

Abstract
Background
Bone morphogenetic protein receptor 2 (BMPR2) is an important transmembrane serine/threonine kinase that involves oncogenic processes in multiple cancers. However, the role of BMPR2 and its regulatory mechanism in pancreatic ductal adenocarcinoma (PDAC) remain unknown.
Methods
We performed a tissue array to explore the expression of BMPR2 in PDAC tissues. The Cell Counting Kit-8 (CCK-8) and colony formation assays were used to measure PDAC cells’ proliferation. Proteomics and mass spectrometry technology was applied to analyze the BMPR2-regulating proteins. Flow cytometry was used to analyze the cell cycle distribution of PDAC cells. Orthotopic pancreatic cancer (PC) and patient-derived xenograft (PDX) models were used for in vivo experiments.
Results
This study revealed the over-expression of BMPR2 in PDAC tissues and its proliferation-promoting role in PDAC cells. By carrying out protein mass spectrometry technique as well as bioinformatics analysis, we identified that BMPR2 regulated the growth factor receptor-bound protein 2/phosphatidylinositol 3-kinase/protein kinase B (GRB2/PI3K/AKT) signaling pathway, and further in vitro experiments showed that inhibition of BMPR2 resulted in suppressing proliferation and G2/M arrest by inhibiting the GRB2/PI3K/AKT signaling pathway in PDAC cells. The inhibition of BMPR2 by LDN193189 showed similar results in PDAC cells, orthotopic PC, and PDX models, which revealed that inhibition of BMPR2 significantly suppressed tumor growth by suppressing the GRB2/PI3K/AKT axis.
Conclusions
Inhibition of BMPR2 suppresses PDAC growth by regulating the GRB2/PI3K/AKT axis and is a promising PDAC treatment strategy.
Keywords: Bone morphogenetic protein receptor 2 (BMPR2), pancreatic ductal adenocarcinoma (PDAC), proliferation, GRB/PI3K/AKT axis
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of the most common malignant tumors with a barely improved 5-year survival rate despite great improvements in surgery and adjuvant therapies in the past decades (1). It has been reported that PDAC will become the second leading cause of cancer-associated death by 2030 in the United States (2). Therefore, more efforts should be made to understand the molecular pathology of PDAC further. Bone morphogenetic proteins (BMPs) are growth factors belonging to the tumor growth factor-β superfamily, which initiate intracellular BMP signaling by binding to type 1 and 2 BMP receptors (3). Signaling transduction mediated by BMP/BMPR has been proven in multiple biological processes, such as self-renewal and stemness maintenance during embryonic development (4). Recently, aberrant expression of bone morphogenetic protein receptor 2 (BMPR2) has been detected in breast and gastric cancer, and abnormal expression of BMPR2 has been proven to involve in proliferation, differentiation, and migration of tumor cells (5-7). However, the role of BMPR2 and its regulating mechanism in PDAC remains unknown.
Our study identified that BMPR2 over-expressed in PDAC tumor compared to normal pancreas tissues by immunohistochemistry (IHC) using tumor microarray. Inhibition of BMPR2 resulted in the suppressed proliferation and G2/M arrest in pancreatic cancer (PC) cells. Using proteomic analysis, we found that GRB2 is the potential target of BMPR2, and its oncogenic role was further shown in PC cells. Growth factor receptor-bound protein 2 (GRB2) is an adaptor protein that involves many cellular functions, such as cell survival and proliferation, and is also an important regulator in multiple oncogenic signaling pathways (8,9). The role of GRB2 has been studied widely in many cancers, especially breast cancer (10-12). We performed bioinformatics analysis to explore the potential molecular mechanisms that GRB2 might involve in. The in vitro experiments indicated that BMPR2 regulated PC cell proliferation by regulating growth factor receptor-bound protein 2/phosphatidylinositol 3-kinase/protein kinase B (GRB2/PI3K/AKT) pathway. The BMPR2 inhibitor LDN193189 significantly inhibited BMPR2-induced activation of the GRB2/PI3K/AKT pathway. Using orthotopic PC and patient-derived xenograft (PDX) models, we further proved that inhibition of BMPR2 suppressed PC growth by inhibiting GRB2/PI3K/AKT axis in vivo. Herein, we have revealed the tumorigenic effect of BMPR2 in PDAC, which provides evidence for using BMPR2 inhibitor in the treatment of PDAC.
We present the following article in accordance with the ARRIVE reporting checklist (available at http://dx.doi.org/10.21037/atm-20-2194).
Methods
Participant samples
The clinical specimens were taken from patients diagnosed with PDAC at Peking University First Hospital, Beijing, China. The samples were washed by saline after excision from participants and then transferred in liquid nitrogen and stored at −80 °C. Samples for PDX construction were transferred in the Dulbecco’s Modified Eagle Medium (DMEM) free of fetal bovine serum (FBS), cut into small blocks, and then implanted subcutaneously in NOD/SCID (nonobese diabetic/severe combined immunodeficiency) mice within 2 h. The study was conducted following the Declaration of Helsinki (as revised in 2013). The Ethics Committee approved the study of Peking University First Hospital (No. 201933), and all participants provided informed consent.
IHC and tumor microarray analysis (TMA)
To further investigate the role of BMPR2 in PC and its clinical relevance, we performed an IHC analysis. The TMA was purchased from Zhuohao Pharmaceutical Technology Co. Ltd., Shanghai, China. The TMA slides were dewaxed in xylene and rehydrated in alcohol, followed by antigen retrieval with citrate buffer (0.01 M sodium citrate, pH 6.0) for 2 min under high pressure. Sections were incubated with primary antibodies (anti-BMPR2, 1:100; anti-GRB2, 1:100; anti-Ki-67, 1:1,000) overnight at 4 °C and then with a biotinylated secondary antibody. Sections were counterstained with a standard Mayer’s hematoxylin and eosin (H&E) solution and then mounted with Permount TM Mounting Medium (Solarbio, Beijing, China). All sections were examined and scored independently by 2 investigators, without any knowledge of participants' clinicopathological data. At least 5 fields were randomly chosen for analysis. Protein expression was evaluated according to the ratio of positive cells and the staining intensity per field. The positive cells ratio was scored as 0 for less than 5%, 1 for 5–10%, 2 for 10–50%, and 3 for >50%. Staining intensity was graded as 1 for weak staining and 2 for strong staining. A total score of 0–6 was calculated and used to further divide samples into categories of negative (scores 0–3) or positive (scores 3–6) expression. Images were obtained using a microscope.
Cell lines, reagents, and animals
A normal human pancreatic epithelial cell line (hTERT-HPNE) and human PC cell lines (Mia-Paca-2, PANC-1, Capan-1, and AsPC-1, T3M4) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). The identity of the cell lines was routinely monitored by short tandem repeat profiling. The hTERT-HPNE, Mia-Paca-2, PANC-1, and Capan-1 cells were cultured in DMEM (Gibco, Waltham, MA, USA). The AsPC-1 and T3M4 cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium (Gibco). All culture media were supplemented with 10% FBS (Gibco), and cells were maintained under standard culture conditions (37 °C, 95% humidified air, and 5% CO2).
The anti-BMPR2 antibody was purchased from R&D Systems (Minneapolis, MN, USA), Cell Counting Kit-8 (CCK-8) was from Dojindo (Kumamoto, Japan), and the propidium iodide (PI) Cell Cycle Analysis Kit was from R&D Systems. The highly potent small molecule BMP pathway inhibitor, LDN193189, was purchased from Selleck Chemicals (Shanghai, China), dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich, St Louis, MO, USA), and directly added to the culture medium. Bagg albino (BALB/c) nude mice (4–6 weeks, female, 18–20 g) and NOD/SCID mice (4–6 weeks, female, 18–20 g) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China) and were maintained in a specific pathogen-free laboratory. Experiments were performed under a project license (J201980) granted by the Animal Ethics Committee of Peking University First Hospital, in compliance with national guidelines for the care and use of animals.
RNA isolation, cDNA synthesis, and qPCR
Total RNA was isolated from snap-frozen tissue and PC cell lines, which were lysed directly by TRIzol (Invitrogen, Carlsbad, CA, USA) and further purified by chloroform and alcohol precipitation. Complementary DNA (cDNA) was synthesized from equal amounts of RNA using the ReverTra Ace® quantitative reverse transcription polymerase chain reaction (RT-qPCR) kit (Toyobo, Osaka, Japan). A 2× SYBR mastermix was combined with 1 µM each of a forward and reverse primer in a 20 µL reaction, and amplification was performed for 40 cycles of 95 °C for 10 s and 60 °C for 30 s on an ABI 7500 system (Applied Biosystems, Foster City, CA, USA). Following amplification, melting curve analysis was performed according to the manufacturer’s protocol, and the software provided by the instrument was used to determine relative expression, normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression. Relative gene expression was analyzed in triplicate. The primer sequences were as follows: GRB2: Forward 5'-GAGAGAGGAT CCATGGTTTTTlTGGCAAAATCCCCAGCGCC-3'; Reverse 5'-GAGAGAGAA TTCTCACGGCTGCTGTGGCACCTGTTCTATGTCC-3', GAPDH: Forward 5'-GGACCTGACCTGCCGTCTAG-3' Reverse: 5'-CCACCACCCTGTTGCTGTAG-3'.
Plasmid construction and transfection
A short hairpin RNA (shRNA) for BMPR2 (NM_001204) and a negative control shRNA with a scrambled sequence were constructed by Obio Technology Corp., Ltd. (Shanghai, China). The sequences of the shRNAs targeting the human BMPR2 gene were GCACCGAAGCGAAACTTAA and ATTAGACAAAACCAAAAAAGAAGCT. The scrambled sequence of the control shRNA was TTCTCCGAACGTGTCACGT. The entire coding sequence of the human BMPR2 gene was cloned, and a 3FLAG-tagged BMPR2 vector was generated by Obio Technology Corp., Ltd. using the pLenti-CMV-MCS-3FLAG-PGK-Puro plasmid. Lentiviruses were produced by transfecting 293T cells with plasmids using VigoFect (Vigorous Biotechnology, Beijing, China). Transfected cells were selected with puromycin, according to the manufacturer’s instructions.
Western blotting
Cell lysates were prepared according to previously described methods (Zhang et al.) and were separated by 10% or 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). After electroblotting onto nitrocellulose membranes (Pall, New York, NY, USA), target proteins were probed with corresponding antibodies and detected using Immobilon Western Chemiluminescent HRP Substrate (MA01821; Millipore, Billerica, MA, USA). Either GAPDH or β-actin was used as a loading control. The antibody information is listed in Table 1.
Table 1. Information of antibodies used in this study.
| Antibody name | Manufacturer | Concentration | Catalog number |
|---|---|---|---|
| Anti-BMPR2 | R&D Systems | 1:2,000 | AF811 |
| Anti-GRB2 | Abcam | 1:5,000 | ab32037 |
| Anti-β-actin | Proteintech | 1:10,000 | 60008-1-Ig |
| Anti-PI3K | Abcam | 1:2,000 | ab140307 |
| Anti-p-PI3K | Abcam | 1:1,000 | ab154598 |
| Anti-AKT | Abcam | 1:500 | ab8805 |
| Anti-p-AKT308 | Abcam | 1:1,000 | ab38449 |
| Anti-p-AKT474 | Abcam | 1:1,000 | ab38513 |
| Anti-Cyclin B1 | Cell Signaling Technology | 1:1,000 | 12231 |
| Anti-CDK1 | Proteintech | 1:1,000 | 19532-1-AP |
Cell growth and colony-formation assay
Cell viability was assessed using CCK-8, and cell colony formation capacity was detected using a colony-formation assay. Cells were seeded at a density of 5,000 cells/well in 96-well plates, with 3 replicates for CCK-8 assays, and were seeded into 6-well plates at a destiny of 500 cells/well for colony-formation assay. The subsequent procedures were performed as previously described (13). Briefly, the cells were fixed with 4% paraformaldehyde for and then stained by crystal violet for 10 min. Then the cells were washed and cell colonies were counted.
Flow cytometry
Cells pellets were fixed in 70% ethanol, stained with PI/RNase Staining Buffer (BD Biosciences, San Jose, CA, USA) for 10 min, and then analyzed with a FACSCalibur flow cytometer (BD Biosciences). The acquisition count was set to 20,000 events for each sample. The nuclei population in each phase of the cell cycle was determined and analyzed, as described previously (13).
Sample preparation and label-free quantitative proteomic analysis
An SDT buffer (4% SDS, 100 mM DTT, and 150 mM Tris-HCl pH 8.0) was added to cell pellets. The lysate was sonicated and then boiled for 15 min. After centrifugation at 14,000 ×g for 40 min, the supernatant was quantified using a bicinchoninic (BCA) protein assay kit (Bio-Rad, Hercules, CA, USA). A total of 20 µg of each protein sample were mixed with loading buffer and boiled for 5 min. Proteins were separated on 12.5% SDS-PAGE gels (14 mA constant current, 90 min). Protein bands were visualized by Coomassie Blue R-250 staining.
Protein suspensions were digested with 4 µg of trypsin (Promega, Madison, WI, USA) in 40 µL of 25 mM NH4HCO3 buffer overnight at 37 °C, and the resulting peptides were collected as a filtrate. Each sample’s peptides were desalted on C18 cartridges (Empore™SPE; standard density; bed I.D., 7 mm, volume, 3 mL; Sigma-Aldrich), concentrated by vacuum centrifugation, and reconstituted in 40 µL of 0.1% (v/v) formic acid. The peptide concentration was estimated by UV light spectral density at 280 nm, using an extinction coefficient of 1.1. NanoLC-MS/MS analyzed each fraction. The MS data were analyzed using MaxQuant software version 1.5.3.17 (Max Planck Institute of Biochemistry, Martinsried, Germany), Gene Set Enrichment Analysis (GSEA) software, and R software. The GSEA of differentially expressed genes was performed against the Molecular Signatures Database version 5.2 (MSigDB) hallmark gene sets (H) collection. Enrichment was assessed by hypergeometric testing using the R stats package (http://www.R-project.org/).
Animal models
The luciferase-expressing pancreatic cell line PANC-1 was injected into BALB/c nude mice’s pancreases to construct orthotopic PC models. After tumor initiation, the mice were injected with the BMPR2 inhibitor LDN193189 (3 mg/kg) or with a vehicle intraperitoneally once a day (n=5). The in vivo imaging system IVIS Spectrum (Perkin Elmer, Waltham, MA, USA) was used to monitor orthotopic tumors’ growth. At the end of the treatment, the tumors were excised and photographed. The tumors were then fixed in 4% paraformaldehyde for IHC and frozen at −80 °C for western blot analysis.
For PDX models, the pancreatic tumor excised from PC patients were cut into small blocks and then implanted subcutaneously in NOD/SCID mice according to the previous study to construct PDX models (14,15). The tumor-bearing mice were injected with the BMPR2 inhibitor LDN193189 (3 mg/kg) or with a vehicle intraperitoneally once a day (n=5). The tumors were measured in the treatment process. At the end of the treatment, the tumors were excised and weighted. The PANC-1 cells transfected with shBMPR2 and its control were injected into the pancreas of mice, and 30 days later, the orthotopic tumors were excised and weighted (n=5). All animal experiments were performed at Peking University Health Science Center and were approved by the Ethics Committee for Animal Studies at Peking University. All animals were treated according to the standards prescribed by the “guidelines for the welfare and use of animals in cancer research” (16).
Statistical analyses
Statistical analyses were performed on data collected from at least 3 independent experiments using Prism 7.0 software (GraphPad, San Diego, CA, USA). Comparisons among >2 groups were performed by analysis of variance (ANOVA), with a Tukey’s test. Student’s t-test performed comparisons between 2 groups of unpaired data. Data are shown in dot plots or histograms as mean ± SEM, and P<0.05 was considered statistically significant. Survival curves were plotted with the Kaplan-Meier method and compared using a 2-sided log-rank test.
Results
BMPR2 was overexpressed in PDAC, and inhibition of BMPR2 suppressed proliferation of PC cells
We performed IHC to measure the expression of BMPR2 in PC and normal pancreas tissues. As shown in Figure 1A,B, the BMPR2 was significantly over-expressed in PDAC tissues compared with normal pancreas tissues. By analyzing the expression level of BMPR2 and the clinicopathological characteristics (Table 2), the results showed that high expression of BMPR2 was correlated with tumor node metastasis (TNM) stage and tumor size, which indicated that BMPR2 might involve in the growth of PC. The correlation analysis of the expression level of BMPR2 in PDAC tissues and the overall participant survival showed that patients with higher expression of BMPR2 had lower overall survival rate, which indicated the prognostic role of BMPR2 in PC patients (Figure 1C). We then detected the expression of BMPR2 in multiple frequently-used PC cell lines, including BxPC-3, SW1990, T3M4, AsPC-1, PANC-1, MiaPaCa-2, and Capan-1 and a normal pancreatic ductal epithelial cell line HPNE. The results showed a relatively higher BMPR2 expression in most PC cell lines (Figure 1D). Thus, cell lines MiaPaCa-2 and PANC-1 were selected for further experiments for their highest expression of BMPR2.
Figure 1.

Inhibition of BMPR2 suppressed proliferation of pancreatic cancer cells. (A) Representative images of IHC results. (B) Analysis of BMPR2 expression in normal pancreas and PDAC tissues. (C) Correlation of BMPR2 expression level and overall survival of PDAC patients. (D) Expression level and quantitative analysis of BMPR2 in PDAC cell lines and a normal ductal epithelial cell line, HPNE. (E) Verification of two sequences of shRNA in down-regulating BMPR2 in two PDAC cell lines. (F) Proliferation of MiaPaCa-2 and PANC-1 cells after down-regulation of BMPR2. (G,H) Colony formation assay and quantitative analysis of MiaPaCa-2 and PANC-1 cells after down-regulation of BMPR2. Scale bar: 1 cm. (I) Cell cycle distribution of MiaPaCa-2 and PANC-1 cells after down-regulation of BMPR2. (J) G2/M related proteins detection after knockdown of BMPR2. IHC, immunohistochemistry; BMPR2, bone morphogenic protein receptor 2; PDAC, pancreatic ductal adenocarcinoma. *, P<0.05; **, P<0.01; ***, P<0.001.
Table 2. Relation between the expression level of BMPR2 and patient clinical features.
| Clinical features | Classification | High BMPR2 expression (%) | Low BMPR2 expression (%) | P value |
|---|---|---|---|---|
| Gender | Male | 17 (26.15) | 16 (24.62) | 0.531 |
| Female | 14 (21.53) | 18 (27.69) | ||
| Age | <60 | 13 (20.0) | 21 (32.31) | 0.279 |
| ≥60 | 16 (24.62) | 15 (23.08) | ||
| Tumor grade | Poor | 17 (26.15) | 9 (13.85) | 0.088 |
| Moderate | 11 (16.92) | 6 (9.23) | ||
| Well | 8 (12.31) | 14 (21.54) | ||
| TNM stage | I-II | 8 (12.31) | 19 (29.23) | <0.0001 |
| III-IV | 28 (43.08) | 10 (15.38) | ||
| Vascular invasion | Yes | 15 (23.08) | 13 (20.0) | 0.635 |
| No | 22 (33.85) | 15 (23.08) | ||
| Tumor size | <3 cm | 11 (16.92) | 19 (29.23) | <0.0001 |
| ≥3 cm | 29 (44.62) | 6 (9.23) | ||
| Nerve invasion | Yes | 13 (20.0) | 18 (27.69) | 0.761 |
| No | 13 (20.0) | 21 (32.31) | ||
| CA19-9 | <39 | 18 (27.69) | 11 (16.92) | 0.063 |
| ≥39 | 14 (21.54) | 22 (33.85) | ||
| Tissue type | pancreatic cancer tissue | 47 (72.31) | 18 (27.69) | <0.0001 |
| Adjacent normal tissue | 9 (13.85) | 56 (86.15) |
TNM, tumor node metastasis; BMPR2, bone morphogenetic protein receptor 2.
To evaluate the role of BMPR2 in PC cells, we transfected the MiaPaCa-2 and PANC-1 cells with 2 sequences of shRNA to down-regulate the expression of BMPR2. As shown in Figure 1E, the 2 sequences of shRNA significantly down-regulated the expression of BMPR2. We then measured the proliferation of these 2 cell lines after inhibition of BMPR2, and the CCK-8 results showed that the proliferation was significantly suppressed after down-regulation of BMPR2 (Figure 1F). Similarly, the colony formation capacities of MiaPaCa-2 and PANC-1 were inhibited as well (Figure 1G,H). By flow cytometry, we analyzed the change of cell cycle distribution after inhibition of BMPR2 in MiaPaCa-2 and PANC-1 cells, and the results showed that the cells were arrested at the G2/M stage after inhibition of BMPR2 (Figure 1I). We further detected the expression of 2 cG2/M stage-related proteins Cyclin B1 and CDK1, and the results indicated that Cyclin B1 and CDK1 were inhibited significantly after knockdown of BMPR2 (Figure 1J). These results confirmed that BMPR2 plays an important role in promoting PC growth.
GRB2 was up-regulated in PDAC and was a potential target regulated by BMPR2
To understand the mechanism of BMPR2 in the proliferation of PC cells, we performed a quantitative proteomic analysis to compare the proteomic change in MiaPaCa-2 cells. As shown in Figure 2A,B, the inhibition of BMPR2 led to significant protein expression variation in MiaPaCa-2 cells. Further protein-protein interaction (PPI) network analysis showed that GRB2 was a potential target regulated by BMPR2 (Figure 2C). We then analyzed the expression of GRB2 in PC and paired normal pancreas tissues in mRNA and protein levels. The results showed that the GRB2 was significantly over-expressed in PDAC tissues in mRNA level measured by quantitative PCR (qPCR) (Figure 2D) as well as protein level measured by western blot (Figure 2E,F). The Kaplan-Meier survival analysis was performed according to the expression level of GRB2. As shown in Figure 2G, a higher expression level of GRB2 had a shorter overall survival rate. We analyzed the correlation between BMPR2 and GRB2 using the data from the Center for Applied Genomics (TCAG) database. The results showed that the expression level of BMPR2 and GRB2 was positively correlated in PDAC (Figure 2H). We then measured the expression of GRB2 in PC cells after the down-regulation of BMPR2. The results showed that GRB2 was significantly down-regulated after BMPR2 inhibition in MiaPaCa-2 and PANC-1 cells (Figure 2I,J). Therefore, as a signaling transduction membrane protein, BMPR2 may exert its proliferation-promoting role by regulating GRB2.
Figure 2.

GRB2 was regulated by BMPR2 in PDAC. (A) Heat map of protein spectrum analysis. (B) Volcano diagram analysis of protein spectrum analysis. (C) PPI network analysis after down-regulation of BMPR2 in MiaPazCa-2 cells. (D) Expression of GRB2 mRNA in PDAC tissues (n=35). (E) Representative western blot analysis of expression of GRB2 in normal pancreas and PDAC tissues. (F) Analysis of expression of GRB2 in normal pancreas and PDAC tissues (n=35). (G) Overall survival rates of pancreatic cancer patients with low or high expression of GRB2. (H) Correlation of BMPR2 and GRB2 in PDAC tissues (data from TCGA database). (I,J) Expression and quantitative analysis of GRB2 after inhibition of BMPR2 in MiaPaCa-2 and PANC-1 cells. ***, P<0.001. PPI, Protein-protein interaction; BMPR2, bone morphogenic protein receptor 2; PDAC, pancreatic ductal adenocarcinoma
GRB2 promoted proliferation of PC cells and reversed BMPR2-inhibition induced proliferation suppression
The above results indicated that GRB2 as a potential downstream target of BMPR2. We, therefore, analyzed the role of GRB2 in the proliferation of PC cells. The CCK-8 results showed that the down-regulation of GRB2 inhibited the growth of MiaPaCa-2 and PANC-1 cells (Figure 3A,B). The colony formation assays showed that the down-regulation of GRB2 inhibited the colony formation capabilities of MiaPaCa-2 and PANC-1 cells (Figure 3C,D,E). Furthermore, we detected the cell cycle distribution changes after the down-regulation of GRB2. The flow cytometry results showed that MiaPaCa-2 and PANC-1 cells were significantly arrested in the G2/M stage (Figure 3F). We further detected the expression of Cyclin B1 and CDK1 by western blot analysis, which showed that these 2 G2/M stage-related proteins were inhibited significantly (Figure 3G). These results indicated the oncogenesis role of GRB2 in PC.
Figure 3.
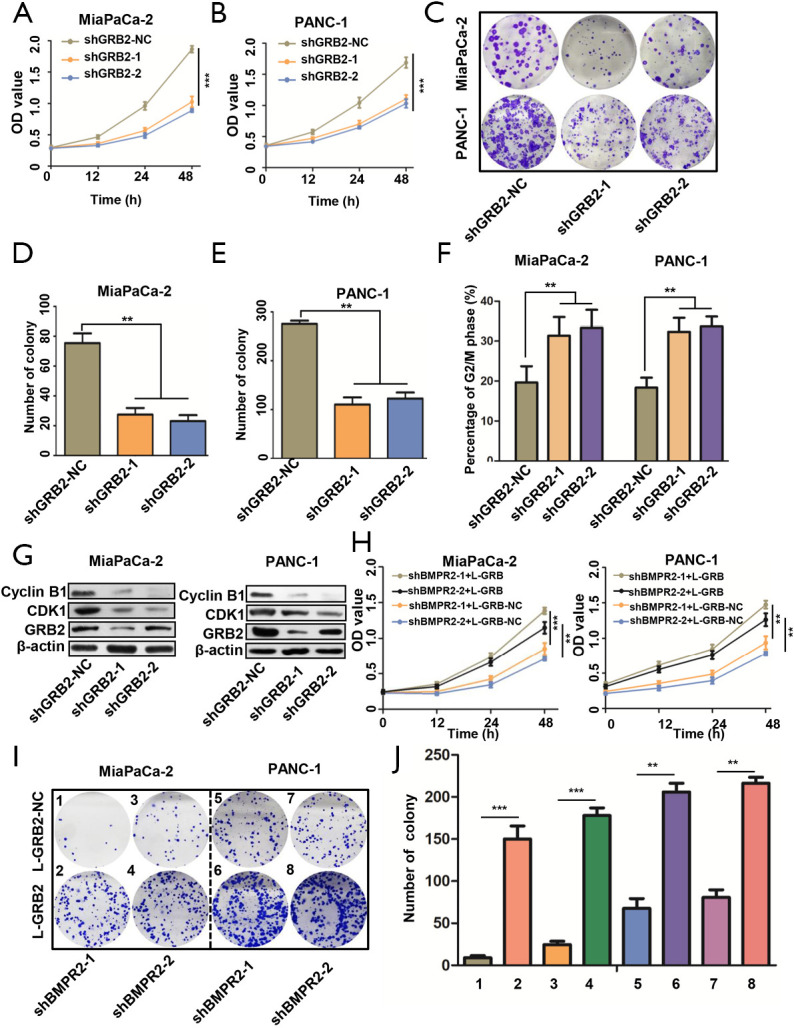
GRB2 reversed down-regulation of BMPR2 induced proliferation suppression. (A,B) Proliferation of MiaPaCa-2 and PANC-1 cells after down-regulation of GRB2. (C,D,E) Colony formation assay and quantitative analysis of MiaPaCa-2 and PANC-1 cells after down-regulation of GRB2. (F) Cell cycle distribution of MiaPaCa-2 and PANC-1 cells after down-regulation of GRB2. (G) G2/M related proteins detection after knockdown of GRB2. (H) GRB2 reversed proliferation of MiaPaCa-2 and PANC-1 cells inhibited by down-regulation of BMPR2. (I,J) GRB2 reversed colony formation capacity of MiaPaCa-2 and PANC-1 cells inhibited by down-regulation of BMPR2. **, P<0.01; ***, P<0.001. BMPR2, bone morphogenic protein receptor 2.
We then performed rescue experiments in CCK-8 and colony formation assays by up-regulation of GRB2 in BMPR2 down-regulated PC cells. The CCK-8 results showed that the up-regulation of GRB2 reversed the proliferation-suppressing induced by inhibition of BMPR2 (Figure 3H). The colony formation assay showed similar results (Figure 3I,J). These results demonstrated that BMPR2 exerts its proliferation-promoting role in PC by regulating GRB2.
GRB2 regulated the PI3K/AKT signaling pathway in PC
Based on the proteomics analysis, we further analyzed the signaling pathway change in BMRP2 down-regulated MiaPaCa-2 cells. As shown in Figure 4A, the most obvious varied signaling pathway was the PI3K/AKT signaling pathway. The G2/M checkpoint signaling pathway also varied significantly, which was coincident with the above cell cycle distribution results. We then performed enrichment analysis, which further confirmed a significant change in the PI3K/AKT signaling pathway (Figure 4B).
Figure 4.

Down-regulation of BMRP2 induced suppression of PI3K/AKT signaling pathway. (A) Bubble chart of protein spectrum analysis. (B) Enrichment plot analysis of PI3K/AKT signaling pathway. (C) Level of p-PI3K and p-AKT in pancreatic cancer cells after down-regulation of BMPR2. (D) Level of p-PI3K and p-AKT in pancreatic cancer cells after down-regulation of GRB2.
To verify these bioinformatics analysis results, we down-regulated BMPR2 in MiaPaCa-2 and PANC-1 cells and detected the level of PI3K, p-PI3K, AKT, and p-AKT. The western blot analysis indicated that the level of p-PI3K and p-AKT were significantly down-regulated in MiaPaCa-2 and PANC-1 cells (Figure 4C). We then detected the level of PI3K, p-PI3K, AKT, and p-AKT after down-regulating GRB2; the results showed that p-PI3K, p-AKT308, and p-AKT474 were inhibited after knockdown of BMPR2 in both PC cell lines. To further verify the regulation of GRB2 on PI3K/AKT, we detected the level of PI3K, p-PI3K, AKT, and p-AKT, and the results indicated that p-PI3K, p-AKT308, and p-AKT474 were inhibited significantly (Figure 4D). These results suggested that BMPR2 regulates the phosphorylation of the PI3K/AKT signaling pathway and promotes the proliferation of PC cells by regulating the expression of GRB2.
LDN193189 inhibited proliferation and induced G2/M arrested in PC cells
After confirming the tumorigenic roles of BMPR2 in PC, we applied the BMPR2 inhibitor LDN193189 to analyze the effect of pharmacological inhibition of BMPR2 on PC cells. As shown in Figure 5A,B, the LDN193189 significantly inhibited proliferation of MiaPaCa-2 and PANC-1 cells, which was concentration-dependent. We further analyzed the cell cycle distribution after treatment with LDN193189 in MiaPaCa-2 and PANC-1 cells, and the results showed that LDN193189 induced G2/M phase arrest in PC cells, which was coincident with the above results. The G2/M phase arrest induced by LDN193189 was concentration-dependent (Figure 5C,D,E,F). We also performed western blot analysis to detect the inhibition of LDN193189 on expression BMPR2 and GRB2, which revealed the concentration-dependent inhibition on BMPR2 and GRB2 in MiaPaCa-2 cells (Figure 5G,H). These results demonstrated that LDN193189 serves as an effective anti-proliferation drug in PC cells by inhibiting BMPR2.
Figure 5.
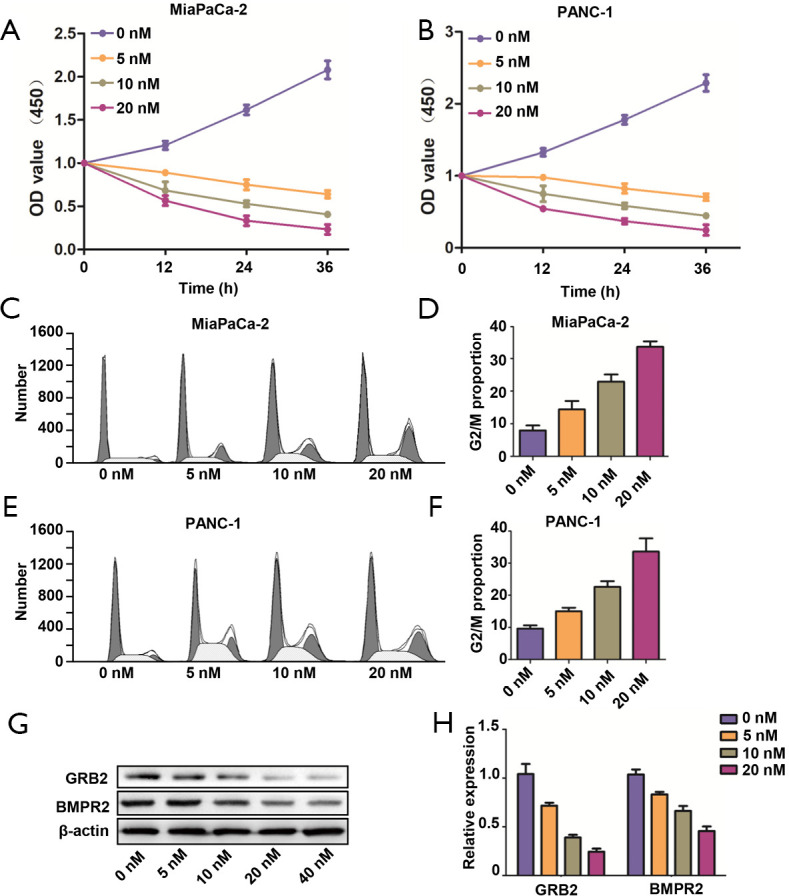
LDNA193189 inhibited proliferation of PDAC cells. (A,B) Proliferation MiaPaCa-2 and PANC-1 cells in different concentration of LDN193189. (C,D) Cell cycle distribution and G2/M proportion analysis of MiaPaCa-2 cells after treatment with different concentrations of LDN193189. (E,F) Cell cycle distribution and G2/M proportion analysis of PANC-1 cells after treatment with different concentrations of LDN193189. (G,H) Expression of BMPR2 and GRB2 in MiaPaCa-2 cells. PDAC, pancreatic ductal adenocarcinoma.
LDN193189 inhibited PC growth by suppressing the BMPR2/GRB2/PI3K/AKT axis
To confirm the inhibition of LDN193189 on PC in vivo, we used 2 persuasive PC models in the animal experiments. After the tumor initiation, in the orthotopic PC models, LDN193189 or the control group saline was injected intraperitoneally once every day. During the treatment, the fluorescence intensities of each were monitored by in vivo imaging system, and the results showed that tumor growth was significantly inhibited by LDN193189 (Figure 6A,B). At the end of the treatment, the orthotopic tumors were excised photographed (Figure 6C). We detected the down-regulated expression of BMPR2, GRB2, and Ki67 in the tumors (Figure 6D). Additionally, using western blot analysis, we showed that BMPR2 and GRB2 were significantly down-regulated in the xenograft tumors, and the level of p-PI3K and p-AKT was obviously decreased (Figure 6E). These results indicated that inhibition of BMPR2 suppresses PC growth by inhibiting the GRB2/PI3K/AKT pathway in vivo.
Figure 6.

LDN193189 inhibited PDAC growth in vivo. (A) Representative bioluminescent images of orthotopic pancreatic cancer models during the treatment. (B) Bioluminescent densities during the treatment. (C) Tumor images at the end of the treatment, n=5. (D) Representative IHC images showing the expression of BMPR2, GRB2 and Ki67 of tumors. (E) Western blot analysis of the orthotopic tumors. (F) Tumor growth curves of PDX during the treatment. (G) Image of tumors excised from PDX mouse models at the end of the treatment, n=5. (H) Tumor weight analysis of PDX models. (I) Western blot analysis of the PDX tumors. (J,K) Tumor growth after knockdown of BMPR2 by shRNA (n=5). Scale bar represents 1 cm. The data are shown as the mean ± SD. ***, P<0.001. IHC, immunohistochemistry; BMPR2, bone morphogenic protein receptor 2; PDX, patient-derived xenograft; PDAC, pancreatic ductal adenocarcinoma.
To further confirm these findings, we repeated this experiment in a PDX model. The tumor volume was measured during treatment (Figure 6F). At the end of treatment, the tumors were excised and weighted. The results showed that LDN193189 inhibited PDX growth significantly (Figure 6G,H). We also detected the level of BMPR2, GRB2, p-PI3K, and p-AKT by western blot analysis in these tumors. Like the orthotopic PC models, the above proteins’ level was reduced (Figure 6I). As we know, LDN193189 is not a specific inhibitor of BMPR2. To verify that knockdown of BMPR2 directly leads to inhibition of PC growth, we downregulated BMPR2 using shRNA in PANC-1 cells and performed in vivo experiments. The results showed that tumors were much smaller in the shBMPR2 group than the control group (Figure 6J,K). Therefore, we demonstrated that down-regulation of BMPR2 in PC inhibits tumor growth by suppressing the GRB2/PI3K/AKT axis.
Discussion
As a BMPR family member, BMPR2 plays a crucial role in regulating the BMP signaling pathway, which functions as an important regulator in the physiological process and tumor progression (4,7,17-19). Binding BMP ligands to BMPRs phosphorylates the type 1 receptor by activating type 2 receptors, resulting in phosphorylation of intracellular signaling proteins (20,21). The most studied BMP-regulated intracellular proteins are SMADs. Activated SMADs complexes translocate to the nucleus and promote transcription of related genes (22). Although BMPR2 is associated with the development of multiple cancers, there has been no relevant research on BMPR2 and its regulatory mechanism in PDAC.
In our current study, we first performed a tissue array to show the expression of BMPR2 in PDAC tissues was higher than that in normal pancreas tissues, and participants with higher BMPR2 expression had worse overall survival. We then revealed that downregulating BMPR2 suppressed PDAC cell proliferation and induced G2/M phase arrest. By protein mass spectrometry technique, bioinformatics analysis, and in vitro cell experiments, we identified that BMPR2 promoted PDAC cell proliferation by regulating the GRB2/PI3K/AKT signaling pathway. By using the BMPR2 inhibitor LDN193189, we confirmed that the inhibition of BMPR2 significantly suppressed PDAC growth in orthotopic PC and PDX models.
The GRB2 protein is part of the GRB family of docking proteins that act as platforms for signaling transduction components (23-26). It was initially discovered as the connecting molecule between the epidermal growth factor receptor (EGFR) and the Ras-mitogen-activated protein kinase (MAPK) pathway (27,28). The GRB2-mediated receptor dimerization inside cells leads to transphosphorylation of the kinase domains of target proteins (29). Recently, GRB2 is involved in the PI3K/AKT signaling pathway in colorectal cancer (30). However, no specific mechanism of GRB2 regulating the PI3K/AKT signaling pathway was found in this study. Our work revealed that the level of the phosphorylation level of PI3K was significantly decreased. Therefore, GRB2 may involve in the phosphorylation process of PI3K and then affect downstream molecules. Although our results suggested that GRB2 is a mediator in transducing signaling from BMPR2 to the PI3K/AKT pathway, the mechanism by which BMPR2 regulates GRB2 requires further study. In our work, we used LDN193189 to treat 2 representative PC models, which provides evidence for applying the BMPR2 inhibitor in PDAC treatment.
Supplementary
The article’s supplementary files as
Acknowledgments
We thank Jing Zhu from the Department of General Surgery, Peking University First Hospital, for her management of participant specimens.
Funding: This work was supported in part by grants from The National Natural Science Foundation of China (81672353, 81871954).
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. The sample collection was conducted in accordance with the Declaration of Helsinki (as revised in 2013), which was approved by the Ethics Committee of Peking University First Hospital (No. 201933) and informed consent was taken from all the patients. Animal experiments were performed under a project license (No. J201980) granted by the Animal Ethics Committee of Peking University First Hospital, in compliance with national guidelines for the care and use of animals.
Reporting Checklist: The authors have completed the ARRIVE reporting checklist. Available at http://dx.doi.org/10.21037/atm-20-2194
Data Sharing Statement: Available at http://dx.doi.org/10.21037/atm-20-2194
Peer Review File: Available at http://dx.doi.org/10.21037/atm-20-2194
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at http://dx.doi.org/10.21037/atm-20-2194). The authors have no conflicts of interest to declare.
(English Language Editor: J. Jones and J. Chapnick)
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin 2020;70:7-30. 10.3322/caac.21590 [DOI] [PubMed] [Google Scholar]
- 2.Rahib L, Smith BD, Aizenberg R, et al. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 2014;74:2913-21. 10.1158/0008-5472.CAN-14-0155 [DOI] [PubMed] [Google Scholar]
- 3.Feng XH, Derynck R. Specificity and versatility in tgf-beta signaling through Smads. Annu Rev Cell Dev Biol 2005;21:659-93. 10.1146/annurev.cellbio.21.022404.142018 [DOI] [PubMed] [Google Scholar]
- 4.Jiao G, Guo W, Ren T, et al. BMPR2 inhibition induced apoptosis and autophagy via destabilization of XIAP in human chondrosarcoma cells. Cell Death Dis 2014;5:e1571. 10.1038/cddis.2014.540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beppu H, Mwizerwa ON, Beppu Y, et al. Stromal inactivation of BMPRII leads to colorectal epithelial overgrowth and polyp formation. Oncogene 2008;27:1063-70. 10.1038/sj.onc.1210720 [DOI] [PubMed] [Google Scholar]
- 6.Groeneveld EH, Burger EH. Bone morphogenetic proteins in human bone regeneration. Eur J Endocrinol 2000;142:9-21. 10.1530/eje.0.1420009 [DOI] [PubMed] [Google Scholar]
- 7.Pouliot F, Blais A, Labrie C. Overexpression of a dominant negative type II bone morphogenetic protein receptor inhibits the growth of human breast cancer cells. Cancer Res 2003;63:277-81. [PubMed] [Google Scholar]
- 8.Giubellino A, Burke TR, Jr, Bottaro DP. Grb2 signaling in cell motility and cancer. Expert Opin Ther Targets 2008;12:1021-33. 10.1517/14728222.12.8.1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mitra P, Kalailingam P, Tan HB, et al. Overexpression of GRB2 Enhances Epithelial to Mesenchymal Transition of A549 Cells by Upregulating SNAIL Expression. Cells 2018;7:97. 10.3390/cells7080097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Al-Husseinawi E, Bui MM, Ahmed AA. Grb2-associated binding protein-1 as a biomarker in bone and soft tissue sarcomas. Pathology 2019;51:610-4. 10.1016/j.pathol.2019.05.003 [DOI] [PubMed] [Google Scholar]
- 11.Lv J, Zhang S, Wu H, et al. Deubiquitinase PSMD14 enhances hepatocellular carcinoma growth and metastasis by stabilizing GRB2. Cancer Lett 2020;469:22-34. 10.1016/j.canlet.2019.10.025 [DOI] [PubMed] [Google Scholar]
- 12.Xu ZH, Yao TZ, Liu W. miR-378a-3p sensitizes ovarian cancer cells to cisplatin through targeting MAPK1/GRB2. Biomed Pharmacother 2018;107:1410-7. 10.1016/j.biopha.2018.08.132 [DOI] [PubMed] [Google Scholar]
- 13.Wang Y, Guo H, Zhang D, et al. Overexpression of SOX18 correlates with accelerated cell growth and poor prognosis in human pancreatic ductal adenocarcinoma. Biochem Biophys Res Commun 2016;479:510-6. 10.1016/j.bbrc.2016.09.099 [DOI] [PubMed] [Google Scholar]
- 14.Chen Q, Wei T, Wang J, et al. Patient-derived xenograft model engraftment predicts poor prognosis after surgery in patients with pancreatic cancer. Pancreatology 2020;20:485-92. 10.1016/j.pan.2020.02.008 [DOI] [PubMed] [Google Scholar]
- 15.Jimeno A, Feldmann G, Suarez-Gauthier A, et al. A direct pancreatic cancer xenograft model as a platform for cancer stem cell therapeutic development. Mol Cancer Ther 2009;8:310-4. 10.1158/1535-7163.MCT-08-0924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Workman P, Aboagye EO, Balkwill F, et al. Guidelines for the welfare and use of animals in cancer research. Br J Cancer 2010;102:1555-77. 10.1038/sj.bjc.6605642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim IY, Lee DH, Lee DK, et al. Restoration of bone morphogenetic protein receptor type II expression leads to a decreased rate of tumor growth in bladder transitional cell carcinoma cell line TSU-Pr1. Cancer Res 2004;64:7355-60. 10.1158/0008-5472.CAN-04-0154 [DOI] [PubMed] [Google Scholar]
- 18.Owens P, Pickup MW, Novitskiy SV, et al. Disruption of bone morphogenetic protein receptor 2 (BMPR2) in mammary tumors promotes metastases through cell autonomous and paracrine mediators. Proc Natl Acad Sci U S A 2012;109:2814-9. 10.1073/pnas.1101139108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Voorneveld PW, Kodach LL, Jacobs RJ, et al. Loss of SMAD4 alters BMP signaling to promote colorectal cancer cell metastasis via activation of Rho and ROCK. Gastroenterology 2014;147:196-208.e13. 10.1053/j.gastro.2014.03.052 [DOI] [PubMed] [Google Scholar]
- 20.Miyazawa K, Shinozaki M, Hara T, et al. Two major Smad pathways in TGF-beta superfamily signalling. Genes Cells 2002;7:1191-204. 10.1046/j.1365-2443.2002.00599.x [DOI] [PubMed] [Google Scholar]
- 21.Wrana JL, Attisano L, Wieser R, et al. Mechanism of activation of the TGF-beta receptor. Nature 1994;370:341-7. 10.1038/370341a0 [DOI] [PubMed] [Google Scholar]
- 22.Lavery K, Swain P, Falb D, et al. BMP-2/4 and BMP-6/7 differentially utilize cell surface receptors to induce osteoblastic differentiation of human bone marrow-derived mesenchymal stem cells. J Biol Chem 2008;283:20948-58. 10.1074/jbc.M800850200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gavert N, Ben-Ze'ev A. Epithelial-mesenchymal transition and the invasive potential of tumors. Trends Mol Med 2008;14:199-209. 10.1016/j.molmed.2008.03.004 [DOI] [PubMed] [Google Scholar]
- 24.Papageorgis P. TGFbeta Signaling in Tumor Initiation, Epithelial-to-Mesenchymal Transition, and Metastasis. J Oncol 2015;2015:587193. 10.1155/2015/587193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thiery JP, Acloque H, Huang RY, et al. Epithelial-mesenchymal transitions in development and disease. Cell 2009;139:871-90. 10.1016/j.cell.2009.11.007 [DOI] [PubMed] [Google Scholar]
- 26.Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol 2006;7:131-42. 10.1038/nrm1835 [DOI] [PubMed] [Google Scholar]
- 27.Buday L, Downward J. Epidermal growth factor regulates p21ras through the formation of a complex of receptor, Grb2 adapter protein, and Sos nucleotide exchange factor. Cell 1993;73:611-20. 10.1016/0092-8674(93)90146-H [DOI] [PubMed] [Google Scholar]
- 28.Lowenstein EJ, Daly RJ, Batzer AG, et al. The SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell 1992;70:431-42. 10.1016/0092-8674(92)90167-B [DOI] [PubMed] [Google Scholar]
- 29.Ijaz M, Wang F, Shahbaz M, et al. The Role of Grb2 in Cancer and Peptides as Grb2 Antagonists. Protein Pept Lett 2018;24:1084-95. 10.2174/0929866525666171123213148 [DOI] [PubMed] [Google Scholar]
- 30.Ding C, Tang W, Wu H, et al. The PEAK1-PPP1R12B axis inhibits tumor growth and metastasis by regulating Grb2/PI3K/Akt signalling in colorectal cancer. Cancer Lett 2019;442:383-95. 10.1016/j.canlet.2018.11.014 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The article’s supplementary files as