

Abstract
The majority of genome-wide association studies have been conducted using samples with a broadly European genetic background. As a field, we acknowledge this limitation and the need to increase the diversity of populations studied. A major challenge when designing and conducting such studies is to assimilate large samples sizes so that we attain enough statistical power to detect variants associated with disease, particularly when trying to identify variants with low and rare minor allele frequencies. In this review, we aimed to illustrate the benefits to genetic characterization of Alzheimer’s disease, in researching currently understudied populations. This is important for both fair representation of world populations and the translatability of findings. To that end, we conducted a literature search to understand the contributions of studies, on different populations, to Alzheimer’s disease genetics. Using both PubMed and Alzforum Mutation Database, we systematically quantified the number of studies reporting variants in known disease-causing genes, in a worldwide manner, and discuss the contributions of research in understudied populations to the identification of novel genetic factors in this disease. Additionally, we compared the effects of genome-wide significant single nucleotide polymorphisms across populations by focusing on loci that show different association profiles between populations (a key example being APOE). Reports of variants in APP, PSEN1 and PSEN2 can initially determine whether patients from a country have been studied for Alzheimer’s disease genetics. Most genome-wide significant associations in non-Hispanic white genome-wide association studies do not reach genome-wide significance in such studies of other populations, with some suggesting an opposite effect direction; this is likely due to much smaller sample sizes attained. There are, however, genome-wide significant associations first identified in understudied populations which have yet to be replicated. Familial studies in understudied populations have identified rare, high effect variants, which have been replicated in other populations. This work functions to both highlight how understudied populations have furthered our understanding of Alzheimer’s disease genetics, and to help us gauge our progress in understanding the genetic architecture of this disease in all populations.
Keywords: Alzheimer’s, genetics, understudied populations, diversity
The majority of genome-wide association studies in Alzheimer’s disease have used samples with a European genetic background. Dehghani et al. discuss the translatability of genome-wide associations between different populations, and highlight how familial studies in understudied populations can reveal novel genetic factors.
Introduction
The purpose of this review is to highlight key findings on the genetics of Alzheimer’s disease in understudied populations. We define such populations in two distinct ways: (i) as countries where there have been few reported variants when discussing studies focused on specific genes; and (ii) as ethnicities with few studies or no representation to date in genome-wide association studies (GWAS). We broadly use the term ‘population’ throughout this review to address both geographical ancestry when patients originate from a particular country, and ethnicity when patients have mixed ancestries but identify as one particular social group. APP, PSEN1 and PSEN2 are the three genes harbouring pathogenic mutations typically causing early-onset Alzheimer’s disease (EOAD) (age at onset <65 years) in an autosomal dominant manner. Five to ten per cent of EOAD is due to mutations in these genes. Over 30 loci have been significantly associated with risk for late-onset Alzheimer’s disease (LOAD); when considering linkage disequilibrium regions, these loci contain up to 400 genes, including ABCA7, BIN1, CLU, CR1, SORL1 and APOE.1 The ε4 allele of APOE is currently the strongest genetic risk factor for LOAD.2 The heritability of Alzheimer’s disease is estimated to be 79% from studies on the Swedish Twin Registry, in a model that considers non-shared environmental influences, with this being higher for EOAD3; however, the single nucleotide polymorphism (SNP) heritability of LOAD (defined as the phenotypic variance attributable to common genetic variants), is predicted to be ∼31%; this was calculated using data from the Alzheimer’s Disease Genetics Consortium.4 This suggests that a large proportion of Alzheimer’s disease heritability is still to be identified and could be due to rare variants.
Approximately 78% of all genetic studies from the NHGRI-EBI GWAS catalogue have been conducted on individuals of European origin.5 Note that the labels ‘European,’ ‘non-Hispanic white’ (NHW) and ‘Caucasian’ are commonly used interchangeably in the literature. In this review, we refer to these individuals as described in the papers referenced and use the term NHW to broadly refer to both Americans (from the USA and Canada only) and Europeans. Not only are there few studies on other individuals, but many variants identified by large genetic studies on NHWs have not been replicated across different populations. This may in part be due to smaller sample sizes used in the replication experiments, or it may suggest that Alzheimer’s disease has a different genetic architecture, and perhaps even different mechanism(s) of disease in different populations. Although it has been suggested that the same molecular pathways are disrupted in Alzheimer’s disease across populations, differences may occur in the genes involved and the specific impinging points in these pathways. Studying different populations is, thus, critical to our better understanding of the genetics of Alzheimer’s disease. This review aims to highlight our progress in studying Alzheimer’s disease across the world by discussing the key findings from such studies and corroborating insights and best practices to lead us through the next phase of human Alzheimer’s disease genetic studies.
Literature search and methods
We first conducted a thorough literature search on 26 October 2019 by programmatically searching PubMed for the following terms: ‘Alzheimer* AND (PS1 OR PS2 OR PSEN1 OR APP OR PSEN2 OR presenilin OR amyloid precursor protein OR S182 OR E5-1) AND (mutation* or variant*) AND + country name’. To interpret the 7781 results, we filtered our results based on the metrics shown in Fig. 1.
Figure 1.
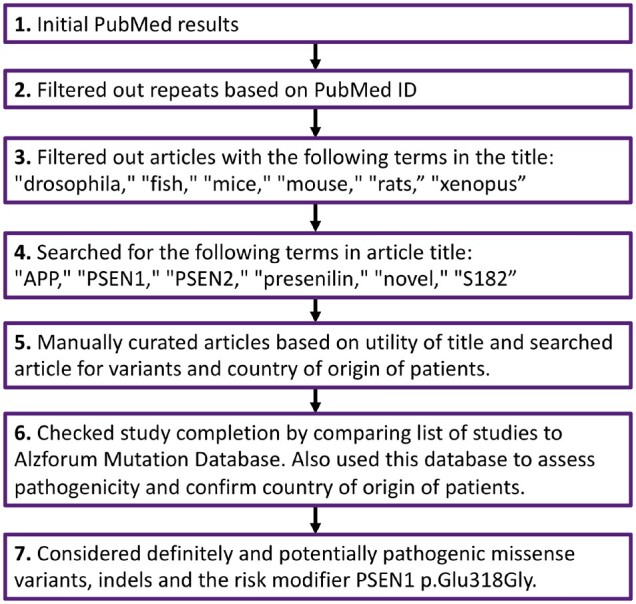
Study flow chart. Flow chart to depict the steps taken for interpreting the literature on Alzheimer’s disease genetics collected through our PubMed search.
We next used a similar approach to determine which populations were lagging in terms of identifying common genetic variability associated with Alzheimer’s disease. To this end, and to explore the representation of different populations in Alzheimer’s disease GWAS, we used information from all available studies, with corresponding ancestry data, from the NHGRI-EBI GWAS catalogue (16 January 2020 release).
In an attempt to compare the effect sizes of GWAS findings in NHW cohorts with other populations, we present odds ratios for the lead SNPs identified in the two most recent Alzheimer’s disease GWAS, which have also been tested in other populations (Supplementary Table 5). We used the results presented by Kunkle et al.1 and Jansen et al.6 to represent NHW cohorts and searched the NHGRI-EBI GWAS catalogue for studies of those same SNPs in other populations. We recorded (i) odds ratios from genome-wide significant SNPs; and (ii) odds ratios for variants in the same locus as these, from other populations. Variants from other populations were not required to reach genome-wide significance (GWS) to be recorded. We also performed a literature search to identify SNPs reaching GWS for association with Alzheimer’s disease first reported in understudied populations (Table 1) and explored if these have been replicated in any subsequent studies.
Table 1.
Table of novel SNPs that were initially identified by reaching genome-wide significance in GWAS performed in understudied populations
| Chromosome | SNP-effect allele | Nearest gene | Population | Source |
|---|---|---|---|---|
| 3 | rs7431992-A | CACNA2D3 | Caribbean Hispanics | Tosto et al.7 |
| 5 | rs75002042-A | FBXL7 | Caribbean Hispanics | Tosto et al.7 |
| 7 | rs112404845-T | COBL | African American | Mez et al.8 |
| 13 | rs16961023-G | SLC10A2 | African American | Mez et al.8 |
| 19 | rs115553053-T | HMHA1 | African American | Reitz et al.9 |
Since these associations were reported, there have been no further reports of associations between Alzheimer’s disease and FBXL7, COBL, SLC10A2 or HMHA1.
Finally, we discuss how familial studies in understudied populations have the power to identify rare, high effect variants, and how reproducible these findings are in other populations to date.
Results
Alzheimer’s disease Mendelian genetics around the world
From an initial 7781 articles, we considered 377 studies. From this search, patients from 47 countries had variants reported in at least one of the three genes APP, PSEN1 and PSEN2 (Fig. 2). Figure 3 shows the number of definitely and potentially pathogenic missense variants or indels in each gene reported in patients from each country from 377 studies. We used these results as a proxy for how much Alzheimer’s disease genetics has been researched across populations. However, this is not a perfect metric, and may be influenced by different factors such as publication bias, for example. This approach assumes screening for Mendelian genes as an indication of research being conducted in a population. We also attempted to consider other confounding factors as detailed below.
Figure 2.

World map with number of studies reporting variants in APP, PSEN1 or PSEN2 in patients per country. Variants included are the risk modifier PSEN1:p.Glu318Gly and missense and indels reported in Alzheimer’s disease cases as definitely or potentially pathogenic. Grey depicts countries with no studies on patients reporting variants in APP, PSEN1 or PSEN2. The number of studies reported ranges from one study in countries depicted in yellow (Brazil, Cuba, Hungary, Israel, Peru, Serbia, Czech Republic, South Africa, Uruguay, Slovenia, Ireland, Slovakia, Taiwan and India) to 46 studies reported in Italy depicted in black, closely followed by 43 studies in Japan and 33 studies in China. References are provided in Supplementary Table 1.
Figure 3.

APP, PSEN1 and PSEN2 variants reported in patients from each country or population. Variants included are the risk modifier PSEN1:p.Glu318Gly and missense and indels reported in Alzheimer’s disease cases as definitely or potentially pathogenic. We can clearly see that variants in PSEN1 are the most common, with patients from the majority of countries reporting between 1 and 15 unique variants in this gene. No reports were found describing PSEN1 variants in patients from Hungary or Taiwan, which is unusual given that pathogenic variants in this gene are the most frequent Mendelian cause of Alzheimer’s disease. This may be accounted for by the fact that, if the patient’s country was not specifically reported, then the study was not included in this analysis. Patients from China, France, Germany, Italy, Japan, Korea, Spain, UK and USA report over this range. Morocco stands out as having patients with more reports of APP variants compared to PSEN1 variants; these are all frameshift variants and may not have been reported in other studies if they focused on missense, although, we can see from gnomAD that indels in APP are rare. In addition to patients from specific countries, there are also single study reports of variants in patients from distinct populations (North American Aboriginal Kindred, Ashkenazi Jewish, African American and Caribbean Hispanic), which have also been included as these encompass different countries. References are provided in Supplementary Table 1.
France is the country with the most variants reported, followed by China. When crossing the data derived from Fig. 2 with that of Fig. 3, it is clear that there are many more studies reporting few or individual variants in Chinese cohorts, as opposed to a small number of large studies in France reporting many variants. Many of the variants in the publications originating from China are novel and not replications of previous findings. Of a total of 63 variants reported in China, 34 of these are unique to China as they have not been reported in any other countries to date (Supplementary Table 2). It is interesting to note that Southern Europe has a considerable number of studies originating from Spain and Italy, but that Portugal is an understudied population for Alzheimer’s disease genetics. This is an important fact that can be missed if this population is combined with the Spanish and broadly referred to as Iberian.
In Colombia, two reported variants include: PSEN1:p.Ile416Thr, which originated on an African haplotype,10 and the widely reported PSEN1:p.Glu280Ala variant, which segregates in a large community of over 5000 individuals from Medellín, Colombia.11 In this understudied, yet highly informative population, it has been shown that elevated levels of tau deposition measured by PET imaging precede clinical onset of Alzheimer’s disease by ∼6 years. Moreover, clinical deterioration can be detected around two decades before dementia onset in such mutation carriers.12 This is important, since the ability to identify at-risk individuals prior to any symptom onset could be used to develop improved clinical trial designs for new therapies for Alzheimer’s disease. Accordingly, there is an ongoing phase II clinical trial (NCT01998841), involving presymptomatic carriers of the PSEN1:p.Glu280Ala variant, which tests the drug crenezumab to target amyloid-β plaques. Even in such a unique population, composed of a very large number of individuals with the same mutation, and thus, very predictable disease course, positive results from this clinical trial could potentially impact patients across the whole world. In addition to the unique ability to develop genetically informed clinical trials in this extended family, the finding of the same mutation in such a large number of individuals also enables the study of genetic modifiers of the disease. Recently it was found that the ε2 allele of APOE delays the age at onset of disease in cases carrying the PSEN1:p.Glu280Ala mutation,13 and the DAOA:p.Arg30Lys variant was found to be significantly associated (P = 1.94 × 10−4) with age at onset.14 More recently, one individual from this kindred demonstrated high resilience to Alzheimer’s disease with a delay of over 30 years on their age at onset of mild cognitive impairment (MCI), despite high amyloid-β plaque load and carrying PSEN1:p.Glu280Ala. They were identified as homozygous for the rare APOE E3 Christchurch variant p.Arg136Ser (equivalent to APOE:p.Arg154Ser). Heterozygous carriers of this variant who also carry PSEN1:p.Glu280Ala progress to MCI at the mean age of 45.15 Additional reports suggest that the PSEN1 Glu280 residue is important in normal protein function for many populations. At the same protein residue, there is a report of another variant, PSEN1:p.Glu280Lys, in three siblings with EOAD from a Malaysian family.16 Ten additional family members (six with neuropsychiatric symptoms and four asymptomatic) were screened and were negative for this variant, suggesting segregation with the disease in that family. Two additional PSEN1 variants, PSEN1:p.Glu280Gly17 and PSEN1:p.Glu280Gln,18 have also been reported in apparent NHW Alzheimer’s disease cases.
With ∼65% protein sequence similarity to PSEN1, the highly homologous PSEN2 gene also harbours mutations that cause EOAD. These two proteins are components of the gamma-secretase complex, which cleaves APP, and in addition to sequence, also share high structural similarity. Despite these similarities, mutations in PSEN2 are rarer (59 in PSEN1, 36 in PSEN2 as per ClinVar, assessed March 2020). The most common mutation in PSEN2 is p.Asn141Ile, first discovered in the Volga Germans. Interestingly, it was noted that seizures were reported in about one-third of Volga German cases with this PSEN2 variant.19,20 This is intriguing and, in a similar way to the work described in the Colombian kindred, opens the possibility of using the Volga population to identify genetic modifiers of Alzheimer’s disease that could be responsible for the seizures in this population. The cohort of 146 affected cases from 11 Volga German families, with seizures reported in 20 of 64 of these cases, is a useful resource for such genotype-phenotype associations.21 Detecting if a patient is susceptible to developing seizures is important for close monitoring and prescription of medications.
Although there are no reports of variants in APP, PSEN1 and PSEN2 causing Alzheimer’s disease in Iceland, there is one report of an APP:p.Ala673Thr variant as protective against Alzheimer’s disease and cognitive decline in an elderly Icelandic population.22 This variant has since been identified in a Finnish individual who lived to 104.8 years.23 There are also reports of North Americans with this variant; some of whom are unaffected or have LOAD.24 All but one (who has Russian ancestry) of these reported individuals have broadly Scandinavian ancestry. These findings clearly exemplify the benefit of studying isolated and/or genetically distinct populations. The reduced genetic heterogeneity within such cohorts allows for the comparison of low-frequency variants between cases and controls; much larger sample sizes are required for sufficient detection power in populations with higher degrees of heterogeneity. It is interesting to note that at the same amino acid position in APP, there is one example of a recessive variant (APP:p.Ala673Val), which has been identified in two Italian siblings, one with early-onset dementia and the other with MCI; family members heterozygous for this variant were reported as unaffected.25
The absence of pathogenic mutations in the three known Alzheimer’s disease genes in a given population can suggest different possibilities: (i) the absence of genetic testing in that population; (ii) the possibility of other causative genes underlying the disease in that population; and/or (iii) the presence of rare high risk genetic variants altering the susceptibility of individuals to Alzheimer’s disease in the population.
In conclusion, one can extrapolate if a population has been studied for Alzheimer’s disease genetics by searching for reports of variants in the genes known to harbour pathogenic variants that are implicated in Mendelian inheritance of Alzheimer’s disease. The prediction of mutational damage to human genes should include population-specific analyses, as the predictions from current tools likely vary depending on ethnic background and the demographic history of the population.26 From our analysis, it is clear that there is wide variability not only in the number of studies across the globe, but also in the number of variants identified in each country. This has implications for future studies in the genetics of Alzheimer’s disease at a global level as it identifies populations to whom resources should be provided so that genetic studies can be performed. Even in the absence of effective therapies to prevent or delay Alzheimer’s disease, knowing the genetic status of an individual or family gives the opportunity for genetic counselling, informed preparation of life affairs (including reproductive options), and may contribute to achieve a more accurate in-life diagnosis.27
Alzheimer’s disease genetic risk in different populations
Representation of other populations in GWAS
After filtering the studies for those focusing on Alzheimer’s disease, we identified 76 studies. In these, there was a clear over-representation of cohorts defined as broadly European (Fig. 4). It is indisputable that large-scale genetic studies have revolutionized our understanding of the genetics of Alzheimer’s disease; however, this over-representation clearly highlights the need to study other populations in order for new genetic risk factors to be identified. To emphasize this for diseases in general, the NHGRI-EBI GWAS catalogue has released the GWAS Diversity Monitor, which allows quasi-real-time monitoring of ancestries represented in GWAS studies.28
Figure 4.

Broad ancestry represented in the 76 Alzheimer’s disease GWAS that are part of the NHGRI-EBI GWAS catalogue. By including the different ancestries within each stage of a study (Supplementary Table 3), this plot provides a clearer view of the ancestries represented in Alzheimer’s disease GWAS to date. We can see that the representation of European ancestry is greater in Alzheimer’s disease GWAS compared to all GWAS.5 Alzheimer’s disease GWAS appear to have a greater representation of African ancestry compared to GWAS overall, but there is still a large percentage of studies with ancestry not reported. References and counts are provided in Supplementary Tables 3 and 4.
Translatability of known Alzheimer’s disease GWAS hits to date
When we compare odds ratios from Alzheimer’s disease GWAS across populations, we see that in most cases, only the odds ratios from the NHW data reached GWS; however, there is one notable exception in APOE. The APOE ε4 haplotype is the strongest genetic risk factor for LOAD and its dose-dependent effects were first discovered in 1993.29 It was subsequently found that the APOE ε2 isoform is protective against Alzheimer’s disease.30 The large increase in risk of LOAD conferred by the ε4 allele in NHWs is not observed in the African American population where the risk conferred by this allele is smaller.31 This disparity has also been described in studies analysing molecular biomarkers such as CSF total and phosphorylated tau in APOE ε4-positive African American Alzheimer’s disease cases.31 Many studies report APOE allele frequencies in different populations and, compared to Europeans, both Africans and Oceanians have higher APOE ε4 allele frequencies, with Asians having the lowest allele frequency.32 The decreased risk observed with APOE ε4 in African Americans is due to local genomic African ancestry, with an estimated odds ratio of 2.34, as opposed to African Americans with local genomic European ancestry in which the odds ratio (OR) is estimated to be 3.05.33 A similar situation has also been shown in Puerto-Ricans (OR = 1.26 on African background, OR = 4.49 on European background)33 and may suggest that the cases in the Caribbean Hispanic GWAS have a stronger African background accounting for their lower odds ratio when compared to other populations in Fig. 5. APOE ε4 carrier frequency in Alzheimer’s disease patients from South America/Mexico (57.3%) is reported to be similar to that of USA/Canada (55.8%).34 This extends our understanding from APOE ε4 or ε2 in conferring risk or protection, respectively, to appreciating how population genetic background, and even ancestry local to the APOE locus, as defined by Rajabli et al.33 can modify such effects.
Figure 5.

Odds ratios in different populations for 10 GWAS loci discussed in the review that were significant in at least one population. Forest plots represent odds ratios (OR) and confidence intervals for SNPs in loci that have been (i) significantly associated with risk of Alzheimer’s disease in at least one population; and (ii) have been tested in at least one other population, for comparison. Loci are named according to the reported closest gene. We have condensed the list of SNPs reported near the same gene in GWAS by using LDproxy [defining linkage disequilibrium (LD) as R2 > 0.7]. If there were multiple odds ratios for the same population (and either all or none reached GWS) then we present the odds ratio from the study reporting the smallest P-value. These differences may be attributable to population-specific LD structures. Using the Wald testa, there are no statistically significant differences when comparing odds ratios for NHWs to odds ratios for SNPs near the same gene in other populations. b = beta; n = sample size; SE = standard error. Specific SNPs for each panel, references and exact values for all 23 GWAS loci significant in at least one population are provided in Supplementary Table 5. aIf > 1.96, P < 0.05.
Despite not always reaching GWS, CLU rs9331896-C is associated with lower risk of Alzheimer’s disease in most populations, with the exception, with an odds ratio close to 1, of African Americans. In line with this finding, Tycko et al.35 failed to identify any CLU polymorphisms associated with Alzheimer’s disease in African Americans. Moreover, a meta-analysis of 14 cohorts (total of 7070 Alzheimer’s disease cases and 8169 control subjects) concluded that rs11136000 in CLU is only associated with Alzheimer’s disease in NHWs (OR = 0.91), and not in African American, Arab or Caribbean Hispanic populations.36 A study on a small South Indian cohort (243 cases and 164 control subjects) also reported no association between rs11136000 and Alzheimer’s disease, following genotyping of this polymorphism.37 In the Chinese population, conflicting reports describing meta-analyses have been published with some reporting a significant association between rs11136000 and LOAD,38 while others concluded that there was no effect.39 More recently, in a large meta-analysis including 24 individual studies, Almeida et al.40 showed rs11136000 to be protective; an association that remained statistically significant for the Caucasian as well as the mixed samples (from China, Japan, India, and Turkey). As expected, there were high levels of genetic heterogeneity in the mixed samples, highlighting the need to study associations in larger cohorts of individual populations.
In the BIN1 locus two SNPs have been reported with disparate associations in different populations, suggesting that independent signals exist in these populations (Fig. 5 and Supplementary Table 5). The first, rs6733839, has been widely reported as a risk variant in various populations; however, in African Americans there was no strong evidence of association.41 The second, rs744373, was identified as a risk variant in Japanese with no evidence for association in Korean or Turkish populations.42 Following targeted sequencing and follow-up genotyping, Vardarajan et al.43 found BIN1:p.Lys358Arg (rs138047593) in eight Caucasian and six Hispanic LOAD patients; segregation was shown in two of six Caribbean Hispanic families. Segregation was not tested in Caucasians because seven of eight of them were from the Toronto LOAD study where there were no reported families.43 Although one Caucasian patient was from the NIA-LOAD study that includes families, segregation was not tested because the allele frequency of this variant for Caucasians in the ExAC database was similar to that observed in the LOAD cohort, suggesting that it was not associated with LOAD.43 Taken together, these data show that BIN1 variants have a similar effect in Asian and NHW populations,44,45 and that large Caribbean Hispanic families have enabled the finding of rare variants that seem to segregate with the disease. In line with this, although there are no additional studies of the EPHA1 lead GWAS SNPs in other populations, Vardarajan et al.43 reported segregation of the EPHA1:p.Pro460Leu variant (rs202178565) in four affected LOAD individuals and absence in three unaffected family members of a Caribbean Hispanic family from the Dominican Republic. The same variant showed no evidence of association in the Caucasian population studied.43 The authors suggested that rs202178565 may play a stronger role in Alzheimer’s disease risk in the Caribbean Hispanic population; however, further studies in larger datasets are needed to confirm this hypothesis.
At the CNTNAP2 locus, Jansen et al.6 found a significant signal for rs114360492. This locus was previously reported in both African Americans as well as in Japanese, however, the direction of effect was opposite in these two populations (risk and protection, respectively); it should be noted that these associations did not reach GWS in these populations.41,46 More recently, a GWAS in a Spanish cohort reported a significant association with rs117834366 near CNTNAP2 in a vascular dementia subcohort at the discovery stage; this had a very high odds ratio of 6.03 (3.22–11.2).47 This finding indicates the possibility that the results observed by Jansen et al.6 could be due to the inclusion of vascular dementia cases in their Alzheimer’s disease cohort. However, the two variants show low frequency in the European population, which increases the likelihood of erroneous results due to low statistical power in smaller cohorts.
The ECHDC3 locus was recently reported in NHWs.1 Interestingly, prior to this finding, the same locus had been reported in a trans-ethnic GWAS where rs7920721-G reached significance when samples from all populations (European ancestry, African Americans, Israeli-Arabs and Japanese) were combined.48 The significance was lost when populations were analysed separately, although the direction of effect was maintained. These findings show the power of using trans-ethnic approaches to identify novel loci when the effects are concordant.
Figure 5 shows that PICALM rs10792832-A presented a protective effect in all populations tested with a GWAS approach. In both South and East Asians, rs3851179 is in linkage disequilibrium (R2 > 0.7) with rs10792832; however, when looking at studies that tested the specific variant outside of a GWAS framework, discordant results were found regarding association of rs3851179 and Alzheimer’s disease in the Asian population.49,50 It is plausible that this may be related to pooling Chinese and Japanese populations together to increase sample size, with small differences in genetic background leading to less clear results. It is also possible that PICALM variants may not affect both populations equally, as a recent study on the Han Chinese population reported no association between variants in PICALM and LOAD.51 Finally, in the same South Indian cohort described previously, there was no association found between PICALM rs3851179 and Alzheimer’s disease.37
The ABCA7 locus is one of the strongest risk loci in African Americans with rs115550680 showing an odds ratio of 1.79 and reaching GWS.9 Although the authors mentioned that the SNP is in linkage disequilibrium with the European risk variant (rs4147929), they have very different allele frequencies in both populations (leading to a high D′ and low R2). This, again, emphasizes the distinct genetic backgrounds among populations that may lead to population-specific genetic architectures of disease. These must be taken into consideration, for example, when translating the finding of risk alleles for polygenic risk scores in different populations. Whether ABCA7 plays a role in Alzheimer’s disease risk in the Asian population is not clear with rs3764650 having been reported to be associated with Alzheimer’s disease in a Han Chinese population52 and no association between ABCA7 variants and Alzheimer’s disease being reported in two other studies on the same population.53,54
A GWAS meta-analysis conducted using data from East Asian, North American and European populations (for a total of 31 106 cases and 55 653 control subjects) investigated the association of CD33 rs3865444 with Alzheimer’s disease susceptibility.55 The authors reported no significant association in the East Asian population (Chinese, Japanese and Korean); however, an odds ratio of 1.7 (P = 0.0036) was reported for the major allele rs3865444-C in the Chinese subgroup with lower heterogeneity compared with the entire East Asian cohort.55 This is in line with the protective effect conferred by rs3865444-A in NHWs6 and suggested in the Spanish, African Americans and Caribbean Hispanics.7,41,48
Many of the SNPs reaching significance in NHWs have been investigated in additional populations. When these are studied under a GWAS framework, the odds ratios in other populations usually do not reach significance, which is due, in all likelihood, to the smaller sample sizes being used. One example of this is the Israeli-Arab population where only 51 cases and 64 controls were used to calculate the odds ratios for the SNPs presented in Fig. 5 and Supplementary Table 5. Data from this population also suggest that both CR1 and FERMT2 have an opposite direction of effect in Israeli-Arabs; however, it should be noted that there are very large confidence intervals associated with these odds ratios due to sample size. Given the clearly reduced statistical power in these smaller cohorts, results tend to focus on either a particular SNP or locus. This may account for the lack of odds ratios reported for Caribbean Hispanics in Fig. 5 and Supplementary Table 5, compared to reports that we have mentioned. Nonetheless, key differences between populations can be observed for variants reaching significance, namely, the lower risk conferred by APOE in African Americans, which is countered by the increased risk conferred by ABCA7. A valuable tool in this space are trans-ethnic GWAS, not only for comparison of odds ratios between populations, but also for aggregating enough samples to detect signals, which may then be replicated in other cohorts. The SNP near ECHDC3 is a good example, where a novel significant association was initially reported in a trans-ethnic GWAS before being replicated in the most recent GWAS in NHWs. Importantly, although this study aggregated samples of multiple ancestries, they also present results for separate populations, which enables future studies in these populations to build upon these population-specific results. Two studies could not be included in this review because they combined NHW and Caribbean Hispanic samples56 and Caucasian and African American samples.57 Lastly, it is important to note that post-GWAS analyses to investigate the potential effects of disease-associated SNPs are very informative and the field should gradually move its resources to systematically perform these for all nominated loci. The CELF1/SPI1 locus is a great example of this, where rs10838725-C had previously been associated with increased risk of Alzheimer’s disease in NHWs (OR 1.08) and recent work has shown that CELF1 rs1057233-G is associated with lower expression of SPI1 in monocytes and macrophages, and delayed Alzheimer’s disease onset.58 It is important to test such associations in cells with different ancestral backgrounds; both the National Centralized Repository for Alzheimer’s Disease and Related Dementias and the Coriell Human Variation Panel may be a helpful resource with cell lines and DNA currently available from populations including African American, Middle Eastern, Italian and Greek.
Discovery of new Alzheimer’s disease risk variants in understudied populations
Five SNPs were first reported in understudied populations to reach GWS for association with Alzheimer’s disease (Table 1); four of these have no reports of follow-up, highlighting a need to replicate these studies. While this manuscript was under review, an additional four novel associations at the EDEM1, ALCAM, GPC6 and VRK3 loci, approaching GWS, have been reported in a meta-analysis of African American Alzheimer’s disease cases and controls, with this analysis also implicating kidney involvement as a potential mechanism of disease.59 A single study identified a 665 kb duplication spanning CACNA2D3 in post-mortem diagnosed LOAD from the Brain Bank of the Brazilian Aging Brain Study Group.60 Although this is not a confirmatory report, it is nonetheless interesting that a rare structural change in a post-mortem case overlapped a gene previously found to be associated with the same disease. There was also one report of statistically significant overexpression of Cacna2d3 in the hypothalamus of aggressive male rats compared to domesticated rats.61 It would be interesting to study such changes in carriers of the risk SNP at this locus since some behavioural changes have also been reported in patients with Alzheimer’s disease.
Prior to the International Genomics of Alzheimer’s Project GWAS by Lambert et al.,62 evidence for association at the SORL1 locus had been obtained in a Japanese case-control study and subsequently replicated in a combined Japanese, Korean and Caucasian cohort.63 A protective effect seems to be present in other populations too (Supplementary Table 5), with the exception of African Americans where, so far, there is no evidence of association. A study in a Caribbean Hispanic familial and sporadic LOAD cohort, found 17 exonic variants significantly associated with Alzheimer’s disease and three shown to segregate with disease in families under a dominant model.64 The association of SORL1 variants with Alzheimer’s disease was first reported in 2007 with data from Israeli-Arab and North American case-control datasets and a Caribbean Hispanic familial cohort.65 To note, SORL1 Alzheimer’s disease risk ranges from modest to causal, with loss-of-function SORL1 variants detected in EOAD.66
An interesting understudied population is the Wadi Ara, where there is a high prevalence of Alzheimer’s disease, low frequency of APOE ε4 and high degrees of consanguinity. In this population, Sherva et al.67 performed a GWAS and homozygosity mapping to identify new genes and risk loci for Alzheimer’s disease. Although they failed to find any significant SNPs in both the standard GWAS and the GWAS utilizing homozygous regions, they identified genes, such as APBA1 or AGER, with Alzheimer’s disease-related biological functions, which were located within stretches of homozygosity nominally associated with Alzheimer’s disease. The lack of significant results likely stems from the fact that this was a study conducted in a small cohort of samples (124 cases and 142 controls). Focusing on the homozygous regions is an interesting approach to studying populations with high levels of consanguinity.
GWAS mostly identify variants that convey small alterations in risk, whereas familial studies have the power to identify rare, high effect variants. The homozygous TREM2: p.Gln33X, p.Thr66Met and p.Tyr38Cys variants were first identified associated with dementia in a report of three separate Turkish frontotemporal dementia-like families.68 These mutations, when homozygous, were previously known to cause polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy, also known as Nasu Hakola disease. The Turkish cases, however, displayed no apparent bone involvement in their phenotype with no history of joint pain or bone cysts. Since this initial finding in frontotemporal dementia-like families, Guerreiro et al.69 demonstrated that the heterozygous TREM2:p.Arg47His (rs75932628) is significantly associated with Alzheimer’s disease—a result that was found independently in the Icelandic population70 and has since been widely replicated, with significant associations reported for Spanish71,72 and French populations.73 Conversely, there are also studies reporting no significant association between rs75932628 and Alzheimer’s disease in Japanese,74 African American,75 Chinese,76-79 and Iranian80 populations, suggesting a population-specific effect. In some populations not only is there no association, but the p.Arg47His locus also seems to be monomorphic, as is the case for the East Asian population in the latest version of gnomAD. Other TREM2 variants have been identified in specific populations, such as the p.Gly55Arg variant reported in an Iranian cohort,80 and the p.Ala130Val in a Han Chinese LOAD patient.76 The non-coding variant rs7748513 is in linkage disequilibrium with p.Arg47His and was reported to be associated with increased risk of Alzheimer’s disease in African Americans.81 In addition to p.Arg47His, the p.Leu211Pro has also been shown to be associated with Alzheimer’s disease in African Americans (P = 0.01).75
Two rare variants in AKAP9 were initially reported as nominally associated with Alzheimer’s disease in African Americans.82 These two variants were also identified in individual Caribbean Hispanic LOAD cases.83 One of these two variants, the p.Arg434Trp, was reported in affected individuals from two large Caribbean Hispanic LOAD families.83 It is important to note that segregation was not shown for this variant, with absence in some affected individuals and presence in potentially presymptomatic individuals who were younger than the usual age at onset of other family members. There was one exception of a single family member aged 78 who was unaffected but identified as a heterozygous carrier.83 Taken together, these data argue against a fully penetrant mode for AKAP9 variants in this Caribbean Hispanic family. Moreover, four other AKAP9 variants were reported in Caribbean Hispanic families, with three of them present in the same family.83 Two different AKAP9 variants have subsequently been reported in LOAD families of European ancestry, although segregation was not shown for either of them.84 These AKAP9 variants are listed in Table 2.
Table 2.
Variants in AKAP9 reported in different populations
| Variant | Amino acid change | Population/s | Source |
|---|---|---|---|
| rs144662445 | p.Ile2546Met | African American, Caribbean Hispanic | Logue et al.82 Vardarajan et al.83 |
| rs149979685 | p.Ser3767Leu | African American, Caribbean Hispanic | Logue et al.82 Vardarajan et al.83 |
| rs144888041a | p.Glu170Asp | Caribbean Hispanic | Vardarajan et al.83 |
| rs771608420 | p.Arg434Trp | Caribbean Hispanic | Vardarajan et al.83 |
| chr7:91706306-A-Ca | p.Glu2250Asp | Caribbean Hispanic | Vardarajan et al.83 |
| rs144054367 | p.Ile3558Val | Caribbean Hispanic | Vardarajan et al.83 |
| rs34101758a | p.Arg3742Pro | Caribbean Hispanic | Vardarajan et al.83 |
| rs202091548 | p.Glu744Lys | European ancestry | Cukier et al.84 |
| rs146797353 | p.Arg1276Gln | European ancestry | Cukier et al.84 |
Variants present in the same family. Variant positions are in hg19. Protein sequence variant is as reported in the Ensembl canonical transcript.
Association of CASP7 with Alzheimer’s disease was first reported in a Caribbean Hispanic haplotype association study.85 This haplotype association study also reported significant association between LRP1B, TNFRSF1A, CDH1, and TG, with Alzheimer’s disease susceptibility in Caribbean Hispanic individuals.85 Since this report, a likely loss-of-function CASP7 variant (rs10553596) has been reported to reduce the risk of Alzheimer’s disease in APOE ε4 homozygotes.86 Finally, the CASP7 rs116437863 variant has been significantly associated with familial LOAD in individuals of European ancestry.87
NOTCH3 variants have been associated with Alzheimer’s disease risk relatively recently and thus the impact of such variants is still not entirely clear. The NOTCH3:p.Arg1231Cys (rs201680145-A) mutation was first reported in one Caucasian patient with cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL).88 There have since been several reports of this mutation in CADASIL patients from central Italy,89 in one Russian CADASIL patient (a compound heterozygote with NOTCH3:p.Phe984Cys)90 and in a Turkish Alzheimer’s disease case.91 This variant is not reported in unrelated Chinese cases diagnosed with CADASIL.92 Since the report linking NOTCH3 mutations with Alzheimer’s disease, there has been growing evidence supporting this association. Following exome sequencing, a missense NOTCH3 variant (rs149307620, p.Ala284Thr) was reported in 10 Alzheimer’s disease cases (confirmed in eight), and not present in controls of European ancestry.93 Screening of whole genome sequence (WGS) data from the Alzheimer’s Disease Sequencing Project (ADSP) and Alzheimer’s Disease Neuroimaging Initiative (ADNI) revealed this variant in one Alzheimer’s disease case, one MCI case and no control subjects. Moreover, two half-first cousins from a Utah high-risk pedigree both had rare missense variants in NOTCH3 which were not identified in the WGS samples from ADSP and ADNI. A study on the Han Chinese population reported NOTCH3:p.Glu585Ala in one probable Alzheimer’s disease case (out of 210 Alzheimer’s disease cases) with familial EOAD, which was absent from 160 population-matched control subjects.94 Despite these results, studies in more populations and in larger sample sizes are required to fully establish the role of NOTCH3 variants in Alzheimer’s disease.
In summary, several variants and genes were initially identified and associated with Alzheimer’s disease in understudied populations that were later found to also have an effect in NHW individuals. This highlights the importance of performing family and cohort studies in diverse populations.
Discussion
In this review we have identified understudied populations as countries with no reported APP, PSEN1 or PSEN2 variants in patients; and ethnicities that either have absent or underpowered genetic association studies to date. Despite these apparent shortcomings, we have highlighted interesting population differences related to APOE and ABCA7 variants, and also hits from GWAS in understudied populations that have not yet been replicated. Moreover, we have shown that such patient cohorts do have the power to reveal rare high effect variants; the implicated gene pathways can potentially provide insights into novel disease mechanisms at play. In regard to current issues moving forward, the human reference genome is not representative of an average genome for all ethnicities, instead it is largely representative of NHW individuals only. As our understanding of human genetics continues to evolve, this creates important limitations. It is also possible that regions encompassing damaging variants in some populations are not mapped in the current reference genome and thus will not be detected. Efforts have been made to create ethnicity-specific reference genomes and there is some interest in designing a pan-genome to account for all genetic variability. Recent work to create a pan-genome of African individuals highlighted that there is over 296.5 Mb of DNA that is not accounted for in the current human genome reference.95
There are disparities in gene variants associated with Alzheimer’s disease between populations, both from an effect size as well as direction of effect perspectives. These may be due to early human migration out of Africa and subsequent founder effects. In Alzheimer’s disease, as in all other complex diseases, substantially more NHW samples are used for initial discovery in genetic studies. Interestingly, in regard to populations used for replication studies, samples of Asian ancestry are currently the most commonly used after NHW samples.96 The smaller sample sizes of Alzheimer’s disease cases obtained for other populations compared to NHWs may be related to stigmatization of Alzheimer’s disease in some cultures, but also to funding availability for large-scale genetic research in those populations. However, it was recently shown that between 1990 and 2016 there has been an increase in both prevalence and burden of dementias in Eastern Mediterranean region countries,97 which may be due to a range of factors, such as an ageing population or improved diagnostic methodologies. Based both on key past discoveries, and the potential for future findings, funding to facilitate the study of understudied populations is imperative. The immediate need to address ethnic and racial disparities in Alzheimer’s disease and related dementias has been the focus of a recent white paper,98 which developed a series of recommendations for future strategies. These included targeted study recruitment and retention of diverse ethno-racial populations, generalization of instruments and analytic methods across research groups, ensuring that ethno-racial subgroup data are reported, creation of methods to reduce impact of small sample size on observations, and development of statistical models of risk and protective factors relevant to ethno-racial groups, to better refine prevalence and incidence.98 Funding, both in terms of access to training and conducting research appears to be the biggest barrier to genetic research in understudied populations. Potential strategies for overcoming this barrier may include grants for trans-national collaborations, and established laboratories hosting researchers from understudied populations with the aim to equip them with knowledge and tools to conduct such research upon return to home institutions. It is important, however, that these efforts occur in full coordination and cooperation with researchers from understudied populations, and that established laboratories do not use such efforts to unilaterally produce data without an active involvement from researchers from understudied populations.99,100
Data from the UK Biobank, which is largely European-based, has been used in Alzheimer’s disease GWAS by taking advantage of proximal Alzheimer’s disease status, where an individual is considered as Alzheimer’s disease-by-proxy if they report Alzheimer’s disease in one of their parents, while individuals with parents without history of Alzheimer’s disease are considered by-proxy controls.6,101 This is an approach that could be used to increase the sample sizes of understudied populations and thus the power for detecting associations. As detailed above, it is critically important to have a better understanding of the allele frequencies across populations, something that population variant databases such as ExAC and gnomAD have enabled us to do, to a certain degree, for some populations. For Middle Eastern populations, at a smaller scale and with no information on family history, one available resource is the Greater Middle East Variome (whole exome sequence data on 1111 unrelated subjects from populations including Northwest and Northeast Africa, the Turkish Peninsula, the Syrian Desert, the Arabian Peninsula, Persia and Pakistan).102 Other population-specific databases include the GenomeAsia 100K Project, the Han Chinese genome database, the Japanese Human Genetic Variation Database and the Genome of the Netherlands.103–106 Additionally, studies on populations with large genetic bottlenecks, such as Finland, where there is increased frequency of extremely rare alleles, can increase the power for finding associations, without needing to drastically increase sample size.107
Large cohorts with the same mutation, such as the Colombian cohort, have demonstrated the power to study genetic disease modifiers, clinical biomarkers and therapeutics with the potential to translate findings to other populations. Related to the clinical setting, polygenic risk scores take into account SNPs across the genome that are associated with modulating risk for disease. The best predictors of risk are derived from the largest GWAS as these will be the best powered studies to identify risk loci and estimate their effect size. Currently, the largest GWAS are on NHWs, which raises the question of how valid these polygenic risk scores are for prediction of Alzheimer’s disease risk in other populations. If greater diversity is not prioritized in genetic studies, current polygenic risk scores may be irrelevant for the majority of the world’s populations and may exacerbate health disparities.108
The vast majority of genetic findings in complex disease in general and Alzheimer’s disease in particular have been made in broadly NHW populations. We have highlighted here how important findings from other populations have been reported over the years and how they have improved our understanding of the genetic bases of this disease. Although we focus on Alzheimer’s disease in this review, the situation is similar for other neurodegenerative diseases including Parkinson’s disease109 and frontotemporal dementia,110 with the genetics of dementia with Lewy bodies still relatively understudied.111 A shift in funding strategies and a broader collaborative mindset is required to allow us to create the next step-change that is needed to move the field to a complete understanding of the genetic architecture of Alzheimer’s disease across populations.
Supplementary Material
Acknowledgements
We thank the Van Andel Institute Bioinformatics and Biostatistics Core, especially Emily Wolfrum and Zach Madaj, for their statistical expertise.
Funding
Research reported in this publication was supported by the National Institute on Aging of the National Institutes of Health under Award Number R01AG067426. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Competing interests
The authors report no competing interests.
Supplementary material
Supplementary material is available at Brain online.
Glossary
- EOAD
early-onset Alzheimer’s disease;
- GWAS
genome-wide association studies;
- GWS
genome-wide significance;
- LOAD
late-onset Alzheimer’s disease;
- NHW
non-Hispanic white;
- SNP
single nucleotide polymorphism
References
- 1. Kunkle BW, Grenier-Boley B, Sims R, et al. ; Alzheimer Disease Genetics Consortium (ADGC). Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet. 2019;51:414-430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Guerreiro R, Hardy J.. Genetics of Alzheimer’s disease. Neurotherapeutics. 2014;11:732-737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gatz M, Reynolds CA, Fratiglioni L, et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry. 2006;63:168-174. [DOI] [PubMed] [Google Scholar]
- 4. Ridge PG, Hoyt KB, Boehme K, et al. Assessment of the genetic variance of late-onset Alzheimer’s disease. Neurobiol Aging. 2016;41:200.e13-200.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Morales J, Welter D, Bowler EH, et al. A standardized framework for representation of ancestry data in genomics studies, with application to the NHGRI-EBI GWAS Catalog. Genome Biol. 2018;19:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jansen IE, Savage JE, Watanabe K, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet. 2019;51:404-413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tosto G, Fu H, Vardarajan BN, et al. F-box/LRR-repeat protein 7 is genetically associated with Alzheimer’s disease. Ann Clin Transl Neurol. 2015;2:810-820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mez J, Chung J, Jun G, et al. ; Alzheimer's Disease Genetics Consortium. Two novel loci, COBL and SLC10A2, for Alzheimer’s disease in African Americans. Alzheimers Dement. 2017;13:119-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Reitz C, Jun G, Naj A, et al. Variants in the ATP-binding cassette transporter (ABCA7), apolipoprotein E ϵ4,and the risk of late-onset Alzheimer disease in African Americans. JAMA. 2013;309:1483-1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ramirez Aguilar L, Acosta-Uribe J, Giraldo MM, et al. Genetic origin of a large family with a novel PSEN1 mutation (Ile416Thr). Alzheimers Dement. 2019;15:709-719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lopera F, Ardilla A, Martínez A, et al. Clinical features of early-onset Alzheimer disease in a large kindred with an E280A presenilin-1 mutation. JAMA. 1997;277:793-799. [PubMed] [Google Scholar]
- 12. Acosta-Baena N, Sepulveda-Falla D, Lopera-Gómez CM, et al. Pre-dementia clinical stages in presenilin 1 E280A familial early-onset Alzheimer’s disease: A retrospective cohort study. Lancet Neurol. 2011;10:213-220. [DOI] [PubMed] [Google Scholar]
- 13. Vélez JI, Lopera F, Sepulveda-Falla D, et al. APOEE2 allele delays age of onset in PSEN1 E280A Alzheimer’s disease. Mol Psychiatry. 2016;21:916-924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vélez JI, Rivera D, Mastronardi CA, et al. A mutation in DAOA modifies the age of onset in PSEN1 E280A Alzheimer’s disease. Neural Plast. 2016;2016:1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Arboleda-Velasquez JF, Lopera F, O’Hare M, et al. Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: A case report. Nat Med. 2019;25:1680-1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ch’ng G-S, An SSA, Bae SO, et al. Identification of two novel mutations, PSEN1 E280K and PRNP G127S, in a Malaysian family. Neuropsychiatr Dis Treat. 2015;11:2315-2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alzheimer’s Disease Collaborative Group. The structure of the presenilin 1 (S182) gene and identification of six novel mutations in early onset AD families. Nat Genet. 1995;11:219-222. [DOI] [PubMed] [Google Scholar]
- 18. Rogaeva E, Bergeron C, Sato C, et al. PS1 Alzheimer’s disease family with spastic paraplegia: The search for a gene modifier. Neurology. 2003;61:1005-1007. [DOI] [PubMed] [Google Scholar]
- 19. Levy-Lahad E, Wasco W, Poorkaj P, et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science. 1995;269:973-977. [DOI] [PubMed] [Google Scholar]
- 20. Rogaev EI, Sherrington R, Rogaeva EA, et al. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer's disease type 3 gene. Nature. 1995;376:775-778. [DOI] [PubMed] [Google Scholar]
- 21. Jayadev S, Leverenz JB, Steinbart E, et al. Alzheimer’s disease phenotypes and genotypes associated with mutations in presenilin 2. Brain. 2010;133:1143-1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jonsson T, Atwal JK, Steinberg S, et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature. 2012;488:96-99. [DOI] [PubMed] [Google Scholar]
- 23. Kero M, Paetau A, Polvikoski T, et al. Amyloid precursor protein (APP) A673T mutation in the elderly Finnish population. Neurobiol Aging. 2013;34:1518.e1-1518.e3. [DOI] [PubMed] [Google Scholar]
- 24. Wang L-S, Naj AC, Graham RR, et al. Rarity of the Alzheimer disease-protective APP A673T variant in the United States. JAMA Neurol. 2015;72:209-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Di Fede G, Catania M, Morbin M, et al. A recessive mutation in the APP gene with dominant-negative effect on amyloidogenesis. Science. 2009;323:1473-1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hussin JG, Hodgkinson A, Idaghdour Y, et al. Recombination affects accumulation of damaging and disease-associated mutations in human populations. Nat Genet. 2015;47:400-404. [DOI] [PubMed] [Google Scholar]
- 27. Cohn-Hokke PE, Elting MW, Pijnenburg YAL, et al. Genetics of dementia: Update and guidelines for the clinician. Am J Med Genet B Genet. 2012;159B:628-643. [DOI] [PubMed] [Google Scholar]
- 28. Mills MC, Rahal C.. The GWAS Diversity Monitor tracks diversity by disease in real time. Nat Genet. 2020;52:242-243. [DOI] [PubMed] [Google Scholar]
- 29. Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921-923. [DOI] [PubMed] [Google Scholar]
- 30. Corder EH, Saunders AM, Risch NJ, et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet. 1994;7:180-184. [DOI] [PubMed] [Google Scholar]
- 31. Morris JC, Schindler SE, McCue LM, et al. Assessment of racial disparities in biomarkers for Alzheimer disease. JAMA Neurol. 2019;76:264-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kamboh MI. Apolipoprotein E polymorphism and susceptibility to Alzheimer’s disease. Hum Biol. 1995;67:195-215. [PubMed] [Google Scholar]
- 33. Rajabli F, Feliciano BE, Celis K, et al. Ancestral origin of ApoE ε4 Alzheimer disease risk in Puerto Rican and African American populations. PLoS Genet. 2018;14:e1007791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cummings JL, Atri A, Ballard C, et al. Insights into globalization: Comparison of patient characteristics and disease progression among geographic regions in a multinational Alzheimer’s disease clinical program. Alzheimers Res Ther. 2018;10:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tycko B, Feng L, Nguyen L, et al. Polymorphisms in the human apolipoprotein-J/clusterin gene: Ethnic variation and distribution in Alzheimer’s disease. Hum Genet. 1996;98:430-436. [DOI] [PubMed] [Google Scholar]
- 36. Jun G, Naj AC, Beecham GW, et al. Meta-analysis confirms CR1, CLU, and PICALM as Alzheimer disease risk loci and reveals interactions with APOE genotypes. Arch Neurol. 2010;67:1473-1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shankarappa BM, Kota LN, Purushottam M, et al. Effect of CLU and PICALM polymorphisms on AD risk: A study from south India. Asian J Psychiatr. 2017;27:7-11. [DOI] [PubMed] [Google Scholar]
- 38. Ma J-F, Liu L-H, Zhang Y, et al. Association study of clusterin polymorphism rs11136000 with late onset Alzheimer’s disease in Chinese Han population. Am J Alzheimers Dis Other Demen. 2011;26:627-630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Han Z, Qu J, Zhao J, Zou X.. Analyzing 74,248 samples confirms the association between CLU rs11136000 polymorphism and Alzheimer’s disease in Caucasian but not Chinese population. Sci Rep. 2018;8:11062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Almeida JFF, Dos Santos LR, Trancozo M, et al. Updated meta-analysis of BIN1, CR1, MS4A6A, CLU, and ABCA7 variants in Alzheimer’s disease. J Mol Neurosci. 2018;64:471-477. [DOI] [PubMed] [Google Scholar]
- 41. Logue MW, Schu M, Vardarajan BN, et al. A comprehensive genetic association study of Alzheimer disease in African Americans. Arch Neurol. 2011;68:1569-1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kaya G, Gündüz E, Acar M, et al. Potential genetic biomarkers in the early diagnosis of Alzheimer disease: APOE and BIN1. Turk J Med Sci. 2015;45:1058-1072. [PubMed] [Google Scholar]
- 43. Vardarajan BN, Ghani M, Kahn A, et al. Rare coding mutations identified by sequencing of Alzheimer disease genome-wide association studies loci. Ann Neurol. 2015;78:487-498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liu G, Zhang S, Cai Z, et al. BIN1 gene rs744373 polymorphism contributes to Alzheimer’s disease in East Asian population. Neurosci Lett. 2013;544:47-51. [DOI] [PubMed] [Google Scholar]
- 45. Zhu R, Liu X, He Z.. The bridging integrator 1 gene polymorphism rs744373 and the risk of Alzheimer’s Disease in Caucasian and Asian populations: An updated meta-analysis. Mol Neurobiol. 2017;54:1419-1428. [DOI] [PubMed] [Google Scholar]
- 46. Hirano A, Ohara T, Takahashi A, et al. A genome-wide association study of late-onset Alzheimer’s disease in a Japanese population. Psychiatr Genet. 2015;25:139-146. [DOI] [PubMed] [Google Scholar]
- 47. Moreno-Grau S, de Rojas I, Hernández I, et al. ; GR@ACE consortium. Genome-wide association analysis of dementia and its clinical endophenotypes reveal novel loci associated with Alzheimer’s disease and three causality networks: The GR@ACE project. Alzheimers Dement. 2019;15:1333-1347. [DOI] [PubMed] [Google Scholar]
- 48. Jun GR, Chung J, Mez J, et al. Transethnic genome-wide scan identifies novel Alzheimer’s disease loci. Alzheimers Dement. 2017;13:727-738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liu G, Zhang L, Feng R, et al. Lack of association between PICALM rs3851179 polymorphism and Alzheimer’s disease in Chinese population and APOEε4-negative subgroup. Neurobiol Aging. 2013;34:1310.e9-1310.e10. [DOI] [PubMed] [Google Scholar]
- 50. Liu G, Zhang S, Cai Z, et al. PICALM gene rs3851179 polymorphism contributes to Alzheimer’s disease in an Asian population. Neuromol Med. 2013;15:384-388. [DOI] [PubMed] [Google Scholar]
- 51. Jiang T, Yu J-T, Tan M-S, et al. Genetic variation in PICALM and Alzheimer’s disease risk in Han Chinese. Neurobiol Aging. 2014;35:934.e1-934.e3. [DOI] [PubMed] [Google Scholar]
- 52. Liao Y-C, Lee W-J, Hwang J-P, et al. ABCA7 gene and the risk of Alzheimer’s disease in Han Chinese in Taiwan. Neurobiol Aging. 2014;35:2423.e7-2423.e13. [DOI] [PubMed] [Google Scholar]
- 53. Liu L-H, Xu J, Deng Y-L, et al. A complex association of ABCA7 genotypes with sporadic Alzheimer disease in Chinese Han population. Alzheimer Dis Assoc Disord. 2014;28:141-144. [DOI] [PubMed] [Google Scholar]
- 54. Tan L, Yu J-T, Zhang W, et al. Association of GWAS-linked loci with late-onset Alzheimer’s disease in a northern Han Chinese population. Alzheimers Dement. 2013;9:546-553. [DOI] [PubMed] [Google Scholar]
- 55. Li X, Shen N, Zhang S, et al. CD33 rs3865444 polymorphism contributes to Alzheimer’s disease susceptibility in Chinese, European, and North American Populations. Mol Neurobiol. 2015;52:414-421. [DOI] [PubMed] [Google Scholar]
- 56. Wijsman EM, Pankratz ND, Choi Y, et al. ; The NIA-LOAD/NCRAD Family Study Group. Genome-wide association of familial late-onset Alzheimer’s disease replicates BIN1 and CLU and nominates CUGBP2 in interaction with APOE. PLoS Genet. 2011;7:e1001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hollingworth P, Sweet R, Sims R, et al. ; the GERAD Consortium. Genome-wide association study of Alzheimer’s disease with psychotic symptoms. Mol Psychiatry. 2012;17:1316-1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Huang K-L, Marcora E, Pimenova AA, et al. ; The International Genomics of Alzheimer's Project. A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer’s disease. Nat Neurosci. 2017;20:1052-1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kunkle BW, Schmidt M, Klein H-U, et al. ; Writing Group for the Alzheimer’s Disease Genetics Consortium (ADGC). Novel Alzheimer disease risk loci and pathways in African American individuals using the African Genome Resources Panel: A meta-analysis [Internet]. JAMA Neurol. 2021;78:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Villela D, Suemoto CK, Pasqualucci CA, Grinberg LT, Rosenberg C.. Do copy number changes in CACNA2D2, CACNA2D3, and CACNA1D constitute a predisposing risk factor for Alzheimer’s disease? Front Genet. 2016;7:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Oshchepkov D, Ponomarenko M, Klimova N, et al. A rat model of human behavior provides evidence of natural selection against underexpression of aggressiveness-related genes in humans. Front Genet. 2019;10:1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lambert J-C, Ibrahim-Verbaas CA, Harold D, . et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet. 2013;45:1452-1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Miyashita A, Koike A, Jun G, et al. ; The Alzheimer Disease Genetics Consortium. SORL1 is genetically associated with late-onset Alzheimer’s disease in Japanese, Koreans and Caucasians. PLoS One. 2013;8:e58618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Vardarajan BN, Zhang Y, Lee JH, et al. Coding mutations in SORL1 and Alzheimer disease. Ann Neurol. 2015;77:215-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Rogaeva E, Meng Y, Lee JH, et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet. 2007;39:168-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Pottier C, Hannequin D, Coutant S, et al. ; PHRC GMAJ Collaborators. High frequency of potentially pathogenic SORL1 mutations in autosomal dominant early-onset Alzheimer disease. Mol Psychiatry. 2012;17:875-879. [DOI] [PubMed] [Google Scholar]
- 67. Sherva R, Baldwin CT, Inzelberg R, et al. Identification of novel candidate genes for Alzheimer’s disease by autozygosity mapping using genome wide SNP data. J Alzheimers Dis. 2011;23:349-359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Guerreiro RJ, Lohmann E, Brás JM, et al. Using exome sequencing to reveal mutations in TREM2 presenting as a frontotemporal dementia-like syndrome without bone involvement. JAMA Neurol. 2013;70:78-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Guerreiro R, Wojtas A, Bras J, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med. 2013;368:117-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Jonsson T, Stefansson H, Steinberg S, et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med. 2013;368:107-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Benitez BA, Cooper B, Pastor P, et al. TREM2 is associated with the risk of Alzheimer’s disease in Spanish population. Neurobiol Aging. 2013;34:1711.e15-1711.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ruiz A, Dols-Icardo O, Bullido MJ, et al. Assessing the role of the TREM2 p.R47H variant as a risk factor for Alzheimer’s disease and frontotemporal dementia. Neurobiol Aging. 2014;35:444.e1-444.e4. [DOI] [PubMed] [Google Scholar]
- 73. Pottier C, Wallon D, Rousseau S, et al. ; GMAJ/COMAJ orators. TREM2 R47H variant as a risk factor for early-onset Alzheimer’s disease. J Alzheimers Dis. 2013;35:45-49. [DOI] [PubMed] [Google Scholar]
- 74. Miyashita A, Wen Y, Kitamura N, et al. Lack of genetic association between TREM2 and late-onset Alzheimer’s disease in a Japanese population. J Alzheimers Dis. 2014;41:1031-1038. [DOI] [PubMed] [Google Scholar]
- 75. Jin SC, Carrasquillo MM, Benitez BA, et al. TREM2 is associated with increased risk for Alzheimer’s disease in African Americans. Mol Neurodegener. 2015;10:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Jiao B, Liu X, Tang B, et al. Investigation of TREM2, PLD3, and UNC5C variants in patients with Alzheimer’s disease from mainland China. Neurobiol Aging. 2014;35:2422.e9-2422.e11. [DOI] [PubMed] [Google Scholar]
- 77. Ma J, Zhou Y, Xu J, et al. Association study of TREM2 polymorphism rs75932628 with late-onset Alzheimer’s disease in Chinese Han population. Neurol Res. 2014;36:894-896. [DOI] [PubMed] [Google Scholar]
- 78. Wang P, Guo Q, Zhou Y, et al. Lack of association between triggering receptor expressed on myeloid cells 2 polymorphism rs75932628 and late-onset Alzheimer’s disease in a Chinese Han population. Psychiatr Genet. 2018;28:16-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Yu J-T, Jiang T, Wang Y-L, et al. Triggering receptor expressed on myeloid cells 2 variant is rare in late-onset Alzheimer’s disease in Han Chinese individuals. Neurobiol Aging. 2014;35:937.e1-937.e3. [DOI] [PubMed] [Google Scholar]
- 80. Mehrjoo Z, Najmabadi A, Abedini SS, et al. Association study of the TREM2 gene and identification of a novel variant in exon 2 in Iranian patients with late-onset Alzheimer’s disease. Med Princ Pract. 2015;24:351-354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Reitz C, Mayeux R; Alzheimer’s Disease Genetics Consortium. TREM2 and neurodegenerative disease. N Engl J Med. 2013;369:1564-1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Logue MW, Schu M, Vardarajan BN, et al. ; Alzheimer Disease Genetics Consortium. Two rare AKAP9 variants are associated with Alzheimer’s disease in African Americans. Alzheimers Dement. 2014;10:609-618.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Vardarajan BN, Barral S, Jaworski J, et al. ; The Alzheimer's Disease Sequencing Project. Whole genome sequencing of Caribbean Hispanic families with late-onset Alzheimer’s disease. Ann Clin Transl Neurol. 2018;5:406-417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Cukier HN, Kunkle BK, Hamilton KL, et al. Exome sequencing of extended families with Alzheimer’s disease identifies novel genes implicated in cell immunity and neuronal function. J Alzheimers Dis Parkinsonism. 2017;7:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Shang Z, Lv H, Zhang M, et al. Genome-wide haplotype association study identify TNFRSF1A, CASP7, LRP1B, CDH1 and TG genes associated with Alzheimer’s disease in Caribbean Hispanic individuals. Oncotarget. 2015;6:42504-42514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ayers KL, Mirshahi UL, Wardeh AH, et al. A loss of function variant in CASP7 protects against Alzheimer’s disease in homozygous APOE ε4 allele carriers. BMC Genomics. 2016;17:445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Zhang X, Zhu C, Beecham G, et al. ; Alzheimer's Disease Sequencing Project. A rare missense variant of CASP7 is associated with familial late-onset Alzheimer’s disease. Alzheimers Dement. 2019;15:441-452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Joutel A, Vahedi K, Corpechot C, et al. Strong clustering and stereotyped nature of Notch3 mutations in CADASIL patients. Lancet. 1997;350:1511-1515. [DOI] [PubMed] [Google Scholar]
- 89. Bianchi S, Zicari E, Carluccio A, et al. CADASIL in central Italy: A retrospective clinical and genetic study in 229 patients. J Neurol. 2015;262:134-141. [DOI] [PubMed] [Google Scholar]
- 90. Abramycheva N, Stepanova M, Kalashnikova L, et al. New mutations in the Notch3 gene in patients with cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy (CADASIL). J Neurol Sci. 2015;349:196-201. [DOI] [PubMed] [Google Scholar]
- 91. Guerreiro RJ, Lohmann E, Kinsella E, et al. Exome sequencing reveals an unexpected genetic cause of disease: NOTCH3 mutation in a Turkish family with Alzheimer’s disease. Neurobiol Aging. 2012;33:1008.e17-1008.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Liu X, Zuo Y, Sun W, et al. The genetic spectrum and the evaluation of CADASIL screening scale in Chinese patients with NOTCH3 mutations. J Neurol Sci. 2015;354:63-69. [DOI] [PubMed] [Google Scholar]
- 93. Patel D, Mez J, Vardarajan BN, et al. ; for the Alzheimer’s Disease Sequencing Project. Association of rare coding mutations with Alzheimer disease and other dementias among adults of European ancestry. JAMA Netw Open. 2019;2:e191350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Wang G, Zhang D-F, Jiang H-Y, et al. Mutation and association analyses of dementia-causal genes in Han Chinese patients with early-onset and familial Alzheimer’s disease. J Psychiatr Res. 2019;113:141-147. [DOI] [PubMed] [Google Scholar]
- 95. Sherman RM, Forman J, Antonescu V, et al. Assembly of a pan-genome from deep sequencing of 910 humans of African descent. Nat Genet. 2019;51:30-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Mills MC, Rahal C.. A scientometric review of genome-wide association studies. Commun Biol. 2019;2:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Fereshtehnejad S-M, Vosoughi K, Heydarpour P, et al. ; Global Burden of Disease Study 2016 Eastern Mediterranean Region Collaborators – Neurological Diseases Section. Burden of neurodegenerative diseases in the Eastern Mediterranean Region, 1990-2016: Findings from the Global Burden of Disease Study 2016. Eur J Neurol. 2019;26:1252-1265. [DOI] [PubMed] [Google Scholar]
- 98. Babulal GM, Quiroz YT, Albensi BC, et al. ; on behalf of the International Society to Advance Alzheimer's Research and Treatment, Alzheimer's Association. Perspectives on ethnic and racial disparities in Alzheimer’s disease and related dementias: Update and areas of immediate need. Alzheimers Dement. 2019;15:292-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Claw KG, Anderson MZ, Begay RL, et al. ; Summer internship for INdigenous peoples in Genomics (SING) Consortium. A framework for enhancing ethical genomic research with Indigenous communities. Nat Commun. 2018;9:2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Jackson L, Kuhlman C, Jackson F, et al. Including vulnerable populations in the assessment of data from vulnerable populations. Front Big Data. 2019;2:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Marioni RE, Harris SE, Zhang Q, et al. GWAS on family history of Alzheimer’s disease. Transl Psychiatry. 2018;8:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Scott EM, Halees A, Itan Y, et al. ; Greater Middle East Variome Consortium. Characterization of greater middle eastern genetic variation for enhanced disease gene discovery. Nat Genet. 2016;48:1071-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Gao Y, Zhang C, Yuan L, et al. ; The Han100K Initiative. PGG.Han: The Han Chinese genome database and analysis platform. Nucleic Acids Res. 2020;48:D971-D976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.GenomeAsia100K Consortium. The GenomeAsia 100K Project enables genetic discoveries across Asia. Nature. 2019;576:106-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Genome of the Netherlands Consortium. Whole-genome sequence variation, population structure and demographic history of the Dutch population. Nat Genet. 2014;46:818-825. [DOI] [PubMed] [Google Scholar]
- 106. Higasa K, Miyake N, Yoshimura J, et al. Human genetic variation database, a reference database of genetic variations in the Japanese population. J Hum Genet. 2016;61:547-553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Locke AE, Steinberg KM, Chiang CWK, et al. ; FinnGen Project. Exome sequencing of Finnish isolates enhances rare-variant association power. Nature. 2019;572:323-328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Martin AR, Kanai M, Kamatani Y, et al. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat Genet. 2019;51:584-591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Bandres-Ciga S, Diez-Fairen M, Kim JJ, et al. Genetics of Parkinson’s disease: An introspection of its journey towards precision medicine. Neurobiol Dis. 2020;137:104782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Sirkis DW, Geier EG, Bonham LW, et al. Recent advances in the genetics of frontotemporal dementia. Curr Genet Med Rep. 2019;7:41-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Guerreiro R, Gibbons E, Tábuas-Pereira M, et al. Genetic architecture of common non-Alzheimer’s disease dementias. Neurobiol Dis. 2020;142:104946. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.