulting in expression of the luciferase. Dual reporter RT4 cells were transiently transfected with cDNAs expressing both wild‐type (WT-CADM3-HA) and mutant (Y172C-CADM3-HA) CADM3. Results showed that both firefly luciferase and nano luciferase levels in the mutant were significantly different to those obtained from transfections with the wild‐type CADM3 (P < 0.001, two-tailed t-test) (Fig. 3A). This result suggests that a large fraction of the protein is retained in the endoplasmic reticulum triggering the UPR. Cells transfected with the mutant construct have similar CADM3 protein expression levels compared to cells transfected with the wild-type construct, as shown by western blot (Supplementary Fig. 5). To analyse the subcellular localization of CADM3, cells were transfected with both wild-type and mutant CADM3 plasmids and immunofluorescence labelled with the endoplasmic reticulum marker, GRP78-BiP. Confocal images show that both wild-type and mutant CADM3 cytoplasmic staining co-localizes in the endoplasmic reticulum in U2OS transfected cells (Fig. 3B). When U2OS cells are co-transfected with both CADM3 and CADM4 (CADM4-myc) plasmids, a fraction of the CADM3 protein (both wild-type and mutant) is targeted to the membrane at cell-cell contact sites, where it co-localized with CADM4, although a large fraction remains retained in the cytoplasm (Fig. 3C). In contrast, CADM4 appears more enriched at the membrane with minimum retention at the cytoplasm.
Figure 3.

Overexpression of CADM3 mutant activates the unfolded protein response. (A) Dual reporter luciferase RT4 cells were transiently transfected with plasmids encoding the human wild‐type (WT) and mutant (Y172C) CADM3. Increased levels of firefly and nano luciferase were observed in the mutant CADM3. (B) Immunofluorescence of U2OS transfected cells shows co-localization of both wild-type and mutant CADM3 with the endoplasmic reticulum marker GPR78-BiP. (C) U2OS cells co-transfected with CADM3 and CADM4 show CADM3 co-localization with CADM4 at cell-cell contact sites.
CADM3 mutant reaches the cell surface and binds CADM4
We have previously shown in rodents that glial CADM4 preferentially bind axonal CADM3 and CADM2.3,8 To examine whether the human mutation in CADM3 affects its ability to interact with CADM4, we mutated the corresponding residue in the rat Cadm3 cDNA (Y170C). The difference in the position of the mutation between rodents (Y170; XP_006250385.1) and humans (Y172; NP_067012.1) is due to the presence of two additional amino acids at position 5 of the human cDNA. Wild-type and mutant CADM3 proteins carrying extracellular HA and intracellular Myc tags, were expressed in HEK293T transfected cells and as expected, were enriched in the cell membrane fraction (Fig. 4A and Supplementary Fig. 6). However, in contrast to the HA-tag antibody, which recognized both the wild-type and the mutated CADM3, an antibody directed to a short sequence in the extracellular region of CADM3, which contains the mutated tyrosine (QKPIITGYKSSLREKETATL)3 showed preferential recognition of the wild-type protein (Fig. 4B). Nevertheless, immunolabelling of non-permeabilized COS7 cells expressing the doubly tagged CADM3 and Cadm3-Y170C (Fig. 4C), as well as a cell surface biotinylation experiment (Fig. 4D), demonstrated that both the wild-type and the mutant proteins reached the cell surface. Furthermore, a soluble protein containing the extracellular domain of CADM4 fused to human Fc (Cadm4-Fc) bound to HEK293 cells expressing either wild-type CADM3 or Cadm3-Y170C (Fig. 4E), demonstrating that the mutant CADM3 is still able to interact with its glial CADM4 ligand. Likewise, co-immunoprecipitation experiments in cells transfected with WT-CADM3-HA and Y172C-CADM3-myc shows homodimer interaction between wild-type and mutant CADM3 molecules (Supplementary Fig. 7).
Figure 4.

CADM3 mutant reaches the cell surface of transfected cells and interacts with CADM4. (A) Western blot analysis of total cell lysate (Total), cytoplasmic and membrane preparation showing the expression of HA-tagged (tag located extracellularly) and Myc-tagged (tag located intracellularly) of rat CADM3 (wild-type, WT) and CADM3 mutant (Y170C) in transfected HEK293 cells, using an antibody to HA or to GAPDH as indicated. Cells transfected with green fluorescent protein (GFP) were used as control. Molecular size markers (kDa) are shown on the right. (B) Western blot analysis of membrane preparations from HEK293 cells expressing the same constructs as in A, using a polyclonal antibody directed to the extracellular domain of CADM3 or an antibody to HA-tag as indicated. Note that the CADM3 antibody recognize the wild-type but not the mutant CADM3 protein. Molecular size markers (kDa) are shown on the right. (C) Both wild-type and mutant proteins are present at the cell surface. Immunolabelling of HEK293 cells transfected with the double-tagged versions of wild-type or mutant CADM3 was carried out using either live (− Tx100) or permeabilized (+ Tx100) cells. The single channels and merged images are shown as indicated. (D) Surface biotinylation of HEK293 cells transfected with wild-type (WT), mutant (Y170C), or GFP, followed by immunoprecipitation (IP) with an antibody to Myc and immunoblot using streptavidin-HRP (Strep-HRP) or an anti-HA antibody as indicated. Molecular size markers (kDa) are shown on the right. (E) The mutant CADM3 interacts with the extracellular domain of CADM4. COS7 cells expressing Myc-tagged wild-type (WT) or mutant (Y170C) CADM3 were incubated with medium containing a soluble extracellular domain of CADM4 fused to the Fc region of human IgG. Expression of CADM3 and binding of Cadm4-Fc was monitored using antibodies to human Fc (Fc-Binding) and Myc tag, as indicated. DAPI labelling (blue) of cell nuclei is shown in the merged images. Scale bars = 20 μm.
The Tyr172Cys CADM3 variant shows reduced co-localization with CADM4 at cellular contact sites in the plasma membrane
To investigate whether the mutation affects CADM3 cell localization, we performed immunofluorescence co-localization experiments. We used L-cells, which lack cell-cell adhesion molecules, stably expressing the human encoding CADM3 plasmids, WT-CADM3-GFP or Y172C-CADM3-GFP and co-transfected with CADM4-Myc. Cells imaged by conventional confocal microscopy show CADM4 co-localized with both wild-type and mutant CADM3 (Fig. 3C). However, the spatial resolution of confocal microscopy is limited to 200 nm, which is well above the scale of the plasma membrane, therefore it cannot distinguish between adjacent molecules. To investigate a more precise localization of CADM3 in cells, we performed STORM imaging, which has a spatial resolution of 20 nm23 (Fig. 5E). While conventional confocal microscopy shows perfect co-localization between CADM3 and CADM4, STORM shows that CADM3 and CADM4 are located in close opposition at their respective plasma membranes (Fig. 5A and B) with overlapping points predominantly localized between cell-cell contact sites (Fig. 5B). In contrast, the mutant Y172C-CADM3 appears more disorganized with significantly decreased co-localization with CADM4 at contact sites (Fig. 5C, D and F), and in addition, it showed predominant localization in the cytoplasm (Fig. 5D). We cannot conclude based on that experiment whether the loss of co-localization is due to a decreased ability of the mutant CADM3 to reach the membrane or due to impaired molecular interaction between CADM3 and CADM4. However, the decreased physical interaction between CADM3 and CADM4 could potentially compromise the axon-glia interaction.
Figure 5.

Super-resolution microscopy shows CADM3 co-localization with CADM4 at plasma membrane contact sites. (A and B) STORM imaging reveals close proximity between WT-CADM3 (red) and CADM4 (green) at cell-cell contact sites in L-cells, which lack cell-cell adhesion molecules. A representative image shows two distinct colour lines formed at cell contact sites with some co-localization overlapping between red and green. (B) Magnified views from regions demarcated by purple boxes. (C and D) Mutant Y172C-CADM3 shows less specific localization to the membrane and diminished co-localization with CADM4. (D) Magnified views from regions demarcated by purple boxes. (E) Illustration of different results obtained with conventional confocal microscopy and STORM imaging. Adjacent CADM molecules cannot be distinguished by conventional microscopy, which shows co-localization (yellow), while STORM is able to distinguish the molecules (red and green). (F) Quantification of co-localization between CADM4 and CADM3. Twenty images from each group were obtained to calculate the results (t-test, P < 0.005).
Knock-in mouse model carrying the mutant allele
To study the cellular and molecular mechanisms that underlie the pathogenicity of the human CADM3 mutation in vivo, we generated knock-in mice carrying the corresponding mutation (Cadm3Y170C). Cadm3Y170C mice were born at the expected Mendelian frequency, were viable, and did not display any overt neurological abnormality. To examine the expression of the mutant allele we performed RT-PCR on RNA isolated from brains of wild-type, heterozygous and homozygous Cadm3Y170C mice. This analysis showed that the mutant mRNA is expressed in both heterozygous and homozygous mice (Fig. 6A and Supplementary Fig. 8). Western blot analysis using a CADM3 antibody that recognizes both the wild-type and the mutant protein revealed the presence of Cadm3-Y170C mutant in membrane protein extract prepared from 3-month-old homozygous mice brains (Fig. 6B). The expression of the mutant Cadm3-Y170C protein in these mice brains was similar to that of Cadm3 in wild-type mice. In contrast, western blot analysis using the same antibody carried out on sciatic nerve extract isolated from 3-month-old homozygous Cadm3Y170C mice revealed that the expression of the mutant protein in the nerve was lower than the expression of CADM3 protein in nerves from wild-type mice of the same age (Fig. 6C and D). However, and in line with the lack of clear neurological phenotype, Cadm3Y170C mice exhibit nerve conduction velocity similar to their wild-type control littermates (Fig. 6E).
Figure 6.

Evaluation of mouse Cadm3Y170C mutation. (A) RT-PCR analysis using wild-type and mutant-specific primers showing the presence of the wild-type and mutant CADM3 variants in the mice. (B) Western blot analysis showing the expression of CADM3 in wild-type and the knock-in homozygous mice brains. (C) Western blot of sciatic nerves isolated from the indicated genotypes using antibodies to CADM3 or GAPDH. Note the reduction in the expression of the mutant protein in sciatic nerves compared to wild-type nerves. (D) Quantification of C. (E) Mutant (knock-in) and wild-type mice show similar nerve conduction velocity. (F–H) Cadm3-Y170C mutant sciatic nerve exhibits normal myelin. (F) Toluidine blue-stained cross-sections of sciatic nerves isolated from 1.5-month-old mouse of the indicated genotype. (G and H) Electron microscopy images of sciatic nerves showing normal myelin morphology in wild-type (WT) and mutant (Cadm3Y170C) nerves. Scale bars: F = 10 μm; G = 2 μm; H = 0.4 μm. *P-value = 0.01, unpaired t-test, n = 3, mean ± standard deviation (SD): wild-type 0.84 ± 0.13, knock-in 0.49 ± 0.04.
Cadm3Y170C mice display abnormal axonal organization
To analyse Cadm3Y170C mice, we focused on potential molecular and structural abnormalities in their myelin unit. Toluidine blue stained sections (Fig. 6F), as well as electron microscopy analysis of cross-sections (Fig. 6G and H) of sciatic nerves isolated from wild-type and homozygous Cadm3Y170C mice showed no major difference in peripheral myelin except a few individual fibres with thickened myelin, reminiscent of tomacula seen in the patient sural nerve biopsy (Fig. 6F). The number of myelinated axons was similar between the two genotypes (wild-type: 2.78 ± 0.11, knock-in: 2.71 ± 0.31 axons per 100 μm2; total of 5600 and 7647 axons counted in three wild-type and knock-in mice, respectively). We also examined whether the mutation affects the molecular organization of the nerve, which is critical to its function. Immunofluorescence analysis of teased sciatic nerve preparations from homozygous Cadm3Y170C mice using antibodies to Caspr (i.e. paranodal junction marker), and Kv1.2 (i.e. juxtaparanodal marker) and NrCAM (i.e. nodal marker), revealed abnormal accumulation of Kv1.2 channels around the juxtaparanodes, as well as in patches along the internodes (knock-in 48 ± 4.34 sites versus 6.67 ± 1.53 sites detected in the knock-in of 300 nodes counted), and some aberrant appearance of Caspr at the edges of the paranodes (Fig. 7A–D). The abnormal localization of Kv1.2 channels was mostly seen along larger calibre axons. We also compared the localization of Kv1.2 channels of Cadm3Y170C mice to mice lacking either Cadm3 (Cadm3−/−) or Cadm4 (Cadm4−/−).7 We noticed a striking aberrant distribution of Kv1.2 channels in Cadm3Y170C compared to wild-type, but surprisingly, completely absent in Cadm3−/− nerves (Fig. 7D) suggesting a gain-of-function effect. Interestingly, we observed a similar abnormal histological phenotype in Cadm4−/− mice, which develop a peripheral neuropathy (Fig. 7D).
Figure 7.
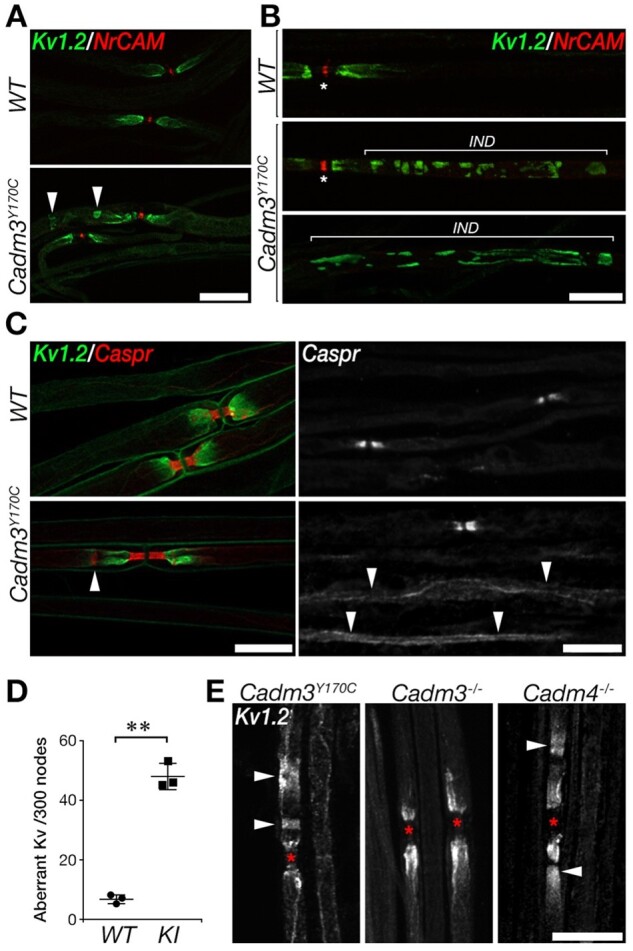
Cadm3Y170C mutant nerves exhibit abnormal axonal organization. Teased sciatic nerves were immunolabelled using antibodies to Kv1.2, NrCAM and Caspr, as indicated. (A and B) Abnormal (arrowheads) distribution of Kv1.2 potassium channels. The location of the internodes (IND) is marked in B and the nodes are marked by asterisks. (C) Mild abnormalities in Caspr (line marked by an arrowhead in lower left panel), as well as abnormal distribution of Caspr along the internodes (arrowheads in lower right panel). (D) Quantification of the number of sites containing abnormal Kv1 clusters in wild-type and mutant (knock-in) nerves. Number of sites is shown per 300 nodes; **P < 0.0001, unpaired t-test, n = 3, 300 nodes per mouse were counted, mean ± SD. (E) Kv1.2 abnormalities are present in the knock-in (left) but not in Cadm3 null mice (middle) and are similar (although less severe) than Cadm4 null mice (right). The location of the nodes is marked by asterisks. Scale bars = 20 μm (A, B, C left, D), 10 μm (C right).
Discussion
The present study identifies CADM3 as a novel CMT disease gene. CADM3 encodes for a neuronal specific cell-adhesion molecule localized to the internodal domain of myelinated fibres. It plays a major role in axon-glia interaction and domain organization maintenance in the internodes. Several studies have indicated that loss of axon-glia interactions leads to neurological abnormalities in mice due to axonal and myelination dysfunction. Mice lacking the paranodal junctional component Caspr exhibit tremor, ataxia and motor paresis.9,11,24 Mice lacking Cadm4, the Cadm3 interaction partner located in Schwann cells, results in a phenotype that resembles CMT, such as impaired motor function and reduced nerve conduction velocity.7 In humans, biallelic mutations in CNTNAP1, which encodes a paranodal cell-adhesion molecule, the contactin-associated protein (Caspr), causes severe phenotypes including congenital hypomyelinating neuropathy-325 and lethal congenital contracture syndrome.26 Finally, biallelic mutations in neurofascin (NFASC) were also recently identified to cause neurodevelopmental impairment and peripheral demyelination.27 Thus, the paranodal region is of recognized importance to the pathogenicity of CMT2; yet, we demonstrate this first direct genetic evidence for a cell adhesion molecule causing axonal CMT in patients. The clinical features we observed in the CADM3 patients are in keeping with early onset autosomal dominant CMT2 with marked upper limb involvement and, in one family, brisk reflexes suggesting upper motor neuron involvement. There was phenotypic heterogeneity among our patients ranging from severe early onset (Family 1 Patient II:1) to a milder adult onset (Family 3 Patient I:2). Nerve conduction studies suggested a predominantly axonal neuropathy but with motor slowing in some patients. Unusually, all patients analysed carry the same missense variant, Tyr172Cys, located in the extracellular domain adjacent to the second Ig loop. Mass spectrometry analysis studies revealed the formation of a new disulphide bond created by the mutated cysteine residue, indicating structural protein rearrangement. CADM proteins mediate specific cell-cell contact through homophilic and heterophilic interactions within the family of proteins. Trans interactions in this family occur through the first Ig-like domain, whereas the second Ig-like domain is necessary for cis interactions. Ig-like domains are stabilized by disulphide bonds, which maintain their structure. In the mutant CADM3, Tyr172 was changed to a cysteine residue. A missense variation that forms a new cysteine may perturb an existing disulphide bond and create an alternative bond with a nearby cysteine. The disulphide bond in the second Ig-like domain of CADM3 occurs between Cys186 and Cys243. The mutation is located in the beginning of the second Ig-like domain, thus the new Cys172 may interfere with the second disulphide bond, which could likely be disturbing the cis and not trans interactions of the protein.
Overexpression of the mutated CADM3 in RT4 Schwann cells resulted in activation of the UPR, suggesting protein misfolding as a potential pathological mechanism. We also explored the spatial resolution of the cell-cell contact site at the nanoscale level with STORM microscopy to more precisely evaluate CADM3 interaction with CADM4. STORM images revealed significantly decreased co-localization between CADM3 and CADM4 at cell-cell contact sites, indicating axon-glia interface disturbance as a potential pathological mechanism of the disease. Interestingly, a sural nerve biopsy from Family 1 revealed the presence of tomaculous swellings, further supporting abnormal axon-glia contacts. Similar abnormal structures were observed in Cadm4 knock-out mice resulting in a CMT-like phenotype.7
Our knock-in mouse model carrying the human Tyr172Cys mutation showed weaker expression of the mutant protein in sciatic nerve compared to Cadm3 wild-type mice. This result may indicate that the targeting of mutant CADM3 to the axonal membrane or its stabilization at the axonal membrane is impaired. This result is in accordance with the observed reduction in its co-localization with CADM4 as observed by super resolution microscopy in L-cells. Our in vivo analysis of teased sciatic nerve fibres from homozygous mice revealed abnormal accumulation of Kv1.2 channels around the juxtaparanodes and in patches along the internodes, as well as mild aberrant appearance of Caspr at the paranodes. We have previously shown that the absence of Cadm4 in mice Schwann cells leads to abnormal organization of the underlying axonal membrane, whereas mice lacking Cadm3 have normal peripheral nervous system myelin.7 It was suggested that the difference between these two mutants reflects compensatory mechanism by other axonal CADM proteins. The presence of axonal organization abnormalities in Cadm3Y170C mice may suggest that this mutation interferes not only with the interaction between CADM3 and CADM4, but also with the interactions between CADM3 and other axonal CADMs, or even other axoglial CAMs, as recently was demonstrated in the CNS for CADM4, MAG and Caspr.9,28 Humans, in contrast to the knock-out mice, which can survive without CADM3, appear to be intolerant to loss-of-function mutations in CADM3, since it is highly constraint for loss-of-function mutations in gnomAD (pLI >0.9). In gnomAD the expected number of loss-of-function variants for CADM3 is 21.9, and the observed number is one variant for the canonical transcript, however that only allele is flagged as ‘low call’. Likewise, we did not observe any loss-of-function mutation in our GENESIS database, which contains 9569 exomes/genome. This observation suggests that the human CADM3 is less redundant than the mouse gene and consequently a compensatory mechanism by other CADMs might be less effective.
The clinical and molecular findings of three independent families with axonal CMT, marked upper limb involvement, and brisk reflexes, harbouring the same pathogenic variant in CADM3, unveils a new pathological mechanism involving axon-glial interaction abnormalities in patients with CMT.
Supplementary Material
Acknowledgements
We are very grateful to the family members for their participation and collaboration in this study.
Funding
U54NS065712 (M.S.) and R01NS105755 (S.Z.) provided by NINDS/NCATS-ORD with support also from the MDA, CMT-Association, Hereditary Neuropathy Foundation, The Genesis Project foundation, the Israel Science Foundation, and the Dr Miriam and Sheldon G. Adelson Medical Research Foundation. E.P. is the Incumbent of the Hanna Hertz Professorial Chair for Multiple Sclerosis and Neuroscience. A.C. thanks the Medical Research Council (MR/T001712/1), Fondazione CARIPLO (2019-1836) and the Italian Ministry of Health - Ricerca Corrente for grant support.
Competing interests
The authors report no competing interests.
Supplementary material
Supplementary material is available at Brain online.
Glossary
- CMT
Charcot-Marie-Tooth disease
- STORM
stochastic optical reconstruction microscopy
- UPR
unfolded protein response
References
- 1. Rasband MN, Peles E. Mechanisms of node of Ranvier assembly.. Nat Rev Neurosci. 2021;22:7-20. [DOI] [PubMed] [Google Scholar]
- 2. Maurel P, Einheber S, Galinska J, et al. Nectin-like proteins mediate axon Schwann cell interactions along the internode and are essential for myelination. J Cell Biol. 2007;178:861-874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Spiegel I, Adamsky K, Eshed Y, et al. A central role for Necl4 (SynCAM4) in Schwann cell-axon interaction and myelination. Nat Neurosci. 2007;10:861-869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kakunaga S, Ikeda W, Itoh S, et al. Nectin-like molecule-1/TSLL1/SynCAM3: a neural tissue-specific immunoglobulin-like cell-cell adhesion molecule localizing at non-junctional contact sites of presynaptic nerve terminals, axons and glia cell processes. J Cell Sci. 2005;118:1267-1277. [DOI] [PubMed] [Google Scholar]
- 5. Sukhanov N, , Vainshtein A, , Eshed-Eisenbach Y, et al. Differential Contribution of Cadm1-Cadm3 Cell Adhesion Molecules to Peripheral Myelinated Axons. J Neurosci. 2021;41(7):1393-1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen MS, Kim H, Jagot-Lacoussiere L, Maurel P.. Cadm3 (Necl-1) interferes with the activation of the PI3 kinase/Akt signaling cascade and inhibits Schwann cell myelination in vitro. Glia. 2016;64:2247-2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Golan N, Kartvelishvily E, Spiegel I, et al. Genetic deletion of Cadm4 results in myelin abnormalities resembling Charcot-Marie-Tooth neuropathy. J Neurosci. 2013;33:10950-10961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Elazar N, Vainshtein AGolan N, et al. Axoglial Adhesion by Cadm4 Regulates CNS Myelination. Neuron. 2019;101:224-231.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elazar N, Vainshtein A, Rechav K, Tsoory M, Eshed-Eisenbach Y, Peles E. Coordinated internodal and paranodal adhesion controls accurate myelination by oligodendrocytes. J Cell Biol. 2019;218(9):2887-2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bai Y, Wu X, Brennan KM, et al. Myelin protein zero mutations and the unfolded protein response in Charcot Marie Tooth disease type 1B. Ann Clin Transl Neurol. 2018;5:445-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gollan L, Salomon D, Salzer JL, Peles E. Caspr regulates the processing of contactin and inhibits its binding to neurofascin. J Cell Biol. 2003;163:1213-1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Feinberg K, Eshed-Eisenbach Y, Frechter S, et al. A glial signal consisting of gliomedin and NrCAM clusters axonal Na+ channels during the formation of nodes of Ranvier. Neuron. 2010;65:490-502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lu S, Fan SB, Yang B, et al. Mapping native disulfide bonds at a proteome scale. Nat Methods. 2015;12:329-331. [DOI] [PubMed] [Google Scholar]
- 14. Rhodes KJ,, Strassle BW, Monaghan MM, et al. Association and colocalization of the Kvbeta1 and Kvbeta2 beta-subunits with Kv1 alpha-subunits in mammalian brain K+ channel complexes. J Neurosci. 1997;17:8246-8258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Peles E,, Nativ M, Lustig M, et al. Identification of a novel contactin-associated transmembrane receptor with multiple domains implicated in protein-protein interactions. EMBO J. 1997;16:978-988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Horresh I,, Bar V, Kissil JL, et al. Organization of myelinated axons by Caspr and Caspr2 requires the cytoskeletal adapter protein 4.1B. J Neurosci. 2010;30:2480-2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gonzalez M, Falk MJ, Gai X, Postrel R, Schule R, Zuchner S.. Innovative genomic collaboration using the GENESIS (GEM.app) platform. Human mutation. 2015;36:950-956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cooper GM, Stone EA, Asimenos G, et al. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15:901-913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rentzsch P, Witten D, Cooper GM, et al. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47:D886-D894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Murphy SM,, Herrmann DN, McDermott MP, et al. Reliability of the CMT neuropathy score (second version) in Charcot-Marie-Tooth disease. J Peripher Nerv Syst. 2011;16:191-198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhou Y, Du G, Hu X, et al. Nectin-like molecule 1 is a protein 4.1N associated protein and recruits protein 4.1N from cytoplasm to the plasma membrane. Biochim Biophys Acta. 2005;1669:142-154. [DOI] [PubMed] [Google Scholar]
- 23. Rust MJ, Bates M, Zhuang X.. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat Methods. 2006;3:793-795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bhat MA, Rios JC, Lu Y, et al. Axon-glia interactions and the domain organization of myelinated axons requires neurexin IV/Caspr/Paranodin. Neuron. 2001;30:369-383. [DOI] [PubMed] [Google Scholar]
- 25. Hengel H, Magee A, Mahanjah M, et al. CNTNAP1 mutations cause CNS hypomyelination and neuropathy with or without arthrogryposis. Neurol Genet. 2017;3:e144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lakhani S, Doan R, Almureikhi M, et al. Identification of a novel CNTNAP1 mutation causing arthrogryposis multiplex congenita with cerebral and cerebellar atrophy. Eur J Med Genet. 2017;60:245-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Efthymiou S,, Salpietro V, Malintan N, et al. Biallelic mutations in neurofascin cause neurodevelopmental impairment and peripheral demyelination. Brain. 2019;142:2948-2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Djannatian M,, Timmler S, Arends M, et al. Two adhesive systems cooperatively regulate axon ensheathment and myelin growth in the CNS. Nat Commun. 2019;10:4794. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data presented in this study are available from the corresponding author upon request.