

Abstract
Objective –
Hyperleptinemia, hallmark of obesity, is a putative pathophysiologic trigger for atherosclerosis. We previously reported a stimulatory effect of leptin on thrombospondin-1 (TSP-1) expression, a proatherogenic matricellular protein implicated in atherogenesis. However, a causal role of TSP-1 in leptin-driven atherosclerosis remains unknown.
Approach and Results –
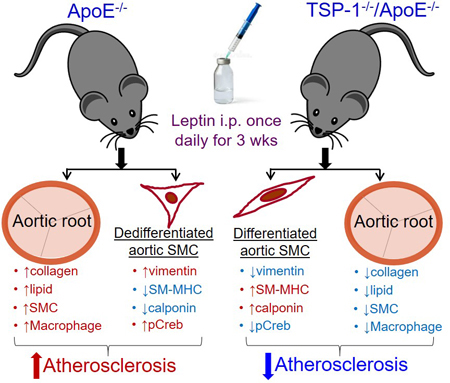
17-wks-old ApoE−/− and TSP-1−/−/ApoE−/− double knockout (dKO) mice, on normocholesterolemic diet, were treated with or without murine recombinant leptin (5µg/g bwt, IP) once daily for three weeks. Using aortic root morphometry and en-face lesion assay, we found that TSP-1 deletion abrogated leptin-stimulated lipid-filled lesion burden, plaque area and collagen accumulation in aortic roots of ApoE−/− mice, shown via Oil red O, H & E and Masson’s trichrome staining, respectively. Immunofluorescence microscopy of aortic roots showed that TSP-1 deficiency blocked leptin-induced inflammatory and smooth muscle cell abundance as well as cellular proliferation in ApoE−/− mice. Moreover, these effects were concomitant to changes in VLDL-triglyceride and HDL-cholesterol levels. Immunoblotting further revealed reduced vimentin and pCREB accompanied with augmented SM-MHC expression in aortic vessels of leptin-treated dKO vs. leptin-treated ApoE−/−; also confirmed in aortic SMCs from the mice genotypes, incubated ± leptin in vitro. Finally, TSP-1 deletion impeded plaque burden in leptin-treated ApoE−/− on western diet, independent of plasma lipid alterations.
Conclusions –
The present study provides evidence for a protective effect of TSP-1 deletion on leptin-stimulated atherogenesis. Our findings suggest a regulatory role of TSP-1 on leptin-induced VSMC phenotypic transition and inflammatory lesion invasion. Collectively, these results underscore TSP-1 as a potential target of leptin-induced vasculopathy.
Keywords: leptin, thrombospondin-1, atherosclerosis, vascular smooth muscle cells, SMC phenotypic switching
Graphical Abstract
Introduction
Obesity is a major risk factor for cardiovascular complications1, accounting for increased morbidity and mortality in obese individuals. Increasing adipose tissue mass is linked to elevated body mass index in obesity, which accompanies the secretion of a number of peptide hormones collectively termed ‘adipokines’. One such protein is the 16-kDa hormone, leptin, produced by the white adipose tissue2. Previous studies3 have reported that elevated plasma leptin levels associate with the carotid artery intima-media thickness, an early marker of atherosclerosis. Multiple lines of evidence indicate that hyperleptinemia, a hallmark of obesity, is a putative pathophysiological trigger for atherosclerotic vascular disease4, 5. Clinical and animal studies have shown increased expression of leptin receptor in atherosclerotic plaques; interaction of leptin with its receptor constitutes the first step in leptin-induced atheroma formation6. While recombinant leptin administration increased lesion area in ApoE−/− mice7, a classical mouse model of atherosclerosis, leptin-deficient hyperlipidemic ob/ob;ApoE−/− mice developed significantly reduced lesions8. Collectively, these data support the notion that leptin promotes atherosclerosis. However, mechanism(s) responsible for leptin-induced atherosclerosis are incompletely understood.
Numerous in vivo and in vitro studies have documented multiple proatherogenic effects of leptin, including increased migration and proliferation of vascular smooth muscle cells (VSMC)9, 10, endothelial cell dysfunction11, enhanced platelet aggregation12 and augmented reactive oxygen species formation9. Previous studies have also shown that leptin modulates the expression of several vascular genes associated with atherosclerosis and abnormal angiogenesis, including cytokines, growth factors and extracellular matrix proteins13–15. We previously reported that leptin, at pathophysiological concentrations relevant to obesity, upregulates a potent proatherogenic and anti-angiogenic protein thrombospondin-1 (TSP-1) expression in human and mouse aortic SMC16.
TSP-1 is a multifunctional glycoprotein belonging to a family of matricellular proteins. It is well appreciated that TSP-1 interaction with cell-surface receptors and matrix-binding partners modulates numerous biological processes including cell adhesion, migration, proliferation, angiogenesis, inflammation and atherosclerosis17, 18. Earlier studies have shown increased TSP-1 expression in response to vascular injury and in atherosclerotic lesions19, 20, with elevated expression in VSMC21. The current state of knowledge about the TSP protein family in atherosclerosis is based upon previous genetic association studies linking single nucleotide polymorphisms in TSP genes with coronary artery disease and myocardial infarction22. Growing evidence indicate that diabetic and obese patients and animals exhibit increased TSP-1 expression in the plasma and visceral adipose tissue, promoting adipose inflammatory response, insulin resistance and metabolic dysfunction23–25. Accumulating literature further demonstrate augmented TSP-1 expression in the heart, vasculature and kidneys of diabetic and obese patients and rodent models26–28. Despite these associations, a causal role of TSP-1 in leptin-induced atherosclerosis remains unexplored.
The objective of the current study was to investigate whether TSP-1 plays a direct role in development of atherosclerotic lesions triggered by exogenous leptin in ApoE−/− mice. Our data demonstrate a protective role of TSP-1 deletion in leptin-stimulated atherogenesis.
Material and Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Mice
All animal treatments and euthanasia procedures were performed in accordance with protocols annually approved by the Institutional Animal Care and Use Committee (IACUC) at Northeast Ohio Medical University (NEOMED). ApoE−/− (stock # 002052) and TSP-1−/− (stock # 006141) mice on C57BL/6J background purchased from The Jackson Laboratory (Bar Harbor, ME) were cross-bred in our animal facility. TSP-1−/−/ApoE−/− double knockout (dKO) mice in F2 generation were utilized in this study. ApoE−/− and TSP-1−/− genotypes were confirmed by PCR, per established protocols (JAX). Animals were housed in pathogen-free environment and kept on 12h:12h light/dark cycle.
Study Design
The design of the present study including procedures for lesion measurement and quantification adhered to the guidelines for experimental atherosclerosis described in the AHA Statement29. Male and female age-matched ApoE−/− and TSP-1−/−/ApoE−/− littermates, weaned at 4 weeks, were maintained on standard laboratory diet (Purina LabDiet 5008) ad libitum until 20 weeks age (study endpoint); animals were weighed weekly. Beginning at 17 weeks of age, mice received IP injections of murine recombinant leptin (5 µg/g bwt, R&D systems) or PBS (vehicle control), once daily for three consecutive weeks. In a concurrent study, 6 weeks old male mice genotypes were placed on Western diet (TD 88137, Teklad, Harlan) until 12 weeks age (study endpoint). Beginning at 9 weeks, leptin or PBS was injected IP once daily for three weeks. Mice were euthanized at study endpoints using fatal plus, per institutionally approved methods; blood and tissue samples were collected and utilized as described in the expanded Methods section in Data Supplement. Briefly, plasma was isolated from blood samples and used to measure total cholesterol and total triglyceride levels using standard enzymatic methods30; cholesterol and triglyceride distribution among plasma lipoproteins was measured using fast-performance liquid chromatography (FPLC)31. The aorta was processed for en-face atherosclerotic lesion assay; aortic root serial sections derived from each mouse were used for morphometric and immunohistochemistry studies. Additionally, vascular lesions were monitored non-invasively using the Vevo 770 High-frequency Ultrasound Imaging System (VisualSonics, Inc. Toronto, Canada). Further, aortic tissue lysates prepared from harvested aortae were used in immunoblotting30. The dosage of leptin was selected based on published findings32, including our previous work16.
Statistical Analyses
Results are expressed as Mean ± SEM. Statistical analyses were performed using GraphPad Prism 6 and Excel’s Real Statistics Resource pack. Prior to analyses, each data set was tested for normality (Shapiro-Wilks; D’Agostino & Pearson; Kolmogorov-Smirnov; Cramer-von Mises; Ryan-Joiner) and homoscedasticity (Levene; Bartlett) on the original scale or after logarithmic or square root data transformation. Parametric methods were applied when assumption of normality was met, whereas non-parametric methods were employed if the assumption was violated. Ordinary one-way ANOVA was used for normally distributed data with similar variance followed by Tukey HSD post-hoc test. For normally distributed data with unequal variance, Welch ANOVA was applied followed by Games-Howell post-hoc test. Mann-Whitney and Kruskal-Wallis were used as the non-parametric counterparts of student’s t-test and one-way ANOVA, respectively; Kruskal-Wallis was followed by the Dunn’s post-hoc test. Statistical significance was considered at p ≤ 0.05.
For further information, please see the Major Resources Table provided in the Data Supplement. In addition, an expanded Methods Section is included in the Data Supplement.
Results
TSP-1 knockout inhibits leptin-induced lesion burden and collagen accumulation in ApoE−/− mice
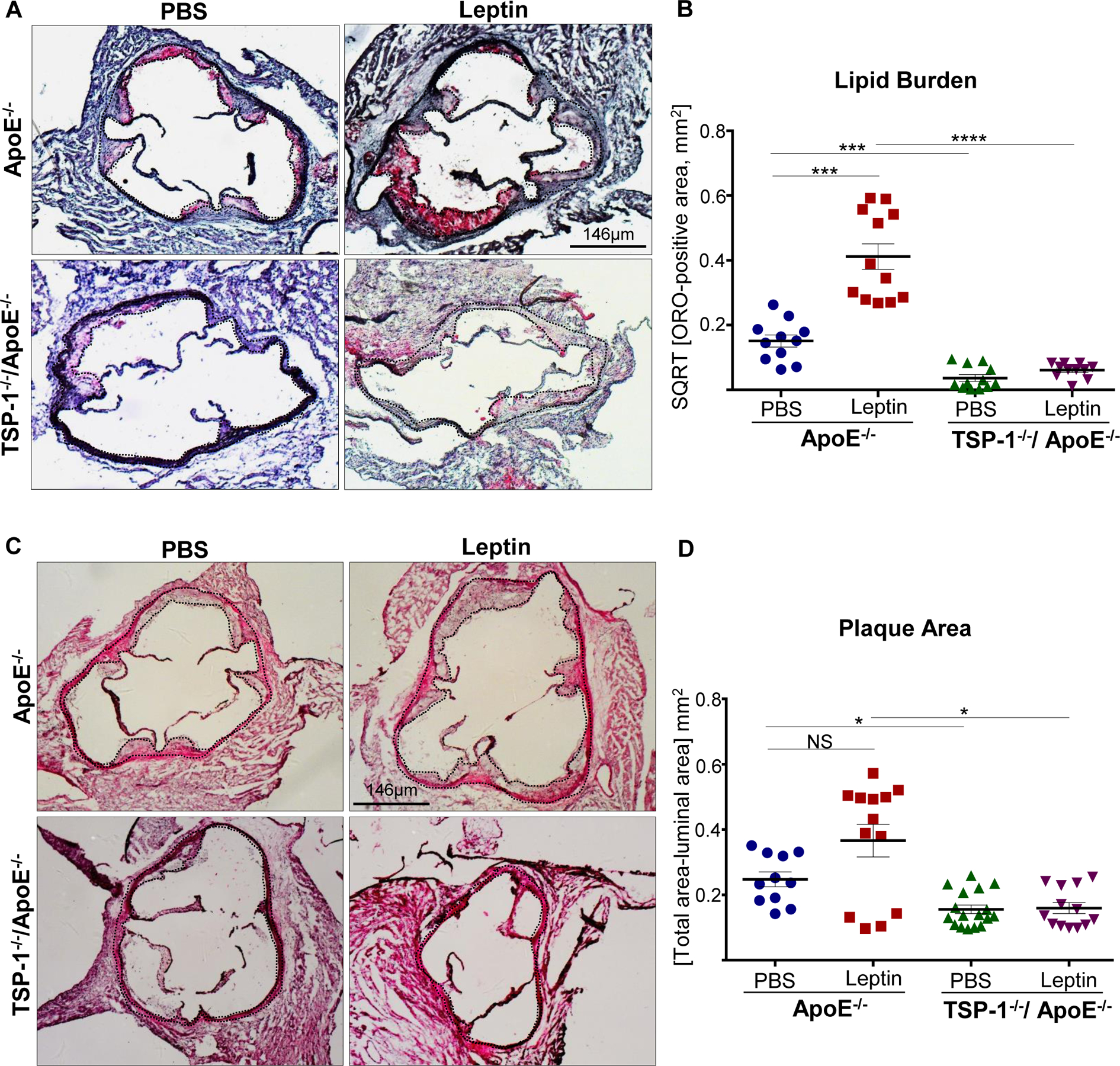
Using aortic root morphometry, we examined atherosclerotic lesion formation in ApoE−/− and TSP-1−/−/ApoE−/− mice following leptin (or PBS) administration. ORO staining of aortic root sections revealed robust lipid-filled lesions in leptin-treated ApoE−/− mice. In contrast, lipid burden was significantly attenuated in root sections of leptin-treated TSP-1−/−/ApoE−/− mice (Figure 1A). Quantification of ORO-positive area showed that exogenous leptin increased lipid burden in both male and female ApoE−/− mice (2.3- and 7.9-fold respectively vs. PBS-treated ApoE−/−), whereas lipid-filled lesions were significantly reduced (>65%) in leptin-treated dKOs; this effect was observed in both sex (Figure 1B and 1C). Interestingly, while lipid burden was decreased in untreated male TSP-1−/−/ApoE−/− (74% vs. untreated ApoE−/− mice), lack of TSP-1 had no effect on lipid burden in female ApoE−/− under basal conditions. Importantly, only untreated ApoE−/− mice revealed sex-specific differences in ORO-positive lipid area (p<0.0001). Next, we assessed plaque area using H & E staining. Leptin-treated ApoE−/− showed increased plaque area compared to PBS-treated ApoE−/− mice. On the other hand, aortic root lesion area was much smaller in TSP-1−/−/ApoE−/− mice treated with leptin (Figure 2A). Specifically, while we found a significant increase in plaque area in both male and female ApoE−/− treated with leptin (p<0.017), these values were markedly diminished in leptin-treated dKOs (p<0.005), irrespective of sex (Figure 2B and 2C). Further, evaluation of aortic root collagen content using Masson’s trichrome staining revealed sex-specific differences between the mice genotypes. Specifically, leptin increased collagen accumulation in ApoE−/− mice (1.4-fold vs. PBS-treated ApoE−/−) while there was a significant decrease in collagen deposition in leptin-treated TSP-1−/−/ApoE−/− (19%, p=.0015). Notably, these differences in collagen content were observed only in the male genotypes (Figure 3). Together, these data clearly demonstrate that TSP-1 deficiency impedes aortic root atherosclerotic lesion formation induced by exogenous leptin administration in ApoE−/− mice.
Figure 1. TSP-1 deletion prevents leptin-induced lipid burden in ApoE−/− mice.
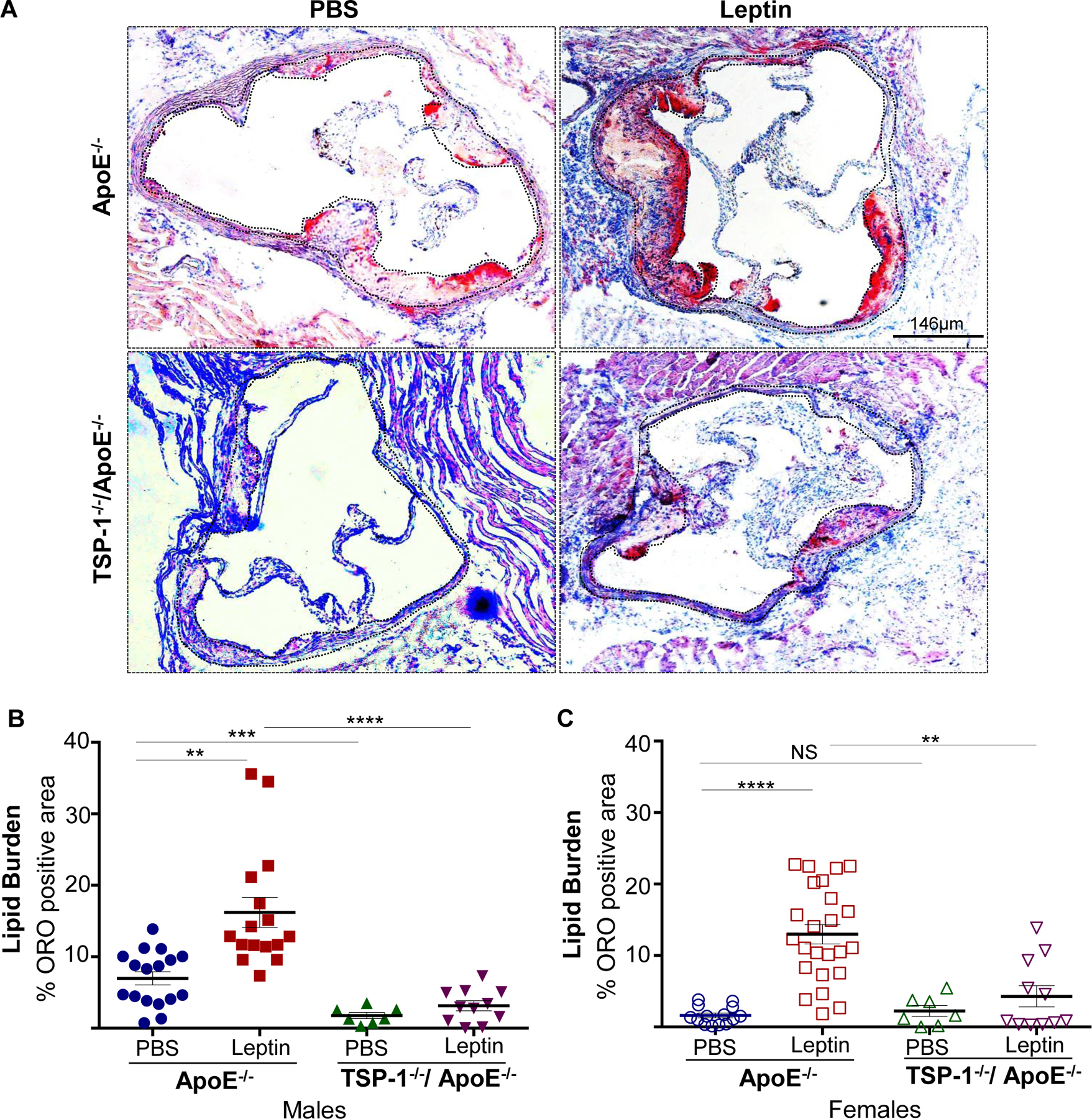
Male and female age-matched ApoE−/− and TSP-1−/−/ApoE−/− littermate mice weaned at 4wks were maintained on standard laboratory diet until 20wks of age. Beginning at 17 wks, mice received murine recombinant leptin (5µg/g) or PBS (vehicle control) IP once daily for three consecutive weeks. Serial sections of equivalent regions of the aortic root were stained with 0.5% ORO followed by hematoxylin counterstaining. All images were captured at 4X magnification. Total lesion area and ORO-positive area (measured in µm2) was quantified using Image J; lipid burden depicted via %ORO positive area. Shown are A) representative ORO-stained aortic root sections and summary data for %ORO positive area in B) male and C) female mice. (PBS-treated ApoE−/−: n=6; Leptin-treated ApoE−/−: n=6; PBS-treated TSP1−/−/ApoE−/−: n=3; Leptin-treated-TSP-1−/−/ApoE−/−: n=4; 2–6 sections/aortic root/group). Dotted line depicts region used for quantification; values denote lipid burden of aortic root per section in each study group; all values are expressed as mean ± SEM. **p<0.005; ***p<0.0005; ****p<0.0001; NS: not significant.
Figure 2. TSP-1 deletion inhibits leptin-induced increase in plaque area in ApoE−/− mice.
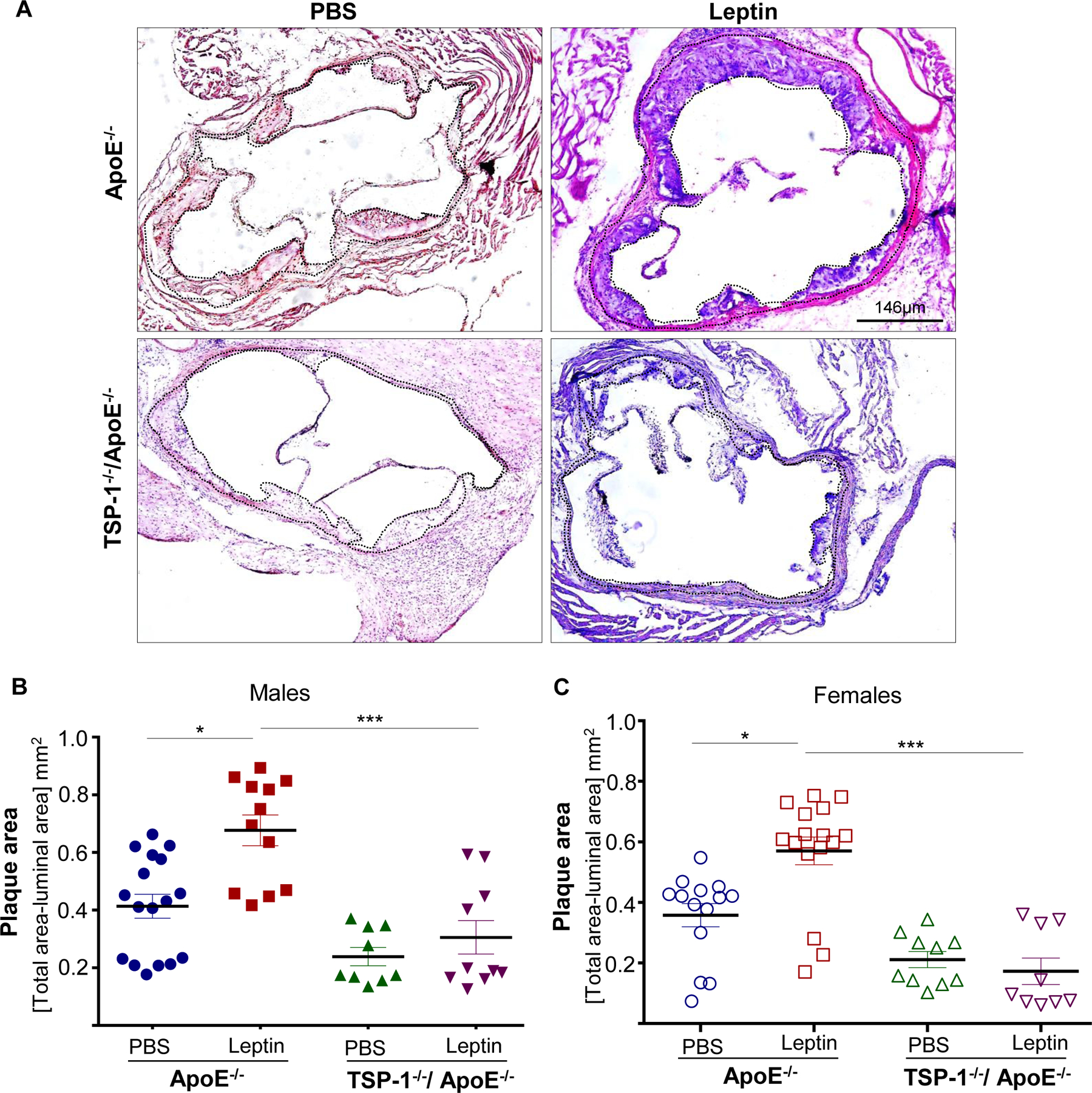
Mice were treated as described in Figure legend 1. Aortic root serial sections were stained with H & E. Aortic root luminal area (in mm2) and total aortic root area (i.e. area enclosed by the internal elastic lamina, in mm2) were measured using Image J; difference between total root area and luminal area denotes plaque area. Shown are A) representative images for H & E-staining (4X magnification) and summary data for plaque area in B) male and C) female mice genotypes. n=4 mice/sex per study group, 3–5 sections per aortic root. Black dotted line marks region used for quantification. Values denote plaque area of aortic root per section in each study group, expressed as mean ± SEM; For C) log-transformed data used for statistical analyses. *p≤0.05; ***p<0.0005
Figure 3. TSP-1 deletion reduces leptin-induced collagen deposition in ApoE−/− mice.
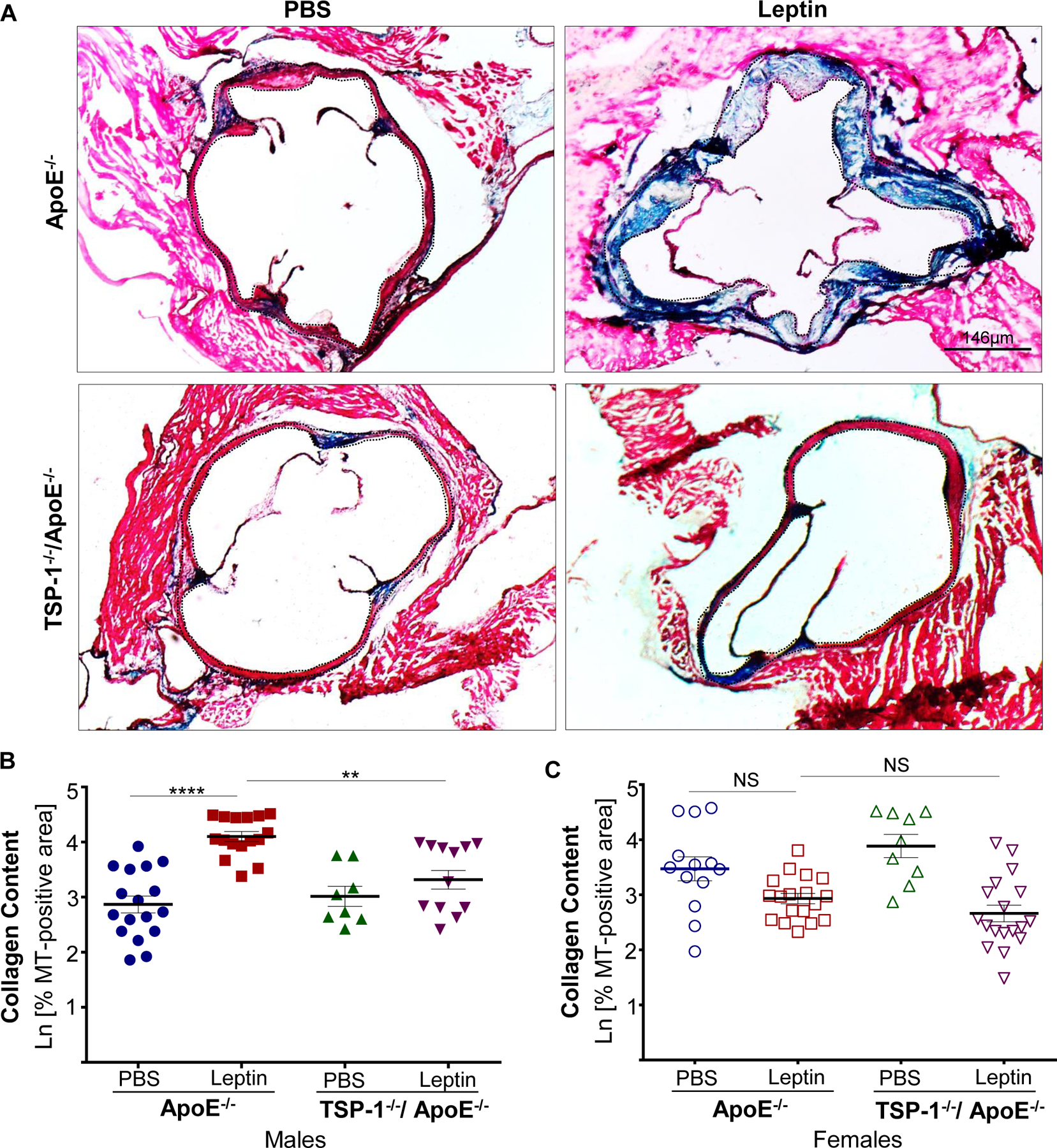
Mice were treated as described in Figure legend 1. Aortic root serial sections were stained with Masson’s trichrome followed by hematoxylin counterstaining. Total lesion area and MT-positive area within lesions (measured in sq. pixels) was quantified using Image J (4X magnification); collagen content is depicted as %MT positive area shown in natural log scale. Shown are A) representative images of MT-staining and summary data for %MT positive area in B) male and C) female mice (n=3–4 mice/sex per study group, 3–6 sections per aortic root). Dotted line depicts region used for quantification. Values denotes collagen content of aortic root per section in each study group; all values are expressed as mean ± SEM. **p<0.005; ****p<0.0001; NS: not significant.
TSP-1 deletion reduces leptin-induced plaque burden in aortic vasculature of ApoE−/− mice
Next, we investigated lesion formation using en face atherosclerotic lesion assay. Shown via ORO staining, we observed a remarkable increase in lipid deposition in the whole aorta of leptin-treated ApoE−/− mice. Notably, lipid-filled lesions were most abundant within the aortic arch of the vessel wall. On the contrary, lipid accumulation was profoundly reduced in leptin-treated TSP-1−/−/ApoE−/− mice. Specifically, quantitation of ORO-positive staining revealed ~4-fold increase in en-face lipid burden in leptin-treated ApoE−/− (p<0.05 vs. PBS-treated ApoE−/− mice). In contrast, lipid buildup was significantly attenuated in the aortic vasculature of leptin-treated dKOs (~86% vs. leptin-treated ApoE−/−, Supplemental Figure IA and IB). Of note, in the absence of exogenous leptin, TSP-1 deficiency had no effect on aortic arch plaque burden in ApoE−/− mice.
Lesion progression was further monitored non-invasively using High-frequency Ultrasound Imaging. Leptin reduced the internal diameters of the ascending aorta and transaortic arch in ApoE−/− mice. Specifically, aortic vessel internal diameter measured at 20 wks age (study endpoint) decreased by 15% in leptin-treated ApoE−/− (p≤0.05 vs. PBS-treated ApoE−/− mice). However, these effects were considerably blunted in leptin-treated dKOs (Supplemental Figure IC and ID). In line with these findings, ultrasound imaging of right common carotid artery (RCCA) revealed pronounced vascular lesions in leptin-treated ApoE−/− resulting in significant reduction in the vessel internal diameter, while leptin-treated TSP-1−/−/ApoE−/− mice developed relatively smaller carotid lesions. Specifically, at 20 wks age leptin-treated ApoE−/− showed reduced RCCA internal diameter vs. PBS-treated ApoE−/− (16%, p<0.05). On the contrary, RCCA internal diameter was greater in leptin-treated TSP-1−/−/ApoE−/− vs. leptin-treated ApoE−/−; notably, this increase in vessel diameter was reflective of a growth-related enhancement noted in dKO mice, compared with baseline measurements (i.e. prior to leptin at 17 wks age), that was abrogated in leptin-treated ApoE−/− (Supplemental Figure II). Taken together, these data clearly demonstrate that TSP-1 deficiency prevents atherosclerotic lesion formation triggered by leptin administration in ApoE−/− mice.
Effect of leptin in vivo on plasma lipid levels in ApoE−/− vs. TSP-1−/−/ApoE−/− mice
To interrogate the role of lipids in reduced lesion burden observed in leptin-treated TSP-1−/−/ApoE−/− mice, we measured plasma lipid profiles in the mice genotypes. Regardless of sex, we observed an upward trend in total cholesterol levels in leptin-treated ApoE−/− mice (22–32% vs. PBS-treated ApoE−/−), whereas total cholesterol was reduced by >35% in leptin-treated dKOs; however, these changes did not reach statistical significance (p>0.09, Figure 4A and 4B). Leptin increased total triglyceride content in both male and female ApoE−/− mice (2.8- and 1.7-fold respectively). On the contrary, triglyceride levels were markedly attenuated in leptin-treated TSP-1−/−/ApoE−/− mice (>50% vs. leptin-treated ApoE−/−); also, these effects were noted in both sex (Figure 4D and 4E). To examine the effect of TSP-1 deletion on plasma lipoprotein distribution, we performed FPLC plasma fractionation studies. Both VLDL- and LDL-cholesterol were minimally affected in leptin-treated TSP-1−/−/ApoE−/− vs. leptin-treated ApoE−/− mice. Moreover, while leptin increased VLDL-triglyceride in ApoE−/− mice, VLDL-triglyceride levels were profoundly reduced in leptin-treated dKOs (Figure 4C and 4F). To our surprise, HDL-cholesterol was elevated in leptin-treated ApoE−/− compared to PBS-treated genotypes as well as leptin-treated dKO mice (Figure 4C). Together, these results demonstrate that TSP-1 deletion modulates leptin-induced lipid distribution in ApoE−/− mice.
Figure 4. Effect of leptin on plasma cholesterol and triglyceride levels in ApoE−/− vs. TSP-1−/−/ApoE−/− mice.
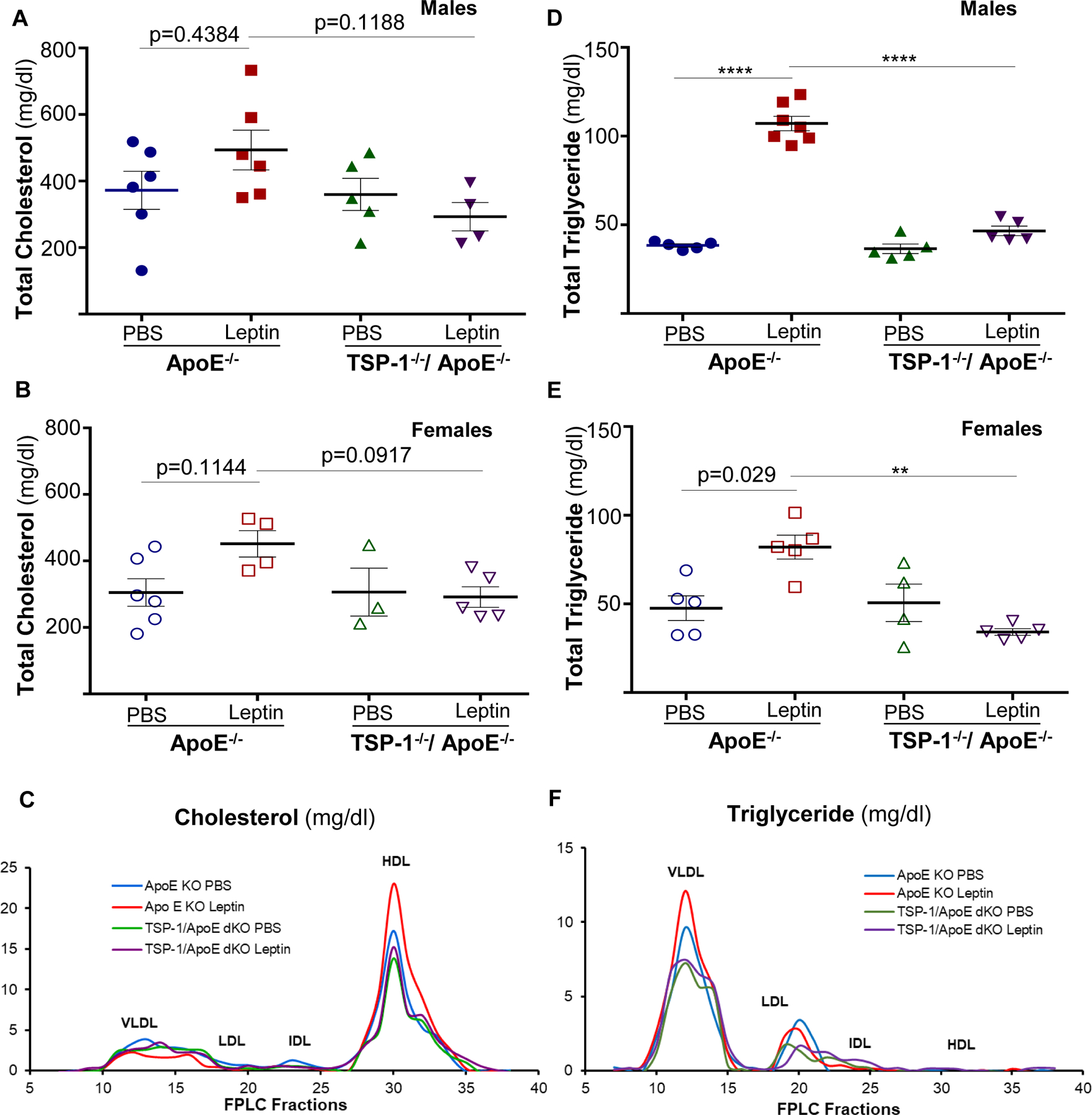
Mice were treated as described in Figure 1. At 20 wks age, plasma samples were utilized for measurement of total cholesterol (A, B), total triglyceride (D, E) and lipoprotein distribution (C, F) via FPLC fractionation (measured in pooled plasma from mice within each study group). For A, B, D and E: n=3–7 mice/sex in each study group) **p<0.005; ****p<0.0001
TSP-1 deficiency ameliorates leptin-induced increase in inflammatory and smooth muscle cell lesion burden in ApoE−/− mice
Immunohistochemistry demonstrated enhanced macrophage and leukocyte infiltration in aortic root lesions of leptin-treated ApoE−/− mice, depicted via CD68 and CD45 staining, respectively. On the other hand, inflammatory cell burden was lower in leptin-treated TSP-1−/−/ApoE−/− mice. Specifically, CD68 and CD45 expression were reduced by 25% and 18% in leptin-treated dKOs (p<0.0001 vs. leptin-treated ApoE−/−), while expression of these inflammatory markers increased >1.2-fold (p<0.0001) in leptin-treated ApoE−/− mice; these changes were observed in both male and female genotypes (Figure 5). To determine the impact of TSP-1 deletion on leptin-induced cell proliferation and SMC lesion abundance, we performed aortic root immunohistochemistry using PCNA and α-SMA antibodies. There was a robust increase in PCNA and α-SMA expression in aortic root lesions of leptin-treated ApoE−/− mice, while PCNA and α-SMA expression diminished in leptin-treated TSP-1−/−/ApoE−/− (Figure 6A). Specifically, immunofluorescence quantification revealed >2-fold increase in PCNA and α-SMA expression in leptin-treated ApoE−/− mice (p<0.002 vs. PBS-treated ApoE−/−). On the contrary, both PCNA and α-SMA expression were attenuated in leptin-treated dKOs (>65%, p<0.0001 vs. leptin-treated ApoE−/−), depicting lower proliferative response and reduced SMC content (Figure 6B and 6C). Importantly, co-immunostaining illustrated enhanced co-localization of α-SMA and PCNA in aortic root lesions of leptin-treated ApoE−/−, while these co-expression patterns were blunted in leptin-treated TSP-1−/−/ApoE−/− mice (Figure 6A-merged). Moreover, the observed changes in SMC abundance and cellular proliferation occurred in both male and female genotypes. Similar to our findings with collagen content and plaque area, effect of TSP-1 deletion on lesion inflammatory and SMC infiltration was noted only in the presence of leptin. In each case, parallel tissue sections incubated without corresponding primary antibodies showed negligible background staining, confirming the staining specificity for each antibody (Supplemental Figure III). Collectively, these data clearly demonstrate that TSP-1 deficiency abrogates leptin-induced inflammatory and SMC lesion invasion in ApoE−/− mice.
Figure 5. TSP-1 deletion abrogates leptin-induced inflammatory cell infiltration in ApoE−/− mice.
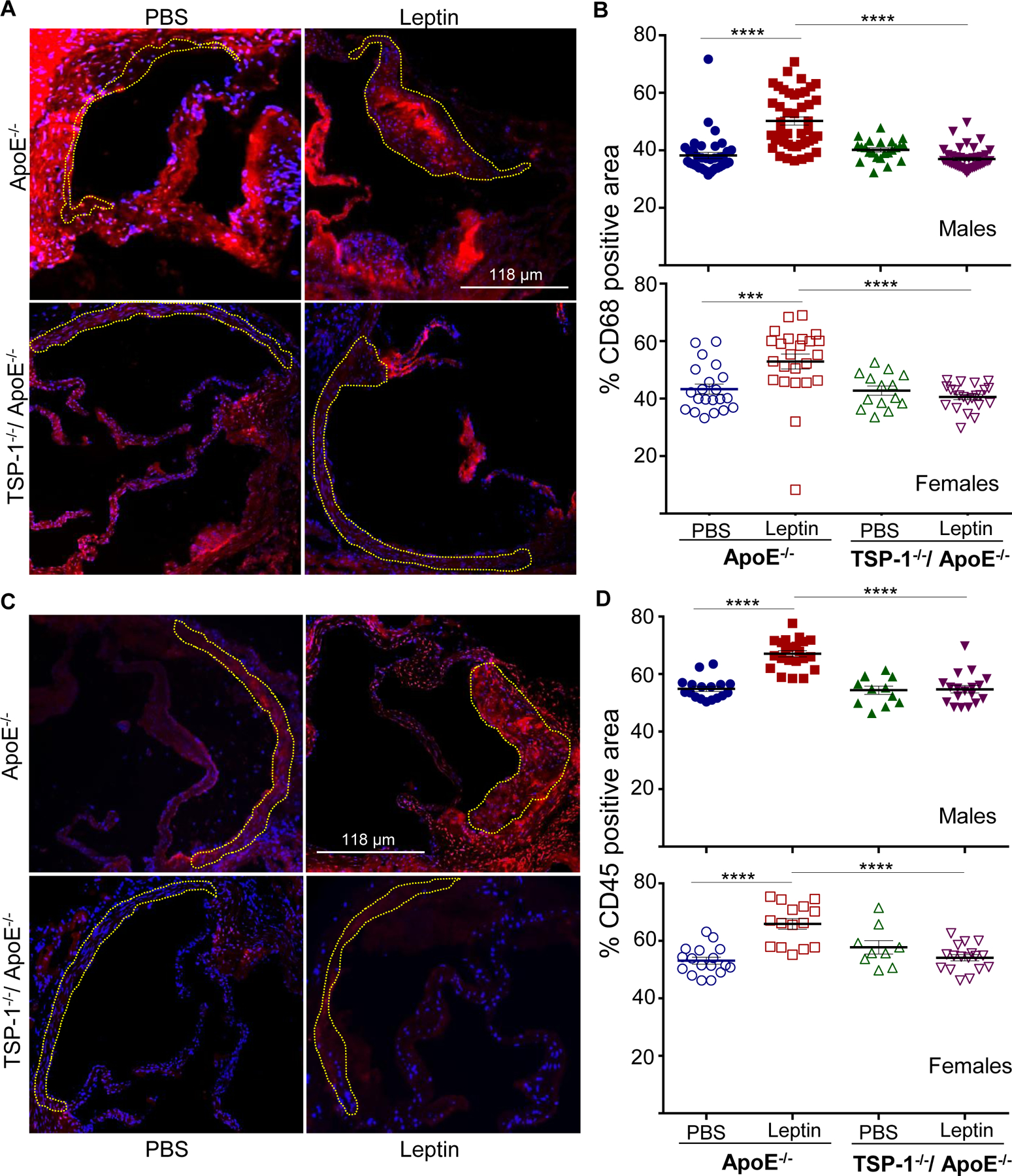
Mice were treated as described in Figure 1. Aortic root sections were subjected to immunohistochemistry using CD68 and CD45 antibodies. Total lesion area and positive staining area of lesions (in sq. pixels) was quantified in each section using Image J. Shown are representative images (10X magnification) and summary graphs for A and B) macrophage and C and D) leukocyte content (n=3–6 mice/sex in each study group; 3–7 sections/aortic root). Yellow dotted line marks region used for quantification. Shown are %CD68 or %CD45 positive area of total lesion per section in each study group; all values are expressed as mean ± SEM; ***p<0.0005; ****p<0.0001
Figure 6. TSP-1 deficiency blocks leptin-induced increase in smooth muscle cell and proliferative cell abundance in ApoE−/− mice.
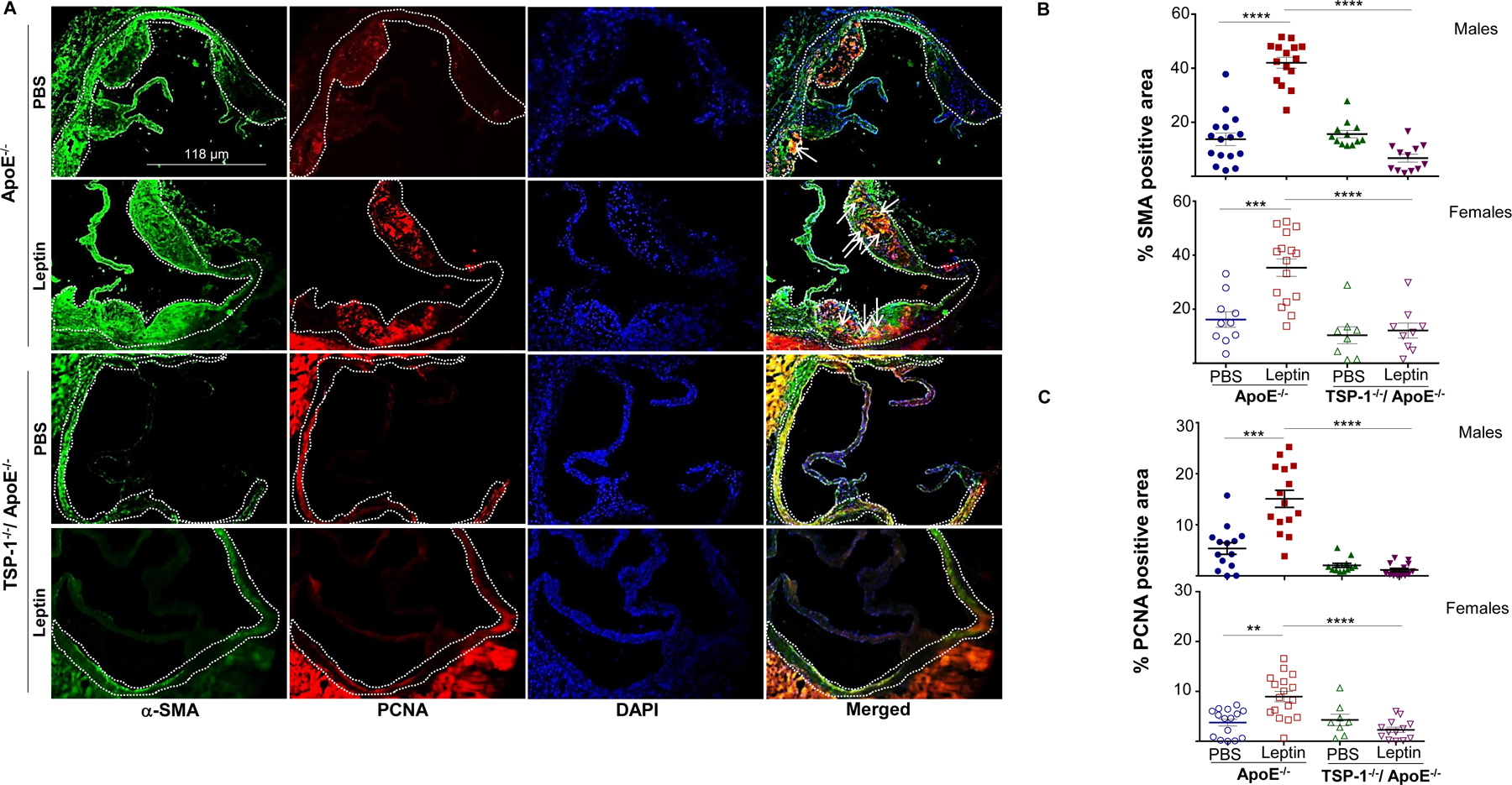
Mice were treated as described in Figure 1. Aortic root sections were used in double immunohistochemistry with α-SMA (SMC marker) and PCNA (proliferation marker) antibodies. A) Shown are representative images for α-SMA (green) and PCNA (red) staining; merge (yellow) shows α-SMA and PCNA co-localization; DAPI denotes nuclei staining (magnification 10X). White dotted line marks region used in quantification; arrows indicate PCNA-positive SMC (yellow). Summary data for B) SMC and C) proliferative cell content are shown (n=3–6 mice/sex in each study group; 3–7 sections/aortic root). Data represents %SMA or %PCNA positive area of total lesion per section in each study group; values are expressed as mean ± SEM; **p<0.005; ***p<0.0005; ****p<0.0001
TSP-1 deletion blocks leptin-induced VSMC phenotypic switching in ApoE−/− mice
To elucidate the role of TSP-1 in VSMC phenotypic switching in response to leptin, we next analysed aortic tissue lysates from the mice genotypes using immunoblotting (Figure 7). Consistent with earlier findings16, leptin in vivo significantly increased TSP-1 expression (>7-fold) in aortic vessels of ApoE−/− mice (p=0.023 vs. PBS-treated ApoE−/−). As expected, TSP-1 expression was undetected in aortic lysates of TSP-1−/−/ApoE−/− mice treated with or without leptin. Elevated TSP-1 expression was accompanied with augmented PCNA expression in leptin-treated ApoE−/− (5-fold, p=0.06). In addition, leptin significantly enhanced vimentin expression (~5-fold) in the aortic vasculature of ApoE−/− mice. In contrast, vimentin expression was reduced (~56%, p=0.05) accompanied with augmented SM-MHC expression (1.8-fold, p=0.015) in leptin-treated TSP-1−/−/ApoE−/−. These results were further validated in vitro using aSMC primary cultures isolated from ApoE−/− and TSP-1−/−/ApoE−/− mice. Specifically, in aSMC from ApoE−/−, incubation with leptin (at concentrations mimicking hyperleptinemia) increased TSP-1 expression (>3-fold, p=0.01) accompanied with enhanced vimentin expression (>2-fold, p=0.04) vs. untreated cells. On the other hand, lack of TSP-1 resulted in elevated calponin and attenuated vimentin expression in leptin-treated aSMC from dKO mice (240%, p=0.05 and 80%, p=0.01 respectively vs. leptin-treated cells from ApoE−/−; Supplemental Figure IV). Additionally, leptin in vivo increased pCREB expression in ApoE−/− mice (2-fold vs. PBS-treated ApoE−/−), while pCREB expression was abrogated in leptin-treated dKOs (36%, p<0.0005 vs. leptin-treated ApoE−/−). Interestingly, in the absence of exogenous leptin, TSP-1 deletion had no significant effect on PCNA, vimentin, SM-MHC or pCREB expression in aortic lysates of ApoE−/− mice. A similar trend in protein expression was also observed in female mice (Supplemental Figure V). Finally, pNfkB expression was unaffected in the mice genotypes treated with or without leptin. Similar results were attained for other inflammatory cytokines (TNF-α, IL-1β), with somewhat reducing trend in IL-1β expression noted in leptin-treated dKOs vs. leptin-treated ApoE−/− (Supplemental Figure VI). Collectively, these results demonstrate that TSP-1 deletion inhibits leptin-induced SMC de-differentiation in ApoE−/− mice.
Figure 7. TSP1 deletion attenuates leptin-induced vimentin and pCREB expression accompanied with augmented SM-MHC expression in ApoE−/− mice.
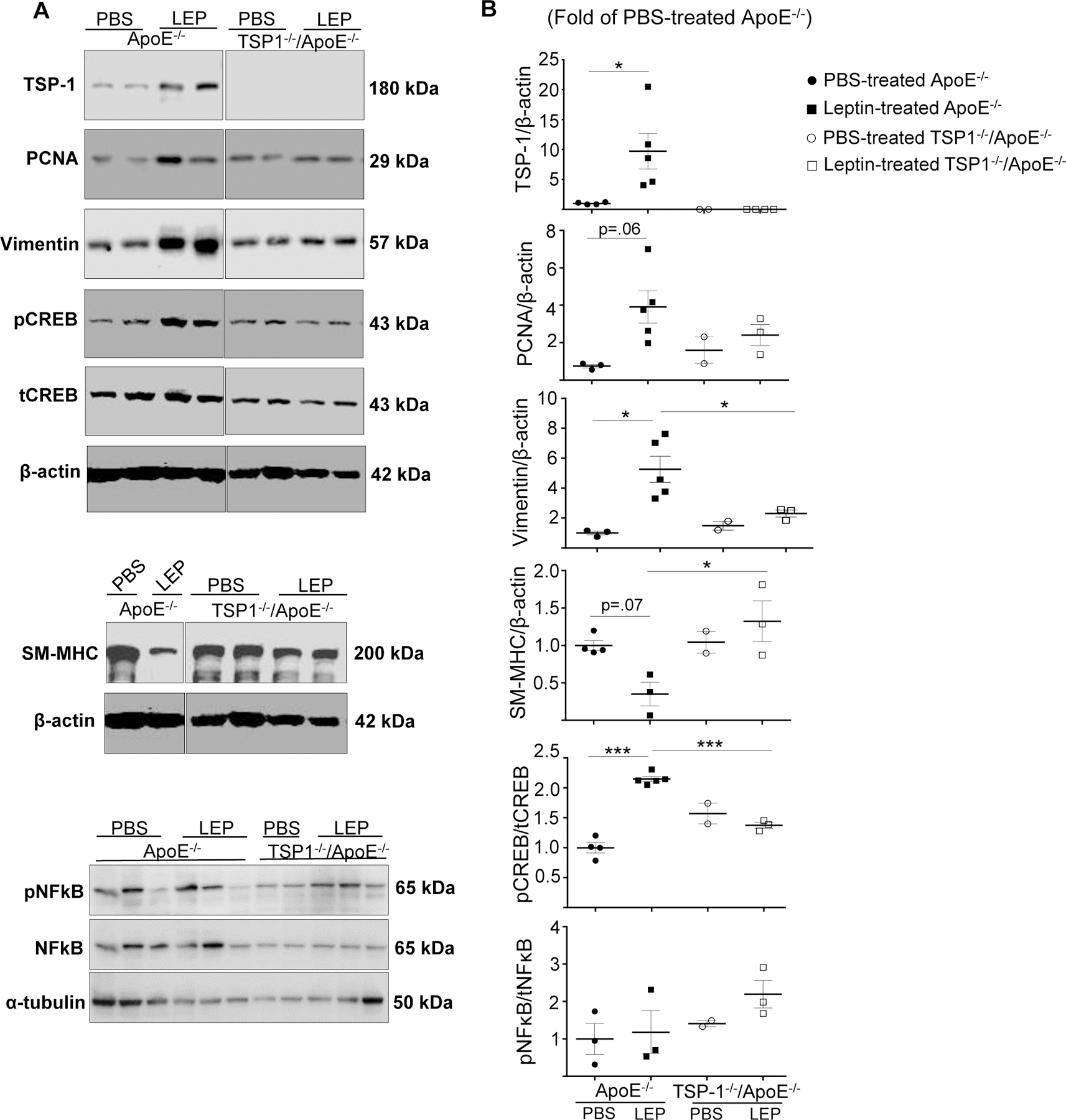
Mice were treated as in Figure 1. Aortic lysates were subjected to western blotting as outlined in Methods. Shown are A) representative western blot images and B) summary graphs for densitometric quantification of immunoblots in male mice; β-actin or α-tubulin used as loading control. Results are presented as fold of PBS-treated ApoE−/−; values are expressed as mean ± SEM (PBS-treated ApoE−/−: n=2; other groups: n=3–5). For A), each lane denotes an individual mouse. Please note, all lane images show proteins loaded and detected on a single immunoblot; however, lanes were re-arranged to improve clarity of presentation. *p≤0.05; ***p<0.0005
TSP-1 deficiency impedes leptin-induced plaque burden in ApoE−/− mice on Western diet, independent of lipid alterations
Following 6 weeks of western diet feeding, leptin-treated male ApoE−/− mice demonstrated robust lipid-filled aortic root lesions compared with PBS-treated ApoE−/−; specifically, lipid burden increased >2-fold under these conditions (p<0.0001, Figure 8). Congruent to earlier observations (Figure 1 and 2), both lipid content and plaque area were significantly reduced in leptin-treated TSP-1−/−/ApoE−/− mice maintained on western diet (>2-fold, p<0.05 vs. leptin-treated ApoE−/−). Moreover, these effects of TSP-1 deletion were noted both in presence and absence of exogenous leptin. Contrary to lipid differences with normocholesterolemic diet (Figure 4), plasma lipid profiles were unchanged in western diet-fed mice genotypes, regardless of leptin administration (p=0.999; Supplemental Figure VII). Together, these results confirm a direct role of TSP-1 in leptin-induced plaque formation under hypercholesterolemia, independent of lipoprotein modifications.
Figure 8. TSP-1 deletion inhibits leptin-induced increase in plaque area in ApoE−/− mice on western diet.
Six weeks old male ApoE−/− and age-matched TSP-1−/−/ApoE−/− littermate mice were maintained on Western diet until 12 wks of age. Beginning at 9 wks, mice were treated with leptin or PBS (similar to Figure 1) for three weeks. Aortic root serial sections were stained with A) 0.5% ORO followed by hematoxylin counterstaining and C) H & E. Shown are representative images (4X magnification) and summary data for A and B) ORO-positive lipid burden and C and D) plaque area (in sq. mm). Dotted line marks region used for quantification. Data depicts lipid burden and plaque area of aortic root per section in each study group; values expressed as mean ± SEM (n=3–4 mice/study group; 3–7 sections/aortic root). For B) SQRT-transformed data used for statistical analyses. *p≤0.05; ***p<0.0005; ****p<0.0001
Discussion
The present study provides novel evidence for a causal role of TSP-1 in leptin-driven atherogenesis. Although earlier work33 has reported a role of TSP-1 in atherosclerosis under basal conditions, the conceptual role of TSP-1 in leptin-driven lesion pathogenesis has not been previously explored. In this report, we provide the first demonstration supporting a TSP-1-dependent mechanism for leptin-stimulated atherosclerosis. Importantly, our data suggest a regulatory role of TSP-1 in leptin-induced VSMC de-differentiation.
Elevated plasma leptin levels correlate with increasing adipose tissue mass in obesity34. A growing body of literature suggest that while centrally-mediated leptin signalling is impaired in obesity, peripheral leptin effects on the heart and vasculature are preserved, attributing to cardiovascular dysfunction35, 36. While appetite-suppressant functions of leptin are well-established, there exist conflicting data on the relative contribution of leptin to vascular dysfunction. Previous studies using mouse models of vascular injury and atherosclerosis have reported that leptin, at concentrations mimicking obesity, promotes vascular remodelling, neointimal growth, vascular calcification and atherosclerotic lesion formation37–39. Specifically, while leptin augmented lesion area and thickness in brachiocephalic and carotid arteries of ApoE−/− mice7, leptin-deficient ob/ob;ApoE−/− mice developed relatively smaller lesions8. Moreover, lesion progression from fatty streaks to fibrous plaques was markedly suppressed in ob/ob;ApoE−/− mice8, supporting the proatherogenic attributes of leptin. On the contrary, few other studies have shown conflicting data. Jun et al. reported that daily low-dose leptin for 12 consecutive weeks substantially reduced lesion area in the aortic arch of Ins2+/Akita;ApoE−/− mice, an atherosclerotic mouse model of Type 1 diabetes, accompanied with diminished plasma LDL-cholesterol40, possibly due to impaired lipoprotein clearance. A later study showed diminished plaques in aortic root and brachiocephalic arteries of ob/ob;LDLR−/− mice41 treated with subphysiological to physiological doses of leptin; these anti-atherogenic effects were linked to improved glucose homeostasis and lipid metabolism as well as adiponectin upregulation. In line with the earlier findings, our data confirm lesion promoting effects of leptin, strengthening the concept that ‘supraphysiological’ leptin levels exert potent proatherogenic effects. Leptin concentrations used in the current study did not produce any adverse phenotypic effects. Although leptin had no effect (p=0.3580) on body weights of ApoE−/− mice, leptin-treated TSP-1−/−/ApoE−/− showed reduced body weight (13.8%) vs. PBS-treated dKOs (p=0.002, Supplemental Figure VIII). These results support the weight-reducing properties of leptin. Moreover, TSP-1 deficiency marginally affected weights of leptin-treated ApoE−/− (p=0.047). These latter results are consistent with an earlier report showing lack of effect of TSP-1 deletion on development of obesity42.
In the present study, we show that absence of the matricellular protein TSP-1 has a protective outcome on atherosclerotic lesion formation triggered by exogenous leptin. We previously reported a direct stimulatory effect of leptin on TSP-1 expression in human and mouse aortic SMC16. Specifically, we have shown that leptin upregulates TSP-1 expression at the level of transcription, mediated via JAK2- and MAPK- signalling mechanisms. Moreover, increased TSP-1 expression was detected in aortic vasculature of leptin-treated wild-type mice, with enhanced accumulation in medial layers of the vessel wall16. Congruent to these findings, the current work demonstrates that genetic deletion of TSP-1 obviates lesion formation prompted by leptin. Specifically, root morphometry revealed diminished plaque burden and lipid-filled area in leptin-treated dKOs vs. leptin-treated ApoE−/−. Similarly, leptin administration led to large lipid-filled en-face lesions in ApoE−/− mice, while considerably smaller-sized lesions developed in leptin-treated dKOs. These results were corroborated with ultrasound imaging revealing a significant reduction in vessel internal diameter of leptin-treated ApoE−/− that was nullified in leptin-treated dKOs; accuracy of such measurements have been validated in an earlier work30 depicting a significant correlation between ultrasound imaging and histological assessment. Collectively, these data support the previously reported role of TSP-1 in promoting neointimal hyperplasia and medial wall thickening43. These results further align with our earlier report illustrating a protective effect of TSP-1 loss on hyperglycemia-induced atherogenesis30. Numerous studies have indicated that collagen accumulation, particularly during initial stages of lesion development, may significantly contribute to plaque growth and luminal narrowing44. Collagen, a major component of the extracellular matrix found in atherosclerotic lesions, is known to promote lesion volume and diverse cellular responses central to plaque growth. Previous studies have reported a regulatory role of TSP-1 on collagen homeostasis via interaction with different matrix metalloproteinases (MMPs) as well as TGF-β-mediated signaling pathways45, 46. Recent studies further indicate that TSP-1 can directly interact with fibrillar collagen and attenuate collagen cross-linking mechanisms resulting in reduced collagen content47. Consistent with these earlier reports, immunoblotting of aortic lysates from TSP1−/−/ApoE−/− mice showed a reduced trend in TGF-β protein expression coupled with increased trend in MMP-13 expression vs. ApoE−/−, under PBS and leptin-treated conditions (Supplemental Figure IX). A large fraction of collagen found in lesions originate from smooth muscle cells. Previous studies have suggested that vascular collagen modulates signalling pathways that mediate VSMC and macrophage phenotypic plasticity48. Growing literature indicate a positive correlation between collagen abundance and VSMC phenotypic transition triggering lesion progression. Our findings that collagen accumulation is attenuated in aortic root of leptin-treated TSP-1−/−/ApoE−/− mice lend additional support to a vasculoprotective role of TSP-1 deficiency under leptin-stimulated conditions. Cogent to our previous findings30, the present work underscores a TSP-1-dependent mechanism, specific to high glucose or high leptin, that drives hyperglycemia- or hyperleptinemia-induced atherosclerosis.
It is worth mentioning that under basal conditions, i.e., in the absence of leptin, we observed a significant reduction in aortic root lesion area in TSP-1−/−/ApoE−/− vs ApoE−/− mice. In an earlier study by Moura et al.33, it was shown that ApoE−/− and TSP-1−/−/ApoE−/− dKO mice developed comparable lesion areas following 6–9 months of normocholesterolemic diet. Although our results may appear somewhat different from that of Moura et al., it is important to note that in the two studies, lesions were examined in mice cohorts at two different ages (20 vs. 36 wks) depicting differing lesion stages. Indeed, in this same study, Moura et al., also reported age- and lesion-stage specific TSP-1 effects that were analogous to our findings. Interestingly, while root morphometry revealed diminished lipid burden in untreated dKOs vs ApoE−/−, both genotypes developed comparable en-face lesions in the aortic vasculature. The observed discrepancy may be related to variable proatherogenic indices along different arterial branches (aorta vs. aortic root).
Multiple studies highlight the contribution of lipids and lipoprotein distribution to the etiology of atherosclerosis. In the present work, plasma total cholesterol in leptin-treated TSP-1−/−/ApoE−/− trended downwards (albeit not statistically significant) compared to leptin-treated ApoE−/−; also, TSP-1 deficiency significantly attenuated leptin-stimulated VLDL- and LDL-triglyceride content in ApoE−/− mice. We speculate that this lipoprotective response of TSP-1 deletion may be due to reduced de novo lipogenesis, reduced VLDL synthesis and/or enhanced VLDL clearance. TG-rich lipoproteins are an important source of fatty acids. Previous studies indicate that oxidized fatty acids generated via lipoprotein lipase-mediated TG hydrolysis may contribute to several pro-inflammatory and cytotoxic effects in the arterial wall (reviewed in49). TG-rich lipoproteins are also known to have large amounts of cholesterol; macrophage endocytic uptake of TG-rich lipoproteins, including VLDL and other remnants, followed by TG catabolism via lysosomal degradative pathway may further exacerbate foam cell sterol accumulation50. We postulate that TSP-1 deficiency may block leptin-stimulated generation of oxidized fatty acid, in turn suppressing proinflammatory responses and macrophage cytotoxicity in the vasculature. Although no significant difference in plasma β-hydroxybutyrate, measured as a surrogate marker of β-fatty acid oxidation, was observed between the treatment groups (Supplemental Figure XA), future studies are needed to assess changes in fatty acid oxidation in aortic tissues. Interestingly, our data indicate a positive correlation between lesion burden and HDL-cholesterol levels. Specifically, while plaque burden and HDL-cholesterol were augmented in leptin-treated ApoE−/−, there was a profound decrease in lesion burden and HDL-cholesterol in leptin-treated dKO mice. It should be noted that plasma HDL-cholesterol was somewhat raised in all our animals, in direct association with relatively low plasma cholesteryl ester transfer protein (CETP) activity (key enzyme in HDL metabolism that mediates transfer of cholesteryl ester from HDL to VLDL/LDL). Notably, while TSP-1 deletion reduced HDL-cholesterol in ApoE−/− mice, under both PBS and leptin-treated conditions (Fig. 4C), no statistically significant difference in CETP activity was detected between the groups (Supplemental Figure XB). Growing literature support the notion that HDL functionality, rather than HDL amount, is a key determinant of cardiovascular risks and outcomes51. Accordingly, our data prompt us to speculate that TSP-1 may modulate HDL function and turnover contributing to leptin’s lesion promoting effects. It is likely that TSP-1 deficiency may promote the anti-inflammatory and atheroprotective properties of HDL during leptin stimulation, despite decreased HDL levels. Additional work is required to interrogate this possibility and will be pursued in future studies. Interestingly, TSP-1 deficiency had no effect on plasma lipid levels in the absence of leptin. In a previous study42, TSP-1−/−/ApoE−/− mice manifested reduced proatherogenic lipoproteins (VLDL, LDL) under low fat and high fat-fed conditions, without noticeable effects on HDL distribution. These differences in results may be due to varying experimental conditions in the two studies, e.g., type of diet and study endpoint. Regardless, our results posit an important regulatory role of TSP-1 on leptin-driven lipoprotein distribution. The underlying mechanism is currently unknown and will be assessed in a future work. It is noteworthy that genetic TSP-1 deletion produced strikingly different effects on plasma lipids when mice were kept on western diet; although TSP-1 deficiency attenuated leptin-induced lipid burden and plaque area, plasma lipid levels were unaltered under these conditions. We propose that under hypercholesterolemic conditions characteristic of western diet, saturation of the enzyme kinetics that mediate cholesterol and triglyceride metabolism may account for lack of effect on lipoprotein distribution with leptin and TSP-1 deletion. Accordingly, our results showing different lesion sizes in the absence of systemic changes in lipid levels with hypercholesterolemic feeding would suggest a direct role of TSP-1 on leptin-stimulated atherosclerosis, independent of lipoprotein modifications.
Atherosclerosis is a multifactorial disease involving numerous cell types including endothelial cells, VSMC and macrophages. Leptin stimulates peritoneal macrophage activation and promotes macrophage foam cell formation52. Earlier studies have shown that leptin upregulates proinflammatory mediators in the vessel wall accounting for accelerated plaque formation53, 54. We previously reported that macrophage and leukocyte lesion invasion was attenuated in hyperglycemic TSP-1−/−/ApoE−/− mice vs. hyperglycemic ApoE−/−30. Consistent with these findings, the current work demonstrates that TSP-1 deficiency inhibits inflammatory lesion infiltration triggered by leptin. Of note, inflammatory cell abundance was unchanged in the mice genotypes basally. A previous study has reported increased inflammatory burden in TSP-1−/−/ApoE−/− following 36 weeks of normocholesterolemic diet33. The observed discrepancy may be related to differential TSP-1 response in early (20 wks) vs. late-stage (36 wks) lesions. Reassuringly, our data support the notion that TSP-1 exerts counterbalancing effects on early vs. late-phase atherosclerotic lesions55. Notably, these results emphasize leptin-specific TSP-1 effect on lesion inflammatory milieu.
In healthy vessels, VSMCs typically reside within medial layers of the vessel wall and are responsible for regulation of the vascular tone. However, exposure to proatherogenic stimuli promotes VSMC migration and proliferation into the intimal vascular wall, events central to the evolution of atherosclerosis. Growing literature indicate that VSMC de-differentiation from quiescent ‘contractile’ to synthetic ‘proliferative’ phenotype is a key player in initiation and progression of atherosclerosis. Specifically, de-differentiated SMCs are characterized by reduced SMC contractile gene expression, enhanced migratory and proliferative response and augmented expression of extracellular matrix proteins56. Previous in vivo and in vitro studies have shown that leptin promotes neointimal growth and vessel remodelling, likely attributed to increased VSMC hypertrophy, migration and proliferation38, 57. Multiple vascular cell types including endothelial cells and VSMC are capable of producing TSP-127. Earlier studies suggest that TSP-1 stimulates VSMC growth and activation, augmenting neointimal formation following vascular injury43, 58. We previously reported that TSP-1, at least in part, mediates VSMC migration and proliferation triggered by leptin. Specifically, we have shown that incubation of primary HASMC cultures with anti-TSP-1 blocking antibody inhibits leptin-induced SMC migration and proliferation compared to cells treated with leptin alone16. We further reported that lack of TSP-1 abrogates leptin-induced PCNA expression in wild type mice16. The present findings that TSP-1 deletion blocks leptin-induced proliferative SMC lesion abundance validate our central hypothesis. Moreover, our data that TSP-1 deficiency increases SMC contractile-specific markers (SM-MHC, calponin) while reducing expression of SMC synthetic marker (vimentin) and pro-mitogenic regulator of VSMC proliferation (pCREB) in leptin-treated ApoE−/− (in vivo and in vitro) suggest a novel regulatory role of TSP-1 in leptin-induced VSMC phenotypic transition. Overall, our data support the idea that augmented TSP-1 levels in response to exogenous leptin accounts for increased SMC activation with accompanying collagen deposition. Similar to lesion size and inflammatory cell abundance, lesion SMC invasion and SMC de-differentiation were comparable in the mice genotypes under basal conditions. It should be noted that while α-SMA expression is typically linked with contractile SMC function, α-SMA is more reliable as SMC lineage marker and less ‘phenotype-specific’. Moreover, there is increasing evidence for SMC diversity and SMC heterogeneity in the vessel wall, raising the likelihood for an intermediate SMC phenotype between the contractile and synthetic states. Earlier studies have suggested that SMCs are capable of bi-directional phenotypic switch, wherein de-differentiated cells may retain certain attributes of differentiation56, 59. Recent studies strongly support distinct lineage-based VSMC populations that are both lesion stage- and region-specific60. Cogent to these reports, our data showing a positive correlation between α-SMA fluorescence intensity and plaque burden illustrate increased SMC lesion infiltration, characteristic of lesion progression. It would be remiss not to mention that while we did not find significant sex-specific differences in the present study, leptin-treated male ApoE−/− revealed higher collagen build-up than leptin-treated female ApoE−/−, suggesting a possible protective response in female mice; the underlying mechanism is however currently unknown.
While the current work clearly suggests VSMC as a putative target of TSP-1, additional in vitro experiments with different vascular cell types and cell-specific TSP-1 KOs are needed. Further, to elucidate the mechanistic role of TSP-1 in leptin-induced SMC transdifferentiation, SMC lineage tracing approaches are warranted. In the absence of such experiments, several questions, e.g. whether de-differentiated SMCs transform into proliferating macrophages or foam cells as well as the spatiotemporal patterns of SMC fate and phenotypic plasticity in response to leptin, and most notably, the regulatory role of TSP-1 in these processes, remain unresolved. While such studies are beyond the scope of the current manuscript, future work (currently underway) will employ SMC fate mapping strategies using hyperleptinemic ApoE−/− mice with SMC-specific TSP-1 deletion.
In conclusion, the present study uncovers a protective outcome of TSP-1 deletion on leptin-induced atherosclerosis. Our findings underscore a fundamental link between leptin and TSP-1 in lesion pathogenesis and highlight TSP-1 as a potential target of leptin-induced vasculopathy. Given the multifunctional properties of TSP-1 relevant to its multidomain structure and cell- and tissue-specific effects, additional studies are warranted to interrogate cell-specific (EC, macrophages) TSP-1 effects on hyperleptinemia-induced atherosclerosis.
Supplementary Material
Highlights.
TSP-1 deletion prevents leptin-induced increase in lipid burden, plaque area and collagen accumulation.
TSP-1 deletion blocks leptin-induced inflammatory and SMC lesion invasion.
TSP-1 deletion inhibits leptin-induced vimentin and pCREB expression while augmenting SM-MHC and calponin expression.
TSP-1 deletion modulates leptin-induced lipoprotein distribution.
Acknowledgements
We thank Dr. Yanqiao Zhang, NEOMED for technical assistance with FPLC studies.
Sources of Funding
This work was supported by American Heart Association Scientist Development Grant 0835190N, Diabetes Action Research and Education Foundation, 1R15HL147245–01 and NEOMED start-up funds to Priya Raman.
Non-standard Abbreviations and Acronyms
- VSMC
vascular smooth muscle cell
- TSP-1
thrombospondin-1
- ORO
oil red O
- H&E
hematoxylin and eosin
- MT
Masson’s trichrome
- PCNA
proliferating cell nuclear antigen
- SM-MHC
smooth muscle-myosin heavy chain
- VLDL
very LDL
- LDL
low-density lipoprotein
- HDL
high-density lipoprotein
- RCCA
right common carotid artery
- α-SMA
alpha-smooth muscle actin
- HASMC
human aortic SMC
- TG
triglyceride
- dKO
double knockout
Footnotes
Disclosures
None.
References
- 1.Marinou K, Tousoulis D, Antonopoulos AS, Stefanadi E, Stefanadis C. Obesity and cardiovascular disease: from pathophysiology to risk stratification. Int J Cardiol. 2010; 138:3–8. [DOI] [PubMed] [Google Scholar]
- 2.Beltowski J. Leptin and atherosclerosis. Atherosclerosis. 2006; 189:47–60. [DOI] [PubMed] [Google Scholar]
- 3.Ciccone M, Vettor R, Pannacciulli N, Minenna A, Bellacicco M, Rizzon P, Giorgino R, De Pergola G. Plasma leptin is independently associated with the intima-media thickness of the common carotid artery. Int J Obes Relat Metab Disord. 2001; 25:805–810. [DOI] [PubMed] [Google Scholar]
- 4.Reilly MP, Iqbal N, Schutta M, Wolfe ML, Scally M, Localio AR, Rader DJ, Kimmel SE. Plasma leptin levels are associated with coronary atherosclerosis in type 2 diabetes. J Clin Endocrinol Metab. 2004; 89:3872–3878. [DOI] [PubMed] [Google Scholar]
- 5.Fantuzzi G, Mazzone T. Adipose tissue and atherosclerosis: exploring the connection. Arterioscler Thromb Vasc Biol. 2007; 27:996–1003. [DOI] [PubMed] [Google Scholar]
- 6.Kang SM, Kwon HM, Hong BK, Kim D, Kim IJ, Choi EY, Jang Y, Kim HS, Kim MS, Kwon HC. Expression of leptin receptor (Ob-R) in human atherosclerotic lesions: potential role in intimal neovascularization. Yonsei Med J. 2000; 41:68–75. [DOI] [PubMed] [Google Scholar]
- 7.Bodary PF, Gu S, Shen Y, Hasty AH, Buckler JM, Eitzman DT. Recombinant leptin promotes atherosclerosis and thrombosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2005; 25:e119–122. [DOI] [PubMed] [Google Scholar]
- 8.Chiba T, Shinozaki S, Nakazawa T, Kawakami A, Ai M, Kaneko E, Kitagawa M, Kondo K, Chait A, Shimokado K. Leptin deficiency suppresses progression of atherosclerosis in apoE-deficient mice. Atherosclerosis. 2008; 196:68–75. [DOI] [PubMed] [Google Scholar]
- 9.Li L, Mamputu JC, Wiernsperger N, Renier G. Signaling pathways involved in human vascular smooth muscle cell proliferation and matrix metalloproteinase-2 expression induced by leptin: inhibitory effect of metformin. Diabetes. 2005; 54:2227–2234. [DOI] [PubMed] [Google Scholar]
- 10.Huang F, Xiong X, Wang H, You S, Zeng H. Leptin-induced vascular smooth muscle cell proliferation via regulating cell cycle, activating ERK1/2 and NF-kappaB. Acta Biochim Biophys Sin (Shanghai). 2010; 42:325–331. [DOI] [PubMed] [Google Scholar]
- 11.Payne GA, Tune JD, Knudson JD. Leptin-induced endothelial dysfunction: a target for therapeutic interventions. Curr Pharm Des. 2014; 20:603–608. [DOI] [PubMed] [Google Scholar]
- 12.Konstantinides S, Schafer K, Koschnick S, Loskutoff DJ. Leptin-dependent platelet aggregation and arterial thrombosis suggests a mechanism for atherothrombotic disease in obesity. J Clin Invest. 2001; 108:1533–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quehenberger P, Exner M, Sunder-Plassmann R, Ruzicka K, Bieglmayer C, Endler G, Muellner C, Speiser W, Wagner O. Leptin induces endothelin-1 in endothelial cells in vitro. Circ Res. 2002; 90:711–718. [DOI] [PubMed] [Google Scholar]
- 14.Park HY, Kwon HM, Lim HJ, Hong BK, Lee JY, Park BE, Jang Y, Cho SY, Kim HS. Potential role of leptin in angiogenesis: leptin induces endothelial cell proliferation and expression of matrix metalloproteinases in vivo and in vitro. Exp Mol Med. 2001; 33:95–102. [DOI] [PubMed] [Google Scholar]
- 15.Raso GM, Pacilio M, Esposito E, Coppola A, Di Carlo R, Meli R. Leptin potentiates IFN-gamma-induced expression of nitric oxide synthase and cyclo-oxygenase-2 in murine macrophage J774A.1. Br J Pharmacol. 2002; 137:799–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chavez RJ, Haney RM, Cuadra RH, Ganguly R, Adapala RK, Thodeti CK, Raman P. Upregulation of thrombospondin-1 expression by leptin in vascular smooth muscle cells via JAK2- and MAPK-dependent pathways. Am J Physiol Cell Physiol. 2012; 303:C179–191. [DOI] [PubMed] [Google Scholar]
- 17.Bornstein P Thrombospondins as matricellular modulators of cell function. J Clin Invest. 2001; 107:929–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adams JC, Lawler J. The thrombospondins. Int J Biochem Cell Biol. 2004; 36:961–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raugi GJ, Mullen JS, Bark DH, Okada T, Mayberg MR. Thrombospondin deposition in rat carotid artery injury. Am J Pathol. 1990; 137:179–185. [PMC free article] [PubMed] [Google Scholar]
- 20.Sajid M, Hu Z, Guo H, Li H, Stouffer GA. Vascular expression of integrin-associated protein and thrombospondin increase after mechanical injury. J Investig Med. 2001; 49:398–406. [DOI] [PubMed] [Google Scholar]
- 21.Riessen R, Kearney M, Lawler J, Isner JM. Immunolocalization of thrombospondin-1 in human atherosclerotic and restenotic arteries. Am Heart J. 1998; 135:357–364. [DOI] [PubMed] [Google Scholar]
- 22.Stenina OI, Topol EJ, Plow EF. Thrombospondins, their polymorphisms, and cardiovascular disease. Arterioscler Thromb Vasc Biol. 2007; 27:1886–1894. [DOI] [PubMed] [Google Scholar]
- 23.Matsuo Y, Tanaka M, Yamakage H, Sasaki Y, Muranaka K, Hata H, Ikai I, Shimatsu A, Inoue M, Chun TH, Satoh-Asahara N. Thrombospondin 1 as a novel biological marker of obesity and metabolic syndrome. Metabolism. 2015; 64:1490–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kong P, Gonzalez-Quesada C, Li N, Cavalera M, Lee DW, Frangogiannis NG. Thrombospondin-1 regulates adiposity and metabolic dysfunction in diet-induced obesity enhancing adipose inflammation and stimulating adipocyte proliferation. Am J Physiol Endocrinol Metab. 2013; 305:E439–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Inoue M, Jiang Y, Barnes RH 2nd, Tokunaga M, Martinez-Santibanez G, Geletka L, Lumeng CN, Buchner DA, Chun TH. Thrombospondin 1 mediates high-fat diet-induced muscle fibrosis and insulin resistance in male mice. Endocrinology. 2013; 154:4548–4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gonzalez-Quesada C, Cavalera M, Biernacka A, Kong P, Lee DW, Saxena A, Frunza O, Dobaczewski M, Shinde A, Frangogiannis NG. Thrombospondin-1 induction in the diabetic myocardium stabilizes the cardiac matrix in addition to promoting vascular rarefaction through angiopoietin-2 upregulation. Circ Res. 2013; 113:1331–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stenina OI, Krukovets I, Wang K, Zhou Z, Forudi F, Penn MS, Topol EJ, Plow EF. Increased expression of thrombospondin-1 in vessel wall of diabetic Zucker rat. Circulation. 2003; 107:3209–3215. [DOI] [PubMed] [Google Scholar]
- 28.Wahab NA, Schaefer L, Weston BS, Yiannikouris O, Wright A, Babelova A, Schaefer R, Mason RM. Glomerular expression of thrombospondin-1, transforming growth factor beta and connective tissue growth factor at different stages of diabetic nephropathy and their interdependent roles in mesangial response to diabetic stimuli. Diabetologia. 2005; 48:2650–2660. [DOI] [PubMed] [Google Scholar]
- 29.Daugherty A, Tall AR, Daemen M, Falk E, Fisher EA, Garcia-Cardena G, Lusis AJ, Owens AP 3rd, Rosenfeld ME, Virmani R, American Heart Association Council on Arteriosclerosis T, Vascular B, Council on Basic Cardiovascular S. Recommendation on Design, Execution, and Reporting of Animal Atherosclerosis Studies: A Scientific Statement From the American Heart Association. Arterioscler Thromb Vasc Biol. 2017; 37:e131–e157. [DOI] [PubMed] [Google Scholar]
- 30.Ganguly R, Sahu S, Ohanyan V, Haney R, Chavez RJ, Shah S, Yalamanchili S, Raman P. Oral chromium picolinate impedes hyperglycemia-induced atherosclerosis and inhibits proatherogenic protein TSP-1 expression in STZ-induced type 1 diabetic ApoE−/− mice. Sci Rep. 2017; 7:45279–45293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu Y, Zalzala M, Xu J, Li Y, Yin L, Zhang Y. A metabolic stress-inducible miR-34a-HNF4alpha pathway regulates lipid and lipoprotein metabolism. Nat Commun. 2015; 6:7466–7476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Malik NM, Carter ND, Murray JF, Scaramuzzi RJ, Wilson CA, Stock MJ. Leptin requirement for conception, implantation, and gestation in the mouse. Endocrinology. 2001; 142:5198–5202. [DOI] [PubMed] [Google Scholar]
- 33.Moura R, Tjwa M, Vandervoort P, Van Kerckhoven S, Holvoet P, Hoylaerts MF. Thrombospondin-1 deficiency accelerates atherosclerotic plaque maturation in ApoE−/− mice. Circ Res. 2008; 103:1181–1189. [DOI] [PubMed] [Google Scholar]
- 34.Yun JE, Kimm H, Jo J, Jee SH. Serum leptin is associated with metabolic syndrome in obese and nonobese Korean populations. Metabolism. 2010; 59:424–429. [DOI] [PubMed] [Google Scholar]
- 35.Tune JD, Considine RV. Effects of leptin on cardiovascular physiology. J Am Soc Hypertens. 2007; 1:231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martin SS, Qasim A, Reilly MP. Leptin resistance: a possible interface of inflammation and metabolism in obesity-related cardiovascular disease. J Am Coll Cardiol. 2008; 52:1201–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schafer K, Halle M, Goeschen C, Dellas C, Pynn M, Loskutoff DJ, Konstantinides S. Leptin promotes vascular remodeling and neointimal growth in mice. Arterioscler Thromb Vasc Biol. 2004; 24:112–117. [DOI] [PubMed] [Google Scholar]
- 38.Bodary PF, Shen Y, Ohman M, Bahrou KL, Vargas FB, Cudney SS, Wickenheiser KJ, Myers MG Jr., Eitzman DT. Leptin regulates neointima formation after arterial injury through mechanisms independent of blood pressure and the leptin receptor/STAT3 signaling pathways involved in energy balance. Arterioscler Thromb Vasc Biol. 2007; 27:70–76. [DOI] [PubMed] [Google Scholar]
- 39.Zeadin M, Butcher M, Werstuck G, Khan M, Yee CK, Shaughnessy SG. Effect of leptin on vascular calcification in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2009; 29:2069–2075. [DOI] [PubMed] [Google Scholar]
- 40.Jun JY, Ma Z, Pyla R, Segar L. Leptin treatment inhibits the progression of atherosclerosis by attenuating hypercholesterolemia in type 1 diabetic Ins2(+/Akita):apoE(−/−) mice. Atherosclerosis. 2012; 225:341–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoffmann A, Ebert T, Kloting N, Dokas J, Jeromin F, Jessnitzer B, Burkhardt R, Fasshauer M, Kralisch S. Leptin dose-dependently decreases atherosclerosis by attenuation of hypercholesterolemia and induction of adiponectin. Biochim Biophys Acta. 2016; 1862:113–120. [DOI] [PubMed] [Google Scholar]
- 42.Maimaitiyiming H, Clemons K, Zhou Q, Norman H, Wang S. Thrombospondin1 deficiency attenuates obesity-associated microvascular complications in ApoE−/− mice. PLoS One. 2015; 10:e0121403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moura R, Tjwa M, Vandervoort P, Cludts K, Hoylaerts MF. Thrombospondin-1 activates medial smooth muscle cells and triggers neointima formation upon mouse carotid artery ligation. Arterioscler Thromb Vasc Biol. 2007; 27:2163–2169. [DOI] [PubMed] [Google Scholar]
- 44.Rekhter MD. Collagen synthesis in atherosclerosis: too much and not enough. Cardiovasc Res. 1999; 41:376–384. [DOI] [PubMed] [Google Scholar]
- 45.Tan K, Lawler J. The interaction of Thrombospondins with extracellular matrix proteins. J Cell Commun Signal. 2009; 3:177–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murphy-Ullrich JE. Thrombospondin 1 and Its Diverse Roles as a Regulator of Extracellular Matrix in Fibrotic Disease. J Histochem Cytochem. 2019; 67:683–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rosini S, Pugh N, Bonna AM, Hulmes DJS, Farndale RW, Adams JC. Thrombospondin-1 promotes matrix homeostasis by interacting with collagen and lysyl oxidase precursors and collagen cross-linking sites. Sci Signal. 2018; 11:eaar2566 [DOI] [PubMed] [Google Scholar]
- 48.Adiguzel E, Ahmad PJ, Franco C, Bendeck MP. Collagens in the progression and complications of atherosclerosis. Vasc Med. 2009; 14:73–89. [DOI] [PubMed] [Google Scholar]
- 49.Linton MRF, Yancey PG, Davies SS, Jerome WG, Linton EF, Song WL, Doran AC, Vickers KC. The Role of Lipids and Lipoproteins in Atherosclerosis. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dungan K, et al. , eds. Endotext: South Dartmouth (MA) 2000. [Google Scholar]
- 50.Talayero BG, Sacks FM. The role of triglycerides in atherosclerosis. Curr Cardiol Rep. 2011; 13:544–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Toth PP, Barter PJ, Rosenson RS, Boden WE, Chapman MJ, Cuchel M, D’Agostino RB Sr., Davidson MH, Davidson WS, Heinecke JW, Karas RH, Kontush A, Krauss RM, Miller M, Rader DJ. High-density lipoproteins: a consensus statement from the National Lipid Association. J Clin Lipidol. 2013; 7:484–525. [DOI] [PubMed] [Google Scholar]
- 52.Maya-Monteiro CM, Almeida PE, D’Avila H, Martins AS, Rezende AP, Castro-Faria-Neto H, Bozza PT. Leptin induces macrophage lipid body formation by a phosphatidylinositol 3-kinase- and mammalian target of rapamycin-dependent mechanism. J Biol Chem. 2008; 283:2203–2210. [DOI] [PubMed] [Google Scholar]
- 53.Dubey L, Hesong Z. Role of leptin in atherogenesis. Exp Clin Cardiol. 2006:11:269–275. [PMC free article] [PubMed] [Google Scholar]
- 54.Hongo S, Watanabe T, Arita S, Kanome T, Kageyama H, Shioda S, Miyazaki A. Leptin modulates ACAT1 expression and cholesterol efflux from human macrophages. Am J Physiol Endocrinol Metab. 2009; 297:E474–482. [DOI] [PubMed] [Google Scholar]
- 55.Stenina OI, Plow EF. Counterbalancing forces: what is thrombospondin-1 doing in atherosclerotic lesions? Circ Res. 2008; 103:1053–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Beamish JA, He P, Kottke-Marchant K, Marchant RE. Molecular regulation of contractile smooth muscle cell phenotype: implications for vascular tissue engineering. Tissue Eng Part B Rev. 2010; 16:467–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zeidan A, Purdham DM, Rajapurohitam V, Javadov S, Chakrabarti S, Karmazyn M. Leptin induces vascular smooth muscle cell hypertrophy through angiotensin II- and endothelin-1-dependent mechanisms and mediates stretch-induced hypertrophy. J Pharmacol Exp Ther. 2005; 315:1075–1084. [DOI] [PubMed] [Google Scholar]
- 58.Helkin A, Maier KG, Gahtan V. Thrombospondin-1, −2 and −5 have differential effects on vascular smooth muscle cell physiology. Biochem Biophys Res Commun. 2015; 464:1022–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pfaltzgraff ER, Bader DM. Heterogeneity in vascular smooth muscle cell embryonic origin in relation to adult structure, physiology, and disease. Dev Dyn. 2015; 244:410–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu M, Gomez D. Smooth Muscle Cell Phenotypic Diversity. Arterioscler Thromb Vasc Biol. 2019; 39:1715–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.