

Abstract
Congenital prothrombin deficiency is an extremely rare, autosomal recessive bleeding disorder with a prevalence of 1 in 2 million individuals. Here, we report a case of congenital prothrombin deficiency with two concurrent mutations in the prothrombin gene (F2), affecting the heavy B chain. The patient presented with a history of multiple bleeding events in his youth that are mostly trauma associated, with a family history of prothrombin deficiency. Laboratory analysis showed a prolonged activated partial thromboplastin time and a prothrombin activity level of 5%. Genetic analysis of the F2 gene identified two heterozygous variants; one is a previously reported pathogenic deletion (c.1814_1815del; p.His605Argfs*13), and the other is a novel missense variant (c.1147C>T; p.Arg383Trp). In silico analysis predicted that p.Arg383Trp is likely to be disease causing, as it affects one of the anion‐binding exosites‐I of the B chain. This case highlights the significance of molecular findings in confirming the diagnosis of patients with congenital prothrombin deficiency.
Keywords: bleeding disorder, dysprothrombinemia, hypoprothrombinemia, prothrombin deficiency, prothrombin mutation
Essentials.
Congenital prothrombin deficiency is a rare bleeding disorder.
We report a patient with congenital prothrombin deficiency.
We identified two genetic variants (one novel) in the prothrombin gene.
This case highlights the relevance of molecular analysis in rare bleeding disorders.
1. INTRODUCTION
Congenital prothrombin deficiency is a rare, autosomal recessive bleeding disorder with a prevalence of 1 in 2 million individuals. 1 It can be classified as hypoprothrombinemia or “true deficiency” (type 1), dysprothrombinemia (type 2), or combined forms. A new classification adds a new category: dysprothrombinemia without bleeding but with thrombosis (type 3). 2 A low level of prothrombin activity is always detectable, as complete prothrombin deficiency is incompatible with life. Although a prothrombin coagulant activity <10% is usually associated with severe bleeding manifestations 3 (including mucosal bleeding, subcutaneous and muscle hematoma, and prolonged bleeding episodes following injury), there is a discrepancy between the bleeding severity and the observed values of coagulation tests. 1 Patients with prothrombin deficiency are either homozygous or compound heterozygous for mutations in the F2 gene, also known as prothrombin. Patients with true deficiency (with homozygous mutations or compound heterozygous mutation) have <10% normal prothrombin levels. Heterozygous mutation carriers usually have prothrombin levels ranging from 30% to 60%, and >70% levels are reported as normal. 1 To the best of our knowledge, <50 cases of hypoprothrombinemia or dysprothrombinemia have been characterized from a molecular genetic perspective. 2 , 4 , 5 , 6 Herein, we report a case of congenital prothrombin deficiency resulting from two concurrent mutations in the F2 gene, affecting the heavy B chain.
2. CASE REPORT
A 30‐year‐old man, originally from Iran, was referred to our clinic with a history of prothrombin deficiency, which was documented during childhood. The patient reported multiple bleeding events in his youth related to trauma, consisting of hemarthrosis of both knees and right elbow. The average frequency of events was one to two bleeds per year, needing treatment with self‐administered FEIBA (factor VIII inhibitor bypassing activity) and/or tranexamic acid. His worst bleeding episode occurred at 18 years of age, when he had a traumatic thigh hemorrhage leading to compartment syndrome necessitating surgical evacuation. No other history of surgical interventions was reported. No gastrointestinal bleeds were reported. However, a single episode of hematuria related to strenuous exercise was documented. In the year before his assessment, he noticed occurrence of more frequent soft‐tissue bleeding events while playing sports. He had been maintained on tranexamic acid and FEIBA or prothrombin complex concentrate as required. Physical examination was unremarkable. Coagulation tests showed a prothrombin time of 19.9 seconds (normal range, 11‐15) and a partial thromboplastin time of 42 seconds (normal range, 20‐29). Prothrombin activity was measured on two separate occasions using factor II–deficient plasma (HemosIL, Instrumentation Laboratory, Bedford, MA, USA) on an ACL Top 500 instrument (Instrumentation Laboratory), and it was reported at 0.05 U/mL (normal range, 0.50‐2.0). Activity of factors V, VII, VIII, IX, X, and XI was reported within normal range. The proband’s sister and one paternal uncle had a documented prothrombin deficiency, as shown in the pedigree (Figure 1). Neither of the proband’s parents had a documented history of bleeding, and it was unknown if they were related by consanguinity.
FIGURE 1.
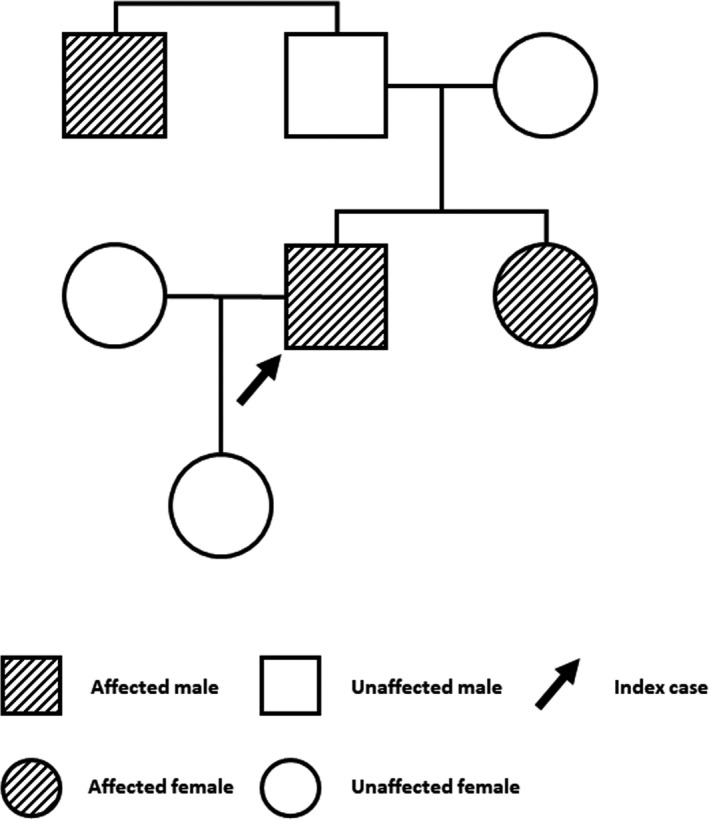
Family pedigree
Since the present report meets the criteria for a case review as established by Western University’s Research Ethics Board, no formal approval was required. After obtaining consent from the patient in accordance with our institutional guidelines, DNA was isolated from peripheral blood leukocytes using standard methods with the MagNA Pure system (Roche Diagnostics, Laval, QC, Canada). The DNA sample was quantified by measuring absorbance with a DTX 880 Multimode Detector (Beckman Coulter, Brea, CA, USA). Coding regions and the flanking intronic regions (up to ±20 base pairs) of the prothrombin gene were polymerase chain reaction (PCR) amplified. Purified PCR products were sequenced using the BigDye Terminator version 1.1 cycle sequencing kit (Life Technologies, Rockville, MD, USA) and subjected to capillary electrophoresis on an ABI 3730 (Life Technologies). Chromatograms obtained after sequencing were analyzed with Mutation Surveyor version 4.0.7 software (SoftGenetics, LLC, State College, PA, USA). Molecular analysis and variant classification were performed by a clinical molecular geneticist at the Molecular Genetics Laboratory of our institution. Variants were interpreted on the basis of the American College of Medical Genetics guidelines. 7 In silico variant analysis was performed using the Alamut Visual Software (https://www.interactive‐biosoftware.com/alamut‐visual/) including Polymorphism Phenotyping‐2 8 (PolyPhen‐2, http://genetics.bwh.harvard.edu/pph2/ index.shtml), SIFT 9 (http://sift.jcvi.org/), and Mutation Taster 10 (http://www.mutationtaster.org/).
3. DISCUSSION
Prothrombin (factor II) is a vitamin K–dependent glycoprotein encoded by a gene on chromosome 11p11‐q1212 in the centromere region, and it contains 14 coding exons. 1 , 11 Prothrombin is synthesized in the liver and is usually present in plasma at concentrations of 1 to 2 μM. The protein contains five functional domains: the propeptide leader sequence, the γ‐carboxyglutamic acid (Gla) domain, two Kringle areas, and the serine protease catalytic domain. 11 Prothrombin is activated by factor Xa in the presence of phospholipids, calcium, and factor Va to form active α‐thrombin, which cleaves fibrinogen into fibrin to form a blood clot. Thrombin is also involved in the activation of other coagulation factors (V, VIII, and XIII) and has a role in platelet activation and aggregation. 12 In addition, thrombin participates in the anticoagulation process as it activates protein C. It plays a role in inflammation, cell proliferation, and tissue repair mechanisms. 13 For an unclear reason, prothrombin mutations are more prevalent in patients from a Latin/Hispanic origin (Puerto Rico, Spain, France, etc.). 2 , 11 Prothrombin deficiency is classified into severe (when factor level <5%), moderate (factor level 5%‐10%), and mild (factor level >10%). 1 In patients with severe deficiency, the condition is always associated with a significant bleeding tendency, usually with prolonged postinjury bleeding and subcutaneous and muscle hematomas. This is similar to the presentation in the proband, though he technically has a moderate deficiency with a level of 5%.
Until recently, dysprothrombinemia was thought of as a cause for bleeding only. Although most patients with dysprothrombinemia have been reported to exhibit a bleeding phenotype, some cases have been reported in which the mutations led to thrombotic episodes. 2 Therefore, dysprothrombinemia can be classified as a hemorrhagic‐thrombotic disorder, depending on the type of the mutation present. 2
Missense mutations (80%) are more common in the prothrombin gene compared to other types of variants including insertions/deletions (10%) and nonsense (6%) and splice site variants (4%). 1 As prothrombin deficiency is a very rare bleeding disorder, identification of new mutations is important to improve our understanding of this condition.
In this patient, we found two heterozygous genetic variants in the prothrombin gene that occurred concurrently. The genetic analysis detected four genetic variations in the prothrombin gene (Table 1). Two of those were benign polymorphisms with a reported allele frequency of 20.5% and 59.3%. The other two variants have been reported at a very low frequency in normal control databases (gnomAD over all allele frequency 0.001%; https://gnomad.broadinstitute.org/).
TABLE 1.
Genetic variants identified in F2 gene in the patient
| Variant (nucleotide position) (NM_000506.3; GRCh38) | Variant (amino acid position) | Overall allele frequency reported in GnomAD, a n (%) | In silico tools b predicting the variant as damaging | ACMG classification |
|---|---|---|---|---|
| c.1814_1815del | p.(His605Argfs*13) | 0.00001 (0.001) | ‐ | ACMG #2 likely pathogenic |
| c.1147C>T | p.(Arg383Trp) | 0.00001 (0.001) |
SIFT (score: 0.02) Mutation Taster (prob, 0.853) Polyphen2 (score, 1.00) |
ACMG #3 variant of unknown significance |
| c.494C>T | p.(Thr165Met) | 0.205 (20.5) | None | ACMG #5 benign polymorphism |
| c.423‐7G>C | ‐ | 0.593 (59.3) | None | ACMG #5 benign polymorphism |
ACMG, American College of Medical Genetics and Genomics. 7
GnomAd frequency (https://gnomad.broadinstitute.org/).
The first variant is a novel, missense, likely pathogenic variant (c.1147C>T) responsible for the substitution of amino acid arginine by tryptophan at the 383 amino acid position in the prothrombin protein p.(Arg383Trp). This mutation results in changing one of the anion‐binding recognition domains, namely, anion‐binding exosite‐I (at Arg20) (Figure 2). These sites are involved in interacting with substrates, cofactors, and inhibitors and are critical for functions like fibrinogen cleavage. 14 , 15 , 16 , 17 , 18 Exosite‐I has been shown to be important for recognition of substrates such as protease activator receptor‐1, factor V (FV), and factor VIII (FVIII); cofactors such as thrombomodulin and glycoprotein Ibα (GPIbα) and inhibitors such as heparin cofactor II. Therefore, considering the significance of these sites, an amino acid change at this residue may interfere with recognition and interaction with substrate and/or binding proteins. This variant is predicted to be disease‐causing by in silico pathogenicity prediction tools (Mutation Taster (prob, 0.853), Polyphen2 (score, 1.00), SIFT (score, 0.02)). Although in silico assessment supports the pathogenic nature of this variant, functional studies need to be performed to evaluate the confirmed consequential impact of this variant at a biochemical level.
FIGURE 2.
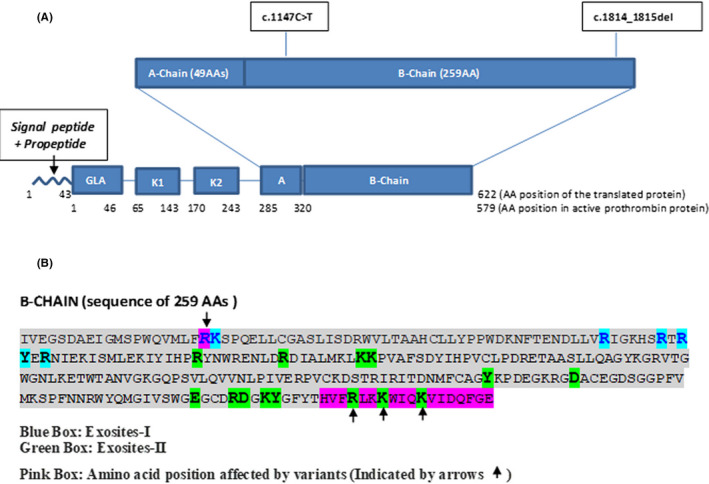
Structure of prothrombin protein and effect of variants on protein. (A) This figure shows the domain architecture of the prothrombin protein with location of the two variants (identified in our patient) in the B‐chain region of the protein. (B) Amino acid sequence of the B chain with highlighted (blue and green) exosite‐I and II positions and pink boxes highlighting the exosite‐I/II positions affected by these two variants
The second variant is a pathogenic deletion of two base pairs (c.1814_1815del), which changes the amino acid reading frame after the 605 amino acid position and results in protein termination after 13 amino acid residues. This deletion causes loss of three important exosite‐II positions falling in this region (Figure 2). Exosite‐II is known to interact with sulfated glycosaminoglycans present in heparin, a cofactor for the inhibition of thrombin by antithrombin. Functional and structural studies have shown that exosite‐II is also involved in the recognition and cleavage of FV and FVIII and also in the association of thrombin with GPIbα. 14 The variant described here causes the loss of three of the five residues that form exosite‐II, likely leading to significant impairment of the functions of thrombin. This deletion (c.1814_1815del) has recently been reported in the heterozygous state in a patient with a coagulation disorder. 19
Collectively, these results indicate that the patient is heterozygous for two genetic variants in the prothrombin gene: one novel missense mutation predicted to be associated with the disease by in silico assessment, and a second deletion mutation that causes loss of a vital region of the gene. Unfortunately, we were not able to get genetic testing done in the proband’s parents or other family members to confirm the compound heterozygous nature of these two variants, yet the positive family history of prothrombin deficiency indicates that these are likely inherited mutations and might be of value during investigation of other cases of prothrombin deficiency. This case highlights the utility of molecular genetic testing in confirming the diagnosis of patients with congenital prothrombin deficiency.
RELATIONSHIP DISCLOSURE
The authors have no conflicts of interest to declare.
AUTHOR CONTRIBUTIONS
EMM, LL, and AL‐L were involved with the care of this patient. PB, AS, and BS were involved in the genetic analysis and interpretation. All authors contributed to the drafting and approval of the final manuscript.
ACKNOWLEDGMENT
We thank Dr. Ian Chin‐Yee for his support, comments, and input.
Handling Editor: Pantep Angchaisuksiri
Contributor Information
Eman M. Mansory, @EmanMansory.
Alejandro Lazo‐Langner, Email: alejandro.lazolangner@lhsc.on.ca, @alejandrolazo2.
REFERENCES
- 1. Lancellotti S, Basso M, De Cristofaro R. Congenital prothrombin deficiency: an update. Semin Thromb Hemost. 2013;39(6):596–606. [DOI] [PubMed] [Google Scholar]
- 2. Girolami A, Ferrari S, Cosi E, Girolami B, Lombardi AM. Congenital prothrombin defects: they are not only associated with bleeding but also with thrombosis: a new classification is needed. Hematology. 2018;23(2):105–110. [DOI] [PubMed] [Google Scholar]
- 3. Peyvandi F, di Michele D, Bolton‐maggs PHB, Lee CA, Tripodi A, Srivastava A. Classification of rare bleeding disorders (RBDs) based on the association between coagulant factor activity and clinical bleeding severity. J Thromb Haemost. 2012;10(9):1938–1943. [DOI] [PubMed] [Google Scholar]
- 4. Bafunno V, Bury L, Tiscia GL, et al. A novel congenital dysprothrombinemia leading to defective prothrombin maturation. Thromb Res. 2014;134(5):1135–1141. [DOI] [PubMed] [Google Scholar]
- 5. Pasmant E, Dumont B, Lacapere JJ, Dautzenberg MD, Bezeaud A. A severe neonatal presentation of factor II deficiency. Eur J Haematol. 2011;87(5):464–466. [DOI] [PubMed] [Google Scholar]
- 6. Seki M, Koh K, Inoue T, et al. Prophylactic administration of prothrombin complex concentrates for congenital prothrombin deficiency with a novel frameshift mutation, prothrombin saitama. Pediatr Blood Cancer. 2013;60(3):503–505. [DOI] [PubMed] [Google Scholar]
- 7. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012;40(W1):W452‐W457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep‐sequencing age. Nat Methods. 2014;11(4):361–362. [DOI] [PubMed] [Google Scholar]
- 11. Girolami A, Santarossa L, Scarparo P, Candeo N, Girolami B. True congenital prothrombin deficiency due to a “new” mutation in the pre‐propeptide (ARG‐39 GLN). Acta Haematol. 2008;120:82–86. [DOI] [PubMed] [Google Scholar]
- 12. Tamary H, Surrey S, Augustine J, Shalmon L, Schwartz E, Rappaport EF. Molecular analysis of a compound heterozygote for hypoprothrombinemia and dysprothrombinemia (‐G 7248/7249 and ARG 340 TRP). Blood Coagul Fibrinolysis. 1997;8(6):337–343. 10.1097/00001721-199709000-00003. [DOI] [PubMed] [Google Scholar]
- 13. Kankan SU, Jin Y, Miao Z, Cheng X, Yang L, Wang M. Phenotypic and genetic analysis of dysprothrombinemia due to a novel homozygous mutation. Hematology. 2017;22(6):380–385. 10.1080/10245332.2017.1287332. [DOI] [PubMed] [Google Scholar]
- 14. Davie EW, Kulman JD. An overview of the structure and function of thrombin. Semin Thromb Hemost. 2006;32(suppl 1):3–15. [DOI] [PubMed] [Google Scholar]
- 15. Chahal G, Thorpe M, Hellman L. The importance of exosite interactions for substrate cleavage by human thrombin. PLoS One. 2015;10(6):e0129511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hall SW, Nagashima M, Zhao L, Morser J, Leung LL. Thrombin interacts with thrombomodulin, protein C, and thrombin‐activatable fibrinolysis inhibitor via specific and distinct domains. J Biol Chem. 1999;274(36):25510–25516. [DOI] [PubMed] [Google Scholar]
- 17. Tsiang M, Jain AK, Dunn KE, Rojas ME, Leung LL, Gibbs CS. Functional mapping of the surface residues of human thrombin. J Biol Chem. 1995;270(28):16854–16863. [DOI] [PubMed] [Google Scholar]
- 18. Bode W, Mayr I, Baumann U, Huber R, Stone SR, Hofsteenge J. The refined 1.9 A crystal structure of human alpha‐thrombin: interaction with D‐Phe‐Pro‐Arg chloromethylketone and significance of the Tyr‐Pro‐Pro‐Trp insertion segment. EMBO J. 1989;8(11):3467–3475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Downes K, Megy K, Duarte D, et al. Diagnostic high‐throughput sequencing of 2396 patients with bleeding, thrombotic, and platelet disorders. Blood. 2019;134(23):2082–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]