

Summary
The Akt family of kinases exert many of their cellular effects via the activation of the mammalian target of rapamycin (mTOR) kinase through a series of intermediary proteins. Multiple lines of evidence have identified the Akt family of kinases as candidate schizophrenia and bipolar disorder genes. Although dysfunction of the prefrontal cortex (PFC) is a key feature of both schizophrenia and bipolar disorder, no studies have comprehensively assessed potential alterations in Akt-mTOR pathway activity in the PFC of either disorder. Here, we examined the activity and expression profile of key proteins in the Akt-mTOR pathway in bipolar disorder and schizophrenia homogenates from two different PFC subregions. Our findings identify reduced Akt-mTOR PFC signaling in a subset of bipolar disorder subjects. Using a reverse-translational approach, we demonstrated that Akt hypofunction the PFC is sufficient to give rise to key cognitive phenotypes that are paralleled by alterations in synaptic connectivity and function.
eTOC blurb
The biochemical changes that contribute to bipolar disorder pathophysiology remain largely unknown. Cahill et al. reveal a reduction in the activation of the Akt-mTOR pathway in the prefrontal cortex of bipolar disorder subjects. Cahill et al. demonstrate that reduced activity of this pathway causes cognitive and synaptic phenotypes.
Introduction
Bipolar disorder and schizophrenia individually afflict approximately 1% of the population (McGrath et al., 2008; Merikangas et al., 2007; Rowland and Marwaha, 2018; Sham et al., 1994; Simeone et al., 2015). Bipolar disorder is characterized by cyclic elevations in mood state (e.g., hypomania/mania) that are commonly interspersed with depressive episodes. Psychotic episodes characterized by delusions and/or hallucinations are present in about half of all bipolar disorder patients (Dunayevich and Keck, 2000; Keck et al., 2003; Mazzarini et al., 2010; van Bergen et al., 2019). Cognitive impairments of a severity deemed clinically relevant occur in the majority of bipolar disorder subjects, and persist during periods without mania or depression (Bortolato et al., 2015; Bourne et al., 2013; Robinson et al., 2006; Sole et al., 2017). Schizophrenia is characterized by altered affect, disordered perceptions of reality, disorganization of thought, and cognitive impairment (Kahn et al., 2015). fMRI studies have identified dorsolateral prefrontal cortex (DLPFC) hypoactivation in both schizophrenia and bipolar disorder, and reduced DLPF engagement is theorized to contribute to the cognitive impairments typical of each disorder (Cremaschi et al., 2013; McKenna et al., 2014; Townsend et al., 2010; Yoon et al., 2008a).
Multiple converging lines of evidence have identified the Akt family of serine/threonine kinases as top candidate genes for schizophrenia and bipolar disorder. There are three Akt isoforms (Akt1, Akt2, and Akt3) encoded from different genes, all of which share a common mechanism of activation and regulate a common cluster of downstream effectors. Notably, Akt1-3 activity, through a series of intermediary proteins, augments the activity of the mammalian target of rapamycin (mTOR) kinase (Xu et al., 2020). AKT1 variants have been identified in schizophrenia and in bipolar disorder subjects with and without psychosis, and haplotype transmission analysis has identified AKT1 as a candidate bipolar disorder gene (Bajestan et al., 2006; Karege et al., 2012; Karege et al., 2010; Mathur et al., 2010; Schwab et al., 2005). Multiple genome wide association studies (GWAS) have implicated AKT3 variants in contributing to schizophrenia, and at present AKT3 is considered a top schizophrenia candidate gene (Goes et al., 2015; Ikeda et al., 2019; Li et al., 2017; Pardinas et al., 2018; Ripke et al., 2013; Schizophrenia Working Group of the Psychiatric Genomics, 2014). In addition, pathway analyses performed on GWAS studies have implicated AKT3 variants as contributing to bipolar disorder (Network and Pathway Analysis Subgroup of Psychiatric Genomics, 2015). Finally, GWAS have identified associations of AKT3 with specific psychiatric disorder-relevant intermediate phenotypes in the general population, including risk-taking behavior, educational attainment, and key aspects of cognitive ability (Karlsson Linner et al., 2019; Lee et al., 2018).
Akt1-3 activity fosters the inhibition of the tuberous sclerosis proteins 1 and 2 (TSC1 and TSC2), and TSC1/2 inhibition, in turn, facilitates Rheb small GTPase-mediated activation of mTOR. It is through these mechanisms that Akt activity commonly culminates in the activation of mTOR. mTOR occurs in two distinct protein complexes referred to as mTORC1 and mTORC2, dictated in part by the interaction of mTOR with the Raptor (mTORC1) or Rictor (mTORC2) proteins (Dibble and Cantley, 2015; Jhanwar-Uniyal et al., 2019; Laplante and Sabatini, 2009). Akt1-3 share a common mechanism of phosphorylation-mediated activation. Notably, phosphoinositide 3-kinase (PI3K) activation converts phospholipid PIP2 to PIP3, resulting in translocation of Akt1-3 to the cellular membrane where it can be phosphorylated at the T308 residue by PDK1 (Dibble and Cantley, 2015; Hemmings and Restuccia, 2012; Pearce et al., 2010). Although T308 phosphorylation is sufficient to activate Akt1-3, phosphorylation of the S473 residue further augments Akt kinase activity. Akt S473 phosphorylation is not mediated by PDK1, but rather by feedback phosphorylation from the mTORC2 (and not mTORC1) complex (Dibble and Cantley, 2015; Pearce et al., 2010).
Despite genomic associations of AKT with bipolar disorder and schizophrenia, few to no studies have assessed possible alterations in the protein activity of the Akt-mTOR pathway in the bipolar disorder or schizophrenia forebrain. Knowledge of this is important given that although no single AKT variant is likely to contribute to the majority of cases, it is possible that alterations in the protein activity of the Akt pathway are prevalent within individual forebrain regions in bipolar disorder and/or schizophrenia subjects. Further, only protein profiling studies can assess changes in Akt pathway activity, thus having a direct link to functional consequences. To this end, we assessed the activity of the Akt pathway in two prefrontal cortical regions in a large cohort of schizophrenia, bipolar disorder, and unaffected control subjects. Our findings identify a reduction in Akt-mTOR prefrontal cortical signaling in bipolar disorder subjects without psychosis, and we demonstrate that this reduction is sufficient to contribute to key neural and behavioral hallmarks of bipolar disorder.
Results
Akt-mTOR pathway aberrations identified in the bipolar disorder dorsolateral prefrontal cortex
First, we assessed PI3K and Akt activity levels in dorsolateral prefrontal cortex (DLPFC) homogenates (Brodmann Area 46) from 35 schizophrenia, 32 bipolar disorder, and 35 control subjects from the Stanley Medical Research Institute via SDS-PAGE followed by Western blotting (Table S1 for cohort details). We performed separate male and female analyses for each condition, and bipolar disorder homogenates were divided into those with psychosis (BP-psychosis) and those without psychosis (BP-no psychosis). Protein phosphorylation levels in human homogenates were normalized to GAPDH, which did not itself differ between diagnostic groups.
PI3K activity is increased by phosphorylation of the Y199 residue (Yu et al., 2009), and in male subjects, levels of phospho-PI3K Y199 (p-PI3K Y199) in BP-psychosis and schizophrenia did not differ from unaffected controls. However, p-PI3K Y199 levels were significantly decreased in male BP-no psychosis relative to male controls and relative to male BP-psychosis, with no corresponding alterations in female subjects (Figures 1A–1C and Figures S1A–C). We found nearly identical changes in levels of phospho-Akt T308 (p-Akt T308) (Figure 1D and Figures S1A–C). Further, while direct comparisons revealed a decrease in p-Akt S473 in male BP-no psychosis relative to male controls and male BP-psychosis, these differences did not survive multiple comparison corrections (Figure 1E and Figures S1A–C). Finally, total levels of Akt did not differ between any groups (Figure 1F and Figures S1A–C).
Figure 1. PI3K-Akt-mTOR pathway protein levels and activity in the DLPFC of bipolar disorder and schizophrenia subjects.
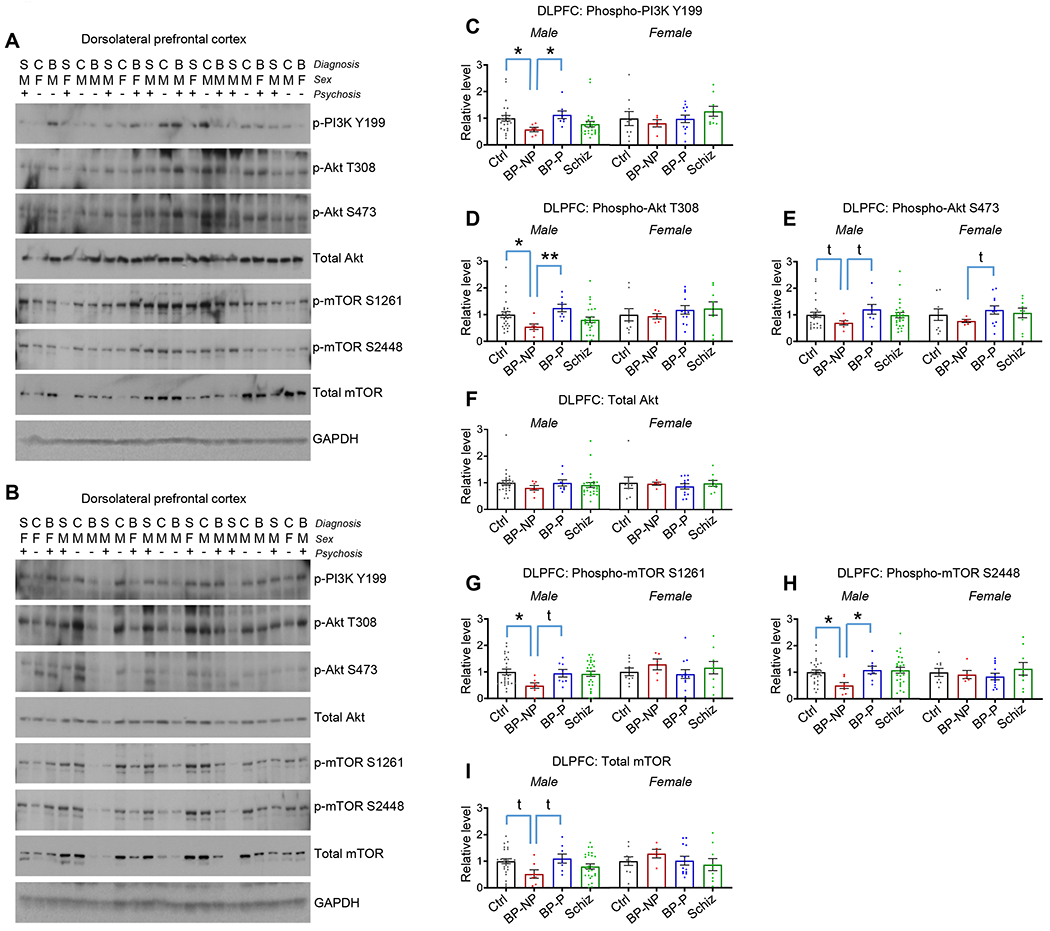
A and B. Blots show phospho and total levels of PI3K, Akt, and mTOR in DLPFC homogenates. Diagnosis (S=schizophrenia, C=control, B=bipolar disorder), sex (M=male, F=female), and absence (−) or presence (+) of psychosis indicated. Figure S1 for the remaining blots.
C. p-PI3K (Y199) in DLPFC homogenates. *p<0.05 with multiple comparison correction. Table S2 for statistical details.
D. p-Akt (T308) in DLPFC homogenates. *p<0.05, **p<0.01 with multiple comparison correction. Table S2 for statistical details.
E. p-Akt (S473) in DLPFC homogenates. t=p<0.05 with direct comparison. Table S2 for statistical details.
F. Total Akt in DLPFC homogenates. No significant differences detected. Table S2 for statistical details.
G. p-mTOR (S1261) in DLPFC homogenates. *p<0.05 with multiple comparison correction; t=p<0.05 with direct comparison. Table S2 for statistical details.
H. p-mTOR (S2448) in DLPFC homogenates. *p<0.05 with multiple comparison correction. Table S2 for statistical details.
I. Total mTOR in DLPFC homogenates. t=p<0.05 with direct comparison. Table S2 for statistical details.
Summary data are the mean+SEM. n=35 control (26 male, 9 female), 12 BP-no psychosis (7 male, 5 female), 20 BP-psychosis (8 male, 12 female), 35 schizophrenia (26 male, 9 female)
Rheb activity resulting from upstream Akt-mediated inhibition of TSC1/2 results in the phosphorylation of the mTOR S1261 residue. Phosphorylation at this residue, in turn, fosters mTOR’s kinase activity to a variety of downstream effectors (Acosta-Jaquez et al., 2009). We found that mTOR S1261 phosphorylation was decreased in male BP-no psychosis relative to male controls. While direct comparison between male BP-no psychosis and male BP-psychosis revealed a significance difference, this effect was not maintained following multiple comparison correction. No changes between female diagnostic groups and controls were detected for p-mTOR S1261 levels (Figure 1G and Figures S1A–C). mTOR activity is also commonly assessed by examining levels of phosphorylation at the S2448 residue (Hoeffer and Klann, 2010; Rosner et al., 2010). Phosphorylation at this site occurs as a feedback loop from downstream mTOR kinase targets such that when activated by mTOR, these kinases augment phosphorylation at this S2448 site (Chiang and Abraham, 2005; Holz and Blenis, 2005). We found a significant decrease in mTOR S2448 phosphorylation in male BP-no psychosis subjects relative to male controls and relative to male BP-psychosis, with no differences between male schizophrenia and male controls. Again, no analogous differences were detected in female BP-no psychosis subjects (Figure 1H and Figures S1A–C). Finally, no differences in total mTOR levels survived multiple comparison correction between any groups among male or female subjects (Figure 1I and Figures S1A–C).
When activated, mTOR controls the activity of numerous kinases, including ULK1, p70 S6 kinase (p70S6K), and eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1), via established phosphorylation sites (Saxton and Sabatini, 2017). Starting first with ULK1, we assessed two phosphorylation sites, one of which is mediated by mTOR activation (S757), and one which is independent of mTOR (S777) (Kim et al., 2011). We found a significant decrease in levels of ULK1 phosphorylated at the S757 site in male BP-no psychosis relative to male BP-psychosis. While direct comparison between male BP no-psychosis and male controls revealed a significant deficit, this effect did not retain significance following multiple comparison corrections (Figures 2A–2C and Figures S2A–C). No significant differences in phosphorylation at the S777 site survived multiple comparison, although direct comparison revealed a deficit in male BP-no psychosis relative to BP-psychosis (Figure 2D and Figures S2A–C). No corresponding differences in either S757 or S777 phosphorylation were detected in female subjects (Figures 2C and 2D and Figures S2A–C), and no differences in total ULK1 were detected in male or female diagnostic groups (Figure 2E and Figures S2A–C).
Figure 2. Protein levels and activity of downstream mTOR effectors in bipolar disorder and schizophrenia DLPFC.
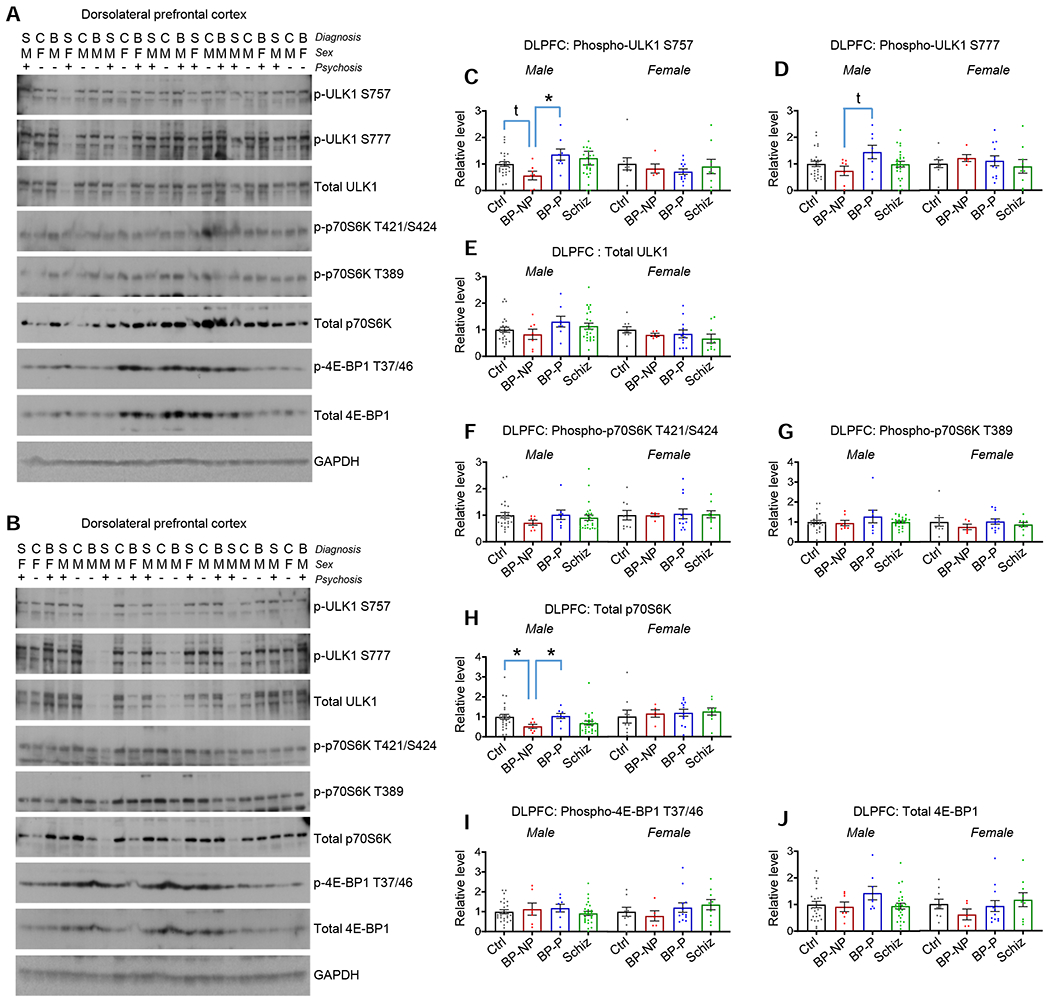
A and B. Blots show phospho and total levels of ULK1, p70S6K, and 4E-BP1 in DLPFC homogenates. Diagnosis (S=schizophrenia, C=control, B=bipolar disorder), sex (M=male, F=female), and absence (−) or presence (+) of psychosis indicated. Figure S2 for the remaining blots.
C. p-ULK1 (S757) in DLPFC homogenates. *p<0.05 with multiple comparison correction; t=p<0.05 with direct comparison. Table S2 for statistical details.
D. p-ULK1 (S777) in DLPFC homogenates. t=p<0.05 with direct comparison. Table S2 for statistical details.
E. Total ULK1 in DLPFC homogenates. No differences between groups detected. Table S2 for statistical details.
F. Phospho-p70S6K (T421/S424) in DLPFC homogenates. No differences between groups detected. Table S2 for statistical details.
G. Phospho-p70S6K (T389) in DLPFC homogenates. No differences between groups detected. Table S2 for statistical details.
H. Total p70S6K in DLPFC homogenates. *p<0.05 with multiple comparison correction. Table S2 for statistical details.
I. p-4E-BP1 (T37/46) in DLPFC homogenate. No differences between groups detected. Table S2 for statistical details.
J. Total 4E-BP1 in DLPFC homogenates. No differences between groups detected. Table S2 for statistical details.
Summary data are the mean+SEM. n=35 control (26 male, 9 female), 12 BP-no psychosis (7 male, 5 female), 20 BP-psychosis (8 male, 12 female), 35 schizophrenia (26 male, 9 female)
Next, we examined the phosphorylation status of the T421/S424 and T389 residues of p70S6K, which represent mTOR-independent (T421/S424) and mTOR-dependent (T389) means of facilitating p70S6K activation (Burnett et al., 1998; Iijima et al., 2002; Pearson et al., 1995). Surprisingly, we detected no differences in any diagnostic groups relative to controls for phosphorylation at the T421/S424 or T389 residues (Figures 2F and 2G and Figures S2A–C). Of note, we detected a significant reduction in total p70S6K in male BP-no psychosis relative to male control and male BP-psychosis; no such differences were detected in homogenates from female subjects (Figure 2H and Figures S2A–C). We also examined the activity of 4E-BP1 by examining phosphorylation levels at the T37/T46 residues (Gingras et al., 1999), and detected no differences between any groups, and no differences in total 4E-BP1 were detected either (Figures 2I and 2J and Figures S2A–C).
In addition to the mTOR pathway, Akt activity regulates the function of multiple additional pathways, most notably GSK3. Akt inhibits GSK3α and GSK3ß via direct phosphorylation of the S21 and S9 residues, respectively (Doble and Woodgett, 2003). Interestingly while we detected a slight yet significant decrease in the GSK3α and GSK3ß phosphorylation in male BP-no psychosis relative to male BP-psychosis, no changes relative to male controls were detected (Figures S3A–G). These findings suggest that alterations in mTOR phosphorylation rather than GSK3 signaling are more closely associated with the deficient Akt signaling in male BP-no psychosis.
Akt-mTOR pathway aberrations identified in the bipolar disorder ventrolateral prefrontal cortex
To determine if the PI3K-Akt-mTOR pathway is altered among these diagnostic groups in other prefrontal cortical regions, we also examined the ventrolateral prefrontal cortex (VLPFC; Brodmann Area 45) in the same cohort of subjects. In VLPFC homogenate, we detected a significant decrease in p-PI3K Y199, p-Akt T308 and p-Akt S473 in male BP-no psychosis relative to male controls, and these Akt phosphorylation alterations were also significantly reduced relative to male BP-psychosis (Figures 3A–3E and Figures S4A–C). Interestingly, we also detected a slight, albeit significant, decrease in total Akt levels in male BP-no psychosis relative to male controls (Figure 3F and Figures S4A–C). For phospho-mTOR in the VLPFC, we detected a significant reduction in S2448 phosphorylation in male BP-no psychosis relative to male controls, with no corresponding change in S1261 phosphorylation (Figures 3G and 3H and Figures S4A–C). Finally, no differences in total mTOR were detected in any other diagnostic groups (Figure 3I and Figures S4A–C).
Figure 3. PI3K-Akt-mTOR pathway protein levels and activity in the VLPFC of bipolar disorder and schizophrenia subjects.
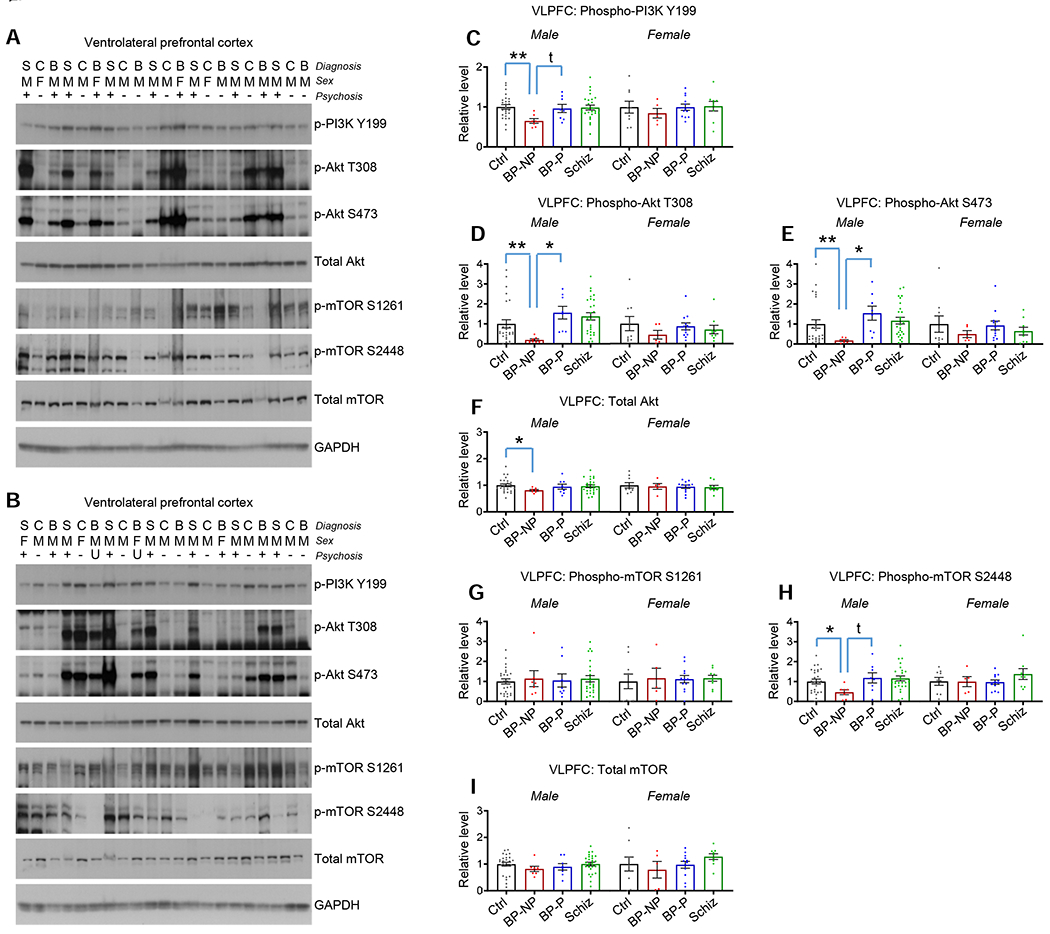
A and B. Blots show phospho and total PI3K, Akt, and mTOR in the VLPFC homogenates. Diagnosis (S=schizophrenia, C=control, B=bipolar disorder), sex (M=male, F=female), and absence (−) or presence (+) of psychosis indicated above each lane (U=psychosis status unknown). Figure S4 for the remaining blots.
C. p-PI3K (Y199) in VLPFC homogenates. **p<0.01 with multiple comparison correction; t=p<0.05 with direct comparison. Table S2 for statistical details.
D. p-Akt (T308) in VLPFC homogenates. *p<0.05, **p<0.01 with multiple comparison correction. Table S2 for statistical details.
E. p-Akt (S473) in VLPFC homogenates. *p<0.05, **p<0.01 with multiple comparison correction. Table S2 for statistical details.
F. Total Akt in VLPFC homogenates. *p<0.05 with multiple comparison correction. Table S2 for statistical details.
G. p-mTOR (S1261) in VLPFC homogenates. No differences between groups detected. Table S2 for statistical details.
H. p-mTOR (S2448) in VLPFC homogenates. *p<0.05 with multiple comparison correction; t=p<0.05 with direct comparison. Table S2 for statistical details.
I. Total mTOR in VLPFC homogenates. No differences between groups detected. Table S2 for statistical details.
Summary data are the mean+SEM. n=35 control (26 male, 9 female), 12 BP-no psychosis (7 male, 5 female), 20 BP-psychosis (8 male, 12 female), 35 schizophrenia (26 male, 9 female)
See also Table S2 and Figures S3 and S4
In examining the activity level of downstream mTOR targets in VLPFC homogenates, no alterations in phospho or total ULK1 between groups remained significant following multiple comparisons (Figures 4A–4E and Figures S5A–C). Conversely, male BP-no psychosis VLPFC homogenates exhibited reductions in the phosphorylation status of the p70S6K T421/S424 and T389 residues relative to male controls, and in the case of T421/T424, also relative to male BP-no psychosis (Figures 4F and 4G and Figures S5A–C). Further, male BP-no psychosis also showed a reduction in total p70S6K relative to male controls (Figure 4H and Figures S5A–C). Finally, pertaining to 4E-BP1, we detected a significant reduction in T37/T46 phosphorylation in male BP-no psychosis relative to male controls and relative to male BP-psychosis (Figure 4I and Figures S5A–C). No changes in in total 4E-BP1 were detected between bipolar disorder subjects and controls; however, we detected a significant total 4E-BP1 reduction in male schizophrenia subjects (Figure 4J and Figures S5A–C).
Figure 4. Protein levels and activity of downstream mTOR effectors in bipolar disorder and schizophrenia VLPFC.
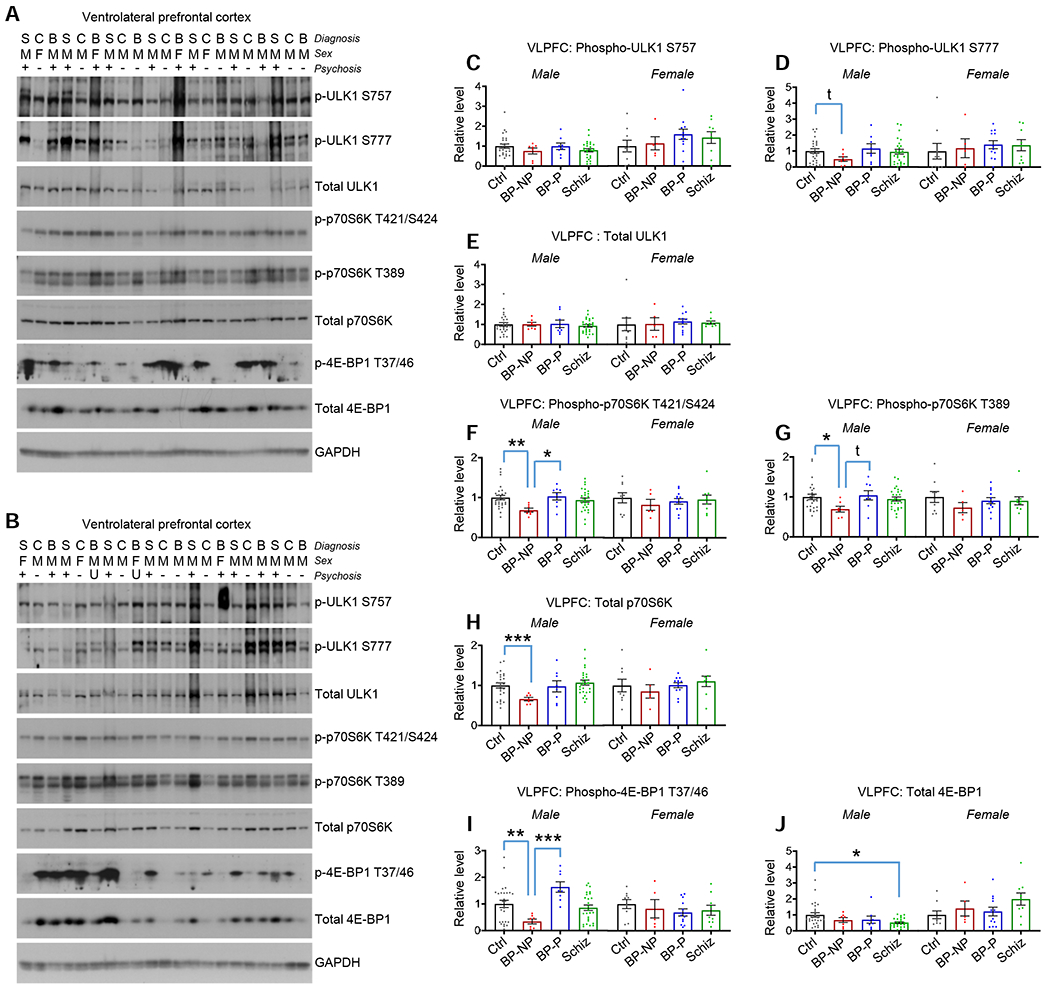
A and B. Blots show phospho and total levels of ULK1, p70S6K, and 4E-BP1 in VLPFC homogenaes. Diagnosis (S=schizophrenia, C=control, B=bipolar disorder), sex (M=male, F=female), and absence (−) or presence (+) of psychosis indicated above each lane (U=psychosis status unknown). Figure S5 for the remaining blots.
C. p-ULK1 (S757) in VLPFC homogenates. No differences between groups detected. Table S2 for statistical details.
D. p-ULK1 (S777) in VLPFC homogenates. t=p<0.05 with direct comparison. Table S2 for statistical details.
E. Total ULK1 in VLPFC homogenates. No differences between groups detected. Table S2 for statistical details.
F. Phospho-p70S6K (T421/S424) in VLPFC homogenates. *p<0.05, **p<0.01 with multiple comparison correction. Table S2 for statistical details.
G. Phospho-p70S6K (T389) in VLPFC homogenates. No differences between groups detected. *p<0.05 with multiple comparison correction; t=p<0.05 with direct comparison. Table S2 for statistical details.
H. Total p70S6K in VLPFC homogenates. ***p<0.001 with multiple comparison correction. Table S2 for statistical details.
I. p-4E-BP1 (T37/46) in VLPFC homogenates. **p<0.01, ***p<0.001 with multiple comparison correction. Table S2 for statistical details.
J. Total 4E-BP1 in VLPFC homogenates. *p<0.05 with multiple comparison correction. Table S2 for statistical details.
Summary data are the mean+SEM. n=35 control (26 male, 9 female), 12 BP-no psychosis (7 male, 5 female), 20 BP-psychosis (8 male, 12 female), 35 schizophrenia (26 male, 9 female)
See also Table S2 and Figures S5 and S6
Finally, we found no differences that survived multiple comparisons between any diagnostic groups and control subjects for GSK3α and GSK3ß phosphorylation levels in the VLPFC (Figures S6A–G). Similar to our DLPFC findings, the lack of major alterations in GSK3 activity in the VLPFC further highlights the specificity of altered mTOR pathway activity as a key downstream consequence of aberrant Akt activity in male BP-no psychosis.
Lack of influence of antipsychotic treatment on Akt or mTOR activity
The lack of differences in the phosphorylation status of Akt or mTOR in BP-psychosis and schizophrenia could potentially be due to the greatly increased lifetime intake of antipsychotic medication in these groups relative to subjects with BP-no psychosis (Table S1). However, DLPFC and VLPFC regression analysis failed to detect significant relationships between antipsychotic intake and Akt or mTOR phosphorylation status in schizophrenia or BP-psychosis (Table S2). This lack of relationship suggests that antipsychotic intake is not masking disease-related changes in Akt or mTOR phosphorylation in BP-psychosis or schizophrenia.
Akt activity disruption in the PFC gives rise to specific cognitive phenotypes in males
Next, we wanted to determine if disruption of Akt activity is sufficient to give rise to PFC-based cognitive phenotypes. While non-primate mammals do not have a dorsolateral or ventrolateral prefrontal cortex, the medial prefrontal cortex (mPFC) is considered the best rodent homologue to multiple human prefrontal cortical areas, including the DLPFC (Farovik et al., 2008; Uylings et al., 2003). Indeed, many of the functions of the DLPFC and VLPFC in humans, including visuospatial memory, executive functions, and attentional processing, are subsumed by the rodent mPFC (Barker et al., 2007; Bissonette et al., 2013; Lalonde, 2002; Yang et al., 2014; Yoon et al., 2008b).
We used herpes simplex virus (HSV) to overexpress dominant-negative Akt co-expressing green fluorescent protein (DN-Akt-GFP) in the mPFC of male mice in vivo (Figure 5A), and examined the resulting effects on mPFC-dependent behaviors 4-5 days post-viral infusion; HSV-GFP was used as a control. DN-Akt contains a K179A mutation, which fully inhibits Akt activity (Andjelkovic et al., 1997); validation of HSV-DN-Akt GFP is shown in Figure 5B–D. HSVs confer neuron-specific transgene expression within 24 hours of infusion into the brain, with maximal expression 2-6 days post-infusion, and expression ceasing ~8 days post-infusion (Neve et al., 2005; Penrod et al., 2015). Separate mice were used for each behavioral test to prevent learning carryover effects. While the mPFC is not critical for all aspects of rodent episodic memory, this region is particularly critical for recalling the location of objects relative to each other and for recalling the environment in which previously explored objects are located, which can be assessed using object-in-place and object-context tests, respectively (Barker et al., 2017; Barker et al., 2007).
Figure 5. The effects of DN-Akt in the mPFC on cognitive phenotypes.
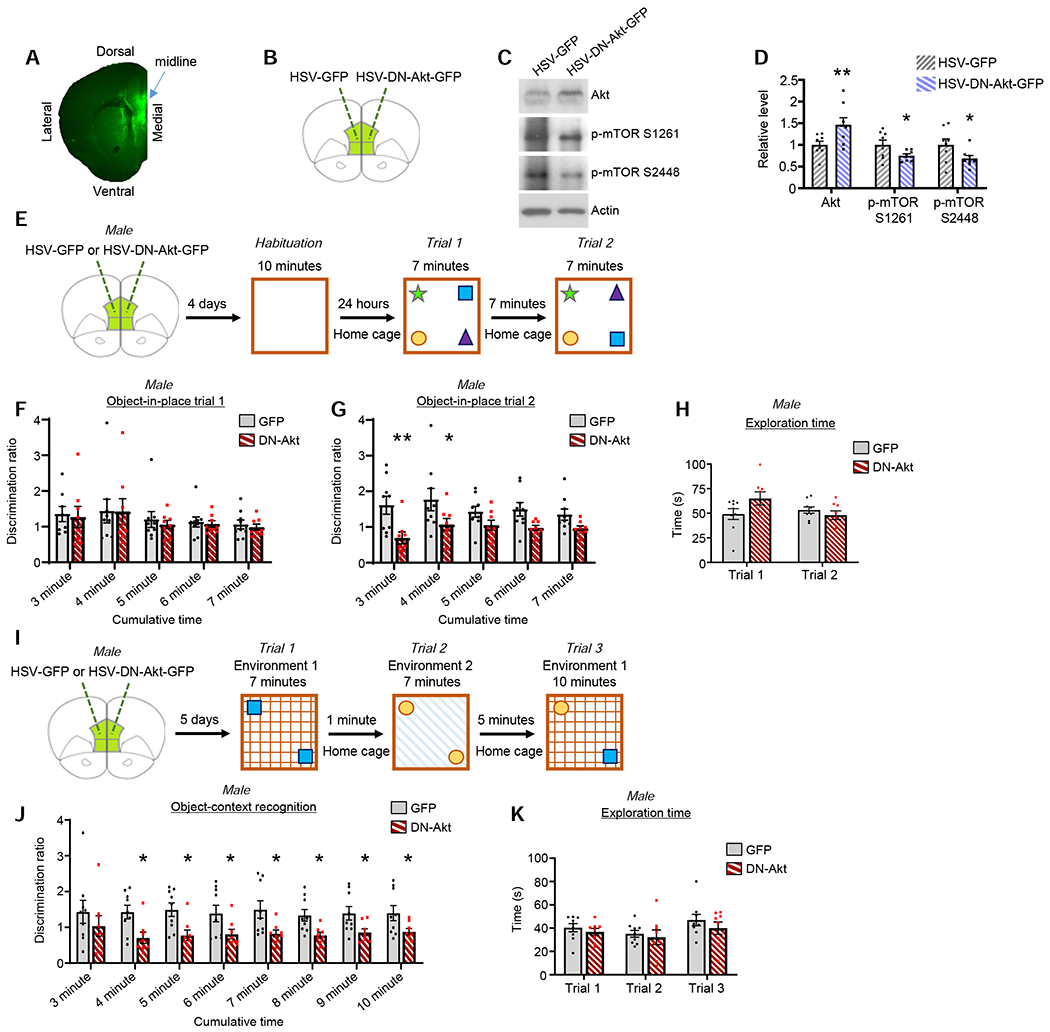
A. Image depicts one cerebral hemisphere. Targeting of HSV-DN-Akt-GFP to the mPFC is shown.
B. Schematic of virus validation approach. One mPFC hemisphere infused with HSV-GFP and the other with HSV-DN-Akt-GFP. 5 days post-infusion, the mPFC was dissected.
C. Western blot showing total Akt, p-mTOR S1261 and p-mTOR S2448 from HSV-GFP and HSV-DN-Akt-GFP mPFC homogenates.
D. HSV-DN-Akt-GFP mPFC infusion increases total Akt (p=0.0014), and reduces phospho-mTOR S1261 (p=0.0370) and phospho-mTOR S2448 (p=0.0215). n=8 GFP and 8 DN-Akt
E. Object-in-place testing schematic. During trial 2, mice should spend more time investigating the objects that switched locations.
F. Object-in-place discrimination ratios for trial 1. As expected, no differences between groups detected. n=9 GFP, 8 DN-Akt
G. Object-in-place discrimination ratios for trial 2. A significant discrimination ratio decrease was detected for the DN-Akt group at the 3 minute and 4 minute cumulative time bins. 2-way repeated measures ANOVA with multiple comparison correction post-hoc (3 min, q=0.0029; 4 min q=0.0165; 5 min q=0.1010; 6 min q=0.0566; 7 min q=0.1010). n=9 GFP, 8 DN-Akt
H. Total time exploring objects during both trials of the object-in-place test. No group differences detected. n=9 GFP, 8 DN-Akt
I. Object-context recognition schematic. During trial 3, mice should spend more time investigating the object that has not been previously explored in that environment (yellow circle in the illustration).
J. Object-context discrimination ratios for trial 3. Significant decrease in discrimination ratio for the DN-Akt group at all cumulative time bins except the 3 minute time bin. 2-way repeated measures ANOVA with multiple comparison correction post-hoc (3 min q=0.1122; 4 min q=0.0253; 5 min q=0.0253; 6 min q=0.0446; 7 min q=0.0258; 8 min q=0.0446; 9 min q=0.0446; 10 min q=0.0446). n=9 GFP, 8 DN-Akt
K. Total time exploring object during each trial. No group differences detected. n=9 GFP, 8 DN-Akt
Summary data are the mean+SEM
See also Figures S7, S8, and S9
First, we assessed male mice in an object-in-place memory test, which assess the ability of mice to recognize a change in the location of objects that had been previously explored, and is mPFC-dependent (Barker et al., 2007) (Figure 5E). This recognition manifests as an increase in the proportion of time spent investigating objects that swapped location as compared to objects whose location was fixed between successive trials. We assessed the cumulative recognition ratio for the first three minutes of trial 1 and trial 2 and for each minute thereafter. As expected, there were no differences between the GFP and DN-Akt groups across trial 1 (Figure 5F). For trial 2, across all time bins spanning from 3 to 7 minutes we detected a significant main effect for DN-Akt expression [F(1, 15)=11.51, p=0.0040], and post-hoc testing revealed a significant recognition ratio deficit in the DN-Akt group at the 3 and 4 minute time points (Figure 5G). Raw time data used to calculate recognition ratios are shown in Figure S7A. Importantly, DN-Akt did not affect the total time spent investigating objects during either trial, indicating that the object-in-place deficit does arise due to a reduced interest in the motivation to explore objects (Figure 5H).
Next, we assessed behaviorally naïve mice in an object-context recognition test known to be sensitive to mPFC disruption (Figure 5I). This test requires that mice recall the environmental context in which novel objects were initially explored (Bekinschtein et al., 2013; Chao et al., 2016). The cumulative recognition ratio during trial 3 was assessed starting at the 3 minute time point and every minute thereafter. We found a significant main effect of DN-Akt expression on recognition ratio across all time bins [F(1, 15)=7.036, p=0.0181]. Post-hoc analysis detected a significant deficit in the DN-Akt group at all time bins except the 3 minute bin (Figure 5J). Raw time data used to calculate recognition ratios are shown in Figure S7B. Importantly, DN-Akt did not affect the total combined time spent investigating objects during any trial (Figure 5K), indicating that the recognition deficit is not the result of a diminished interest in exploration.
In addition to specific types of episodic memory, the rodent mPFC is important for spatial working memory, including spontaneous alternation behavior (Kermath et al., 2020a; Lalonde, 2002; Yang et al., 2014). Interestingly, DN-Akt in the mPFC did not affect performance in a Y-maze spontaneous alternation assessment of spatial working memory (Figures S7C and S7D). This suggests that DN-Akt does not equally affect all aspects of mPFC-based cognition.
Lack of effect of Akt activity impairment in the PFC on non-cognitive behaviors
Converging evidence indicates that the mPFC is also important for mouse sociability and social novelty (Cao et al., 2018; Lee et al., 2016). To determine if DN-Akt affects non-cognitive behaviors that are sensitive to mPFC manipulation, we assessed male mice in a three-chamber social approach/social novelty task (Figure 6A). For social approach behavior, no main effect of DN-Akt on performance was identified (Figure 6B); raw time data used to calculate approach ratios are shown in Figure S7E. After social approach, preference for social novelty was assessed (Figure 6A). Interestingly, we found that mice in the GFP and DN-Akt groups showed a similar level of preference for interaction with the novel mouse (Figure 6C). Raw time data used to calculate approach ratios are shown in Figure S7F.
Figure 6. The effects of DN-Akt in the mPFC on social and coping behaviors.
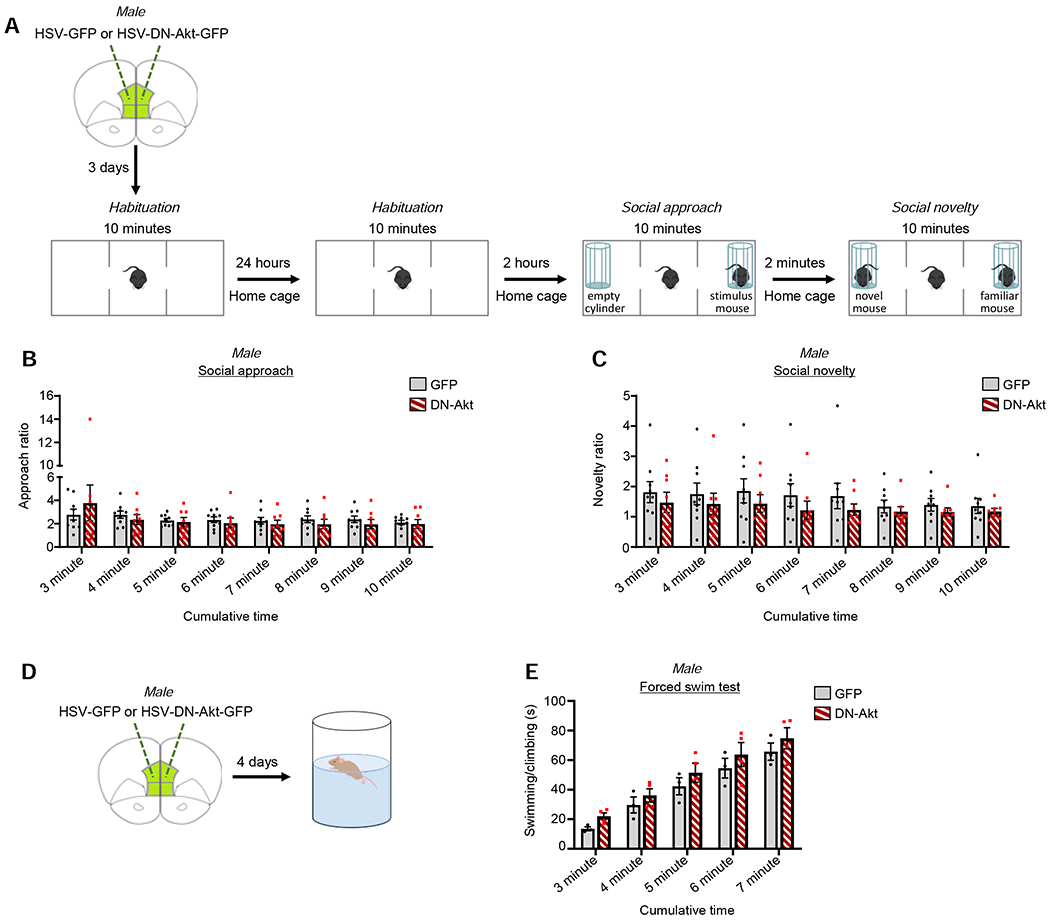
A. Social behavior schematic.
B. Social approach ratio. Post-hoc analysis revealed no significant differences between groups at any cumulative time points (p>0.05). 2-way repeated measures ANOVA with multiple comparison correction post-hoc. n=9 GFP, 8 DN-Akt
C. Social novelty ratio. Post-hoc analysis revealed no differences between the GFP and DN-Akt groups at any time points (p>0.05). n=9 GFP, 8 DN-Akt
D. Schematic of the forced swim test.
E. Cumulative time spent mobile (swimming/climbing) during the forced swim test. No group differences detected (p>0.05). 2-way repeated measures ANOVA with multiple comparison correction post-hoc. n=3 GFP, 4 DN-Akt
Summary data are the mean+SEM
See also Figures S7, S8, and S9
The impairment in episodic memory yet simultaneous lack of deficit in social behavior is suggestive of a cognitive-specificity to the effects of DN-Akt delivery to the mPFC. To further assess this possibility, we assessed mice in a forced swim test of stress coping behavior which has been shown to be sensitive to mPFC manipulation (Hamani et al., 2010) (Figure 6D). We found that when mice were subjected to room temperature water, the cumulative time spent active across the 7 minute testing period was similar in GFP and DN-Akt mice across all time points (Figure 6E). These data are suggestive of a lack of effect of mPFC DN-Akt on coping responses.
Akt activity disruption in the PFC also impairs cognition in female mice
As Akt activity disruption in the mPFC impairs specific aspects of cognition, we wanted to determine if this disruption has equivalent effects on female mice. For object-in-place testing in female mice (Figure S8A), we found similar levels of object pair preference in the GFP and DN-Akt groups during trial 1, as expected (Figure S8B). For trial 2, we found a main effect for DN-Akt expression across all time bins [F(1, 18)=4.719, p=0.0434], with post-hoc testing identifying a significant recognition ratio deficit in the DN-Akt group at the 3 minute time point (Figure S8C). Further, differences in object exploration motivation cannot account for the impaired DN-Akt group performance (Figure S8D). For object-context recognition memory in female mice (Figure S8E), we detected a main effect for DN-Akt overexpression across the time bins [F(1, 19)=4.419, p=0.0491], with post-hoc testing identifying a decreased discrimination ratio in the DN-Akt group at the 3 minute time bin (Figure S8F) that was not due to a motivational deficit (Figure S8G).
Wildtype Akt overexpression in the PFC does not impact cognition
It is conceivable that manipulation of frank Akt levels rather than alterations in Akt engagement per se are sufficient to impair mPFC-mediated cognition. To assess this possibility, we used a HSV to overexpress wildtype Akt (HSV-WT-Akt-GFP) into the mPFC and assessed object-in-place performance (Figure S9A); virus validation in Figure S9B and C. We found that WT-Akt mPFC overexpression did not impact object-in-place memory (no main effect of WT-Akt during trial 2, p=0.3002) (Figure S9D–F), and in separate mice, WT-Akt did not affect object-context recognition memory (no main effect of WT-Akt, p=0.8654) (Figure S9G–I). Collectively, these findings indicate that wildtype Akt infusion fails to recapitulate the cognitive impairments of DN-Akt.
Disruption of Akt activity attenuates dendritic spine maintenance
One of the key, consistent pathological hallmarks of bipolar disorder is a loss of dendritic spines on basal dendrites of pyramidal neurons in the DLPFC (Konopaske et al., 2014; Tobe et al., 2017). To determine if disruption of Akt activity is capable of causing dendritic spine aberrations, we infused HSV-GFP or HSV-DN-Akt-GFP into the mPFC of mice, and examined basal dendritic spine density and morphology 4 days post-viral infusion (Figures 7A–C). Attention was devoted to pyramidal neurons of the infralimbic subregion of the mPFC whose somas were located in layers 2/3. Dendritic spines were semi-automatically classified into three groups – thin, stubby, or mushroom – using our established methods (Cahill et al., 2016; Cahill et al., 2018). We found that DN-Akt-GFP decreased total dendritic spine density on mPFC pyramidal neuron basal dendrites as compared to HSV-GFP (Figures 7D and 7E), and spine subtype analysis indicates that DN-Akt reduced the density of all spine subytpes (Figures 7F–H). DN-Akt did not impact mean spine head diameters within the thin, stubby, or mushroom subtypes (Figures 7I–7K).
Figure 7. The effects of DN-Akt and mutant ULK1 on dendritic spine phenotypes.
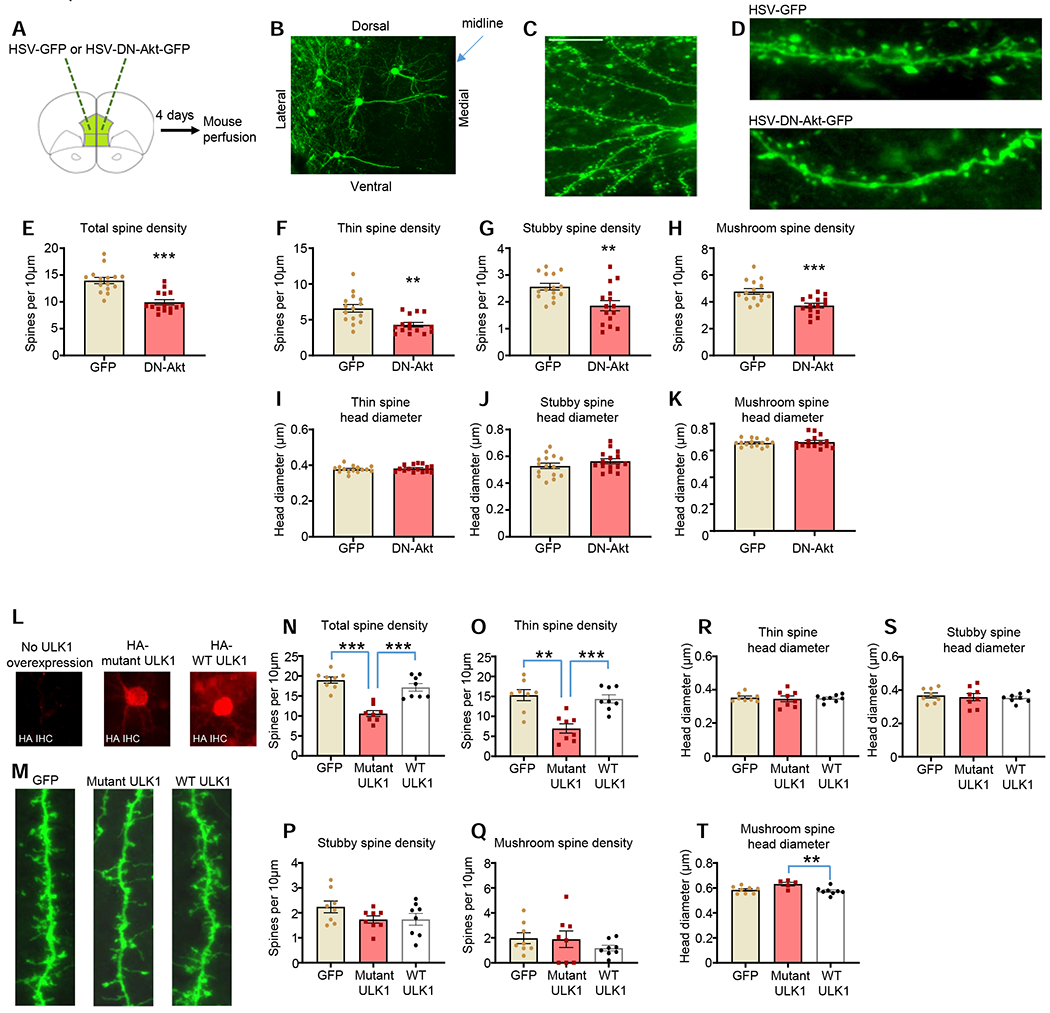
A. Experimental summary. HSV-GFP or HSV-DN-Akt-GFP infused into the mPFC of male mice. 4 days later mice were perfused and prepared for spine imaging.
B. Low magnification image of mPFC pyramidal neurons infected with HSV-GFP. Scale bar=100μm
C. Image of basal dendrite tree from HSV-infected mPFC pyramidal neuron. Scale bar=20μm
D. Representative basal dendrite segments from GFP and DN-Akt-GFP neurons. Scale bar=10μm
E. DN-Akt reduces total basal dendritic spine density in mPFC pyramidal neurons [t(28)=5.327, p<0.0001]. n=15 neurons from 3 mice (GFP) and 15 neurons from 3 mice (DN-Akt)
F-H. DN-Akt reduces the density of thin dendritic spines (F) [t(28)=3.660, p=0.001], stubby dendritic spines (G) [t(28)=3.129, p=0.0041], and mushroom dendritic spines (H) [t(28)=3.833, p=0.0007]. n=15 neurons from 3 mice (GFP) and 15 neurons from 3 mice (DN-Akt)
I-K. DN-Akt does not affect the mean head diameter of thin (I) [t(28)=0.5958, p=0.5561], stubby (J) [t(28)=1.253, p=0.2207], or mushroom spine subtypes (K) [t(28)=0.4714, p=0.6410].
L. DIV24 cultured cortical neurons were transfected with GFP alone or GFP in combination with HA-tagged mutant ULK1 or wildtype ULK1, and neurons fixed at DIV28. HA labeling identified neurons overexpressing ULK1 constructs. Scale bar=20μm
M. Representative basal dendrite segments (dendrites shown=40μm) for neurons expressing GFP alone or GFP in combination with ULK1 constructs.
N. Mutant ULK1 reduced total spine density relative to GFP and WT-ULK1 conditions. Welch one-way ANOVA: GFP vs. mutant ULK1, p<0.0001; mutant ULK1 vs. wildtype ULK1, p=0.0004; GFP vs. wildtype ULK1, p=0.4064. n=8 neurons per condition.
O. Mutant ULK1 overexpression reduced thin spine density relative to GFP and WT-ULK1 conditions. Welch one-way ANOVA: GFP vs. mutant ULK1, p=0.0011; mutant ULK1 vs. wildtype ULK1, p=0.0008; GFP vs. wildtype ULK1, p=0.9240. n=8 neurons per condition
P and Q. Neither ULK1 construct affected the density of stubby (P) or mushroom spines (Q). p>0.05 for all comparisons n=8 neurons per condition.
R and S. Neither ULK construct affected the head diameter of thin spines (R) or stubby spines (S). p>0.05 for all comparisons. n=8 neurons per condition
T. Wildtype ULK1 showed a slight, yet significant decrease in mean mushroom spine head diameter relative to mutant ULK1. No other differences between groups detected. Welch one-way ANOVA: GFP vs. mutant ULK1, p=0.0772; mutant ULK1 vs. wildtype ULK1, p=0.0377; GFP vs. wildtype ULK1, p=0.8793. n=8 neurons per condition.
Summary data are the mean+SEM
Our human DLPFC findings identify a reduction in ULK1 S757 phosphorylation in male BP-psychosis as compared to male BP-psychosis. To determine if blocking phosphorylation at the ULK1 S757 residue impacts spine stability, in mature cultured cortical neurons we co-transfected GFP and a ULK1 construct in which the S757 residue is mutated to alanine to render it phospho-incompetent (mutant ULK1, S757A); construct described previously (Kim et al., 2011). Comparisons were made to GFP transfection alone conditions and to neurons co-transfected with GFP and wildtype ULK1. Immunohistochemistry against the HA tag on the ULK1 constructs identified overexpressing neurons (Figure 7L). We found that 4 day ULK1 S757A overexpression, but not wildtype ULK1 overxpreression, reduced total basal dendritic spine density relative to the GFP alone condition (Figure 7M and N). Subtype analysis indicates that this reduction in total spine density by ULK1 S757A arises due to a significant reduction in the density of thin spines (Figure 7O–7Q). Overexpression of ULK1 S757A had minimal effects on the head diameter of the different spine subtypes (Figures 7R–T). Collectively, these findings indicating that obstructing the engagement of the S757 residue on ULK1 has pronounced effects on spine density with comparatively modest effects on head diameter.
Aberrant Akt activity reduces AMPA mEPSCs and immediate early gene induction
Next, we wanted to determine if the reduction in dendritic spine density in response to HSV-DN-Akt-GFP overexpression is associated with alterations in synaptic function. To this end, we assessed AMPA mEPSC in layer 2/3 mPFC (infralimbic subregion) pyramidal neurons following the overexpression of HSV-DN-Akt-GFP or HSV-GFP (Figure 8A). We found normal resting potential, membrane capacitance, and membrane resistance in the DN-Akt and GFP neurons (Figures 8B–D). DN-Akt reduced mEPSC mean peak amplitude relative to GFP neurons (Figure 8E), and also resulted in a significant leftward shift in the cumulative amplitude curve (Figure 8F). Further, DN-Akt reduced the mean frequency of mEPSCs compared to GFP (Figure 8G), and deep analysis indicates that DN-Akt reduced mEPSC frequency in the three amplitude bins spanning from 5-20 pA (Figure 8H).
Figure 8. The effects of DN-Akt on AMPA mEPSCs and novelty-mediated induction of IEGs.
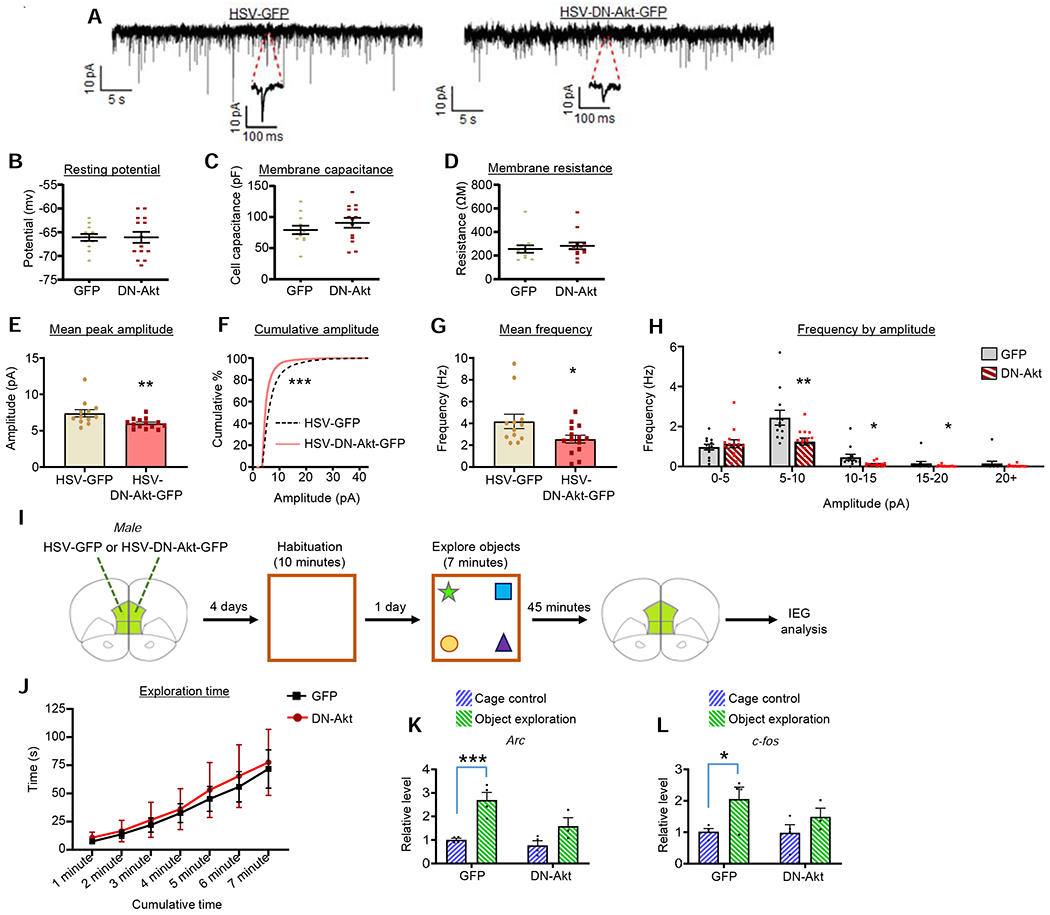
A. Representative AMPA mEPSC traces from mPFC (infralimbic subregion) layer 2/3 pyramidal neurons of young adult mice infused with HSV-GFP or HSV-DN-Akt-GFP.
B-D. No differences in resting potential (B) (p=0.99), membrane capacitance (C) (p=0.29), or membrane resistance (D) (p=0.30) were detected between groups. n=12 neurons from 5 mice (GFP) and 14 neurons from 4 mice (DN-Akt)
E. DN-Akt-GFP reduced the mean peak amplitude of mEPSCs in mPFC neurons. Two-tailed t-test, p=0.0037. n=12 neurons from 5 mice (GFP) and 14 neurons from 4 mice (DN-Akt)
F. Cumulative amplitude curve shows a significant leftward shift in the curve in the DN-Akt group. ROC curve analysis, p=0.0003
G. DN-Akt-GFP reduced mean mEPSC frequency in mPFC neurons. Two-tailed t-test, p=0.0271. n=12 neurons from 5 mice (GFP) and 14 neurons from 4 mice (DN-Akt)
H. mEPSC frequency as a function on amplitude bin. DN-Akt-GFP reduced the frequency of mEPSCs in the 5-10 pA (p=0.008), 10-15 pA (p=0.021), and 15-20 pA (p=0.0485) amplitude bins, with no effect on the 0-5 pA (p=0.99) or 20+ pA (p=0.5015) amplitude bins.
I. Experimental summary.
J. Line graph depicts cumulative time spent exploring objects across the 7 minute assessment period. No differences between groups detected. 2-way ANOVA with multiple comparison post-hoc. n=4 GFP, 3 DN-Akt
K. Arc in cage control and object exploration groups in the GFP and DN-Akt mice. No group differences in baseline (cage control) Arc levels detected. In response to object exploration, GFP, but not DN-Akt, mice show a significant increase in Arc over baseline levels. 2-way ANOVA with Bonferroni post-hoc [GFP, p=0.0009; DN-Akt, p=0.0986]. n=4 GFP cage, 4 GFP objects, 4 DN-Akt cage, 3 DN-Akt objects
L. c-fos in cage control and object exploration groups in the GFP and DN-Akt mice. No group differences in baseline (cage control) c-fos levels detected. In response to object exploration, GFP, but not DN-Akt, mice show a significant increase in c-fos over baseline levels. 2-way ANOVA with Bonferroni post-hoc [GFP, p=0.0367; DN-Akt, p=0.4706]. n=4 GFP cage, 4 GFP objects, 4 DN-Akt cage, 3 DN-Akt objects
Summary data are the mean+SEM
The cognitive impairments resulting from DN-Akt infusion into the mPFC could occur due to reduced responsiveness of this region during novelty-mediated exploration. To assess this possibility, we infused HSV-DN-Akt-GFP or HSV-GFP into the mPFC of mice, and following habituation to an open field, allowed mice to explore four novel objects for 7 minutes. 45 minutes following the start of object exploration, the mPFC was dissected and levels of the Arc and c-fos immediate early genes (IEGs) assessed (Figure 8I). IEGs are rapidly upregulated in populations of activated neurons, and as such are a commonly used indirect, yet highly quantifiable, means of assessing the molecular hallmarks of neuronal activity both in vitro and in vivo (Gallo et al., 2018; Kubik et al., 2007; Minatohara et al., 2015). Cage control mice that underwent viral infusion and open field habituation but were not exposed to the novel objects were used for baseline comparisons. First, among the object exploration groups, we found no differences in total object exploration time across the 7 minute assessment period between the GFP and DN-Akt mice (Figure 8J). For Arc levels, we found that among cage control mice there were no differences between the GFP or DN-Akt groups, suggesting the DN-Akt does not affect baseline mPFC Arc. However, following object exploration we found that GFP, but not DN-Akt, mice show an increase in Arc compared to their cage control counterparts (Figure 8K). Similar findings were detected for c-fos (Figure 8L). Collectively, these findings indicate that DN-Akt in the mPFC diminishes the responsivity of neurons during novelty exploration.
Discussion
Here we identified decreased Akt-mTOR signaling in the DLPFC and VLPFC of human bipolar disorder subjects and demonstrate that reproducing this Akt signaling impairment in the PFC of mice is sufficient in and of itself to cause cognitive disruption, dendritic spine loss, reduced synaptic function, and PFC hypoengagement. Mimicking the full spectrum and complexities of human bipolar disorder cognitive impairment in mice is not possible considering that many bipolar disorder cognitive aberrations span the verbal domain. However, that Akt activity disruption in the PFC of mice is able to at least partially impair the function of this brain region is consistent with these Akt aberrations potentially attenuating cognitive function in human subjects as well.
That bipolar disorder with and without psychosis show disparate alterations in Akt-mTOR activity is in keeping with accumulating evidence showing that genetic susceptibility loci for these bipolar disorder subtypes show many major differences (Park et al., 2004), and that these differences extend to signal transduction pathways (Kermath et al., 2020b). Further, gene profiling has found that the risk loci overlap between schizophrenia and bipolar disorder is stronger for bipolar disorder subjects who experience psychosis (Bipolar et al., 2018; Stahl et al., 2019). While many studies have uncovered protein alterations in bipolar disorder, few studies have examined differences by sex and by psychosis history. Our findings demonstrate a future need to consider these variables for identifying and characterizing signaling alterations in the bipolar disorder forebrain.
Our findings highlight ULK1 as the only aberrantly regulated mTORC1 target in the DLPFC of male BP-no psychosis subjects, while in the VLPFC, the activity of both p70S6K and 4E-BP1 were reduced in male BP-no psychosis with minimal changes in ULK1. With ULK1 we remain cautious in over-interpreting our findings as the deficit in ULK1 S757 phosphorylation in the DLPFC of male BP-no psychosis relative to male controls was significant with direct comparison only; however, the S757 decrease in male BP-no psychosis relative to male BP-psychosis remained significant following multiple comparison correction. ULK1 plays a critical role in autophagy (Zachari and Ganley, 2017), and increased ULK1 S757 phosphorylation occurs in response to increased mTORC1 activation thereby reducing ULK1’s activity and hence reducing autophagy (Kim et al., 2011). Thus, the decrease in ULK1 S757 phosphorylation in the male BP-no psychosis DLPFC is suggestive of increased ULK1-mediated autophagy. Autophagy is one of the primary mechanisms responsible for synapse remodeling in pyramidal neurons, such that increased autophagy facilitates dendritic spine pruning or removal (Nikoletopoulou and Tavernarakis, 2018). Evidence suggests that decreased autophagy via increased mTORC1 activation underlies the characteristic increase in dendritic spine density in the cortex of autism subjects (Tang et al., 2014). In contrast, as bipolar disorder is typified by a loss of prefrontal cortical dendritic spine density (Konopaske et al., 2014; Tobe et al., 2017), it is conceivable that increased autophagy via decreased mTORC1 activation contributes to spine loss in male BP-no psychosis subjects. Consistent with the possible involvement of increased autophagy of dendritic spines, we found that overexpression of ULK1 S757A reduced dendritic spine density in cortical neurons. The S757A mutation would expectedly reduce the ability of basal mTORC1 activity to decrease ULK1 activity, thereby resulting in ULK1 that is more active with increased autophagic activity (Kim et al., 2011).
P70S6K regulates translation initiation via its actions on several molecules, including eIF4B, PDCD4, and eEF2K (Holz et al., 2005; Magnuson et al., 2012). Our findings identified reduced activity of p70S6K in the VLPFC in male BP-no psychosis. The activity of p70S6K is known to be regulated by the phosphorylation of multiple residues, including T389, T421, and T424. Phosphorylation at the T421/T424 residues located in the autoinhibitory domain is a critical initial step in the activation of p70S6K. These residues are phosphorylated by p42/44 MAPK, and have been shown to represent mTOR-independent phosphorylation sites (Iijima et al., 2002). Phosphorylation at the T389 residue, on the other hand, is mTORC1-dependent and is critical for the full activation of p70S6K (Burnett et al., 1998; Pearson et al., 1995). T389 phosphorylation is thought to occur after the prior alleviation of p70S6K autoinhibition (Pullen and Thomas, 1997). The reduction in phosphorylation across the T389 and T421/424 residues in the VLPFC of BP-no psychosis suggests that p70S6K activity is suppressed via attenuation of mTORC1-dependent signaling, as well as through likely non-mTOR pathways. One complicating factor is that we also detected a reduction in total p70S6K in male BP-no psychosis. It is thus possible, perhaps even likely, that reduced p70S6K phosphorylation in male BP-no psychosis results not from a loss in the activity of pathways that regulate its activity per se, but rather are a consequence of less p70S6K being expressed.
Additionally, in the VLPFC of male BP-no psychosis we detected a reduction in phosphorylation of 4E-BP1 at the T37/46 residues, both of which are known mTORC1 targets (Gingras et al., 1999). 4E-BP1 binds to the translation initiation factor eIF4E at the 7-methylguanosine mRNA cap, and this interaction inhibits cap-dependent translation (Modrak-Wojcik et al., 2013; Uttam et al., 2018). Phosphorylation of 4E-BP1, in turn, reduces its binding capabilities to eIF4E thereby facilitating that interaction of eIF4E with another initiation factor, eIF4G, which represents a critical initial step for the recruitment of the 40S ribosomal subunit to the mRNA 5’ end (Showkat et al., 2014; Uttam et al., 2018). Thus, while 4E-BP1 phosphorylation facilitates translation initiation, reduced phosphorylation of 4E-BP1, as occurs in the VLPFC of male BP-no psychosis, would expectedly impede translation initiation.
Cognitive impairments are a core bipolar disorder feature regardless of the presence of psychosis (Burton et al., 2018; Jimenez-Lopez et al., 2017; Sanchez-Morla et al., 2009; Selva et al., 2007), and PFC hypofunction has been repeatedly implicated in these deficits (Cremaschi et al., 2013; McKenna et al., 2014; Townsend et al., 2010). Understanding the biochemical underpinnings of cognitive disruption in bipolar disorder is of great interest as cognitive symptoms are among the best predictors of social disruption severity in patients (Bonnin et al., 2010; Martinez-Aran et al., 2007). Importantly, while the manic and depressive episodes of bipolar disorder are cyclic, cognitive impairments persist even when a patient is neither in a manic nor depressive state (i.e., euthymia) (Elias et al., 2017; Martinez-Aran et al., 2004a; Martinez-Aran et al., 2004b). Finally, there is no established pharmacological regimen for treating cognitive symptoms, unlike altered mood states (Sole et al., 2017).
Episodic memory refers to the what, where, and when aspects of experience, and impaired episodic memory, including visuospatial episodic memory, is a feature of bipolar disorder that persists during euthymia (Deckersbach et al., 2004a; Deckersbach et al., 2004b; Torres et al., 2007; Tsitsipa and Fountoulakis, 2015). Episodic memory dysfunction in bipolar disorder is thought to manifest in part from dysfunction of the prefrontal cortex (Deckersbach et al., 2006). The object-in-place and object-context deficits identified in our study in response to DN-Akt expression in the mPFC pertain to the where aspects of episodic memory. It is interesting that non-cognitive behaviors, such as social approach and forced swim, which are known to either engage or be sensitive to mPFC disruptions (Cao et al., 2018; Hamani et al., 2010; Lee et al., 2016), were not affected by DN-Akt. This is suggestive of a cognitive-specificity to the observed DN-Akt-elicited behavioral phenotypes.
Multiple studies have revealed a reduction of pyramidal neuronal dendritic spine density along PFC basal dendrites in bipolar disorder subjects (Konopaske et al., 2014; Tobe et al., 2017). We found that DN-Akt produces a striking reduction of basal dendritic spine density in the mPFC, and this spine loss occurs across all major spine subtypes. Consistent with a reduction in dendritic spines, we found a reduction in the frequency of AMPA mEPSCs in DN-Akt overexpressing mPFC neurons, and this reduction spanned a broad range of mEPSC amplitude bins. The reduction in dendritic spines by DN-Akt can account for this reduction in mEPSC frequency. Further, we found that DN-Akt reduced mEPSC amplitude in mPFC neurons. As spine head size correlates with functional AMPA receptor content (Noguchi, et al., 2011; Matsuzaki, et al., 2001), the reduction in mushroom spines in response to DN-Akt overexpression can account for the reduction in mEPSC amplitude. The effects of DN-Akt in reducing dendritic spine density and AMPA mEPSC frequency and amplitude likely contributes to the reduced ability of novelty exploration to engage the mPFC as revealed by our IEG induction assessments. This is highly suggestive of the mPFC being “offline” as a consequence of DN-Akt.
AKT variants have been repeatedly identified in bipolar disorder and schizophrenia (Bajestan et al., 2006; Karege et al., 2012; Karege et al., 2010; Li et al., 2017; Mathur et al., 2010; Ripke et al., 2013; Schizophrenia Working Group of the Psychiatric Genomics, 2014; Schwab et al., 2005). While we were surprised to find no effects of schizophrenia on the Akt-mTOR pathway, as our DLPFC homogenates were derived from Bodmann’s Area 46, our findings do not rule out the possibility of Akt-mTOR alterations within other Brodmann Areas that comprise the DLPFC (e.g., BA 9). Interestingly, our recent work using DLPFC homogenates from the same subjects as this study identified an increase in protein activity of a RhoA small GTPase signaling pathway in bipolar disorder subjects with psychosis, with no alteration in bipolar disorder without psychosis or in schizophrenia (Kermath et al., 2020b). Taken together with our Akt findings, these previous RhoA pathway results are further consistent with the presence of multiple distinct intracellular signaling alterations that are chiefly dictated by the presence or absence of psychosis in bipolar disorder. Understanding the mechanisms by which increased RhoA network signaling and decreased Akt-mTOR signaling in the PFC segregate on the presence or absence of psychosis in bipolar disorder, respectively, remains an interesting question for the future.
STAR Methods
RESOURSE AVAILABILITY
Lead contact
Further information and requests for resource and reagents should be directed to and will be fulfilled by the Lead Contact, Michael Cahill (michael.cahill@wisc.edu).
Materials availability
This study did not generate new unique reagents. The HSVs used have been described previously (Cahill et al., 2016), but were repropragated for this study. HSVs for DN-Akt and WT-Akt are available from the lead contact upon request. The ULK1 constructs were provided by Dr. Kun-Liang Guan (University of California, San Diego) and are described in previous work (Kim et al., 2011).
Data and code availability
This study did not generate code, but datasets related to the current study are available from the Lead Contact, Michael Cahill, upon request.
EXPERIMENTAL MODEL AND SUBJECTS DETAILS
Experimental mice
Male and female c57BL/6J mice were used for all experiments (breeders from Jackson Laboratories); the sex of the mouse used for individual experiments is described in the text and clearly indicated in the applicable figures. For most experiments, mice were aged 10-16 weeks at the start of assessment, and age matched between experimental conditions. For electrophysiology experiments, mice were 8-9 weeks of age at the time of viral infusion. For most experiments mice were housed in standard size cage with 4 mice per cage. For female behavioral studies, mice were housed in larger cages in groups of 10 mice per cage. The typical weight of mice used for experiments ranged from 25-31 grams. All animals had continual access for food and water ad libitum. All mice were housed in a conventional vivarium located in the same building where experiments were performed and were on a 12 hour light/dark cycle. Behaviorally naïve mice were used for all studies. Mice were assigned to experimental groups using simple randomization in a manner that assured age matching between experimental conditions. All animal studies were approved by the University of Wisconsin at Madison Animal Care and Use Committee.
Dissociated cortical neuron cultures
The primary cultures of cortical neurons were obtained under sterile conditions from E18 embryos of pregnant Sprague-Dawley rats. Neurons were DIV21 at the time of transfection and DIV26 at the time of fixation.
Human brain homogenates
DLPFC (Brodmann Area 46) and VLPFC (Brodmann Area 45) homogenates were obtained from the Stanley Medical Research Institute (SMRI) array collection (Kensington, MD). The experimenters did not have access to the personal identification of the subjects comprising the array collection thereby assuring anonymity. 35 control, 35 schizophrenia, and 35 bipolar disorder subject homogenates were received from SMRI; the same subjects were the source of the DLPFC and VLPFC samples. Note that at the request of the SMRI, one bipolar disorder subject was dropped from all analyses as this subject had a hereditary stroke disorder [cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) syndrome]. This thus left a total of 34 bipolar disorder homogenates. As biochemical analysis was performed on the presence or absence of psychosis, 2 additional sample were eliminated from analysis as psychosis status was not determined in either individual, leaving a final total of 32 bipolar disorder subjects. Samples were sent with the diagnosis and sex of individual homogenates blinded.
METHOD DETAILS
Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis (SDS-PAGE) and Western blotting
Per homogenate sample, 8 μg of human homogenate was mixed with reducing sample buffer containing sodium dodecyl sulfate (SDS) and dithiothreitol (DTT). Samples were heated for 5 minutes at 95°C and resolved on Bio Rad 4-15% Tris–HCL gels in running buffer containing SDS for 2 hours at 150 V. Proteins were subsequently transferred onto PVDF membranes (Immobilin-P; Millipore) in transfer buffer containing 15% methanol (SDS-free) for 1 hour at 100 V at 4°C. Following transfer, in order long term protein stability PVDF membranes were dried at room temperature for approximately 30 minutes, then briefly rehydrated in methanol prior to beginning antibody-based labeling. Membranes were blocked for 1 hours in tris buffered saline (TBS) containing 0.1 % Tween-20 (TBS-T) and 5% bovine serum albumin (BSA; fraction V). Primary antibody and secondary antibody incubations were also performed in TBS-T containing 5% BSA with primary antibody incubation done overnight at 4°C and secondary antibody incubations done at room temperature for 1 hour. Membranes were developed using chemiluminescent substrate (Thermo Scientific), and protein levels determined using densitometry (Image J) with normalization to GAPDH. GAPDH levels were not affected by diagnosis. Western blots used in this study were originally run as part of previous work by our group examining unrelated proteins (Kermath et al., 2020b); membranes were stripped an re-probed for the proteins indicated throughout the figures, which aside from GAPDH, were not probed for in prior studies and are not related to protein probes in prior studies. Antibody dilutions used: PI3K (all total and phospho antibodies) 1:1500, Akt (total and all phospho) 1:3000, mTOR (total and phospho) 1:3000, ULK1 (total and phospho) 1:2000; p70-S6K (total and phospho) 1:2000, 4E-BP (total and phospho) 1:1500, all phospho-GSK antibodies (1:1500), and GAPDH (1:5000).
Stereotaxic viral infusions into the mPFC
HSV-GFP or HSV-DN-Akt-GFP have been described and validated previously (Cahill et al., 2016; Russo et al., 2007). A Kopf Instruments dual arm stereotaxic mounted with Hamilton 33-guage syringes was used viral infusion. The surgical plane of anesthesia was maintained using ketamine/xylazine. 0.5 μl of virus per hemisphere was infused into the mPFC at an infusion rate of 0.1 μl per minute. Following the cessation of virus infusion, the needles were left undisturbed for an additional 5 minutes to facilitate virus diffusion. The following coordinates, relative to Bregma, were used to specifically target the infralimbic region of the mPFC: 1.95mm anterior; 0.8mm lateral; 2.7mm ventral; at a 15° fixed angle.
mPFC microdissection
Following cervical dislocation, tissue was sectioned at 1 mm in a chilled stainless steel brain matrix. Virally-infected mPFC tissue was microdissected using a 0.2 mm tissue puncher. During dissection, the GFP signal from the virus was detected using a Leica fluorescent stereomicroscope, and this GFP signal used to assure that the only the area in which the virus was present was dissected.
Dendritic spine imaging and analysis
Mice that were infused with HSV-GFP or HSV-Rap1-GFP underwent transcardial perfusion with PBS for 1 minute followed by 4% paraformaldehyde for 7 minutes at a flow rate of 5 mls/minute. Brains were stored in 4% paraformaldehyde overnight at 4°C, then transferred to PBS. Brains were sectioned on a vibratome and 100 μm thick sections collected. mPFC sections were immunostained with GFP (Aves Labs; 1:1000 dilution) and AlexaFluor 488 secondary antibody (Jackson ImmunoResearch) to boost the native GFP signal emanating from the virally-infected neurons. Z-stacks of primary basal dendrites from mPFC neurons were imaged using a Keyence BZ-X700E scanning microscope. A 0.2 μm step size was used for Z-stack collection, and Z-stack images collapsed and deconvolved using BZ analyzer software (full focus algorithm). Pyramidal neurons whose soma was located in layers 2/3 of the infralimbic suregion of the mPFC were imaged.
Dendritic spines were semi-automatically analyzed using NeuronStudio using methods detailed in our previous work (Cahill et al., 2016; Cahill et al., 2018). Approximately 50μm of basal dendrite was analyzed per neuron. The program automatically places a hollow ellipse on each spine head, such that the ellipse diameter corresponds to the diameter of the spine head. In instances in which the ellipse fails to span the spine head width, or over-expands the spine head, manual adjustment to the ellipse diameter can be made. In addition to spine head diameter, spines were classified into thin, stubby, or mushroom subtypes using our previous methods. Any spines that lacked a discernible neck, were classified at stubby. Mushroom spine were spine with a clear neck and spine head diameter greater than or equal to 0.5μm. Thin spines were those with a clear neck and a head diameter of less than 0.5μm. Dendritic spine imaging and analysis were performed blinded to experimental conditions.
Quantitative PCR
RNeasy Plus Micro kit (Qiagen, #74034) was used to isolate RNA from mPFC (infralimbic/prelimbic) punches. A cDNA reverse transcription kit containing RNA inhibitor (Applied Biosystems, #4374966) was used to convert 200ng of RNA to single-stranded cDNA, following the instructions of the manufacturer. Real-time PCR reactions were performed in triplicate via TaqMan primer and probe sets from Applied Biosystems (c-fos, Mm00487425_m1; Arc, Mm01204954_g1; Gapdh, Mm99999915_g1; Tubulin, Mm00495806_g1). The following run parameters on an ABI StepOne machine were used: 50°C for 2 mins, 95°C for 20 secs, 40 cycles of 95°C for 1 sec, 60°C for 20 sec. Each gene’s relative expression was determined using the −2(ΔΔCt) method. Levels were normalized to those of Tubulin, which did not itself differ between groups. Relative changes were normalized to the GFP no object control group.
Cortical cell culture
Rat fetuses were removed from euthanized timed-pregnant dames via caesarean section and placed into a 60mm sterile petri dish containing cold hanks’ balanced salt solution (HBSS; Gibco) supplemented with 1% 100mM sodium pyruvate (Invitrogen), 0.1% D+ glucose (Sigma), 10mM 1M HEPES (Gibco). Fetuses were then decapitated and their brains were isolated. The meninges were removed and cortical neurons were then isolated from 6-8 cortical hemispheres. The cortical hemispheres were placed in 1.5mL tubes with 1mL of HBSS. The cortices were digested with 110μL of trypsin at 37°C for 30 minutes. Cells were washed twice in a plating media: Dulbecco’s Modified Eagle’s Medium (DMEM; Gibco) containing 1X penicillin/streptomycin (P/S; Invitrogen), 1X 200mM L-glutamine (Gibco) and 10% fetal bovine serum (FBS; Gibco).
1mL of DMEM/cortical cells were transferred to a 15mL sterile centrifuge tube containing 4mLs of DMEM. The cells were centrifuged at 350 rpm for 7 minutes, and the supernatant was removed. The cells were resuspended in the plating media and the density of cell suspension was adjusted to 5 × 104 cells/mL. The cortical neuron cells were then plated onto glass coverslips in 6 well plates. Prior to plating coverslips were precoated with 0.1 mg/mL poly-D-lysine (Sigma) overnight.
The cultured plates were stored in a 5% CO2 incubator at 37°C for approximately 3 hours. Then cultured media was replaced by neuronal media: Neurobasal media (Gibco) supplemented with 1X B27 (Gibco), 1X P/S, and 1X 200mM L-glutamine. Cortical neurons were treated one time at DIV 2 with cytosine B-D arabinofuranoside (ARA-C; Sigma) to inhibit dividing cells. Starting on DIV 5 the neuronal media supplemented with DL-AP5 (TOCRIS) was refreshed by exchanging half of the volume with fresh media every three days.
On DIV 21, cultured cortical neurons were transfected with cDNA plasmids using Lipofectamine (Invitrogen) according to the manufacturer’s instructions. Neurons were fixed 5 days post transfection (DIV26) with 3.7% formaldehyde / 4% sucrose and incubated for 10 minutes at room temperature (RT). Cultured neuron cells were rinsed three times with 1X PBS. Neurons were then incubated in ice cold methanol for 10 minutes at −20°C and then washed three times in 1X PBS supplemented with 0.3% Triton X-100 (T) before they were incubated in a blocking solution (3% donkey blocking serum (DBS), 0.3% T, and 1X PBS) at RT for 1 hour. The following primary antibodies in blocking solution were then applied to the neurons at 4°C overnight: GFP (Chicken monoclonal, 1:1000 dilution, Aves Labs) and HA-Tag (Rabbit monoclonal, 1:1000, Cell Signaling Technology). Following primary antibody incubations, cells were washed three times in 1X PBS and then incubated in 3% DBS + 1X PBS with the following Alexa Fluor secondary (Jackson Immuoresearch) antibodies for 1 hour at room temperature: donkey anti-chicken 488 and donkey anti-rabbit 555.
Electrophysiology
HSV-GFP or HSV-DN-Akt-GFP were infused into the mPFC of C57BI/6J mice using the coordinates for viral infusion detailed above.
Picrotoxin (Sigma-Aldrich; P1675) was dissolved in 100% DMSO and diluted to 0.1% DMSO in aCSF. Tetrodotoxin citrate (HelloBio; HB1035) and D-AP5 (HelloBio; HB0225) were dissolved in ddH2O. Brains from transcardially perfused mice were sliced into coronal sections (250 μM containing IL PFC) in sucrose aCSF (aCSF; in mM: 234 sucrose, 2.5 KCl, 1.25 NaH2PO4, 10 MgSO4, 0.5 CaCl, 26 NaHCO3, and 11 glucose). Slices were then transferred to an incubation chamber containing aCSF (in mM: 126 NaCl, 2.5 KCl, 1.25 NaH2PO4, 2 MgCl, 2 CaCl, 26 NaHCO3, and 10 glucose; bubbled with 95% O2-5% CO2) and held at 34°C. Recordings were made from a submersion chamber perfused with aCSF (2 ml/min) maintained at 32-34°C.
Borosilicate glass electrodes (3–6 MΩ) were filled with K-gluconate internal solution (in mM: 115 potassium gluconate, 20 KCl, 1.5 MgCl, 10 phosphocreatine-Tris, 10 HEPS, 0.1 EGTA, 2 Mg-ATP, and 0.4 Na-GTP; pH 7.2–7.4, 290 mOsm). GFP-positive neurons in the IL PFC were visualized using an upright microscope (Olympus) using infrared and epifluorescent illumination. Layer 2/3 pyramidal cells were distinguished by location and individual cell morphology. Whole-cell patch-clamp recordings were made from transfected cells using a Multiclamp 700B amplifier and a Digidata 1440A digitizer (Molecular Devices). Whole-cell junction potential was not corrected. Traces were sampled (20 kHz), filtered (Bessel 2 kHz), and digitally stored. Cells with membrane potential greater than −50 mV or series resistance of 30 MΩ were omitted from analysis. Cells were held at −70 mV and miniature EPSCs were recorded for 5 min in the presence of picrotoxin (50 μM), TTX (1 μM), and D-AP5 (50 μM).
Traces were compiled in pClamp 10 (Clampfit). Traces (2 min per cell) were filtered (LoPass Butterworth 500 Hz) and analyzed by Mini Analysis (Synaptosoft). Miniature EPSCs with peak amplitudes lower than 2.5 pA were excluded from the analysis.
General behavioral procedures
All behavioral assessments were performed and analyzed blinded to experimental conditions, and were recorded via an overhead digital zoom camera. Naïve mice were used for each behavioral test in order to prevent any learning carry over effects between tests.
Object-in-place testing
Procedures were adopted from previous studies (Barker et al., 2007), and as described in our previous work (Kermath et al., 2020a). A gray plastic open field box with dimensions of 40cm wide, 40cm long, and a 30cm height was purchased (Maze Engineers), and the box surrounded by distal spatial cues. The floor of the box contained a square textured bottom (16 square pattern). Prior to starting object-in-place assessment, mice were habituated to the empty open field box for 10 minutes. For trial 1, 4 unique plastic objects were placed 15 cm from the box corners, with each object about 8 cm in height. Each object had a distinctive visual and textural components. Mice were allowed to explore objects for 7 minutes, at which time mice were removed from the open field and placed into their home cage for 7 minutes. After trial 1 and prior to start of trial 2, the location of two of the four objects were swapped, thereby resulting in a pair of swapped objects, and a pair on objects whose location was not altered. For trial 2, mice were allowed 7 minutes of object exploration. The recognition ratio assessed was calculated as the proportion of exploration time devoted to the objects that swapped location vs. those that remained in the same location; chance level=1.0. Object exploration was defined as any activity in which the nose of the mouse was directed within 1cm of an object. Importantly, behaviors such as sitting/resting which touching an object, or rare attempts at climbing an object, were not considered object exploration. Testing trials were recorded using an overhead digital zoom camera, and for quantification, recordings were scaled to one-third real-time speed in order to ensure accurate assessments. Investigation time was quanitified via a digital stopwatch with a resolution of 0.01 seconds and 0.0005% accuracy. The test and analysis was performed blinded to experimental conditions.
Object-context recognition
Procedures were adopted from previous work (Wilson et al., 2013). Two distinct boxes, both with dimensions of 40 cm length and width, and 30 cm height were used for object-context recognition. One box was made of gray plastic walls and had a square textured floor (context 1). The other box contained glass walls with an alternating stripped light/dark pattern and a uniformly textured floor (context 2). For trial 1, mice were placed in context 1 and allowed to explore two identical plastic objects located 15 cm from the box corners for 7 minutes. Mice were then placed into their home cage for 1 minute, and for trial 2 were placed into context 2 which contained two identical, yet novel plastic objects, for 7 minutes. Mice were then returned to their home cage for 5 minutes, and for trial 3, mice placed into context 1, which now contained one object from trial 1 and one object from trial 2. The recognition ratio for trial 3 was calculated as the proportion of time spent investigating the context mismatched object vs. the context matched object; chance level=1.0. Criteria for object exploration and methods of analysis were the same as those described for the object-in-place test.
Temporal order testing
Procedures were adopted from previous work (Barker et al., 2007). For temporal order testing, the same open field box as described for the object-in-place test was used. Mice were first habituated to the empty open field box for 10 minutes. For trial 1, mice were placed in the box and allowed to explore two identical plastic objects located 15cm from the corner of the box for 10 minutes. Mice were then returned to their home cages for 1 hour. For trial 2, mice were allowed to explore the open field, which now contained two identical yet novel objects for 10 minutes. Mice were again returned to their home cage for 1 hour, and for trial 3 placed into the open field box which contain one object from trial 1, and one object from trial 2. Criteria for object exploration and methods of analysis were the same as those described for the object-in-place test. The temporal recognition index was calculated as the time spent investigating the less recently encountered object from trial 1 divided by the total time spent investigating both objects; chance level=0.5.
Social motivation and social recognition
Procedures were adopted from previous studies (Moy et al., 2004). Social behavior was performed in a three-chamber apparatus 80 cm long, 40.5 cm wide, and with a height of 40 cm. The chamber was divided using clear plastic partitions into two large chambers and a smaller middle chamber. The day before the start of testing, mice were habituated to the empty three-chambered apparatus via two, 10 minute sessions. The next day the mice were again habituated to the empty apparatus for 10 minutes. For social approach testing, a mesh cylinder was placed in each end chamber. One cylinder contained a novel stimulus mouse (social chamber), while the other cylinder remained empty (non-social chamber). The cylinders allowed for visual and olfactory exchange. Mice were placed in the center chamber and allowed to free explore the entire apparatus for 10 minutes. The amount of time spent directly investigating the cylinders in the social vs. non-social end chamber was quantified. The approach ratio was calculated as time spent investigating the cylinder containing a novel mouse divided by the amount of time spent investigating the empty cylinder.
After social approach testing, mice were placed into their home cages for 2 minutes. During this time, a novel mouse was placed in the previously empty mesh cylinder, while the now familiar mouse from the social approach test remained in the same mesh cylinder. Thus, one end chamber contained a familiar mouse, and one end chamber a novel mouse. The amount of time directly contacting the mesh cylinder of the novel mouse vs. the familiar mouse was then quantified as a ratio of social recognition. The novelty ratio was calculated as time spent investigating the cylinder containing the unfamiliar mouse divided by the amount of time spent investigating the cylinder containing a familiar mouse.
Forced swim test
Forced swim was performed using previously described procedures (Bagot et al., 2016). A glass cylinder filled with room temperature (22°C) water. The water level was such that the tail of the mouse could not touch the bottom of the cylinder and such that the forepaws of the mouse could not reach the top of the cylinder. The amount of time spent active (swimming, attempted climbing) was quantified over a 7 minute period.
Y-maze spontaneous alternation
The Y-maze procedures were performed as described in our previous work (Kermath et al., 2020a). The Y-maze contains three arms 120° from each other (each arm 35 cm x 5 cm). Mice were placed in one arm of the Y-maze. Once a mouse entered each arm once, mice were allowed 10 minutes for free exploration, and arm entry order analyzed. The number of successive three arm entries triplet alternations were divided by the total number of possible three arm entries (total number of arm entries minus 2). This value is the spontaneous alternation percentage.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were analyzed using GraphPad Prism version 8.4.2. ANOVA main effect statistics are reported within the text, with the results of ANOVA post-hoc analyses contained within the figure legends. The n for each comparison group is contained within the figure legends. For human biochemistry analysis, separate Welch one-way ANOVAs for male and female subjects were performed (Welch was used in order to properly control for differences in the variance between groups). Multiple comparisons correction for human biochemistry analysis was performed using Dunnett’s T3 post-hoc for the following comparisons: ctrl vs. BP-NP; ctrl vs. BP-P; BP-NP vs. BP-P, and ctrl vs. schiz. For biochemical studies, in instances in which significance was present for direct Welch t-test comparisons, but lost in response to Dunnett’s T3 multiple-comparison corrections, the symbol ‘t’ was indicated on the graph, and that significance did not hold following comparison corrections indicated in the figure legend. In one instance, a Grubbs’ test outlier in human homogenates was detected and removed from analysis that affected the significance threshold (BP-NP group for p-PI3K Y199). Comparison between two groups were in most instances made using a 2-tailed Student’s t-test, with equal variance confirmed using the F-test and normal distribution confirmed using the D’Agostino & Pearson test. Square root transformation was used in instances of abnormal distribution, and if normality was still not obtained post-transformation, Mann-Whitney tests on non-transformed data were used. IEG data was analyzed using a 2-way ANOVA with Bonferroni post-hoc comparison (repeated measures in the case of virus validation). ULK1 spine analysis was assessed using Welch one-way ANOVA with Dunnett T3 post-hoc for multiple comparison corrections. Behavioral data involving longitudinal assessment across multiple time points was analyzed using a repeated measures 2-way ANOVA, and post-hoc analysis corrected for multiple comparisons by correcting the false discovery rate using the two-stage linear set-up procedure of Benjamini, Krieger and Yekutieli (Q=0.05). On graphs, *p<0.05, **p<0.01, ***p<0.001.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Phospho-pi3k Y199 | Cell Signaling Technology | Cat# 4228, RRID:AB_659940 |
| Phospho-Akt T308 | Cell Signaling Technology | Cat# 4056, RRID:AB_331163) |
| Phospho-Akt S473 | Cell Signaling Technology | Cat# 4060, RRID:AB_2315049 |
| Pan Akt | Cell Signaling Technology | Cat# 4691, RRID:AB_915783 |
| Akt1 | Cell Signaling Technology | Cat# 2938, RRID:AB_915788 |
| Phospho-mTOR S2448 | Cell Signaling Technology | Cat# 2971, RRID:AB_330970 |
| Phospho-mTOR S1261 | Diane Fingar (University of Michigan) | Acosta-Jaquez et al., 2009 |
| Total mTOR | Cell Signaling Technology | Cat# 2983, RRID:AB_2105622 |
| Phospho-ULK1 S757 | Cell Signaling Technology | Cat# 6888, RRID:AB_10829226) |
| Phospho-ULK1 S777 | Millipore | Cat# ABC213, RRID:AB_2861423 |
| Total ULK1 | Cell Signaling Technology | Cat# 8054, RRID:AB_11178668 |
| Phospho-p70s6k T421/S424 | Cell Signaling Technology | Cat# 9204, RRID:AB_2265913) |
| Phospho-p70s6k T389 | Cell Signaling Technology | Cat# 9234, RRID:AB_2269803 |
| Total p70s6k | Cell Signaling Technology | Cat# 2708, RRID:AB_390722 |
| Phospho-4e-bp1 T37/46 | Cell Signaling Technology | Cat# 2855, RRID:AB_560835 |
| Total 4e-bp1 | Cell Signaling Technology | Cat# 9644, RRID:AB_2097841 |
| Phospho-gsk3α S21 | Cell Signaling Technology | Cat# 9316, RRID:AB_659836 |
| p-gsk3ß S9 | Cell Signaling Technology | Cat# 5558, RRID:AB_10013750 |
| GAPDH | Santa Cruz Biotechnology | Cat# sc-47724, RRID:AB_627678 |
| Green fluorescent protein (GFP) | Aves Labs | Cat# GFP-1010, RRID:AB_2307313) |
| HA-Tag | Cell Signaling Technology | Cat# 3724, RRID:AB_1549585 |
| Actin | Thermo Fisher Scientific | Cat# MA5-15739, RRID:AB_10979409) |
| Bacterial and Virus Strains | ||
| HSV-dominant-negative Akt-GFP | This study | |
| HSV-wildtype AKT-GFP | This study | |
| Biological Samples | ||
| Tissue repository Array Collection (105 samples in total) | Stanley Medical Research Institute | |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Alexa Fluor 488 | Jackson Immuno Research | Cat# 703-545-155, RRID:AB_2340375 |
| Lipofectamine 2000 | Invitrogen | Cat# 11668019 |
| Opti-MEM | Gibco | Cat# 31985062 |
| DL-AP5 (for cell culture) | Tocris | Cat# 0105 |
| Vectashield Hardset antifade mounting medium | Vector Laboratories | Cat# SKU: H-1400 |
| Cytosine B-D-arabinofuranoside | Sigma | Cat# C1768 |
| L-glutamine | Invitrogen | Cat# 25030081 |
| Sodium pyruvate | Invitrogen | Cat# 11360070 |
| Penicillin/streptomycin | Invitrogen | Cat# 15140122 |
| Critical Commercial Assays | ||
| RNeasy Plus Micro Kit | Qiagen | Cat# 74034 |
| High-Capacity cDNA Reverse Transcription Kit with RNase Inhibitor | Applied Biosystems | Cat# 4374966 |
| Picrotoxin | Sigma-Aldrich | Cat# P1675 |
| Tetrodotoxin citrate | Hello Bio | Cat# HB1035 |
| D-AP5 (for e-phys) | Hello Bio | Cat# HB0225 |
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| C57BL/6J | Jackson Laboratories | IMSR_JAX:000664 |
| Oligonucleotides | ||
| C-fos primer | Applied Biosystems | Mm00487425_m1 |
| Arc primer | Applied Biosystems | Mm01204954_g1 |
| GAPDH primer | Applied Biosystems | Mm99999915_g1 |
| Tubulin primer | Applied Biosystems | Mm00495806_g1 |
| Recombinant DNA | ||
| Wildtype ULK1 | Kun-Liang Guan | Kim et al., 2011 |
| ULK1 S757A | Kun-Liang Guan | Kim et al., 2011 |
| Software and Algorithms | ||
| Image J FIJI | NIH | https://imagej.net/; RRID:SCR_002285 |
| Graph Pad Prism | Graph Pad | https://www.graphpad.com/scientific-software/prism/;RRID:SCR_002798 |
| BZ Analyzer Software | Keyence | RRID:SCR_017205 |
| NeuroStudio | Icahn School of Medicine at Mount Sinai | RRID:SCR_013798 |
| Mini Analysis | Synaptosoft | RRID:SCR_002184 |
| Other | ||
Highlights.
A reduction in Akt-mTOR signaling occurs the PFC of male bipolar disorder subjects
Akt hypofunction reduces synaptic structural and functional plasticity in the PFC
Akt hypofunction is sufficient to impair PFC-mediated cognition
Acknowledgements
We thank the Stanley Medical Research Institute for the human brain homogenates, and Dr. Diane Fingar (University of Michigan) for the p-mTOR S1261 antibody. ULK1 constructs were provided by Dr. Kun-Liang Guan (University of California, San Diego). This work was supported by a NARSAD Young Investigator Award from the Brain & Behavior Research Foundation (MEC) and by National Institute of Health (National Institute of Mental Health) award R21MH125227 (MEC) and R01MH111604 (AJR).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Acosta-Jaquez HA, Keller JA, Foster KG, Ekim B, Soliman GA, Feener EP, Ballif BA, and Fingar DC (2009). Site-specific mTOR phosphorylation promotes mTORC1-mediated signaling and cell growth. Mol Cell Biol 29, 4308–4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andjelkovic M, Alessi DR, Meier R, Fernandez A, Lamb NJ, Frech M, Cron P, Cohen P, Lucocq JM, and Hemmings BA (1997). Role of translocation in the activation and function of protein kinase B. J Biol Chem 272, 31515–31524. [DOI] [PubMed] [Google Scholar]
- Bagot RC, Cates HM, Purushothaman I, Lorsch ZS, Walker DM, Wang J, Huang X, Schluter OM, Maze I, Pena CJ, et al. (2016). Circuit-wide Transcriptional Profiling Reveals Brain Region-Specific Gene Networks Regulating Depression Susceptibility. Neuron 90, 969–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajestan SN, Sabouri AH, Nakamura M, Takashima H, Keikhaee MR, Behdani F, Fayyazi MR, Sargolzaee MR, Bajestan MN, Sabouri Z, et al. (2006). Association of AKT1 haplotype with the risk of schizophrenia in Iranian population. Am J Med Genet B Neuropsychiatr Genet 141B, 383–386. [DOI] [PubMed] [Google Scholar]
- Barker GR, Banks PJ, Scott H, Ralph GS, Mitrophanous KA, Wong LF, Bashir ZI, Uney JB, and Warburton EC (2017). Separate elements of episodic memory subserved by distinct hippocampal-prefrontal connections. Nat Neurosci 20, 242–250. [DOI] [PubMed] [Google Scholar]
- Barker GR, Bird F, Alexander V, and Warburton EC (2007). Recognition memory for objects, place, and temporal order: a disconnection analysis of the role of the medial prefrontal cortex and perirhinal cortex. J Neurosci 27, 2948–2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekinschtein P, Renner MC, Gonzalez MC, and Weisstaub N (2013). Role of medial prefrontal cortex serotonin 2A receptors in the control of retrieval of recognition memory in rats. J Neurosci 33, 15716–15725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bipolar D, Schizophrenia Working Group of the Psychiatric Genomics Consortium. Electronic address, d.r.v.e., Bipolar D, and Schizophrenia Working Group of the Psychiatric Genomics, C. (2018). Genomic Dissection of Bipolar Disorder and Schizophrenia, Including 28 Subphenotypes. Cell 173, 1705–1715 e1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissonette GB, Powell EM, and Roesch MR (2013). Neural structures underlying set-shifting: roles of medial prefrontal cortex and anterior cingulate cortex. Behav Brain Res 250, 91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnin CM, Martinez-Aran A, Torrent C, Pacchiarotti I, Rosa AR, Franco C, Murru A, Sanchez-Moreno J, and Vieta E (2010). Clinical and neurocognitive predictors of functional outcome in bipolar euthymic patients: a long-term, follow-up study. J Affect Disord 121, 156–160. [DOI] [PubMed] [Google Scholar]
- Bortolato B, Miskowiak KW, Kohler CA, Vieta E, and Carvalho AF (2015). Cognitive dysfunction in bipolar disorder and schizophrenia: a systematic review of meta-analyses. Neuropsychiatr Dis Treat 11, 3111–3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne C, Aydemir O, Balanza-Martinez V, Bora E, Brissos S, Cavanagh JT, Clark L, Cubukcuoglu Z, Dias VV, Dittmann S, et al. (2013). Neuropsychological testing of cognitive impairment in euthymic bipolar disorder: an individual patient data meta-analysis. Acta Psychiatr Scand 128, 149–162. [DOI] [PubMed] [Google Scholar]
- Burnett PE, Barrow RK, Cohen NA, Snyder SH, and Sabatini DM (1998). RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc Natl Acad Sci U S A 95, 1432–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton CZ, Ryan KA, Kamali M, Marshall DF, Harrington G, McInnis MG, and Tso IF (2018). Psychosis in bipolar disorder: Does it represent a more “severe” illness? Bipolar Disord 20, 18–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill ME, Bagot RC, Gancarz AM, Walker DM, Sun H, Wang ZJ, Heller EA, Feng J, Kennedy PJ, Koo JW, et al. (2016). Bidirectional Synaptic Structural Plasticity after Chronic Cocaine Administration Occurs through Rap1 Small GTPase Signaling. Neuron 89, 566–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill ME, Walker DM, Gancarz AM, Wang ZJ, Lardner CK, Bagot RC, Neve RL, Dietz DM, and Nestler EJ (2018). The dendritic spine morphogenic effects of repeated cocaine use occur through the regulation of serum response factor signaling. Mol Psychiatry 23, 1474–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao W, Lin S, Xia QQ, Du YL, Yang Q, Zhang MY, Lu YQ, Xu J, Duan SM, Xia J, et al. (2018). Gamma Oscillation Dysfunction in mPFC Leads to Social Deficits in Neuroligin 3 R451C Knockin Mice. Neuron 97, 1253–1260 e1257. [DOI] [PubMed] [Google Scholar]
- Chao OY, Huston JP, Li JS, Wang AL, and de Souza Silva MA (2016). The medial prefrontal cortex-lateral entorhinal cortex circuit is essential for episodic-like memory and associative object-recognition. Hippocampus 26, 633–645. [DOI] [PubMed] [Google Scholar]
- Chiang GG, and Abraham RT (2005). Phosphorylation of mammalian target of rapamycin (mTOR) at Ser-2448 is mediated by p70S6 kinase. J Biol Chem 280, 25485–25490. [DOI] [PubMed] [Google Scholar]
- Cremaschi L, Penzo B, Palazzo M, Dobrea C, Cristoffanini M, Dell’Osso B, and Altamura AC (2013). Assessing working memory via N-back task in euthymic bipolar I disorder patients: a review of functional magnetic resonance imaging studies. Neuropsychobiology 68, 63–70. [DOI] [PubMed] [Google Scholar]
- Deckersbach T, Dougherty DD, Savage C, McMurrich S, Fischman AJ, Nierenberg A, Sachs G, and Rauch SL (2006). Impaired recruitment of the dorsolateral prefrontal cortex and hippocampus during encoding in bipolar disorder. Biol Psychiatry 59, 138–146. [DOI] [PubMed] [Google Scholar]
- Deckersbach T, McMurrich S, Ogutha J, Savage CR, Sachs G, and Rauch SL (2004a). Characteristics of non-verbal memory impairment in bipolar disorder: the role of encoding strategies. Psychol Med 34, 823–832. [DOI] [PubMed] [Google Scholar]
- Deckersbach T, Savage CR, Reilly-Harrington N, Clark L, Sachs G, and Rauch SL (2004b). Episodic memory impairment in bipolar disorder and obsessive-compulsive disorder: the role of memory strategies. Bipolar Disord 6, 233–244. [DOI] [PubMed] [Google Scholar]
- Dibble CC, and Cantley LC (2015). Regulation of mTORC1 by PI3K signaling. Trends Cell Biol 25, 545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doble BW, and Woodgett JR (2003). GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci 116, 1175–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunayevich E, and Keck PE Jr. (2000). Prevalence and description of psychotic features in bipolar mania. Curr Psychiatry Rep 2, 286–290. [DOI] [PubMed] [Google Scholar]
- Elias LR, Miskowiak KW, Vale AM, Kohler CA, Kjaerstad HL, Stubbs B, Kessing LV, Vieta E, Maes M, Goldstein BI, and Carvalho AF (2017). Cognitive Impairment in Euthymic Pediatric Bipolar Disorder: A Systematic Review and Meta-Analysis. J Am Acad Child Adolesc Psychiatry 56, 286–296. [DOI] [PubMed] [Google Scholar]
- Farovik A, Dupont LM, Arce M, and Eichenbaum H (2008). Medial prefrontal cortex supports recollection, but not familiarity, in the rat. J Neurosci 28, 13428–13434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo FT, Katche C, Morici JF, Medina JH, and Weisstaub NV (2018). Immediate Early Genes, Memory and Psychiatric Disorders: Focus on c-Fos, Egr1 and Arc. Front Behav Neurosci 12, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras AC, Gygi SP, Raught B, Polakiewicz RD, Abraham RT, Hoekstra MF, Aebersold R, and Sonenberg N (1999). Regulation of 4E-BP1 phosphorylation: a novel two-step mechanism. Genes Dev 13, 1422–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goes FS, McGrath J, Avramopoulos D, Wolyniec P, Pirooznia M, Ruczinski I, Nestadt G, Kenny EE, Vacic V, Peters I, et al. (2015). Genome-wide association study of schizophrenia in Ashkenazi Jews. Am J Med Genet B Neuropsychiatr Genet 168, 649–659. [DOI] [PubMed] [Google Scholar]
- Hamani C, Diwan M, Macedo CE, Brandao ML, Shumake J, Gonzalez-Lima F, Raymond R, Lozano AM, Fletcher PJ, and Nobrega JN (2010). Antidepressant-like effects of medial prefrontal cortex deep brain stimulation in rats. Biol Psychiatry 67, 117–124. [DOI] [PubMed] [Google Scholar]
- Hemmings BA, and Restuccia DF (2012). PI3K-PKB/Akt pathway. Cold Spring Harb Perspect Biol 4, a011189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeffer CA, and Klann E (2010). mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci 33, 67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holz MK, Ballif BA, Gygi SP, and Blenis J (2005). mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell 123, 569–580. [DOI] [PubMed] [Google Scholar]
- Holz MK, and Blenis J (2005). Identification of S6 kinase 1 as a novel mammalian target of rapamycin (mTOR)-phosphorylating kinase. J Biol Chem 280, 26089–26093. [DOI] [PubMed] [Google Scholar]
- Iijima Y, Laser M, Shiraishi H, Willey CD, Sundaravadivel B, Xu L, McDermott PJ, and Kuppuswamy D (2002). c-Raf/MEK/ERK pathway controls protein kinase C-mediated p70S6K activation in adult cardiac muscle cells. J Biol Chem 277, 23065–23075. [DOI] [PubMed] [Google Scholar]
- Ikeda M, Takahashi A, Kamatani Y, Momozawa Y, Saito T, Kondo K, Shimasaki A, Kawase K, Sakusabe T, Iwayama Y, et al. (2019). Genome-Wide Association Study Detected Novel Susceptibility Genes for Schizophrenia and Shared Trans-Populations/Diseases Genetic Effect. Schizophr Bull 45, 824–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhanwar-Uniyal M, Wainwright JV, Mohan AL, Tobias ME, Murali R, Gandhi CD, and Schmidt MH (2019). Diverse signaling mechanisms of mTOR complexes: mTORC1 and mTORC2 in forming a formidable relationship. Adv Biol Regul 72, 51–62. [DOI] [PubMed] [Google Scholar]
- Jimenez-Lopez E, Aparicio AI, Sanchez-Morla EM, Rodriguez-Jimenez R, Vieta E, and Santos JL (2017). Neurocognition in patients with psychotic and non-psychotic bipolar I disorder. A comparative study with individuals with schizophrenia. J Affect Disord 222, 169–176. [DOI] [PubMed] [Google Scholar]
- Kahn RS, Sommer IE, Murray RM, Meyer-Lindenberg A, Weinberger DR, Cannon TD, O’Donovan M, Correll CU, Kane JM, van Os J, and Insel TR (2015). Schizophrenia. Nat Rev Dis Primers 1, 15067. [DOI] [PubMed] [Google Scholar]
- Karege F, Meary A, Perroud N, Jamain S, Leboyer M, Ballmann E, Fernandez R, Malafosse A, and Schurhoff F (2012). Genetic overlap between schizophrenia and bipolar disorder: a study with AKT1 gene variants and clinical phenotypes. Schizophr Res 135, 8–14. [DOI] [PubMed] [Google Scholar]
- Karege F, Perroud N, Schurhoff F, Meary A, Marillier G, Burkhardt S, Ballmann E, Fernandez R, Jamain S, Leboyer M, et al. (2010). Association of AKT1 gene variants and protein expression in both schizophrenia and bipolar disorder. Genes Brain Behav 9, 503–511. [DOI] [PubMed] [Google Scholar]
- Karlsson Linner R, Biroli P, Kong E, Meddens SFW, Wedow R, Fontana MA, Lebreton M, Tino SP, Abdellaoui A, Hammerschlag AR, et al. (2019). Genome-wide association analyses of risk tolerance and risky behaviors in over 1 million individuals identify hundreds of loci and shared genetic influences. Nat Genet 51, 245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck PE Jr., McElroy SL, Havens JR, Altshuler LL, Nolen WA, Frye MA, Suppes T, Denicoff KD, Kupka R, Leverich GS, et al. (2003). Psychosis in bipolar disorder: phenomenology and impact on morbidity and course of illness. Compr Psychiatry 44, 263–269. [DOI] [PubMed] [Google Scholar]
- Kermath BA, Vanderplow AM, Bjornson KJ, Seablom EN, Novak AM, Bernhardt CR, and Cahill ME (2020a). The Rap1 small GTPase is a critical mediator of the effects of stress on prefrontal cortical dysfunction. Mol Psychiatry [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kermath BA, Vanderplow AM, and Cahill ME (2020b). Dysregulated Prefrontal Cortical RhoA Signal Transduction in Bipolar Disorder with Psychosis: New Implications for Disease Pathophysiology. Cereb Cortex 30, 59–71. [DOI] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, and Guan KL (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13, 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopaske GT, Lange N, Coyle JT, and Benes FM (2014). Prefrontal cortical dendritic spine pathology in schizophrenia and bipolar disorder. JAMA Psychiatry 71, 1323–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubik S, Miyashita T, and Guzowski JF (2007). Using immediate-early genes to map hippocampal subregional functions. Learn Mem 14, 758–770. [DOI] [PubMed] [Google Scholar]
- Lalonde R (2002). The neurobiological basis of spontaneous alternation. Neurosci Biobehav Rev 26, 91–104. [DOI] [PubMed] [Google Scholar]
- Laplante M, and Sabatini DM (2009). mTOR signaling at a glance. J Cell Sci 122, 3589–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E, Rhim I, Lee JW, Ghim JW, Lee S, Kim E, and Jung MW (2016). Enhanced Neuronal Activity in the Medial Prefrontal Cortex during Social Approach Behavior. J Neurosci 36, 6926–6936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JJ, Wedow R, Okbay A, Kong E, Maghzian O, Zacher M, Nguyen-Viet TA, Bowers P, Sidorenko J, Karlsson Linner R, et al. (2018). Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat Genet 50, 1112–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Chen J, Yu H, He L, Xu Y, Zhang D, Yi Q, Li C, Li X, Shen J, et al. (2017). Genome-wide association analysis identifies 30 new susceptibility loci for schizophrenia. Nat Genet 49, 1576–1583. [DOI] [PubMed] [Google Scholar]
- Magnuson B, Ekim B, and Fingar DC (2012). Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem J 441, 1–21. [DOI] [PubMed] [Google Scholar]
- Martinez-Aran A, Vieta E, Colom F, Torrent C, Sanchez-Moreno J, Reinares M, Benabarre A, Goikolea JM, Brugue E, Daban C, and Salamero M (2004a). Cognitive impairment in euthymic bipolar patients: implications for clinical and functional outcome. Bipolar Disord 6, 224–232. [DOI] [PubMed] [Google Scholar]
- Martinez-Aran A, Vieta E, Reinares M, Colom F, Torrent C, Sanchez-Moreno J, Benabarre A, Goikolea JM, Comes M, and Salamero M (2004b). Cognitive function across manic or hypomanic, depressed, and euthymic states in bipolar disorder. Am J Psychiatry 161, 262–270. [DOI] [PubMed] [Google Scholar]
- Martinez-Aran A, Vieta E, Torrent C, Sanchez-Moreno J, Goikolea JM, Salamero M, Malhi GS, Gonzalez-Pinto A, Daban C, Alvarez-Grandi S, et al. (2007). Functional outcome in bipolar disorder: the role of clinical and cognitive factors. Bipolar Disord 9, 103–113. [DOI] [PubMed] [Google Scholar]
- Mathur A, Law MH, Megson IL, Shaw DJ, and Wei J (2010). Genetic association of the AKT1 gene with schizophrenia in a British population. Psychiatr Genet 20, 118–122. [DOI] [PubMed] [Google Scholar]
- Mazzarini L, Colom F, Pacchiarotti I, Nivoli AM, Murru A, Bonnin CM, Cruz N, Sanchez-Moreno J, Kotzalidis GD, Girardi P, et al. (2010). Psychotic versus non-psychotic bipolar II disorder. J Affect Disord 126, 55–60. [DOI] [PubMed] [Google Scholar]
- McGrath J, Saha S, Chant D, and Welham J (2008). Schizophrenia: a concise overview of incidence, prevalence, and mortality. Epidemiol Rev 30, 67–76. [DOI] [PubMed] [Google Scholar]
- McKenna BS, Sutherland AN, Legenkaya AP, and Eyler LT (2014). Abnormalities of brain response during encoding into verbal working memory among euthymic patients with bipolar disorder. Bipolar Disord 16, 289–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merikangas KR, Akiskal HS, Angst J, Greenberg PE, Hirschfeld RM, Petukhova M, and Kessler RC (2007). Lifetime and 12-month prevalence of bipolar spectrum disorder in the National Comorbidity Survey replication. Arch Gen Psychiatry 64, 543–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minatohara K, Akiyoshi M, and Okuno H (2015). Role of Immediate-Early Genes in Synaptic Plasticity and Neuronal Ensembles Underlying the Memory Trace. Front Mol Neurosci 8, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modrak-Wojcik A, Gorka M, Niedzwiecka K, Zdanowski K, Zuberek J, Niedzwiecka A, and Stolarski R (2013). Eukaryotic translation initiation is controlled by cooperativity effects within ternary complexes of 4E-BP1, eIF4E, and the mRNA 5’ cap. FEBS Lett 587, 3928–3934. [DOI] [PubMed] [Google Scholar]
- Moy SS, Nadler JJ, Perez A, Barbaro RP, Johns JM, Magnuson TR, Piven J, and Crawley JN (2004). Sociability and preference for social novelty in five inbred strains: an approach to assess autistic-like behavior in mice. Genes Brain Behav 3, 287–302. [DOI] [PubMed] [Google Scholar]
- Network, and Pathway Analysis Subgroup of Psychiatric Genomics, C. (2015). Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nat Neurosci 18, 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neve RL, Neve KA, Nestler EJ, and Carlezon WA Jr. (2005). Use of herpes virus amplicon vectors to study brain disorders. Biotechniques 39, 381–391. [DOI] [PubMed] [Google Scholar]
- Nikoletopoulou V, and Tavernarakis N (2018). Regulation and Roles of Autophagy at Synapses. Trends Cell Biol 28, 646–661. [DOI] [PubMed] [Google Scholar]
- Pardinas AF, Holmans P, Pocklington AJ, Escott-Price V, Ripke S, Carrera N, Legge SE, Bishop S, Cameron D, Hamshere ML, et al. (2018). Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat Genet 50, 381–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park N, Juo SH, Cheng R, Liu J, Loth JE, Lilliston B, Nee J, Grunn A, Kanyas K, Lerer B, et al. (2004). Linkage analysis of psychosis in bipolar pedigrees suggests novel putative loci for bipolar disorder and shared susceptibility with schizophrenia. Mol Psychiatry 9, 1091–1099. [DOI] [PubMed] [Google Scholar]
- Pearce LR, Komander D, and Alessi DR (2010). The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol 11, 9–22. [DOI] [PubMed] [Google Scholar]
- Pearson RB, Dennis PB, Han JW, Williamson NA, Kozma SC, Wettenhall RE, and Thomas G (1995). The principal target of rapamycin-induced p70s6k inactivation is a novel phosphorylation site within a conserved hydrophobic domain. EMBO J 14, 5279–5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penrod RD, Wells AM, Carlezon WA Jr., and Cowan CW (2015). Use of Adeno-Associated and Herpes Simplex Viral Vectors for In Vivo Neuronal Expression in Mice. Curr Protoc Neurosci 73, 4 37 31–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullen N, and Thomas G (1997). The modular phosphorylation and activation of p70s6k. FEBS Lett 410, 78–82. [DOI] [PubMed] [Google Scholar]
- Ripke S, O’Dushlaine C, Chambert K, Moran JL, Kahler AK, Akterin S, Bergen SE, Collins AL, Crowley JJ, Fromer M, et al. (2013). Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat Genet 45, 1150–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson LJ, Thompson JM, Gallagher P, Goswami U, Young AH, Ferrier IN, and Moore PB (2006). A meta-analysis of cognitive deficits in euthymic patients with bipolar disorder. J Affect Disord 93, 105–115. [DOI] [PubMed] [Google Scholar]
- Rosner M, Siegel N, Valli A, Fuchs C, and Hengstschlager M (2010). mTOR phosphorylated at S2448 binds to raptor and rictor. Amino Acids 38, 223–228. [DOI] [PubMed] [Google Scholar]
- Rowland TA, and Marwaha S (2018). Epidemiology and risk factors for bipolar disorder. Ther Adv Psychopharmacol 8, 251–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo SJ, Bolanos CA, Theobald DE, DeCarolis NA, Renthal W, Kumar A, Winstanley CA, Renthal NE, Wiley MD, Self DW, et al. (2007). IRS2-Akt pathway in midbrain dopamine neurons regulates behavioral and cellular responses to opiates. Nat Neurosci 10, 93–99. [DOI] [PubMed] [Google Scholar]
- Sanchez-Morla EM, Barabash A, Martinez-Vizcaino V, Tabares-Seisdedos R, Balanza-Martinez V, Cabranes-Diaz JA, Baca-Baldomero E, and Gomez JL (2009). Comparative study of neurocognitive function in euthymic bipolar patients and stabilized schizophrenic patients. Psychiatry Res 169, 220–228. [DOI] [PubMed] [Google Scholar]
- Saxton RA, and Sabatini DM (2017). mTOR Signaling in Growth, Metabolism, and Disease. Cell 169, 361–371. [DOI] [PubMed] [Google Scholar]
- Schizophrenia Working Group of the Psychiatric Genomics, C. (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab SG, Hoefgen B, Hanses C, Hassenbach MB, Albus M, Lerer B, Trixler M, Maier W, and Wildenauer DB (2005). Further evidence for association of variants in the AKT1 gene with schizophrenia in a sample of European sib-pair families. Biol Psychiatry 58, 446–450. [DOI] [PubMed] [Google Scholar]
- Selva G, Salazar J, Balanza-Martinez V, Martinez-Aran A, Rubio C, Daban C, Sanchez-Moreno J, Vieta E, and Tabares-Seisdedos R (2007). Bipolar I patients with and without a history of psychotic symptoms: do they differ in their cognitive functioning? J Psychiatr Res 41, 265–272. [DOI] [PubMed] [Google Scholar]
- Sham PC, MacLean CJ, and Kendler KS (1994). A typological model of schizophrenia based on age at onset, sex and familial morbidity. Acta Psychiatr Scand 89, 135–141. [DOI] [PubMed] [Google Scholar]
- Showkat M, Beigh MA, and Andrabi KI (2014). mTOR Signaling in Protein Translation Regulation: Implications in Cancer Genesis and Therapeutic Interventions. Mol Biol Int 2014, 686984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simeone JC, Ward AJ, Rotella P, Collins J, and Windisch R (2015). An evaluation of variation in published estimates of schizophrenia prevalence from 1990 horizontal line 2013: a systematic literature review. BMC Psychiatry 15, 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sole B, Jimenez E, Torrent C, Reinares M, Bonnin CDM, Torres I, Varo C, Grande I, Valls E, Salagre E, et al. (2017). Cognitive Impairment in Bipolar Disorder: Treatment and Prevention Strategies. Int J Neuropsychopharmacol 20, 670–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl EA, Breen G, Forstner AJ, McQuillin A, Ripke S, Trubetskoy V, Mattheisen M, Wang Y, Coleman JRI, Gaspar HA, et al. (2019). Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat Genet 51, 793–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G, Gudsnuk K, Kuo SH, Cotrina ML, Rosoklija G, Sosunov A, Sonders MS, Kanter E, Castagna C, Yamamoto A, et al. (2014). Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 83, 1131–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobe BTD, Crain AM, Winquist AM, Calabrese B, Makihara H, Zhao WN, Lalonde J, Nakamura H, Konopaske G, Sidor M, et al. (2017). Probing the lithium-response pathway in hiPSCs implicates the phosphoregulatory set-point for a cytoskeletal modulator in bipolar pathogenesis. Proc Natl Acad Sci U S A 114, E4462–E4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres IJ, Boudreau VG, and Yatham LN (2007). Neuropsychological functioning in euthymic bipolar disorder: a meta-analysis. Acta Psychiatr Scand Suppl 17–26. [DOI] [PubMed] [Google Scholar]
- Townsend J, Bookheimer SY, Foland-Ross LC, Sugar CA, and Altshuler LL (2010). fMRI abnormalities in dorsolateral prefrontal cortex during a working memory task in manic, euthymic and depressed bipolar subjects. Psychiatry Res 182, 22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsitsipa E, and Fountoulakis KN (2015). The neurocognitive functioning in bipolar disorder: a systematic review of data. Ann Gen Psychiatry 14, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uttam S, Wong C, Price TJ, and Khoutorsky A (2018). eIF4E-Dependent Translational Control: A Central Mechanism for Regulation of Pain Plasticity. Front Genet 9, 470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uylings HB, Groenewegen HJ, and Kolb B (2003). Do rats have a prefrontal cortex? Behav Brain Res 146, 3–17. [DOI] [PubMed] [Google Scholar]
- van Bergen AH, Verkooijen S, Vreeker A, Abramovic L, Hillegers MH, Spijker AT, Hoencamp E, Regeer EJ, Knapen SE, Riemersma-van der Lek RF, et al. (2019). The characteristics of psychotic features in bipolar disorder. Psychol Med 49, 2036–2048. [DOI] [PubMed] [Google Scholar]
- Wilson DI, Langston RF, Schlesiger MI, Wagner M, Watanabe S, and Ainge JA (2013). Lateral entorhinal cortex is critical for novel object-context recognition. Hippocampus 23, 352–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F, Na L, Li Y, and Chen L (2020). Roles of the PI3K/AKT/mTOR signalling pathways in neurodegenerative diseases and tumours. Cell Biosci 10, 54. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Yang ST, Shi Y, Wang Q, Peng JY, and Li BM (2014). Neuronal representation of working memory in the medial prefrontal cortex of rats. Mol Brain 7, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JH, Minzenberg MJ, Ursu S, Ryan Walter BS, Wendelken C, Ragland JD, and Carter CS (2008a). Association of dorsolateral prefrontal cortex dysfunction with disrupted coordinated brain activity in schizophrenia: relationship with impaired cognition, behavioral disorganization, and global function. Am J Psychiatry 165, 1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon T, Okada J, Jung MW, and Kim JJ (2008b). Prefrontal cortex and hippocampus subserve different components of working memory in rats. Learn Mem 15, 97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Anjum R, Kubota K, Rush J, Villen J, and Gygi SP (2009). A site-specific, multiplexed kinase activity assay using stable-isotope dilution and high-resolution mass spectrometry. Proc Natl Acad Sci U S A 106, 11606–11611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachari M, and Ganley IG (2017). The mammalian ULK1 complex and autophagy initiation. Essays Biochem 61, 585–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate code, but datasets related to the current study are available from the Lead Contact, Michael Cahill, upon request.