

Abstract
In vitro‒in vivo correlation (IVIVC) of solid dosage forms should be established basically between in vitro and in vivo dissolution of active pharmaceutical ingredients. Nevertheless, in vivo dissolution profiles have never been accurately portrayed. The current practice of IVIVC has to resort to in vivo absorption fractions (Fa). In this proof-of-concept study, in vivo dissolution of a model poorly water-soluble drug fenofibrate (FNB) was investigated by fluorescence bioimaging. FNB crystals were first labeled by near-infrared fluorophores with aggregation-caused quenching properties. The dyes illuminated FNB crystals but quenched immediately and absolutely once been released into aqueous media, enabling accurate monitoring of residual drug crystals. The linearity established between fluorescence and crystal concentration justified reliable quantification of FNB crystals. In vitro dissolution was first measured following pharmacopoeia monograph protocols with well-documented IVIVC. The synchronicity between fluorescence and in vitro dissolution of FNB supported using fluorescence as a measure for determination of dissolution. In vitro dissolution correlated well with in vivo dissolution, acquired by either live or ex vivo imaging. The newly established IVIVC was further validated by correlating both in vitro and in vivo dissolution with Fa obtained from pharmacokinetic data.
KEY WORDS: In vivo dissolution, Fenofibrate, Fluorescence, Aggregation-caused quenching, Bioimaging, IVIVC
Graphical abstract
Authentic in vitro‒in vivo correlation should be established between in vitro dissolution and in vivo dissolution which however has never been determined accurately. This study provides proof of concept of in vivo dissolution based on live imaging of fluorescently hybridized crystals of a model poorly water-soluble drug fenofibrate.
1. Introduction
Active pharmaceutical ingredients (APIs) intended for oral administration are preferably formulated as solid dosage forms, which are principally comprised of drug particles−in either crystalline or amorphous form−and excipients, such as tablets and capsules, owing to stability considerations and ease of manufacturing, transportation, storage and administration. Solid dosage forms, except those meant for sustained or controlled release, are generally designed to disintegrate rapidly in the gastrointestinal tract (GIT) to liberate drug particles, which further dissolve to present the drugs in a solubilized state1. Drug molecules are thereafter absorbed across the enteric epithelia via different mechanisms—passive diffusion, facilitated diffusion or energy-consuming active transport2. Drug particles per se—generally in the size range of a few to a few tens of microns—are unable to be absorbed via the enteric epithelia. Despite recent reports of oral absorption of minute amount of drug particles with a size of several hundred nanometers3, 4, 5, 6, a majority of drugs are absorbed in a molecularly dissolved state. The dissolution of drug particles following disintegration of solid dosage forms is one of the rate-limiting steps that determine the overall absorption rate and content7, 8, 9.
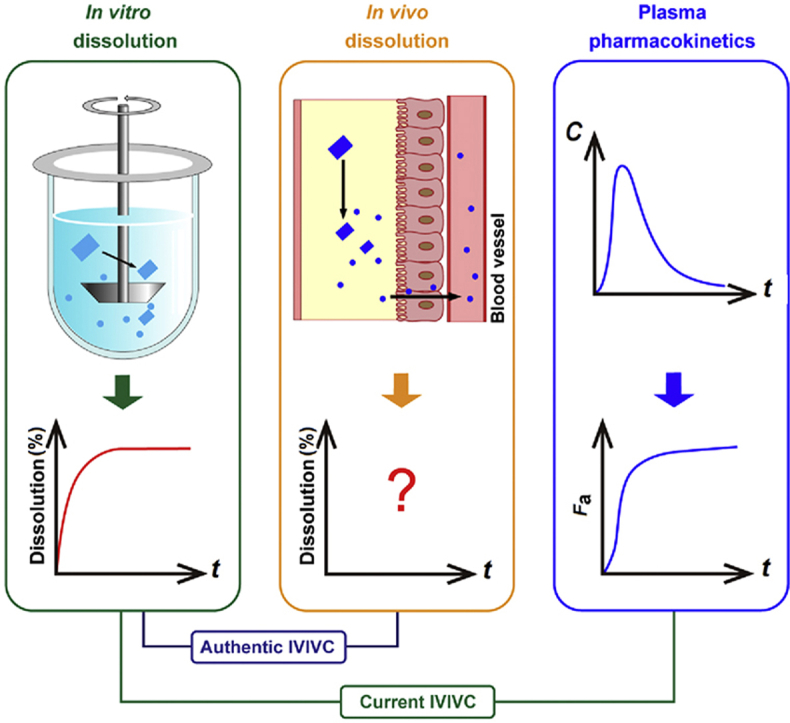
The exploration of in vivo dissolution digs information crucial for the understanding of drug absorption. It can be employed to guide not only the establishment of in vitro dissolution protocols but also the development of formulations. Unfortunately, the virtual in vivo dissolution profile of any drugs has never been accurately determined due to a lack of functional tools to monitor changes in total amount of dissolved drugs or residual drug particles continuously in the GIT. A great deal of time and money has been wasted on tedious formulation optimization without the guidance of in vivo dissolution10. The current in vitro dissolution protocols, which should stem from in vivo dissolution, have been established merely on the basis of in vitro observations and experience. Monograph in vitro dissolution test so far serves well as a measure to inspect formulation conformity but perform awkwardly to predict in vivo performance. In order to elevate the eligibility of in vitro dissolution test, bio-relevant media have been widely utilized to mimic in vivo conditions11, 12, 13, 14. However, in vitro‒in vivo correlation (IVIVC) should be established to justify in vitro dissolution test15, 16, 17. Paradoxically, the establishment of IVIVC calls for in vivo dissolution data18, 19, 20. Since in vivo dissolution cannot be measured accurately at the moment, previous establishment of IVIVC has to resort to in vivo absorption fractions (Fa)20, 21, 22, presuming that certain correlation between Fa and in vivo dissolution can be built. Fig. 1 is a schematic demonstration of the correlation among in vitro dissolution, in vivo dissolution and Fa. The calculation of Fa is based on deconvolution of plasma pharmacokinetic data by the Wagner–Nelson or Riegelman‒Loo method17,23, 24, 25. The deconvolution approach assumes linear absorption into and elimination from the circulation, which however is often biased, especially for poorly permeable drugs such as Class III and Class IV compounds under Biopharmaceutics Classification System (BCS), compromising the establishment of IVIVC26. So far, the overall acceptance rate of all IVIVC studies is very low. For instance, only 40% for all submissions to U.S. Food and Drug Administration (FDA)17,27, 28, 29. The European Innovative Medicines Initiative and FDA launched major research projects—the OrBiTo (oral biopharmaceutical tools) and the 21st Century BA/BE, respectively—to establish in vitro/in silico tools to predict in vivo drug behaviors so as to elevate the success rate of IVIVC10,13,30,31. The complex and variable gastrointestinal conditions compromise the accuracy of these tools32, 33, 34.
Figure 1.

Schematic demonstration of correlation among in vitro dissolution, in vivo dissolution and absorption fraction of drug crystals (A), and the rationale of the ACQ-based fluorescent bioimaging of fenofibrate crystals (B). Current IVIVC is established between in vitro dissolution and Fa as a makeshift. Nevertheless, authentic IVIVC should be established between in vitro and in vivo dissolution.
Basically, IVIVC should be established between in vitro and in vivo dissolution. It is because in vivo dissolution data are not available that Fa has been employed as a makeshift substitute to establish IVIVC. Difficult as it is, measurement of in vivo dissolution has been tried before. An intraluminal sampling technique utilizing a long multichannel tube (Loc-I-Gut) was developed to detect drug levels directly in the GIT35, 36, 37. Although the technique enables simultaneous aspiration of both gastric and intestinal fluids, its drawbacks are equally apparent. Unlike in in vitro dissolution media, drugs are non-uniformly distributed in the GIT. It is impractical to get a panoramic view of drug dissolution by sampling at a fixed position. Dissolution of undissolved drug particles continues during separation of the aspirated fluids as well, while the processing—by for example centrifugation and ultrafiltration—may accelerate dissolution38. In addition, the invasive sampling catheter may cause duodenogastric reflux39. All of the disadvantages compromise the accuracy of the sampling strategy. As a result, only changes in drug concentrations are recorded and it is impossible to draw an in vivo dissolution profile based on the intraluminal sampling technique. Instead of measuring dissolved drugs, undissolved drug particles can be measured following intraluminal sampling in human GIT, too40. However, the same drawbacks as measuring dissolved drugs exist.
In this proof-of-concept study, a novel strategy is implemented to measure in vivo drug dissolution by real-time monitoring of residual particles in the GIT based on fluorescence live imaging in rats. In the previous studies, we made the first step to identify crystalline or amorphous drug nanoparticles in vivo after labeling with near-infrared fluorophores using the hybridized drug crystallization technique5,6. The dyes bear a BODIPY or an aza-BODIPY parent structure and emit fluorescence when molecularly dispersed (“on”)41,42. Unlike conventional fluorophores43, 44, 45, 46, 47, the fluorescence of this kind of dyes quenches immediately and completely (“off”) upon nanocrystal dissolution and dye release into aqueous media, as dye molecules aggregate via π‒π stacking, a phenomenon commonly known as aggregation-caused quenching (ACQ) in fluorescence bioimaging48, 49, 50. Therefore, the fluorescence signals can be used to represent undissolved nanocrystals. By exploiting the “on-to-off” signal switching feature of ACQ dyes, the in vivo fate of a series of nanocarriers has been explored via different administration routes51, 52, 53.
Herein, fenofibrate (FNB), a Class II compound under BCS, was utilized as a model drug to testify the concept of in vivo dissolution, because of well-established IVIVC22,54, 55, 56. An ACQ dye with an aza-BODIPY parent structure, P2, was embedded into the lattices of FNB crystals to prepare fluorescently hybridized FNB crystals (FNB–HCs). In vivo dissolution was investigated by real-time live imaging and correlated to in vitro dissolution. To validate the in vivo dissolution model, in vivo Fa calculated from pharmacokinetic data was employed to perform correlation with both in vitro and in vivo dissolution.
2. Materials and methods
2.1. Materials
FNB was purchased from Xuzhou Enhua pharmaceutical Co., Ltd. (Xuzhou, China). HPMC-E5 was provided by Shanghai Colorcon Coating Technology Co., Ltd. (Shanghai, China). ACQ dye P2 (λabs/λem = 708 nm/732 nm) was synthesized in our laboratory according to previous procedures57,58. Powders for reconstitution of biorelevant gastrointestinal fluids were obtained from Biorelevant. Com Ltd. (Croydon, UK). Isoflurane was obtained from Shandong Keyuan Pharmaceutical Co., Ltd. (Jinan, China). Deionized water was prepared by a Milli-Q® (Millipore, Molsheim, France) water purification system, filtered through 0.2 μm membrane and used in all experiments.
Male Sprague–Dawley (SD) rats, weighing 200 ± 20 g, were obtained from Shanghai Laboratory Animal Center (Shanghai, China). All experimental procedures involving use of animals were approved by the Institutional Animal Care and Use Committee at School of Pharmacy, Fudan University, China.
2.2. Preparation of FNB-HCs
FNB-HCs were prepared using an anti-solvent crystallization method with modifications59. The effects of stirring speed, solvent volume and drug concentration on particle size and span of size distribution were screened (Supporting Information Fig. S1). Briefly, both P2 (80 μg) and FNB (60 mg) were first dissolved in 1 mL ethanol. The ethanol solution was then poured rapidly into 50 mL HPMC-E5 aqueous solution (0.04%, w/v) under stirring and stirred continuously at a speed of 700 rpm for 5 min (IKA® RW20 Digital, Staufen, Germany). The suspensions obtained were filtered through a 220 nm polycarbonate membrane (Sigma–Aldrich, St. Louis, MO, USA), while the precipitates were rinsed with 50 mL water to remove residual HPMC-E5. FNB-HCs were collected after drying the precipitates at room temperature under vacuum.
2.3. Characterization of FNB-HCs
For measurement of particle size, span and fluorescence intensity, FNB-HCs were suspended in water under ultrasonication (Ningbo Xinzhi biotechnology Co., Ltd., China) to an FNB concentration of 12 mg/mL. The particle size and span were determined by a Malvern Mastersizer 3000 laser particle analyzer (Malvern instrument, Malvern, UK) at room temperature. The operating conditions were set as follows: refractive index 1.52; absorption rate 0.1; density 1 g/cm3; shading limit 0.5%–5%. The FNB-HC suspensions (200 μL) were instilled into 96-well plates for measurement of fluorescence intensity using IVIS Spectrum Live Imaging System (PerkinElmer, Waltham, MA, USA) by a region of interest (ROI) quantification method49,50.
For measurement of the encapsulation efficiency (EE, %) and drug loading (DL, %) of P2, 10 mg FNB-HCs were dissolved in 3 mL ethanol, and the fluorescence intensity of P2 was measured by a Cary Eclipse fluorescence spectrophotometer (Agilent, Santa Clara, CA, USA). The loading amount of P2 was measured by a pre-established calibration curve based on the fluorescence intensity of P2 measured in ethanol. The EE (%) and DL (%) were calculated by the following Eqs. (1), (2):
| (1) |
| (2) |
The morphology of FNB-HCs, as well as FNB raw materials, was observed by an SU8000 scanning electron microscopy (SEM, Hitachi, Tokyo, Japan). The solid samples were sputter coated with a conductive layer of gold palladium and observed at an accelerating excitation voltage of 10 kV. The differential scanning calorimetry (DSC) thermograms were collected using a 204A/G thermal analyser (Netzsch, Bühl, Germany) at a heating speed of 5 °C/min from 50 to 165 °C under a nitrogen gas flow of 30 mL/min. The powder X-ray diffractometry (PXRD) patterns of the same samples were recorded by a D/MAX-2500/PC X-ray diffractometer (Rigaku, Tokyo, Japan) with a Cu source of radiation over the 2θ range from 7° to 45°, with a step angle of 0.02° and counting frequency of 2°/min.
2.4. Fluorescence stability of FNB-HCs
The fluorescence stability of FNB-HCs in water, buffers of different pHs and simulated physiological media was investigated to assess dye leakage or premature water infiltration into the crystals lattices, all of which may result in interference to live imaging5,6,42. During the experiment, FNB-HCs were suspended in 10 mL of the medium to a concentration of 2.4 mg/mL and incubated at 37 °C. Samples were withdrawn at 0, 0.5, 1.0, 1.5, 2.0, 3.0, 4.0, 6.0, 8.0 and 12.0 h, respectively, and measured for fluorescence intensity by a fluorescence spectrophotometer.
2.5. Fluorescence quenching sensitivity to water
In the previous study48, the ACQ effect of the dye P2 was investigated by observing the fluorescence intensity as a function of water content in a binary acetonitrile/water system. In this study, the same experiment was performed on FNB-HCs, but using an ethanol/water system instead to make sure that FNB crystals could dissolve quickly enough. A series of aqueous ethanol with varied water contents in the range of 1%–99% (v/v) were prepared. Then, 10 μL FNB-HC suspension (12 mg/mL) were dissolved in 40 mL aqueous ethanol. The fluorescence spectra were scanned by a fluorescence spectrophotometer at an excitation wavelength of 690 nm and emission wavelength range of 710–800 nm. The spectral slits were set to 10 nm.
2.6. Quantification of FNB-HCs in dissolution media
A calibration curve was established between fluorescence intensity and concentration of FNB-HCs. The fluorescence intensity of FNB-HC suspensions with a series of concentrations were measured using a fluorescence spectrophotometer. In parallel, 1 mL of the suspensions was dissolved in methanol and diluted to 50 mL to determine the concentration of FNB by Agilent 1260 series HPLC system (Agilent). The correlation between fluorescence intensity and concentration of FNB-HCs was established. The precision and accuracy of the assay were evaluated at high, medium and low levels of FNB-HCs, respectively (Supporting Information Fig. S2, Tables S1 and S2).
2.7. In vitro dissolution
In vitro dissolution of FNB-HCs was studied using a ZRS-8G dissolution tester (Tianjin, China) following ChP II (the paddle method) monograph procedures for evaluation of FNB formulations, which have been validated with good IVIVC (in vitro dissolution vs. Fa)60. Sodium dodecyl sulfate aqueous solution (0.05 mol/L, 1000 mL) was utilized as the dissolution media to set a sink condition. The rotation speed was set to 50 rpm, while samples equivalent to 12 mg of FNB (1 mL suspension) were directly instilled into the beaker. At predetermined intervals, 2 mL samples were withdrawn and equal volume of blank media was compensated immediately. The fluorescence intensity of the samples was measured immediately using a fluorescence spectrophotometer. The percentage of residual fluorescence intensity was obtained by setting the initial fluorescence intensity as 100%. The fluorescence intensity of each sample was also substituted into the calibration curve to calculate the residual percentage of FNB-HCs (corrected). In the meantime, the dissolution samples were filtered through 0.22 μm PES membrane (Fine Scientific Ltd., Toronto, Canada) and assayed for FNB by HPLC. The accumulated percentage of dissolved FNB was subtracted from 100% to get the percentage of residual FNB-HCs. For ease of comparison, in vitro dissolution profiles were displayed as the percentages of residual FNB-HCs vs. time plots.
2.8. In vivo dissolution
Before the experiment, depilatory cream was applied to remove the hair on the abdominal side to reduce hair-derived auto-fluorescence. Since FNB exhibits high inter-individual variation under fasted state, the formulation is administered after a meal due to the pronounced food effects61. The animals were placed in individual cages and allowed free access to food and water before experiment. The animals were randomly divided into two groups, and treated with FNB-HCs and quenched P2 dispersion as a control, respectively. Suspension equivalent to 12 mg FNB (1 mL) was administered to the rats by gavage. Epi-Illumination mode was used for IVIS scanning with excitation/emission wavelengths set to 710 nm/760 nm to capture P2 signals post administration. An on-line gas anesthetizing system using isoflurane was used to anaesthetize the rats during the imaging process. Fluorescence intensity measured instantly after administration was set as 100%, while the percentages of residual fluorescence intensity vs. time was plotted to depict the in vivo dissolution profiles.
Owing to the poor penetration of fluorescence, the whole GIT was dissected and observed ex vivo using the IVIS system. In brief, the animals were raised and administered with the test samples following the same procedures as for live imaging. At each time point, three animals were sacrificed by cervical dislocation and the gastrointestinal tissues were collected and imaged. The percentage of residual fluorescence intensity in ex vivo tissues was recorded. The dissolution profiles obtained ex vivo were compared with the in vivo profiles.
2.9. Validation by Fa-based IVIVC
To study the pharmacokinetics of FNB-HCs, 1 mL suspension equivalent to 12 mg FNB was administered to the rats by gavage. Blood samples (500 μL) were withdrawn into heparinized test tubes from the eye socket vein pre-dosing (0 h) and at time intervals of 0.083, 0.167, 0.333, 0.5, 0.75, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 16 and 24 h post-dosing. Plasma was collected after centrifugation (Anke TGL-16G, Shanghai, China) of the blood samples at 3000 rpm for 5 min and stored at −20 °C for subsequent analysis. Since FNB is rapidly metabolized into fenofibric acid in blood, the pharmacokinetics of FNB was evaluated based on monitoring of fenofibric acid62. Fenofibric acid in rat plasma was extracted by liquid–liquid extraction following procedures established in a previous study63. The method development and validation details are shown in the Supporting Information. Pharmacokinetic parameters were calculated by DAS 2.0 (Shanghai Bioguider Medicinal Technology Co., Ltd., Shanghai, China). In addition to pharmacokinetic parameters such as Cmax, tmax and AUC, Fa was calculated by deconvolution using the Wagner–Nelson method for a one-compartmental model in Eq. (3):
| (3) |
where Ct is the drug plasma concentration at time t, AUC is the area under the curve, and k is the elimination rate constant.
A point-to-point correlation between Fa and in vivo dissolution or in vitro dissolution, as obtained in Sections 2.6 and 2.7, was investigated. For ease of comparison, all data were presented as percent of residual FNB-HCs.
3. Results and discussion
3.1. Characterization of FNB-HCs
The physicochemical properties of FNB-HCs are summarized in Table 1. The D90 of FNB-HCs is 7.33 ± 0.64 μm, which is similar to the commercial FNB preparation Lipanthyl® (<10 μm)64. The EE (%) and DL (%) of P2 in FNB-HCs is 25.98 ± 0.32% and 0.02 ± 0.01%, respectively. The fluorescence intensity of FNB-HCs is strong enough for in vivo imaging owing to the high quantum yield of P257,58. Owing to the extremely low loading of the dye, the morphology and crystallographic properties of FNB do not change significantly (Fig. 2). FNB-HCs present as bulk crystals with a size much smaller than that of FNB raw crystals (Fig. 2A and B). DSC thermograms show an endothermic peak at 84 °C for both FNB and FNB-HCs (Fig. 2D), representing the melting point of FNB65,66. The PXRD diffractograms show characteristic diffraction peaks at 2°, 11°, 20°, 21°, 25°, 30° and 34° for FNB-HCs, which is consistent with FNB raw crystals (Fig. 2E). It is concluded that FNB-HCs retain physical properties similar to FNB raw crystals except particle size.
Table 1.
Physicochemical properties of FNB-HCs and FNB raw crystals (mean ± SD, n = 3).
| Size (μm) |
Span | EE (%) | DL (%) | TRE [(p/s)/(μW/cm2) × 10−9] | |||
|---|---|---|---|---|---|---|---|
| D10 | D50 | D90 | |||||
| FNB-HC | 3.2 ± 0.2 | 4.8 ± 0.4 | 7.3 ± 0.6 | 0.8 ± 0.03 | 26.98 ± 0.32 | 0.02 ± 0.01 | 4.58 ± 0.17 |
| FNB | 80.3 ± 33.5 | 131.5 ± 28.5 | 215.3 ± 30.6 | 1.1 ± 0.3 | |||
D10, D50 and D90 refer to the diameter below which there are 10%, 50% and 90% of particles.
Figure 2.

SEM photographs of FNB raw material (A) and FNB-HCs (B); size distribution of FNB raw crystals and FNB-HCs (C); DSC thermograms (D); powder X-ray diffractograms (E).
3.2. Fluorescence stability and sensitivity to water
To precisely and sensitively monitor the dissolution of FNB-HCs, the dyes should be securely embedded inside the crystal lattices, while the fluorescence should quench instantly and completely upon dissolution of the crystals. The fluorescence stability of FNB-HCs and the sensitivity to water were thus validated.
The very good fluorescence stability of FNB-HCs in pure water and all buffers of different pHs (Fig. 3A) indicates a lack of dye leakage or water infiltration into the crystal lattices, both of which could lead to false signals owing to premature quenching41,67. With regard to fasted-state simulated small intestine fluid (FaSSIF) and fed-state simulated small intestine fluid (FeSSIF), the fluorescence decreases to 90% and 75% of the original after 12 h (Fig. 3B), respectively. Nevertheless, it does not mean dye leakage or water infiltration. It is mainly due to the presence of surfactants—generally phospholipids and bile salts—in both FaSSIF and FeSSIF that solubilize FNB-HCs and thereby lead to gradual decrease of fluorescence. As FeSSIF is fortified with more surfactants than FaSSIF, more FNB-HCs dissolve in FeSSIF than in FaSSIF, resulting in lower levels of residual fluorescence in FeSSIF (Fig. 3B). The fact of much stable fluorescence in simulated gastric fluid (SGF) serves as a counter-evidence to support the observations in FeSSIF because SGF does not contain any surfactants. Simultaneous monitoring of FNB gives synchronous dissolution behaviors (Fig. 3C and D). The fluorescent quenching should be ascribed to the dissolution of FNB-HCs, rather than dye leakage or water infiltration. Therefore, fluorescence quenching is justified as a measure to determine dissolution of FNB-HCs.
Figure 3.

In vitro fluorescent stability of FNB-HCs in buffers of different pHs (ABS; acetate buffered saline; PBS: phosphate buffered saline) and pure water (A) and in different bio-relevant fluids (B). SGF, simulated gastric fluid; FeSSIF, fed-state simulated small intestinal fluid; FaSSIF, fasted-state simulated small intestinal fluids. Validation of dissolution by measuring the drug: in buffers and water (C); in different bio-relevant fluids (D). Data are expressed as mean ± SD (n = 3). Fluorescent spectra of FNB-HCs in aqueous ethanol (E) and the plot of fluorescent intensity vs. water content (F).
The fluorescence spectra of FNB-HCs dissolved in aqueous ethanol is shown in Fig. 3E. The fluorescence decreases as a function of water content and quenches completely at a turning point of 70% (Fig. 3F). It stands to reason that the fluorescence of FNB-HCs is able to quench absolutely upon dissolution in the aqueous biological environment.
3.3. In vitro dissolution
In vitro dissolution was first determined by measuring FNB in the dissolution medium following conventional monograph protocol. To facilitate comparison with fluorescence quenching, the dissolution profile is expressed as undissolved residual drug vs. time by deducting the percentages of dissolved FNB from 100% (Fig. 4A, red line), rather than a routine display of accumulative dissolved percentages. The residual amount of FNB-HCs was quantified based on residual fluorescence according to a calibration curve (Supporting Information Fig. S2). Linearity was observed between fluorescence intensity and undissolved FNB-HCs within the concentration range of 2.10–135 μg/mL (r = 0.9994). The accuracy was between 95% and 105% for samples of low, medium and high concentrations, and the precision was less than 6% for both within-day and between-day assays (Supporting Information Tables S1 and S2). The fluorescence-based dissolution profile is shown in Fig. 4A (blue line). Good correlation (r = 0.9824) is observed between the two in vitro dissolution profiles measured by monitoring either dissolved FNB or fluorescence (Fig. 4B).
Figure 4.

In vitro dissolution of FNB-HCs determined by monitoring either FNB (red line) or fluorescence (A, blue line) and correlation (B) that highlight synchronicity between drug dissolution and fluorescence quenching. Data are expressed as mean ± SD (n = 3).
The quantitative correlation is based on stable embedment of ACQ dyes in the FNB crystalline lattices and sensitive water-quenching capability. Along with dissolution of FNB-HCs, the ACQ dyes are released into an aqueous environment and quench absolutely due to self-aggregation, while the embedded ACQ dyes in residual FNB-HCs still remain emissive. As revealed in the stability study, no dye leakage and water infiltration that could result in false signals are recorded. The fluorescence signals can be used to identify and measure FNB-HCs.
The finding here, together with that observed in the stability study, proves the synchronicity between FNB dissolution and fluorescence quenching, which establishes the basis for further investigation of dissolution by real-time monitoring of residual particle-associated fluorescence.
3.4. In vivo dissolution
Fig. 5A shows the live images of SD rats after oral administration of FNB-HCs by gavage. Almost no fluorescence signals discriminable from the background were found in rats treated with pre-quenched P2 dispersion, indicating little fluorescence reillumination (Supporting Information Fig. S3). For the rats treated with FNB-HCs, the signals mainly locate to the abdominal region. The fluorescence shows a declining trend and disappears at 60 min post administration, indicating gradual dissolving of FNB-HCs. In view that the fluorescence represents residual FNB-HCs, the in vivo dissolution profile is thus delineated from the residual percentage of fluorescence with the initial value set to 100% (Fig. 5B, blue line) and compared with the in vitro dissolution profile (Fig. 5B, red line). A not-too-bad correlation is established with a correlation coefficient of 0.90 (Fig. 5C).
Figure 5.

In vivo dissolution based on live imaging of residual FNB-HCs. Live images of SD rats after oral administration of FNB-HCs gavage (A). Comparison of in vivo dissolution profile obtained from residual percentage of fluorescent intensity (blue line) with the in vitro dissolution profile (B, red line) and IVIVC established between them (C). Data are expressed as mean ± SD (n = 3).
Considering the limited tissue penetration of fluorescence, the whole GIT were dissected for IVIS imaging after sacrificing the animals at each time point (Fig. 6A). The pre-quenched P2 control exhibits little fluorescence as well, reconfirming negligible interference due to reillumination (Supporting Information Fig. S4). The fluorescence of residual FNB-HCs resides mainly in the stomach during the period of observation and attenuates with time. At 60 min, the fluorescence almost disappears due to fast dissolution of micronized FNB crystals68. The ex vivo results comply with that obtained by live imaging (Fig. 5A). Similarly, the residual percentage of fluorescence intensity in the stomach vs. time is employed to plot the in vivo dissolution profiles (Fig. 6B, blue line). Very good correlation is observed between in vivo and in vitro dissolution profile (Fig. 6B, red line) with a correlation coefficient of 0.95 (Fig. 6C). Although ex vivo imaging confirms the results of live imaging, the latter is however preferred for ease of handling, monitoring in real time and not needing to sacrifice a large number of animals.
Figure 6.

In vivo dissolution based on ex vivo imaging of residual FNB-HCs. Ex vivo images of the whole isolated GI segments after oral administration of FNB-HCs by gavage (A). Comparison of the in vivo dissolution profile obtained from residual percentage of fluorescent intensity in the stomach (blue line) and the in vitro dissolution (B, red line) and IVIVC established between them (C). Data are expressed as mean ± SD (n = 3).
3.5. Validation by IVIVC with Fa
Since FNB is rapidly metabolized into fenofibric acid and unmeasurable in plasma, pharmacokinetics of FNB were evaluated based on plasma concentration of fenofibric acid62. The plasma fenofibric acid concentration vs. time profile after oral administration of FNB-HCs is shown in Fig. 7A. The plasma fenofibric acid concentration increases rapidly and reaches a maximum at around 1 h post administration due to the quick dissolution of FNB in the stomach (Figure 5, Figure 6). The main pharmacokinetic parameters of oral FNB-HCs are shown in Table 2. Results indicate that the plasma levels of fenofibric acid increase quickly after administration by gavage, with a Cmax of 13.22 μg/mL at 1.17 h. The value of Tmax is comparable to that of the commercial product Lipanthyl®69,70.
Figure 7.

Mean plasma concentration of fenofibric acid vs. time plot in rats post administration of FNB-HCs by gavage (A); normalized values of Favs. time (B); correlation between Fa and in vitro dissolution (C); correlation between Fa and in vivo dissolution (D). Data are expressed as mean ± SD (n = 5).
Table 2.
Main pharmacokinetic parameters of fenofibric acid post oral administration of FNB-HCs by gavage.
| PK parameter | Valuea |
|---|---|
| Cmax (μg/mL) | 13.22 ± 1.30 |
| Tmax (h) | 1.17 ± 0.29 |
| AUC0‒∞ (μg⋅h/mL) | 184.72 ± 54.11 |
| K (1/h) | 0.07 ± 0.01 |
Data are mean ± SD, n = 5.
The in vivo Fa of FNB-HCs was calculated via Eq. (3) and plotted vs. time (Fig. 7B). One compartmental model was determined based on the criterion of minimum AIC given by the pharmacokinetic software (DAS 2.0, Shanghai, China). Therefore, deconvolution by the Wagner–Nelson method for one-compartment model is applicable for the calculation of Fa. Under fed state, Fa of FNB-HCs reaches a maximum of 114% in approximately 60 min, and the Fa profile correlates very well with the in vitro dissolution profile with a correlation coefficient of 0.94 (Fig. 7C), confirming a level-A IVIVC according to FDA guidelines71,72. Being a BCS Class II drug with good permeability, Fa reflects the in vivo dissolution of FNB. Therefore, the in vivo dissolution profile also correlates very well with the Fa profile with a correlation coefficient of 0.97 (Fig. 7D). The good correlation among in vitro dissolution, in vivo dissolution and in vivo Fa strengthens the applicability of in vivo dissolution for establishment of IVIVC.
4. General discussion
In vivo dissolution is not a new concept. Both administrative agencies and academics have called on investigations into it, aiming to realize authentic IVIVC18, 19, 20. However, previous endeavors failed due to a lack of workable tools to monitor either dissolved drugs or residual drug particles as a whole in real time, based on which the in vivo dissolution percentages could be calculated. This article is probably the first report on in vivo dissolution profiles based on measurement of residual drug particles, as well as the realization of IVIVC between in vitro and in vivo dissolution. The high correlation coefficient (r = 0.8996) observed in this study between in vitro and in vivo dissolution stands for a Level A point-to-point IVIVC according to FDA guidelines18,19. The newly established IVIVC is further validated by the conventional IVIVC protocol—correlation with Fa—calculated from the pharmacokinetic profiles. Level A IVIVC has been established for FNB formulations between in vitro dissolution and Fa by previous studies22,54, 55, 56 and the current study confirms previous findings. The fact that good IVIVC exists between in vivo dissolution and Fa strengthens the newly established IVIVC between in vitro and in vivo dissolution.
It is not hard to understand that ready absorption of the APIs in the GIT is a prerequisite for a conventional Fa-based IVIVC study. Apparently, it is impractical to expect high-level IVIVC from Class III and IV compounds because of limited and irregular absorption owing to poor permeability. Although Fa of poorly permeable drugs can be measured as well, the value is too limited to be utilized to establish IVIVC. Therefore, the overall success rate for Class III and IV drugs is very low. The conclusions of this study suggest that future IVIVC may be established between in vitro and in vivo dissolution profiles instead of in vitro dissolution and Fa. The newly proposed protocol may help evade the absorption uncertainties associated with poorly permeable drugs. IVIVC should reflect both pharmaceutic and biopharmaceutic behaviors of drugs in the GIT, but not only its absorption. It is envisioned that the acceptance rate of IVIVC for Class III and IV drugs would be highly increased if in vivo dissolution is employed to investigate IVIVC by administrative agencies.
FNB is chosen as the model drug because it is a highly permeable drug (Class II) with well-established IVIVC22,54, 55, 56, a feather that can be utilized to validate the in vitro/in vivo dissolution correlation hypothesis. On the other hand, the poor water-solubility of FNB grants temporal flexibility for the measurement of in vivo dissolution based on fluorescence. We also tested propranolol, a Class I drug, that is highly water-soluble and permeable with well-established IVIVC (see Supporting Information). Unfortunately, the drug dissolves so quickly in the GIT that we failed to capture sufficient data because data collection demands more time than actually allowed. In this study, we just showcase the proof of concept of in vivo dissolution by using FNB. It is highly expectable to expand the concept to more model drugs ranging from Class I to Class IV.
In this study, the proof of concept of in vivo dissolution as well as the IVIVC based on it has been validated, but there is still a long way ahead to put the concepts into practical use. There are a few obstacles yet to be overcome. It should be noted that all experiments have been carried out in rats. There are so far no similar or alternative methods available for human studies. On the other hand, in live imaging, the light must penetrate the body tissues first, which may absorb a majority of the energy and possess auto-fluorescence that interferes with live imaging. To reduce such interference, NIR region II dyes (1000–1700 nm) with higher penetration and less tissue auto-fluorescence might be a better choice to replace NIR region I dyes in future studies73,74.
The investigation of in vivo dissolution is meant to provide guidance for the screening of in vitro dissolution protocols based on establishment of IVIVC. In this study, we did not perform screening of in vitro dissolution conditions but employed pharmacopeia method with well-established and documented IVIVC. On the contrary, the current IVIVC is used to test in vivo dissolution as a measure to establish authentic IVIVC. Although the pharmacopeia method has been developed according to human data, it is well replicated in the current rat model. It is expected that in the future in vivo dissolution profiles can be used as a standard to screen in vitro dissolution conditions. It is also very interesting to notice that the efficient of in vivo dissolution vs. Fa (r = 0.9691) correlation is higher than that of in vitro dissolution vs. Fa (r = 0.9375), which implies that the current in vitro dissolution protocols for FNB might not be an ideal one and there are still margins for improvement.
The currently running IVIVC protocols acceptable in regulatory agencies is solely based on a makeshift correlation between in vitro dissolution and Fa. As Fa is subject to various factors that influence the absorption and systemic exposure of APIs, IVIVC of conventional practice can be established only when proportional correlation exists between virtual in vivo dissolution and Fa. Nevertheless, irregular or erroneous absorption, which is common for BCS III and IV drugs, hinders the successful establishment of IVIVC. The current low acceptance rate bespeaks the bottleneck of current IVIVC practices. If in vivo dissolution is able to be measured with accuracy, regulatory agencies would like to accept the new in vitro‒in vivo dissolution IVIVC protocol because it is direct and authentic correlation between dissolution profiles. Owing to limitations with the bioimaging strategy used in this study, we only provide the proof of concept of in vivo dissolution-based IVIVC. The procedures employed in this study are not meant to be used universally for other models but calls for more investigation on the concepts and protocols involved. The application of in vivo dissolution concept in IVIVC is expectable if accurate measurement of in vivo dissolution in humans comes into truth in the future.
5. Conclusions
The concept of in vivo dissolution is proved with FNB as a model for poorly water-soluble drug based on real-time monitoring of residual or undissolved drug crystals in GIT. In order to identify and measure FNB crystals, an NIR fluorophore, P2, suitable for non-invasive live imaging is utilized to label FNB crystals through recrystallization. The complete and instant quenching of fluorescence upon dissolving and release into aqueous media justifies the accurate identification of FNB-HCs both in vitro and in vivo. The fluorescence stability of FNB-HCs in buffers and bio-relevant media indicates no dye leakage and water infiltration, which may compromise the accuracy of bioimaging. Linearity between crystal-borne fluorescence and residual crystal amount is established (r = 0.9824) for the quantification of FNB-HCs. In vitro dissolution is studied under pharmacopeia conditions by monitoring either FNB or fluorescence, and very good correlation exist between dissolution profiles acquired by the two approaches, confirming the reliability of using fluorescence to monitor crystal dissolution. In vivo dissolution is first investigated by live imaging and results indicate fast and complete dissolution in the stomach within 60 min. Correlation of in vivo with in vitro dissolution profile gives a coefficient of 0.8996, implying a level A point-to-point correlation according to FDA guidelines. Elevated correlation levels with a coefficient of 0.9454 are observed between in vivo dissolution acquired in ex vivo GIT and in vitro dissolution. Both live and ex vivo imaging results of in vivo dissolution validate the correctness of the current pharmacopeia in vitro dissolution protocols. The correlation between Fa and in vitro dissolution (r = 0.9375) confirms the well established Level A IVIVC, whereas the higher correlation coefficient of 0.9691 between Fa and in vivo dissolution validates the potential application of in vivo dissolution for the establishment of authentic IVIVC.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Nos. 81973247, 81872815, 81872826 and 81690263) and Science and Technology Commission of Shanghai Municipality (19XD1400300, China). We are grateful for Prof. Dr. Yu-Kyoung Oh of Seoul National University Nano Biodrug Delivery Lab for the illuminating comments she provided.
Author contributions
Wei Wu and Yi Lu designed the research. Yinqian Yang carried out the experiments and performed data analysis. Yongjiu Lv, Chengying Shen, Tingting Shi and Haisheng He participated in part of the experiments. Weili Zhao and Xiaochun Dong provided experimental drugs and quality control. Yinqian Yang wrote the manuscript. Wei Wu, Yi Lu and Jianping Qi revised the manuscript. All of the authors have read and approved the final manuscript.
Conflicts of interest
The authors have no conflicts of interest to declare.
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Appendix A. Supporting information
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2020.08.002.
Appendix ASupplementary data
The following is the Supplementary data to this article:
Multimedia component 1
References
- 1.Allen L.V., Jr., Popovich N.G., Aansel H.C. 8th ed. Lippincott Williams & Wilkins; Baltimore: 2013. Ansel's pharmaceutical dosage forms and drug delivery systems. [Google Scholar]
- 2.Shargel L., Yu A.B.C. 7th ed. McGraw-Hill Education; New York: 2016. Applied biopharmaceutics & pharmacokinetics. [Google Scholar]
- 3.Guo M., Wei M., Li W., Guo M., Guo C., Ma M. Impacts of particle shapes on the oral delivery of drug nanocrystals: mucus permeation, transepithelial transport and bioavailability. J Control Release. 2019;307:64–75. doi: 10.1016/j.jconrel.2019.06.015. [DOI] [PubMed] [Google Scholar]
- 4.Fu Q., Sun J., Ai X., Zhang P., Li M., Wang Y. Nimodipine nanocrystals for oral bioavailability improvement: role of mesenteric lymph transport in the oral absorption. Int J Pharm. 2013;448:290–297. doi: 10.1016/j.ijpharm.2013.01.065. [DOI] [PubMed] [Google Scholar]
- 5.Xie Y., Shi B., Xia F., Qi J., Dong X., Zhao W. Epithelia transmembrane transport of orally administered ultrafine drug particles evidenced by environment sensitive fluorophores in cellular and animal studies. J Control Release. 2018;270:65–75. doi: 10.1016/j.jconrel.2017.11.046. [DOI] [PubMed] [Google Scholar]
- 6.Shen C., Yang Y., Shen B., Xie Y., Qi J., Dong X. Self-discriminating fluorescent hybrid nanocrystals: efficient and accurate tracking of translocation via oral delivery. Nanoscale. 2017;10:436–450. doi: 10.1039/c7nr06052a. [DOI] [PubMed] [Google Scholar]
- 7.Sugano K., Terada K. Rate- and extent-limiting factors of oral drug absorption: theory and applications. J Pharm Sci. 2015;104:2777–2788. doi: 10.1002/jps.24391. [DOI] [PubMed] [Google Scholar]
- 8.Shi Q., Moinuddin S.M., Cai T. Advances in coamorphous drug delivery systems. Acta Pharm Sin B. 2019;9:19–35. doi: 10.1016/j.apsb.2018.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cook J.A. A technique to estimate in vivo dissolution profiles without data from a solution. AAPS J. 2012;14:433–436. doi: 10.1208/s12248-012-9355-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kostewicz E.S., Abrahamsson B., Brewster M., Brouwers J., Butler J., Carlert S. In vitro models for the prediction of in vivo performance of oral dosage forms. Eur J Pharmaceut Sci. 2014;57:342–366. doi: 10.1016/j.ejps.2013.08.024. [DOI] [PubMed] [Google Scholar]
- 11.Bou-Chacra N., Melo K.J.C., Morales I.A.C., Stippler E.S., Kesisoglou F., Yazdanian M. Evolution of choice of solubility and dissolution media after two decades of Biopharmaceutical Classification System. AAPS J. 2017;19:989–1001. doi: 10.1208/s12248-017-0085-5. [DOI] [PubMed] [Google Scholar]
- 12.Charalabidis A., Sfouni M., Bergstrom C., Macheras P. The biopharmaceutics classification system (BCS) and the biopharmaceutics drug disposition classification system (BDDCS): beyond guidelines. Int J Pharm. 2019;566:264–281. doi: 10.1016/j.ijpharm.2019.05.041. [DOI] [PubMed] [Google Scholar]
- 13.Hens B., Sinko P.D., Job N., Dean M., Al-Gousous J., Salehi N. Formulation predictive dissolution (fPD) testing to advance oral drug product development: an introduction to the US FDA funded ‘21st Century BA/BE' project. Int J Pharm. 2018;548:120–127. doi: 10.1016/j.ijpharm.2018.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaur N., Narang A., Bansal A.K. Use of biorelevant dissolution and PBPK modeling to predict oral drug absorption. Eur J Pharm Biopharm. 2018;129:222–246. doi: 10.1016/j.ejpb.2018.05.024. [DOI] [PubMed] [Google Scholar]
- 15.Emami J. In vitro‒in vivo correlation: from theory to applications. J Pharm Pharmaceut Sci. 2006;9:169–189. [PubMed] [Google Scholar]
- 16.Ruiz Picazo A., Martinez-Martinez M.T., Colon-Useche S., Iriarte R., Sanchez-Dengra B., Gonzalez-Alvarez M. In vitro dissolution as a tool for formulation selection: telmisartan two-step IVIVC. Mol Pharm. 2018;15:2307–2315. doi: 10.1021/acs.molpharmaceut.8b00153. [DOI] [PubMed] [Google Scholar]
- 17.Suarez-Sharp S., Li M., Duan J., Shah H., Seo P. Regulatory experience with in vivo in vitro correlations (IVIVC) in new drug applications. AAPS J. 2016;18:1379–1390. doi: 10.1208/s12248-016-9966-2. [DOI] [PubMed] [Google Scholar]
- 18.U.S. Food and Drug Administration Guidance for industry-extended release oral dosage forms-development, evaluation and application of in vitro‒in vivo correlations. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/extended-release-oral-dosage-forms-development-evaluation-and-application-vitroin-vivo-correlations Available from: [DOI] [PubMed]
- 19.U.S. Food and Drug Administration Waiver on in vivo bioavailability and bioequivalence studies for immediate-release solid oral dosage forms based on a Biopharmaceutics classification systems. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/waiver-vivo-bioavailability-and-bioequivalence-studies-immediate-release-solid-oral-dosage-forms Available from:
- 20.Lu Y., Kim S., Park K. In vitro‒in vivo correlation: perspectives on model development. Int J Pharm. 2011;418:142–148. doi: 10.1016/j.ijpharm.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kytariolos J., Dokoumetzidis A., Macheras P. Power law IVIVC: an application of fractional kinetics for drug release and absorption. Eur J Pharmaceut Sci. 2010;41:299–304. doi: 10.1016/j.ejps.2010.06.015. [DOI] [PubMed] [Google Scholar]
- 22.Xu H., Shi Y., Vela S., Marroum P., Gao P. Developing quantitative in vitro‒in vivo correlation for fenofibrate immediate-release formulations with the biphasic dissolution-partition test method. J Pharm Sci. 2018;107:476–487. doi: 10.1016/j.xphs.2017.06.018. [DOI] [PubMed] [Google Scholar]
- 23.Wagner J.G. Estimation of theophylline absorption rate by means of the Wagner‒Nelson equation. J Allergy Clin Immunol. 1986;78:681–688. doi: 10.1016/0091-6749(86)90046-1. [DOI] [PubMed] [Google Scholar]
- 24.Wagner J.G. Effect of using an incorrect elimination rate constant in application of the Wagner‒Nelson method to theophylline data in cases of zero order absorption. Biopharm Drug Dispos. 1984;5:75–83. doi: 10.1002/bdd.2510050110. [DOI] [PubMed] [Google Scholar]
- 25.Ostrowski M., Wilkowska E., Baczek T. The influence of averaging procedure on the accuracy of IVIVC predictions: immediate release dosage form case study. J Pharm Sci. 2010;99:5040–5045. doi: 10.1002/jps.22209. [DOI] [PubMed] [Google Scholar]
- 26.Wang Y., Nedelman J. Bias in the Wagner-Nelson estimate of the fraction of drug absorbed. Pharm Res (N Y) 2002;19:470–476. doi: 10.1023/a:1015195612726. [DOI] [PubMed] [Google Scholar]
- 27.Cardot J.M., Davit B.M. In vitro-in vivo correlations: tricks and traps. AAPS J. 2012;14:491–499. doi: 10.1208/s12248-012-9359-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaur P., Jiang X., Duan J., Stier E. Applications of in vitro‒in vivo correlations in generic drug development: case studies. AAPS J. 2015;17:1035–1039. doi: 10.1208/s12248-015-9765-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nguyen M.A., Flanagan T., Brewster M., Kesisoglou F., Beato S., Biewenga J. A survey on IVIVC/IVIVR development in the pharmaceutical industry—past experience and current perspectives. Eur J Pharmaceut Sci. 2017;102:1–13. doi: 10.1016/j.ejps.2017.02.029. [DOI] [PubMed] [Google Scholar]
- 30.Lennernas H., Lindahl A., Van Peer A., Ollier C., Flanagan T., Lionberger R. In vivo predictive dissolution (IPD) and biopharmaceutical modeling and simulation: future use of modern approaches and methodologies in a regulatory context. Mol Pharm. 2017;14:1307–1314. doi: 10.1021/acs.molpharmaceut.6b00824. [DOI] [PubMed] [Google Scholar]
- 31.Roudier B., Davit B., Schutz H., Cardot J.M. Impact of data base structure in a successful in vitro‒in vivo correlation for pharmaceutical products. AAPS J. 2015;17:24–34. doi: 10.1208/s12248-014-9680-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brouwers J., Augustijns P. Resolving intraluminal drug and formulation behavior: gastrointestinal concentration profiling in humans. Eur J Pharmaceut Sci. 2014;61:2–10. doi: 10.1016/j.ejps.2014.01.010. [DOI] [PubMed] [Google Scholar]
- 33.Hens B., Corsetti M., Spiller R., Marciani L., Vanuytsel T., Tack J. Exploring gastrointestinal variables affecting drug and formulation behavior: methodologies, challenges and opportunities. Int J Pharm. 2017;519:79–97. doi: 10.1016/j.ijpharm.2016.11.063. [DOI] [PubMed] [Google Scholar]
- 34.Butler J., Hens B., Vertzoni M., Brouwers J., Berben P., Dressman J. In vitro models for the prediction of in vivo performance of oral dosage forms: recent progress from partnership through the IMI OrBiTo collaboration. Eur J Pharm Biopharm. 2019;136:70–83. doi: 10.1016/j.ejpb.2018.12.010. [DOI] [PubMed] [Google Scholar]
- 35.Knutson P.F.T., Ahlström H., Magnusson A., Tannergren C., Lennerna H. Increased understanding of intestinal drug permeability determined by the LOC-I-GUT approach using multislice computed tomography. Mol Pharm. 2008;6:2–10. doi: 10.1021/mp800145r. [DOI] [PubMed] [Google Scholar]
- 36.Petri N., Tannergren C., Holst B., Mellon F.A., Bao Y., Plumb G.W. Absorption/metabolism of sulforaphane and quercetin, and regulation of phase II enzymes, in human jejunum in vivo. Drug Metab Dispos. 2003;31:805–813. doi: 10.1124/dmd.31.6.805. [DOI] [PubMed] [Google Scholar]
- 37.Brouwers J., Ingels F., Tack J., Augustijns P. Determination of intraluminal theophylline concentrations after oral intake of an immediate- and a slow-release dosage form. J Pharm Pharmacol. 2005;57:987–996. doi: 10.1211/0022357056631. [DOI] [PubMed] [Google Scholar]
- 38.Koenigsknecht M.J., Baker J.R., Wen B., Frances A., Zhang H., Yu A. In vivo dissolution and systemic absorption of immediate release ibuprofen in human gastrointestinal tract under fed and fasted conditions. Mol Pharm. 2017;14:4295–4304. doi: 10.1021/acs.molpharmaceut.7b00425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bergstrom C.A., Holm R., Jorgensen S.A., Andersson S.B., Artursson P., Beato S. Early pharmaceutical profiling to predict oral drug absorption: current status and unmet needs. Eur J Pharmaceut Sci. 2014;57:173–199. doi: 10.1016/j.ejps.2013.10.015. [DOI] [PubMed] [Google Scholar]
- 40.Bermejo M., Paixão P., Hens B., Tsume Y., Koenigsknecht M.J., Baker J.R. Linking the gastrointestinal behavior of ibuprofen with the systemic exposure between and within humans—part 1: fasted state conditions. Mol Pharm. 2018;15:5454–5467. doi: 10.1021/acs.molpharmaceut.8b00515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu Y., Lv Y., Li T. Hybrid drug nanocrystals. Adv Drug Deliv Rev. 2019;143:115–133. doi: 10.1016/j.addr.2019.06.006. [DOI] [PubMed] [Google Scholar]
- 42.Qi J., Hu X., Dong X., Lu Y., Lu H., Zhao W. Towards more accurate bioimaging of drug nanocarriers: turning aggregation-caused quenching into a useful tool. Adv Drug Deliv Rev. 2019;143:206–225. doi: 10.1016/j.addr.2019.05.009. [DOI] [PubMed] [Google Scholar]
- 43.Wang L., Liu M., Chen Y. Carbon dots and terbium co-enhanced fluorescence of europium nanoparticles for cell Imaging. J Biomed Nanotechnol. 2018;14:1898–1905. doi: 10.1166/jbn.2018.2630. [DOI] [PubMed] [Google Scholar]
- 44.Wang B., Lv P., Cai H., Li Y., Zhu H., Lui S. Enzyme-responsive copolymer as a theranostic prodrug for tumor in vivo imaging and efficient chemotherapy. J Biomed Nanotechnol. 2019;15:1897–1908. doi: 10.1166/jbn.2019.2833. [DOI] [PubMed] [Google Scholar]
- 45.Hu D., Chen L., Qu Y., Peng J., Chu B., Hao Y. Oxygen-generating hybrid polymeric nanoparticles with encapsulated doxorubicin and chlorin e6 for trimodal imaging-guided combined chemo-photodynamic therapy. Theranostics. 2018;8:1558–1574. doi: 10.7150/thno.22989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Y., Zhang Y., Wang J., Liang X.J. Aggregation-induced emission (AIE) fluorophores as imaging tools to trace the biological fate of nano-based drug delivery systems. Adv Drug Deliv Rev. 2019;143:161–176. doi: 10.1016/j.addr.2018.12.004. [DOI] [PubMed] [Google Scholar]
- 47.Chen T., He B., Tao J., He Y., Deng H., Wang X. Application of Förster resonance energy transfer (FRET) technique to elucidate intracellular and in vivo biofate of nanomedicines. Adv Drug Deliv Rev. 2019;143:177–205. doi: 10.1016/j.addr.2019.04.009. [DOI] [PubMed] [Google Scholar]
- 48.Hu X., Zhang J., Yu Z., Xie Y., He H., Qi J. Environment-responsive aza-BODIPY dyes quenching in water as potential probes to visualize the in vivo fate of lipid-based nanocarriers. Nanomedicine. 2015;11:1939–1948. doi: 10.1016/j.nano.2015.06.013. [DOI] [PubMed] [Google Scholar]
- 49.Hu X., Fan W., Yu Z., Lu Y., Qi J., Zhang J. Evidence does not support absorption of intact solid lipid nanoparticles via oral delivery. Nanoscale. 2016;8:7024–7035. doi: 10.1039/c5nr07474f. [DOI] [PubMed] [Google Scholar]
- 50.Hu X., Dong X., Lu Y., Qi J., Zhao W., Wu W. Bioimaging of nanoparticles: the crucial role of discriminating nanoparticles from free probes. Drug Discov Today. 2017;22:382–387. doi: 10.1016/j.drudis.2016.10.002. [DOI] [PubMed] [Google Scholar]
- 51.Ahmad E., Feng Y., Qi J., Fan W., Ma Y., He H. Evidence of nose-to-brain delivery of nanoemulsions: cargoes but not vehicles. Nanoscale. 2017;9:1174–1183. doi: 10.1039/c6nr07581a. [DOI] [PubMed] [Google Scholar]
- 52.Xia F., Fan W., Jiang S., Ma Y., Lu Y., Qi J. Size-dependent translocation of nanoemulsions via oral delivery. ACS Appl Mater Interfaces. 2017;9:21660–21672. doi: 10.1021/acsami.7b04916. [DOI] [PubMed] [Google Scholar]
- 53.He H., Zhang J., Xie Y., Lu Y., Qi J., Ahmad E. Bioimaging of intravenous polymeric micelles based on discrimination of integral particles using an environment-responsive probe. Mol Pharm. 2016;13:4013–4019. doi: 10.1021/acs.molpharmaceut.6b00705. [DOI] [PubMed] [Google Scholar]
- 54.Pestieau A., Lebrun S., Cahay B., Brouwers A., Streel B., Cardot J.M. Evaluation of different in vitro dissolution tests based on level A in vitro‒in vivo correlations for fenofibrate self-emulsifying lipid-based formulations. Eur J Pharm Biopharm. 2017;112:18–29. doi: 10.1016/j.ejpb.2016.10.030. [DOI] [PubMed] [Google Scholar]
- 55.Zuo B., Sun Y., Li H., Liu X., Zhai Y., Sun J. Preparation and in vitro/in vivo evaluation of fenofibrate nanocrystals. Int J Pharm. 2013;455:267–275. doi: 10.1016/j.ijpharm.2013.07.021. [DOI] [PubMed] [Google Scholar]
- 56.Hens B., Brouwers J., Corsetti M., Augustijns P. Gastrointestinal behavior of nano- and microsized fenofibrate: in vivo evaluation in man and in vitro simulation by assessment of the permeation potential. Eur J Pharmaceut Sci. 2015;77:40–47. doi: 10.1016/j.ejps.2015.05.023. [DOI] [PubMed] [Google Scholar]
- 57.Zhao W., Carreira E.M. Conformationally restricted aza-bodipy: a highly fluorescent, stable, near-infrared-absorbing dye. Angew Chem Int Ed Engl. 2005;44:1677–1679. doi: 10.1002/anie.200461868. [DOI] [PubMed] [Google Scholar]
- 58.Zhao W., Carreira E.M. Conformationally restricted aza-BODIPY: highly fluorescent, stable near-infrared absorbing dyes. Chemistry. 2006;12:7254–7263. doi: 10.1002/chem.200600527. [DOI] [PubMed] [Google Scholar]
- 59.Zhu W., Romanski F.S., Meng X., Mitra S., Tomassone M.S. Atomistic simulation study of surfactant and polymer interactions on the surface of a fenofibrate crystal. Eur J Pharmaceut Sci. 2011;42:452–461. doi: 10.1016/j.ejps.2011.01.009. [DOI] [PubMed] [Google Scholar]
- 60.O'Shea J.P., Faisal W., Ruane-O'Hora T., Devine K.J., Kostewicz E.S., O'Driscoll C.M. Lipidic dispersion to reduce food dependent oral bioavailability of fenofibrate: in vitro, in vivo and in silico assessments. Eur J Pharm Biopharm. 2015;96:207–216. doi: 10.1016/j.ejpb.2015.07.014. [DOI] [PubMed] [Google Scholar]
- 61.Guichard J.P., Blouquin P., Qing Y. A new formulation of fenofibrate: suprabioavailable tablets. Curr Med Res Opin. 2008;16:134–138. [PubMed] [Google Scholar]
- 62.Hanafy A., Spahn-Langguth H., Vergnault G., Grenier P., Tubic Grozdanis M., Lenhardt T. Pharmacokinetic evaluation of oral fenofibrate nanosuspensions and SLN in comparison to conventional suspensions of micronized drug. Adv Drug Deliv Rev. 2007;59:419–426. doi: 10.1016/j.addr.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 63.Weng T., Qi J., Lu Y., Wang K., Tian Z., Hu K. The role of lipid-based nano delivery systems on oral bioavailability enhancement of fenofibrate, a BCS II drug: comparison with fast-release formulations. J Nanobiotechnol. 2014;12:39. doi: 10.1186/s12951-014-0039-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Curtet B, Teillaud E, Reginault P, inventors. Fournier Innovation et Synergie, assignee. Novel dosage form of fenofibrate. United States patent US No. 4895726. January 23, 1980.
- 65.Yang L., Shao Y., Han H.K. Development of omega-3 phospholipid-based solid dispersion of fenofibrate for the enhancement of oral bioavailability. Eur J Pharmaceut Sci. 2015;78:103–110. doi: 10.1016/j.ejps.2015.07.007. [DOI] [PubMed] [Google Scholar]
- 66.Jadhav N.V., Vavia P.R. Dodecylamine template-based hexagonal mesoporous silica (HMS) as a carrier for improved oral delivery of fenofibrate. AAPS PharmSciTech. 2017;18:2764–2773. doi: 10.1208/s12249-017-0761-x. [DOI] [PubMed] [Google Scholar]
- 67.Yuan H., Zhao W., Wu W. How can aggregation-caused quenching based bioimaging of drug nanocarriers be improved?. Ther Deliv. 2020;11:809–812. doi: 10.4155/tde-2019-0082. [DOI] [PubMed] [Google Scholar]
- 68.Li F., Zheng X., Bao Y., Chen T., Zeng J., Xu X. Fenofibrate modified-release pellets with lag phase and high oral bioavailability. Drug Des Dev Ther. 2019;13:141–151. doi: 10.2147/DDDT.S179266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bahloul B., Lassoued M.A., Seguin J., Lai-Kuen R., Dhotel H., Sfar S. Self-emulsifying drug delivery system developed by the HLB-RSM approach: characterization by transmission electron microscopy and pharmacokinetic study. Int J Pharm. 2015;487:56–63. doi: 10.1016/j.ijpharm.2015.04.018. [DOI] [PubMed] [Google Scholar]
- 70.Juenemann D., Jantratid E., Wagner C., Reppas C., Vertzoni M., Dressman J.B. Biorelevant in vitro dissolution testing of products containing micronized or nanosized fenofibrate with a view to predicting plasma profiles. Eur J Pharm Biopharm. 2011;77:257–264. doi: 10.1016/j.ejpb.2010.10.012. [DOI] [PubMed] [Google Scholar]
- 71.González-García I., Mangas-Sanjuán V., Merino-Sanjuán M., Bermejo M. In vitro in vivo correlations general concepts methodologies and regulatory applications. Drug Dev Ind Pharm. 2015;41:1935–1947. doi: 10.3109/03639045.2015.1054833. [DOI] [PubMed] [Google Scholar]
- 72.Cardot J.M., Beyssac E., Alric M. In vitro–in vivo correlation: importance of dissolution in IVIVC. Dissolution Technol. 2007;14:15–19. [Google Scholar]
- 73.Antaris A.L., Chen H., Cheng K., Sun Y., Hong G., Qu C. A small-molecule dye for NIR-II imaging. Nat Mater. 2016;15:235–242. doi: 10.1038/nmat4476. [DOI] [PubMed] [Google Scholar]
- 74.Cai Y., Wei Z., Song C., Tang C., Han W., Dong X. Optical nano-agents in the second near-infrared window for biomedical applications. Chem Soc Rev. 2019;48:22–37. doi: 10.1039/c8cs00494c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Multimedia component 1