

Abstract
Objective
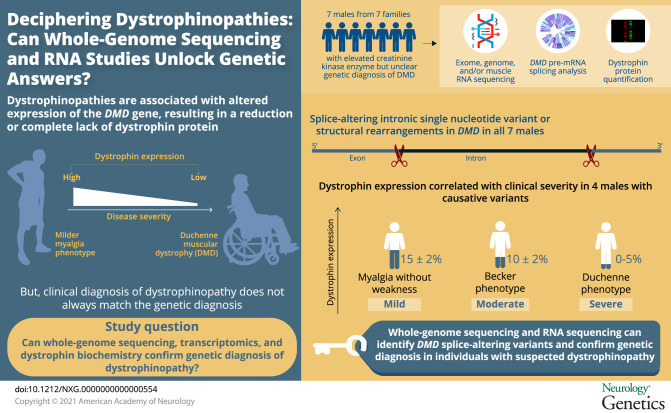
To describe the diagnostic utility of whole-genome sequencing and RNA studies in boys with suspected dystrophinopathy, for whom multiplex ligation-dependent probe amplification and exomic parallel sequencing failed to yield a genetic diagnosis, and to use remnant normal DMD splicing in 3 families to define critical levels of wild-type dystrophin bridging clinical spectrums of Duchenne to myalgia.
Methods
Exome, genome, and/or muscle RNA sequencing was performed for 7 males with elevated creatine kinase. PCR of muscle-derived complementary DNA (cDNA) studied consequences for DMD premessenger RNA (pre-mRNA) splicing. Quantitative Western blot was used to determine levels of dystrophin, relative to control muscle.
Results
Splice-altering intronic single nucleotide variants or structural rearrangements in DMD were identified in all 7 families. Four individuals, with abnormal splicing causing a premature stop codon and nonsense-mediated decay, expressed remnant levels of normally spliced DMD mRNA. Quantitative Western blot enabled correlation of wild-type dystrophin and clinical severity, with 0%–5% dystrophin conferring a Duchenne phenotype, 10% ± 2% a Becker phenotype, and 15% ± 2% dystrophin associated with myalgia without manifesting weakness.
Conclusions
Whole-genome sequencing relied heavily on RNA studies to identify DMD splice-altering variants. Short-read RNA sequencing was regularly confounded by the effectiveness of nonsense-mediated mRNA decay and low read depth of the giant DMD mRNA. PCR of muscle cDNA provided a simple, yet informative approach. Highly relevant to genetic therapies for dystrophinopathies, our data align strongly with previous studies of mutant dystrophin in Becker muscular dystrophy, with the collective conclusion that a fractional increase in levels of normal dystrophin between 5% and 20% is clinically significant.
Duchenne muscular dystrophy (DMD) is a severe X-linked disorder primarily affecting approximately 1 in 5,000 male births.1–3 DMD shows a relentlessly progressive course, resulting in loss of ambulation in teens, and early mortality due to cardiac or respiratory involvement.4,5 Dystrophinopathies range clinically from the severe DMD to asymptomatic hyperCKemia.5–12 DMD is associated with the absence of dystrophin in muscle due to loss-of-function variants in the DMD gene encoding dystrophin,5,6 whereas Becker muscular dystrophy (BMD) is associated with variants in DMD that result in reduced levels of (mutated) dystrophin.5,6
The DMD gene is the largest gene in the human genome, with numerous enormous introns.13,14 One-third of pathogenic DMD variants are de novo,15,16 with most affected individuals bearing insertions or deletions (indels) of coding exons.15,17 Pathogenic DMD missense variants are rare,6,15,18 and noncoding variants are emerging as an important rare cause of dystrophinopathy.15,17,19,20 Approximately 5% of patients clinically diagnosed with DMD do not have a genetic diagnosis after mutational analysis.5
Herein, we show the diagnostic application of whole-genome sequencing, transcriptomics, and dystrophin protein biochemistry to secure a genetic diagnosis for 13 affected males from 7 families with elevated creatine kinase (CK) who remained undiagnosed following multiplex ligation-dependent probe amplification (MLPA) and exomic sequencing. Importantly, we identify 3 families with DMD splicing variants who produce varying levels of mis-spliced transcripts that encode a premature stop codon and are targeted by nonsense-mediated decay, though express varying levels of remnant, normally spliced DMD mRNA. Therefore, quantitative Western blot (WB) of muscle biopsy specimens from these 3 dystrophin hypomorphs has uniquely enabled specific correlation of levels of wild-type (WT) dystrophin with clinical severity.
Methods
Standard Protocol Approvals, Registrations, and Patient Consents
This study was approved by the Children's Hospital at Westmead Human Research Ethics Committee (Biospecimen Bank_10/CHW/45) with informed, written consent from all participants.
We describe a retrospective cohort of boys diagnosed with DMD variants from genomic and RNA studies, who had elevated CK and dystrophic muscle biopsies, and were undiagnosed after MLPA and exomic parallel sequencing.
Immunohistochemistry and Western Blotting
Immunohistochemistry21 and Western blotting22 were performed as previously described; WB used NuPAGE 3%–8% Tris-Acetate precast gels (Invitrogen by Thermo Fisher Scientific, NSW, Australia). Antibodies: for immunohistochemistry, muscle fiber membranes were stained with anti-dystrophin DYS1, DYS2, DYS3, and anti-spectrin SPEC1 (Leica Biosystems, VIC, Australia); with anti-mouse Alexa Fluor 555 secondary antibody, membranes were counterstained with wheat germ agglutinin-AF488 (WGA), and nuclei were stained with DAPI (Invitrogen Thermo Fisher Scientific). WBs were probed with DYS1 (Leica Biosystems), rabbit polyclonal dystrophin antibody (Rb-DMD; ab15277; Abcam), α-actinin-2 (4A3, gift from A. Beggs, Children's Hospital Boston, Boston, MA), sarcomeric actin (clone 5C5, A2172; Sigma-Aldrich), and the anti-mouse or anti-rabbit IgG light chain HRP-conjugated secondary antibodies (GE Healthcare, NSW, Australia). The rabbit polyclonal dystrophin antibody (Rb-DMD; ab15277; Abcam) detects a 10-fold serial dilution, whereas DYS2 is less sensitive (detects a 4-fold serial dilution). Therefore, ab15277 was selected due to provision of a more informative standard curve for semiquantification of dystrophin levels in the probands. ImageJ23 was used to measure the densities of the patient and serially diluted controls bands to create a standard curve, as previously described.19 Semiquantitation of dystrophin levels was performed by comparing densities of the dystrophin band in patient sample relative to the standard curves of dystrophin in 2 age- and sex-matched controls across 3 experimental replicates.
Massively Parallel Sequencing
Whole-exome sequencing (probands and AI:1, AI:2, and AII:2), PCR-free whole-genome sequencing (probands from families A and B, D–G), and RNA sequencing (RNA-seq; probands from families A, B, D, E, and G) were performed at the Broad Institute of Harvard and MIT as previously described.20 RNA-seq was performed for CII:2 at PathWest Laboratory Medicine WA as previously described for the fetal samples in reference 24.
Sanger Sequencing and RT-PCR
RNA was extracted, and reverse transcription PCR (RT-PCR) was performed as previously described.25 Primers used for AII:1 have been previously described.20 The remaining primer details are as follows: Ex42F 5′-CAATGCTCCTGACCTCTGTG-3′; Ex43/44R 5′-CTGTCAAATCGCCCTTGTCG-3′; LINC00251Ex3R 5′-CTGAAATGGGTGGGATGAAG-3′; LINC00251Ex2F 5′-GATGCCCCTTAACCAAGGAC-3′; Ex26F 5′-GATGCACGAATGGATGACAC-3′; Ex27R 5′-TGTGCTACAGGTGGAGCTTG-3′; Ex26/27F 5′-GCAGTTGAAGAGATGAAGAGAGC-3′; Ex29R 5′-TGGGTTATCCTCTGAATGTCG-3′; In26PF 5′-AAA-CTTAGTTCGGCCCCATG-3′; Ex48F 5′-GTTAAATCATCTGCTGCTGTGG-3′; Ex54R 5′-ACTGGCGGAGGTCTTTGG-3′; Ex49/52F 5′-ACTCAGCCAGTGA-AGGCAAC-3′; Ex53R 5′-TCCTAAGACCTGCTCAGCT-TC-3′; Ex51F 5′-CGACTGGCTTTCTCTGCTTG-3′; Ex50/52F 5′-CAAATCCTGCATTGTTGCAGG-3′; GAPDHEx3F 5′-TCACCAGGGCTGCTTTTAAC-3′; and GAPDHEx6R 5′-GGCAGAGATGATGACCCTTT-3′. Confirmation and segregation analysis of DMD variants was performed by Sanger sequencing,21 except for family F in which DNA was not available. Primers used for families A, D, E, and G have been previously described.20 The remaining primer details are as follows: family B—In43F 5′-TTTAGTTTCCAGCCACTCCTGTC-3′ with chr8R 5′-TAGCAGGGGCAAGG-GTTG-3′ and chr8F 5′-TGCCTCTCCAGAATGAGGAC-3′ with In43R 5′-CGGGGAACATCACACACC-3′ to confirm insertion breakpoints; family C—In26F 5′-CGAAGGAAAC-TGGTATGTAG-3′ with In26R 5′-AAAGCCGTATGACAGATTCG-3′ to determine causative variant. PCR conditions were 5 minutes 95°C; 35 cycles—30 seconds 95°C, 30 seconds 58°C, and 1 minute 72°C; 8 minutes 72°C; or as described in reference 20.
Whole-Genome Sequencing Analysis
PCR-Free whole-genome sequencing was performed on an Illumina HiSeq X Ten using 2 × 150 paired end reads at 30× mean coverage. The sequencing reads were aligned to the GRCh37 genome reference and single nucleotide variants (SNVs), small insertions and deletions (indels) were detected using methods previously described in reference 20. A reanalysis of rare (Genome Aggregation Database [gnomAD] AF < 0.005) SNVs and indels revealed no pathogenic DMD variants. The Manta tool from Illumina (PMID: 26647377) was used to identify structural variants or split read abnormalities within the DMD gene. Putative structural variants were manually inspected within Integrative Genomic Browser (IGV) to validate and resolve exact breakpoints of structural rearrangements.
RNA-seq Analysis
RNA-seq analysis was performed as described in reference 20. Briefly, all samples were jointly processed and aligned with the Genotype-Tissue Expression Consortium (GTEx)26 to identify spliced reads only seen in patients or groups of patients and missing in controls. In addition, given the nature of the previously suspected diagnosis of a dystrophinopathy, in cases in which this approach did not lead to a diagnosis, exonic read depth was mapped in each patient and compared with controls and sashimi plots of patients were manually inspected using the IGV for the DMD gene. In cases, in which RNA-seq identified a mis-splicing event, patient exome and genomes were manually evaluated, depending on availability.
Data Availability
Data not published within this article are available by request from any qualified investigator.
Results
Clinical Presentation
Four families have been described previously in reference 20: AII:1 as N33; DII:1 as C3; EII:1 as C4; and GII:1 as C2. Clinical presentation, DMD variants, and dystrophin WB results are summarized in the table. Briefly, AII:1 presented at 15 years with muscle pain, fatigue, and episodes of myoglobinuria with exercise and elevated serum CK (CK 1,400–7,500 U/L, normal range <200 U/L). He has 2 affected brothers with myalgia and elevated serum CK (300–14,700 U/L (figure 1A). Family B is a 4-generation family with an X-linked muscular dystrophy with cardiomyopathy. BIII:2 was diagnosed with dilated cardiomyopathy in his 20s, underwent cardiac transplantation at age 29 years, and died of transplant-related complications at age 31 years. BIII:7 was diagnosed with BMD in his mid-teens. He has no known history of cardiomyopathy and remains ambulant in his 40s (figure 1B). BIV:1 showed elevated serum CK 9,964 U/L at age 6 months. Now age 5 years, he has proximal muscle weakness, bilateral calf hypertrophy, and normal echocardiogram. CII:2 presented at age 9 years with progressive limb-girdle weakness, requiring intermittent use of a wheelchair from age 13 years and nocturnal bilateral positive airway pressure (BiPAP) from age 28 years. He has normal cardiac function with serum CK of 420 U/L at age 31 years (figure 1C). DII:1 presented at age 3.5 years with proximal weakness, calf hypertrophy, positive Gowers sign, and serum CK of 14,500 U/L. He required use of a wheelchair from age 13 years. Echocardiogram at age 17 years showed reduced contractility (ejection fraction 30%–35%) with normal left ventricle size (figure 1D). EII:1 presented at age 6 years with muscle weakness, enlarged calves, and serum CK of 18,889 U/L. He required use of a wheelchair at age 9 years and has no known cardiac or respiratory involvement. EII:1 has a similarly affected brother (figure 1E). FII:6 presented at age 3.5 years with proximal muscle weakness, positive Gowers sign, prominent calves, and serum CK of 24,000 U/L. He remains ambulant, but is toe walking at age 9 years. He has no known cardiac or respiratory involvement. FII:6's mother (FI:6) reports muscle pain and has elevated serum CK of ∼500 U/L (figure 1F). GII:1 presented at age 5 years with waddling gait, calf hypertrophy, positive Gowers sign, and serum CK levels of >12,000 U/L. He required use of a wheelchair from age 7 years. Echocardiogram at age 9 years showed borderline increase in heart size, and he died at age 10 years from cardiac complications (figure 1G).
Table.
Clinical Presentation, DMD Variants, and Dystrophin Western Blot

Figure 1. Pedigree of Families A–G.

Index patient for each family denoted with black arrow. Affected members colored in red, and carriers part colored in red.
DMD Diagnostic Genetic Testing
DMD MLPA and Sanger sequencing were performed and reported normal for AII:1, BIV:1, CII:2, DII:1, EII:1, and GII:1. DMD MLPA performed for FII:6 revealed duplications of exons 31–37 and 43–44, which were predicted to be in-frame and therefore considered inconsistent with his severe Duchenne-like phenotype, though with high clinical suspicion of causality. A genetic basis could not be identified via whole-exome sequencing (AII:1, BIV:1, DII:1, EII:1, FII:6, and GII:1, with duplications of exons 31–37 and 43–44 confirmed for FII:6) or massively parallel sequencing of a targeted neuromuscular gene panel (CII:2).
Immunohistochemistry Demonstrates Dystrophin Abnormalities in Skeletal Muscle Biopsies
Skeletal muscle immunohistochemistry for AII:1, BIV:1, DII:1, EII:1, FII:6, and GII:1 confirms abnormalities in dystrophin (figure e-1, links.lww.com/NXG/A367). Using 3 anti-dystrophin antibodies, AII:1 and BIV:1 showed reduced dystrophin staining, whereas DII:1, EII:1, FII:6, and GII:1 showed absent staining (figure e-1 absent dystrophin staining shown only for GII:1). WGA outlines the myofibers and labels the endomysium in patient and control skeletal muscle samples.
Correlation of Splicing Analyses With Whole-Genome Sequencing Identifies Pathogenic Intronic and Structural Variants Inducing Abnormal DMD Splicing
Six individuals (AII:1, BIV:1, DII:1, EII:1, FII:6, and GII:1) were subject to whole-genome sequencing, 6 individuals were subject to RNA-seq (AII:1, BIV:1, CII:2, DII:1, EII:1, and GII:1), and 4 individuals (AII:1, BIV:1, CII:2, and DII:1) were analyzed by RT-PCR of muscle-derived mRNA. Scrutiny of DMD transcripts (NM_004006.2, 11,058 nucleotides [nt] in length) shows typical 3′ bias in read depth (vastly more reads at the 3′ end compared with the 5′ end of DMD transcripts). Acknowledging 3′ bias, an abnormal profile of DMD transcript read depth was apparent for BIV:1, DII:1, EII:1, and GII:1 (figure 2A), relative to multiple muscle controls from the GTEx consortium.26
Figure 2. Muscle RNA Studies of DMD in Patients.

(A) RNA-seq read coverage of DMD exons in muscle RNA from AII:1, BIV:1, DII:1, EII:1, and GII:1 and 2 GTEx controls. Red arrows indicate the reduction in read depth, which corresponds with the location of DMD structural variants for BIV:1, DII:1, EII:1, and GII:1. (B–G) RT-PCR studies of muscle-derived RNA of patients with splicing abnormalities and 3 male controls (C1, quadriceps, 6.5 years; C2, vastus lateralis, 17 years; C3, unknown, 20 years). Primers used are listed at the bottom right of each gel image and are labeled according to their location (exon; Ex, intron; In, pseudoexon; P) and orientation (forward; F, reverse; R). Bridging primers span a splice junction and are denoted by X/Y, where X and Y are exons the primer spans. All results were confirmed by Sanger sequencing. (B) RT-PCR showing reduced levels of correctly spliced DMD transcript (exons 43 and 44) in AII:1 and BIV:1 compared with controls. AII:1 shows the inclusion of a 128-bp pseudoexon. (C) Primers specific to the 128 bp pseudoexon revealed that the inclusion is specific to AII:1 (Sanger sequencing showed that the faint bands in C1 were non-DMD sequences). Sequencing reveals that faint bands in AII:1 correspond to multiple pseudoexons in DMD incorporated into a minority of DMD transcripts. (D) Various chr8 pseudoexons and LINC00251 exons are included in DMD transcripts as a result of the chr8 insertion in BIV:1. The lowest band detected in all samples in the top gel corresponds to non-DMD sequences. (E) RT-PCR confirms the inclusion of a 84-bp pseudoexon in CII:2 in the majority of DMD transcripts. Normal splicing can only be detected in very low levels in CII:2 by bridging primers. The 92 bp pseudoexon is absent in control samples. (F) RT-PCR of DII:1 confirms that exon 51 is absent from all DMD transcripts. A bridging primer indicates that skipping of both exons 50 and 51 is a low frequency event observed in both controls and DII:1. (G) GAPDH loading controls to indicate that similar concentrations of complementary DNA were used for both control and patient samples. DMD = Duchenne muscular dystrophy; DMD = DMD gene or transcript; RT-PCR = reverse transcription PCR.
Standard variant filtering approaches of genomic sequencing failed to identify most causal variants. RNA-seq identified abnormal pseudoexon inclusion into DMD transcripts for families A and C. The remaining pathogenic variants were identified only through the combination of whole-genome sequencing, bioinformatics, and RNA analyses.
A genetic diagnosis in AII:1 was identified in a previous study20 with a deep intronic pathogenic variant GRCh37:ChrX:32274692G>A; c.6290+30954C>T inducing partial mis-splicing of DMD. The DMD c.6290+30954C>T variant creates a cryptic donor 5′ splice site resulting in inclusion of a variant-activated pseudoexon of 128 nt inserted between exon 43 and exon 44, which encodes 59 missense amino acids and effects a frameshift, resulting in a premature termination codon encoded by exon 44 (figure 3A). RT-PCR confirmed abnormal inclusion of the variant-activated pseudoexon and residual normal splicing of DMD exons 42-43-44-45 (figure 2, B and C).
Figure 3. Schematics of Variants Identified in Families A–G.

(A) Family A: intronic c.6290+30954C>T (black arrow) creates a cryptic donor splice site, leading to inclusion of a 128-bp pseudoexon (red, within DMD intron 43) into the DMD mRNA, causing a frameshift and stop codon (red arrow) encoded by exon 44 (ex44). Gene direction is demonstrated by gray arrows. Reading frame between exons is shown by shape complementarity. (B) Family B: insertion of 116,284 bp of chr8 (red sequence) into DMD intron 43. The insertion includes LINC00251 exons 1–3 (black outlined exons). A 124-bp sequence of intron 43 of DMD (chrX:32,276,895-32,277,018) is duplicated as part of the structural rearrangement and now flanks the chr8 insertion. In addition, there is an insertion of 13 bp (insGCCTTTGCCCACA, shown in green) adjacent to 1 copy of the 124-bp duplication. mRNA studies show evidence for numerous, different abnormal splicing events from DMD exon 43 to various pseudoexons (red exons) and LINC00251 exons (red exons with black outlines) within the chr8 insertion. Low levels of normal DMD splicing (from exons 43 and 44; blue exons) are also observed. Frame of splicing in pseudoexons and LINC00251 exons not shown. (C) Family C: intronic c.3603+820G>T (black arrow) increases the strength of the polypyrimidine tract leading to use of a cryptic acceptor splice site (3/5 algorithms within Alamut Visual biosoftware predictions; MaxEntScan, NNSPLICE, and GeneSplicer) leading to inclusion of a 84-bp pseudoexon (red, within DMD intron 26) into the DMD mRNA, encoding a stop codon (red arrow) 39 nucleotides into the pseudoexon. Gene direction is demonstrated by gray arrows. Reading frame between exons is shown by shape complementarity. (D) Family D: inversion of DMD exon 51 and flanking adjacent intronic sequence. Flanking the structural rearrangement are 2 intronic deletions (orange 3.5 kb and purple 44 bp) and an insertion of CCAATA (green). mRNA studies show exon 51 skipping, causing a frameshift and a premature stop codon (TAG, encoded by exon 52; red arrow). (E) Family E: A 2.6-Mb inversion on the X chromosome between 2 breakpoints; A in intron 45 of CFAP47, 1.9 Mb upstream of exon 1 of DMD (GRCh37:chrX:35,180,364) and B in intron 18 of DMD (GRCh37:chrX:32,521,892, NM_004006.2). This reverses the orientation of exons 1–18 of DMD, which are now joined to CFAP47 sequences upstream of exon. The DMD gene is in blue, exons dark blue, and introns light blue. Intergenic sequence (non-DMD ChrX in green). (F) Family F: A 4.1-Mb inversion on the X chromosome between 2 breakpoints; A is 3.8 Mb upstream of exon 1 of DMD (GRCh37:chrX:36236087) and B in intron 44 of DMD (GRCh37:chrX:32122714). This reverses the orientation of exons 1–44 of DMD, which are now joined to intergenic sequence upstream of exon 1. This is accompanied by duplication of exons 31–37 (orange) and exons 43 and 44 (purple) around the breakpoint. (G) Family G: A 119.8-Mb inversion on the X chromosome between 2 breakpoints; A in an intergenic region on the q arm of the X chromosome, 118 Mb upstream of exon 1 of DMD (GRCh37:chrX:151,194,962), and B in intron 60 of DMD (GRCh37:chrX:31,379,010, NM_004006.2). This reverses the orientation of exons 1–60 of DMD, which are now joined to intergenic sequence upstream of exon 1. In addition, 2 deletions were identified at these breakpoints; an intronic 63 bp deletion (orange, GRCh37:chrX:31,378,947-31,379,009) and an intergenic 18 bp deletion (purple, GRCh37:chrX:151,194,963-151,194,980). X chromosome displayed in unusual orientation with q arm to the left, so the DMD gene is presented with exons in order. DMD = DMD gene or transcript. mRNA = messenger RNA.
RNA-seq for BIV:1 showed low levels of DMD transcripts, with a distinct drop in reads from exon 44 onward (figure 2A, arrow). Bespoke realignment and analyses of WGS data identified insertion of ∼118,000 nt of chromosome 8 (chr8) sequences within DMD intron 43, encompassing the LINC00251 gene locus. RT-PCR showed that the chr8 insertion induced abnormal splicing of the DMD gene (figure 3B). Multiple adverse events were detected that involved splicing from exon 43 of DMD to various pseudoexons and LINC00251 exons within the chr8 insertion. Sanger sequencing with bespoke PCR over the breakpoints on gDNA confirmed the chr8 inclusion in intron 43 and provided a diagnostic assay that confirmed segregation of the insertion within the family pedigree. Normal splicing of DMD exons 42-43-44-45 was observed as a low-frequency event (figure 2, B and D).
For CII:2, manual analysis of RNA-seq data identified abnormal inclusion of 84 nt from intron 26 into a majority of DMD transcripts (figure 3C). Sanger sequencing of the genomic region in gDNA from CII:2 identified a deep intronic variant GRCh37:ChrX:32471959C>A, c.3603+820G>T that was absent in gnomAD. The DMD c.3603+820G>T variant in intron 26 disrupts an AG, creating an AG-exclusion zone between an available consensus lariat branch point and 3′ splice site.27 Spliceosomal use of a naturally occurring consensus 5′ splice site sequence and this strengthened 3′ splice site result in the inclusion of a variant-activated pseudoexon into a majority of DMD transcripts, encoding 19 missense amino acids followed by a stop codon (figure 3C). RT-PCR confirmed abnormal inclusion of the variant-activated pseudoexon into DMD transcripts and residual, low levels of DMD transcripts with normal splicing of exons 25-26-27 (figure 2E). Sanger sequencing confirmed that the c.3603+820G>T variant was de novo in CII:2.
For DII:1, RNA-seq in a previous study20 showed low levels of DMD transcripts with exon 51 skipping, inducing a frameshift and premature stop codon encoded by exon 52 (r.7310_7542del, p.Ser2437Cysfs*33, figure 3D). Interrogation of WGS determined presence of a DMD structural rearrangement rendering DMD exon 51 in the reverse orientation and unable to be spliced into the DMD mRNA, confirmed by Sanger sequencing. RT-PCR confirms exon 51 skipping as the predominant mis-splicing event in DII:1, with skipping of exons 50 and 51 a low-frequency, in-frame event observed in both DII:1 and controls (figure 2F). Low levels of exon 50 and 51 skipping are consistent with low levels of dystrophin detected by WB analysis (figure 4C).
Figure 4. Western Blot Panel for All Patients.

(A.a) Western blot was performed on skeletal muscle from index patients from families A and B (AII:1 and BIV:1) against DYS1 (rod domain epitope) and Rb-DMD (C-terminal epitope) with serial dilutions (1/2, 3/4, 5/6, and 9/10) human control skeletal muscle. Muscle lysate derived from an individual with Duchenne muscular dystrophy and undetectable levels of dystrophin by Western blot (DMD control; deltoid, 14-year-old boy, GRCh37:chrX:32364116G>A, NM_004006.2:c.5530C>T, p.Arg1844*) were added to diluted controls to normalize total protein loading in each lane of the gel. Loading controls: a-actinin-2 and myosin (coomassie). (A.b) Image J23 was used to measure the densities of the patient and serially diluted controls bands to create a standard curve. Quantification of relative dystrophin levels was performed by comparing patient sample densities to the control standard curves across the 3 gels shown. AII:1 demonstrates 15.5% ± 1.9% levels of dystrophin protein relative to controls. BIV:1 demonstrates 9.6% ± 1.7% levels of dystrophin protein relative to control. (B) Western blot analysis on skeletal muscle from patient CII:2 against DYS1 shows undetectable levels of dystrophin compared with controls. Loading controls: myosin (coomassie). (C) Western blot analysis on skeletal muscle from patients DII:1, EII:1, FII:6, and GII:1 against DYS1 compared with human control skeletal muscle. DII:1 shows very low levels of dystrophin. EII:1 FII:6, and GII:1 show undetectable levels of dystrophin. Loading controls: a-actinin-2 and sarcomeric actin. Male controls used: C1, tibialis anterior, 16 years; C2, unknown, 5.5 years; C3, unknown, 14 years; C4, quadriceps, 4.5 years. DMD = Duchenne muscular dystrophy.
RNA-seq showed an abrupt loss of transcripts after exon 18 in EII:1, as previously described in reference 20 (figure 2). WGS showed evidence for an inversion within the DMD gene reversing the orientation of exons 1–18 of DMD, which are now joined to intergenic sequences upstream of exon 1, explaining the presence of abruptly terminating exon 1–18 transcripts transcribed from the DMD promoter (figure 3E). The 1.9 Mb intergenic region included in the inversion contains FAM47A, FAM47B, and TMEM47 genes. Sanger sequencing of genomic DNA over the breakpoints confirmed the inversion.
FII:6 with in-frame duplications of exons 31–37 and 43–44 identified on DMD MLPA, was shown by WGS to have a larger, more complex structural rearrangement (figure 3F), which reverses the orientation of exons 1–44 of DMD which are now joined to intron 45 of CFAP47, upstream of exon 1. Expression of CFAP47 is likely to be disrupted. However, the clinical significance of loss of CFAP47 expression is unknown.
In GII:1, RNA-seq in a previous study20 showed low read count for DMD transcripts, with evidence for even fewer reads from exon 60. Closer scrutiny of whole-genome sequencing data identified a structural rearrangement reversing the orientation of exons 1–60 of DMD, which are now joined to intergenic sequences upstream of exon 1 (figure 3G). Sanger sequencing of genomic DNA over the breakpoints confirmed the inversion.
WB Analyses Define the Threshold of WT Dystrophin Conferring Clinical Phenotypes of Duchenne to Myalgia
Our splicing studies reveal that AII:1, BIV:1, and CII:2 each have residual levels of normally spliced DMD transcripts, with abnormal splicing events apparently targeted for degradation by nonsense-mediated decay (figure 2, B–E). Therefore, these individuals uniquely provide an opportunity to quantify levels of WT dystrophin and correlate with clinical phenotype. Quantitative WB (figure 4) using skeletal muscle biospecimens reveals (1) ∼15% ± 2% normal dystrophin levels in AII:1, correlating with a myalgia phenotype without apparent weakness; (2) ∼10% ± 2% levels of dystrophin in BIV:1 (figure 4A) with Becker muscular dystrophy, mild weakness, and cardiac phenotype; and (3); 0%–5% levels of dystrophin in affected individuals who present with a severe Becker (CII:2) or Duchenne phenotype (DII:1, EII:1, FII:6, and GII:1) (figure 4, B and C).
Discussion
Our study further substantiates DMD splicing variants as an important causal basis for males presenting with symptoms consistent with a dystrophinopathy, for whom exomic sequencing approaches or MLPA return negative findings. A causal splicing variant in DMD was identified in all 7 families within our dystrophinopathy cohort and includes 13 affected males presenting with hyperCKemia with pain and/or muscle weakness and/or cardiac involvement.
Importantly, identification of the causative variant in DMD within this hard-to-diagnose cohort required deployment of WGS, RNA-seq and/or bespoke RT-PCR studies of mRNA isolated from skeletal muscle. For example, for CII:2, RNA-seq was crucial to identify the inclusion of an 84 base pair (bp) pseudoexon encoding a frameshift which prompted Sanger sequencing of this region, which lead to the identification of the casual intron 26 c.3603+820G>T variant, which was undetectable by gene panel testing, Sanger sequencing of the individual exons or MLPA. Although multiple genetic investigations are costly and not available currently to many diagnostic laboratories, costs incurred through muscle biopsy, WGS, or RNA studies are insignificant relative to the cost burden to health services for dystrophinopathy cases, for example, the heart transplantation for family B. A precise genetic diagnosis for an X-linked disorder has important and wide-reaching implications for genetic, prenatal, and prognostic counseling across the wider family unit and can inform reproductive decision making. In addition, a genetic diagnosis could enable future customizable treatments such as splice-modulating antisense oligonucleotide drugs,28 which would theoretically be applicable to families A, C, and D.
Although WGS and RNA-seq bring powerful adjunct tests to clinical genomics, shortcomings of short read massively parallel sequencing were clearly observed in this study. As human exons are typically 100–150 bp in length, short-read RNA-seq is limited in that a single read does not effectively bridge multiple exons. Most significantly, RNA-seq for BIV:1, DII:1, EII:1, and GII:1 was confounded by the effectiveness of nonsense-mediated decay, an innate surveillance mechanism that degrades mRNA bearing a premature stop codon.15,17 We suspect that the reason we do not see a profound reduction in read depth for AII:1 (figure 2A) is due in part to higher read depth across the transcriptome, including DMD, and in part to the residual normal splicing of a significant proportion of DMD transcripts (27%). Nonsense-mediated decay amplifies inherent challenges associated with RNA-seq of very large mRNAs, where mRNA capture and sequencing library construction result in a characteristic bias in read depth, with vastly more reads at the 3′ end than the 5′ end of a very long mRNA. Notably, common disease genes in neuromuscular disorders are among the largest coding mRNAs in humans, with DMD mRNA ∼14,000 nt, NEB mRNA ∼50,000 nt, and TTN mRNA ∼100,000 nt. Therefore, ribosomal RNA depletion and/or long read RNA-seq approaches, which display reduced 3′ bias, may be more effective for diagnosing neuromuscular disorders.
Regular data filtering approaches of genomic sequencing failed to identify most of the causal variants (excluding families A and C found on RNA-seq). This is likely due to the nature of the variants themselves (noncanonical splice affecting variants or structural variants), small read lengths, and mapping restrictions against the reference sequence. The structural rearrangement within DMD intron 43 of family B took extensive bioinformatic analysis to delineate, even when our RT-PCR (data not shown) and RNA-seq studies had indicated intron 43 as the likely location of the problem. Although (in retrospect) the copy number variation of the duplicated region of chr8 is evident, informatics approaches to map split reads to precisely define the breakpoints were challenging and ultimately required both informatics and Sanger sequencing of PCR amplicons to fully resolve. Of note, the bespoke PCR uniquely identifying the DMD intron 43 structural rearrangement was clinically preferred as the diagnostic test for segregation and carrier testing due to its greater specificity relative to the microarray to detect the chr8 copy number variation. The availability of a validated bespoke PCR also means that carrier females in this family could have prenatal diagnosis of male pregnancies.
Although families B, D, and G have cardiac involvement that is common in dystrophinopathy, families A, C, E, and F do not have reported cardiac symptoms and are being monitored for possible development of cardiac symptoms. The profound cardiac involvement in family B raises suspicion of potential differences in DMD pre-mRNA mis-splicing between cardiac and skeletal muscle activated by the insertion of 118 kb of Chr8 sequences containing the LINC00251 gene. It is plausible that the severe cardiac involvement in family B is due to more fully penetrant DMD mis-splicing in cardiac tissue compared with skeletal muscle. Unfortunately, no stored cardiac specimens were available for mRNA studies from other affected family members who had undergone transplant surgery. It is also possible that levels of inclusion of the frameshifting pseudoexon in family C may differ between skeletal muscle (and potentially between different skeletal muscles) and cardiac muscle.
In conclusion, we highlight DMD splicing variants as an important causal basis in individuals with a suspected dystrophinopathy who remain undiagnosed after exomic sequencing or MLPA approaches. Causative DMD variants identified in AII:1, BIV:1, and CII:2 that induce partial mis-splicing of DMD mRNA provided us with a unique opportunity; each affected individual produced varying levels of remnant, normally spliced DMD mRNA, with all mis-spliced transcripts encoding a premature stop codon and targeted by nonsense-mediated decay. Therefore, we were able to use quantitative WB to correlate levels of WT dystrophin with clinical severity. We establish a steep therapeutic range of WT dystrophin protein levels (figure 4A); with ∼15% WT dystrophin associated with myalgia without apparent weakness, ∼10% levels of WT dystrophin associated with Becker muscular dystrophy, mild weakness, and cardiac phenotype, and <5% WT dystrophin associated with a severe Becker or Duchenne-like phenotype. Our findings broadly concur with previous studies correlating levels of mutated dystrophin in BMD with clinical severity,7,29–31 supporting the notion of a functional redundancy within the spectrin-like repeats of the dystrophin rod domain. Of great relevance to international efforts to develop genetic therapies in DMD, our data provide compelling evidence that with early intervention, only fractional increases in levels of dystrophin are likely to result in clinical improvement.
Acknowledgment
The authors thank the families for their invaluable contributions to this research and the clinicians and health care workers involved in their assessment and management. For cytogenomic analysis on family B, the authors thank the laboratory of Artur P. Darmanian, Sydney Genome Diagnostics, within the Children's Hospital at Westmead, NSW, Australia. For bespoke PCR segregation analysis on extended family members from family B, the authors thank Dr. Bruce Bennetts, Dr. Gladys Ho, and Sydney Genome Diagnostics, Western Sydney Genetics Program, The Children's Hospital at Westmead, NSW, Australia. For histopathology on family C, the authors thank the Department of Anatomical Pathology, South Eastern Area Laboratory Services, Sydney Children's Hospital, Randwick, NSW, Australia. The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the NIH (commonfund.nih.gov/GTEx) and by the NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The datasets used for the analyses described in this manuscript were obtained from dbGaP at http://www.ncbi.nlm.nih.gov/gap through dbGaP accession number phs000424.v7.p2.
Glossary
- bp
base pair
- CK
creatine kinase
- DMD
Duchenne muscular dystrophy
- gnomAD
Genome Aggregation Database
- GTEx
Genotype-Tissue Expression
- IGV
Integrative Genomic Browser
- MLPA
multiplex ligation-dependent probe amplification
- mRNA
messenger RNA
- nt
nucleotide
- RNA-seq
RNA sequencing
- RT-PCR
reverse transcription PCR
- SNV
single nucleotide variant
- WB
Western blot
- WGA
wheat germ agglutinin
- WT
wild type
Appendix. Authors
Footnotes
Editorial, page e529
Contributor Information
Leigh B. Waddell, Email: leigh.waddell@health.nsw.gov.au.
Samantha J. Bryen, Email: samantha.bryen@health.nsw.gov.au.
Beryl B. Cummings, Email: berylc@broadinstitute.org.
Adam Bournazos, Email: adam.bournazos@health.nsw.gov.au.
Frances J. Evesson, Email: frances.evesson@health.nsw.gov.au.
Himanshu Joshi, Email: himanshu.joshi@sydney.edu.au.
Jamie L. Marshall, Email: jmarshal@broadinstitute.org.
Taru Tukiainen, Email: taru.tukiainen@gmail.com.
Elise Valkanas, Email: valkanas@broadinstitute.org.
Ben Weisburd, Email: weisburd@broadinstitute.org.
Simon Sadedin, Email: simon.sadedin@vcgs.org.au.
Mark R. Davis, Email: mark.davis@health.wa.gov.au.
Fathimath Faiz, Email: fathimath.faiz@health.wa.gov.au.
Rebecca Gooding, Email: rebecca.gooding@health.wa.gov.au.
Sarah A. Sandaradura, Email: sarah.sandaradura@health.nsw.gov.au.
Gina L. O'Grady, Email: ginao@adhb.govt.nz.
Michel C. Tchan, Email: michel.tchan@health.nsw.gov.au.
David R. Mowat, Email: david.mowat@health.nsw.gov.au.
Emily C. Oates, Email: e.oates@unsw.edu.au.
Michelle A. Farrar, Email: michelle.farrar@health.nsw.gov.au.
Hugo Sampaio, Email: hugo.sampaio@health.nsw.gov.au.
Alan Ma, Email: alan.ma@health.nsw.gov.au.
Katherine Neas, Email: katherine.neas@ccdhb.org.nz.
Min-Xia Wang, Email: minxia.wang@sydney.edu.au.
Amanda Charlton, Email: acharlton@adhb.govt.nz.
Charles Chan, Email: charles.chan2@health.nsw.gov.au.
Diane N. Kenwright, Email: diane.kenwright@otago.ac.nz.
Nicole Graf, Email: nicole.graf@health.nsw.gov.au.
Susan Arbuckle, Email: susan.arbuckle@health.nsw.gov.au.
Nigel F. Clarke, Email: nnnjournal@neurology.org.
Daniel G. MacArthur, Email: dgmacarthur@gmail.com.
Kristi J. Jones, Email: kristi.jones@health.nsw.gov.au.
Monkol Lek, Email: monkol.lek@yale.edu.
Study Funding
This study was supported by the National Health and Medical Research Council of Australia (APP1048816 and APP1136197 S.T.C., APP1080587 S.T.C., D.G.M.). S.J. Bryen is supported by a Muscular Dystrophy New South Wales PhD scholarship. E.C. Oates is supported by NHMRC ECR: GNT1090428. WES, WGS, and RNA-seq was provided by the Broad Institute of MIT and Harvard Center for Mendelian Genomics (Broad CMG) and was funded by the National Human Genome Research Institute, the National Eye Institute, and the National Heart, Lung and Blood Institute's NIH grant UM1 HG008900 to D.G.M. and Heidi Rehm.
Disclosure
S.T. Cooper is director of Frontier Genomics Pty Ltd. (Australia). Frontier Genomics has not traded (as of September 25, 2020). Frontier Genomics Pty Ltd. (Australia) has no existing financial relationships that will benefit from publication of these data. N.F. Clarke is deceased; disclosures are not included for this author. The remaining coauthors do not have any relationships, financial or otherwise, that may result in a perceived conflict of interest. Go to Neurology.org/NG for full disclosures.
References
- 1.Moat SJ, Bradley DM, Salmon R, Clarke A, Hartley L. Newborn bloodspot screening for Duchenne muscular dystrophy: 21 years experience in Wales (UK). Eur J Hum Genet 2013;21:1049–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Helderman-Van Den Enden ATJM, Madan K, Breuning MH, et al. An urgent need for a change in policy revealed by a study on prenatal testing for Duchenne muscular dystrophy. Eur J Hum Genet 2013;21:21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mendell JR, Shilling C, Leslie ND, et al. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann Neurol 2012;71:304–313. [DOI] [PubMed] [Google Scholar]
- 4.Emery AEH. The muscular dystrophies. Lancet 2002;359:687–695. [DOI] [PubMed] [Google Scholar]
- 5.Flanigan KM. Duchenne and Becker muscular dystrophies. Neurol Clin 2014;32:671–688. [DOI] [PubMed] [Google Scholar]
- 6.Flanigan KM, Dunn DM, Von Niederhausern A, et al. Mutational spectrum of DMD mutations in dystrophinopathy patients: application of modern diagnostic techniques to a large cohort. Hum Mutat 2009;30:1657–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bushby KMD, Gardner-Medwin D. The clinical, genetic and dystrophin characteristics of Becker muscular dystrophy: I. Natural history. J Neurol 1993;240:98–104. [DOI] [PubMed] [Google Scholar]
- 8.Yazaki M, Yoshida K, Nakamura A, et al. Clinical characteristics of aged Becker muscular dystrophy patients with onset after 30 years. Eur Neurol 1999;42:145–149. [DOI] [PubMed] [Google Scholar]
- 9.Heald A, Anderson LVB, Bushby KMD, Shaw PJ. Becker muscular dystrophy with onset after 60 years. Neurology 1994;44:2388–2390. [DOI] [PubMed] [Google Scholar]
- 10.Bushby KMD, Cleghorn NJ, Curtis A, et al. Identification of a mutation in the promoter region of the dystrophin gene in a patient with atypical Becker muscular dystrophy. Hum Genet 1991;88:195–199. [DOI] [PubMed] [Google Scholar]
- 11.Minetti C, Tanji K, Chang HW, et al. Dystrophinopathy in two young boys with exercise-induced cramps and myoglobinuria. Eur J Pediatr 1993;152:848–851. [DOI] [PubMed] [Google Scholar]
- 12.Melis MA, Cau M, Muntoni F, et al. Elevation of serum creatine kinase as the only manifestation of an intragenic deletion of the dystrophin gene in three unrelated families. Eur J Paediatr Neurol 1998;2:255–261. [DOI] [PubMed] [Google Scholar]
- 13.Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol 2003;2:731–740. [DOI] [PubMed] [Google Scholar]
- 14.Ferlini A, Neri M, Gualandi F. The medical genetics of dystrophinopathies: molecular genetic diagnosis and its impact on clinical practice. Neuromuscul Disord 2013;23:4–14. [DOI] [PubMed] [Google Scholar]
- 15.Bladen CL, Salgado D, Monges S, et al. The TREAT-NMD DMD global database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat 2015;36:395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laing NG. Molecular genetics and genetic counselling for Duchenne/Becker muscular dystrophy. Mol Cel Biol Hum Dis Ser 1993;3:37–84. [DOI] [PubMed] [Google Scholar]
- 17.Juan-Mateu J, Gonzalez-Quereda L, Rodriguez MJ, et al. DMD mutations in 576 dystrophinopathy families: a step forward in genotype-phenotype correlations. PLoS One 2015;10:e0135189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tuffery-Giraud S, Béroud C, Leturcq F, et al. Genotype-phenotype analysis in 2,405 patients with a dystrophinopathy using the UMD-DMD database: a model of nationwide knowledgebase. Hum Mutat 2009;30:934–945. [DOI] [PubMed] [Google Scholar]
- 19.Jones HF, Bryen SJ, Waddell LB, et al. Importance of muscle biopsy to establish pathogenicity of DMD missense and splice variants. Neuromuscul Disord 2019;29:913–919. [DOI] [PubMed] [Google Scholar]
- 20.Cummings BB, Marshall JL, Tukiainen T, et al. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci Transl Med 2017;9:eaal5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Waddell LB, Tran J, Zheng XF, et al. A study of FHL1, BAG3, MATR3, PTRF and TCAP in Australian muscular dystrophy patients. Neuromuscul Disord 2011;21:776–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cooper ST, Lo HP, North KN. Single section Western blot: improving the molecular diagnosis of the muscular dystrophies. Neurology 2003;61:93–97. [DOI] [PubMed] [Google Scholar]
- 23.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012;9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bryen SJ, Ewans L, Pinner J, et al. Recurrent TTN metatranscript‐only c.39974‐11T>G splice variant associated with autosomal recessive arthrogryposis multiplex congenita and myopathy. Hum Mutat 2020;41:403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bryen SJ, Joshi H, Evesson FJ, et al. Pathogenic abnormal splicing due to intronic deletions that induce biophysical space constraint for spliceosome assembly. Am J Hum Genet 2019;105:573–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat Genet 2013;45:580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wimmer K, Schamschula E, Wernstedt A, et al. AG‐exclusion zones revisited: lessons to learn from 91 intronic NF1 3′ splice site mutations outside the canonical AG‐dinucleotides. Hum Mutat 2020;41:1145–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim J, Hu C, El Achkar CM, et al. Patient-customized oligonucleotide therapy for a rare genetic disease. N Engl J Med 2019;381:1644–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muntoni F. Is a muscle biopsy in Duchenne dystrophy really necessary? Neurology 2001;57:574–575. [DOI] [PubMed] [Google Scholar]
- 30.Hoffman EP, Kunkel LM, Angelini C, Clarke A, Johnson M, Harris JB. Improved diagnosis of Becker muscular dystrophy by dystrophin testing. Neurology 1989;39:1011–1017. [DOI] [PubMed] [Google Scholar]
- 31.Van Den Bergen JC, Wokke BH, Janson AA, et al. Dystrophin levels and clinical severity in Becker muscular dystrophy patients. J Neurol Neurosurg Psychiatry 2014;85:747–753. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data not published within this article are available by request from any qualified investigator.