

Abstract
Objective
Pathogenic variants in TNNT3, the gene encoding fast skeletal muscle troponin T, were first described in autosomal dominant distal arthrogryposis type 2B2. Recently, a homozygous splice site variant, c.681+1G>A, was identified in a patient with nemaline myopathy and distal arthrogryposis. Here, we describe the second individual with congenital myopathy associated with biallelic TNNT3 variants.
Methods
Clinical exome sequencing data from a patient with molecularly undiagnosed congenital myopathy underwent research reanalysis. Clinical and histopathologic data were collected and compared with the single reported patient with TNNT3-related congenital myopathy.
Results
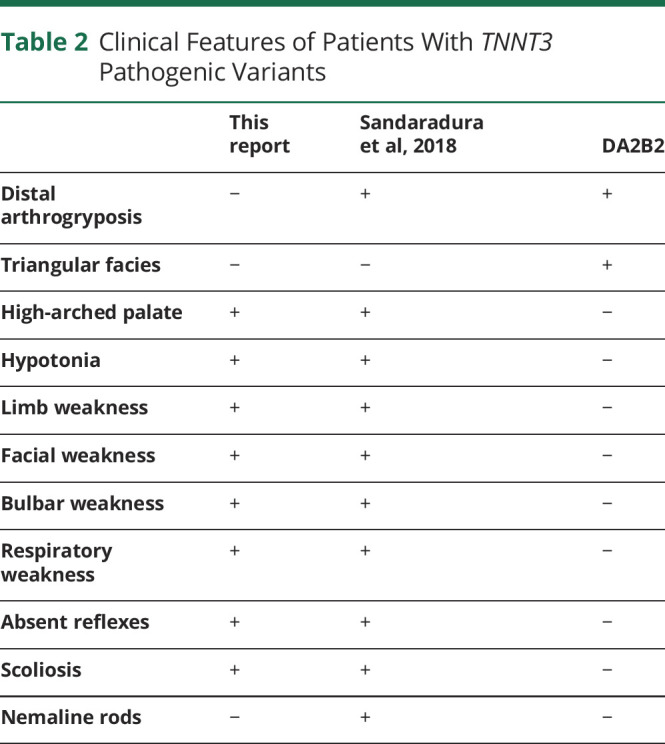
A homozygous TNNT3 variant, c.481-1G>A, was identified. This variant alters a consensus splice acceptor and is predicted to affect splicing by multiple in silico prediction tools. Both the patient reported here and the previously published patient exhibited limb, bulbar, and respiratory muscle weakness from birth, which improved over time. Other shared features include history of polyhydramnios, hypotonia, scoliosis, and high-arched palate. Distal arthrogryposis and nemaline rods, findings reported in the first patient with TNNT3-related congenital myopathy, were not observed in the patient reported here.
Conclusions
This report provides further evidence for the association of biallelic TNNT3 variants with severe recessive congenital myopathy with or without nemaline rods and distal arthrogryposis. TNNT3 sequencing and copy number analysis should be incorporated into the workup of congenital myopathies.
Pathogenic heterozygous variation in TNNT3, the gene encoding fast skeletal muscle troponin T, was first identified in families with autosomal dominant distal arthrogryposis type 2B2 (DA2B2, MIM #618435).1-3 Clinical features of DA2B2 include distal arthrogryposis and triangular facies. As a component of the troponin complex, troponin T plays an integral role in skeletal muscle contraction.4 There are 3 paralogs encoding troponin T with tissue-specific expression patterns: TNNT1 (slow skeletal), TNNT2 (cardiac), and TNNT3 (fast skeletal).4 To date, all DA2B2-associated TNNT3 variants involve a single arginine residue, p.Arg63 (p.Arg63Ser, p.Arg63Cys, and p.Arg63His).1-3 The TNNT3 variant p.Arg63His increased contractile force in rabbit psoas muscle fibers, suggesting that DA2B2 results from a gain-of-function (GoF) mechanism.5 More recently, a neonate with nemaline myopathy, distal arthrogryposis, and a homozygous TNNT3 splice variant, c.681+1G>A, was reported.6 Here, we describe the second patient with biallelic variation at the locus; a novel homozygous TNNT3 canonical splice site variant associated with congenital myopathy.
Methods
The patient was identified by local collaborators and was recruited through the Baylor-Hopkins Center for Mendelian Genomics initiative as part of an adult and pediatric neuromuscular disease cohort.
Standard Protocol Approvals, Registrations, and Patient Consents
Written informed consent including consent to publish photographs was obtained under Baylor College of Medicine Institutional Review Board Protocol H-29697.
Clinical exome sequencing (ES) was performed by Baylor Genetics (Houston, TX). The resulting FASTQ file was transferred to the Baylor-Hopkins Center for Mendelian Genomics for reanalysis through our in-house pipeline as previously described.7-9 Absence of heterozygosity (AOH) was determined via BafCalculator and based on calculated B-allele frequency from unphased exome data as described.8 As the AOH region size cutoff for BafCalculator is >1.5 Mb, BAM files were manually inspected using Integrative Genomics Viewer (software.broadinstitute.org/software/igv/) for evidence of smaller AOH regions.
Data Availability
Anonymized data not published within this article will be made available by request from any qualified investigator.
Results
The male proband was born full term to unrelated Latino parents (figure 1, A and B). Prenatal complications included decreased fetal movement and polyhydramnios. Birth was complicated by respiratory failure requiring noninvasive ventilatory support. The patient ultimately required tracheostomy for persistent respiratory failure and gastrostomy placement for dysphagia before hospital discharge at age 3 months. He also had excessive drooling, which required treatment with glycopyrrolate. Although developmentally delayed, the patient was able to sit independently at age 1 year and by age 3 years could stand with support. Examination at 3 years demonstrated limb, truncal, and neck flexion weakness, myopathic facies, axial and appendicular hypotonia, absent reflexes, ligamentous laxity, scoliosis, pectus carinatum, high-arched palate, and bitemporal narrowing (figure 1C). He exhibited antigravity or greater strength in all muscle groups examined without any difference between proximal or distal muscle groups. Echocardiogram and ECG were normal. He required continuous positive airway pressure at night but no respiratory support during the day. Biopsy of the right quadriceps was performed at age 4 years (figure 1D). Marked fiber size variability was observed. Poor fiber type differentiation was appreciated with the myosin ATPase reaction (data not shown). No necrotic, degenerating, or regenerating fibers were appreciated. There was significant fatty infiltration, increase in perimysial and endomysial connective tissue, and thickening of fibrous septa. Scattered fibers with increased internal nuclei were detected (data not shown). No central cores or nemaline rods were detected. At 4 years, he was accidentally disconnected from his trach collar and had catastrophic hypoxic-ischemic brain injury.
Figure 1. Homozygous Splice Site TNNT3 Variants Cause a Severe Congenital Myopathy.

(A) Family pedigree. The proband is the only affected child of 2 unrelated parents of Latin American descent (father's origin: Mexico; mother's origin: Argentina). Exome sequencing identified a homozygous TNNT3 c.481-1G>A variant in the proband. His parents were not available for testing. (B) Sanger sequencing confirming the genotype determined by exome sequencing in the proband. (C) X-ray showing severe neuromuscular scoliosis. (D) Muscle histology of the proband at age 4 years. H&E staining (D.a) demonstrates fiber size variability, fatty infiltration, and increased perimysial and endomysial connective tissue. Gomori trichrome staining (D.b, D.c) failed to identify nemaline rods. (D.a, D.b) 100× magnification, (D.c) 200× magnification.
Research reanalysis of ES data identified a homozygous splice variant in TNNT3 (NM_006757.4):c.481-1G>A at position GRCh37/hg19: chr11:1955775 (figure 1B and table 1). The variant was absent in our internal database of >14,000 exomes and has a gnomAD minor allele frequency of 1.655 x 10−5 (4 heterozygotes). Three of the 4 heterozygotes in gnomAD are Latino, and the remaining heterozygote is categorized as “Other”. Consistent with the lack of known parental consanguinity, B-allelic frequency analysis detected a total autosomal AOH size of 60.3 Mb. Manual inspection of the patient's BAM file revealed a 565.2 Kb AOH block surrounding the variant (figure S1, links.lww.com/NXG/A413). Taken together with population data, these AOH data from ES suggest that TNNT3: c.481-1G>A may represent a Clan Genomics derived founder mutation in the Latino population.10,11 The nucleotide is well conserved (figure 2B), and the variant is predicted damaging by multiple variant annotation tools (MutationTaster: Disease causing; CADD score: 33) and splicing predictors (table 1).
Table 1.
Summary of TNNT3 Pathogenic Alleles

Figure 2. TNNT3 Variant Details and Model of TNNT3-Related Disease.

(A) Exon structure of TNNT3 (NM_006757.4). TNNT3 is located on chromosome 11p15.5. Variants linked to autosomal recessive congenital myopathy are indicated in red, whereas variants associated with autosomal dominant DA2B2 are in blue. (B) Location and conservation of TNNT3 splice variants c.481-1G>A and c.681+1G>A. Intronic sequences are indicated in lowercase. Exonic sequences are indicated in upper case. Consensus splice sites are indicated in bold. The variant location in highlighted in orange. (C) Predicted impact of c.481-1G>A on splicing and final protein product. Consensus splice sites are indicated in bold. The variant location is highlighted in orange. Normal splicing and translation of wild-type TNNT3 mRNA (NM_006757.4) results in a 258 amino acid protein. The c.481-1G>A substitution is predicted to cause loss of the consensus splice acceptor and result in the creation of a new splice acceptor shifted +1 bp in the 3′ direction. This new splice acceptor should result in a −1 frameshift and therefore is predicted to cause a prematurely truncated protein, p.Ala161LeufsTer14. (D) TNNT3 disease model. Genotype is indicated above the colored box, and phenotype is displayed below. Monoallelic LoF TNNT3 variants are tolerated (green), whereas biallelic LoF TNNT3 variants (blue) cause severe congenital myopathy with respiratory and bulbar muscle weakness and scoliosis. In contrast, gain-of-function missense variants increasing contractility (red) result in autosomal dominant DA2B2.
Discussion
Although TNNT3 was first associated with autosomal dominant DA2B2 (MIM #618435),1-3 the severe phenotype of Tnnt3−/− mice suggests that human biallelic loss of function (LoF) might cause a more severe neuromuscular disease than previously recognized.12 Although crosses of wild-type and Tnnt3+/− mice produced offspring with a normal mendelian distribution, no adult Tnnt3−/− mice resulted from Tnnt3+/− x Tnnt3+/− crosses.12 However, Tnnt3−/− embryos were detected at embryonic day 18.5, and dead Tnnt3−/− pups were found shortly after birth. As troponin T3 is robustly expressed in the diaphragm and Tnnt3−/− pups had no heart or lung abnormalities, perinatal mortality was attributed to respiratory failure. These observations are consistent with troponin T3's known role as a critical component of the type 2 fiber troponin complex required for muscle contraction.4
This report details the second patient with congenital myopathy associated with biallelic TNNT3 variants and the fifth pathogenic TNNT3 variant (figure 2A). Both patients with congenital myopathy had homozygous variants involving consensus splice sites (Sandaradura et al,6 c.681+1G>A; this report, c.481-1G>A; figure 2B). c.681+1G>A is predicted to cause loss of a canonical splice donor site by multiple in silico algorithms (e.g., SpliceAI, MMSp, Human Splice Finder, table 1). cDNA from the first patient's muscle biopsy showed exon 14 skipping and increased intron 14 retention, which are predicted to cause either premature truncation or exonization.6 Western blot demonstrated severe reduction in troponin T3 protein with no truncated or elongated protein products.6 Thus, any abnormal splicing products not subject to nonsense mediated decay were likely unstable and degraded. The variant reported here, c.481-1G>A, is predicted to cause acceptor loss by SpliceAI, MMSp and Human Splice Finder and acceptor gain by SpliceAI and Human Splice Finder (table 1). Therefore, this substitution should shift the splice acceptor site +1 bp in the 3′ direction, resulting in a −1 frameshift and a prematurely truncated protein product, p.Ala161LeufsTer14 (figure 2C). As the resulting premature termination codon falls within the 13th of 16 exons, the mutant mRNA should be subject to nonsense-mediated decay.13 c.481-1G>A is therefore likely to represent a LoF allele, but this cannot be definitively established without additional studies examining its impact on TNNT3 expression and function. Another possibility would be that c.481-1G>A could cause GoF. For example, a recurrent de novo dominantly acting splice gain variant in COL6A1 was recently identified in patients with collagen VI–related dystrophy. The variant, COL6A1(NM_001848.3):c.930+189C>T, recurs due to a CpG dinucleotide and results in the inclusion of an additional 24 amino acids within a G-X-Y repeat region. Disruption of such G-X-Y repeat regions is a well-known mechanism of dominant negative disease in collagen disorders. If TNNT3:c.481-1G>A does cause GoF, it must influence TNNT3 function in a manner distinct from the known TNNT3 GoF variant c.188G>A (p.Arg63His), as neither of the patient's parents had DA2B2.
Shared clinical features between the 2 patients include bulbar, respiratory, and limb weakness, history of polyhydramnios, scoliosis, and high-arched palate (table 2). Both required gastrotomy due to bulbar weakness. The patient reported here has a persistent requirement for ventilatory support at night, whereas the patient described by Sandaradura et al6 required lifelong noninvasive ventilation. In contrast, Sandaradura et al's6 patient exhibited distal arthrogryposis and nemaline rods, whereas our patient did not. Arthrogryposis results from fetal akinesia and can be a variable finding in many congenital neuromuscular disorders.14 Similarly, nemaline rods are not always present in nemaline myopathy or may only manifest with age.15 Neither patient has the triangular facies seen in DA2B2.1-3
Table 2.
Clinical Features of Patients With TNNT3 Pathogenic Variants
TNNT1, the TNNT3 paralog encoding slow skeletal muscle troponin T, is responsible for nemaline myopathy 5, Amish type (NEM5; MIM #605355).16-18 First described in consanguineous Amish families in which a homozygous nonsense variant in exon 11 of TNNT1 segregated with disease, NEM5 has subsequently been reported in multiple ethnic groups (Dutch, Hispanic, Palestinians, Ashkenazi, and French) due to other biallelic TNNT1 variants (e.g., exonic deletions, other nonsense variants, and splice variants).15 NEM5 has neonatal onset and a progressive, fatal course with muscle weakness and atrophy, contractures, and chest wall rigidity causing pectus carinatum. In a large cohort of children with NEM5 of Amish ancestry, median survival was 18 months.17 In contrast, both patients with TNNT3-related congenital myopathy exhibited gradual improvement in limb strength.6 These observations suggest differential capacity for postnatal adaptation to TNNT1 vs TNNT3 deficiency in humans. As both patients with TNNT3-related congenital myopathy had improving limb strength but persistent respiratory weakness and respiratory-related complications (i.e., mortality or hypoxic-ischemic brain injury), there may also be differential capacity for postnatal adaptation to TNNT3 deficiency between muscle groups. The comparison between NEM5 and TNNT3-related congenital myopathy may also suggest the existence of milder TNNT3-related myopathies. An example of this is the recent identification of a homozygous TNNT1 missense variant, which segregated with slowly progressive limb-girdle muscle weakness, spine rigidity, contractures, and restrictive lung disease in a French-Canadian family.19
In conclusion, this report describes the second patient with biallelic TNNT3 variants and severe congenital myopathy associated with respiratory and bulbar muscle weakness and scoliosis. Although this does not definitively establish TNNT3 as a cause of congenital myopathy, these 2 case reports combined with animal model data provide compelling evidence for the existence of TNNT3-related congenital myopathy. TNNT3 sequencing and copy number analysis should therefore be incorporated into the workup of congenital myopathies. Moreover, all patients with congenital myopathy with unsolved clinical exomes should undergo reanalyses of extant ES data and consider WGS studies and perhaps RNA-seq on muscle biopsy.20-22
Glossary
- AOH
absence of heterozygosity
- ES
exome sequencing
- GoF
gain of function
- LoF
loss of function
Appendix. Authors

Contributor Information
Daniel G. Calame, Email: daniel.calame@bcm.edu.
Jawid Fatih, Email: jawid.fatih@bcm.edu.
Isabella Herman, Email: isabellg@bcm.edu.
Zeynep Coban Akdemir, Email: zeynep.cobanakdemir@bcm.edu.
Haowei Du, Email: haoweid@bcm.edu.
Shalini N. Jhangiani, Email: jhangian@bcm.edu.
Richard A. Gibbs, Email: agibbs@bcm.edu.
Dana Marafi, Email: dana.marafi@bcm.edu.
Davut Pehlivan, Email: pehlivan@bcm.edu.
Jennifer E. Posey, Email: jennifer.posey@bcm.edu.
Timothy Lotze, Email: tlotze@bcm.edu.
Pedro Mancias, Email: pedro.mancias@uth.tmc.edu.
Meenakshi Bidwai Bhattacharjee, Email: meenakshi.b.bhattacharjee@uth.tmc.edu.
Study Funding
This study was supported in part by the US National Human Genome Research Institute (NHGRI) and National Heart Lung and Blood Institute (NHLBI) to the Baylor-Hopkins Center for Mendelian Genomics (BHCMG, UM1 HG006542, J.R.L.) and NHGRI grant to Baylor College of Medicine Human Genome Sequencing Center (U54HG003273 to R.A.G.), US National Institute of Neurological Disorders and Stroke (NINDS) (R35NS105078 to J.R.L.), and Muscular Dystrophy Association (MDA) (512848 to J.R.L.). D.M. is supported by a Medical Genetics Research Fellowship Program through the United States National Institutes of Health (T32 GM007526-42). D.P. is supported by a Clinical Research Training Scholarship in Neuromuscular Disease partnered by the American Academy of Neurology (AAN), American Brain Foundation (ABF), and Muscle Study Group (MSG) and International Rett Syndrome Foundation (IRSF grant #3701-1). J.E.P. was supported by NHGRI K08 HG008986.
Disclosure
J.R. Lupski has stock ownership in 23andMe; is a paid consultant for Regeneron Genetics Center; and is a coinventor on multiple United States and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, and bacterial genomic fingerprinting. The Department of Molecular and Human Genetics at Baylor College of Medicine receives revenue from clinical genetic testing conducted at Baylor Genetics (BG) Laboratories. The other authors report no disclosures relevant to the manuscript. Go to Neurology.org/NG for full disclosures.
References
- 1.Sung SS, Brassington AM, Krakowiak PA, Carey JC, Jorde LB, Bamshad M. Mutations in TNNT3 cause multiple congenital contractures: a second locus for distal arthrogryposis type 2B. Am J Hum Genet. 2003;73(1):212–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhao N, Jiang M, Han W, et al. A novel mutation in TNNT3 associated with Sheldon-Hall syndrome in a Chinese family with vertical talus. Eur J Med Genet. 2011;54(3):351–353. [DOI] [PubMed] [Google Scholar]
- 3.Beck AE, McMillin MJ, Gildersleeve HI, et al. Spectrum of mutations that cause distal arthrogryposis types 1 and 2B. Am J Med Genet A. 2013;161A(3):550–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wei B, Jin JP. Troponin T isoforms and posttranscriptional modifications: evolution, regulation and function. Arch Biochem Biophys. 2011;505(2):144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robinson P, Lipscomb S, Preston LC, et al. Mutations in fast skeletal troponin I, troponin T, and beta-tropomyosin that cause distal arthrogryposis all increase contractile function. FASEB J. 2007;21(3):896–905. [DOI] [PubMed] [Google Scholar]
- 6.Sandaradura SA, Bournazos A, Mallawaarachchi A, et al. Nemaline myopathy and distal arthrogryposis associated with an autosomal recessive TNNT3 splice variant. Hum Mutat. 2018;39(3):383–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karaca E, Harel T, Pehlivan D, et al. Genes that affect brain structure and function identified by rare variant analyses of mendelian neurologic disease. Neuron. 2015;88(3):499–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eldomery MK, Coban-Akdemir Z, Harel T, et al. Lessons learned from additional research analyses of unsolved clinical exome cases. Genome Med. 2017;9(1):26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karaca E, Posey JE, Coban Akdemir Z, et al. Phenotypic expansion illuminates multilocus pathogenic variation. Genet Med. 2018;20(12):1528–1537.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lupski JR, Belmont JW, Boerwinkle E, Gibbs RA. Clan genomics and the complex architecture of human disease. Cell. 2011;147(1):32–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gonzaga-Jauregui C, Yesil G, Nistala H, et al. Functional biology of the Steel syndrome founder allele and evidence for clan genomics derivation of COL27A1 pathogenic alleles worldwide. Eur J Hum Genet. 2020;28(9):1243–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ju Y, Li J, Xie C, et al. Troponin T3 expression in skeletal and smooth muscle is required for growth and postnatal survival: characterization of Tnnt3(tm2a(KOMP)Wtsi) mice. Genesis. 2013;51(9):667–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coban-Akdemir Z, White JJ, Song X, et al. Identifying genes whose mutant transcripts cause dominant disease traits by potential gain-of-function alleles. Am J Hum Genet. 2018;103(2):171–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pehlivan D, Bayram Y, Gunes N, et al. The genomics of arthrogryposis, a complex trait: candidate genes and further evidence for oligogenic inheritance. Am J Hum Genet. 2019;105(1):132–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sewry CA, Laitila JM, Wallgren-Pettersson C. Nemaline myopathies: a current view. J Muscle Res Cell Motil. 2019;40(2):111–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnston JJ, Kelley RI, Crawford TO, et al. A novel nemaline myopathy in the Amish caused by a mutation in troponin T1. Am J Hum Genet. 2000;67(4):814–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fox MD, Carson VJ, Feng HZ, et al. TNNT1 nemaline myopathy: natural history and therapeutic frontier. Hum Mol Genet. 2018;27(18):3272–3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Géraud J, Dieterich K, Rendu J, et al. Clinical phenotype and loss of the slow skeletal muscle troponin T in three new patients with recessive TNNT1 nemaline myopathy. J Med Genet. 2020. doi: 10.1136/jmedgenet-2019-106714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pellerin D, Aykanat A, Ellezam B, et al. Novel recessive TNNT1 congenital core-rod myopathy in French Canadians. Ann Neurol. 2020;87(4):568–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cummings BB, Marshall JL, Tukiainen T, et al. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci Transl Med. 2017;9(386):eaal5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gonorazky HD, Naumenko S, Ramani AK, et al. Expanding the boundaries of RNA sequencing as a diagnostic tool for rare mendelian disease. Am J Hum Genet. 2019;104(5):1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murdock DR, Dai H, Burrage LC, et al. Transcriptome-directed analysis for Mendelian disease diagnosis overcomes limitations of conventional genomic testing. J Clin Invest. 2020;131:e141500. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data not published within this article will be made available by request from any qualified investigator.