

SUMMARY
Development of blood-forming (hemogenic) endothelial cells that give rise to hematopoietic stem and progenitor cells (HSPCs) is critical during embryogenesis to generate the embryonic and postnatal hematopoietic system. We previously demonstrated that the specification of murine hemogenic endothelial cells is promoted by retinoic acid (RA) signaling and requires downstream endothelial cell cycle control. Whether this mechanism is conserved in human hemogenic endothelial cell specification is unknown. Here, we present a protocol to derive primordial endothelial cells from human embryonic stem cells and promote their specification toward hemogenic endothelial cells. Furthermore, we demonstrate that RA treatment significantly increases human hemogenic endothelial cell specification. That is, RA promotes endothelial cell cycle arrest to enable RA-induced instructive signals to upregulate the genes needed for hematopoietic transition. These insights provide guidance for the ex vivo generation of autologous human hemogenic endothelial cells that are needed to produce human HSPCs for regenerative medicine applications.
In Brief
Retinoic acid signaling and cell-cycle control are known to promote mouse hemogenic endothelial cell specification. Qiu et al. present a protocol to derive endothelial cells from human stem cells and promote their hemogenic specification. These insights provide guidance for generating human hemogenic endothelial cells for regenerative medicine applications.
Graphical Abstract
INTRODUCTION
Mammalian hematopoiesis occurs in two phases during embryogenesis: primitive and definitive hematopoiesis. In the mouse, definitive hematopoiesis takes place mainly within the yolk sac and the aorta-gonad-mesonephros (AGM) region (de Bruijn et al., 2000; Medvinsky and Dzierzak, 1996; Müller et al., 1994), and is marked by the generation of multipotent hematopoietic stem and progenitor cells (HSPCs) from specialized blood-forming (or hemogenic) endothelial cells. Transplantation experiments have shown that cells isolated from the AGM region as early as embryonic day (E) 10.5 can confer long-term hematopoietic repopulation in lethally irradiated recipient mice (de Bruijn et al., 2000; Medvinsky and Dzierzak, 1996; Müller et al., 1994); these cells are referred to as adult repopulating HSPCs. Thus, the HSPCs produced by hemogenic endothelial cells are the foundation of the adult hematopoietic hierarchy, providing the continuous production of blood cells throughout life. Therefore, understanding the mechanisms by which human hemogenic endothelial cells are specified is a critical first step toward generating human HSPCs for the treatment of hematopoietic disorders.
Several regulators have been previously implicated in the specification of murine hemogenic endothelial cells in the yolk sac and AGM, including retinoic acid (RA), c-Kit, Notch, and p27 (Bohnsack et al., 2004; Chanda et al., 2013; Goldie et al., 2008; Lai et al., 2003; Marcelo et al., 2013b; Niederreither et al., 1999). It is believed that RA promotes a signaling cascade, mediating the downstream Kit-Notch-p27 signaling axis (reviewed in Marcelo et al., 2013a). p27 functions as a cyclin-dependent kinase (CDK) inhibitor that restricts cell cycle progression at G1 phase (Hitomi et al., 2006), and we (Marcelo et al. (2013b) have previously shown the necessity of p27-mediated cell cycle arrest in the process of murine hemogenic endothelial cell specification. In addition, a number of studies have demonstrated the link between the cell cycle state of stem cells and cell fate decisions (Gonzales et al., 2015; Pauklin and Vallier, 2013; Singh et al., 2013) and suggest that cells preferably make cell fate decisions during G1 phase (Dekker, 2014; Tumbar and Belmont, 2001). Furthermore, there are molecular differences between early G1 and late G1 that provide distinct windows of opportunity for extrinsic signals to promote the differentiation of distinct phenotypes (Pauklin and Vallier, 2013). Whether hemogenic specification of endothelial cells occurs specifically in G1 phase and whether early versus late G1 state is more permissive for this process remain to be determined, as does the potential role of RA in the regulation of human hemogenic endothelial cell specification. We investigate these unanswered questions in this study.
Elucidating the mechanism(s) by which endothelial cells acquire blood-forming potential is a critical first step toward understanding the establishment of the hematopoietic system. Such studies provide insights into the production of cells that can ultimately generate human HSPCs ex vivo from potentially autologous human pluripotent stem cells, which can be used for the treatment of hematopoietic disorders. Toward this goal, we established an in vitro model of human hemogenic endothelial cell development that is feeder-free, requires few growth factors, and can be performed in 1 week. Thus, unlike other systems using feeder cells or embryoid bodies, our system minimizes potential confounding effects introduced by mouse cells or the concurrent development of other organ systems. This highly controlled culture environment allows us to dissect the regulation of human endothelial cell development and their hemogenic specification. Using this system, we discovered the essential roles of RA and the cell cycle state in the process of human hemogenic endothelial cell specification. We believe that this system will serve as a critical tool for further studying human hemogenic endothelial cell development, which is the first step toward optimizing the production of transplantable blood cells for therapeutic purposes.
RESULTS
Simple In Vitro System to Derive Human Hemogenic Endothelial Cells from Human Embryonic Stem Cells (hESCs)
A number of protocols were developed to differentiate hESCs toward hematopoietic cells (Krassowska et al., 2006; Lengerke et al., 2009; Niwa et al., 2011); however, these protocols require either usage of feeder cells, formation of embryoid bodies, and/or addition of numerous growth factors, and can take up to 3 weeks. Sriram et al. (2015) previously established a feeder-free protocol to differentiate hESC into endothelial cells. We discovered that treating hESC-derived endothelial cells with RA can promote specification toward hemogenic endothelial cells that give rise to differentiated hematopoietic lineages (Figure 1A). This protocol requires no feeder cells or formation of embryoid bodies. In addition, it requires only 4 growth factors and takes ~1 week from start to finish.
Figure 1. Simple In Vitro System to Derive Human Hemogenic Endothelial Cells from Human Embryonic Stem Cells (hESCs).

(A) Schematic representation of differentiation protocol, partially adapted from Sriram et al. (2015).
(B) qPCR analysis of markers associated with pluripotency (SOX2), primitive streak/early mesoderm (TBXT), mesoderm (HAND1), endothelium (CDH5), arterial endothelium (EFNB2), venous endothelium (EPHB4), and hematopoiesis (RUNX1); n = 3 for all groups. Student’s t test: p < 0.0001 (SOX2), p = 0.0008 (TBXT), p = 0.0253 (HAND1), p = 0.0028 (CDH5), p = 0.0018 (EFNB2), p = 0.0049 (EPHB4), p = 0.0029 (RUNX1).
(C) Flow cytometry result showed generation of endothelial cells (CD31+CD45−) and blood cells (CD45+) on day 8 (D8).
(D) Giemsa staining revealed generation of all blood cell lineages. Original magnification, 40×. Scale bar, 25 μm.
(E) Flow cytometry result showed generation of multi-lineage blood cells.
Data are represented as means ± SDs.
To validate the protocol, we performed qPCR analysis of markers associated with cell fate transitions (Figure 1B). We found that on day 1 (D1) of differentiation, the level of pluripotency marker SOX2 significantly decreased, accompanied by the upregulation of primitive streak marker TBXT. Primitive streak cells were then differentiated into mesoderm cells marked by elevated HAND1 expression on D3. Endothelial cell marker CDH5 peaked on D5, with elevated expression of both arterial endothelial cell marker EFNB2 and venous endothelial cell marker EPHB4. Upon treatment with 0.5 μM RA, we observed gradually increased expression of RUNX1 mRNA, which marks the transition toward hematopoietic cell fates.
We then performed flow cytometry on D8 to isolate adherent endothelial cells (CD31+CD45−) and blood cells (CD45+), and confirmed their phenotypes on the protein level (Figure 1C). We also observed 40%–45% live cells in the culture supernatant (Figure S2A); using Giemsa staining to better evaluate their phenotype, we identified blood cells of all lineages, including erythrocytes, lymphocytes, macrophages, monocytes, megakaryocytes, neutrophils, and basophils (Figure 1D). Lastly, we confirmed the generation of blood cells of multiple lineages using flow cytometry, using markers of lymphoid cells (CD20 and CD3), myeloid cells (CD14, CD66b, CD41, and CD235ab), and leukocytes (CD11b and CD45) (Figures 1E, S1, and S2). These data demonstrate that our protocol promotes the efficient differentiation of hESCs into endothelial cells and hematopoietic cells.
Characterization of the Phenotype of Human Hemogenic Endothelial Cells on Population and Single-Cell Levels
We next sought to identify the subset of endothelial cells that presumably generated the hematopoietic cells, and define their phenotype. The phenotype of murine hemogenic endothelial cells has been previously defined as Flk-1+Kit+CD45−Side Population (SP) cells (Goldie et al., 2008; Marcelo et al., 2013b; Nadin et al., 2003; Wulf et al., 2003). Here, we used flow cytometric separation of subpopulations of cells and a methylcellulose-based blood-forming assay to define the phenotype of human hemogenic endothelial cells as CD31+KDR+KIT+CD34+CDH5−CD45− cells, as outlined in Figure 2A and described further below.
Figure 2. Characterization of the Phenotype of Human Hemogenic Endothelial Cells on a Population Level.

(A) Schematic representation of blood-forming assay.
(B) Representative photographs showing morphology of BFU-E erythroid colony, CFU-E erythroid colony, CFU-GM granulocyte-monocyte colony, and CFU-GEMM multipotent hematopoietic progenitor colony. Scale bars, 100 μm.
(C) Among non-HSPC (CD45−), hematopoietic potential is enriched within the CD31+ population; n = 3 for all groups. Student’s t test: p = 0.03. Solid bars represent positive groups. Checkered bars represent negative groups. Striped bars represent parental groups.
(D) Among CD31+CD45− cells, hematopoietic potential is enriched within the CDH5− and CD34− populations; n = 3 for all groups. Student’s t test: p = 0.005 (CDH5), p = 0.04 (CD34). Solid bars represent positive groups. Checkered bars represent negative groups. Striped bars represent parental groups.
(E) Among CD31+CDH5−CD45− cells, hematopoietic potential is enriched within the KIT+ and CD34+ populations; n = 3 for all groups. Student’s t test: p = 0.03 (KIT), p = 0.003 (CD34). Solid bars represent positive groups. Checkered bars represent negative groups. Striped bars represent parental groups.
(F) Among CD31+KIT+CD34+CDH5−CD45− cells, hematopoietic potential is enriched within the KDR+ population (~85% CFU-E, 2% BFU-E, 11% CFU-GM, 2% CFU-GEMM); n = 3 for all groups. Student’s t test: p = 0.03. Unless labeled otherwise, solid bars represent positive groups. Checkered bars represent negative groups. Striped bars represent parental groups.
Data are represented as means ± SDs.
Initially, we tested the ability of our whole-cell population to generate multiple hematopoietic cell colony types, including multilineage CFU-GEMM (colony-forming unit-granulocyte/erythrocyte/monocyte/megakaryocyte), CFU-GM (colony-forming unit-granulocyte/monocyte), BFU-E (burst-forming unit-erythroid) and CFU-E (colony-forming unit-erythroid) colonies (Figure 2B). To begin to characterize the phenotype of the blood-forming endothelial cells within this population, we excluded other cells with blood-forming potential, specifically CD45+ HSPCs, by isolating CD45− cells using fluorescence-activated cell sorting (FACS). We then further separated the CD45− cells into various subgroups, based on the expression of known endothelial- or hematopoietic-enriched cell surface proteins. Among the CD45− cells, hematopoietic potential was enriched within the CD31+ population (Figures 2C and S3A).
We then isolated CD31+CD45− cells, known to be an enriched population of endothelial cells, and similarly separated them into additional subgroups based on the expression of specific proteins enriched in endothelial or hematopoietic cells. Among the CD31+CD45− endothelial cells, hematopoietic potential was enriched within the CDH5− and CD34− populations (Figures 2D and S3B). Since CD34 marks definitive human hematopoietic progenitors but not primitive hematopoietic progenitors (Dege and Sturgeon, 2017), we further isolated CD31+CD45−CDH5− cells. Among them, hematopoietic potential (colony-forming ability) was enriched within the KIT+ and CD34+ populations (Figures 2E and S3C). Lastly, we isolated CD31+CD45−CDH5−KIT+CD34+ cells and found that hematopoietic potential was enriched within the kinase insert domain receptor-positive (KDR+) population (Figures 2F and S3D–S3H). Thus, the CD31+KDR+KIT+CD34+CDH5−CD45− population of hESC-derived cells is highly enriched for human hemogenic endothelial cells (Figure S3H).
Upon characterizing these cells on a population level, we further evaluated their hematopoietic potential on a clonal level (Figure 3A). Single human hemogenic endothelial cells (CD31+KDR+KIT+CD34+CDH5−CD45−) were FACS sorted into 96-well plates on D0 (Figure 3Bi). By D3, small endothelial cell colonies formed. These cells not only demonstrated the typical cobblestone morphology (Figure 3Bii) but also the functionality of acetelyated, fluorescently labeled low-density lipoprotein (DiI AcLDL) uptake (Figure 3Biii) and the expression of endothelial nitric oxide synthase (eNOS) (Data S1, Data S2, and Data S3), which are characteristics of endothelial cells. Although some mature blood cells, such as monocytes or macrophages, can also take up DiI AcLDL, they were FACS excluded from the population tested in this assay, which contained only CD45− cells. Thus, these data show that human hemogenic endothelial cells derived from hESCs are able to proliferate in culture and maintain the characteristics of endogenous vascular endothelial cells. By D14, endothelial cells could still be observed among the hematopoietic colonies (Figure 3Biv and v). We further confirmed the identity of various hematopoietic colonies that single hESC-derived hemogenic endothelial cells were capable of giving rise to using immunostaining. These colonies include BFU-E erythroid colonies marked by CD235ab expression (Figure 3Ci and ii), CFU-E erythroid colonies marked by CD235ab expression (Figure 3Ciii and iv), CFU-GM colonies marked by CD66b and CD14 expression (Figure 3D), and CFU-GEMM multipotent hematopoietic progenitor colonies marked by the expression of CD66b, CD235ab, CD14, and CD41 (Figure 3E). Overall, we demonstrated that these hESC-derived CD31+KDR+KIT+CD34+CDH5−CD45− human hemogenic endothelial cells exhibit multi-lineage hematopoietic potential on both population and single-cell levels.
Figure 3. Characterization of the Phenotype of Human Hemogenic Endothelial Cells on a Single-Cell Level.

(A) Schematic representation of single-cell blood-forming assay.
(B) Representative photographs showing single human hemogenic endothelial cell on D0 (i), hemogenic endothelial cell colony on D3 (ii), DiI AcLDL uptake of hemogenic endothelial cells on D3 (iii), and adherent hemogenic endothelial cells among blood cells on D14 (iv and v). Scale bars, 35 μm for (i), (ii), and (iii). Scale bars, 170 μm for (iv) and (v).
(C) Representative photographs showing BFU-E (i and ii) and CFU-E (iii and iv) on D8, marked by CD235ab. Scale bars, 35 μm.
(D) Representative photographs showing CFU-GM on D14, marked by CD66b and CD14. Scale bars, 35 μm.
(E) Representative photographs showing CFU-GEMM on D14, marked by CD66b, CD235ab, CD14, and CD41. Scale bars, 70 μm.
RA Promotes Early G1 Cell Cycle State of hESC-Derived Endothelial Cells and the Enrichment of Hemogenic Endothelial Cells
To determine whether RA could enhance the specification of human hemogenic endothelial cells, we compared untreated hESC-derived endothelial cells (control group) to similar cells treated at D5 with vehicle (DMSO group) or RA (RA group), and quantified hemogenic endothelial cell generation on D8 of differentiation (Figure 4A). We used both control and DMSO groups because DMSO was previously shown to increase pluripotent stem cell differentiation (Chetty et al., 2013). We found that after 72 h of treatment, RA significantly increased the differentiation of hESC-derived human hemogenic endothelial cells, whereas DMSO had no effect (Figures 4B and S4A). When cells were treated with both RA and a RA receptor (RAR) pan-antagonist (RAi), RA-induced human hemogenic endothelial cell specification was largely blocked (Figures 4B and S4A).
Figure 4. RA Promotes Early G1 Cell Cycle State of hESC-Derived Endothelial Cells and Their Specification toward Hemogenic Endothelial Cells.

(A) Schematic representation of experimental design.
(B) RA induces differentiation of hemogenic endothelial cells; n = 3 for all groups. Student’s t test: p = 0.0068 (DMSO versus RA), p = 0.0005 (DMSO versus RA+RAi), p = 0.0218 (RA versus RA+RAi).
(C) Schematic representation of the cell cycle states observable using the FUCCI system.
(D) RA induces early G1 cell cycle enrichment in endothelial cells. Cell cycle distribution was measured by expression of the FUCCI reporter construct using flow cytometry; n = 3 for all groups. (i) Student’s t test: p = 0.0012 (control versus RA), p = 0.0019 (DMSO versus RA). (ii) Student’s t test: p = 0.0122 (DMSO versus RA), p = 0.0102 (DMSO versus RA+RAi). (iii) Student’s t test: p = 0.0002 (control versus RA), p = 0.0389 (DMSO versus RA). (iv) Student’s t test: p = 0.0355 (control versus RA), p = 0.0084 (DMSO versus RA).
(E) In E10.5 mouse AGM, significantly more hemogenic endothelial cells were in late G1 phase compared to non-blood-forming endothelial cells; n = 4 for all groups. Student’s t test: p < 0.0001.
(F) Picture of H9-FUCCI-hESC-derived endothelial cells. Original magnification, 20×.
(G) In hESC culture on D8, significantly more hemogenic endothelial cells were in early and late G1 compared to non-blood-forming endothelial cells; n = 3 for all groups. Student’s t test: p = 0.0005 (early G1), p < 0.0001 (late G1), p < 0.0001 (G1/S), p = 0.0016 (S/G2/M).
Data are represented as means ± SDs.
We previously found that cell cycle state plays a critical role in murine arterial and hemogenic endothelial cell specification in vivo (Fang et al., 2017; Marcelo et al., 2013b). Here, we investigated the effect of RA on cell cycle distribution in vitro using H9-hESCs carrying the fluorescent ubiquitination-based cell cycle indicator (FUCCI) (H9-FUCCI-hESC reporter cell line) (Pauklin and Vallier, 2013). The FUCCI system uses antiphase oscillating proteins to mark distinct cell cycle states with fluorescent indicators, and cell cycle distribution can be analyzed via flow cytometry (Figure 4C) (Sakaue-Sawano et al., 2008). We found that RA treatment significantly increased the proportion of hESC-derived endothelial cells in early G1 phase and decreased the proportion transitioning from G1 to S phase (Figures 4D, S4B, and S4C). We also found that DMSO significantly increased the proportion of cells in late G1 phase (data not shown), which is consistent with previous findings (Chetty et al., 2013). When cells were treated with both RA and RAi, RA-induced changes in the cell cycle state were suppressed. These data show that RA promotes the differentiation and enrichment of hemogenic endothelial cells from hESCs, and this is accompanied by an increased proportion of cells in early G1 phase and a decreased proportion transitioning from G1 to S phase.
Mouse and Human Hemogenic Endothelial Cells Are Predominantly in G1 Cell Cycle State In Vivo and In Vitro
Our previous studies of murine yolk sac and AGM endothelial cells (Marcelo et al., 2013b), and the studies above in hESC-derived endothelial cells, demonstrate that in both model systems, RA promotes hemogenic specification, as well as cell cycle arrest. To further define the cell cycle state of murine hemogenic endothelial cells versus non-blood-forming endothelial cells in vivo during definitive hematopoiesis, we used FUCCI-red mice, carrying mKO fluorescent protein that marks cells in G1 phase (Sakaue-Sawano et al., 2008). Using flow cytometry, we isolated murine E10.5 AGM endothelial cells (CD31+CD45−SP−) and hemogenic endothelial cells (Flk-1+Kit+CD45−SP) and found that the proportion of hemogenic endothelial cells in G1 phase was significantly higher than in non-blood-forming endothelial cells in the AGM (Figures 4E and S5A). To further define the cell cycle state of hESC-derived human hemogenic endothelial cells, we used the H9-FUCCI-hESCs described above (shown in Figure 4F) (Pauklin and Vallier, 2013), which can clearly delineate cells in early G1, late G1, G1-to-S, and S/G/M phases (depicted in Figure 4C). We found that compared to total human endothelial cells (CD31+CD45−), a significantly greater proportion of human hemogenic endothelial cells (CD31+KDR+KIT+CD34+CDH5−CD45−) were also in G1 phase and a significantly lower proportion were actively cycling (Figures 4G and S5B). Overall, these studies implicate a role for cell cycle state in human hemogenic endothelial cell specification and highlight a potentially conserved developmental paradigm in mice and humans.
Early G1 Cell Cycle State Is Necessary but Not Sufficient to Enable Hemogenic Endothelial Cell Specification
We found that hemogenic endothelial cells in vivo and in vitro were predominantly in G1 phase, and G1 arrest is required for murine hemogenic endothelial cell specification (Marcelo et al., 2013b). In addition, RA treatment, which promoted human hemogenic endothelial cell development, specifically increased the proportion of hESC-derived endothelial cells in early G1 phase, and concomitantly decreased the proportion of cells exiting G1 phase. Therefore, we specifically focused on the role of G1 state in hemogenic specification, and tested whether early G1 versus late G1 cell cycle state is necessary for hemogenic specification. To do so, we treated endothelial cells with a CDK4/6 inhibitor (CDK4/6i), which causes an accumulation of cells specifically in late G1 phase (Schmidt and Sebastian, 2018), and tested their response to RA (Figure 5A). As expected, RARACDK4/6i treatment increased the proportion of endothelial cells in late G1 phase (Figures 5Bii and S6Aii). In contrast, RA treatment alone increased the proportion of endothelial cells in early G1 phase (Figures 5Bi and S6Ai), consistent with the studies described above. The effects of RA and CDK4/6i on G1/S and S/G2/M phases were comparable (Figures 5Biii, 5Biv, S6Aiii, and S6Aiv). When hESC-derived endothelial cells were arrested in late G1 phase, as opposed to early G1 phase, hemogenic endothelial cell specification was suppressed even in the presence of RA (Figures 5C and S6B). Thus, the early G1 cell cycle state appears to be necessary for RA-induced hemogenic endothelial cell specification. Of note, we did not test the potential role of S/G2/M states for several reasons: a low proportion (<10%, data not shown) of hemogenic endothelial cells reside in this state in vivo and in vitro, we cannot verify arrest in S versus G2 versus M using this FUCCI reporter cell line, and the drugs available to arrest cells in these phases are associated with confounding effects such as cytotoxicity, increased apoptosis, and chromosome damage.
Figure 5. Early G1 Cell Cycle State of Endothelial Cells Is Necessary but Not Sufficient to Enable Hemogenic Specification.

(A) Schematic representation of experimental design.
(B) When cells were treated with both RA and CDK4/6i, CDK4/6i overrides effect of RA on cell cycle state and enriches endothelial cells in late G1 phase. Cell cycle distribution was measured by expression of the FUCCI reporter construct using flow cytometry; n = 3 for all groups. (i) Student’s t test: p = 0.0130 (DMSO versus RA), p = 0.0423 (DMSO versus RA+CDK4/6i), p = 0.0497 (RA versus RA+CDK4/6i). (ii) Student’s t test: p = 0.0205 (DMSO versus RA+CDK4/6i), p = 0.0253 (RA versus RA+CDK4/6i). (iii) Student’s t test: p = 0.0010 (DMSO versus RA), p = 0.0016 (DMSO versus RA+CDK4/6i). (iv) Student’s t test: p = 0.0046 (DMSO versus RA), p < 0.0001 (DMSO versus RA+CDK4/6i), p = 0.0015 (RA versus RA+CDK4/6i).
(C) In the absence of early G1 cell cycle arrest in endothelial cells, RA+CDK4/6i could not induce hemogenic endothelial cell specification; n = 3 for all groups. Student’s t test: p = 0.0233 (DMSO versus RA), p = 0.0059 (RA versus RA+CDK4/6i).
(D) Schematic representation of experimental design.
(E) Similar to RA, CDK2i induces early G1 cell cycle arrest in endothelial cells. Cell cycle distribution was measured by expression of the FUCCI reporter constructusing flow cytometry; n = 3 for all groups. (i) Student’s t test: p < 0.0001 (DMSO versus RA), p = 0.0196 (DMSO versus CDK2i), p = 0.0007 (DMSO versus CDK4/6i). (ii) Student’s t test: p = 0.0115 (DMSO versus CDK2i), p = 0.0007 (DMSO versus CDK4/6i). (iii) Student’s t test: p = 0.0005 (DMSO versus CDK4/6i). (iv) Student’s t test: p = 0.0148 (DMSO versus CDK2i), p = 0.0014 (DMSO versus CDK4/6i).
(F) Unlike RA, CDK2i could not induce hemogenic endothelial cell specification; n = 3 for all groups. Student’s t test: p = 0.0016 (DMSO versus RA), p = 0.0051 (DMSO versus CDK4/6i).
(G) CDK2i did not artificially suppress cell growth or cell differentiation in culture. Student’s t test: p = 0.0077 (DMSO versus RA), p = 0.0015 (DMSO versusRA+CDK2i).
Data are represented as means ± SDs
To test whether the early G1 cell cycle state in endothelial cells is sufficient for hemogenic specification, hESC-derived endothelial cells were treated with either RA, CDK4/6i, or CDK2 inhibitor (CDK2i), which arrests cells specifically in early G1 phase of the cell cycle (Chao et al., 2019) (Figure 5D). The CDK2i (CVT-313) that we used is a cell-permeable purine analog that acts as a potent and selective ATP-competitive inhibitor of CDK2, which inhibits other kinases only at much higher concentrations. As expected, and similar to RA, CDK2i increased the proportion of endothelial cells in early G1 phase (Figures 5Ei and S7Ai); in contrast, CDK4/6i increased the proportion of endothelial cells in late G1 phase (Figures 5Eii and S7Aii). We found that arresting endothelial cells in either early G1 or late G1 alone (in the absence of RA treatment) was not sufficient to promote hemogenic specification (Figures 5F and S7B). In addition, when hESC-derived endothelial cells were treated with both RA and CDK2i, there was a trend toward an increase in hemogenic endothelial cell specification (Figures 5G and S7C). Thus, early G1 cell cycle arrest of human endothelial cells is necessary but not sufficient to promote hemogenic specification.
Bulk RNA Sequencing (RNA-Seq) Reveals Candidates of Additional Signals That RA Provides to Promote Hemogenic Endothelial Cell Specification
To gain further mechanistic insight into the role of RA during human hemogenic endothelial cell specification, we performed bulk RNA-seq of H1-hESC-derived endothelial cells (CD31+CD45−) treated with or without RA (Figure 6A). Both control (Figure 6Bi) and RA-treated (Figure 6Bii) endothelial cells (CD31+CD45−) were FACS isolated on D8 and subjected to bulk RNA-seq.
Figure 6. Bulk RNA Sequencing Reveals Candidate Mediators of RA Regulation of Human Endothelial Cell Cycle Control and Hemogenic Specification.

(A) Schematic representation of experimental design.
(B) Endothelial cells (CD31+CD45−) treated with (i) n/a or (ii) RA were collected on D8 for RNA sequencing using FACS sorting.
(C) Gene set enrichment analysis was performed for genes exclusively downregulated in RA-treated endothelial cells. Relevant and significant (p < 0.05) terms are shown.
(D) Gene set enrichment analysis was performed for genes exclusively upregulated in RA-treated endothelial cells. Relevant and significant (p < 0.05) terms are shown.
(E) Selected markers demonstrate upregulation of genes associated with hematopoietic cell fate in endothelial cells by RA.
We identified 2,547 genes significantly downregulated and 2,208 genes significantly upregulated in RA-treated endothelial cells, and performed gene set enrichment analysis of the datasets, respectively. We found that genes downregulated by RA were strongly associated with the vascular endothelial growth factor (VEGF) signaling pathway, which plays an essential role in endothelial cell proliferation and arterial specification (Bernatchez et al., 1999; Lawson et al., 2002) (Figure 6C). Transcription factors associated with these genes include ETV2, which is necessary for endothelial cell differentiation (De Val and Black, 2009), and KLF5, which promotes endothelial cell proliferation via the regulation of VEGF (Gao et al., 2015; Nagai et al., 2005). In addition, our analysis revealed a shift away from an endothelial phenotype upon RA treatment, demonstrated by the downregulation of genes associated with KDR expression and a human umbilical vein endothelial cell (HUVEC) phenotype. Lastly, the gene set enrichment analysis results further confirmed the role of RA in cell cycle regulation. Genes downregulated by RA are linked to transcription factor E2F1, which promotes the G1-to-S phase transition (Poppy Roworth et al., 2015).
In contrast, we found that genes upregulated by RA treatment were strongly associated with RA signaling (as expected), as well as Notch, Wnt and Hedgehog (HH) signaling. Notch is known to promote murine hemogenic specification downstream of RA (Marcelo et al., 2013b), and these results suggest conserved molecular regulation of mouse and human hemogenic endothelial cell specification. Wnt signaling has long been recognized as an important regulator of hematopoiesis. More specifically, canonical Wnt signaling plays an important role in maintaining hematopoietic stem cell (HSC) self-renewal, and has also been shown to interact with other signaling pathways, including HH and Notch (Staal and Luis, 2010) (Figure 6D). SMAD factors are also significantly upregulated with RA treatment. They play a critical role in regulating endothelial cell growth suppression (Bohnsack et al., 2004), as well as the fate of HSPCs during development and postnatally, by mediating transforming growth factor β (TGF-β) signaling (Larsson and Karlsson, 2005). Lastly, genes upregulated by RA are also associated with SOX2 and TRIM28 activities, and both have been shown to support the early stages of blood cell development (Hosoya et al., 2013; Sarkar and Hochedlinger, 2013).
We further examined RA regulation of genes known to be involved in vascular development and hemogenic specification (Figure 6E). Similar to our findings above, RA treatment led to a shift away from endothelial phenotype and toward a hematopoietic phenotype. More specifically, RA suppressed the expression of VEGF receptors KDR and Fms-related receptor tyrosine kinase 1 (FLT1), which are important for endothelial cell proliferation and maintenance (Melincovici et al., 2018). In contrast, RA upregulated the expression of a number of genes associated with murine hemogenic endothelial cells and HSPCs, including RAR, KIT, NOTCH, CDKN1B, GATA2 and RUNX1. In addition, the expression of a number of SMAD family members was upregulated, as were ligands of the HH and Wnt signaling pathways, and homeobox A1 (HOXA1) genes that are known to play a role in hematopoietic development (Alsayegh et al., 2019). These results are consistent with the gene set enrichment analyses, and suggest that there is potential crosstalk among the RA/HOX/SMAD/HH/Wnt signaling pathways that instructs the hemogenic specification of endothelial cells and concomitant downregulation of endothelial cell identity.
RNA-Seq Analysis Provides Insights That Link Endothelial Cell Cycle State to Hemogenic Specification
To gain further mechanistic insight into the role of cell cycle regulation in human hemogenic endothelial cell specification, we performed bulk RNA-seq of H9-FUCCI-hESC-derived endothelial cells (CD31+CD45−) in distinct cell cycle states (Figure 7A). Cells in early G1, late G1, G1/S, and S/G2/M phases were FACS isolated and subjected to bulk RNA-seq (Figure 7B). We identified genes exclusively upregulated in early G1 phase (1,854 genes) and late G1 phase (2,387 genes) and performed gene set enrichment analysis. Genes exclusively upregulated in early G1 were predominantly associated with hematopoietic development, and hESC-derived endothelial cells in early G1 phase showed high resemblance to cell types such as MOLT-4 cell (T lymphoblast), erythroid cell, B lymphoblast, and cord blood (Figure 7C). In contrast, genes exclusively upregulated in late G1 were predominantly linked to vascular development and angiogenesis, while some genes were associated with heart, ventricle, atrium, placenta, whole blood, and monocytes (Figure 7C).
Figure 7. Bulk RNA Sequencing Provides Insights That Link Endothelial Cell Cycle State to Hemogenic Specification.

(A) Schematic representation of experimental design.
(B) Endothelial cells (CD31+CD45−) in different cell cycle phases (early G1, late G1, G1/S, S/G2/M) were collected on D8 for RNA sequencing using FACS sorting. (C) Gene set enrichment analysis was performed for genes exclusively upregulated in early and late G1 cell cycle phases. Relevant and significant (p < 0.05) terms are shown.
(D) Gene marker expression in various cell cycle phases.
(E) Selected markers demonstrate enrichment of genes associated with hematopoiesis in early G1 phase.
(F) Selected markers demonstrate enrichment of genes associated with vascular development and arterial endothelium in late G1 phase.
(G) Schematic representation of working model.
Data are represented as the highest/lowest value within 1.5x interquartile range (IQR) above/below the 75th/25th percentile.
To further investigate the link between endothelial cell cycle state and cell fate decisions, we examined the differential expression of specific genes associated with vascular development, hemogenic specification, and arterial specification in endothelial cells in distinct cell-cycle states (Figure 7D). We found an enrichment of genes associated with hemogenic specification in early G1 phase (e.g., CDKN1B, ESAM, HES1, GATA2, TAL1, LMO2) (Figure 7E). In contrast, we observed an enrichment of genes associated with arterial specification in late G1 phase (e.g., GJA4, GJA5, NRP1, EFNB2, DLL4, TGFBR2) (Figure 7F), which is consistent with our previous findings in murine arterial endothelial cell specification (Fang et al., 2017).
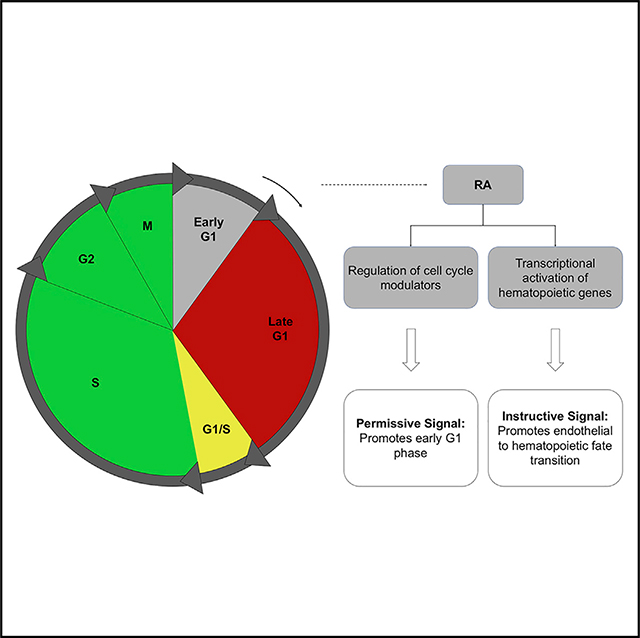
Our studies suggest that RA plays a dual role in human hemogenic endothelial cell specification. That is, it promotes endothelial cell arrest in early G1 phase, which is necessary to enable hemogenic specification (Figure 7G), and also induces signaling pathways that are necessary for altering gene expression, specifically in early G1 state, to promote a hemogenic phenotype and suppress endothelial cell identity.
DISCUSSION
Previous studies have demonstrated the endothelial origin of blood cells in vivo (Marcelo et al., 2013a; Medvinsky and Dzierzak, 1996; Taoudi and Medvinsky, 2007). Further mechanistic studies have shed light on how a subset of murine endothelial cells acquires hematopoietic potential, highlighting the critical role of the RA/Kit/Notch/p27 signaling pathway and cell cycle regulation (Goldie et al., 2008; Marcelo et al., 2013b). However, whether a specific cell cycle state is necessary for hemogenic specification of endothelial cells or whether this mechanism is conserved in humans was unknown. In these studies, we established a simple in vitro system to derive human endothelial and hemogenic endothelial cells and delineate their mechanism(s) of development.
Compared to existing systems that can be used to derive endothelial and hematopoietic cells from hESCs, our system has several advancements. For example, it does not require the use of mouse feeder cells or the formation of embryoid bodies, and is therefore less technically challenging to adopt. In addition, our system reduces the presence of other cell types and/or the development or other organ systems to provide a more controlled environment and consistency for molecular mechanistic studies. Our system also requires fewer growth factors and less time and is therefore more economical. Using this system, we showed that RA plays a critical role in the specification of human hemogenic endothelial cells, which we defined on a clonal level.
We propose that RA has two roles during hemogenic specification—promoting cell cycle arrest of endothelial cells in early G1 phase (permissive signal) and activating transcriptional programs (instructive signal) that enable the transition from endothelial to hemogenic endothelial cells, specifically in early G1. Thus, we hypothesize that arrest in early G1 state provides a distinct “window of opportunity” for endothelial cells to respond to external stimuli that promotes their specification toward a hemogenic endothelial cell phenotype. In addition, RA extends the window of time for hemogenic specification by enriching endothelial cells in early G1; RA also activates signaling pathways and transcriptional regulators that promote hemogenic specification in early G1. Consistent with this idea, others have shown that the cell cycle state determines the propensity for hESCs to commit to mesodermal versus endodermal and ectodermal lineages (Pauklin and Vallier, 2013).
We demonstrated that both of these functions of RA are required for hemogenic specification. To test the cell-cycle-regulation role of RA, we showed that chemically enriching endothelial cells in a different cell cycle phase (late G1 versus early G1 that is induced by RA) prevented hemogenic specification despite the presence of RA. To test the transcriptional role of RA, we demonstrated that chemically enriching endothelial cells in early G1 phase with CDK2i was not capable of inducing hemogenic specification in the absence of RA. These data show that both the cell cycle regulation and the transcriptional role of RA are necessary for hemogenic specification. Although hemogenic specification occurs while endothelial cell cycle progression is arrested, after specification, hemogenic endothelial cells must progress into S/G2/M states to divide and give rise to HSPCs. Thus, further understanding of cell cycle control throughout this endothelial-to-hematopoietic transition process is needed.
Our RNA-seq analyses revealed signaling pathway(s) and transcriptional regulators that RA may activate to enable cell cycle arrest and an endothelial to hemogenic endothelial cell fate transition. Interestingly, some of the same RA-induced pathways that regulate murine hemogenic specification, such as Notch and KIT, appear to be conserved in this process in human endothelial cells. Our analyses also revealed crosstalk with other pathways, including SMAD, Wnt, and HH signaling. Thus, it will be important in future studies to elucidate the exact roles of and potential interactions among these factors that may function downstream of RA to regulate the human endothelial cell cycle state and hemogenic specification.
It is not clear why hemogenic specification occurs in early G1 state of the endothelial cell cycle. Our RNA-seq data revealed enrichment of genes associated with hemogenic specification in early G1 phase and genes associated with arterial specification in late G1 phase, consistent with our previous studies (Fang et al., 2017). However, we also observed an enrichment of genes associated with hemogenic endothelial cells (e.g., NOTCH1, DLL4, HEY1, KIT, RUNX1) in late G1 phase. Thus, we propose that hemogenic specification initiates in early G1 and continues into late G1. Similarly, our data also suggest that arterial specification initiates in late G1 and continues into G1/S; that is, an enrichment of genes associated with arterial specification (e.g., BMP2, BMP4, BMP7, WNT2, WNT5B, WNT8A) was observed in G1/S phase in addition to late G1 phase. Other studies showed that expression of developmental regulators such as HOXD1, TBX5, and SOX11 is specifically upregulated in both early and late G1 phases (Singh et al., 2013). In contrast, human stem cells in S and G2 phases have been shown to possess an intrinsic tendency toward the pluripotent state (Gonzales et al., 2015). Despite recent discoveries linking cell cycle state to cell fate decisions in stem cells, understanding the molecular role of the cell cycle state during endothelial cell differentiation and hemogenic specification requires further investigation.
Finally, although our study focused on the process of hemogenic endothelial cell specification, cell cycle control has also been previously implicated in murine arterial endothelial cell specification (Fang et al., 2017), and our RNA-seq data suggest that this is conserved in human endothelial cell development. The use of emerging technologies such as single-cell RNA-seq will enable us to further delineate the heterogeneity within and phenotypic continuum among endothelial cell subtypes and their molecular regulators during their development. Such insights will improve our ability to apply developmental advances to the generation of distinct endothelial cell subtypes for tissue engineering and regenerative medicine. In addition, our system could likely be developed further to optimize the generation of transplantable HSPCs from human hemogenic endothelial cells for clinical therapies.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Karen K. Hirschi (kkh4yy@virginia.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The accession number for the RNA-seq data reported in this paper is Gene Expression Omnibus (GEO): GSE159694.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
H1-hESC were obtained from Yale Stem Cell Center and used up to passage 90. H9-Fucci-hESC were obtained from Dr. Ludovic Vallier’s lab at Cambridge Stem Cell Institute and used up to passage 90 (Pauklin and Vallier, 2013). All hESC were cultured in mTeSR™ medium (STEMCELL Technologies cat# 85850) on Matrigel-coated plates (Corning® Matrigel® Growth Factor Reduced (GFR) Basement Membrane Matrix, LDEV-free, 5 mL, Corning cat# 356230) as described in Fu et al. (2011). Importantly, 50 μg/ml G-418 solution (Sigma cat# 4727878001) was added to the culture medium for H9-Fucci-hESC to eliminate non-transduced cells. For passage, 160 μl/ml Dispase (STEMCELL Technologies cat# 07913) was added to cell culture. Cells were incubated with Dispase at 37°C for 45min. Afterward, cells were transferred to Falcon tubes containing DMEM:F12 medium (GIBCO cat# 11320–033) for washing. 12ml of DMEM:F12 medium was used per 1ml of cell culture medium. Let tubes stand at room temperature for 5–10min or until all cell pellets settle to the bottom of the tubes. Remove supernatant and cells were resuspended in mTeSR™ medium, dispersed into small clumps, and re-plated onto Matrigel-coated plates.
Animals
All animals used were on a C57BL/6 background (female, 8 – 12 weeks old) and all studies were conducted in compliance with Yale’s Institutional Animal Care and Usage Committee guidelines. FUCCI-red animals (B6.Cg-Tg(FucciG1)#596Bsi, Riken, cat# RBRC02707) were crossed with either FUCCI-red animals or wild-type C57BL/6J animals (JAX, cat# 000664).
METHOD DETAILS
Differentiation protocol
H1-hESC or H9-Fucci-hESC were passaged using the method described above and re-plated onto Matrigel-coated plates. These hESC colonies were maintained in mTeSR for 24 hours, after which the cells were gently washed with DMEM:F12 and differentiated in chemically defined, serum-free, animal component-free medium STEMdiffAPEL2 (STEMCELL Technologies cat# 05270) supplemented with Protein-Free Hybridoma Medium (PFHM-II, GIBCO, cat# 12040077) by adding 5 mL of PFHM to 100 mL of STEMdiffAPEL2 medium. Growth factors depicted in Figure 1A were added sequentially (Stemolecule CHIR99021, 5 μM, Stemgent cat# 04–0004; Recombinant Human FGF basic/FGF2/bFGF (146 aa) Protein, 50 ng/ml, R&D systems cat# 233-FB-025; Recombinant Human BMP-4 Protein, 25 ng/ml, R&D systems cat# 314-BP-010; Recombinant Human VEGF165, 50 ng/ml, PeproTech, cat# 100–20) as described in Sriram et al. (2015). For certain experiments, cells were treated with RA (0.5 μM, Sigma-Aldrich, cat# R2625–50MG), RAi (AGN 194310, 6nM, MedChemExpress, cat# HY-16681), CDK4/6i (PD 0332991 isethionate, 2nM, Sigma-Aldrich, cat# PZ0199–5MG), and/or CDK2i (CVT-313, 0.5 μM, Santa Cruz Biotechnology, cat# 199986–75-9) every 24hrs starting Day5.
qPCR
Transcriptomes were prepared from cultured hESC/hESC-derived cells using RNeasy Plus Micro Kit (QIAGEN, cat# 74034) and 5X All-In-One RT MasterMix (abm, cat# G490). Resulting cDNA libraries were probed by qPCR using published qPCR primers (PrimerBank, sequences listed in Table S1) and miScript SYBR® Green PCR Kit (QIAGEN, cat# 218073).
Giemsa stain
1ml of supernatant was taken on Day8 of differentiation and dispersed onto a microscope slide using a slide centrifuge. Slides were then fixed in methanol for 5min. Upon air drying the slides, Giemsa Stain (Sigma-Aldrich, cat# GS500–500ml) was diluted 1:20 with deionized water and slides were stained for 60min at room temperature. Afterward slides were briefly rinsed with deionized water, air-dried, and mounted to be evaluated under a microscope.
Flow cytometry analysis for cell culture
Buffer list: sorting buffer (HBSS, GIBCO, cat# 14175–095; 1%FBS, Gemini Bio, cat# 100–106; supplemented with 1:500 diluted Normocin - Antimicrobial Reagent, InvivoGen, cat# ant-nr-1); antibody staining buffer (HBSS; 10%FBS; supplemented with 1:500 diluted Normocin); Hoechst staining buffer (DMEM:F12-high glucose, GIBCO, cat# 11965–092; 10%FBS; supplemented with 1:500 diluted Normocin).
Antibody list: Pacific Blue anti-human CD31 Antibody (BioLegend, cat# 303113), FITC anti-human CD31 Antibody (BioLegend, cat# 303104), Alexa Fluor® 700 anti-human CD45 Antibody (BioLegend, cat# 304024), APC/Cyanine7 anti-human CD45 Antibody (BioLegend, cat# 304014), PE/Cy7 anti-human CD309 (VEGFR2) Antibody (BioLegend, cat# 359911), APC anti-human CD117 (c-kit) Antibody (BioLegend, cat# 313206), PE anti-human CD144 (VE-Cadherin) Antibody (BioLegend, cat# 348506), Pacific Blue anti-human CD34 Antibody (BioLegend, cat# 343512), FITC anti-human CD20 Antibody (BioLegend, cat# 302303), Brilliant Violet 421 anti-human CD3 Antibody (BioLegend, cat# 317343), Brilliant Violet 421 anti-human CD14 Antibody (BioLegend, cat# 325627), Alexa Fluor® 594 anti-human CD66b Antibody (BioLegend, cat# 392907), Alexa Fluor® 488 anti-human CD41 Antibody (BioLegend, cat# 303723), APC anti-human CD235ab Antibody (BioLegend, cat# 306607), PE anti-human CD11b Antibody (BioLegend, cat# 301305), Alexa Fluor® 700 anti-human CD45 Antibody (BioLegend, cat# 304024), Zombie NIR Fixable Viability Kit (BioLegend, cat# 423105).
If side population staining was needed, prewarmed Hoechst staining buffer in 37°C water bath for 15min. Differentiated cells were harvested with Accutase (STEMCELL Technologies, cat# 7920) and spun 5min at 1,000xrcf. at 4°C to pellet. Cells were then washed once with DMEM:F12 and spun 5min at 1,000xrcf. at 4°C to pellet. Cells were resuspended in prewarmed Hoechst staining buffer such that cells were at a final concentration of 1×106 cells/ml. At least 100 μL of sample (1×105 cells) was aliquoted into fresh 1.5 mL tubes for control. The following sample and necessary control tubes were included: Unstained; Hoechst 33342 only; Hoechst 33342 only + Verapamil; all single-color antibody controls; Sample (received all colors, except for Verapamil). Verapamil diluted in 95% ethanol was added to the “Hoechst 33342 only + Verapamil” control tube to a final concentration of 50 μM. All tubes were incubated for 5 minutes at 37°C. (CAUTION: Verapamil is a potent calcium-channel blocking agent and is extremely toxic. Handle with gloves). Hoechst 33342 was then added to the “Hoechst 33342 only” control, to the Hoechst 33342 + Verapamil only control, and to the “Sample” tube to a final concentration of 5 μg/ml. All tubes were incubated for 1 hour at 37°C, protected from light. Tubes were gently mixed through inversion every 15 minutes. (CAUTION: Hoechst 33342 is a toxic nuclear dye and should be handled with gloves). Tubes were spun 5min at 1,000xrcf. at 4°C to pellet. Supernatant was removed and cell pellets were resuspended at a concentration of 1×105 cells/ml in cold antibody staining buffer. Fluorescently-conjugated antibodies were added to appropriate antibody-only control tubes, and to the “Sample” tube. Dilutions need to be optimized for different antibodies and FACS machines. Tubes were incubated on ice for 30 minutes, protected from light. Afterward tubes were spun 5min at 1,000xrcf. at 4°C to pellet. Supernatant was removed and cell pellets were resuspended in 600 μL ice-cold sorting buffer. Samples were strained through mesh filter cap of 5 mL round bottom polystyrene FACS tube (Corning, cat# 352235) and stored on ice, protected from light, for immediate FACS.
Methylcellulose-based blood forming assay
For population level assay, cells were sorted using FACS and seeded onto fibronectin coated dishes (Fibronectin Human Protein, Plasma, 4 μg/cm2, GIBCO, cat# 33016015) as described in Dege and Sturgeon (2017), with MethoCult H4435 Enriched (STEMCELL Technologies, cat# 4435). For single-cell level assay, cells were directly sorted into fibronectin coated 96-well plates using FACS. Afterward, a P1000 tip with the tip trimmed to widen its bore was used to gently resuspend each well of the seeded methylcellulose media in a tissue culture hood to ensure optimal growth.
CFU-E erythroid colony and BFU-E erythroid colony were analyzed 8 days after plating. CFU-GM granulocyte-monocyte colony and CFU-GEMM multipotent hematopoietic progenitor colony were analyzed 14 days after plating.
Dil AcLDL uptake
To demonstrate endothelial phenotypic function of hESC-derived hemogenic endothelial cells in culture, active uptake of DiI AcLDL was measured. On Day3 of culturing of hESC-derived hemogenic endothelial cells in MethoCult H4435 Enriched medium (STEMCELL Technologies, cat# 4435), medium was removed and cells were washed 3 times with 1xPBS (GIBCO, cat# 10010031) to remove the residual methylcellulose medium. 1ml of endothelial cell medium (PromoCell, cat# C-22011) was added to the dish and cells were stained with 10 μg/ml Dil AcLDL (DiI-Ac-LDL Kit, Cell Applications, cat# 022K) for 4hrs at 37°C and then imaging with fluorescence microscope.
Immunostaining of hematopoietic colonies
Antibody list: Alexa Fluor® 488 anti-human CD41 Antibody (Biolegend, cat# 303723), APC anti-human CD235ab Antibody (Biolegend, cat# 306607), CD66b-Alexa594 (Biolegend, cat# 392907), CD14-BV421 (Biolegend, cat# 325627).
Single hemogenic endothelial cell was seeded in fibronectin coated 96-well plates (Cellvis, cat# P96–1.5P) as described above. Cells were cultured in 100 μL of methylcellulose medium (STEMCELL Technologies, cat# 4435) per well for 8 days before staining for CFU-E and BFU-E, for 14 days before staining for CFU-GM and CFU-GEMM. For staining, antibodies were 1:100 diluted in IMDM (GIBCO, cat# 12440046). 30 μL of diluted antibodies was added to each well on top of the methylcellulose medium (pipette slowly so as not to disturb the hematopoietic colonies). Incubate plates at 37°C overnight before imaging with fluorescence microscope.
Flow cytometry analysis for animal tissue
Flow cytometry was performed as described in Fang et al. (2016). The list of antibodies used can also be found in Fang et al. (2016).
QUANTIFICATION AND STATISTICAL ANALYSIS
RNA sequencing
All mRNA samples were isolated using RNeasy Plus Micro Kit (QIAGEN, cat# 74034). Next-generation whole transcriptome Illumina sequencing was performed with the Yale Center for Genome Analysis. Fastq-files were generated with bcl2fastq2_v2.19.0 and then uploaded to the usegalaxy.org platform (Afgan et al., 2018) for quality control and trimming (Galaxy ToolShed v 1.0.2; FASTQ/A short-reads pre-processing tools: http://hannonlab.cshl.edu/fastx_toolkit/) (Blankenberg et al., 2010). Sequences were aligned to the hg38 genome using HISAT2 (v 2.1.0+galaxy4) (Kim et al., 2015) and aligned sequences were quantified with featureCounts (v 1.6.3+galaxy2) (Liao et al., 2014). Differential gene expression analysis was performed with DESeq2 (R package v 1.24.0) (Love et al., 2014) using unmanipulated count data. Heatmaps were generated with ComplexHeatmap (R package v 2.0.0) using data transformed with DESeq2 and batch-corrected with limma (R package v 3.40.6) (Phipson et al., 2016).
Statistical analysis
Two-tailed Student’s t tests were performed using Prism 7 for MacOSX software for all analyses. p values < 0.05 were considered as significant: *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001. Standard deviations are depicted in all graphs.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Pacific Blue anti-human CD31 Antibody | BioLegend | Cat#303113; RRID:AB_1877196 |
| FITC anti-human CD31 Antibody | BioLegend | Cat#303104; RRID:AB_314330 |
| Alexa Fluor® 700 anti-human CD45 Antibody | BioLegend | Cat#304024; RRID:AB_493761 |
| APC/Cyanine7 anti-human CD45 Antibody | BioLegend | Cat#304014; RRID:AB_314402 |
| PE/Cy7 anti-human CD309 (VEGFR2) Antibody | BioLegend | Cat#359911; RRID:AB_2563551 |
| APC anti-human CD117 (c-kit) Antibody | BioLegend | Cat#313206; RRID:AB_314985 |
| PE anti-human CD144 (VE-Cadherin) Antibody | BioLegend | Cat#348506; RRID:AB_2077978 |
| Pacific Blue anti-human CD34 Antibody | BioLegend | Cat#343512; RRID:AB_1877197 |
| FITC anti-human CD20 Antibody | BioLegend | Cat#302303; RRID:AB_314251 |
| Brilliant Violet 421 anti-human CD3 Antibody | BioLegend | Cat#317343; RRID:AB_2565848 |
| Brilliant Violet 421 anti-human CD14 Antibody | BioLegend | Cat#325627; RRID:AB_2561342 |
| Alexa Fluor® 594 anti-human CD66b Antibody | BioLegend | Cat#392907 |
| Alexa Fluor® 488 anti-human CD41 Antibody | BioLegend | Cat#303723; RRID:AB_2566302 |
| APC anti-human CD235ab Antibody | BioLegend | Cat#306607; RRID:AB_314625 |
| PE anti-human CD11b Antibody | BioLegend | Cat#301305; RRID:AB_314157 |
| Alexa Fluor® 700 anti-human CD45 Antibody | BioLegend | Cat#304024; RRID:AB_493761 |
| Zombie NIR Fixable Viability Kit | BioLegend | Cat#423105 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Stemolecule CHIR99021 | Stemgent | Cat#04–0004 |
| Recombinant Human FGF basic/FGF2/ bFGF (146 aa) Protein | R&D systems | Cat#233-FB-025 |
| Recombinant Human BMP-4 Protein | R&D systems | Cat#314-BP-010 |
| Recombinant Human VEGF165 | PeproTech | Cat#100–20 |
| Retinoic acid | Sigma-Aldrich | Cat#R2625–50MG |
| AGN 194310 | MedChemExpress | Cat#HY-16681 |
| PD 0332991 isethionate | Sigma-Aldrich | Cat#PZ0199–5MG |
| CVT-313 | Santa Cruz Biotechnology | Cat#199986–75-9 |
| Giemsa stain, modified | Sigma-Aldrich | Cat#GS500–500ml |
| Normocin - Antimicrobial Reagent | InvivoGen | Cat#ant-nr-1 |
| Fibronectin Human Protein, Plasma | GIBCO | Cat#33016015 |
| Corning® Matrigel® Growth Factor Reduced (GFR) Basement Membrane Matrix, LDEV-free, 5 mL | Corning | Cat#356230 |
| Critical Commercial Assays | ||
| RNeasy Plus Micro Kit | QIAGEN | Cat#74034 |
| 5X All-In-One RT MasterMix | abm | Cat#G490 |
| miScript SYBR® Green PCR Kit | QIAGEN | Cat#218073 |
| Dil-Ac-LDL Kit | Cell Applications | Cat#022K |
| Deposited Data | ||
| RNA-seq-1 | This paper | GSE159694 |
| RNA-seq-2 | This paper | GSE159694 |
| Experimental Models: Cell Lines | ||
| H1-hESC | Yale Stem Cell Center | N/A |
| H9-Fucci-hESC | Pauklin and Vallier, 2013 | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: B6.Cg-Tg(FucciG1)#596Bsi | Riken | Cat#RBRC02707 |
| Mouse: C57BL/6J | JAX | Cat#000664 |
| Oligonucleotides | ||
| See Table S1 for oligonucleotide sequences | N/A | N/A |
| Software and Algorithms | ||
| Prism 7 | Graphpad Software | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| bcl2fastq2_v2.19.0 | Illumina | https://support.illumina.com/sequencing/sequencing_software/bcl2fastq-conversion-software.html |
| Galaxy | Afgan et al., 2018 | https://usegalaxy.org/ |
| DESeq2 | Love et al., 2014 | N/A |
| ComplexHeatmap | Phipson et al., 2016 | N/A |
| limma | Phipson et al., 2016 | N/A |
Highlights.
A simple and efficient protocol to derive human hemogenic endothelial cells is described
Functional human hemogenic endothelial cells are characterized on a clonal level
Retinoic acid signaling is required for human hemogenic endothelial specification
Cell-cycle state is a critical determinant of human endothelial cell specification
ACKNOWLEDGMENTS
The authors would like to thank Dr. L. Vallier for providing H9-FUCCI-hESC. This work was funded by NIH grants HL128064 and U2EB017103 and the CT Innovations 15-RMB-YALE-04 grant to K.K.H.; NIH grants R01-AI130471 and R01-CA228019 and Merit Review award I01BX002919 from the Department of Veterans Affairs, BLR&D Service, to E.C.B.; and the Swedish Society for Medical Research and the Stanford Dean’s Fellowship to S.N. The contents of this publication do not represent the views of the Department of Veterans Affairs or the United States government.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108465.
REFERENCES
- Afgan E, Baker D, Batut B, van den Beek M, Bouvier D, Cech M, Chilton J, Clements D, Coraor N, Grüning BA, et al. (2018). The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 46 (W1), W537–W544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsayegh K, Cortés-Medina LV, Ramos-Mandujano G, Badraiq H, and Li M (2019). Hematopoietic Differentiation of Human Pluripotent Stem Cells: HOX and GATA Transcription Factors as Master Regulators. Curr. Genomics 20, 438–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernatchez PN, Soker S, and Sirois MG (1999). Vascular endothelial growth factor effect on endothelial cell proliferation, migration, and platelet-activating factor synthesis is Flk-1-dependent. J. Biol. Chem 274, 31047–31054. [DOI] [PubMed] [Google Scholar]
- Blankenberg D, Gordon A, Von Kuster G, Coraor N, Taylor J, and Nekrutenko A; Galaxy Team (2010). Manipulation of FASTQ data with Galaxy. Bioinformatics 26, 1783–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnsack BL, Lai L, Dolle P, and Hirschi KK (2004). Signaling hierarchy downstream of retinoic acid that independently regulates vascular remodeling and endothelial cell proliferation. Genes Dev. 18, 1345–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanda B, Ditadi A, Iscove NN, and Keller G (2013). Retinoic acid signaling is essential for embryonic hematopoietic stem cell development. Cell 155, 215–227. [DOI] [PubMed] [Google Scholar]
- Chao HX, Fakhreddin RI, Shimerov HK, Kedziora KM, Kumar RJ, Perez J, Limas JC, Grant GD, Cook JG, Gupta GP, and Purvis JE (2019). Evidence that the human cell cycle is a series of uncoupled, memoryless phases. Mol. Syst. Biol 15, e8604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chetty S, Pagliuca FW, Honore C, Kweudjeu A, Rezania A, and Melton DA (2013). A simple tool to improve pluripotent stem cell differentiation. Nat. Methods 10, 553–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruijn MF, Speck NA, Peeters MC, and Dzierzak E (2000). Definitive hematopoietic stem cells first develop within the major arterial regions of the mouse embryo. EMBO J. 19, 2465–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Val S, and Black BL (2009). Transcriptional control of endothelial cell development. Dev. Cell 16, 180–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dege C, and Sturgeon CM (2017). Directed Differentiation of Primitive and Definitive Hematopoietic Progenitors from Human Pluripotent Stem Cells. J. Vis. Exp (129), 55196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker J (2014). Two ways to fold the genome during the cell cycle: insights obtained with chromosome conformation capture. Epigenetics Chromatin 7, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang JS, Gritz EC, Marcelo KL, and Hirschi KK (2016). Isolation of Murine Embryonic Hemogenic Endothelial Cells. J. Vis. Exp (112), 54150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang JS, Coon BG, Gillis N, Chen Z, Qiu J, Chittenden TW, Burt JM, Schwartz MA, and Hirschi KK (2017). Shear-induced Notch-Cx37-p27 axis arrests endothelial cell cycle to enable arterial specification. Nat. Commun 8, 2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X, Toh WS, Liu H, Lu K, Li M, and Cao T (2011). Establishment of clinically compliant human embryonic stem cells in an autologous feeder-free system. Tissue Eng. Part C Methods 17, 927–937. [DOI] [PubMed] [Google Scholar]
- Gao Y, Wu K, Chen Y, Zhou J, Du C, Shi Q, Xu S, Jia J, Tang X, Li F, et al. (2015). Beyond proliferation: KLF5 promotes angiogenesis of bladder cancer through directly regulating VEGFA transcription. Oncotarget 6, 43791–43805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldie LC, Lucitti JL, Dickinson ME, and Hirschi KK (2008). Cell signaling directing the formation and function of hemogenic endothelium during murine embryogenesis. Blood 112, 3194–3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzales KA, Liang H, Lim YS, Chan YS, Yeo JC, Tan CP, Gao B, Le B, Tan ZY, Low KY, et al. (2015). Deterministic Restriction on Pluripotent State Dissolution by Cell-Cycle Pathways. Cell 162, 564–579. [DOI] [PubMed] [Google Scholar]
- Hitomi M, Yang K, Guo Y, Fretthold J, Harwalkar J, and Stacey DW (2006). p27Kip1 and cyclin dependent kinase 2 regulate passage through the restriction point. Cell Cycle 5, 2281–2289. [DOI] [PubMed] [Google Scholar]
- Hosoya T, Clifford M, Losson R, Tanabe O, and Engel JD (2013). TRIM28 is essential for erythroblast differentiation in the mouse. Blood 122, 3798–3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, and Salzberg SL (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krassowska A, Gordon-Keylock S, Samuel K, Gilchrist D, Dzierzak E, Oostendorp R, Forrester LM, and Ansell JD (2006). Promotion of haematopoietic activity in embryonic stem cells by the aorta-gonad-mesonephros microenvironment. Exp. Cell Res 312, 3595–3603. [DOI] [PubMed] [Google Scholar]
- Lai L, Bohnsack BL, Niederreither K, and Hirschi KK (2003). Retinoic acid regulates endothelial cell proliferation during vasculogenesis. Development 130, 6465–6474. [DOI] [PubMed] [Google Scholar]
- Larsson J, and Karlsson S (2005). The role of Smad signaling in hematopoiesis. Oncogene 24, 5676–5692. [DOI] [PubMed] [Google Scholar]
- Lawson ND, Vogel AM, and Weinstein BM (2002). Sonic hedgehog and vascular endothelial growth factor act upstream of the Notch pathway during arterial endothelial differentiation. Dev. Cell 3, 127–136. [DOI] [PubMed] [Google Scholar]
- Lengerke C, Grauer M, Niebuhr NI, Riedt T, Kanz L, Park IH, and Daley GQ (2009). Hematopoietic development from human induced pluripotent stem cells. Ann. N Y Acad. Sci 1176, 219–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcelo KL, Goldie LC, and Hirschi KK (2013a). Regulation of endothelial cell differentiation and specification. Circ. Res 112, 1272–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcelo KL, Sills TM, Coskun S, Vasavada H, Sanglikar S, Goldie LC, and Hirschi KK (2013b). Hemogenic endothelial cell specification requires c-Kit, Notch signaling, and p27-mediated cell-cycle control. Dev. Cell 27, 504–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvinsky A, and Dzierzak E (1996). Definitive hematopoiesis is autonomously initiated by the AGM region. Cell 86, 897–906. [DOI] [PubMed] [Google Scholar]
- Melincovici CS, Boşxca AB, Şxuşxman S, Mărginean M, Mihu C, Istrate M, Moldovan IM, Roman AL, and Mihu CM (2018). Vascular endothelial growth factor (VEGF) - key factor in normal and pathological angiogenesis. Rom. J. Morphol. Embryol 59, 455–467. [PubMed] [Google Scholar]
- Müller AM, Medvinsky A, Strouboulis J, Grosveld F, and Dzierzak E (1994). Development of hematopoietic stem cell activity in the mouse embryo. Immunity 1, 291–301. [DOI] [PubMed] [Google Scholar]
- Nadin BM, Goodell MA, and Hirschi KK (2003). Phenotype and hematopoietic potential of side population cells throughout embryonic development. Blood 102, 2436–2443. [DOI] [PubMed] [Google Scholar]
- Nagai R, Suzuki T, Aizawa K, Shindo T, and Manabe I (2005). Significance of the transcription factor KLF5 in cardiovascular remodeling. J. Thromb. Haemost 3, 1569–1576. [DOI] [PubMed] [Google Scholar]
- Niederreither K, Subbarayan V, Dollé P, and Chambon P (1999). Embryonic retinoic acid synthesis is essential for early mouse post-implantation development. Nat. Genet 21, 444–448. [DOI] [PubMed] [Google Scholar]
- Niwa A, Heike T, Umeda K, Oshima K, Kato I, Sakai H, Suemori H, Nakahata T, and Saito MK (2011). A novel serum-free monolayer culture for orderly hematopoietic differentiation of human pluripotent cells via mesodermal progenitors. PLOS ONE 6, e22261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauklin S, and Vallier L (2013). The cell-cycle state of stem cells determines cell fate propensity. Cell 155, 135–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phipson B, Lee S, Majewski IJ, Alexander WS, and Smyth GK (2016). Robust Hyperparameter Estimation Protects against Hypervariable Genes and Improves Power to Detect Differential Expression. Ann. Appl. Stat 10, 946–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poppy Roworth A, Ghari F, and La Thangue NB (2015). To live or let die - complexity within the E2F1 pathway. Mol. Cell. Oncol 2, e970480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, et al. (2008). Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 132, 487–498. [DOI] [PubMed] [Google Scholar]
- Sarkar A, and Hochedlinger K (2013). The sox family of transcription factors: versatile regulators of stem and progenitor cell fate. Cell Stem Cell 12, 15–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M, and Sebastian M (2018). Palbociclib-The First of a New Class of Cell Cycle Inhibitors. Recent Results Cancer Res. 211, 153–175. [DOI] [PubMed] [Google Scholar]
- Singh AM, Chappell J, Trost R, Lin L, Wang T, Tang J, Matlock BK, Weller KP, Wu H, Zhao S, et al. (2013). Cell-cycle control of developmentally regulated transcription factors accounts for heterogeneity in human pluripotent cells. Stem Cell Reports 1, 532–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriram G, Tan JY, Islam I, Rufaihah AJ, and Cao T (2015). Efficient differentiation of human embryonic stem cells to arterial and venous endothelial cells under feeder- and serum-free conditions. Stem Cell Res. Ther 6, 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staal FJ, and Luis TC (2010). Wnt signaling in hematopoiesis: crucial factors for self-renewal, proliferation, and cell fate decisions. J. Cell. Biochem 109, 844–849. [DOI] [PubMed] [Google Scholar]
- Taoudi S, and Medvinsky A (2007). Functional identification of the hematopoietic stem cell niche in the ventral domain of the embryonic dorsal aorta. Proc. Natl. Acad. Sci. USA 104, 9399–9403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumbar T, and Belmont AS (2001). Interphase movements of a DNA chromosome region modulated by VP16 transcriptional activator. Nat. Cell Biol 3, 134–139. [DOI] [PubMed] [Google Scholar]
- Wulf GG, Luo KL, Jackson KA, Brenner MK, and Goodell MA (2003). Cells of the hepatic side population contribute to liver regeneration and can be replenished with bone marrow stem cells. Haematologica 88, 368–378. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA-seq data reported in this paper is Gene Expression Omnibus (GEO): GSE159694.