

Abstract
Objective:
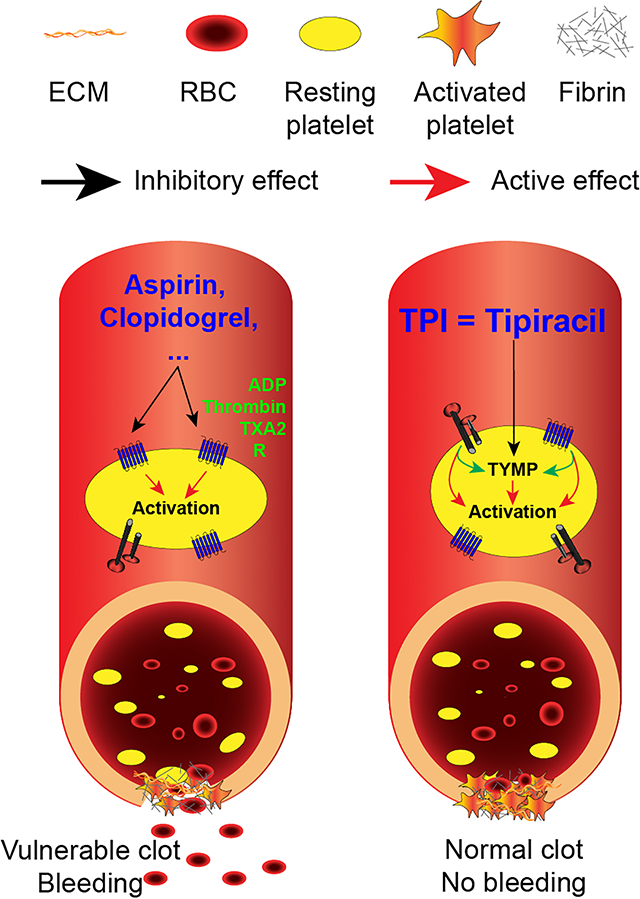
Current antiplatelet medications increase the risk of bleeding, which leads to a clear clinical need in developing novel mechanism-based antiplatelet drugs. Thymidine phosphorylase (TYMP), a cytoplasm protein that is highly expressed in platelets, facilitates multiple agonist-induced platelet activation and enhances thrombosis. Tipiracil hydrochloride (TPI), a selective TYMP inhibitor, has been approved by the FDA for clinical use. We tested the hypothesis that TPI is a safe antithrombotic medication.
Approach and Results:
By co-expression of TYMP and Lyn, glutathione S-transferase tagged Lyn-SH3 domain, or Lyn-SH2 domain, we showed the direct evidence that TYMP binds to Lyn through both SH3 and SH2 domains, and TPI diminished the binding. TYMP deficiency significantly inhibits thrombosis in vivo in both sexes. Pretreatment of platelets with TPI rapidly inhibited collagen- and ADP-induced platelet aggregation. Under either normal or hyperlipidemic conditions, treating wild type (WT) mice with TPI via intraperitoneal injection, intravenous injection, or gavage feeding dramatically inhibited thrombosis without inducing significant bleeding. Even at high doses, TPI has a lower bleeding side effect compared to aspirin and clopidogrel. Intravenous delivery of TPI alone or combined with tissue plasminogen activator dramatically inhibited thrombosis. Dual administration of a very low dose of aspirin and TPI, which had no antithrombotic effects when used alone, significantly inhibited thrombosis without disturbing hemostasis.
Conclusions:
This study demonstrated that inhibition of TYMP, a cytoplasmic protein, attenuated multiple signaling pathways that mediate platelet activation, aggregation, and thrombosis. TPI can be used as a novel antithrombotic medication without the increase in risk of bleeding.
Keywords: thymidine phosphorylase, tipiracil, platelet activation
Subject terms: Platelets, Thrombosis, Vascular Disease
Graphical Abstract
Introduction
Thrombotic events are a major cause of morbidity and mortality both in the US and worldwide, and, therefore, they remain as a significant area of interest for clinical research focusing on the discovery of new mechanisms and development of novel therapies.1–3 While, the reasons are complex, the lack of suitable targets is a key factor as to why new antithrombotic agents are not being developed.4 Platelet activation and hyper-aggregation at the site of vascular injury is the primary pathogenic component of thrombosis, which can lead to vessel occlusion resulting in myocardial infarction and ischemic stroke. In this context, in addition to the glycoprotein (GP) Ib-XI-V binding to von willebrand factor (vWF)/collagen complexes,5 other platelet surface receptors, including α2β1 integrin and GPVI, also confer both platelet adhesion and activation in response to exposed collagens at the site of vascular injury.6–8 The adhered and activated platelets release soluble agonists, including adenosine diphosphate (ADP), thrombin, and thromboxane A2 (TXA2), that activate additional platelets locally via G protein coupled receptors (GPCRs), notably the ADP-receptor P2Y12 and thrombin receptor PAR1.7, 9 Consequently, various antiplatelet drugs, such as aspirin (COX inhibitor), clopidogrel (P2Y12 inhibitor), and vorapaxar (PAR1 inhibitor), have been developed and used clinically for the primary or secondary prevention of platelet-mediated thrombotic events.10–13 However, due to the systemic effects of their targets, these drugs present major side effects including injury to the gastrointestinal mucosa, thrombocytopenia, and systemic hemorrhage.2, 10–13 Permanent inhibition of platelet function is also problematic for patients who need emergency surgery.14 In addition, some patients do not respond to the current antiplatelet regimens and still have a high incidence of vascular thrombosis.2 Therefore, there is a significant clinical interest in elucidating unique molecular mechanisms of platelet-mediated thrombus formation that can lead to the development of a novel antiplatelet therapy with minimized systemic risks.2, 15 Currently, the majority of antiplatelet drugs are targeted at platelet surface receptors and drugs that safely inhibit platelet function by targeting intracellular proteins have yet to be developed.
Human thymidine phosphorylase (TYMP) was first isolated from amniochorion16 and later from human platelets,17 thus it is also known as platelet-derived endothelial cell growth factor.18 TYMP is mainly found inside the cell due to the lack of an amino-terminal hydrophobic leader sequence required for cell secretion.17 TYMP belongs to the family of pyrimidine nucleoside phosphorylases, and its primary function is to drive the salvage pathway of pyrimidine nucleosides.19–21 In the presence of inorganic phosphate, TYMP reversely catalyzes thymidine to thymine and 2-D-deoxyribose-1-phosphate, and the latter is further degraded to 2-D-deoxyribose.21, 22 TYMP also has deoxyribosyl transferase activity and can transfer the deoxyribosyl moiety from a pyrimidine nucleoside to another pyrimidine base that results in the formation of a new pyrimidine nucleoside.21, 23 Functional TYMP acts as a homodimer and plays a key role in pyrimidine nucleoside metabolism with the purpose of maintaining a pool of pyrimidine nucleotides that can be used for DNA repair and replication. However, the role of TYMP in platelets, a non-nucleated cell, remains unclear. Our recent study demonstrated that TYMP can act as a signaling protein and it may potentially modulate signaling via protein-protein interactions between its N-terminal proline-rich region and the SH3 domain of its partner protein.21, 24 TYMP deficiency dramatically inhibited platelet response to the conventional agonists: collagen, ADP, and thrombin.24 Inhibition of TYMP activity with KIN59, a novel, but not selective, TYMP inhibitor,25 dramatically inhibited platelet activation in vitro and thrombosis in vivo.24 These data suggest that TYMP may be a targetable intracellular protein in platelets and its inhibition is antithrombotic. The potent and selective TYMP inhibitor, tipiracil hydrochloride (TPI), is an auxiliary component of a novel anticancer drug, Lonsurf, which contains trifluridine and TPI at a molar ratio of 1:0.5. Lonsurf was recently approved by the FDA for clinical use, suggesting that TPI could be repositioned as a novel antiplatelet and antithrombotic drug. In this study, we examined the role of TPI on platelet activation and thrombosis, using both in vitro and in vivo studies, and provided evidence that TPI-mediated TYMP inhibition can be repurposed as a safe and effective antithrombotic therapy. TPI may be suitable for use as an antithrombotic therapy in patients at high risk of developing thrombotic cardiovascular diseases. The addition of low dose TPI to low dose aspirin could also be used as a new dual antiplatelet therapy.
Materials and Methods
The authors declare that all supporting data are available within the article and its online supplementary files.
Materials
Platelet agonists, including ADP, collagen, and thrombin as well as CHRONO-LUME® were purchased from Chrono-log (Havertown, PA). Collagen related peptide (CRP) was a gift from Dr. Peter Newman (Blood Research Institute, WI) as mentioned before.24 Antibodies to phosphorylated AKT (4060S), pan-AKT (2920S), TYMP (4307S), and GST (2624S) were purchased from Cell Signaling Technology (Danvers, MA). Antibodies to TYMP (SC-56584), Lyn (SC-7274), and Actin (sc-8432) were purchased from Santa Cruz Biotechnology (Dallas, TX). Antibody to TYMP was also purchased from abcam (ab180783). FITC-conjugated P-selectin antibody (Cat#553744) and the isotype control IgG were purchased from BD Biosciences (San Jose, CA). Tipiracil hydrochloride (TPI) (T2366) was purchased from TargetMol (Boston, MA). All other chemical reagents were purchased from Sigma (St. Louis, MO) except where specifically indicated.
Animals
Tymp–/– mouse strain was generated by Dr. Hirano and has been back-crossed into C57BL/6J background more than 10 times.24, 26 Wild type (WT) C57BL/6J mice breeders were purchased from The Jackson Laboratory (Bar Harbor, ME). Mice had ad libitum access to Laboratory Rodent Diet (Cat# 5001, LabDiet) and water. Breeding mice were genotyped for TYMP before mating and all mice were given identity using ear tags at the time of weaning. Mice in 8 to 12 weeks old in both sexes were used in this study. All procedures and manipulations of animals have been approved by the Institutional Animal Care and Use Committee of Marshall University (IACUC#: 1033528).
Murine carotid artery thrombosis model and tail bleeding assay
The ferric chloride (FeCl3) induced carotid artery thrombosis model has been described before in detail.27, 28 Briefly, the mouse was anesthetized with ketamine and xylazine (100/10 mg/kg). Vascular injury was induced by topically applying a piece of filter paper (1 × 2 mm) saturated with 7.5% FeCl3 solution to the carotid artery for 1 min. Thrombus formation was observed in real-time using an intravital microscope (Leica DM6 FS) and video images were captured with a 14-bit Retiga R1 CCD color digital camera (Teledyne QImaging, Surrey, BC, Canda) and a STP7-S-STDT Streampix 7 software (Norpix, Montreal, Canada). The endpoints were set as: 1) blood flow has ceased for > 30 seconds; or 2) occlusion is not seen after 30 min of FeCl3 injury. In this case, 30 min was assigned to that mouse for the purpose of statistical analysis.
In some experiments, a jugular vein catheter prepared with P-10 tubing was placed and used for drug administration. In this case, platelets were labeled by bolus injection of 50 μL Rhodamine 6G solution through the jugular vein catheter and then thrombosis was initiated as mentioned above. TPI (62.5 – 1,000 μg/kg), tPA (125–1,000 μg/kg), or combination of different doses of TPI and tPA were delivered to mice at 5 min after thrombus initiation. Thrombus formation was continually monitored using endpoints as mentioned above.
Platelet isolation and aggregation assay
Mice were anesthetized with ketamine/xylazine (100/10 mg/kg) and whole blood was drawn from the inferior vena cava (IVC) using 0.109 M sodium citrate as an anticoagulant.24, 29, 30 Modified Tyrode’s buffer (in mM: 137 NaCl, 2.7 KCl, 12 NaHCO3, 0.4 NaH2PO4, 5 HEPES, 0.1% glucose and 0.35% BSA, pH 7.2), 0.7 volumes of the whole blood, was added, and platelet-rich plasma (PRP) was separated by centrifugation at 100 g for 10 min, and top 2/3 of the PRP transferred into a new tube. The buffer coat and red blood cells sedation were further centrifuged at 1,000 g for 6 minutes to obtain platelet-poor-plasma (PPP). Platelets number were counted with a hemocytometer, adjusted to a concentration of 2.5 × 108/ml using PPP. 400 μL of this PRP was used for platelet aggregation assay monitored by an Aggregometer (Model 700, Chrono-log, Havertown, PA) and data were acquisted using Aggrolink8 version 1.29.29–31 Sample were stirred constantly at 1,200 RPM at 37 °C. CaCl2/MgCl2 was added to the reaction to a final concentration of 1 mM immediately before adding the selected agonist, as indicated in the Results. Light transmission was monitored over time and aggregation was quantified with 100% aggregation corresponding to 100% light transmission. In some experiments, platelets were pretreated with different concentrations of TPI or vehicle solution (DMSO or saline) for 2 minutes, unless stated otherwise, before the initiation of aggregation.
Isolation of total RNA from circulating nucleated cells
Mouse whole blood were drawn through the IVC puncture, added 0.7 volume of Modified Tyrode’s buffer, and centrifugated for PRP isolation as mentioned above. The bottom 1/3 of the PRP and the buffy coat were pooled from 6 mice in each strain, treated with red blood cell lysis buffer (ab204733, Cambridge, MA), and then nucleated cells were pelleted. RNA isolation was conducted using a Qiagen RNeasy Plus Universal Kit (Cat# 73404) based on the introduction of the manufacture. cDNA was constructed using the SuperScript VILO cDNA synthesis kit (Invitrogen, Carlsbad, CA). Primer pairs mTPU2 (CGC GGT GAT AGA TGG AAG AGC A) and mTPL2 (CCG CTG ATC ATT GGC ACC TTA C), as well as mGADPHU2 (CAG CAA CTC CCA CTC TTC CAC CTT CG) and mGADPHL2 (GGC CTC TCT TGC TCA GTG TCC TTG CT) were used for mouse TYMP and GAPDH amplification, which generated 258 bp (TYMP) and 196 bp (GAPDH) PCR products, respectively. The Platinum™ SuperFi II PCR Master Mix (Invitrogen, Carlsbad, CA) was used for PCR reaction.
Cellix flow chamber-based platelet adhesion and aggregation assay
Cellix flow chambers were overnight coated with collagen 100 μg/mL in PBS and blocked with 0.1% BSA in PBS for 30 min at room temperature before usage. Whole blood was drawn from mouse as above mentioned, stained with Rhodamine 6G (final concentration of 0.1 μg/μL), repleted with CaCl2/MgCl2 in a final concentration of 1 mM, and immediately perfused through the flow chamber with a flow shear at 65 dyn/cm2. In some experiments, the whole blood from one mouse was divided into two parts (450 μL/part). One part was treated with TPI in a final concentration of 50 μM for 5 min and another part was treated with saline and used as control. Platelet accumulation on the coated chamber surface was monitored and recorded in real time for 3 min with an Invert Microscope (Leica DM8) with a video camera operated by the CaptaVision+ v2.1 software (ACCU-SCOPE®, Commack, NY). The flow chambers were washed with PBS for additional 3 min to remove the free blood cells and then images at the six marker-sites designed by the manufacture were taken and fluorescent intensity was measured using Image J (Fiji, NIH).
Compare the therapeutic effect of TPI to aspirin and clopidogrel
8 to 10-week-old WT mice was gavage fed with TPI (0.06, 0.17, 0.5 and 1 mg/Kg), Aspirin (0.5, 1, and 2.5 mg/Kg), or clopidogrel (1, 2.5, and 5 mg/Kg), or the combination of aspirin 0.5 mg/kg and TPI 50 μg/kg, once daily for one week, and then subjected to the thrombosis model. WT mice also received intraperitoneal injection of TPI 17 mg/kg or vehicle for 3 days, or gavage feeding with TPI (0.06 mg/Kg/day) for 14 to 30 days, and then were subjected to the thrombosis model and tail bleeding assay.
Tail bleeding assay was conducted in some mice immediately after the thrombosis study as well as in some mice that were not used for the in vivo thrombosis study. Briefly, the tail was cut at the site of 1 cm from the tip with a sharp razor blade and immediately immersed into warm saline (37 °C) and cessation time of bleeding was recorded.32 If bleeding showed no stoppage after 9 min, which is about three times longer than the average bleeding time that we observed in the normal WT mice, 9 min was assigned to that mouse for statistical analysis.
Examine the effect of TYMP inhibition on thrombosis under hyperlipidemia
Male WT mice were fed a western diet (WD, TD.88137) for 3 weeks, and then randomly divided into two groups. One group was continually fed with WD and another group was fed with a customized WD (TD.190501), which has the same component as TD.88137 with the addition of 10.7 mg/kg TPI, for one more week. Mice fed with TD.190501 received TPI at a dose of approximately 1 mg/kg body weight/day. These mice were then subjected to the 7.5% FeCl3 induced thrombosis model. Age-matched mice on normal laboratory diet were used as control.
Evaluation of platelet signaling activation
Platelets in PRP were pooled from 8–10 mice, and platelet concentration was adjusted to 2.5 × 108/ml with PPP and then divided into 4 equal parts. CaCl2/MgCl2 in a final concentration of 1 mM was added immediately before platelets activation was initiated with 2.5 μM ADP. Platelet activation was stopped by adding an EDTA/PGE1 (2 mM and 1μg/ml final concentration, respectively) mixture at 1, 3, and 5 min. No ADP-treated platelets were used as resting platelet control (0 minute). Platelets were pelleted immediately by centrifugation and lysed in radioimmunoprecipitation assay (RIPA) buffer (ThermoFisher, Waltham, MA) containing proteinase/phosphatase inhibitor cocktail (ThermoFisher). Thirty micrograms of total proteins were separated in SDS-PAGE, transferred to a PVDF membrane, and blotted with antibodies as indicated in the Result. Membranes were stripped and re-blotted for a total protein or a housekeeping protein as loading control.
Flowcytometry assay of platelet activation
Platelets in PRP were adjusted to 2.5 × 108/ml with PPP and then divided into 8 equal parts. CaCl2/MgCl2 in a final concentration of 1 mM was added immediately before platelets activation was initiated with 2.5 μM ADP. Platelet activation was stopped by adding the EDTA/PGE1 mixture at 1, 3, and 5 min. No ADP-treated platelets were used as resting platelet control (0 minute). Platelets were then fixed with 2% paraformaldehyde, stained with a FITC conjugated rat anti-mouse P-selectin antibody (BD pharmingen), and P-selectin exposure on the platelet surface was analyzed by flowcytometry.
Generation of fusion proteins to determine that TYMP binds to LYN through the SH3 domain
We previously showed that TYMP binds to Lyn, Fyn and Yes, and this binding may be via the proline-rich N-terminus of TYMP to the SH3 domain in the SFKs.24 To further test this hypothesis, the human Lyn SH3 domain nucleotides (hLynSH3, amino acids 63–123) was amplified by PCR using primer pairs 5’-ATC GTG GTC GCC CTG TAC-3’ and 5’-ATT CAG CTT AGC CAC GTA GTT AGA T-3’, and Lyn SH2 domain nucleotides (hLynSH2, amino acids 126–226) was amplified using primer pairs 5’-TCT TAA GGA TCC TGG TTC TTT AAG GAC ATT ACC AG-3’ and 5’-TTC AAG GCG GCC GCA CAA GCC TTT TCC AGT CTC C-3’, and a codon optimized human Lyn encoding plasmid pEGFP-N1-human lyn-GFP (Addgene, plasmid # 35958) as a template. The hLynSH3 products were cloned into a pCR2.1 TA cloning vector (Invitrogen, Carlsbad CA), transformed into One Shot TOP10 E. coli (Invitrogen), and screened with blue and white selection. The insert orientation was confirmed by sequencing with the M13 forward primer, 5’-GTA AAA CGA CGG CCA GT-3’, and a reverse primer 5’-ATC GTG GTC GCC CTG TAC-3’. The SH3 domain cut out from the pCR2.1 using BamHI and NotI, and the SH2 PCR products digested with BamHI and NotI were purified with Qiagen Gel Extraction kit, and then cloned into a mammalian expression vector pEBG (Addgene, plasmid # 22227) with an N-terminal glutathione S-transferase (GST) affinity tag between the restriction sites BamHI and NotI, and a new plasmid pEBG-GST-hLynSH3 and pEBG-GST-hLynSH2 were generated.
We had previously constructed a pCDNA6B/his-hTYMP plasmid vector.33 pEBG-GST-hLynSH3, pEBG-GST-hLynSH2, or pEBG-GST vector were co-transfected with pCDNA6B/his-hTYMP plasmid into COS-7 cells using X-tremeGENE 9 DNA transfection reagent (Roche, Mannheim Germany), and at same time, the transfected cells were treated with or without 50 μM TPI. The cells were harvested 36 hours later and lysed in a Pierce™ IP Lysis Buffer (ThermoFisher Scientific). One milligram of total protein was added to Glutathione MagBeads (GenScript, Piscataway, NJ) and purified by affinity chromatography based on the manufacture’s introduction. The GST, GST-hLynSH3 or GST-hLynSH2 eluates were used for western blot assay to confirm the presence of TYMP.
pEGFP-N1-human Lyn-GFP and pCDNA6B/his-hTYMP plasmids were also co-transfected to the COS-7 cells. TYMP was pulled down using immobilized Ni+ on magnetic sepharose beads (GE Healthcare Life Sciences) for His-tagged protein purification. To do this, the cell lysates were adjusted to contain (in mM) 5 imidazole, 200 Na3PO4, and 500 NaCl, then adjusted to a pH of 7.4 before added to the His Mag Sepharose Ni beads. His-hTYMP eluates were used for western blot assay for Lyn.
Statistical analyses
Data are expressed as mean ± SEM. The data have been analyzed for normality and equal variance as a justification for using the 2-tailed Student’s t test, Mann Whitney test, or 1-way ANOVA with Bonferroni post-hoc test for multiple comparisons. In some cases, data were analyzed by Log-rank test using the Kaplan-Meier survival curve. GraphPad Prism (version 8.3.1) was used for data analysis and a p ≤ 0.05 was considered statistically significant.
Results
TYMP binds to Lyn SH3 and SH2 domains
Our previous study demonstrated that TYMP is a novel signaling protein and it binds to the SH3 domain-containing proteins, potentially through its N-terminal proline-rich region.24 This finding was confirmed by overexpressing full length human TYMP and Lyn in Cos-7 cells. By pulling down His-Tagged TYMP using the lysates prepared from the co-transfected cells, we found that both endogenous and overexpressed Lyn were present in the eluate (Fig. 1A), which corroborates our previous finding using human and mouse platelet lysates.24 To further show direct evidence, we generated GST-SH3 and GST-SH2 fusion proteins and examined their binding to human TYMP by GST pull-down assay. As shown in Fig. 1B, pulling down GST-SH3 or GST-SH2 also pulled down human TYMP, suggesting that both Lyn SH2 and SH3 domains can mediate the TYMP/Lyn binding. TPI treatment prevented TYMP binding to the Lyn SH2 or SH3 domains, suggesting that the bindings are TYMP specific. These data demonstrate that TYMP can bind to its partner proteins through their SH3 or SH2 domain and, therefore, acts as a signaling protein.
Fig. 1. TYMP binds to its partner through their SH3 or SH2 domain.

A. pCDNA6/his-hTYMP plasmid vector, either alone or combined with pEGFP-N1-hLyn-GFP vector, was transfected into Cos-7 cells, and His-Tagged TYMP was pulled down using His Mag Sepharose Ni beads. Inputs and elutes were blotted using anti-Lyn antibody. B. pEBG-GST-SH3(hLyn), pEBG-GST-SH2(hLyn), or pEBG-GST empty vector were co-transfected with pCDNA6/his-hTYMP into Cos-7 cells with or without TPI (50 μM) in culture media. The cell lysates were used for GST pull-down assays and eluted TYMP was detected by western blot using an anti-human TYMP antibody. In both panels, I: input; B: blank (2x Laemmli sample buffer only), E: elute. Blots represents 2–3 repeats.
TYMP deficiency or inhibition attenuates platelet aggregation to collagen-coated surfaces
Adhesion to the site of vessel wall injury is an essential function for platelets and is the first step in primary hemostasis. During these events, specific membrane receptors on the platelet surface bind to the exposed proteins of the subendothelial matrix.34 Under high shear conditions, the initial adhesion to the vessel wall in response to injury is mediated by GPIb-XI-V binding to vWF/collagen complexes,5 which is then followed by the binding of integrin α2β1 and GPVI to collagen.35 In our previous study, we found that TYMP deficiency may not affect the initial platelet adhesion in the in vivo thrombosis model.24 To solidify this finding, we coated Cellix Vena8 Fluoro+ chamber with type I collagen36, 37 and perfused the chambers with fluorescently labeled mouse whole blood. As shown in Fig. 2A, we found that TYMP-deficiency did not affect platelet binding to the type I collagen-coated surface. However, TYMP-deficiency dramatically decreased platelet aggregation at the end of the three-minute observation. Inhibition of TYMP with 50 μM TPI also significantly reduced platelet aggregation to the collagen-coated surface (Fig. 2B). These data suggest that TYMP plays a functional role in GPVI signal transduction, which primarily mediates collagen-induced platelet activation and aggregation;8, 38 however, TYMP deficiency or inhibition does not affect initial platelet adhesion to collagen that is primarily mediated by the α2β1 receptor39–41.
Fig. 2. Cellix Vena8 Fluoro+ biochip-based platelet aggregation assay.

The flow chambers were overnight coated with 100 μg/ml collagen in PBS. A. Whole blood drew from male WT and Tymp–/– mice were stained with Rhodamine 6G and perfused into the flow chamber at a shear rate 65 dynes/cm2. B. WT whole blood treated with 50 μM TPI in saline for 5 min or saline alone were perfused into the chamber at a shear rate 65 dynes/cm2. Graphs show areas covered by aggregated platelets at 3 minutes after perfusion. Images were analyzed by Image J.
Tipiracil hydrochloride, a potent and selective TYMP inhibitor, attenuates platelet activation
TPI, an FDA-approved selective TYMP inhibitor that recently became commercially available, was used to examine effects of TYMP inhibition on platelet activation and thrombosis. In order to determine if TYMP in circulating nucleated cells could affect hemostasis, we examined TYMP expression in mouse peripheral white blood cells by RT-PCR. Unexpectedly, we found that TYMP was not expressed in these nucleated cells (Fig. 3A). However, TYMP was detected in WT platelets, and TYMP haplodeficiency dramatically reduced platelet TYMP expression (Fig. 3B). TPI dose-dependently (62.5, 125, and 250 μM) inhibited 0.5 μg/ml CRP-induced platelet shape change (not shown) and 50 μM completely blocked CRP-induced platelet aggregation (Fig. 3C). In line with the findings in Fig. 2B, pretreatment of WT platelets with TPI dose-dependently inhibited collagen-induced platelet aggregation (Fig. 3D and E). TPI (50 μM) had no effect on collagen induced Tymp–/– platelet aggregation (Fig. 3F). TPI also attenuated ADP-induced WT platelet aggregation but had no effect on Tymp–/– platelets (Fig. 3G). These data suggest that the inhibitory effects of TPI on platelet activation are mediated by TYMP. This hypothesis is further supported by examination of ADP-induced AKT phosphorylation, which has been used as a marker of platelet activation in many studies.24, 42 As shown in Fig. 3H, we found that ADP-induced phosphorylation of AKT at S473 was bi-phasic. TYMP deficiency may have had either no effect on or slightly increased AKT activation within the first minute after ADP stimulation, but dramatically delayed the second phase of AKT phosphorylation. TPI also dramatically reduced ADP-stimulated AKT phosphorylation, especially during the second phase (Fig. 3I). Since Lyn has been reported to play a role in platelet granular section,43 we further examined the role of TYMP on granular release. TYMP deficiency likely has no effect on dense granular release in response to collagen stimulation (Supplemental Figure I). However, TPI significantly attenuated ADP-stimulated P-selectin exposure on the platelet surface (Fig. 3J).
Fig. 3. Inhibition of TYMP in vitro inhibits platelet activation.

A. Whole blood were drawn from 6 WT and 6 Tymp–/– male mice and platelet-rich plasma (PRP) was separated by centrifugation. The bottom 1/3 of the PRP and the buffy coat were pooled together, treated with red blood cell lysis buffer, and the nucleated cells were collected and used for RNA extraction. RT-PCR was used to examine TYMP expression in the mRNA level. Liver RNAs were used as positive control. Leu. = leukocytes. B. TYMP expression in the WT and Tymp+/− platelets were examined by western blot. Integrin β3 and actin were reblotted as loading control. C. WT and Tymp–/– platelets in PRP were treated with 50 μM TPI for 2 min and then CRP-induced platelet aggregation was assessed. D & E. WT platelets in PRP were treated with different concentration of TPI for 2 min and then collagen (1 μg/ml) induced platelet activation was assessed. F. Tymp–/– platelets in PRP were treated with saline or 50 μM TPI in saline for 2 min and then collagen-induced platelet aggregation was assessed. N=5 in each condition. G. WT and Tymp−/− platelets in PRP were treated with 50 μM TPI for 2 min and then 2.5 μM ADP-induced platelet aggregation was assessed. H. WT and Tymp–/– platelets in PRP were treated with 2.5 μM ADP for the indicated times and then AKT activation were evaluated. Blot represents two repeats. I. WT platelets in PRP pooled from 10 mice were divided into 8 parts. Four parts were treated with 50 μM TPI and another 4 parts were treated with saline as controls before they were treated with 2.5 μM ADP for the indicated times. Platelet lysates were used for assessing AKT phosphorylation. J. WT platelet in PRP were divided into 8 groups, treated with saline or 50 μM TPI, and then treated with 2.5 μM ADP for the indicated times. Platelet surface p-selectin exposure was analyzed by flow cytometry and data were shown as ratio of control (0 minute). N=3.
TYMP deficiency results in significant antithrombotic effects
We previously reported that TYMP haplodeficiency or complete deletion significantly inhibited thrombosis in male mice.24 We have now found that TYMP deficiency in female mice also significantly inhibits thrombosis (Fig. 4A and Supplemental Figure IIA). When both male and female mice data were combined, we found that cessation of blood flow within 30 minutes was seen in all WT mice (n=17) with an average vessel occlusion time of 9.81 ± 2.25 min (Fig. 4C and Supplemental Figure IIB). 10 of the 18 Tymp+/− mice and 7 of the 16 Tymp–/– mice showed flow cessation within the 30 min observation period, with average occlusion times >20 min. These data suggest that TYMP, a cytosolic protein, plays a mechanistic role in platelet activation and thrombosis in both sexes. The antithrombotic effect of TYMP deficiency may be more effective in males than in females. Partial deficiency of TYMP is enough to achieve a significant antithrombotic effect.
Fig. 4. TYMP deficiency in vivo inhibits thrombosis.

8–10 weeks WT, Tymp+/–, and Tymp–/– mice in both sexes were subjected to the 7.5% FeCl3-induced thrombosis model. Log-rank test was used to compare role of TYMP deficiency on thrombosis. A. Frequency of time to occlusion in female mice. B. Frequency of time to occlusion in both male and female mice.
TPI inhibits thrombosis under both normal and hyperlipidemic conditions
Having shown that inhibition of TYMP with TPI significantly inhibits CRP-, collagen-, and ADP-induced platelet aggregation in vitro, we further examined its effect on in vivo thrombosis. Intraperitoneal injection of TPI once per day for three days at a dose of 17 mg/kg completely inhibited occlusive thrombus formation in the carotid arteries induced by 7.5% FeCl3 vessel injury (Fig. 5A). Direct intravenous injection of TPI at doses of 1.7 and 0.17 mg/kg also significantly inhibited thrombosis (Fig. 5B) when compared to mice receiving saline injection alone. Interestingly, gavage feeding of TPI at a lower dose of 60 μg/kg/day for 3 days also significantly inhibited in vivo thrombosis (Fig. 5C & Supplemental Figure IIIA). These data strongly suggest that inhibition of TYMP by TPI may be an effective antithrombotic therapy.
Fig. 5. TPI inhibits thrombosis under both normal and hyperlipidemia conditions without disturbing hemostasis.

Male WT mice were treated with TPI by intraperitoneal (IP) injection (A) or intravenous injection (B), or oral administration (C) at the indicated doses and then subjected to the FeCl3 induced thrombosis model. D and E, male WT mice fed with a western diet (WD, TD.88137) for 3 weeks, then divided into two groups. One group continually received WD and another group received a customized WD, TD. 190501, for additional one week. Mice were then subjected to the 7.5% FeCl3 induced thrombosis model. Age-matched male WT mice on normal laboratory diet (NLD) were used as control.
Patients consuming a high-fat, high-calorie meal can have a decrease by about 40% in TPI absorption.44 Most of the patients with atherothrombotic vascular diseases have comorbid hyperlipidemia, which can lead to platelet hyperactivity.31 We thus tested the antithrombotic effect of TPI in mice under WD-feeding. Feeding WT mice with a WD for 4 weeks is sufficient to shorten the thrombosis time when compared to age-matched mice fed with a normal laboratory diet.45 As shown in Fig. 5D & E, Supplemental Figure IIIB, and online supplemental video I–III, feeding mice with WD for three weeks followed by feeding TD.190501 for one week is sufficient to reduce hyperlipidemia-enhanced thrombosis.
Compare the side effects of TPI vs. aspirin and clopidogrel
Current antiplatelet and antithrombotic drugs require chronic dosing to be effective and can have significant bleeding side effects.2, 15, 46 Aspirin and clopidogrel are the most frequently used anti-thrombotic drugs in a clinical setting. We thus compared their therapeutic and side effects with TPI. WT mice were gavage fed with different doses of aspirin and clopidogrel for one week and then subjected to the 7.5% FeCl3-induced thrombosis model. We found that 1 mg/kg/day aspirin and 2.5 mg/kg/day clopidogrel were the lowest effective doses that achieved a significant antithrombotic effect when compared with WT mice without any treatment (Fig. 6A and supplementary video IV and V). Although we have found that gavage feeding of TPI, as low as 60 μg/kg/day, significantly inhibited thrombosis in vivo (Fig. 5D), we chose two higher doses of TPI, 0.5 and 1 mg/kg/day, in order to explore any potential side effects associated with TPI usage. Both doses of TPI feeding significantly inhibited thrombosis (Fig. 6A & B, and supplementary video VI). Although 1 mg/kg TPI slightly prolonged tail bleeding time, 0.5 mg/kg TPI-treated mice had same level of bleeding time when comparted to the WT mice and was dramatically shorter than the bleeding time in the mice that received the effective dose of aspirin or clopidogrel (Fig. 6C).
Fig. 6. Comparison the therapeutic and side effect of TPI with aspirin and clopidogrel.

A. Male WT mice were gavage fed with different doses of TPI, aspirin, and clopidogrel in saline for one week and then subjected to the 7.5% FeCl3-induced thrombosis model. WT male mice received saline were used as control. B. Representative video images for mice received 1 mg/kg TPI, 1 mg/kg aspirin, and 2.5 mg/kg clopidogrel are shown. C. Bleeding times in mice received aspirin (1.0 mg/kg), clopidogrel (2.5 mg/kg), and TPI (0.5 and 1 mg/kg) were compared with WT mice without ant treatment. * p<0.05, ** p<0.005, and **** p<0.0001 vs. WT. † p<0.005 vs. TPI 0.5 mg/kg.
TPI is a quick-acting antithrombotic drug and co-administration with tissue plasminogen activator (tPA) reduces the tPA dose needed for effective thrombolysis without disturbing hemostasis.
The window for treating patients with acute myocardial infarction or ischemic stroke is narrow, and tPA (Alteplase) is the only FDA-approved clot-busting drug. However, intravenous perfusion of tPA has a high risk of systemic coagulopathies and bleeding complications.47 By using the FeCl3-induced carotid artery thrombosis model, we have found that bolus injection of tPA (Alteplase, 1 mg/kg) 5 min after initiation of vascular injury effectively lysed the established thrombi.28 However, due to de novo platelet activation, we also observed persistent formation of new platelet-rich thrombi after tPA administration; thus, thrombi underwent repeated size variations.28 By bolus injection of 1 mg/kg TPI to mice 5 min after 7.5% FeCl3-induced vessel injury, we found that thrombosis formation was either significantly prolonged or that there was no blood flow cessation within the 30 minutes of observation (Fig. 7A). These data suggest that TPI may be a quick-acting antithrombotic drug. We thus further narrowed down the minimal effective dose needed for TPI to inhibit on-site thrombus growth and found that the lowest effective dose is between 75–100 μg/kg. By using the same model, we also found that the lowest effective dose of tPA for thrombolysis is between 0.25 to 0.5 mg/kg (Fig. 7B). Interestingly, co-administration of TPI 75 μg/kg and tPA 0.25 mg/kg, as shown in Fig. 7C and 7D, as well as supplementary video VII, significantly inhibited the growth of the on-site thrombi compared to either drug used separately. These data suggest that co-administration of TPI reduces the tPA dose required for thrombolysis. A high dose TPI alone or a low dose of TPI in combination with a low dose of tPA could be a novel strategy for treating patients with acute myocardial infarction or ischemic stroke without causing coagulopathy.
Fig. 7. TPI is a quick-acting anti-thrombotic drug that reduces the effective dose of tPA on preventing occlusive thrombi formation.

Thrombosis in female WT mice were initiated with 7.5% FeCl3 and 5 min later, TPI (A), tPA (B), or the combination of TPI and tPA (C) in saline at the indicated doses were bolus injected into mice through a jugular vein catheter, and thrombosis times were assessed. D. Representative images for mice received 0.25 mg/kg tPA and 0.075 mg/kg TPI bolus injection are shown.
Combination of TPI and aspirin is a new, safe dual antiplatelet therapy.
Dual antiplatelet therapy is commonly used in the first several months after cardiac procedures such as stent placement or transcatheter aortic valve implantation.48, 49 Combination of aspirin and P2Y12 inhibitor greatly increases the risk of bleeding, so there is still a need for a robust, acute antithrombotic treatment that does not significantly disturb hemostasis.50 For this reason, we examined if a combination of aspirin and TPI could be utilized as a new dual antiplatelet therapy with negligible side effects. We chose a dose of 0.5 mg/kg for aspirin and 50 μg/kg for TPI, as treating WT mice with either of these two drugs alone at the selected dose did not affect thrombosis (Fig. 6A and 7A). As shown in Fig. 8A, gavage feeding of WT male mice with this combination significantly prolonged time to occlusive thrombus formation when compared with 0.5 mg/kg aspirin alone. When comparing the tail bleeding time to WT mice without treatment, we found that the combination of these low doses of aspirin and TPI did not significantly increase bleeding (Fig. 8B).
Fig. 8. Combination of low dose of aspirin and TPI is a new, safe dual antiplatelet therapy.

A. WT male mice were gavage fed with either aspirin 0.5 mg/kg/day (same data were shown in Fig. 6A) or a combination of aspirin 0.5 mg/kg/day and TPI 50 μg/kg/day in 200 μL saline for one week and then subjected to the 7.5% FeCl3-induced carotid artery injury thrombosis model. B. Tail bleeding time was assessed in the mice received aspirin 0.5 mg-TPI 50 μg/kg/day after the thrombosis study and compared with age-matched WT mice without any treatment.
Discussion
Vascular thrombosis is the primary event in many life-threatening diseases, such as myocardial infarction or ischemic stroke, and platelet activation and aggregation are major components of thrombosis. Consequently, various antiplatelet medications are used clinically for the primary or secondary prevention of thrombosis. However, most of these drugs irreversibly block platelet surface receptors involved in platelet activation and aggregation, which results in systemic side effects, including thrombocytopenia and hemorrhage. Therefore, there is an urgent need to elucidate unique molecular mechanisms of platelet-mediated arterial thrombus formation that can be modulated to allow for targeted anti-platelet therapy while minimizing systemic risks.2, 15 In addition, to the best of our knowledge, no antiplatelet drugs have been developed to target platelet cytosolic proteins. Aspirin inhibits TXA2 production, which then attenuates thromboxane receptor-mediated platelet activation, and is, therefore, still considered a platelet-receptor mediated effect. By using male mice, we recently discovered that TYMP, a cytoplasmic protein that is highly expressed in platelets, plays important roles in maintaining normal platelet function and is essential for platelet activation induced by multiple agonists, including collagen, ADP, and thrombin.24 We have now found that TYMP also enhances platelet activation and thrombosis in female mice, although TYMP deficiency in the female mice likely had less of an antithrombotic effect compared to the male mice.24 These new data suggest a universal and mechanistic role of TYMP on platelet activation and thrombosis.
In our previous study, we examined the role of KIN59, a non-competitive TYMP inhibitor, on platelet activation and thrombosis.24 KIN59 is not a TYMP-selective inhibitor.25 However, TPI, which inhibits TYMP activity by competing with its substrate thymidine and thymine at the enzyme substrate binding site, is a selective and potent TYMP inhibitor. The binding of substrates or inhibitors to TYMP induces a closed conformational change, which is essential for a TYMP-mediated phosphorolytic reaction to occur.21, 22 In this study, we thoroughly examined the effects of TYMP inhibition in vivo on platelet activation and thrombosis, as well as any side effects resulting from the administration of TPI.
TPI is water soluble (>13.25 mg/ml). These is no published study examining the pharmacokinetics of TPI alone. Administration of a single dose of Lonsurf at 35 mg/m2 generates the absorption rate of TPI 301 ng·h/ml determined by area under the curve analysis. The maximum plasma concentration of TPI is 69 ng/ml, which was observed at 3 h after administration.51 However, since TPI has a volume of distribution (Vd/F) 333 L, it could be quickly distributed into tissue and intracellular compartments, rather than staying in plasma circulation.52 At steady state, TPI half-life is 2.4 h, and around 77% is excreted through feces and urinary with unchanged structure.53 We gave TPI to mice using three different routes, namely intraperitoneal injection, intravenous injection, and oral administration, and examined its effect using different disease models. Our data clearly demonstrated that TPI is an effective and safe antithrombotic compound, even under a hyperlipidemic condition, in mice. The effective dose of TPI could be as low as 60 μg/kg/day without significantly affecting hemostasis. Most importantly, intravenous injections of TPI, either delivered alone or in combination with tPA, also significantly inhibited the growth of the on-site thrombi. Considering this, TYMP inhibition could be a potential treatment option for patients with acute myocardial infarction or stroke. This fast-acting characteristic also indicates that TPI can be used in interventional cardiology procedures, such as percutaneous coronary intervention and transcatheter aortic valve implantation, either alone or combined with other drugs, to temporally inhibit platelet function, as TYMP inhibitors rapidly and reversibly inhibit platelet aggregation.24
Platelets are a major source of TYMP, with each human platelet containing about 5,400 to 11,600 copies of TYMP,54, 55 but the function of TYMP in platelet physiology and function remains unclear. TYMP has been implicated in diseases that have a high risk of thrombosis, such as atherosclerosis,56, 57 cancer,58, 59 and diabetes mellitus.60 In addition, ionizing radiation, which induces significant TYMP expression, has been associated with thrombotic vascular occlusion.61 In the current study, we found that TYMP deficiency or inhibition does not affect initial platelet binding to a collagen-coated surface; however, it dramatically attenuated platelet aggregation on the collagen-coated surface in a flow chamber assay. These data suggest that TYMP likely has no effect on another collagen receptor, α2β1, which primarily plays an adhesive role for platelet deposition to the injury site.38 In combination with our published data,24 these data further demonstrated that TYMP likely plays a more important role in the GPVI signaling-mediated platelet activation. GPVI is found exclusively on platelets and megakaryocytes, and it is the predominant platelet receptor for collagen.62 Deficiency of GPVI in humans and mice is not associated with a strong bleeding diathesis.40 Although some small molecules, such as losartan and honokiol, have been shown to inhibit GPVI signaling, they have an inhibitory effect on other platelet aggregation and thrombus formation pathways, which could increase bleeding risk and other side effects.63 A recent phase I study using Revacept, a dimeric GPVI-Fc fusion protein that blocks collagen/GPVI binding mediated platelet activation, demonstrated that targeting the GPVI signaling is a safe intervention.64 However, Revacept is delivered by intravenous injection which limits it to situational use, such as for percutaneous coronary intervention.65 In contrast, TPI may have more broad indications.
In addition to GPVI signaling, TYMP also participates in GPCR-mediated platelet activation. In our previous study, we showed that TYMP deficiency reduced ADP-induced platelet P-selectin exposure. Here, we further demonstrated that TYMP deficiency or inhibition attenuated ADP-induced AKT phosphorylation in platelets. ADP-induced platelet activation requires concomitant signaling from both P2Y1 and P2Y12 receptors that couple to Gαq and Gαi, respectively. However, ADP-induced AKT activation is predominantly mediated by the platelet P2Y12 receptor.66 While we still do not know how TYMP affects GPCR signaling pathways, TYMP deficiency may put a “brake” on autocrine or paracrine mechanisms of GPCR-mediated platelet activation.
The current study, unexpectedly, also demonstrated that mouse peripheral white blood cells do not express TYMP. While fresh isolated human monocytes express low to undetectable TYMP,67 TYMP is reportedly expressed in human macrophages.67 Furthermore, we also did not detect TYMP mRNA in mouse intraperitoneal macrophages (data not shown). Red blood cells also do not express TYMP. Therefore, findings of the current study should be platelet dependent. These data provide a strong rationale that TPI could be repositioned as a potential antithrombotic medicine. To this end, we compared the prophylactic therapeutic effects and side effects of TPI with aspirin and clopidogrel using the murine FeCl3-induced thrombosis model and tail bleeding assay. We found that the lowest dose for aspirin and clopidogrel in inhibiting mouse thrombosis is 1 mg/kg and 2.5 mg/kg, respectively. As predicted, these doses of aspirin and clopidogrel dramatically prolonged tail bleeding time. Although a high dose of TPI (1mg/kg) occasionally prolonged bleeding time in some mice, mice that received doses of 0.5 mg/kg/day TPI showed no prolonged bleeding time in all cases. As an auxiliary component of the anticancer drug, Lonsurf, TPI has been evaluated in clinical trials 68, 69 and has been shown to be systemically safe. TYMP deficiency does not affect high dose thrombin-stimulated platelet activation.24 These data suggest that TPI may be safer and more beneficial than the traditional antiplatelet drugs. Most importantly, oral administration of very low doses of aspirin and TPI dramatically inhibited thrombosis without inducing any defects in hemostasis, suggesting that this combination could be a novel remedy for patients with a high risk of thrombosis. TPI alone may also be a prophylactic antiplatelet drug for patients who are allergic to aspirin. Clinical trial studies are necessary to demonstrate this hypothesis.
In summary, our study demonstrated that TYMP, a cytosolic protein, plays an important role in platelet activation and thrombosis. TYMP can be safely and rapidly inhibited by TPI. TPI-mediated TYMP inhibition dramatically inhibited platelet activation and thrombosis in both normal and hyperlipidemic conditions, as well as in different disease models, in mice. When given at effective antithrombotic doses, TPI does not cause bleeding disorders, as seen often in patients treated with other antiplatelet drugs.
Supplementary Material
Highlights.
Thymidine phosphorylase, a newly identified platelet cytoplasm signaling protein, is essential for platelet activation and thrombosis.
Tipiracil hydrochloride, a specific thymidine phosphorylase inhibitor, inhibits thrombosis in mice without affecting hemostasis.
Thymidine phosphorylase is rapidly targetable and tipiracil hydrochloride has a broad of application in preventing thrombotic diseases.
Acknowledgements
a) No other persons besides the authors have made substantial contributions to this manuscript.
b) This study is supported by Marshall University Institute Fund (To Dr. Wei Li), Marshall University, School of Medicine and College of Pharmacy Collaborative Grant (PI: Dr. Wei Li), NIH R15HL145573 (PI: Wei Li), NIH R01HL129179 (PI: Anirban Sen Gupta, Co-I: Wei Li), NIH R01HL130090 (PI: Thomas M McIntyre, Co-I Wei Li), WV-INBRE grant P20GM103434 (PI: Gary Rankin), and NASA West Virginia Space Grant Consortium, Training Grant # NNX15AI01H (Adam Belcher). This project described was also supported by the National Institute Of General Medical Sciences, U54GM104942. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
c) None
Non-standard Abbreviations and Acronyms
- GPVI
glycoprotein VI
- ADP
adenosine diphosphate
- GPCRs
G protein coupled receptors
- TYMP
thymidine phosphorylase
- GAPDH
Glyceraldehyde 3-phosphate dehydrogenase
- TPI
tipiracil hydrochloride
- WT
wild type
- FeCl3
ferric chloride
- PRP
platelet-rich plasma
- GST
glutathione S-transferase
- CRP
collagen related peptide
- tPA
tissue plasminogen activator
References
- 1.Day ISCfWT. Thrombosis: A major contributor to the global disease burden. J Thromb Haemost. 2014;12:1580–1590 [DOI] [PubMed] [Google Scholar]
- 2.Jackson SP. Arterial thrombosis--insidious, unpredictable and deadly. Nat Med. 2011;17:1423–1436 [DOI] [PubMed] [Google Scholar]
- 3.Murray CJ, Vos T, Lozano R, et al. Disability-adjusted life years (dalys) for 291 diseases and injuries in 21 regions, 1990–2010: A systematic analysis for the global burden of disease study 2010. Lancet. 2012;380:2197–2223 [DOI] [PubMed] [Google Scholar]
- 4.Metharom P, Berndt MC, Baker RI, Andrews RK. Current state and novel approaches of antiplatelet therapy. Arteriosclerosis, thrombosis, and vascular biology. 2015;35:1327–1338 [DOI] [PubMed] [Google Scholar]
- 5.Andrews RK, Berndt MC. Chapter 10 - the gpib-ix-v complex. In: Michelson AD, ed. Platelets (third edition). Academic Press; 2013:195–213. [Google Scholar]
- 6.Ezumi Y, Shindoh K, Tsuji M, Takayama H. Physical and functional association of the src family kinases fyn and lyn with the collagen receptor glycoprotein vi-fc receptor gamma chain complex on human platelets. The Journal of experimental medicine. 1998;188:267–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Furie BC, Furie B. Tissue factor pathway vs. Collagen pathway for in vivo platelet activation. Blood Cells Mol Dis. 2006;36:135–138 [DOI] [PubMed] [Google Scholar]
- 8.Stegner D, Nieswandt B. Platelet receptor signaling in thrombus formation. J Mol Med (Berl). 2011;89:109–121 [DOI] [PubMed] [Google Scholar]
- 9.Smyth SS, Woulfe DS, Weitz JI, Gachet C, Conley PB, Goodman SG, Roe MT, Kuliopulos A, Moliterno DJ, French PA, Steinhubl SR, Becker RC, Platelet Colloquium P. G-protein-coupled receptors as signaling targets for antiplatelet therapy. Arteriosclerosis, thrombosis, and vascular biology. 2009;29:449–457 [DOI] [PubMed] [Google Scholar]
- 10.Depta JP, Bhatt DL. New approaches to inhibiting platelets and coagulation. Annu Rev Pharmacol Toxicol. 2015;55:373–397 [DOI] [PubMed] [Google Scholar]
- 11.Franchi F, Angiolillo DJ. Novel antiplatelet agents in acute coronary syndrome. Nat Rev Cardiol. 2015;12:30–47 [DOI] [PubMed] [Google Scholar]
- 12.Desai NR, Bhatt DL. The state of periprocedural antiplatelet therapy after recent trials. JACC Cardiovasc Interv. 2010;3:571–583 [DOI] [PubMed] [Google Scholar]
- 13.de Souza Brito F, Tricoci P. Novel anti-platelet agents: Focus on thrombin receptor antagonists. J Cardiovasc Transl Res. 2013;6:415–424 [DOI] [PubMed] [Google Scholar]
- 14.Chassot PG, Delabays A, Spahn DR. Perioperative antiplatelet therapy: The case for continuing therapy in patients at risk of myocardial infarction. Br J Anaesth. 2007;99:316–328 [DOI] [PubMed] [Google Scholar]
- 15.Capodanno D, Ferreiro JL, Angiolillo DJ. Antiplatelet therapy: New pharmacological agents and changing paradigms. J Thromb Haemost. 2013;11 Suppl 1:316–329 [DOI] [PubMed] [Google Scholar]
- 16.Kubilus J, Lee LD, Baden HP. Purification of thymidine phosphorylase from human amniochorion. Biochimica et biophysica acta. 1978;527:221–228 [DOI] [PubMed] [Google Scholar]
- 17.Desgranges C, Razaka G, Rabaud M, Bricaud H. Catabolism of thymidine in human blood platelets: Purification and properties of thymidine phosphorylase. Biochimica et biophysica acta. 1981;654:211–218 [DOI] [PubMed] [Google Scholar]
- 18.Miyazono K, Okabe T, Urabe A, Takaku F, Heldin CH. Purification and properties of an endothelial cell growth factor from human platelets. The Journal of biological chemistry. 1987;262:4098–4103 [PubMed] [Google Scholar]
- 19.Bronckaers A, Aguado L, Negri A, Camarasa MJ, Balzarini J, Perez-Perez MJ, Gago F, Liekens S. Identification of aspartic acid-203 in human thymidine phosphorylase as an important residue for both catalysis and non-competitive inhibition by the small molecule “crystallization chaperone” 5’-o-tritylinosine (kin59). Biochemical pharmacology. 2009;78:231–240 [DOI] [PubMed] [Google Scholar]
- 20.Liekens S, Bronckaers A, Perez-Perez MJ, Balzarini J. Targeting platelet-derived endothelial cell growth factor/thymidine phosphorylase for cancer therapy. Biochemical pharmacology. 2007;74:1555–1567 [DOI] [PubMed] [Google Scholar]
- 21.Li W, Yue H. Thymidine phosphorylase: A potential new target for treating cardiovascular disease. Trends Cardiovasc Med. 2018;28:157–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Norman RA, Barry ST, Bate M, et al. Crystal structure of human thymidine phosphorylase in complex with a small molecule inhibitor. Structure. 2004;12:75–84 [DOI] [PubMed] [Google Scholar]
- 23.Schwartz M Thymidine phosphorylase from escherichia coli. Properties and kinetics. European journal of biochemistry / FEBS. 1971;21:191–198 [DOI] [PubMed] [Google Scholar]
- 24.Li W, Gigante A, Perez-Perez MJ, Yue H, Hirano M, McIntyre TM, Silverstein RL. Thymidine phosphorylase participates in platelet signaling and promotes thrombosis. Circulation research. 2014;115:997–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liekens S, Balzarini J, Hernandez AI, De Clercq E, Priego EM, Camarasa MJ, Perez-Perez MJ. Thymidine phosphorylase is noncompetitively inhibited by 5’-o-trityl-inosine (kin59) and related compounds. Nucleosides, nucleotides & nucleic acids. 2006;25:975–980 [DOI] [PubMed] [Google Scholar]
- 26.Lopez LC, Akman HO, Garcia-Cazorla A, Dorado B, Marti R, Nishino I, Tadesse S, Pizzorno G, Shungu D, Bonilla E, Tanji K, Hirano M. Unbalanced deoxynucleotide pools cause mitochondrial DNA instability in thymidine phosphorylase-deficient mice. Human molecular genetics. 2009;18:714–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li W, McIntyre TM, Silverstein RL. Ferric chloride-induced murine carotid arterial injury: A model of redox pathology. Redox Biol. 2013;1:50–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li W, Nieman M, Sen Gupta A Ferric chloride-induced murine thrombosis models. J. Vis. Exp. 2016;115:e54479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen K, Li W, Major J, Rahaman SO, Febbraio M, Silverstein RL. Vav guanine nucleotide exchange factors link hyperlipidemia and a prothrombotic state. Blood. 2011;117:5744–5750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ghosh A, Li W, Febbraio M, Espinola RG, McCrae KR, Cockrell E, Silverstein RL. Platelet cd36 mediates interactions with endothelial cell-derived microparticles and contributes to thrombosis in mice. The Journal of clinical investigation. 2008;118:1934–1943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen K, Febbraio M, Li W, Silverstein RL. A specific cd36-dependent signaling pathway is required for platelet activation by oxidized low-density lipoprotein. Circulation research. 2008;102:1512–1519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pawlowski CL, Li W, Sun M, Ravichandran K, Hickman D, Kos C, Kaur G, Sen Gupta A. Platelet microparticle-inspired clot-responsive nanomedicine for targeted fibrinolysis. Biomaterials. 2017;128:94–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yue H, Tanaka K, Furukawa T, Karnik SS, Li W. Thymidine phosphorylase inhibits vascular smooth muscle cell proliferation via upregulation of stat3. Biochimica et biophysica acta. 2012;1823:1316–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ruggeri ZM, Mendolicchio GL. Adhesion mechanisms in platelet function. Circulation research. 2007;100:1673–1685 [DOI] [PubMed] [Google Scholar]
- 35.Sarratt KL, Chen H, Zutter MM, Santoro SA, Hammer DA, Kahn ML. Gpvi and alpha2beta1 play independent critical roles during platelet adhesion and aggregate formation to collagen under flow. Blood. 2005;106:1268–1277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gupta N, Li W, Willard B, Silverstein RL, McIntyre TM. Proteasome proteolysis supports stimulated platelet function and thrombosis. Arteriosclerosis, thrombosis, and vascular biology. 2014;34:160–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Srikanthan S, Li W, Silverstein RL, McIntyre TM. Exosome poly-ubiquitin inhibits platelet activation, downregulates cd36 and inhibits pro-atherothombotic cellular functions. J Thromb Haemost. 2014;12:1906–1917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pugh N, Simpson AM, Smethurst PA, de Groot PG, Raynal N, Farndale RW. Synergism between platelet collagen receptors defined using receptor-specific collagen-mimetic peptide substrata in flowing blood. Blood. 2010;115:5069–5079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Siljander PR, Hamaia S, Peachey AR, Slatter DA, Smethurst PA, Ouwehand WH, Knight CG, Farndale RW. Integrin activation state determines selectivity for novel recognition sites in fibrillar collagens. The Journal of biological chemistry. 2004;279:47763–47772 [DOI] [PubMed] [Google Scholar]
- 40.Kato K, Kanaji T, Russell S, Kunicki TJ, Furihata K, Kanaji S, Marchese P, Reininger A, Ruggeri ZM, Ware J. The contribution of glycoprotein vi to stable platelet adhesion and thrombus formation illustrated by targeted gene deletion. Blood. 2003;102:1701–1707 [DOI] [PubMed] [Google Scholar]
- 41.Manganaro D, Consonni A, Guidetti GF, Canobbio I, Visconte C, Kim S, Okigaki M, Falasca M, Hirsch E, Kunapuli SP, Torti M. Activation of phosphatidylinositol 3-kinase beta by the platelet collagen receptors integrin alpha2beta1 and gpvi: The role of pyk2 and c-cbl. Biochimica et biophysica acta. 2015;1853:1879–1888 [DOI] [PubMed] [Google Scholar]
- 42.Woulfe DS. Akt signaling in platelets and thrombosis. Expert Rev Hematol. 2010;3:81–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Z, Zhang G, Liu J, Stojanovic A, Ruan C, Lowell CA, Du X. An important role of the src family kinase lyn in stimulating platelet granule secretion. The Journal of biological chemistry. 2010;285:12559–12570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yoshino T, Kojima T, Bando H, Yamazaki T, Naito Y, Mukai H, Fuse N, Goto K, Ito Y, Doi T, Ohtsu A. Effect of food on the pharmacokinetics of tas-102 and its efficacy and safety in patients with advanced solid tumors. Cancer science. 2016;107:659–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang M, Li W, Harberg C, Chen W, Yue H, Ferreira RB, Wynia-Smith SL, Carroll KS, Zielonka J, Flaumenhaft R, Silverstein RL, Smith BC. Cysteine sulfenylation by cd36 signaling promotes arterial thrombosis in dyslipidemia. Blood Adv. 2020;4:4494–4507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Surin WR, Prakash P, Barthwal MK, Dikshit M. Optimization of ferric chloride induced thrombosis model in rats: Effect of anti-platelet and anti-coagulant drugs. J Pharmacol Toxicol Methods. 2010;61:287–291 [DOI] [PubMed] [Google Scholar]
- 47.Adams HP Jr., Adams RJ, Brott T, del Zoppo GJ, Furlan A, Goldstein LB, Grubb RL, Higashida R, Kidwell C, Kwiatkowski TG, Marler JR, Hademenos GJ, Stroke Council of the American Stroke A. Guidelines for the early management of patients with ischemic stroke: A scientific statement from the stroke council of the american stroke association. Stroke. 2003;34:1056–1083 [DOI] [PubMed] [Google Scholar]
- 48.Ando T, Takagi H, Briasoulis A, Afonso L. Single versus dual anti-platelet therapy post transcatheter aortic valve implantation: A meta-analysis of randomized controlled trials. J Thromb Thrombolysis. 2017;44:448–456 [DOI] [PubMed] [Google Scholar]
- 49.Pirlet C, Legrand V, Nyssen A, Pierard L, Gach O. Duration of dual anti-platelet therapy - state of the art after the dapt and pegasus-timi 54 trials. Acta Cardiol. 2017;72:256–264 [DOI] [PubMed] [Google Scholar]
- 50.Alexander JH, Wojdyla D, Vora AN, Thomas L, Granger CB, Goodman SG, Aronson R, Windecker S, Mehran R, Lopes RD. The risk / benefit tradeoff of antithrombotic therapy in patients with atrial fibrillation early and late after an acute coronary syndrome or percutaneous coronary intervention: Insights from augustus. Circulation. 2020 [DOI] [PubMed] [Google Scholar]
- 51.Cleary JM, Rosen LS, Yoshida K, Rasco D, Shapiro GI, Sun W. A phase 1 study of the pharmacokinetics of nucleoside analog trifluridine and thymidine phosphorylase inhibitor tipiracil (components of tas-102) vs trifluridine alone. Invest New Drugs. 2017;35:189–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Smith DA, Beaumont K, Maurer TS, Di L. Volume of distribution in drug design. Journal of medicinal chemistry. 2015;58:5691–5698 [DOI] [PubMed] [Google Scholar]
- 53.Lee JJ, Seraj J, Yoshida K, et al. Human mass balance study of tas-102 using (14)c analyzed by accelerator mass spectrometry. Cancer chemotherapy and pharmacology. 2016;77:515–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Burkhart JM, Vaudel M, Gambaryan S, Radau S, Walter U, Martens L, Geiger J, Sickmann A, Zahedi RP. The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood. 2012;120:e73–82 [DOI] [PubMed] [Google Scholar]
- 55.Wijten P, van Holten T, Woo LL, Bleijerveld OB, Roest M, Heck AJ, Scholten A. High precision platelet releasate definition by quantitative reversed protein profiling--brief report. Arteriosclerosis, thrombosis, and vascular biology. 2013;33:1635–1638 [DOI] [PubMed] [Google Scholar]
- 56.Ignatescu MC, Gharehbaghi-Schnell E, Hassan A, Rezaie-Majd S, Korschineck I, Schleef RR, Glogar HD, Lang IM. Expression of the angiogenic protein, platelet-derived endothelial cell growth factor, in coronary atherosclerotic plaques: In vivo correlation of lesional microvessel density and constrictive vascular remodeling. Arteriosclerosis, thrombosis, and vascular biology. 1999;19:2340–2347 [DOI] [PubMed] [Google Scholar]
- 57.Boyle JJ, Wilson B, Bicknell R, Harrower S, Weissberg PL, Fan TP. Expression of angiogenic factor thymidine phosphorylase and angiogenesis in human atherosclerosis. The Journal of pathology. 2000;192:234–242 [DOI] [PubMed] [Google Scholar]
- 58.Connolly GC, Khorana AA. Emerging risk stratification approaches to cancer-associated thrombosis: Risk factors, biomarkers and a risk score. Thrombosis research. 2010;125 Suppl 2:S1–7 [DOI] [PubMed] [Google Scholar]
- 59.Demers M, Krause DS, Schatzberg D, Martinod K, Voorhees JR, Fuchs TA, Scadden DT, Wagner DD. Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:13076–13081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hamed EA, Zakary MM, Abdelal RM, Abdel Moneim EM. Vasculopathy in type 2 diabetes mellitus: Role of specific angiogenic modulators. J Physiol Biochem. 2011;67:339–349 [DOI] [PubMed] [Google Scholar]
- 61.Goldin-Lang P, Pels K, Tran QV, Szotowski B, Wittchen F, Antoniak S, Willich T, Witt H, Hummel M, Lenze D, Poller W, Schultheiss HP, Rauch U. Effect of ionizing radiation on cellular procoagulability and co-ordinated gene alterations. Haematologica. 2007;92:1091–1098 [DOI] [PubMed] [Google Scholar]
- 62.Nieswandt B, Watson SP. Platelet-collagen interaction: Is gpvi the central receptor? Blood. 2003;102:449–461 [DOI] [PubMed] [Google Scholar]
- 63.Onselaer M-B, Nagy M, Pallini C, Pike JA, Perrella G, Quintanilla LG, Eble JA, Poulter NS, Heemskerk JWM, Watson SP. Comparison of the gpvi inhibitors losartan and honokiol. Platelets. 2020;31:187–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ungerer M, Rosport K, Bultmann A, Piechatzek R, Uhland K, Schlieper P, Gawaz M, Munch G. Novel antiplatelet drug revacept (dimeric glycoprotein vi-fc) specifically and efficiently inhibited collagen-induced platelet aggregation without affecting general hemostasis in humans. Circulation. 2011;123:1891–1899 [DOI] [PubMed] [Google Scholar]
- 65.Schupke S, Hein-Rothweiler R, Mayer K, et al. Revacept, a novel inhibitor of platelet adhesion, in patients undergoing elective pci-design and rationale of the randomized isar-plaster trial. Thromb Haemost. 2019;119:1539–1545 [DOI] [PubMed] [Google Scholar]
- 66.Kim S, Jin J, Kunapuli SP. Akt activation in platelets depends on gi signaling pathways. The Journal of biological chemistry. 2004;279:4186–4195 [DOI] [PubMed] [Google Scholar]
- 67.Triques K, Stevenson M. Characterization of restrictions to human immunodeficiency virus type 1 infection of monocytes. J Virol. 2004;78:5523–5527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mayer RJ, Van Cutsem E, Falcone A, et al. Randomized trial of tas-102 for refractory metastatic colorectal cancer. N Engl J Med. 2015;372:1909–1919 [DOI] [PubMed] [Google Scholar]
- 69.Yoshino T, Mizunuma N, Yamazaki K, et al. Tas-102 monotherapy for pretreated metastatic colorectal cancer: A double-blind, randomised, placebo-controlled phase 2 trial. The lancet oncology. 2012;13:993–1001 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.