

Abstract
Objective:
Viral macrophage inflammatory protein 2 (vMIP-II/vCCL2) binds to multiple chemokine receptors, and vMIP-II based PET tracer (64Cu-DOTA-vMIP-II: vMIP-II tracer) accumulates at atherosclerotic lesions in mice. Given that it would be expected to react with multiple chemokine receptors on monocytes and macrophages, we wondered if its accumulation in atherosclerosis lesion-bearing mice might correlate with overall macrophage burden or, alternatively, the pace of monocyte recruitment.
Approach and Results:
We employed a mouse model of atherosclerosis regression involving AAV-mApoE treatment of Apoe−/− mice where the pace of monocyte recruitment slows before macrophage burden subsequently declines. Accumulation of 64Cu-DOTA-vMIP-II at Apoe−/− plaque sites was strong but declined with AAV-mApoE-induced decline in monocyte recruitment, before macrophage burden reduced. Monocyte depletion indicated that monocytes and macrophages themselves were not the only target of the 64Cu-DOTA-vMIP-II tracer. Using fluorescence-tagged vMIP-II tracer, competitive receptor blocking with CXCR4 antagonists, endothelial specific Cre-mediated deletion of CXCR4, CXCR4-specific tracer 64Cu-DOTA-FC131, and CXCR4 staining during disease progression and regression, we show endothelial cell expression of CXCR4 is a key target of 64Cu-DOTA-vMIP-II imaging. Expression of CXCR4 was low in non-plaque areas, but strongly detected on endothelium of progressing plaques, especially on proliferating endothelium, where vascular permeability was increased and monocyte recruitment was strongest.
Conclusions:
Endothelial injury status of plaques is marked by CXCR4 expression and that this injury correlates with the tendency of such plaques to recruit monocytes. Furthermore, our findings suggest PET tracers that mark CXCR4 can be used translationally to monitor the state of plaque injury and monocyte recruitment.
Keywords: atherosclerosis, PET tracer, chemokine receptor, vMIP-II, CXCR4, endothelial cells, endothelial injury
Subject terms: Nuclear Cardiology and PET, vascular biology, translational studies
Graphical Abstract
Introduction
Atherosclerotic disease is still one of the leading causes of death all over the world even after several medical technologies such as revascularization therapy, including catheter intervention and surgery, and drug treatment have been developed 1. Several imaging tools including ultrasonography, catheter angiography, computed tomography (CT) and magnetic resonance imaging (MRI) have also been developed to detect and assess atherosclerotic plaques 2. However, while many of these tools can detect differences in plaque burden and stenosis of the vascular lumen, measurement of features that predict plaque rupture are not necessarily met by these tools. That is, atherosclerotic plaque rupture, which causes sudden occlusion of an artery and subsequent ischemic tissue injury downstream of the culprit site, does not always occur at severely stenotic lesions 3,4. Thus, tools that can assess plaque status beyond its overall size are needed 5. Positron emission tomography (PET) is non-invasive nuclear medicine imaging modality using a specific radiolabeled tracer to detect a molecular activity in vivo. It is currently attractive to assess atherosclerotic plaques because of its high sensitivity and its potential for coupling to probes that allow functional evaluation of molecular and cellular aspects. Some specific radioligands are now being used or considered to target the specific pathophysiological process in atherosclerosis 2,6–8. The identification of imaging tools that predict plaque behavior requires not only application of tools designed to assess specific targets, but follow-up studies to characterize how the tools behave in a complex environment in vivo. Given that the recruitment of monocytes is a signature feature of atherosclerotic plaque activity 9–12, tools designed to assess monocyte and macrophage activity may be particularly valuable.
Chemokines are a group of about 50 chemotactic heparin-binding cytokines, which are known to be involved in various inflammatory diseases due to their critical roles in directing the movement of circulating leukocytes to sites of inflammation or injury through corresponding chemokine receptors 13. At least 10 chemokine receptors have been identified or implicated in atherosclerotic plaque 14,15. Viral macrophage inflammatory protein 2 (vMIP-II), also known as vCCL2, is encoded by Kaposi’s sarcoma-associated human herpes virus 8 (HHV-8) and is able to bind to multiple chemokine receptors including CCR1, CCR2, CCR3, CCR5, CCR8, CCR10, XCR1, CX3CR1, CXCR4, and CXCR7 (ACKR3) 16–19, such that it detects collectively the majority of atherosclerosis-related chemokine receptors. Recently, we developed vMIP-II based nanoprobes for PET imaging (64Cu-DOTA-vMIP-II), and we documented its uptake in wire injury-induced atherosclerotic plaques 20. However, we did not fully investigate the underlying basis for the signal intensity of the vMIP-II based PET tracer. In particular, it remained unclear which cell populations and chemokine receptors were the major source of the PET signal.
We set out to address that issue herein, with the goal to take into account the possibility that the tracer may not have full access to deeper areas of plaques that might be rich in macrophages and thus might preferentially react with populations available to the blood circulation, such as monocytes. To that end, we designed a protocol that took advantage of the fact that monocyte recruitment to plaques in Apoe−/− mice is readily modulated by restoring apoE expression using viral vectors 12. Indeed, monocyte recruitment is dramatically suppressed by apoE expression in Apoe−/− mice before macrophage burden decreases. Thus, use of this approach that kinetically separates changes in monocyte recruitment with overall macrophage burden would allow us to ask whether the broad spectrum vMIP-II based PET tracer could be used to read out one or the other of these parameters.
Here, we demonstrate that the PET signal derived from the 64Cu-DOTA-vMIP-II tracer indeed correlates with monocyte recruitment into atherosclerotic plaque rather than macrophage burden. However, in the process of exploring mechanism of action, we uncover the surprising result that 64Cu-DOTA-vMIP-II signal in Apoe−/− mice prominently targeted CXCR4 on endothelium. This finding then opened the door for further experiments that revealed that CXCR4 expression is induced on the endothelium at the margins of plaques where endothelial permeability and proliferation is highest. Through the studies presented herein, we argue that CXCR4 serves as an important marker of injury in the endothelium and that this status is closely linked to monocyte recruitment. Considering that CXCR4 locus in human has been reported to be as a key determinant in coronary heart disease 21, these new findings underscore the possibility that CXCR4-targeted PET tracers may report critical information about plaque status in a clinical setting.
Material and Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Mice
Mice were housed in specific pathogen-free animal facilities, maintained by Washington University School of Medicine. Apoe−/− (B6.129P2-Apoetm1Unc/J, 002052) mice were purchased from Jackson Laboratories. Diet was switched to high fat diet (HFD) (Teklad, TD.88137) containing 21% milk fat and 0.15% cholesterol from 5–6 weeks of age for 10 weeks to accelerate atherosclerotic plaque formation. Tamoxifen-inducible endothelial specific CXCR4 deficient mice were generated by crossing Cxcr4flox/flox (B6.129P2-Cxcr4tm2Yzo/J, 008767) mice purchased from Jackson Laboratories with VE-cadherin-CreERT2 (C57BL/6-Tg(Cdh5-cre/ERT2)1Rha) mice22. To induce atherosclerosis formation, they were injected with 5.0 × 1011 genome copies (GC) of adeno-associated virus 2 vector encoding murine PCSK9 (AAV-mPCSK9 vector, Vector Biolabs) intravenously at 5–6 weeks old, and fed with HFD for 10 weeks. Activation of cre recombinase was induced by oral administration of tamoxifen every 3 days over a 9-day period, 1 week before the PET imaging analysis. All experiment procedures were performed in compliance with guidelines set forth by the NIH Office of Laboratory Animal Welfare and approved by the Institutional Animal Care and Use Committee of Washington University.
Autoradiography of whole thoracic aortas
64Cu-DOTA-vMIP-II tracer was synthesized and radiolabeled as we previously reported 20. Whole thoracic aorta of Apoe−/− on HFD for 10 weeks was collected 3 h after intravenous injection of 64Cu-DOTA-vMIP-II tracer via tail vein and surrounding tissues were cleaned up. Collected aortas were cut, opened, washed with PBS, placed on a charged phosphor screen and exposed overnight. Images were obtained with a GE Typhoon FLA 9500 Variable Mode Laser Scanner at 50 micron resolution. After taking autoradiography images, samples were fixed with 4% paraformaldehyde (PFA) and stained with oil red O (ORO) to assess distribution of atherosclerotic plaques. Specifically, aortas were pretreated with 100% propylene glycol (Sigma-Aldrich) for 15 min, then incubated with ORO solution (Sigma-Aldrich) for 3 h at RT. They were washed with 85% propylene glycol, and then washed 3 times with PBS. After staining, aortas were placed between 2 clear sheets and imaged by M205 FA stereoscope system (Leica).
Positron Emission Tomographic (PET) Imaging and biodistribution of PET tracer
PET imaging: Mice were anesthetized with isoflurane and injected with 3.7 MBq of 64Cu-DOTA-vMIP-II or 64Cu-DOTA-FC131 in 100 µL of saline via the tail vein. Small animal PET scan (40–60 min dynamic scan) were performed on either microPET Focus 220 (Siemens, Malvern, PA) or Inveon PET/CT system (Siemens, Malvern, PA). The microPET images were corrected for attenuation, scatter, normalization, and camera dead time and co-registered with microCT images. All of the PET scanners were cross-calibrated periodically. The microPET images were reconstructed with the maximum a posteriori (MAP) algorithm and analyzed by Inveon Research Workplace. The uptake was calculated as the percent injected dose per gram (%ID/g) of tissue in three-dimensional regions of interest (ROIs) without the correction for partial volume effect.
Post-PET biodistribution:
At 3 h post injection of PET tracer, the mice were anesthetized with inhaled isoflurane prior to euthanasia by cervical dislocation. Organs of interest were collected, weighed, and counted in Beckman 8000 gamma counter (Beckman, Fullterton, CA). Standards of each tracer were prepared and counted with the biodistribution samples together to calculate the percentage of the injected dose per gram of tissue (%ID/g).
Plaque regression model
Atherosclerotic plaque regression was induced in Apoe−/− on HFD for 10 weeks as previously described 12. Briefly, 1.0 × 1012 GC of adeno-associated virus 8 vector encoding murine Apoe (AAV-mApoE vector, Vector Biolabs) was injected intravenously to complement Apoe expression. Only male mice were used in this model because AAV vector is much less effective in transducing female mice 23. Then, PET imaging of 64Cu-DOTA-vMIP-II tracer, biodistribution of the tracer in aortas, monocyte recruitment, macrophage accumulation and atherosclerotic plaque area were assessed at baseline, and 2 or 4 weeks after transduction. After PET imaging, aortas were taken out, weighted and counted in gamma counter to measure biodistribution. Hearts were fixed and embedded in OCT compound for cryosections (10 μm) of aortic sinuses. Monocyte recruitment was assessed by counting bead-labeled monocytes in atherosclerotic plaques. In more detail, Ly6Chi monocytes were labeled by i.v. injection of 1.0 µm Fluoresbrite® Polychromatic Red Microspheres (Polysciences Inc.) 2 days before sacrifice and 3 days after i.v. injection of 200 μL of clodronate-loaded liposomes (Liposoma BV). Approximately 30% of Ly6Chi monocytes are labeled by this method (Supplemental Figure ID). Macrophage accumulation in atherosclerosis was assessed by staining with anti-CD68 mAb (FA-11, Bio-Rad) or MOMA-2 (Bio-Rad). Plaque size was analyzed by ORO staining to provide contrast. Plasma total cholesterol was measured by Cholesterol E kit (Wako) according to the manufacture’s instruction.
Assessment of vascular permeability with Evans Blue
To assess vascular permeability in atherosclerotic aortas, 200 μL of 0.5% Evans Blue solution was injected into Apoe−/− mice intravenously. Then, 30 min later, they were euthanatized by CO2 inhalation and thoracic aortas were collected after perfusion with ice cold PBS. Collected aortas were placed between 2 clear sheets and imaged by M205 FA stereoscope system with Cy5 channel (Leica). CXCR4 co-staining was done after fixation using whole mount staining protocol (Supplemental methods).
Statistics
Statistics were evaluated and data plotted using Prism Graphpad software, version 8. Data are presented as mean ± SEM. Statistical comparisons between 2 groups were performed using Mann-Whitney U test. When comparing 2 populations in the same mouse, paired t tests were used. In dealing with experiments included more than two groups, statistical significance was tested by one-way analysis of variance with the Tukey’s post hoc test.
Results
64Cu-DOTA-vMIP-II signal correlates with monocyte recruitment rather than plaque size or macrophage burden in atherosclerotic plaques
We first confirmed that 64Cu-DOTA-vMIP-II tracer (vMIP-II tracer) accumulates at atherosclerotic plaques by comparing uptake of the tracer assessed by autoradiography with plaque distribution assessed by ORO staining in whole thoracic aortas from Apoe−/− mice fed with high fat diet (HFD) for 10 weeks. As shown in Figure 1A, tracer localization was prominent at the arch region where high lipid deposition was localized, suggesting strong recognition of plaque by the tracer. Next, we used a plaque regression model12 to compare the uptake of 64Cu-DOTA-vMIP-II tracer with several biological parameters related to atherosclerosis and over time. Specifically, we treated Apoe−/− mice on HFD with AAV8 vector encoding murine Apoe (AAV-mApoE vector) to restore Apoe expression or PBS as control. Then, in an experimental design depicted in Figure 1B, PET imaging was carried out at baseline, and 2 or 4 weeks after transduction. In this model, plasma total cholesterol level decreased and reached nadir at ~200 mg/dL 1 week after AAV-mApoE transduction (n=13 per group at baseline, n=7 vs. 8 at 3 days, n=12 per group at 1 week, n=9 vs. 10 at 2 weeks, n=4 per group at 3 and 4 weeks, Supplemental Figure IA). There was no difference in atherosclerotic plaque area in this time period (n=8 at baseline, n=5 vs. 6 at 2 weeks, n=4 per group at 4 weeks, Figure 1C–D), whereas it was significantly decreased 8 weeks after treatment (n=4 per group, Supplemental Figure IB). Monocyte recruitment was assessed by labeling Ly6Chi monocytes with fluorescent polystyrene particles (Supplemental Figure IC–D) 12. Counting of bead-labeled cells in plaques confirmed reduction of monocyte recruitment by 2 weeks after transduction with AAV-mApoE and time points beyond (n=8 at baseline, n=5 vs. 6 at 2 weeks, n=4 per group at 4 weeks, Figure 1E). Macrophage accumulation analyzed by CD68 staining did not change at 2 weeks but decreased at 4 weeks post treatment (n=8 at baseline, n=5 vs. 6 at 2 weeks, n=4 per group at 4 weeks, Figure 1F–G). MOMA-2 staining showed a similar result as that obtained with CD68 staining (n=8 at baseline, n=5 vs. 6 at 2 weeks, n=4 per group at 4 weeks, Supplemental Figure IE). Therefore, in this model, plasma total cholesterol is decreased at first, then monocyte recruitment quells, and finally macrophage accumulation and plaque size are decreased in turn. Uptake of 64Cu-DOTA-vMIP-II tracer at the aortic arch, assessed by PET imaging, was decreased by 2 weeks of AAV-mApoE treatment (n=8 at baseline, n=5 vs. 6 at 2 weeks, n=4 per group at 2 weeks, Figure 1H–I). Biodistribution in aortas was likewise decreased (n=8 at baseline, n=5 vs. 6 at 2 weeks, n=4 vs.3 at 4 weeks, Supplemental Figure IF). Linear regression analysis also showed the positive correlation between PET signal and monocyte recruitment (n=27, Online Figure IG). Thus, reductions in monocyte recruitment but not changes in plaque macrophage burden or plaque area, tracked most closely with the aortic uptake of 64Cu-DOTA-vMIP-II tracer.
Figure 1. PET signal derived from 64Cu-DOTA-vMIP-II tracer correlates with monocyte recruitment into atherosclerotic plaques.
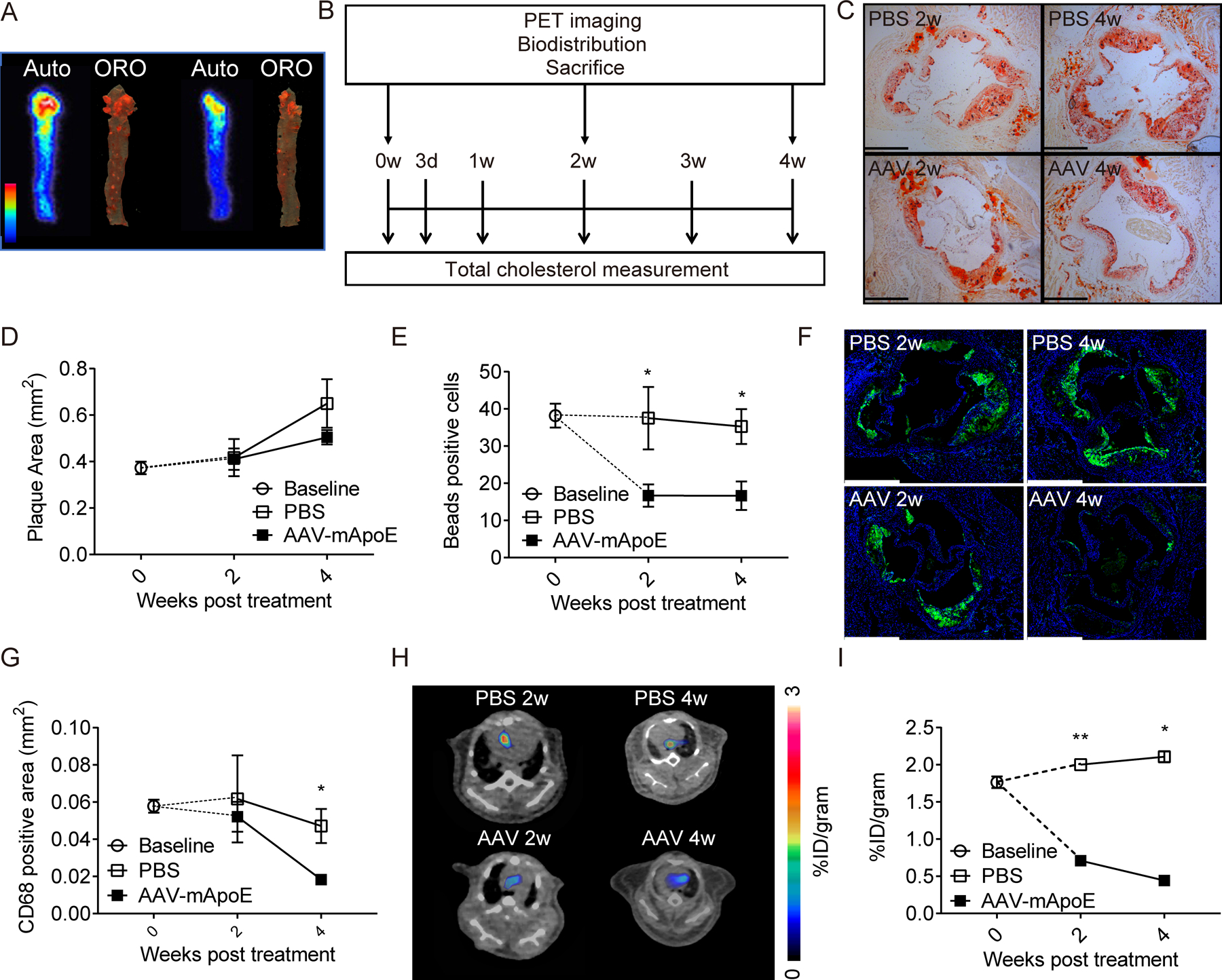
(A) Autoradiography of 64Cu-DOTA-vMIP-II (left: indicated as auto) and corresponding oil red O staining (right: indicated as ORO) of whole thoracic aortas. (B) Scheme of experimental protocol of plaque regression model. Time course of (C-D) plaque areas in aortic sinuses, (E) bead-labeled cells in plaques (F-G) CD68 positive areas, and. (H) Representative 64Cu-DOTA-vMIP-II PET images at aortic arches and (I) quantification of PET signal (n=4–8 per point). All data are shown as means ± SEM. *p<0.05, **p<0.01 by Mann-Whitney U test.
In vivo distribution of 64Cu-DOTA-vMIP-II tracer
To further assess the distribution of 64Cu-DOTA-vMIP-II tracers in vivo, we made a Cy5 -conjugated non-radioactive vMIP-II tracer (Cy5-vMIP-II). Within 1 h after injection, this tracer was found in blood of HFD-fed Apoe−/− mice mainly bound to monocytes (Ly6Chi>Ly6Clo) and neutrophils, with lesser binding to T and B cells in a dose-dependent manner (Figure 2A). Here, most of Cy5-conjugated tracers lined the plaque surface and colocalized with CD31+ endothelial cells (ECs), rather than monocytes/macrophages, in the plaque sections (Figure 2B). Flow cytometric analysis of atherosclerosis-bearing aortas confirmed binding to ECs and recruited monocytes and neutrophils, but not macrophages (Figure 2C, Supplemental Figure IIA) or CD11b− non-myeloid cells (Supplemental Figure IIB). The tracer may have limited access to plaque macrophages, as it may not deeply penetrate into plaques. Another dye-conjugated vMIP-II tracer (CF640R-vMIP-II) also showed its binding capacity for peripheral monocytes and neutrophils (Supplemental Figure IIC), and accumulation at atherosclerotic plaques in whole mount imaging of the thoracic aorta (Supplemental Figure IID) and in aortic ECs (Supplemental Figure IIE). We conclude that there are two likely sources of PET signal arising from the use of vMIP-II tracer in atherosclerotic mice: (1) tracer-bound blood leukocytes, neutrophils or monocytes, recruited into atherosclerotic lesions and (2) direct binding to plaque ECs.
Figure 2. Cy5-conjugated vMIP-II tracer binds to blood monocytes and neutrophils, and to plaque endothelial cells (ECs) in vivo.
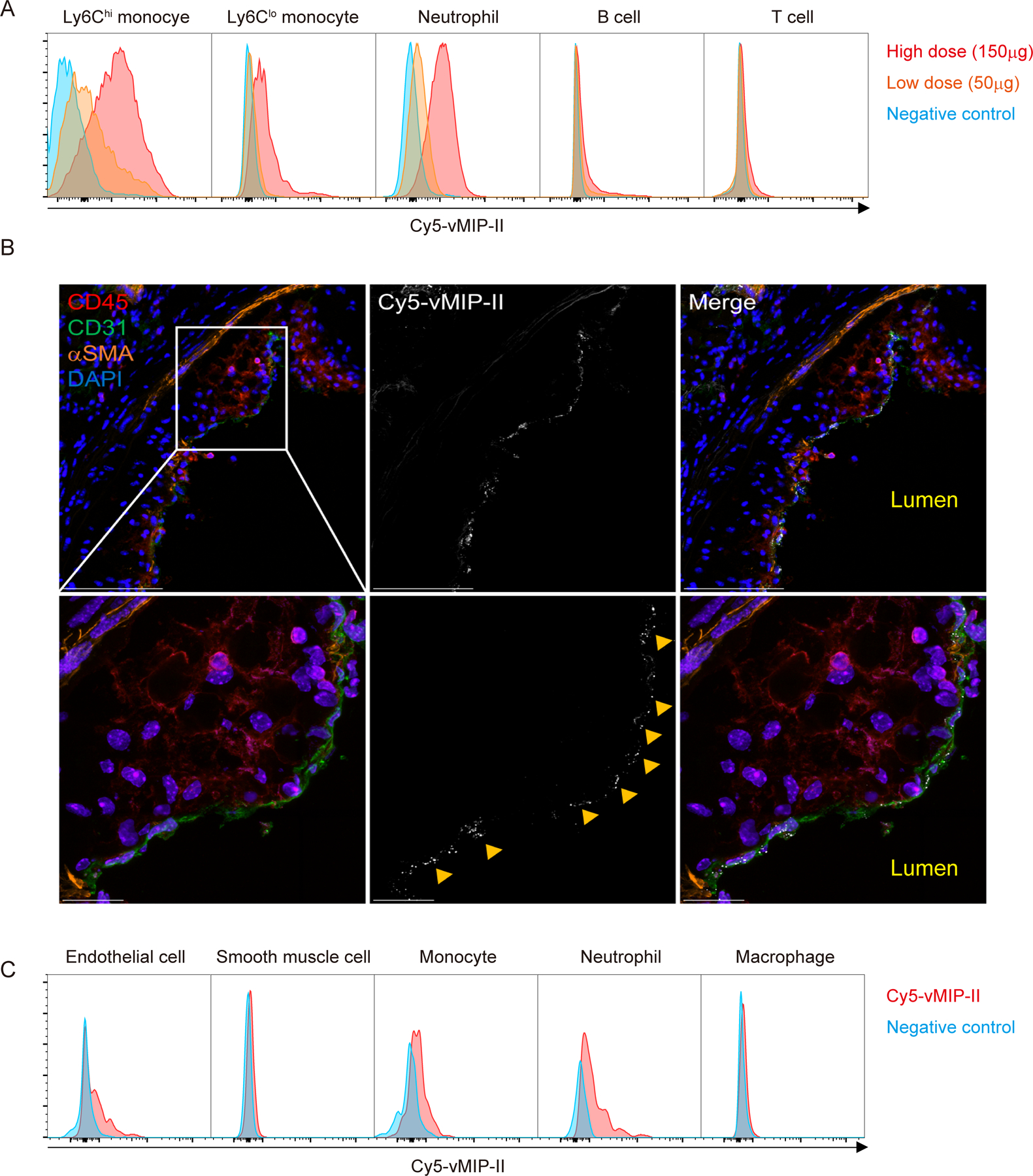
(A) Histogram of Cy5+ leukocyte populations in peripheral blood of Apoe−/− mice 1 h after injection of high dose (150 μg) or low dose (50 μg) of Cy5-vMIP-II tracer. Negative control is each cell population from Apoe−/− mice injected PBS. (B) Distribution of Cy5-vMIP-II tracer (150 μg/mouse) in atherosclerotic plaques examined at low (upper) and high (middle) magnification. Accumulation of the tracer is indicated by yellow arrows. Scale bar: 100 μm (upper) and 20 μm (bottom) (C) Histogram of Cy5+ cell populations in the aorta of Apoe−/− mice 1 h after injection of 150 μg of Cy5-vMIP-II tracer. Negative control is each cell population from Apoe−/− mice injected PBS.
Recruitment of tracer-bound neutrophils and monocytes are not major source of PET signal of 64Cu-DOTA-vMIP-II tracer
To assess the concept that the tracer directly detected monocyte or neutrophil recruitment to plaque, we tried to deplete peripheral monocytes and neutrophils using depleting antibodies in Apoe−/− mice fed with HFD. Although neutrophil depletion using anti-Ly6G was not so effective as recently reported24, both neutrophils and Ly6Chi monocytes were effectively depleted by use of Gr-1 mAb recognizing Ly6C and Ly6G (n=4 per group, Supplemental Figure IIIA–B). Ly6Clo monocytes were not depleted in this experiment. However, more than half of the monocytes recruited to atherosclerotic plaques are Ly6Chi monocytes 25,26, and the preferential binding of dye conjugated-vMIP-II to Ly6Chi monocytes predisposes to detecting this subset (Figure 2A, Supplemental Figure IIC). Approximately 25% reduction of the uptake was observed 24 h after treatment even after both neutrophil and Ly6Chi monocyte were depleted (n=4 in control group, n=3 in Ly6G group, n=3 Gr-1 group, Figure 3A–B). Biodistribution of the tracer in aortas from the same experiment confirmed the partial and modest impact of monocyte and neutrophil depletion (n=4 in control group, n=3 in Ly6G group, n=3 Gr-1 group, Supplemental Figure IIIC). Therefore, despite correlating with monocyte influx, direct recognition of monocytes or neutrophils does not appear to be the prime target of the vMIP-II tracer in regressing plaques.
Figure 3. Recruitment of tracer-bound leukocytes, non-specific binding of the tracer, or change in plaque glycosaminoglycan content do not account for the reduction of PET signal in regressing plaques.
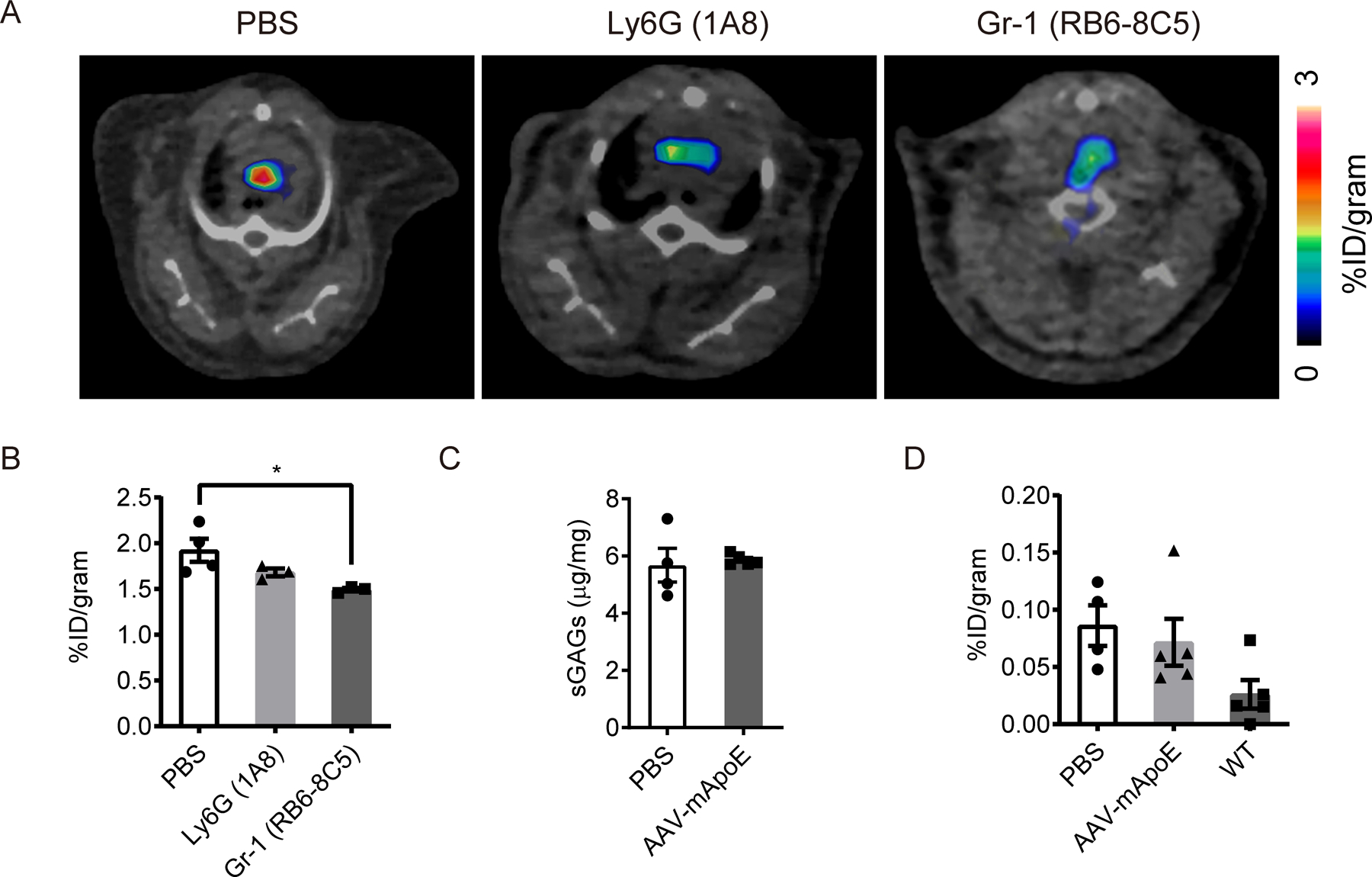
(A) Representative 64Cu-DOTA-vMIP-II PET images and (B) quantification (n=3–4 per group) at aortic arches of Apoe−/− mice on HFD with or without leukocyte depletion. *p<0.05 by 1-way ANOVA with the Turkey post hoc test. (C) Sulfated glycosaminoglycan (sGAG) content in whole thoracic aortas from Apoe−/− mice with or without 2 weeks of AAV-mApoE treatment (n=4–5 per group). (D) Biodistribution of 64Cu-DOTA-PEG in aortas from Apoe−/− mice with or without 2 weeks of AAV-mApoE treatment, or WT mice (n=4–5 per group). All data are shown as means ± SEM.
Neither change of glycosaminoglycan content in atherosclerosis nor nonspecific tracer recruitment into plaques accounts for tracer reactivity
Several chemokines including vMIP-II can also bind to glycosaminoglycans (GAGs) 27,28 which are known to have important roles in leukocyte adhesion and recruitment, and vessel permeability. Moreover, the amount of each type of GAGs changes respectively in atherosclerotic artery 29–31. Thus, we considered the possibility that plaque GAGs that might bind to the 64Cu-DOTA-vMIP-II tracer were increased in atherosclerotic plaques actively recruiting monocytes. To this end, we measured sulfated GAG content in aortas from Apoe−/− mice with or without AAV treatment, but we found no reduction in total GAG content 2 weeks after AAV-mApoE treatment (n=4 vs. 5, Figure 3C). Moreover, the content of subtypes of GAGs (O- and N-sulfated GAGs) also did not change (n=4 vs. 5, Supplemental Figure IIID).
We also considered the possibility that the tracer identity is unimportant and that the greater intensity in progressing plaques reflects higher endothelial permeability to nonspecific substances 32,33. To assess this, we employed a tracer which had the similar molecular weight (~10 KDa) as the vMIP-II tracer but without the chemokine receptor binding activity (64Cu-DOTA-PEG). We then measured biodistribution of this tracer in aortas from Apoe−/− mice with or without AAV treatment, or WT mice as control. Although tracer accumulation in aortas tended to increase in Apoe−/− mice compared with WT mice, the signal was quite low overall and there was no difference between Apoe−/− mice with and without AAV-mApoE treatment (n=4 in PBS group, n=5 in AAV-mApoE group, n=5 in WT group, Figure 3D). Moreover, the uptake of nonspecific 64Cu-DOTA-PEG tracer was negligible compared with 64Cu-DOTA-vMIP-II tracer. We thus conclude that 64Cu-DOTA-vMIP-II tracer uptake in atherosclerotic plaque was mainly due to the binding to chemokine receptors overexpressed on aortic endothelial cells.
64Cu-DOTA-vMIP-II tracer mainly reports CXCR4 expression on plaque ECs that varies with disease activity
Some vascular ECs highly express CXCR4 34,35, a receptor that has binding capacity for vMIP-II 16–18. Given that we visualized tracer binding to endothelium (Figure 2B, Supplemental Figure IIE), it seemed possible that vMIP-II tracer binds to plaque ECs through CXCR4. Staining of aortic sinus from Apoe−/− mice on HFD showed that CXCR4 was expressed in plaque ECs and by a few scattered macrophages and smooth muscle cells (Figure 4A). CXCR4 expression in the aorta highlighted plaque areas (Figure 4B–C; arrows point to raised plaques in both panels, arrowhead in Figure 4C to nonplaque area).
Figure 4. CXCR4 is highly expressed in plaque endothelial cells especially at the edge or the shoulder of the plaque.
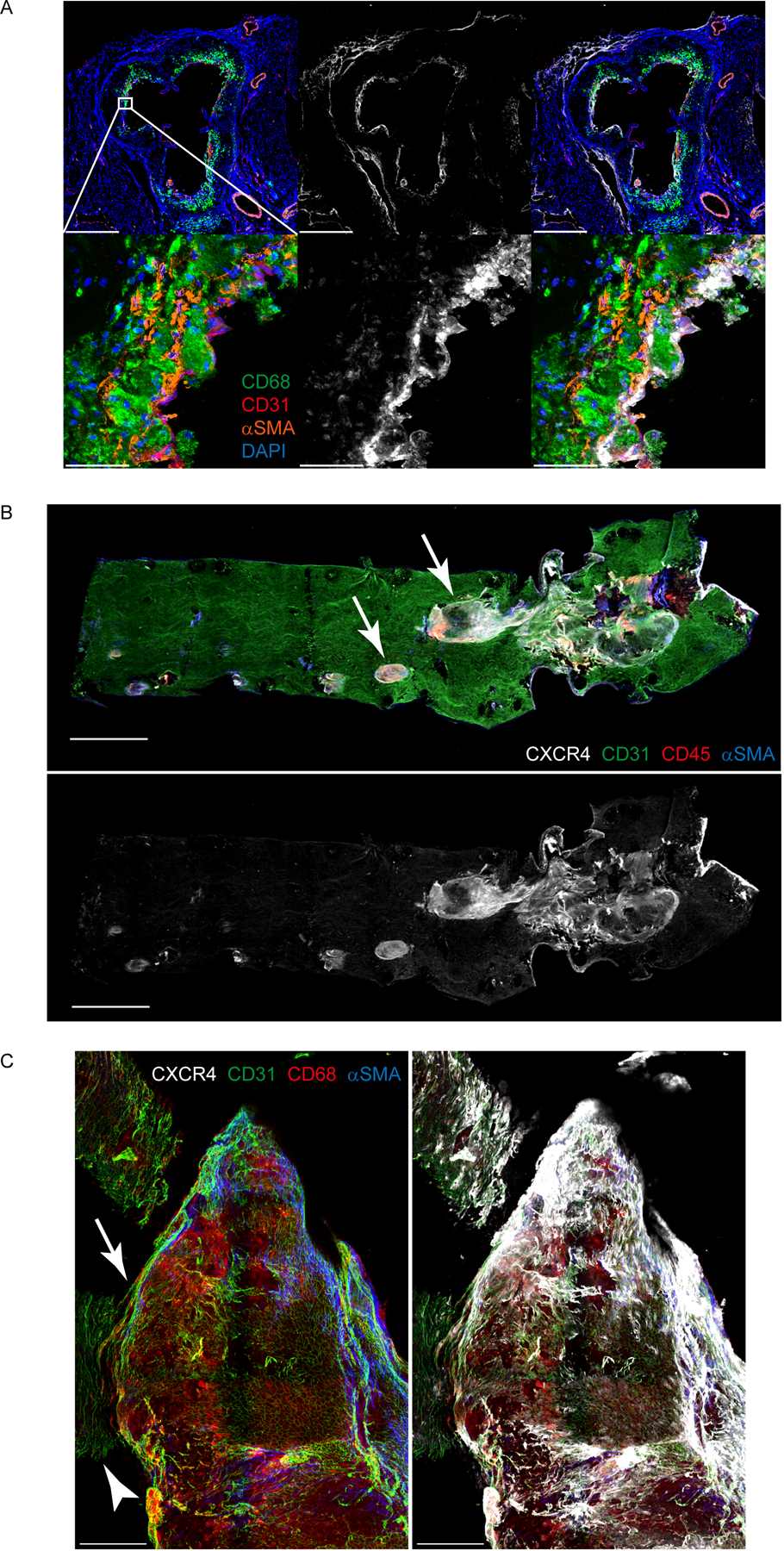
(A) CXCR4 expression in aortic sinus from Apoe−/− mice on HFD. Scale bar: 500 μm (upper) and 50 μm (bottom) (B) CXCR4 distribution in whole thoracic aorta from Apoe−/− mice on HFD. Scale bar: 1 mm (C) CXCR4 distribution in the plaque at the aortic arch from Apoe−/− mice on HFD. Scale bar: 200 μm. Arrow points to raised plaque on a field of surrounding nonplaque area (full background including arrow marked by arrowhead).
If CXCR4 might be a target of vMIP-II tracer, we wondered if CXCR4 expression on plaque endothelium declined during plaque regression. Antibody staining for CXCR4 in aortic plaque sinus sections showed markedly reduced signal in regressing plaques (n=6 per group, Figure 5A–B). Flow cytometric analysis also showed a reduction in CXCR4hi ECs 2 weeks post AAV-mApoE treatment (n=7 per group, Figure 5C–D). The recent literature demonstrated that vMIP-II also has a binding capacity for one of the atypical chemokine receptors CXCR7 (ACKR3)19 which is expressed in ECs36. However, CXCR7 expression on aortic ECs did not change post AAV-mApoE treatment (n=7 per group, Supplemental Figure IVA–B).
Figure 5. Plaque regression is associated with loss of CXCR4 on aortic endothelium.
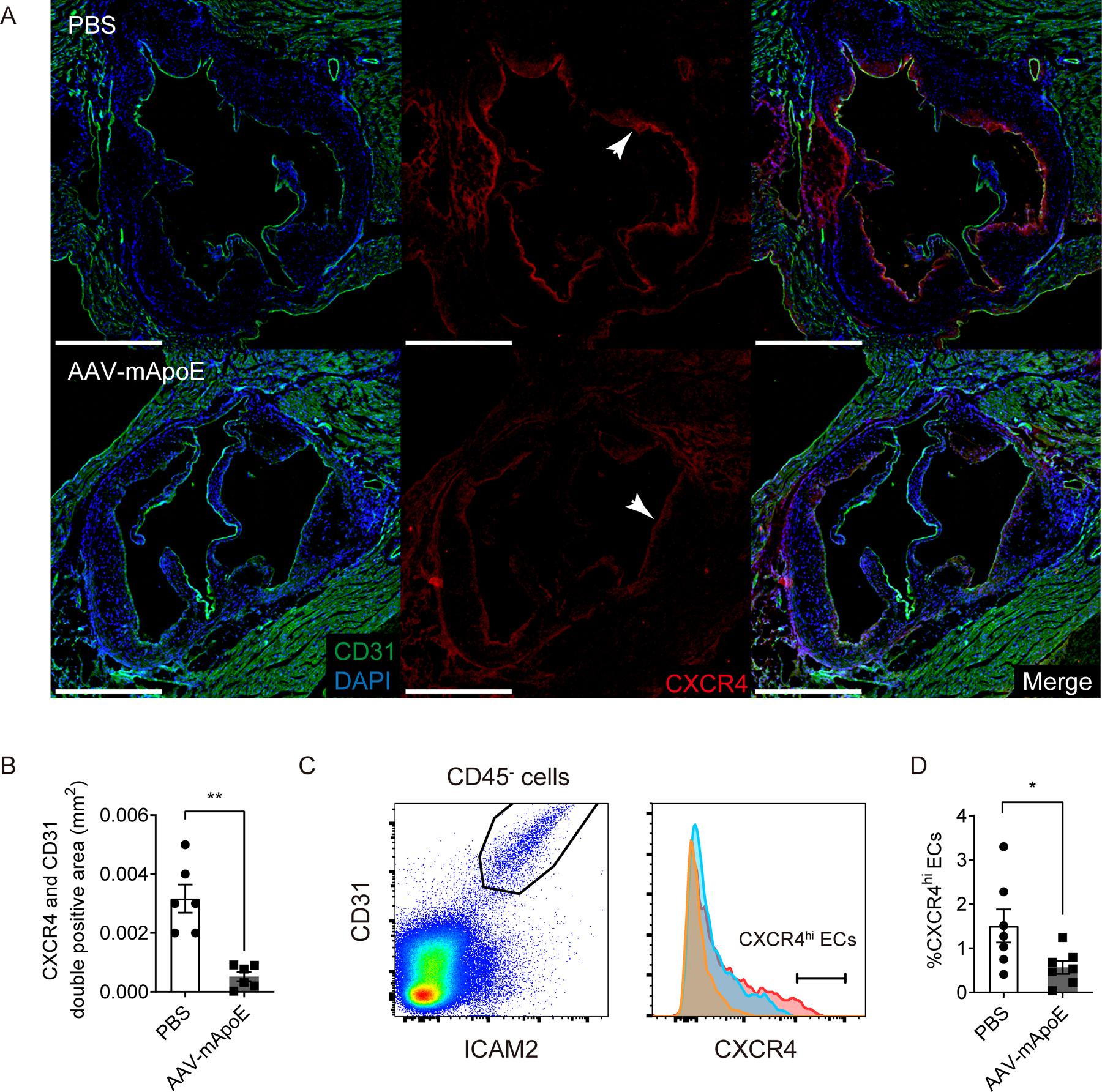
(A) Representative images of CXCR4 expression in aortic sinus from Apoe−/− mice on HFD after 2 weeks of PBS (upper) or AAV-mApoE (bottom) treatment. Scale bar: 500 μm, and (B) quantification of CXCR4+ CD31+ areas (n=6 per group). (C) Representative histogram of CXCR4 expression on aortic ECs from Apoe−/− mice on HFD after 2 weeks of PBS (red) or AAV-mApoE (blue) treatment. Yellow histogram is isotype control. (D) The percentage of CXCR4hi ECs in total aortic ECs with or without 2 weeks of AAV-mApoE treatment (n=7 per group). All data are shown as means ± SEM. *p<0.05, **p<0.01 by Mann-Whitney U test.
Blocking of CXCR4 by CXCR4-selective antagonist FC131 37, which uses the same binding site with vMIP-II 38, dramatically reduced PET signal of vMIP-II tracer at aortic arches of Apoe−/− mice on HFD (n=5 per group, Figure 6A–B), as also observed when analyzed by biodistribution (n=8 per group, Supplemental Figure IVC). Moreover, we generated tamoxifen-inducible endothelial specific CXCR4 deficient mice by crossing CXCR4flox/flox mice with VE-cadherin-CreERT2 mice to further evaluate whether endothelial CXCR4 is a major accessible target for vMIP-II tracer. These mice were treated with AAV-mPCSK9 vector and fed with HFD to induce atherosclerosis formation. Then, CXCR4 deletion was induced by tamoxifen treatment (Figure 6C, Supplemental Figure IVD) before PET imaging analysis. The PET signal of vMIP-II tracer was attenuated, though not eliminated, by endothelial specific CXCR4 deletion (n=9 in control group, n=7 in CXCR4 deletion group, Figure 6D–E, n=3 in control group, n=4 in CXCR4 deletion group, Supplemental Figure IVE). We also demonstrated that our fluorescence-conjugated versions of vMIP-II bound to both human CXCR4 and CXCR7 expressing cells in vitro but displayed a higher affinity for CXCR4 (n=3 per group, Supplemental Figure IVF–G). Altogether, these data illustrate that a major target for the vMIP-II tracer in atherosclerosis-affected aortas is CXCR4 on endothelium.
Figure 6. CXCR4 antagonists interfere with 64Cu-DOTA-vMIP-II detection of atherosclerosis in Apoe−/− mice.
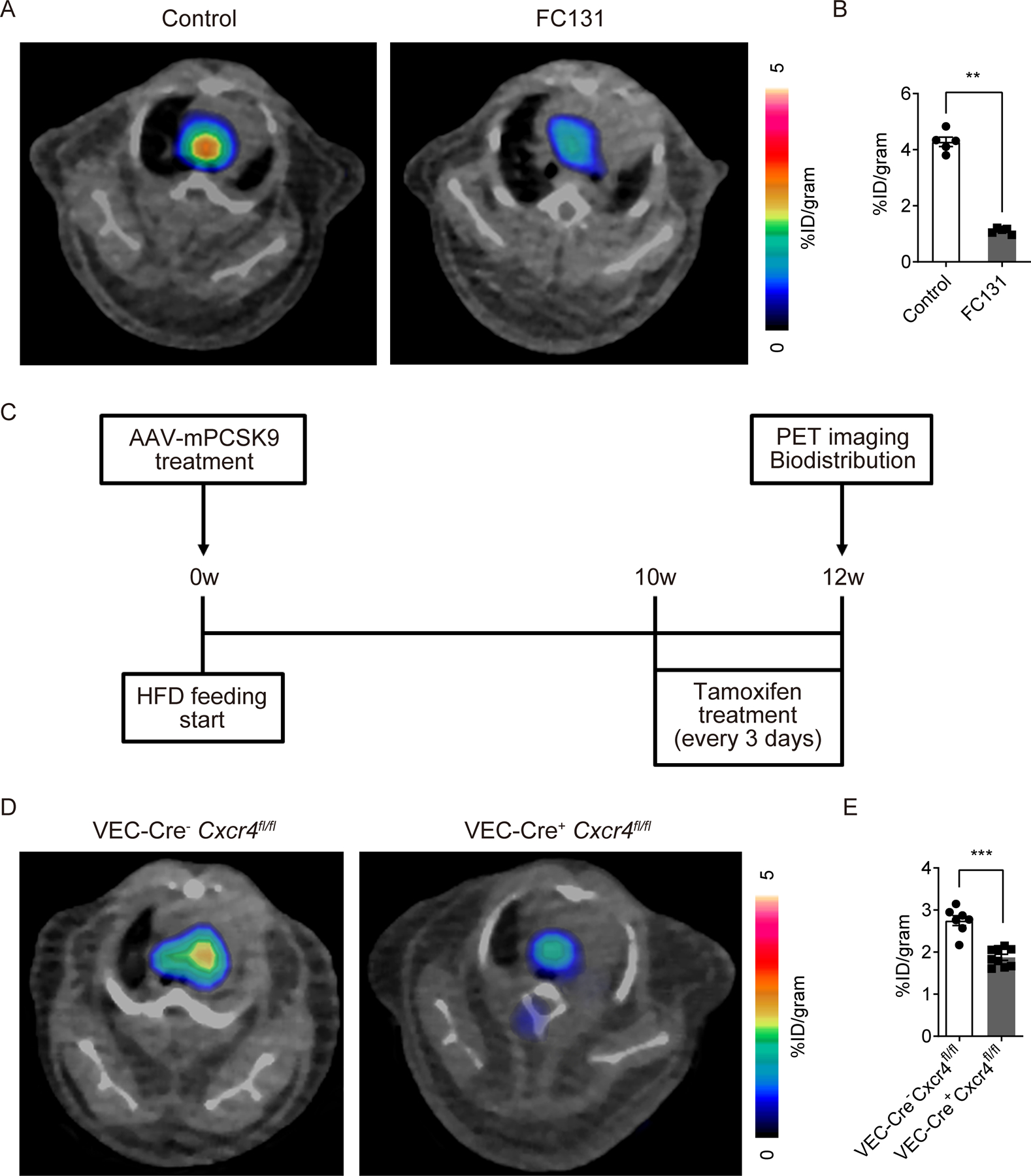
(A) Representative 64Cu-DOTA-vMIP-II PET images and (B) quantification at aortic arches of Apoe−/− mice on HFD with or without CXCR4 blocking by 0.5 mg of FC131 (n=5 per group). (C) Scheme of experimental protocol of endothelial specific CXCR4 deletion model. (D) Representative 64Cu-DOTA-vMIP-II PET images and (E) quantification at aortic arches of Cxcr4flox/flox mice with or without VE-Cadherin-CreERT2 (VEC-Cre) treated with AAV-mPCSK9 (n=7–9 per group). All data are shown as means ± SEM. **p<0.01, ***p<0.001 by Mann-Whitney U test.
CXCR4 expression in atherosclerotic lesions marks sites where endothelial permeability is prominently enhanced
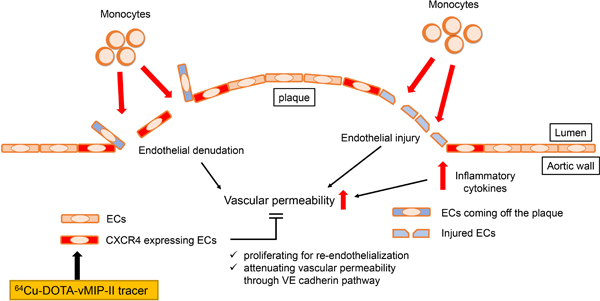
Vascular permeability is known to be increased at atherosclerotic lesions 32,33. Endothelial CXCR4 attenuates vascular permeability through enhancement of VE-cadherin expression and stabilization of junctional VE-cadherin complexes 21. Therefore, we wondered whether endothelial CXCR4 expression is upregulated to compensate for enhanced vascular permeability at atherosclerotic lesions. To examine this hypothesis, we first assessed distribution of permeability in thoracic aorta lesions of Apoe−/− mice on HFD by in vivo injection of Evans Blue dye. As expected, Evans Blue leakage mainly occurred at atherosclerotic lesions but interestingly especially at the edge of plaques (Figure 7A). Indeed, co-staining of CXCR4 demonstrated Evans Blue leakage around CXCR4-expressing endothelial areas (Figure 7B). Since CXCR4 blocking or endothelial specific CXCR4 deficiency was earlier reported to enhance Evans Blue leakage from aortic arches 21, this colocalization likely reflects a compensatory attempt of the endothelium to combat elevated permeability.
Figure 7. Vascular permeability is increased along the margins of atherosclerotic plaques where CXCR4-expressing ECs proliferate.
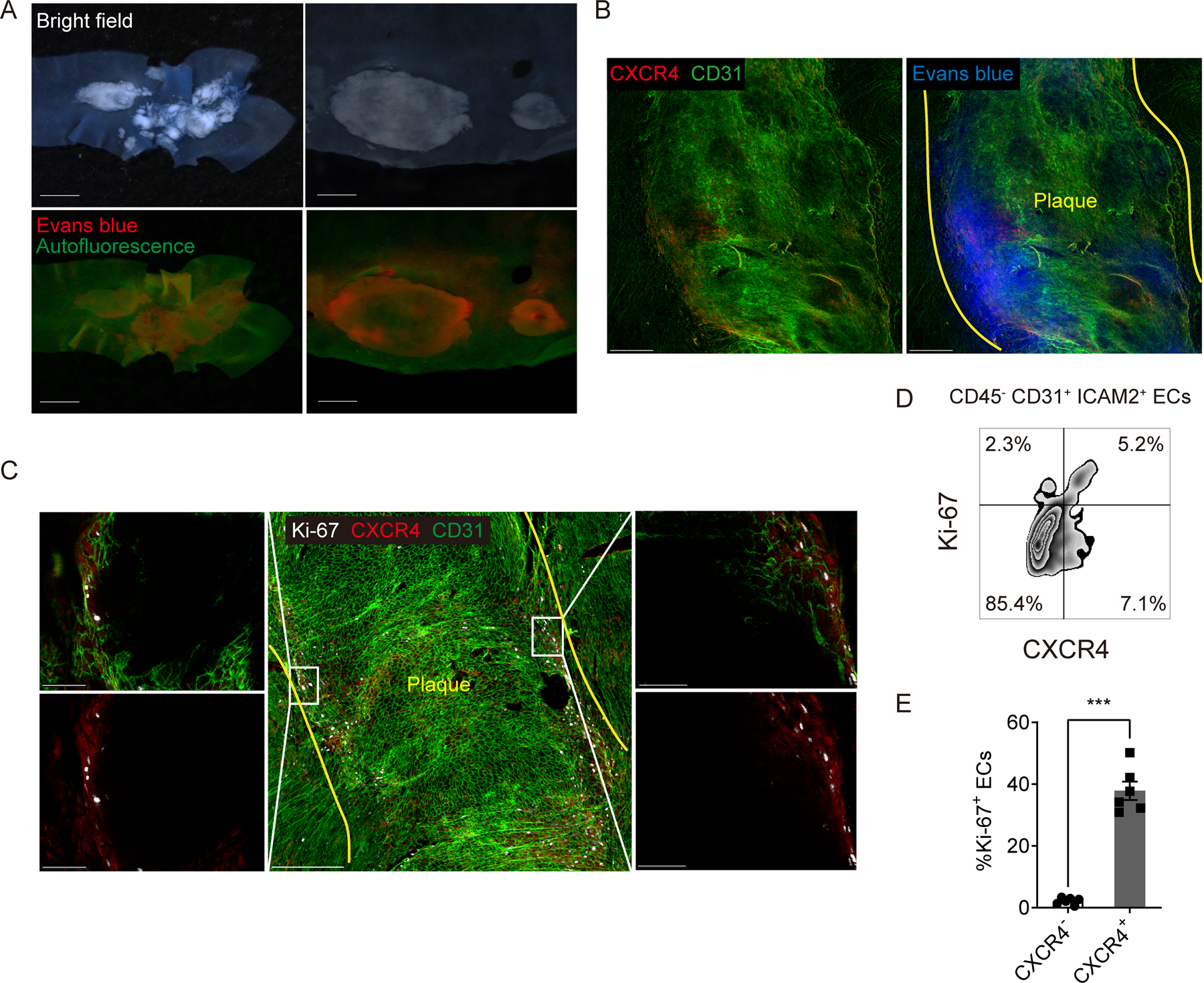
(A) Bright field (upper) and fluorescent (bottom) stereoscope images of an aortic arch (left) and plaques in a descending aorta (right) from Apoe−/− mice injected 200 μL of 0.5% Evans blue. Scale bar: 1 mm (left) and 500 μm (right) (B) CXCR4 distribution in a plaque of Apoe−/− mice injected evans blue. Yellow lines indicate the edged of the plaque. Scale bar: 200 μm (C) Ki-67 and CXCR4 distribution in atherosclerotic plaques at an aortic arch with low (middle) and high (left and right) magnification. Yellow lines indicate the edged of the plaque. Scale bar: 200 μm (middle) and 50 μm (left and right). (D) The scheme for gating of Ki-67 or CXCR4 positive ECs. (E) The percentage of Ki-67 positive cells in CXCR4 positive or negative ECs (n=6 per group). Data are shown as means ± SEM. ***p<0.001 by paired t test.
Analysis of endothelial changes in proliferation and turnover
Endothelial denudation and turnover are promoted at atherosclerotic lesions 39,40. After vascular injury, CXCR4 enhances endothelial proliferation and re-endothelialization while Ki-67+ ECs are reduced in endothelial-specific CXCR4-deficient mice after endothelial injury 41. Therefore, we hypothesized that endothelial CXCR4 may promote recovery from endothelial injury or denudation especially at the edge of plaques by promoting proliferation. To assess this possibility, we stained Ki-67 as a proliferation marker in whole thoracic aorta. Ki-67+ cells were mainly located at the edge of atherosclerotic plaques and were CXCR4+ ECs (Figure 7C, Supplemental Figure IVH). Flow cytometric analysis confirmed that CXCR4+ aortic ECs were more proliferative (n=6 per group, Figure 7D–E). These data are consistent with the previously reported protective role that CXCR4 has in endothelial repair and in combatting heightened permeability 41. Despite the potentially direct protective effects of CXCR4, ECs that have induced CXCR4 would be those facing adverse conditions that lead to heightened permeability and need for repair in the first place and thus CXCR4+ ECs may also express pro-inflammatory markers. Indeed, CXCR4+ HUVEC upregulate MCP-1 (CCL2) 42, a chemokine that can directly induce monocyte recruitment. Consistent with this, MCP1 expression was increased in CXCR4 expressing aortic ECs from Apoe−/− mice on HFD (n=5 per group, Supplemental Figure VA–B). Thus, our data, together with existing literature, would suggest that CXCR4 expression on ECs correlates with endothelial loss of homeostasis and thus appears to mark ECs that actively promote monocyte recruitment, while also having intrinsic protective roles to restore homeostasis.
CXCR4 specific PET tracer also accumulates in plaque ECs and shows a reduction of PET signal in regressing plaques
To further investigate the hypothesis that CXCR4 signal reports the level of endothelial injury in atherosclerosis, we compared 64Cu-DOTA-vMIP-II to 64Cu-DOTA-FC131, a CXCR4 specific tracer. Although PET signals arising from CXCR4-specific tracer is already reported in atherosclerosis 43–46, the plaque characteristics, or biological parameters of plaque activity, that may or may not correlate with its PET signal are still not fully understood. Similar to studies with the vMIP-II tracer (Figure 1), attenuation of CXCR4-specific PET signal and biodistribution in thoracic aortas were observed 2 weeks post AAV-mApoE treatment (n=6 per group, Figure 8A–B, Supplemental Figure VC), when monocyte recruitment was reduced by plaque area and macrophage burden had not changed. We also detected accumulation of the tracer in other organs (Supplemental Figure VC), as expected, where the apoE vector reduced binding only in the aorta for both the CXCR4-specific tracer and the vMIP II tracer (Figure 8B). Dye-conjugated CXCR4 specific tracer (CF640R-FC131) also accumulated in plaque ECs (Figure 8C) and bound to peripheral monocytes and less to neutrophils and B cells (Supplemental Figure VD), which is comparable with CXCR4 expression in peripheral leukocytes (Supplemental Figure VE). These data indicate that CXCR4-specific tracer can be a useful tool to detect endothelial status in plaques, including sufficient levels of endothelial injury to promote monocyte recruitment.
Figure 8. FC131-based CXCR4-specific tracer mirrors the behavior of vMIP-II tracer in regressing versus progressing atherosclerotic plaques.
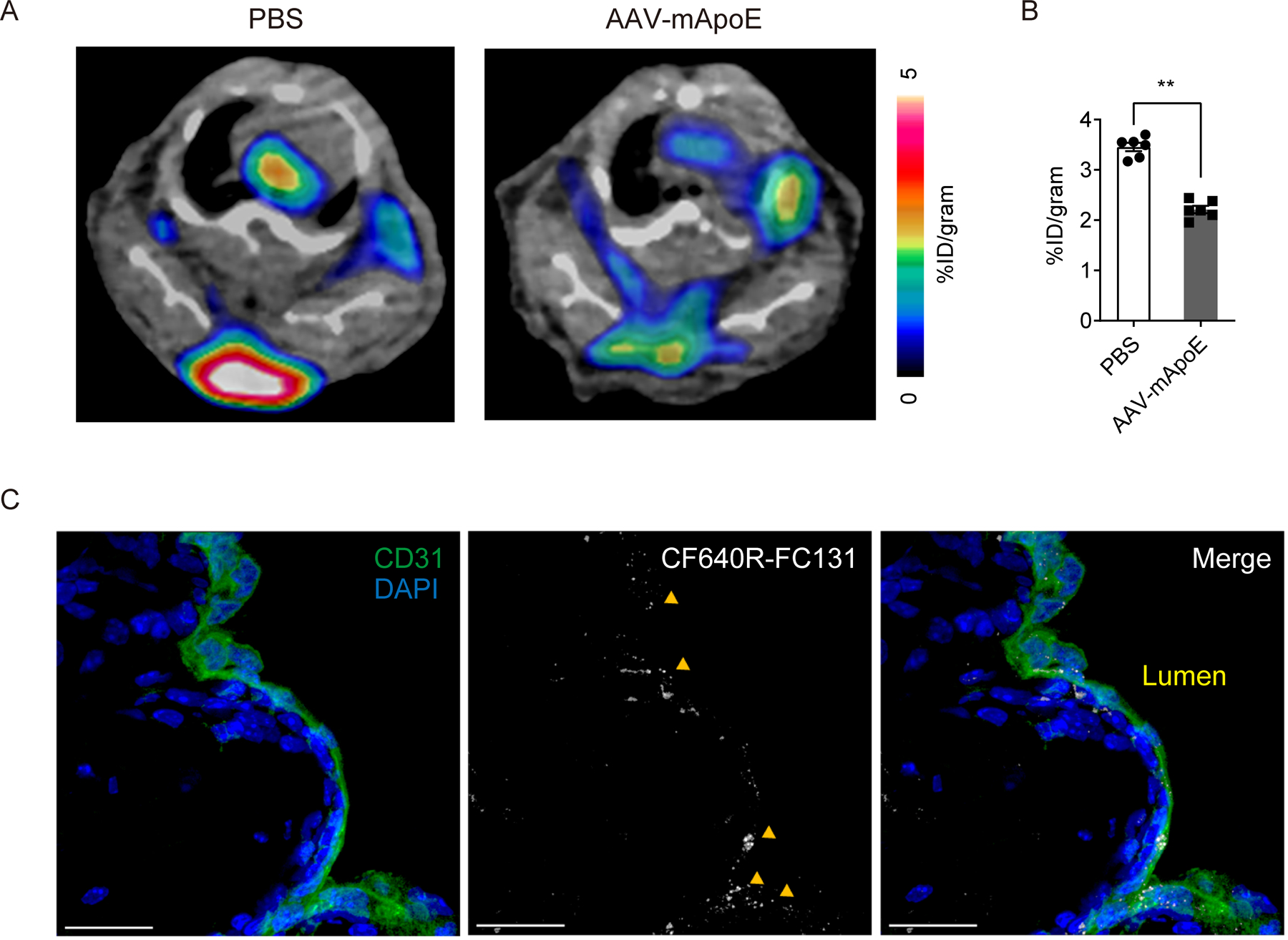
(A) Representative 64Cu-DOTA-FC131 images at aortic arches of Apoe−/− mice on HFD 2 weeks post PBS or AAV-mApoE treatment and (B) quantification of PET signal (n=6 per group). Data are shown as means ± SEM. **p<0.01 by by Mann-Whitney U test. (C) Accumulation of CF640R-FC131 tracer (40 μg/mouse) in an atherosclerotic plaque. Accumulation of the tracer is indicated by yellow arrows. Scale bar: 20 μm
Discussion
We had earlier developed the vMIP-II based PET tracer 64Cu-DOTA-vMIP-II, which accumulates at atherosclerotic lesions in mice 20. Based on the nature of vMIP-II, this tracer can bind to a series of chemokine receptors, including most atherosclerosis-related chemokine receptors 16–18. Initially, it seemed reasonable to expect that this tracer might react broadly with plaque macrophages in mouse models of atherosclerosis. However, we were also cognizant of the possibility that some regions of plaque might be inaccessible to the tracer and thus we set out to determine if the tracer might, in fact, be most accessible to monocytes and thereby serve as a beacon of the relative tendency of monocytes to enter atherosclerotic plaques. Indeed, by employing a model system in which monocyte recruitment can be modulated before changes in plaque burden occur, we were able to determine that vMIP-II tracer accumulation in the artery wall of mice with experimental atherosclerosis is a reliable correlate to monocyte recruitment, which in turn is a strong correlate to disease activity.
We were surprised, however, that CXCR4 expressed on plaque ECs was a major target of vMIP-II tracer. Many of the chemokine receptors that would be targets of the vMIP-II tracer are expressed by monocytes and macrophages, including CXCR4 itself. Yet, our data suggest that macrophages in plaques may be rather inaccessible to our chemokine receptor targeting tracers. We argue that this problem is due to inadequate access of the tracer to the deeper regions of the plaque, whereas the endothelium is quite accessible and thus dominates in signal. Monocytes in the blood are also accessible to the tracer, but the pace of recruitment of monocytes, even during higher levels of recruitment, may also be too low to obscure a key role for endothelial cell binding in overall magnitude.
When our data unexpectedly pointed to the plaque endothelium as a major target, we began to consider endothelial CXCR4 as a dominant ligand for the vMIP-II tracer. Indeed, we were able to block vMIP-II tracer signal with both CXCR4-specific antagonist and endothelial specific CXCR4 deletion. We speculate that the more modest decline of the PET signal in endothelial cell-specific CXCR4 deletion compared to CXCR4 blocking is because some tracers which cannot bind to CXCR4 deficient endothelium may be able to penetrate a plaque and bind to other cells in the absence of the most accessible ligand CXCR4. CXCR4 is known to be expressed in ECs 34,35, albeit not expressed under all conditions. We did not directly demonstrate how endothelial CXCR4 is associated with monocyte recruitment in atherosclerosis. However, based on the literature that re-endothelialization of arteries, endothelial proliferation 41, and reduced atherosclerosis 21 are linked to CXCR4 expression and function, we argue that CXCR4 itself has a protective role. The local regeneration of ECs recovering from endothelial injury in vivo shows enhancement of CXCR4 expression 47. Endothelial CXCR4 is also reported to be athero-protective through attenuating vascular permeability by enhancing VE-cadherin expression and stabilizing VE-cadherin complexes 21. However, despite its likely protective functional role, the induction of its expression is known to be a consequence of endothelial injury 41 and thus its expression correlates with active plaque. Indeed, we found that CXCR4 expressing aortic ECs are more likely to express MCP-1. This expression of MCP-1 does not mean that endothelial CXCR4 signaling per se is athero-promoting, but rather that its expression is associated with a loss of homeostasis that is associated with inflammation, even though the net effect of CXCR4 signaling per se is athero-protective 21.
Overall, we propose that CXCR4 is upregulated in plaque ECs, especially at the edge or shoulder of the plaque, to compensate increased vascular permeability through promotion of re-endothelialization. However, pathophysiological activities which enhance vascular permeability are not completely compensated by the upregulation of CXCR4 and signs of ongoing injury include monocyte recruitment. The potential utility of this tracer in a clinical setting may include detection of very early plaques even before atherosclerosis is formed, which is currently difficult, because monocyte recruitment or increased endothelial permeability is important for the initiation of atherosclerosis 32,48,49. Given that impairment of re-endothelialization is an important factor inducing restenosis after percutaneous coronary intervention 50,51, PET imaging of endothelial CXCR4 targeting tracer may also be useful to predict fatal complications after coronary intervention. Even though nowadays restenosis after stenting is drastically reduced by drug-eluting stent, very late stent thrombosis is arising as a too-often fatal problem instead, and this problem is also caused by delayed re-endothelialization 51–53. Furthermore, this tracer might be useful to detect unstable plaque by recognizing endothelial denudation which is one of the features of plaque vulnerability 54,55 although further human study is necessary because of lack of an animal model of plaque rupture. For whatever such clinical utilization, selected high risk patients would be a target of the PET scanning in terms of the cost and radiation exposure.
It is important to point out that an argument has been that a different tracer targeting CXCR4, 68Ga-Pentixafor, has been reported to have the capacity to penetrate plaques and reach deeper macrophages therein 43. 68Ga-Pentixafor is, in fact, a smaller molecule than our chemokine receptor-targeting tracers and thus may have distinct bioavailability. Thus, a tracer with limited access to deeper area of plaques, like the one we describe here, may behave differently than 68Ga-Pentixafor. Future studies will be needed to address this point.
The source of the ligand for CXCR4, CXCL12, is another interest for future studies. Recently, elevated blood CXCL12 level has been identified as a risk factor of coronary artery disease, 56,57 and endothelial-specific CXCL12 deletion was reported to accelerate plaque progression in murine atherosclerosis 57, seemingly opposite to endothelial CXCR4. However, CXCL12 has another receptor, CXCR7, expressed on ECs 36 which regulates systemic CXCL12 level 58. Considering these complexities, an important future direction will be to investigate the biology of CXCL12 in the endothelial injury response. It may be the case that the local availability of CXCL12 determines the pace at which the injured endothelium returns to normalcy, thereby regulating the period of time that a plaque might be most at risk. In summary, we report that 64Cu-DOTA-vMIP-II PET tracer identifies monocyte recruitment in part by directly binding to monocytes, but also because it identifies areas of injured endothelium through binding to CXCR4. We thus believe that this tracer, or others binding CXCR4, has potentially ideal characteristics to identify plaques that may need clinical attention. Furthermore, because CXCR4 expressing endothelial cells are positioned in key locations, like the border of inflammatory plaques, future efforts to combine CXCR4 targeting with state-of-the-art nanotherapeutic approaches 59 may be an especially appealing way to enrich therapeutics to the most pathologic atherosclerotic lesions.
Supplementary Material
Highlights.
64Cu-DOTA-vMIP-II PET tracer is useful to assess monocyte recruitment or endothelial injury.
The main target of 64Cu-DOTA-vMIP-II tracer is endothelial CXCR4 expressed primarily at the edge or the shoulder of atherosclerotic plaques.
CXCR4 is highly expressed on proliferating plaque endothelial cells especially at the edge or the shoulder where vascular permeability is enhanced.
CXCR4-specific 64Cu-DOTA-FC131 tracer shows similar reactivity with 64Cu-DOTA-vMIP-II tracer.
Acknowledgments
We thank Mary Wohltmann for expert assistance with the mouse colony and Shashi Bala Kumar for assistance with imaging.
Source of Funding
This work was funded by R01 HL125655, R35 HL145212, and P41 EB025815 to YL; by DP1DK109668, R37AI049653, HL118206, and Washington University internal funds to GJR; and Luxembourg Institute of Health (LIH) MESR and Luxembourg National Research Fund (FNR) (PRIDE-11012546 “NextImmune”). OB was the recipient of a one-year fellowship from the Banyu Life Sciences Foundation International. His present address is Kyoto University, Japan. LH was supported by American Heart Association Career Development Award #18CDA34110273. AE was supported by T32 HL07081. His present address is Missouri Baptist University, Saint Louis, MO. Resources from the Diabetes Research Center were also utilized and funded by P30 DK020579.
Non-standard Abbreviations and Acronyms:
- AAV-mApoE vector
Adeno-associated virus 8 vector encoding murine Apoe
- CT
Computed tomography
- ECs
Endothelial cells
- GAGs
Glycosaminoglycans
- HFD
High fat diet
- MRI
Magnetic resonance imaging
- ORO
Oil red O
- PET
Positron emission tomography
- vMIP-II
Viral macrophage inflammatory protein 2
- vMIP-II tracer
64Cu-DOTA-vMIP-II tracer
Footnotes
Disclosures
The authors have declared that no conflict of interest exists.
References
- 1.Tyrovolas S, Lotufo PA, John D, et al. Global, Regional, and National Burden of Cardiovascular Diseases for 10 Causes, 1990 to 2015. J Am Coll Cardiol 2017;70:1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tarkin JM, Dweck MR, Evans NR, Takx RAP, Brown AJ, Tawakol A, Fayad ZA, Rudd JHF. Imaging Atherosclerosis. Circ Res 2016;118:750–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Little WC, Constantinescu M, Applegate RJ, Kutcher M a, Burrows MT, Kahl FR, Santamore WP. Can coronary angiography predict the site of a subsequent myocardial infarction in patients with mild-to-moderate coronary artery disease? Circulation. 1988;78:1157–1166. [DOI] [PubMed] [Google Scholar]
- 4.Ambrose JA, Tannenbaum MA, Alexopoulos D, Hjemdahl-Monsen CE, Leavy J, Weiss M, Borrico S, Gorlin R, Fuster V. Angiographic progression of coronary artery disease and the development of myocardial infarction. J Am Coll Cardiol 1988;12:56–62. [DOI] [PubMed] [Google Scholar]
- 5.Finn AV, Nakano M, Narula J, Kolodgie FD, Virmani R. Concept of vulnerable/unstable plaque. Arterioscler Thromb Vasc Biol 2010;30:1282–1292. [DOI] [PubMed] [Google Scholar]
- 6.Dobrucki W, Dobrucki LW, Sinusas AJ. PET and SPECT in cardiovascular molecular imaging. Nat Rev Cardiol 2009;7:38–47. [DOI] [PubMed] [Google Scholar]
- 7.Evans NR, Tarkin JM, Chowdhury MM, Warburton EA, Rudd JHF. PET Imaging of Atherosclerotic Disease: Advancing Plaque Assessment from Anatomy to Pathophysiology. Curr Atheroscler Rep 2016;18:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Andrews JPM, Fayad ZA, Dweck MR. New methods to image unstable atherosclerotic plaques. Atherosclerosis. 2018;272:118–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nahrendorf M, Swirski FK. Cholesterol, CCR2, and monocyte phenotypes in atherosclerosis. Eur Heart J 2017;38:1594–1596. [DOI] [PubMed] [Google Scholar]
- 10.Nahrendorf M, Frantz S, Swirski FK, Mulder WJM, Randolph G, Ertl G, Ntziachristos V, Piek JJ, Stroes ES, Schwaiger M, Mann DL, Fayad ZA. Imaging systemic inflammatory networks in ischemic heart disease. J. Am. Coll. Cardiol 2015;65:1583–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Williams JW, Huang L-H, Randolph GJ. Cytokine Circuits in Cardiovascular Disease. Immunity. 2019;50:941–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Potteaux S, Gautier EL, Hutchison SB, van Rooijen N, Rader DJ, Thomas MJ, Sorci-Thomas MG, Randolph GJ. Suppressed monocyte recruitment drives macrophage removal from atherosclerotic plaques of Apoe−/− mice during disease regression. J Clin Invest 2011;121:2025–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Charo IF, Ransohoff RM. The Many Roles of Chemokines and Chemokine Receptors in Inflammation. N Engl J Med 2006;354:610–621. [DOI] [PubMed] [Google Scholar]
- 14.Zernecke A, Weber C. Chemokines in the vascular inflammatory response of atherosclerosis. Cardiovasc Res 2010;86:192–201. [DOI] [PubMed] [Google Scholar]
- 15.Van Der Vorst EPC, Döring Y, Weber C. Chemokines. Arterioscler Thromb Vasc Biol 2015;35:e52–e56. [DOI] [PubMed] [Google Scholar]
- 16.Kledal TN, Rosenkilde MM, Coulin F, Simmons G, Johnsen AH, Alouani S, Power CA, Luttichau HR, Gerstoft J, Clapham PR, Clark-Lewis I, Wells TN, Schwartz TW. A broad-spectrum chemokine antagonist encoded by Kaposi’s sarcoma-associated herpesvirus. Science. 1997;277:1656–1659. [DOI] [PubMed] [Google Scholar]
- 17.Lüttichau HR, Johnsen AH, Jurlander J, Rosenkilde MM, Schwartz TW. Kaposi sarcoma-associated herpes virus targets the lymphotactin receptor with both a broad spectrum antagonist vCCL2 and a highly selective and potent agonist vCCL3. J Biol Chem 2007;282:17794–17805. [DOI] [PubMed] [Google Scholar]
- 18.Szpakowska M, Chevigne A. vCCL2/vMIP-II, the viral master KEYmokine. J Leukoc Biol 2016;99:893–900. [DOI] [PubMed] [Google Scholar]
- 19.Szpakowska M, Dupuis N, Baragli A, Counson M, Hanson J, Piette J, Chevigné A. Human herpesvirus 8-encoded chemokine vCCL2/vMIP-II is an agonist of the atypical chemokine receptor ACKR3/CXCR7. Biochem Pharmacol 2016;114:14–21. [DOI] [PubMed] [Google Scholar]
- 20.Liu Y, Pierce R, Luehmann HP, Sharp TL, Welch MJ. PET Imaging of Chemokine Receptors in Vascular Injury–Accelerated Atherosclerosis. J Nucl Med 2013;54:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Döring Y, Noels H, Van Der Vorst EPC, et al. Vascular CXCR4 limits atherosclerosis by maintaining arterial integrity: Evidence from mouse and human studies. Circulation. 2017;136:388–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sörensen I, Adams RH, Gossler A. DLL1-mediated Notch activation regulates endothelial identity in mouse fetal arteries. Blood. 2009;113:5680–5688. [DOI] [PubMed] [Google Scholar]
- 23.Davidoff AM, Ng CYC, Zhou J, Spence Y, Nathwani AC. Sex significantly influences transduction of murine liver by recombinant adeno-associated viral vectors through an androgen-dependent pathway. Blood. 2003;102:480–488. [DOI] [PubMed] [Google Scholar]
- 24.Boivin G, Faget J, Ancey PB, Gkasti A, Mussard J, Engblom C, Pfirschke C, Contat C, Pascual J, Vazquez J, Bendriss-Vermare N, Caux C, Vozenin MC, Pittet MJ, Gunzer M, Meylan E. Durable and controlled depletion of neutrophils in mice. Nat Commun 2020;11:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, Garin A, Liu J, Mack M, van Rooijen N, Lira SA, Habenicht AJ, Randolph GJ. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest 2007;117:185–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, Pittet MJ. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest 2007;117:195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao B, Liwang PJ. Characterization of the interactions of vMIP-II, and a dimeric variant of vMIP-II, with glycosaminoglycans. Biochemistry. 2010;49:7012–7022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuschen GSV, Coulin F, Power CA, Proudfoot AEI, Hubbard RE, Hoogewerf AJ, Wells TNC. Glycosaminoglycans interact selectively with chemokines and modulate receptor binding and cellular responses. Biochemistry. 1999;38:12959–12968. [DOI] [PubMed] [Google Scholar]
- 29.Stevens RL, Colombo M, Gonzales JJ, Hollander W, Schmid K. The glycosaminoglycans of the human artery and their changes in atherosclerosis. J Clin Invest 1976;58:470–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Didangelos A, Mayr U, Monaco C, Mayr M. Novel role of ADAMTS-5 protein in proteoglycan turnover and lipoprotein retention in atherosclerosis. J Biol Chem 2012;287:19341–19345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kunjathoor VV, Chiu DS, O’Brien KD, LeBoeuf RC. Accumulation of biglycan and perlecan, but not versican, in lesions of murine models of atherosclerosis. Arterioscler Thromb Vasc Biol 2002;22:462–468. [DOI] [PubMed] [Google Scholar]
- 32.Gimbrone MA, García-Cardeña G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ Res 2016;118:620–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith A, Protti A, Phinikaridou A, Warley A, Botnar RM, Indermuehle A, Shah A, Andia ME. Noninvasive Magnetic Resonance Imaging Evaluation of Endothelial Permeability in Murine Atherosclerosis Using an Albumin-Binding Contrast Agent. Circulation. 2012;126:707–719. [DOI] [PubMed] [Google Scholar]
- 34.Gupta SK, Lysko PG, Pillarisetti K, Ohlstein E, Stadel JM. Chemokine Receptors in Human Endothelial Cells. J Biol Chem 1998;273:4282–4287. [DOI] [PubMed] [Google Scholar]
- 35.Murdoch C, Monk PN, Finn A. CXC chemokine receptor expression on human endothelial cells. Cytokine [Internet]. 1999;11:704–712. [DOI] [PubMed] [Google Scholar]
- 36.Nibbs RJB, Graham GJ. Immune regulation by atypical chemokine receptors. Nat. Rev. Immunol 2013;13:815–829. [DOI] [PubMed] [Google Scholar]
- 37.Oishi S, Kuroyanagi T, Kubo T, Montpas N, Yoshikawa Y, Misu R, Kobayashi Y, Ohno H, Heveker N, Furuya T, Fujii N. Development of Novel CXC Chemokine Receptor 7 (CXCR7) Ligands: Selectivity Switch from CXCR4 Antagonists with a Cyclic Pentapeptide Scaffold. J Med Chem 2015;58:5218–5225. [DOI] [PubMed] [Google Scholar]
- 38.Kawatkar SP, Yan M, Gevariya H, Lim MY, Eisold S, Zhu X, Huang Z, An J. Computational analysis of the structural mechanism of inhibition of chemokine receptor CXCR4 by small molecule antagonists. Exp Biol Med 2011;236:844–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim DN, Scott RF, Schmee J, Thomas WA. Endothelial cell denudation, labelling indices and monocyte attachment in advanced swine coronary artery lesions. Atherosclerosis. 1988;73:247–57. [DOI] [PubMed] [Google Scholar]
- 40.Foteinos G, Hu Y, Xiao Q, Metzler B, Xu Q. Rapid endothelial turnover in atherosclerosis-prone areas coincides with stem cell repair in apolipoprotein E-deficient mice. Circulation. 2008;117:1856–1863. [DOI] [PubMed] [Google Scholar]
- 41.Noels H, Zhou B, Tilstam PV, Theelen W, Li X, Pawig L, Schmitz C, Akhtar S, Simsekyilmaz S, Shagdarsuren E, Schober A, Adams RH, Bernhagen J, Liehn EA, Döring Y, Weber C. Deficiency of endothelial Cxcr4 reduces reendothelialization and enhances neointimal hyperplasia after vascular injury in atherosclerosis-prone mice. Arterioscler Thromb Vasc Biol 2014;34:1209. [DOI] [PubMed] [Google Scholar]
- 42.Melchionna R, Porcelli D, Mangoni A, Carlini D, Liuzzo G, Spinetti G, Antonini A, Capogrossi MC, Napolitano M. Laminar shear stress inhibits CXCR4 expression on endothelial cells: functional consequences for atherogenesis. FASEB J 2005;19:629–31. [DOI] [PubMed] [Google Scholar]
- 43.Hyafil F, Pelisek J, Laitinen I, et al. Imaging the cytokine receptor CXCR4 in atherosclerotic plaques with the radiotracer 68Ga-pentixafor for positron emission tomography. J Nucl Med 2017;58:499–506. [DOI] [PubMed] [Google Scholar]
- 44.Li X, Heber D, Leike T, Beitzke D, Lu X, Zhang X, Wei Y, Mitterhauser M, Wadsak W, Kropf S, Wester HJ, Loewe C, Hacker M, Haug AR. [68Ga]Pentixafor-PET/MRI for the detection of Chemokine receptor 4 expression in atherosclerotic plaques. Eur J Nucl Med Mol Imaging. 2018;45:558–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weiberg D, Thackeray JT, Daum G, Sohns JM, Kropf S, Wester H-J, Ross TL, Bengel FM, Derlin T. Clinical Molecular Imaging of Chemokine Receptor CXCR4 Expression in Atherosclerotic Plaque using 68 Ga-Pentixafor PET: Correlation with Cardiovascular Risk Factors and Calcified Plaque Burden. J Nucl Med 2018;59:266–272. [DOI] [PubMed] [Google Scholar]
- 46.Derlin T, Sedding DG, Dutzmann J, et al. Imaging of chemokine receptor CXCR4 expression in culprit and nonculprit coronary atherosclerotic plaque using motion-corrected [68Ga]pentixafor PET/CT. Eur J Nucl Med Mol Imaging. 2018;45:1934–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McDonald AI, Shirali AS, Aragón R, Ma F, Hernandez G, Vaughn DA, Mack JJ, Lim TY, Sunshine H, Zhao P, Kalinichenko V, Hai T, Pelegrini M, Ardehali R, Iruela-Arispe ML. Endothelial Regeneration of Large Vessels Is a Biphasic Process Driven by Local Cells with Distinct Proliferative Capacities. Cell Stem Cell. 2018;23:210–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Glass CK, Witztum JL. Atherosclerosis: The road ahead. Cell. 2001;104:503–516. [DOI] [PubMed] [Google Scholar]
- 49.Watkins H, Farrall M. Genetic susceptibility to coronary artery disease: from promise to progress. Nat Rev Genet 2006;7:163–173. [DOI] [PubMed] [Google Scholar]
- 50.Leon MB, Tsapenko M, Kipshidze N, Moses J, Serruys P, Dangas G, Kutryk M. Role of the endothelium in modulating neointimal formation. J Am Coll Cardiol 2004;44:733–739. [DOI] [PubMed] [Google Scholar]
- 51.Vanags LZ, Wong NKP, Nicholls SJ, Bursill CA. High-Density Lipoproteins and Apolipoprotein A-I Improve Stent Biocompatibility. Arterioscler Thromb Vasc Biol 2018;38:1691–1701. [DOI] [PubMed] [Google Scholar]
- 52.Otsuka F, Finn AV, Yazdani SK, Nakano M, Kolodgie FD, Virmani R. The importance of the endothelium in atherothrombosis and coronary stenting. Nat Rev Cardiol 2012;9:439–453. [DOI] [PubMed] [Google Scholar]
- 53.Torrado J, Buckley L, Durán A, Trujillo P, Toldo S, Valle Raleigh J, Abbate A, Biondi-Zoccai G, Guzmán LA. Restenosis, Stent Thrombosis, and Bleeding Complications: Navigating Between Scylla and Charybdis. J Am Coll Cardiol 2018;71:1676–1695. [DOI] [PubMed] [Google Scholar]
- 54.Virmani R, Burke AP, Farb A, Kolodgie FD. Pathology of the Vulnerable Plaque. J Am Coll Cardiol 2006;47:0–5. [DOI] [PubMed] [Google Scholar]
- 55.Stefanadis C, Antoniou CK, Tsiachris D, Pietri P. Coronary atherosclerotic vulnerable plaque: Current perspectives. J Am Heart Assoc 2017;6:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sjaarda J, Gerstein H, Chong M, Yusuf S, Meyre D, Anand SS, Hess S, Paré G. Blood CSF1 and CXCL12 as Causal Mediators of Coronary Artery Disease. J Am Coll Cardiol 2018;72:300–310. [DOI] [PubMed] [Google Scholar]
- 57.Döring Y, Van Der Vorst EPC, Duchene J, Jansen Y, Gencer S, Bidzhekov K, Atzler D, Santovito D, Rader DJ, Saleheen D, Weber C. CXCL12 Derived from endothelial cells promotes atherosclerosis to drive coronary artery disease. Circulation. 2019;139:1338–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Berahovich RD, Zabel BA, Lewén S, Walters MJ, Ebsworth K, Wang Y, Jaen JC, Schall TJ. Endothelial expression of CXCR7 and the regulation of systemic CXCL12 levels. Immunology. 2014;141:111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Binderup T, Duivenvoorden R, Fay F, et al. Imaging-assisted nanoimmunotherapy for atherosclerosis in multiple species. Sci Transl Med 2019;11:eaaw7736. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.