

Abstract
OBJECTIVE:
Atherosclerotic lesions are often characterized by accumulation of oxidized low-density lipoprotein (OxLDL), which is associated with vascular inflammation and lesion vulnerability to rupture. Extracellular apolipoprotein A-I binding protein (AIBP, encoded by APOA1BP gene), when secreted, promotes cholesterol efflux and regulates lipid rafts dynamics, but its role as an intracellular protein in mammalian cells remains unknown. The aim of this work was to determine function of intracellular AIBP in macrophages exposed to OxLDL and in atherosclerotic lesions.
APPROACH AND RESULTS:
Using a novel monoclonal antibody against human and mouse AIBP, which are highly homologous, we demonstrated robust AIBP expression in human and mouse atherosclerotic lesions. We observed significantly reduced autophagy in bone marrow-derived macrophages, isolated from Apoa1bp−/− compared to wild type mice, which were exposed to OxLDL. In atherosclerotic lesions from Apoa1bp−/− mice subjected to Ldlr knockdown and fed a Western diet, autophagy was reduced, whereas apoptosis was increased, when compared with that in wild type mice. AIBP expression was necessary for efficient control of ROS and cell death and for mitochondria quality control in macrophages exposed to OxLDL. Mitochondria-localized AIBP, via its N-terminal domain, associated with E3 ubiquitin-protein ligase Parkin (PARK2), mitofusin (MFN)-1 and MFN2, but not Bcl2/adenovirus E1B 19-kDa-interacting protein-3 (BNIP3), and regulated ubiquitination of MFN1 and MFN2, key components of mitophagy.
CONCLUSIONS:
These data suggest that intracellular AIBP is a new regulator of autophagy in macrophages. Mitochondria-localized AIBP augments mitophagy and participates in mitochondria quality control, protecting macrophages against cell death in the context of atherosclerosis.
Keywords: AIBP, autophagy, mitophagy, macrophage, oxidized LDL, atherosclerosis
Graphical Abstract
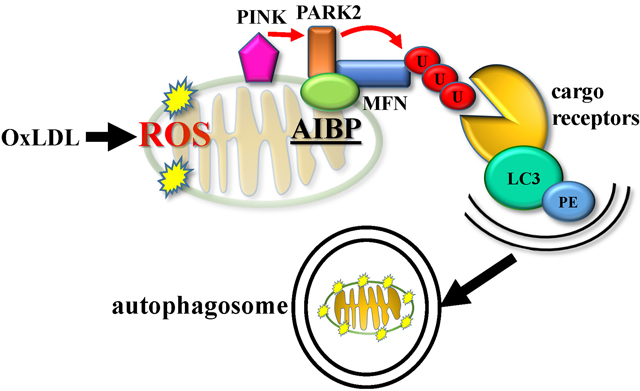
INTRODUCTION
Atherosclerosis is a chronic inflammatory disease of large blood vessels that can cause stroke, heart attack, and peripheral vascular disease.1 Oxidative modification of low-density lipoprotein (LDL) is the early event and a major risk factor in the development of atherosclerosis.2 Oxidation specific epitopes in oxidized LDL (OxLDL) are proinflammatory and proatherogenic, promoting macrophage foam cell formation and the development of atherosclerotic lesions.3 In addition, OxLDL and malondialdehyde modified (MDA)-LDL induce autophagy in macrophages.4, 5
Autophagy is a protective process of degradation of dysfunctional intracellular components to maintain cellular homeostasis.6, 7 Facilitating autophagy has been proposed as a new strategy for treatment of atherosclerosis.8, 9 Macrophages that are differentiated from the monocytes recruited to the vascular wall uncontrollably internalize OxLDL and become lipid-loaded foam cells. Death of foam cells in the vascular wall results in accumulation of inflammatory debris and extracellular lipid, making atherosclerotic lesions vulnerable to rupture. Autophagy helps macrophage survival, and the results from several cellular and animal models suggest that regulation of autophagy plays important roles in both early and advanced atherosclerosis.10, 11
Mitochondrial ATP production is central to cellular homeostasis and survival.12 Mitochondrial dysfunction is emphasized as a leading mechanism of atherosclerosis.13 Therefore, the cellular process of eliminating damaged mitochondria by autophagy, known as mitophagy, is important to keeping cells healthy and preventing disease development.14 Mitophagy plays an important role in age-related disorders and neurodegenerative diseases.15–19 In the context of atherosclerosis, exposure to OxLDL induces impairment of mitochondrial function by reduction in membrane potential, and the dysfunctional mitochondria are removed via mitophagy, which play a critical role in mitochondrial quality control and atheroprotection.20
Apolipoprotein A-I (APOA-I) binding protein (AIBP) was discovered in a yeast two-hybrid screen of proteins that associate with APOA-I.21 Our studies have demonstrated that AIBP facilitates cholesterol efflux from endothelial cells and macrophages to HDL, resulting in the reduced abundance of cholesterol-rich lipid raft microdomains in the plasma membrane.22, 23 In addition, AIBP has been found to exhibit NAD(P)H-hydrate epimerase (NAXE) and ADP-ribosyltransferase activities in vitro and in bacterial systems,24, 25 but no evidence of the importance of these enzymatic functions in mammalian cell has yet been reported. In hypercholesterolemic mouse models, we and others have demonstrated that overexpression of AIBP through delivery with adeno-associated virus (AAV) reduces atherosclerotic lesion development, while Apoa1bp knockout increases atherosclerosis.23, 26 Further, we have reported that intrathecal injection of recombinant AIBP alleviates neuropathic pain states and that inhaled AIBP protein attenuates acute lung injury in mice.27, 28 Although AIBP expressed in transfected cells was in mitochondria,29 there were no studies examining the role of endogenous AIBP in mitochondrial function.
In this study, we demonstrated that AIBP plays an important role in autophagosome formation in macrophages exposed to OxLDL. Moreover, we found that AIBP associated with mitochondrial proteins to promote mitophagy and that hypercholesterolemic Apoa1bp−/− mice had reduced macrophage MAP1LC3/LC3 (further referred to as LC3) expression but increased numbers of TUNEL-positive macrophages in atherosclerotic lesions compared to wild type mice. Our findings suggest that AIBP expression is an important factor in the removal of damaged mitochondria in macrophages.
MATERIALS AND METHODS
The authors will make their data, analytic methods, and study materials available to other researchers upon request.
Animals, diet and assessment of atherosclerosis
All experiments were conducted according to protocols approved by the Institutional Animal Care and Use Committee of the University of California, San Diego. Male C57BL/6J and Apoa1bp−/− mice30 were housed up to 4 per standard cage at room temperature and maintained on a 12:12 hour light:dark cycle, with lights on at 07:00. To knock down the Ldlr gene expression, C57BL/6J and Apoa1bp−/− male mice were given weekly intraperitoneal injections of antisense oligonucleotides (ASO, courtesy of Ionis Pharmaceuticals, Inc.) against mouse Ldlr at 5 mg/kg bodyweight for the first four weeks, after which injections proceeded biweekly for an additional eight weeks. Concomitantly with the ASO intervention, mice were fed for 12 weeks a Western diet (Envigo TD.96121) containing 21% milk fat and 1.25% cholesterol, starting at 10 weeks of age. Both food and water were available ad libitum. Blood was collected as previously described.23 Total cholesterol and triglyceride levels were measured using BioVision’s K603 and EnzyChrom’s ETGA-200 enzymatic assays, respectively. Atherosclerosis was assessed as previously described in our lab31 and in adherence to the AHA guidelines. Briefly, mice were perfused and hearts were fixed and embedded in OCT and serially sectioned. To quantify aortic root atherosclerosis, sections were cut from the aortic valve origin to the last leaflet at 100 μm intervals. Atherosclerotic plaques were stained using Oil Red O, and counterstained using hematoxylin. To quantify plaque necrotic cores, additional slides were stained with Gomori’s trichome satin. Total lesion and necrotic core areas were quantified via computer-assisted image analysis (ImagePro). Sex as a biological variable in atherosclerosis experiments was not studied in this work and will be evaluated in future studies.
Human tissue samples
Human carotid endarterectomy specimens were obtained from patients undergoing carotid endarterectomy with a protocol approved by the University of California, San Diego Human Research Protection Program.
Monoclonal anti-AIBP antibody
Apoa1bp−/− mice were immunized with recombinant human AIBP lacking any tag. After 39 days, serum titers of AIBP-specific antibodies were determined by ELISA against AIBP. Following a recall boost, splenocytes were harvested from immunized mice and fused with a murine myeloma partner (p3X63Ag8.653) using a ClonaCell™-HY Hybridoma kit (StemCell Technologies, Cambridge, MA). Fused cells were resuspended in a semi-solid HAT hybridoma selection medium. Ten-14 days later, visible colonies were transferred into ClonaCell®-HY Growth Medium (DMEM, pre-selected serum, HT, gentamycin, and supplements). Supernatants from resulting clonal hybridomas were subsequently screened by ELISA against AIBP. Positive clones with monoclonal antibodies specific to AIBP were sub-cloned by limiting dilution in semi-solid gel, without HAT, and re-tested by ELISA. The colony designated as BE-1 was expanded and cryopreserved. The BE-1 colony was expanded by BioXCell (West Lebanon, NH) in tissue culture in a stirred tank fermentation with Hybridoma-SFM medium supplemented with 1% Fetal Clone 3 (Life Technologies, Carlsbad, CA) and purified with Protein A/G resin.
Immunohistochemistry of human carotid and mouse tissues
Immunohistochemistry of paraffin-embedded and optimal cutting temperature (OCT)-embedded sections was performed as described in the previous study.28, 32 Briefly, paraffin-embedded human carotid artery cross-sections were deparaffinized, and antigens were retrieved by incubation in a sodium citrate buffer for 30 min at 95°C. Murine aortic root OCT-embedded, frozen-sections were fixed, incubated with 1% sodium dodecyl sulfate (SDS) for antigen retrieval, blocked with TBS containing 5% normal goat serum/1% BSA for 30 min at room temperature, followed by incubation with F(ab) fragment anti-mouse IgG (H+L) (Abcam ab6668) for 1 hour at room temperature. The mouse monoclonal anti-AIBP antibody BE-1 (10 μg/ml diluted with blocking buffer) was used to stain sections in a humidified chamber at 4°C overnight. Sections were then incubated with an anti-mouse IgG -Alkaline Phosphatase (Sigma A3438), diluted with blocking buffer 1:50, for 30 min at room temperature, and visualized with Vector Red substrate (Vector SK-5100). Sections were counterstained with hematoxylin for 30s and mounted with Simpo-Mount (IHC World EO3–18). Stained tissue sections were imaged using a NanoZoomer Slide Scanner (Hamamatsu).
Cell culture
Bone marrow-derived macrophages (BMDM) were cultured as described previously.5 In brief, bone marrow cells isolated from tibias and femurs of C57BL/6 and Apoa1bp−/− male mice were incubated with CSF1/macrophage colony stimulating factor (L929 conditioned medium) following a published protocol.33 Human embryonic kidney 293 (HEK293) and human hepatocellular carcinoma cell line HepG2 were cultured in DMEM (Cellgro, 10–013-CV) supplemented with 10% fetal bovine serum (FBS; Omega Scientific, FB-01) and 50 μg/ml gentamicin (Omega Scientific, GT-10). Platinum-E (Plat-E) retroviral packaging cell line (Cell BioLabs, RV-101)34 were cultured in DMEM supplemented with 10% FBS, 1μg/ml puromycin (InvivoGen, ant-pr-1), 10μg/ml blasticidin (InvivoGen, ant-bl-05), and 50μg/ml gentamicin.
LDL Modification
Human native low-density lipoprotein (LDL) was purchased from Alfa Aesar (BT-603). OxLDL was produced in vitro as previously reported.5 Briefly, human LDL was extensively dialyzed against PBS to remove EDTA and 0.1 mg/ml of human LDL was incubated with 10 μM CuSO4 for 18 hours at 37°C. Thiobarbituric acid reactive substances (TBARS; typically, more than 30 nmol/mg in OxLDL) were measured to confirm LDL oxidation. OxLDL was concentrated to 1 mg/ml using a 100 kDa cut off centrifugal concentrator (Millipore, UFC810024) and sterile filtered (0.22 μm). Acetylated LDL (AcLDL, 360-28-10) was purchased from Lee Biosolutions.
Macrophage lipid accumulation
Cells were incubated for 24 h with PBS or 25 μg/ml OxLDL in L929 conditioned medium supplemented with 10% lipoprotein-deficient serum. To detect intracellular accumulation of neutral lipid, BMDM were stained with Oil Red O (Sigma, O0625) and hematoxylin (Vector Laboratories, H-3404) as previously published.5 Images were captured with NanoZoomer (Hamamatsu). Intracellular Oil Red O was extracted in 0.5 ml isopropanol (Fisher Scientific, A416500) and optical density was measured at 510 nm.
Flow cytometry assay
BMDM were incubated with PBS or 25 μg/ml OxLDL for 24 hours. Cells were fixed with 3.7% formaldehyde for 20 min and blocked with an anti-CD16/CD32 antibody (FcRr blocker, BD Bioscience) for 30 min on ice. Cells were incubated with APC-conjugated anti-CD36 antibody (BioLegend) for 1 hour at 4°C, washed 5 times with PBS, and analyzed using a FACSCanto II (BD Bioscience) flow cytometer.
Measurement of ROS
Total ROS was measured as previously described.35 In brief, BMDM were stimulated with PBS or 25 μg/ml OxLDL for 24 hours and then incubated with 10 μM 2’,7’-dichlorodihydrofluorescein diacetate (DCF-DA, Invitrogen, D399) for 20 min. Mitochondrial ROS was measured using MitoSox Red reagent according to the manufacturer’s protocol (Invitrogen). Cells were analyzed by Airyscan confocal microscope (Carl Zeiss) and FACS (BD Biosciences). Geometric means of FACS histograms were measured and presented as bar graphs.
Antibodies and reagents
The following antibodies were purchased from Cell Signaling Technology: LC3 (#4108 and #12741), β-ACTIN (#4967), SIRT3 (#5490), phospho-ULK1 (#5869), and VDAC (#4661). Antibodies were purchased from Santa Cruz Biotechnology: PARKIN (sc-32282), myc (sc-40), ha (sc-805), ULK1 (sc-390904), Beclin1 (sc-11427), and UBI (sc-8017 HRP). The following antibodies were purchased from Sigma-Aldrich: flag (F3165). The following antibodies were purchased from Abcam: SQSTM1/p62 (ab56416). The following antibodies were purchased from Invitrogen: Alexa Fluor 488-labeled anti-rabbit IgG (A11034), Alexa Fluor 568-labeled anti-mouse IgG (A11031), Alexa Fluor 568-labeled anti-rat IgG (A11077), Alexa Fluor 647-labeled anti-mouse IgG (A21237), and OXPHOS (#45–7999). A rabbit polyclonal antibody against mouse AIBP was a kind gift from Dr. Longhou Fang (Houston Methodist Research Institute). A rabbit polyclonal antibody against LDLR (10785–1-AP) was purchased from Proteintech. Antibodies against CD68 (#137001) and CD36 (#102612) were purchased from BioLegend. An antibody against CD16/CD32 (FcRr blocker, #553141) and CD31 (#562939) were purchased from BD Bioscience and Bafilomycin A1 (#196000) was purchased from Calbiochem.
JC-1 assay
Mitochondrial membrane potential was measured using a Mitoprobe™ JC-1 assay kit (Thermo Fisher Scientific, M34152) according to manufacturer’s instructions. In brief, BMDM were stimulated with either PBS or 25 ug/ml of OxLDL for 24 h. Cells were incubated with 2 μM of JC-1 probe for 30 min at 37°C, washed with PBS, and analyzed for the shift of fluorescence emission from green (529nm) to red (590nm) using a FACSCanto II flow cytometer (BD Bioscience). Mitochondrial membrane depolarization was calculated by a decrease in the red (aggregate)/green (monomer) fluorescence ratio. To disrupt mitochondrial membrane potential, cells were incubated with 10 μM of carbonyl cyanide 3-chlorophenylhydrazone (CCCP) for 24 h at 37°C. Geometric means of FACS histograms were measured and presented as bar graphs.
Retrovirus production
To produce retroviruses expressing mt-Keima, we used Plat-E retroviral packaging cell line. The day before transfection, Plat-E cells were plated and transfected with pCHAC-mt-mKeima (Addgene, plasmid#72342). After 48 hours, the supernatant containing retroviruses expressing mt-Keima was collected, filtered, and stored at -80°C.
Mt-Keima mitophagy assay
To monitor mammalian mitophagy, the mt-Keima assay was performed as previously described.36 BMDM isolated from WT and Apoa1bp−/− mice were infected with retroviruses expressing mt-Keima containing 8μg/ml polybrene (Sigma, H9268). At 24 h post infection, cells were incubated with OxLDL for additional 24 h and analyzed using a flow cytometry and an Airyscan confocal microscope (Carl Zeiss). Mitophagy was quantified in a flow cytometry assay using the ratio of red signal (550nm) to green signal (440nm) from mt-Keima, with a FACSCanto II (BD Bioscience).
Co-immunoprecipitation assay
HEK293 and HepG2 cells were transfected with human flag-tagged AIBP, PARK2-myc, MFN1-myc and/or MFN2-myc using GenJet In Vitro DNA transfection reagent (SignaGen Laboratories). At 2 days after transfection, cells were lysed with an ice-cold lysis buffer (50 mM Tris-HCl, pH 7.5, 1% NP-40, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 5 mM Na3VO4, 1 mM NaF, and a protease inhibitor cocktail from MedChemExpress). Cell lysates were preincubated with protein A/G Sepharose beads (Genesee Scientific, 20–528 and 20–537) for 30 min at 4°C and immunoprecipitated with appropriate primary antibodies overnight at 4°C. Next day, the lysates were further incubated with protein A/G Sepharose beads for additional 1 h at 4°C. Immune complexes were washed five times with lysis buffer, run on a Nu-PAGE Bis-Tris gel (Invitrogen), and antibody-bound proteins were detected by immunoblotting with secondary antibodies.
Western blot analysis
BMDM were plated at 0.5×106 cells/well in a 12-well plate. The next day, cells were stimulated with 25 μg/ml OxLDL for the indicated time. Cells were harvested and the lysates subjected to a 4–12% Nu-PAGE Bis-Tris gel (Invitrogen) electrophoresis and immunoblotted with appropriate primary antibodies overnight. The membranes were washed 2 times with TBS containing 0.1% Tween-20 (T-TBS), incubated with secondary antibodies for 1 hour at room temperature, and washed 5 times with T-TBS. The images were captured using a UVP imaging system.
Quantitative PCR
Total RNA was isolated using Nucleospin RNA columns (Clontech). cDNA was synthesized from isolated RNA using cDNA EcoDry (Clontech), following manufacturer’s instruction. Quantitative PCR was performed using a KAPA SYBR FAST Universal qPCR kit (KAPA Biosystems, KK4602), with primers ordered from Integrated DNA Technologies (IDT), and a Rotor Gene Q thermocycler (Qiagen).
Immunocytochemistry
To visualize the induction of autophagy, cells were stained and imaged as previously described.5 Briefly, cells plated on a coverslip were washed with PBS and fixed with 3.7% formaldehyde for 15 min at 37°C and then permeabilized with 0.5% Triton X-100 for an additional 10 min at room temperature. Cells were washed with PBS and blocked with 5% BSA containing 2% mouse/rabbit normal serum for 30 min at 37°C, incubated with primary antibodies, followed by secondary antibodies. After three washes with 0.1% Triton X-100 in PBS and two washes with PBS, coverslips were mounted on a cover glass with Prolong Gold AntiFade reagent with 4′,6-diamidino-2-phenylindole (DAPI) (Cell Signaling Technology). Fluorescent images were captured using an Airyscan confocal microscope (Carl Zeiss).
Annexin V staining
Annexin V binding was analyzed using an Annexin V-FITC apoptosis detection kit (eBioscience, BMS500FI) according to manufacturer’s protocol. Briefly, BMDM were incubated with PBS or OxLDL for 24 h, then sequentially washed with PBS and binding buffer. Cells were incubated with FITC-conjugated Annexin V for 15 min, washed two times with binding buffer, and then 7-aminoactinomycin D (7-AAD) was added. Annexin V binding was analyzed by FACSCanto II (BD Bioscience). Four regions were selected: viable cells (Annexin V- / 7AAD-), early apoptotic cells (Annexin V+ / 7AAD-), late apoptotic/necrotic cells (Annexin V+/7ADD+), and late necrotic cells (Annexin V- / 7AAD+). Apoptotic cells were calculated by combining early and late apoptotic cells.
Mitochondria isolation
BMDM isolated from C57BL/6J and Apoa1bp−/− mice were stimulated with either PBS or OxLDL for 24 h. Mitochondria were purified using mitochondria isolation kit (ThermoFisher Scientific, 89874) according to manufacturer’s protocol.
TUNEL staining
Detection of apoptosis was performed by TUNEL assay using an In Situ Cell Death Detection Kit, TMR red (Sigma-Aldrich, 12156792910) according to manufacturer’s protocol. In brief, frozen tissue sections were fixed in 3.7% paraformaldehyde for 20 min at room temperature, washed two times with PBS and permeabilized with 0.1% Triton-X 100 in 0.1% sodium citrate for 2 min on ice. Tissue sections were incubated with the TUNEL reaction mixture for 1 h at 37°C, washed five times with PBS, and mounted with Prolong Gold AntiFade reagent with 4′,6-diamidino-2-phenylindole (DAPI) (Cell Signaling Technology). The tissue was counterstained with an anti-CD68 antibody. TUNEL-positive cells were captured by AiryScan confocal microscope (Carl Zeiss).
Statistical analyses
Data are presented as mean±SEM, unless otherwise stated. Data have been analyzed for normality using Shapiro-Wilk test as provided in GraphPad Prism, and the data sets that passed the normality test were subjected to parametric analyses, with equal or unequal variance. For comparison between two groups, statistical analysis was performed using two-tailed Student’s t-test. For multiple group and multi-variable comparisons, we used one-way ANOVA or two-way ANOVA followed by the Bonferroni post hoc test, using GraphPad Prism. In independent experiments with high systemic variation (Figures 3B, 5I, 6E and IC), repeated measures t-test or AOVA, or one-sample t-test were used as explained in figure legends. Differences with a P-value of less than 0.05 were considered statistically significant.”
Figure 3.
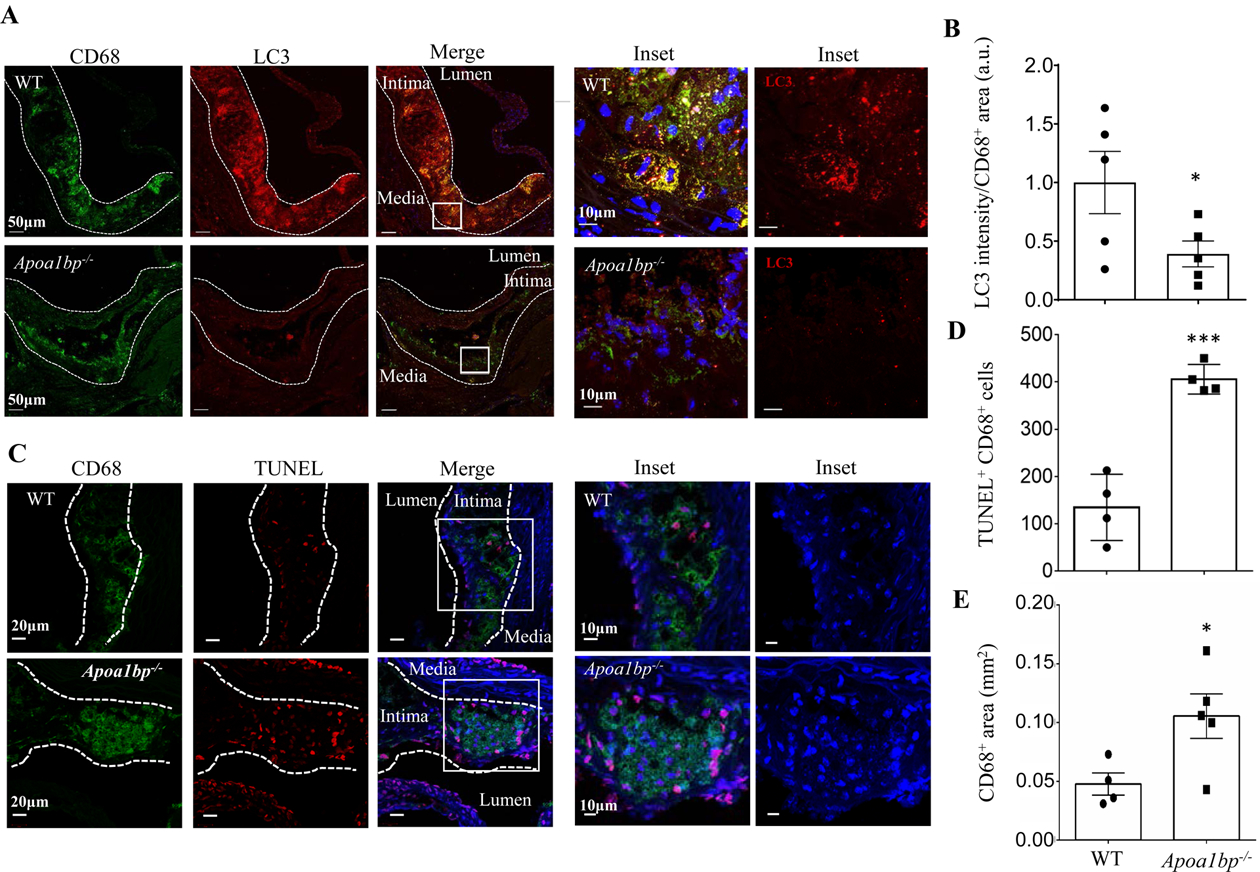
AIBP expression regulates autophagy and cell survival in atherosclerotic lesions of hypercholesterolemic mice. (A and B) Aortic root sections from hypercholesterolemic WT and Apoa1bp−/− mice were stained with anti-LC3 (red) and anti-CD68 (green) antibodies to assay for autophagy in macrophage-rich areas. The LC3 fluorescent intensities were measured in CD68+ areas and specific LC3 intensities per mm2 of CD68+ areas were calculated. Data are from immunohistochemical staining conducted on 5 separate days and because fluorescence intensity varies from day-to-day, repeated measures t-test was conducted to calculate the p-value. (C and D) TUNEL staining showing apoptotic cells in the aortic root sections. The numbers of TUNEL and CD68 double-positive cells were counted. (E) CD68-positive area in each lesion. Mean±SEM; N=4–5. ***, p<0.0005.
Figure 5.
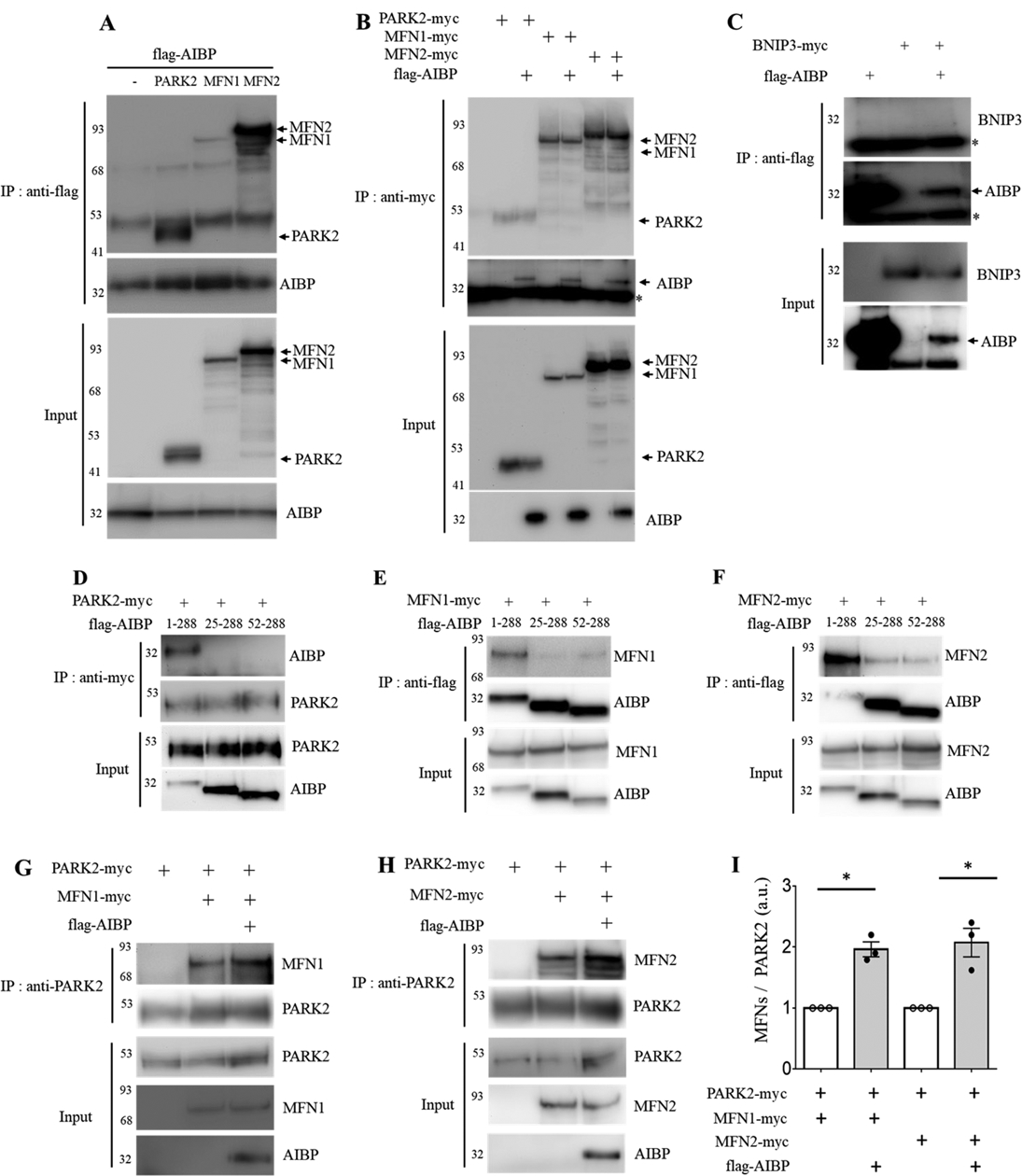
AIBP associates with PARK2, MFN1 and MFN2 and enhances PARK2-MFN1/2 interactions. (A-B) HEK 293 cells were co-transfected with flag-tagged AIBP (1–289 aa) and PARK2-myc, MFN1-myc, or MFN2-myc. (A) AIBP-driven immunoprecipitation with an anti-flag antibody. (B) Immunoprecipitation driven by PARK2, MFN1, or MFN2 with an anti-myc antibody. (C) AIBP does not bind to BNIP3. HEK 293 cells were co-transfected with flag-tagged AIBP and BNIP3-myc. Cell lysates were immunoprecipitated with an anti-flag antibody. *, IgG light chain. (D-F) HEK293 cells were co-transfected with flag-tagged AIBPs (1–289 aa, 25–289 aa, or 52–289 aa) together with (D) PARK2-myc, (E) MFN1-myc, or (F) MFN2-myc. (D) PARK2 and (E and F) AIBPs were immunoprecipitated with an anti-myc or anti-flag antibody, respectively. (G-I) HEK 293 cells were co-transfected with flag-tagged AIBP, PARK2-myc, and MFN1-myc or MFN2-myc. Cell lysates were immunoprecipitated with an anti-PARK2 antibody, and the bound (G) MFN1 and (H) MFN2 were detected by immunoblotting with an anti-myc antibody. (I) Band intensities were measured and MFN/PARK2 ratios calculated. Because of variation in independent transient transfection/pull-down/blot experiments conducted on separate days, experimental samples (with AIBP) were normalized to the control samples in which AIBP was not added. With control samples (no AIBP) set as 1, one-sample t-test was conducted to test the hypothesis whether the experimental group (with AIBP) was different from 1, i.e., whether AIBP significantly increased MFN1 and MFN2 binding to PARK2 compared to no-AIBP controls. Mean±SEM; N=3–4. *, p<0.05; **, p<0.005; ***, p<0.0005; ****, p<0.0001.
Figure 6.
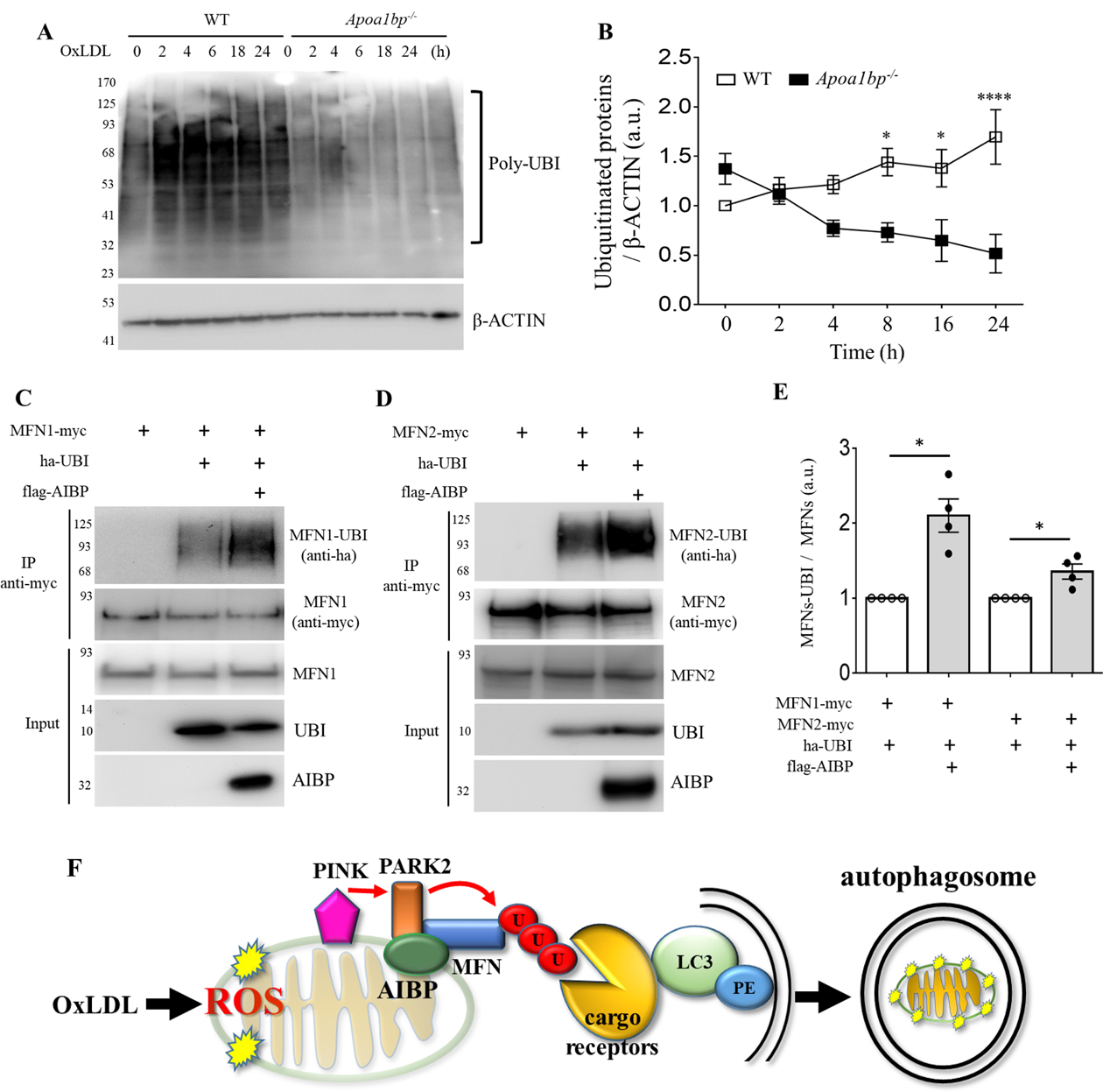
AIBP regulates the ubiquitination pathway. (A and B) BMDM isolated from WT and Apoa1bp−/− mice were incubated with PBS or 25 μg/ml OxLDL for indicated time. Cells were harvested and immunoblotted with anti-UBI and β-ACTIN antibodies. The band intensities were quantified. (C and D) HEK 293 cells were co-transfected with flag-tagged AIBP, ha-ubiquitin, and MFN1-myc or MFN2-myc. Cell lysates were immunoprecipitated with an anti-myc antibody, and ubiquitination was detected by immunoblotting with an anti-ha antibody. (E) Band intensities were measured and MFN-UBI/MFN ratios calculated. Because of variation in independent transient transfection/pull-down/blot experiments conducted on separate days, experimental samples (with AIBP) were normalized to the control samples in which AIBP was not added. With control samples (no AIBP) set as 1, one-sample t-test was conducted to test the hypothesis whether the experimental group (with AIBP) was different from 1, i.e., whether AIBP significantly increased MFN1 and MFN2 ubiquitination compared to no-AIBP controls. Mean±SEM; N=3–4. *, p<0.05; **, p<0.005; ***, p<0.0005; ****, p<0.0001. (F) Schematic overview of the regulation of mitophagy by AIBP. Intracellular AIBP associates with mitochondrial proteins Parkin and MFN1/MFN2 and upregulates ubiquitination of MFN1 and MFN2; in turn ubiquitinated MFN are recognized by cargo receptors interacting with lipidated LC3 anchored in the membrane to form an autophagosome. In the context of atherosclerosis, this regulation is particularly important in removal of damaged mitochondria in macrophages exposed to atherogenic OxLDL.
RESULTS
AIBP expression in atherosclerotic lesions and regulation by OxLDL.
Because mouse and human AIBP have a high degree of homology, in order to generate a monoclonal antibody to AIBP, we immunized an Apoa1bp−/− mouse with recombinant human AIBP from which a His-tag was cleaved. The resulting monoclonal antibody BE-1 (see Methods for details) was used to stain atherosclerotic lesions from WT mice, with lesions from Apoa1bp−/− mice serving as a negative control. AIBP was abundantly expressed in mouse atherosclerotic lesions (Figure 1A) and, importantly, in human carotid specimens (Figure 1B). AIBP staining colocalized predominantly with the macrophage marker CD68 but also partially with alpha SMC actin and CD31 (Figure IA in Data Supplement). These results suggest that AIBP is expressed in multiple vascular cell types. Because OxLDL’s OSE are abundant in blood and atherosclerotic plaques of patients with cardiovascular disease,3, 37 we tested if OxLDL regulates AIBP expression in macrophages. We found that OxLDL treatment increases AIBP protein expression in BMDM (Figure 1C). AcLDL also induced AIBP protein expression in macrophages but not LPS, Pam3CSK4 or TNFα (Figure IB – IE in Data Supplement). These results, showing upregulated AIBP expression in macrophages under experimental atherogenic conditions and in human and mouse atherosclerotic lesions support the importance of gaining mechanistic insights into function of intracellular AIBP.
Figure 1.
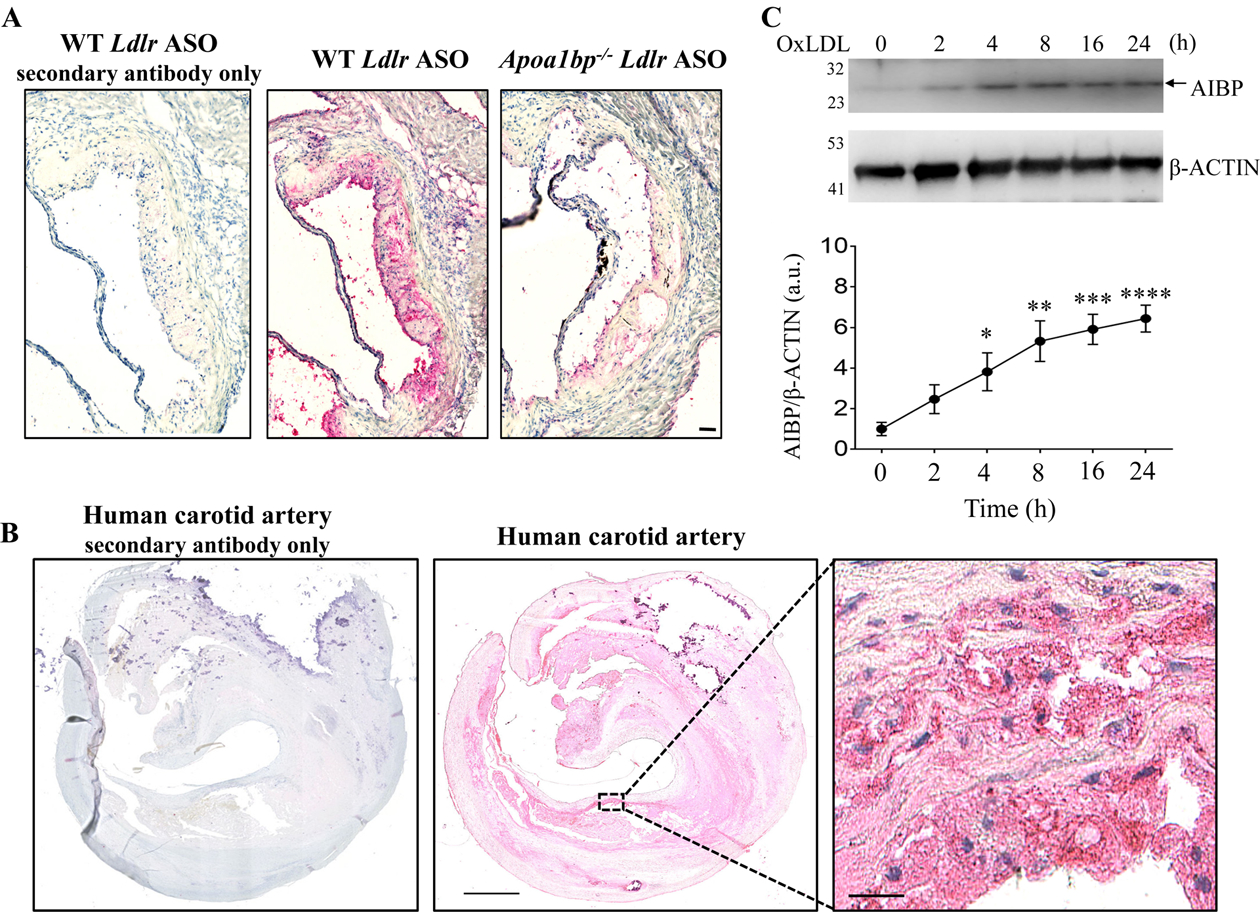
AIBP expression in atherosclerotic lesions and in OxLDL-stimulated macrophages. Sections of the aortic root of hypercholesterolemic WT and Apoa1bp−/− mice (A) and human carotid artery (B) were stained with the monoclonal anti-AIBP antibody BE-1, followed by a secondary antibody or by the secondary antibody only, and counterstained with H&E. Representative images of three specimens tested. (C) BMDM isolated from WT mice were stimulated with 25 μg/ml OxLDL for indicated time. Cells lysates were immunoblotted with anti-AIBP and β-ACTIN antibodies. Band intensities were quantified. Mean±SEM; N=4–5. *, p<0.05; **, p<0.005; ***, p<0.0005; ****, p<0.0001 vs. time zero. Scale bars: 50 μm in A, 1 mm in B, and 25 μm in the zoomed-in region in B.
AIBP deficiency reduces autophagy in macrophages in response to oxidized LDL.
Macrophage autophagy under atherogenic conditions is reported to be a protective mechanism.10, 11 To test whether macrophage AIBP regulates autophagy, BMDM isolated from WT and Apoa1bp−/− mice were incubated with 25 μg/ml of OxLDL. OxLDL-induced accumulation of LC3-II was significantly lower in Apoa1bp−/− compared to WT macrophages (Figures 2A – 2D). Inhibition of autophagosome-lysosome fusion by targeting lysosomes with bafilomycin A1 showed further accumulation of LC3-II in WT macrophages; however, accumulation of LC3-II was reduced in Apoa1bp−/− macrophages (Figures 2E and 2F). Unlike in autophagy induced by other stimuli, in OxLDL-induced macrophage autophagy we did not observe reductions in SQSTM1/p62 expression. This was likely due to OxLDL-induced upregulation of Sqstm1 mRNA and protein expression (Figures IIA–IIC in Data Supplement). There were no significant genotype-dependent or OxLDL-induced changes in mRNA expression of Map1lc3b, Atg5, Atg7, Beclin1, or lysosomal Lamp1 expression. Protein expression of BECN1 or ULK1 did not change, but OxLDL induced phosphorylation of ULK-1 in WT but not Apoa1bp−/− macrophages (Figures IID–IIL in Data Supplement). To exclude the possibility that reduced autophagy in Apoa1bp−/− macrophages was due to impaired OxLDL uptake, we tested expression of CD36, the primary receptor for OxLDL in macrophages, which is also upregulated in response to OxLDL.38, 39 The mRNA and cell surface protein expression of CD36, both in unstimulated and OxLDL-treated cells, were equal in WT and Apoa1bp−/− macrophages (Figures IIIA–IIIC in Data Supplement). Furthermore, intracellular lipid accumulation in Apoa1bp−/− macrophages was slightly higher than in WT (Figures IIID and IIIE in Data Supplement), suggesting that impaired autophagy in Apoa1bp−/− macrophages cannot be explained by reduced OxLDL uptake.
Figure 2.
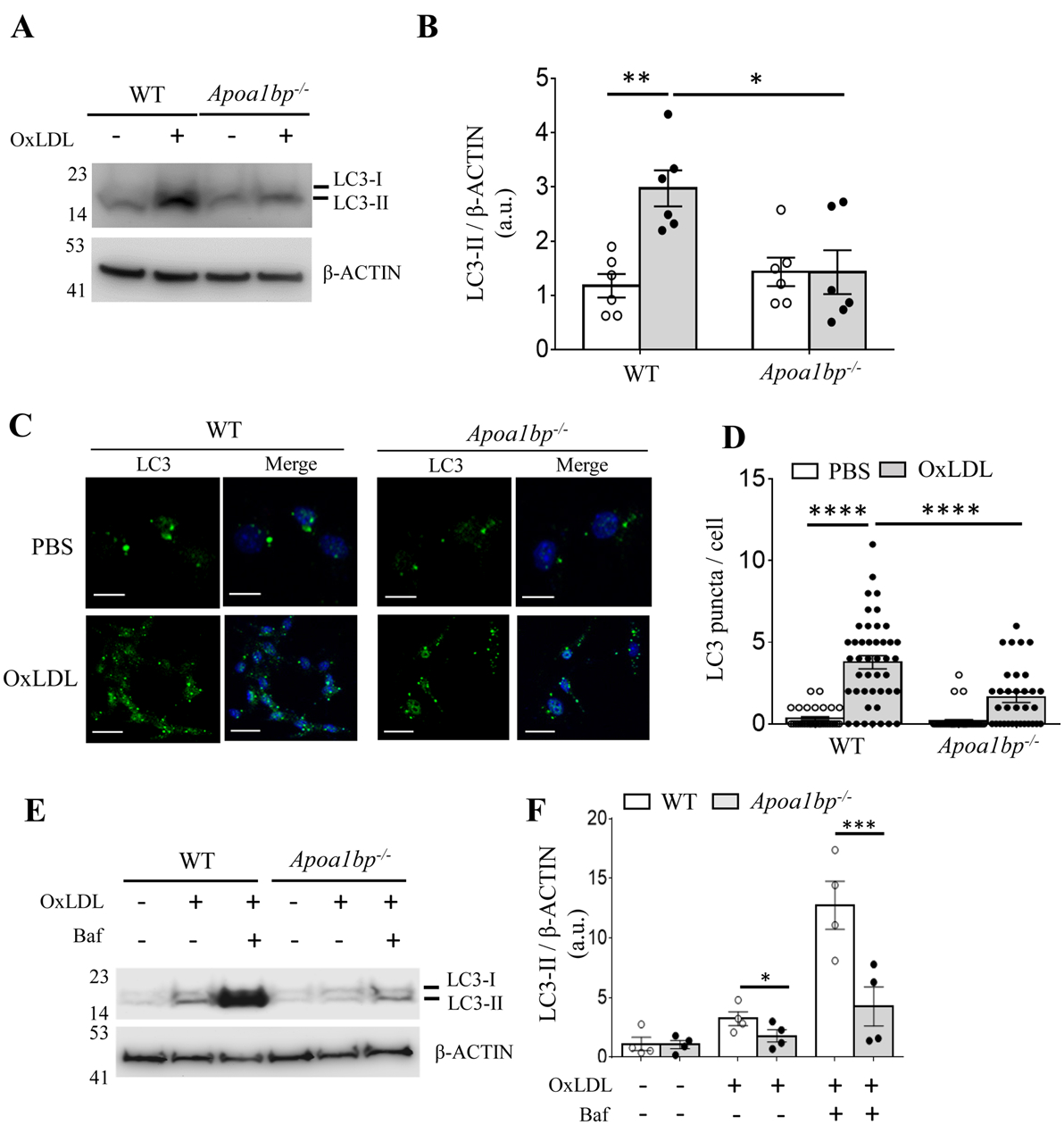
AIBP regulates OxLDL-induced autophagy. (A to D) BMDM isolated from WT and Apoa1bp−/− mice were incubated with PBS or 25 μg/ml OxLDL for 24 h. Cell lysates were immunoblotted with antibodies against LC3 and β-ACTIN, and band intensities were quantified. For imaging, cells were stained with an anti-LC3 antibody and DAPI. The numbers of LC3 puncta per cell were counted from three independent experiments. (E and F) Cells were pretreated with or without 100 nM Baf for 1h and then incubated with 25 μg/ml OxLDL for 24 h. Cell lysates were immunoblotted with indicated antibodies, and band intensities were quantified. Mean±SEM; N=3–5. *, p<0.05; **, p<0.005; ***, p<0.0005; ****; p<0.0001. Scale bar: 20 μm.
Hypercholesterolemic Apoa1bp−/− mice have reduced macrophage LC3 expression and increased apoptosis in atherosclerotic lesions.
To assess the in vivo relevance of our findings that AIBP promotes autophagy, the cellular process that is atheroprotective, we evaluated macrophage LC3 expression and apoptosis in aortic roots of hypercholesterolemic male mice. WT and Apoa1bp−/− mice were injected with antisense oligonucleotides (ASO) against the LDL receptor40 and fed a Western diet for 12 weeks. Both WT and Apoa1bp−/− mice had a similar weight gain over the course of the study. At the end of the study, the LDL protein expression in the liver was largely undetectable or very low (Figures IVA and IVB in Data Supplement). Plasma cholesterol levels reached 2000–3000 μg/dL at 4 weeks and decreased to a 500–700 μg/dL range by 12 weeks (Figure IVC in Data Supplement). Both total cholesterol and triglyceride levels in terminal blood trended higher in Apoa1bp−/− mice (Figures IVC and IVD in Data Supplement). Accordingly, the size of atherosclerotic lesions trended higher in Apoa1bp−/− compared to WT mice but were not significantly different (Figure IVE in Data Supplement), facilitating the comparisons of specific markers. The statistical difference between these atherosclerosis results in Apoa1bp−/− vs. WT mice and those previously reported by us and others23, 26 can be explained by the different Ldlr intervention: here we used Ldlr ASO in C57BL/6J mice, while previous studies employed Ldlr−/− mice. To assess the effect of AIBP on autophagy and apoptosis in vivo, lesions of a similar size in Apoa1bp−/− and WT groups were analyzed for CD68 (macrophages), LC3 and TUNEL. We found that macrophage LC3 expression was significantly decreased (Figures 3A and 3B) and the number of TUNEL positive macrophages was dramatically increased (Figures 3C and 3D) in Apoa1bp−/− compared to WT mice. Accordingly, the size of necrotic cores was significantly larger in the lesions of Apoa1bp−/− compared to WT mice (Figures IVF in Data Supplement). The overall area covered with CD68+ cells was larger in lesions of Apoa1bp−/− compared to WT mice (Figure 3E). These in vivo results indicate that AIBP contributes to the induction of autophagy and inhibition of apoptotic cell death in atherosclerotic lesions.
AIBP deficiency augments OxLDL-induced ROS generation and apoptosis.
It is well appreciated that autophagy inhibits ROS accumulation and apoptosis through the removal of damaged organelles and particularly damaged mitochondria.41–43 To test whether AIBP regulates the intracellular ROS generation and apoptotic cell death, BMDM isolated from WT and Apoa1bp−/− mice were incubated with OxLDL, and ROS levels were determined by confocal microscopy and flow cytometry. We found that OxLDL-induced intracellular ROS levels (Figures 4A, 4B and VA in Data Supplement) and apoptotic cell death (Figure 4C and VC in Data Supplement) were significantly increased in Apoa1bp−/− macrophages.
Figure 4.
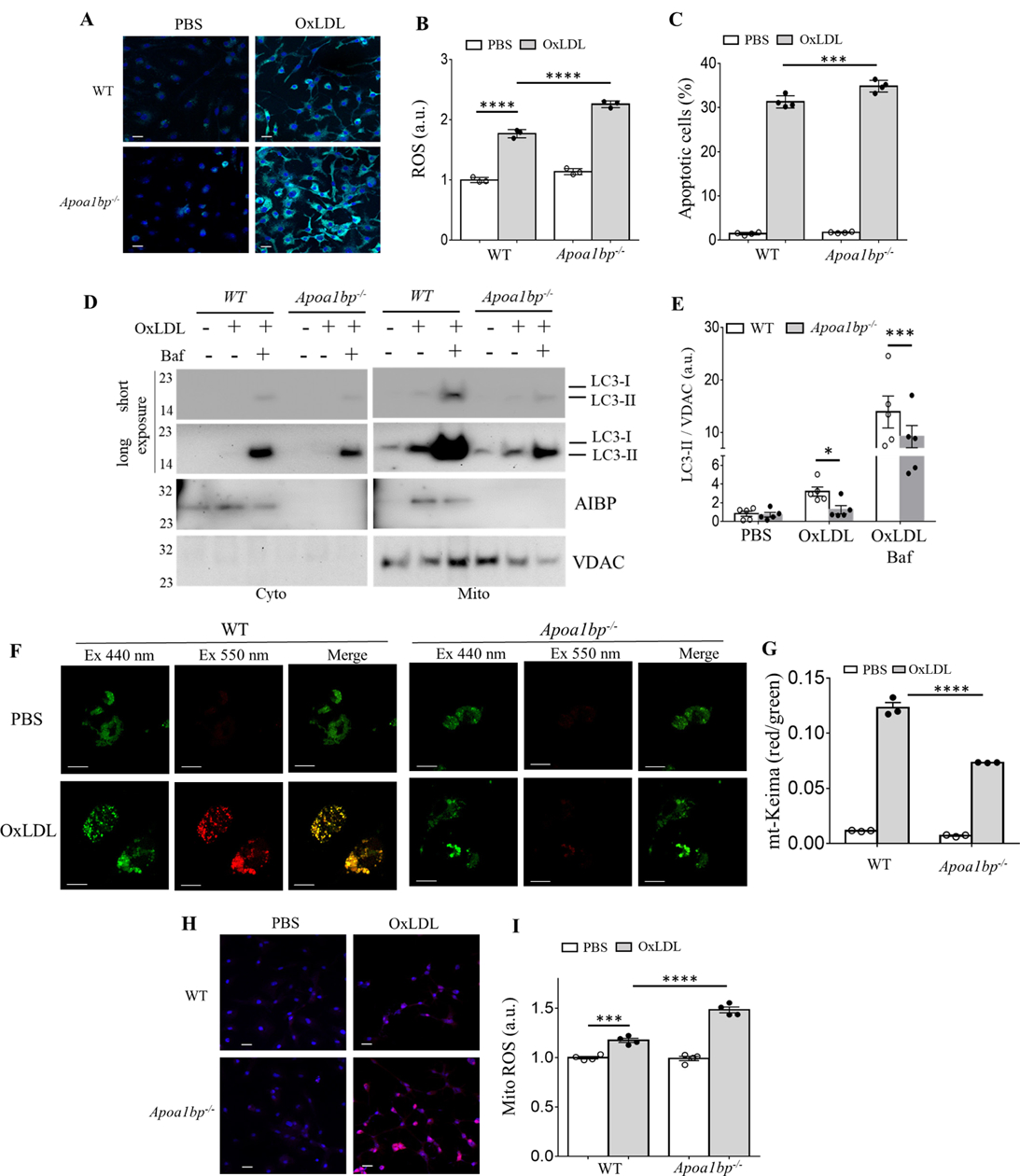
Macrophage AIBP regulates OxLDL-induced ROS, apoptosis and mitophagy. BMDM isolated from WT and Apoa1bp−/− mice were incubated with PBS or 25 μg/ml OxLDL for 24 h and ROS generation was measured using (A) a confocal microscope or (B) flow cytometer. Geometric means of FACS histograms were measured and presented as scattered bar graphs. (C) Apoptotic cells were detected by Annexin V staining using a flow cytometry analysis. (D-E) LC3-II and AIBP expression in isolated mitochondria were measured by immunoblot. (F-G) BMDM were infected with retrovirus encoding mt-Keima for 24 h and incubated with PBS or 25 μg/ml OxLDL for additional 24 h. Mitophagy flux was analyzed by confocal microscopy, capturing images at both 440nm (green, mitochondria) and 550nm (red, lysosome) (F), and flow cytometry (G). Mitochondrial ROS were measured using (H) confocal microscopy and (I) flow cytometry. Mean±SEM; N=3–5. *, p<0.05; **, p<0.005; ***, p<0.0005; ****, p<0.0001. Scale bars, 20μm in A and H, 10μm in F.
AIBP regulates mitophagy and mitochondrial ROS generation.
The elimination of damaged mitochondria by mitophagy leads to decreased ROS generation and cell death.44, 45 To investigate whether AIBP is involved in OxLDL-induced mitophagy, BMDM isolated from WT and Apoa1bp−/− mice were incubated with OxLDL for 24 h, and mitochondria were isolated. As shown in Figures 4D and 4E, OxLDL induced accumulation of LC3-II as well as AIBP in mitochondria. However, the accumulation of LC3-II was significantly decreased in Apoa1bp−/− macrophages. In a separate experiment, we infected BMDMs with a retrovirus encoding mito-Keima, which detects mitochondria fusion with the lysosome, and found significant reductions in mitophagy in Apoa1bp−/− compared to WT macrophages (Figures 4F and 4G).We also found that mitochondrial ROS (MitoROS) were significantly increased in Apoa1bp−/− macrophages in response to OxLDL (Figures 4H, 4I and VB in Data Supplement).
AIBP is important for sustaining mitochondrial function in OxLDL-treated macrophages.
Next, we tested whether AIBP regulates mitochondrial function. AIBP deficiency resulted in statistically significant differences in the membrane depolarization in response to OxLDL, while there was no difference in CCCP-induced mitochondrial depolarization between WT and Apoa1bp−/− macrophages (Figures VIA and VIB in Data Supplement). OxLDL induced a significant increase in the expression of oxidative phosphorylation (OXPHOS) complex I protein in WT but not Apoa1bp−/− macrophages, while the expression of complexes II-V did not change (Figures VIC and VID in Data Supplement). These findings suggest that AIBP plays a role in regulation of mitochondrial function, both in unstimulated macrophages and in response to OxLDL.
AIBP associates with Parkin, Mitofusin 1 and Mitofusin 2.
To elucidate the mechanism of the AIBP effect on mitophagy, we first tested whether AIBP binds to mitochondrial proteins. We found that AIBP associated with Parkin (PARK2), mitofusin (MFN)-1 and MFN2 (Figures 5A and 5B). However, AIBP did not bind BNIP3, which is associated with PARK2-independent mitophagy (Figure 5C). Next, to determine the region in the AIBP molecule responsible for PARK2, MFN1 and MFN2 binding, we generated truncated mutants of AIBP. It has been proposed that the amino acids 1–24 in the AIBP sequence constitute a signal peptide driving protein secretion.46 Another group has suggested that amino acids 1–51 of AIBP constitute a mitochondria localization peptide.29 Thus, we cloned two truncated mutants of AIBP, 25–289 aa, and 52–289 aa. As shown in Figures 5D – 5F and Figures VIIA – VIIC in Data Supplement, only the full-length of AIBP, but not the two truncated mutants colocalized with and pulled down PARK2, MFN1 and MFN2. These results indicate that the 1–24 aa of AIBP is the sequence responsible for mitochondrial targeting and possibly for binding with PARK2, MFN1 and MFN2, however, we cannot exclude the possibility that lack of mitochondrial localization of the 25–289 aa AIBP precludes its interaction with mitochondrial proteins, which may occur via different protein domains.
AIBP enhances PARK2-MFN1/2 interaction and ubiquitination of MFN1/2.
MFN1 and MFN2 are outer membrane proteins involved in mitochondrial fusion.47–49 The PINK1/PARK2 pathway induces ubiquitination of MFN1/2, which is necessary for induction of mitophagy.50–52 To test whether AIBP affects the interaction between PARK2 and MFN1/2, cells were transfected with AIBP, PARK2 and either MFN1 or MFN2. As reported earlier, PARK2 binds to both MFN1 and MFN2.51 These interactions were significantly enhanced by AIBP expression (Figures 5G – 5H). The PARK2-mediated ubiquitination pathway is important for mitochondrial quality control to remove damaged mitochondria. As shown in Figures 6A and 6B, OxLDL induced robust protein ubiquitination in WT but not Apoa1bp−/− macrophage. To test whether AIBP regulates ubiquitination of MFN1/2, cells were transfected with AIBP, myc-tagged MFN1 or myc-tagged MFN2, and with HA-tagged ubiquitin. Results shown in Figures 6C – 6E indicate that ubiquitination of both MFN 1 and MFN2 was enhanced in cells overexpressing AIBP. These results agree with the above data (Figures 5G – 5I) showing that AIBP enhances MFN1 and MFN2 interaction with the ubiquitin ligase PARK2.
DISCUSSION
In earlier studies using two different animal models, zebrafish and mouse, we and others have demonstrated that secreted AIBP reduces lipid rafts formation via augmented cholesterol efflux from endothelial cells and macrophages, resulting in the inhibition of angiogenesis and atherogenesis, reduction of neuropathic pain and acute lung inflammation, regulation of hematopoietic stem and progenitor cell fate, HIV replication, and glaucomatous neuroinflammation.22, 23, 27, 28, 30, 53–55 In addition to the function of extracellular AIBP in regulating cholesterol efflux, the protein has been reported to have an NAD(P)HX epimerase (NAXE) or ADP-ribosyltransferase enzymatic activity, as well as being involved in vitamin B6 metabolism.24, 25, 56 Evolution of AIBP (NAXE) function was discussed in our recent review article.57 Because AIBP (NAXE) expressed in transfected cells was shown to localize to mitochondria,29 in this study, we examined role of AIBP in mitochondria and discovered that it regulates mitophagy. These findings do not exclude a potential role of AIBP in regulation of autophagy of other organelles. Our findings are illustrated in Figure 6F: In the context of atherosclerosis-associated, OxLDL-induced macrophage autophagy, AIBP association with PARK2, MFN1 and MFN2 augments MFN1 and NFN2 ubiquitination and subsequent formation of an autophagosome.
Two mitophagy pathways have been described in mammalian cells, receptor-dependent and receptor-independent, the latter is ubiquitin-mediated. Receptors that regulate mitophagy have been found in both inner and outer mitochondria membrane (IMM and OMM, respectively). Nip3-like protein X (NIX) and BNIP3 are common OMM proteins involved in receptor-mediated mitophagy.58, 59 In our study, AIBP did not bind to BNIP3, suggesting that AIBP does not participate in the receptor-dependent mitophagy pathway. However, we found that AIBP associated with PARK2, the ubiquitin ligase regulating ubiquitination of MFN1 and MFN2. In the absence of AIBP, ubiquitination of MFN1 and MFN2 was reduced and OxLDL-mitophagy significantly inhibited. As a result of deficient mitophagy, mitochondrial ROS were increased, mitochondrial function impaired and the overall cell survival decreased in Apoa1bp−/− compared to WT macrophages. In atherosclerotic lesions, in which OSEs of OxLDL are a major component,60 AIBP deficiency resulted in decreased autophagy and increased apoptosis. In addition to the AIBP-regulated autophagy mechanisms, this phenotype in the lesions of Apoa1bp−/− mice could be due to increased lipid load in macrophages, which has been demonstrated in vitro with Apoa1bp−/− BMDM, and associated cytotoxicity independent of autophagy.
We found that expression of only OXPHOS complex I was significantly reduced in OxLDL-stimulated Apoa1bp−/− macrophages. The complex I is dependent on NADH and its deficiency in patients is associated with respiratory chain defect in childhood61–63 and renal oncocytoma.64 In humans, AIBP (NAXE) loss-of-function mutations confer a lethal neurometabolic phenotype in infants, and fibroblasts from these subjects accumulate toxic NAD(P)HX metabolites.65–67 However, loss of AIBP in E. coli, yeast, plants or mice does not affect viability or growth.57
The ubiquitination pathway in mitophagy controls mitochondrial quality.58, 68 Cells exposed to OxLDL showed activation of the ubiquitin pathway. However, protein ubiquitination was impaired in AIBP-deficient macrophages. MFN1 and MFN2 are OMM proteins that regulate mitochondria fusion. Both MFN1 and MFN2 have an N-terminal GTPase domain, which is important for membrane fusion between either two adjacent mitochondria or between mitochondria and endoplasmic reticulum.69–71 Ubiquitination of MFN1 and MFN2 in a PINK/PARK2-dependent pathway is an early event that facilitates mitophagy.50–52 Indeed, we found that AIBP associated with PARK2 and MFN1/2 and enhanced ubiquitination of MFN1 and MFN2. Intriguingly, our findings show that the amino acids 1–24 of AIBP, designated as a signal peptide,46 are responsible for mitochondrial localization and possibly for binding with PARK2, MFN1 and MFN2. These data suggest that the putative signal peptide of AIBP, possibly dependent on its posttranslational modification, is important for not only the secretion of AIBP but also regulation of removal of damaged mitochondria via activation of the ubiquitin pathway. In a separate study, we found significant changes in the architecture of mitochondria in Müller glia and retinal ganglion cells of Apoa1bp−/− mice,55 which may be the result, at least in part, of defective mitophagy. In addition to the mechanism of AIBP-mediated augmentation of PINK/PARK2-dependent ubiquitination of MFN1 and MFN2, we cannot exclude a connection between AIBP-regulated lipid raft dynamics in the plasma membrane and its effect on mitophagy and mitochondria quality control. Although our study was focused on the role of AIBP in regulation of mitophagy, future studies will test whether AIBP regulates other types of autophagy, e.g. lipophagy, which is also an important pathway in atherosclerosis.
In summary, in this work we demonstrate that intracellular AIBP regulates autophagy in atherosclerosis and mitophagy in macrophages exposed to the atherogenic factor OxLDL. Strategies to increase intracellular AIBP expression and thus augment mitochondria quality control may have therapeutic implications in atherosclerosis.
Supplementary Material
HIGHLIGHTS.
Apolipoprotein A-I binding protein (AIBP) is highly expressed in human and mouse atherosclerotic lesions.
AIBP regulates autophagy in primary macrophages exposed to oxidized LDL.
Macrophages in atherosclerotic lesions from AIBP knockout mice are characterized by reduced autophagy and increased apoptosis.
The mechanism involves association of mitochondria-localized AIBP with PARK2, MFN1 and MFN2 and regulation of ubiquitination of MFN1 and MFN2, the key component in the process of mitophagy.
Acknowledgments
We would like to thank Dr. Youngil Lee (University of West Florida) for providing PARK2-myc, MFN1-myc, MFN2-myc, BNIP3-myc and ha-UBI plasmids and Dr. Longhou Fang (Houston Methodist Research Institute) for providing an anti-AIBP polyclonal antibody. We are grateful to Ionis Pharmaceuticals, Inc. for the gift of Ldlr ASO.
Sources of Funding
This work was supported by National Institute of Health Grants HL135737, HL136275 and NS102432. NINDS P30 NS047101 supports UCSD School of Medicine Microscopy and Histology Core.
Nonstandard Abbreviations and Acronyms
- APOA-I
apolipoprotein A-I
- AIBP
APOA-I binding protein
- ASO
antisense oligonucleotides
- BMDM
bone marrow-derived macrophage
- BNIP3
Bcl2/adenovirus E1B 19-kDa-interacting protein 3
- CD36
cluster of differentiation 36
- LDLR
low-density lipoprotein receptor
- MAP1LC3/LC3
microtubule-associated protein 1 light chain 3
- MFN
mitofusin
- NAXE
NAD(P)H-hydrate epimerase
- OxLDL
oxidized low-density lipoprotein
- PINK1
PTEN-induced kinase 1
- ROS
reactive oxygen species
- SQSTM1/p62
sequestosome 1/p62
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
Footnotes
Disclosures
YIM is a scientific co-founder of Raft Pharmaceuticals LLC. ST is a consultant to Boston Heart Diagnostics and is a co-inventor and receives royalties from patents owned by UCSD on oxidation-specific antibodies and of biomarkers related to oxidized lipoproteins, has a dual appointment at UCSD and Ionis Pharmaceuticals, and is a co-founder and has an equity interest in Oxitope, Inc and Kleanthi Diagnostics, LLC. Although these relationships have been identified for conflict of interest management based on the overall scope of the project and its potential benefit to Oxitope and Kleanthi Diagnostics LLC, the research findings included in this particular publication may not necessarily relate to the interests of Oxitope, Inc and Kleanthi Diagnostics, LLC. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies. Other authors declare that they have no competing interests.
REFERENCES
- 1.Libby P Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:2045–2051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hansson GK, Libby P The immune response in atherosclerosis: A double-edged sword. Nat Rev Immunol. 2006;6:508–519 [DOI] [PubMed] [Google Scholar]
- 3.Miller YI, Choi SH, Wiesner P, Fang L, Harkewicz R, Hartvigsen K, Boullier A, Gonen A, Diehl CJ, Que X, Montano E, Shaw PX, Tsimikas S, Binder CJ, Witztum JL. Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ Res. 2011;108:235–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peng N, Meng N, Wang S, Zhao F, Zhao J, Su L, Zhang S, Zhang Y, Zhao B, Miao J. An activator of mtor inhibits oxldl-induced autophagy and apoptosis in vascular endothelial cells and restricts atherosclerosis in apolipoprotein e(-)/(-) mice. Sci Rep. 2014;4:5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choi SH, Gonen A, Diehl CJ, Kim J, Almazan F, Witztum JL, Miller YI. Syk regulates macrophage MHC-II expression via activation of autophagy in response to oxidized ldl. Autophagy. 2015;11:785–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mizushima N, Komatsu M. Autophagy: Renovation of cells and tissues. Cell. 2011;147:728–741 [DOI] [PubMed] [Google Scholar]
- 7.Kundu M, Thompson CB. Autophagy: Basic principles and relevance to disease. Annu Rev Pathol. 2008;3:427–455 [DOI] [PubMed] [Google Scholar]
- 8.Shao BZ, Han BZ, Zeng YX, Su DF, Liu C. The roles of macrophage autophagy in atherosclerosis. Acta Pharmacol Sin. 2016;37:150–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sergin I, Evans TD, Zhang X, Bhattacharya S, Stokes CJ, Song E, Ali S, Dehestani B, Holloway KB, Micevych PS, Javaheri A, Crowley JR, Ballabio A, Schilling JD, Epelman S, Weihl CC, Diwan A, Fan D, Zayed MA, Razani B. Exploiting macrophage autophagy-lysosomal biogenesis as a therapy for atherosclerosis. Nat Commun. 2017;8:15750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Razani B, Feng C, Coleman T, Emanuel R, Wen H, Hwang S, Ting JP, Virgin HW, Kastan MB, Semenkovich CF. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab. 2012;15:534–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liao X, Sluimer JC, Wang Y, Subramanian M, Brown K, Pattison JS, Robbins J, Martinez J, Tabas I. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab. 2012;15:545–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan DC. Mitochondria: Dynamic organelles in disease, aging, and development. Cell. 2006;125:1241–1252 [DOI] [PubMed] [Google Scholar]
- 13.Madamanchi NR, Runge MS. Mitochondrial dysfunction in atherosclerosis. Circ Res. 2007;100:460–473 [DOI] [PubMed] [Google Scholar]
- 14.Rodger CE, McWilliams TG, Ganley IG. Mammalian mitophagy - from in vitro molecules to in vivo models. FEBS J. 2018;285:1185–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lou G, Palikaras K, Lautrup S, Scheibye-Knudsen M, Tavernarakis N, Fang EF. Mitophagy and neuroprotection. Trends Mol Med. 2019 [DOI] [PubMed] [Google Scholar]
- 16.Miller S, Muqit MMK. Therapeutic approaches to enhance pink1/parkin mediated mitophagy for the treatment of parkinson’s disease. Neurosci Lett. 2019;705:7–13 [DOI] [PubMed] [Google Scholar]
- 17.Fang EF, Hou Y, Palikaras K, Adriaanse BA, Kerr JS, Yang B, Lautrup S, Hasan-Olive MM, Caponio D, Dan X, Rocktaschel P, Croteau DL, Akbari M, Greig NH, Fladby T, Nilsen H, Cader MZ, Mattson MP, Tavernarakis N, Bohr VA. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of alzheimer’s disease. Nat Neurosci. 2019;22:401–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cummins N, Tweedie A, Zuryn S, Bertran-Gonzalez J, Gotz J. Disease-associated tau impairs mitophagy by inhibiting parkin translocation to mitochondria. EMBO J. 2019;38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dai Y, Hu X, Sun X. Overexpression of parkin protects retinal ganglion cells in experimental glaucoma. Cell Death Dis. 2018;9:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Swiader A, Nahapetyan H, Faccini J, D’Angelo R, Mucher E, Elbaz M, Boya P, Vindis C. Mitophagy acts as a safeguard mechanism against human vascular smooth muscle cell apoptosis induced by atherogenic lipids. Oncotarget. 2016;7:28821–28835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ritter M, Buechler C, Boettcher A, Barlage S, Schmitz-Madry A, Orso E, Bared SM, Schmiedeknecht G, Baehr CH, Fricker G, Schmitz G. Cloning and characterization of a novel apolipoprotein a-i binding protein, ai-bp, secreted by cells of the kidney proximal tubules in response to hdl or apoa-i. Genomics. 2002;79:693–702 [DOI] [PubMed] [Google Scholar]
- 22.Fang L, Choi SH, Baek JS, Liu C, Almazan F, Ulrich F, Wiesner P, Taleb A, Deer E, Pattison J, Torres-Vazquez J, Li AC, Miller YI. Control of angiogenesis by aibp-mediated cholesterol efflux. Nature. 2013;498:118–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schneider DA, Choi SH, Agatisa-Boyle C, Zhu L, Kim J, Pattison J, Sears DD, Gordts P, Fang L, Miller YI. Aibp protects against metabolic abnormalities and atherosclerosis. J Lipid Res. 2018;59:854–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marbaix AY, Noel G, Detroux AM, Vertommen D, Van Schaftingen E, Linster CL. Extremely conserved atp- or adp-dependent enzymatic system for nicotinamide nucleotide repair. J Biol Chem. 2011;286:41246–41252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shumilin IA, Cymborowski M, Chertihin O, Jha KN, Herr JC, Lesley SA, Joachimiak A, Minor W. Identification of unknown protein function using metabolite cocktail screening. Structure. 2012;20:1715–1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang M, Zhao GJ, Yao F, Xia XD, Gong D, Zhao ZW, Chen LY, Zheng XL, Tang XE, Tang CK. Aibp reduces atherosclerosis by promoting reverse cholesterol transport and ameliorating inflammation in apoe(−/−) mice. Atherosclerosis. 2018;273:122–130 [DOI] [PubMed] [Google Scholar]
- 27.Woller SA, Choi SH, An EJ, Low H, Schneider DA, Ramachandran R, Kim J, Bae YS, Sviridov D, Corr M, Yaksh TL, Miller YI. Inhibition of neuroinflammation by aibp: Spinal effects upon facilitated pain states. Cell Rep. 2018;23:2667–2677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choi SH, Wallace AM, Schneider DA, Burg E, Kim J, Alekseeva E, Ubags ND, Cool CD, Fang L, Suratt BT, Miller YI. Aibp augments cholesterol efflux from alveolar macrophages to surfactant and reduces acute lung inflammation. JCI Insight. 2018; 3:e120519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marbaix AY, Tyteca D, Niehaus TD, Hanson AD, Linster CL, Van Schaftingen E. Occurrence and subcellular distribution of the nadphx repair system in mammals. Biochem J. 2014;460:49–58 [DOI] [PubMed] [Google Scholar]
- 30.Mao R, Meng S, Gu Q, Araujo-Gutierrez R, Kumar S, Yan Q, Almazan F, Youker KA, Fu Y, Pownall HJ, Cooke JP, Miller YI, Fang L. Aibp limits angiogenesis through gamma-secretase-mediated upregulation of notch signaling. Circ Res. 2017;120:1727–1739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsimikas S, Miyanohara A, Hartvigsen K, Merki E, Shaw PX, Chou MY, Pattison J, Torzewski M, Sollors J, Friedmann T, Lai NC, Hammond HK, Getz GS, Reardon CA, Li AC, Banka CL, Witztum JL. Human oxidation-specific antibodies reduce foam cell formation and atherosclerosis progression. J Am Coll Cardiol. 2011;58:1715–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chou MY, Fogelstrand L, Hartvigsen K, Hansen LF, Woelkers D, Shaw PX, Choi J, Perkmann T, Backhed F, Miller YI, Horkko S, Corr M, Witztum JL, Binder CJ. Oxidation-specific epitopes are dominant targets of innate natural antibodies in mice and humans. J Clin Invest. 2009;119:1335–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sawka-Verhelle D, Escoubet-Lozach L, Fong AL, Hester KD, Herzig S, Lebrun P, Glass CK. Pe-1/mets, an antiproliferative ets repressor factor, is induced by creb-1/crem-1 during macrophage differentiation. J Biol Chem. 2004;279:17772–17784 [DOI] [PubMed] [Google Scholar]
- 34.Morita S, Kojima T, Kitamura T. Plat-e: An efficient and stable system for transient packaging of retroviruses. Gene Ther. 2000;7:1063–1066 [DOI] [PubMed] [Google Scholar]
- 35.Bae YS, Lee JH, Choi SH, Kim S, Almazan F, Witztum JL, Miller YI. Macrophages generate reactive oxygen species in response to minimally oxidized low-density lipoprotein: Toll-like receptor 4- and spleen tyrosine kinase-dependent activation of nadph oxidase 2. Circ Res. 2009;104:210–218, 221p following 218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun N, Malide D, Liu J, Rovira II, Combs CA, Finkel T. A fluorescence-based imaging method to measure in vitro and in vivo mitophagy using mt-keima. Nat Protoc. 2017;12:1576–1587 [DOI] [PubMed] [Google Scholar]
- 37.Miller YI, Tsimikas S. Oxidation-specific epitopes as targets for biotheranostic applications in humans: Biomarkers, molecular imaging and therapeutics. Curr Opin Lipidol. 2013;24:426–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nagy L, Tontonoz P, Alvarez JG, Chen H, Evans RM. Oxidized ldl regulates macrophage gene expression through ligand activation of ppargamma. Cell. 1998;93:229–240 [DOI] [PubMed] [Google Scholar]
- 39.Feng J, Han J, Pearce SF, Silverstein RL, Gotto AM, Jr., Hajjar DP, Nicholson AC. Induction of cd36 expression by oxidized ldl and il-4 by a common signaling pathway dependent on protein kinase c and ppar-gamma. J Lipid Res. 2000;41:688–696 [PubMed] [Google Scholar]
- 40.Basu D, Hu Y, Huggins LA, Mullick AE, Graham MJ, Wietecha T, Barnhart S, Mogul A, Pfeiffer K, Zirlik A, Fisher EA, Bornfeldt KE, Willecke F, Goldberg IJ. Novel reversible model of atherosclerosis and regression using oligonucleotide regulation of the ldl receptor. Circ Res. 2018;122:560–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li Q, Yin Y, Zheng Y, Chen F, Jin P. Inhibition of autophagy promoted high glucose/ros-mediated apoptosis in adscs. Stem Cell Res Ther. 2018;9:289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li L, Tan J, Miao Y, Lei P, Zhang Q. Ros and autophagy: Interactions and molecular regulatory mechanisms. Cell Mol Neurobiol. 2015;35:615–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Basit F, van Oppen LM, Schockel L, Bossenbroek HM, van Emst-de Vries SE, Hermeling JC, Grefte S, Kopitz C, Heroult M, Hgm Willems P, Koopman WJ. Mitochondrial complex i inhibition triggers a mitophagy-dependent ros increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis. 2017;8:e2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fan P, Xie XH, Chen CH, Peng X, Zhang P, Yang C, Wang YT. Molecular regulation mechanisms and interactions between reactive oxygen species and mitophagy. DNA Cell Biol. 2019;38:10–22 [DOI] [PubMed] [Google Scholar]
- 45.Shefa U, Jeong NY, Song IO, Chung HJ, Kim D, Jung J, Huh Y. Mitophagy links oxidative stress conditions and neurodegenerative diseases. Neural Regen Res. 2019;14:749–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jha KN, Shumilin IA, Digilio LC, Chertihin O, Zheng H, Schmitz G, Visconti PE, Flickinger CJ, Minor W, Herr JC. Biochemical and structural characterization of apolipoprotein a-i binding protein, a novel phosphoprotein with a potential role in sperm capacitation. Endocrinology. 2008;149:2108–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rojo M, Legros F, Chateau D, Lombes A. Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane gtpase fzo. J Cell Sci. 2002;115:1663–1674 [DOI] [PubMed] [Google Scholar]
- 48.Eura Y, Ishihara N, Yokota S, Mihara K. Two mitofusin proteins, mammalian homologues of fzo, with distinct functions are both required for mitochondrial fusion. J Biochem. 2003;134:333–344 [DOI] [PubMed] [Google Scholar]
- 49.Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins mfn1 and mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160:189–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW. Mitofusin 1 and mitofusin 2 are ubiquitinated in a pink1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet. 2010;19:4861–4870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Glauser L, Sonnay S, Stafa K, Moore DJ. Parkin promotes the ubiquitination and degradation of the mitochondrial fusion factor mitofusin 1. J Neurochem. 2011;118:636–645 [DOI] [PubMed] [Google Scholar]
- 52.McLelland GL, Goiran T, Yi W, Dorval G, Chen CX, Lauinger ND, Krahn AI, Valimehr S, Rakovic A, Rouiller I, Durcan TM, Trempe JF, Fon EA. Mfn2 ubiquitination by pink1/parkin gates the p97-dependent release of er from mitochondria to drive mitophagy. Elife. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gu Q, Yang X, Lv J, Zhang J, Xia B, Kim JD, Wang R, Xiong F, Meng S, Clements TP, Tandon B, Wagner DS, Diaz MF, Wenzel PL, Miller YI, Traver D, Cooke JP, Li W, Zon LI, Chen K, Bai Y, Fang L. Aibp-mediated cholesterol efflux instructs hematopoietic stem and progenitor cell fate. Science. 2019;363:1085–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dubrovsky L, Ward A, Choi SH, Pushkarsky T, Brichacek B, Vanpouille C, Adzhubei AA, Mukhamedova N, Sviridov D, Margolis L, Jones RB, Miller YI, Bukrinsky M. Inhibition of hiv replication by apolipoprotein a-i binding protein targeting the lipid rafts. mBio. 2020;11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Choi SH, Kim KY, Perkins GA, Phan S, Edwards G, Xia Y, Kim J, Skowronska-Krawczyk D, Weinreb RN, Ellisman MH, Miller YI, Ju WK. Aibp protects retinal ganglion cells against neuroinflammation and mitochondrial dysfunction in glaucomatous neurodegeneration. Redox Biol. 2020;37:101703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Niehaus TD, Elbadawi-Sidhu M, Huang L, Prunetti L, Gregory JF 3rd, de Crecy-Lagard V, Fiehn O, Hanson AD. Evidence that the metabolite repair enzyme nad(p)hx epimerase has a moonlighting function. Biosci Rep. 2018;38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fang L, Miller YI. Regulation of lipid rafts, angiogenesis and inflammation by aibp. Curr Opin Lipidol. 2019;30:218–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kubli DA, Gustafsson AB. Mitochondria and mitophagy: The yin and yang of cell death control. Circ Res. 2012;111:1208–1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu L, Sakakibara K, Chen Q, Okamoto K. Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res. 2014;24:787–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Leibundgut G, Witztum JL, Tsimikas S. Oxidation-specific epitopes and immunological responses: Translational biotheranostic implications for atherosclerosis. Curr Opin Pharmacol. 2013;13:168–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.von Kleist-Retzow JC, Cormier-Daire V, de Lonlay P, Parfait B, Chretien D, Rustin P, Feingold J, Rotig A, Munnich A. A high rate (20%-30%) of parental consanguinity in cytochrome-oxidase deficiency. Am J Hum Genet. 1998;63:428–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kirby DM, Crawford M, Cleary MA, Dahl HH, Dennett X, Thorburn DR. Respiratory chain complex i deficiency: An underdiagnosed energy generation disorder. Neurology. 1999;52:1255–1264 [DOI] [PubMed] [Google Scholar]
- 63.Scaglia F, Towbin JA, Craigen WJ, Belmont JW, Smith EO, Neish SR, Ware SM, Hunter JV, Fernbach SD, Vladutiu GD, Wong LJ, Vogel H. Clinical spectrum, morbidity, and mortality in 113 pediatric patients with mitochondrial disease. Pediatrics. 2004;114:925–931 [DOI] [PubMed] [Google Scholar]
- 64.Mayr JA, Meierhofer D, Zimmermann F, Feichtinger R, Kogler C, Ratschek M, Schmeller N, Sperl W, Kofler B. Loss of complex i due to mitochondrial DNA mutations in renal oncocytoma. Clin Cancer Res. 2008;14:2270–2275 [DOI] [PubMed] [Google Scholar]
- 65.Kremer LS, Danhauser K, Herebian D, Petkovic Ramadza D, Piekutowska-Abramczuk D, Seibt A, Muller-Felber W, Haack TB, Ploski R, Lohmeier K, Schneider D, Klee D, Rokicki D, Mayatepek E, Strom TM, Meitinger T, Klopstock T, Pronicka E, Mayr JA, Baric I, Distelmaier F, Prokisch H. Naxe mutations disrupt the cellular nad(p)hx repair system and cause a lethal neurometabolic disorder of early childhood. Am J Hum Genet. 2016;99:894–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Trinh J, Imhoff S, Dulovic-Mahlow M, Kandaswamy KK, Tadic V, Schafer J, Dobricic V, Nolte A, Werber M, Rolfs A, Munchau A, Klein C, Lohmann K, Bruggemann N. Novel naxe variants as a cause for neurometabolic disorder: Implications for treatment. J Neurol. 2019 [DOI] [PubMed] [Google Scholar]
- 67.Yu D, Zhao FM, Cai XT, Zhou H, Cheng Y. [clinical and genetic features of early-onset progressive encephalopathy associated with naxe gene mutations]. Zhongguo Dang Dai Er Ke Za Zhi. 2018;20:524–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rub C, Wilkening A, Voos W. Mitochondrial quality control by the pink1/parkin system. Cell Tissue Res. 2017;367:111–123 [DOI] [PubMed] [Google Scholar]
- 69.Schrepfer E, Scorrano L. Mitofusins, from mitochondria to metabolism. Mol Cell. 2016;61:683–694 [DOI] [PubMed] [Google Scholar]
- 70.Formosa LE, Ryan MT. Mitochondrial fusion: Reaching the end of mitofusin’s tether. J Cell Biol. 2016;215:597–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Filadi R, Pendin D, Pizzo P. Mitofusin 2: From functions to disease. Cell Death Dis. 2018;9:330. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.