

Abstract
Objective
To explore the clinical efficacy and safety of subcutaneous (SC) rozanolixizumab, an anti-neonatal Fc receptor humanized monoclonal antibody, in patients with generalized myasthenia gravis (gMG).
Methods
In this phase 2a, randomized, double-blind, placebo-controlled, 2-period, multicenter trial (NCT03052751), patients were randomized (1:1) in period 1 (days 1–29) to 3 once-weekly (Q1W) SC infusions of rozanolixizumab 7 mg/kg or placebo. In period 2 (days 29–43), patients were re-randomized to either rozanolixizumab 7 mg/kg or 4 mg/kg (3 Q1W SC infusions), followed by an observation period (days 44–99). Primary endpoint was change from baseline to day 29 in Quantitative Myasthenia Gravis (QMG) score. Secondary endpoints were change from baseline to day 29 in MG–Activities of Daily Living (MG-ADL) and MG-Composite (MGC) scores and safety.
Results
Forty-three patients were randomized (rozanolixizumab 21, placebo 22 [period 1]). Least squares (LS) mean change from baseline to day 29 for rozanolixizumab vs placebo was as follows: QMG (LS mean −1.8 vs −1.2, difference −0.7, 95% upper confidence limit [UCL] 0.8; p = 0.221; not statistically significant), MG-ADL (LS mean −1.8 vs −0.4, difference −1.4, 95% UCL −0.4), and MGC (LS mean −3.1 vs −1.2, difference −1.8, 95% UCL 0.4) scores. Efficacy measures continued to improve with rozanolixizumab 7 mg/kg in period 2. The most common adverse event in period 1 was headache (rozanolixizumab 57%, placebo 14%).
Conclusion
Whereas change from baseline in QMG was not statistically significant, the data overall suggest rozanolixizumab may provide clinical benefit in patients with gMG and was generally well tolerated. Phase 3 evaluation is ongoing (NCT03971422).
Classification of Evidence
This study provides Class I evidence that for patients with gMG, rozanolixizumab is well-tolerated, but did not significantly improve QMG score.
Acquired myasthenia gravis (MG) is an autoimmune disease driven by the presence of pathogenic immunoglobulin G (IgG) autoantibodies that impair synaptic transmission at the neuromuscular junction.1
Therapeutic approaches for MG and other IgG-driven autoimmune diseases are evolving, with an increased interest in more targeted approaches, such as reducing pathogenic IgG autoantibodies by targeting the neonatal Fc receptor (FcRn).2 The physiologic role of FcRn is to maintain IgG and albumin homeostasis.3 When bound to FcRn, IgG is saved from lysosomal degradation and is recycled into the circulation.3,4 Howard et al.2 reported reductions in pathogenic IgG autoantibodies and clinical improvements in patients with MG following treatment with an FcRn antagonist (efgartigimod). Limitations of current treatments (e.g., IV immunoglobulin [IVIg] and plasma exchange [PLEX]) include an uncertain mode of action with IVIg and removal of other plasma proteins besides IgG with PLEX.3,5,6 Targeting FcRn may offer an alternative treatment option for patients with MG vs current treatments, with improved tolerability and a reduced treatment burden.
Rozanolixizumab, a subcutaneously (SC) infused monoclonal antibody that specifically targets FcRn, prevents IgG recycling by inhibiting the interaction of FcRn with IgG; lack of IgG binding results in unbound IgG being eliminated via the natural lysosomal degradation pathway.7 We have previously shown dose-dependent reductions in IgG concentrations following IV and SC infusions of rozanolixizumab in a first-in-human trial in healthy volunteers.8
The study described here explored the dose and frequency of rozanolixizumab SC infusion in patients with moderate to severe generalized MG (gMG) and we report, for the first time, the clinical efficacy and safety of rozanolixizumab in this population.
Methods
Primary Research Question
This phase 2a randomized controlled trial sought to determine the clinical efficacy, safety, tolerability, and pharmacodynamic (PD) effect of rozanolixizumab in patients with gMG. This study intended to provide Class I evidence that, for patients with gMG, rozanolixizumab is well-tolerated and improves Quantitative Myasthenia Gravis (QMG) score.
Trial Design and Patients
This multicenter, phase 2a, randomized, investigator- and patient-blind, placebo-controlled, 2-period, treatment-sequence trial, evaluating the clinical efficacy, safety, and tolerability of rozanolixizumab in patients with moderate to severe gMG, was conducted at 17 sites.
Patients were eligible to participate if they were at least 18 years of age and had a documented diagnosis of gMG with evidence of elevated autoantibodies (anti-acetylcholine receptor [AChR] or anti-muscle-specific kinase [MuSK]) prior to screening. Eligibility required that, in the opinion of the investigator, IVIg or PLEX might be considered as a treatment option. A QMG score of ≥11 at baseline and a serum total IgG concentration of >6 g/L at screening were also required. Patients were excluded if MG affected only the ocular muscles, they were in myasthenic crisis at screening or showing signs of imminent myasthenic crisis, or experiencing severe weakness affecting oropharyngeal or respiratory muscles. In addition, patients were excluded if they had previously received rozanolixizumab treatment, had received another investigational medicinal product within 30 days of screening, or had a known hypersensitivity to any component of rozanolixizumab, including l-proline. Patients with renal impairment (serum creatinine ≥1.4 mg/dL [women], ≥1.5 mg/dL [men]) or >2x upper limit of normal (ULN) for alanine aminotransferase, aspartate aminotransferase, alkaline phosphatase, or > ULN total bilirubin were excluded from the trial. Patients with a family history of primary immunodeficiency, a clinically relevant active infection, a serious infection requiring hospitalization within 6 weeks prior to first dose of rozanolixizumab, or those with clinically relevant ongoing infections were excluded. Patients who were treated with rituximab 6 months prior to the baseline visit (or within 12 months if B cells remained outside of normal range) were excluded. Patients were excluded if they had received the immunosuppressants cyclophosphamide or pimecrolimus or vinca alkaloids within 6 months, 4 weeks, and 12 weeks, respectively, prior to the baseline visit; the biological agents abatacept, belimumab, golimumab, natalizumab, ofatumumab, or veltuzumab within 6 months of the baseline visit; or transmembrane activator and calcium modulator and cyclophilin-ligand interactor (TACI) Ig within 10 months of the baseline visit; received other treatments such as IVIg or SC immunoglobulins (3 months prior to baseline), IPP-201101 (3 months prior to baseline), PLEX, or immunoabsorption (6 weeks prior to baseline visit); or if they had been thymectomized in the past 6 months or had a history of a thymoma requiring chemotherapy or radiotherapy.
Standard Protocol Approvals, Registrations, and Patient Consents
The trial (ClinicalTrials.gov Identifier: NCT03052751) was performed in accordance with the principles of the Declaration of Helsinki and the International Conference on Harmonisation Guidance for Good Clinical Practice. The study protocol (appendix), amendments, and patient-informed consent were reviewed by national, regional, or independent ethics committees or institutional review boards. Written informed consent was obtained from all patients.
Procedures
Adults with moderate to severe gMG were randomized 1:1 in dosing period 1 (days 1–29) to receive 3 once-weekly (Q1W) SC infusions of rozanolixizumab 7 mg/kg or placebo; treatment was administered on days 1, 8, and 15. SC infusions of rozanolixizumab were administered into the abdominal wall, using an infusion pump, at an infusion speed of 20 mL/h over 30 minutes. A treatment-free period of 2 weeks occurred between the last dose in period 1 (day 15) and initiation of period 2 (day 29). Patients were stratified based on the treatment received in period 1 and re-randomized in period 2 (days 29–43); the placebo group and the rozanolixizumab 7 mg/kg group were each re-randomized to 3 Q1W SC infusions of either rozanolixizumab 7 mg/kg or 4 mg/kg in a 1:1 manner. For dosing period 2, treatment was administered on days 29, 36, and 43; days 44–99 constituted an observation period. The dosing period time points were based on PD observations from the first-in-human study8 and subsequent modeling.
The multiple arms of period 2 aimed to determine the sustainability of any clinical effects and the safety of longer treatment with 7 mg/kg rozanolixizumab (7 mg/kg / 7 mg/kg); assess whether the 4 mg/kg dose was sufficient to maintain clinical effects (rozanolixizumab 7 mg/kg / 4 mg/kg); and assess whether the 4 mg/kg dose was sufficient to elicit a beneficial effect (placebo/rozanolixizumab 4 mg/kg).
Interactive response technology, provided by an external vendor, was used to anonymize (5-digit identifier) and randomize patients to treatment groups. Upon re-randomization in dosing period 2, a second unique randomization number was also assigned.
To ensure trial blinding, injections were prepared at the investigational sites by unblinded, designated study-site pharmacists. All other trial personnel remained blinded and did not have access to medication-related information.
Outcomes
The primary objective was to evaluate the clinical efficacy to day 29 of rozanolixizumab as a chronic/intermittent treatment in patients with moderate to severe gMG. Secondary objectives were to evaluate the safety, tolerability, and PD effects of rozanolixizumab. An additional exploratory objective aimed to assess the effect of rozanolixizumab on MG-specific serum autoantibodies (anti-AChR and anti-MuSK).
Change in QMG score from baseline to day 29 (period 1) was the primary endpoint. Secondary endpoints included change in MG–activities of daily living (MG-ADL) and MG–composite (MGC) scores from baseline to day 29 (period 1). Other variables, all assessed from baseline over the entire trial duration (periods 1 and 2), were changes in QMG, MG-ADL, and MGC scores; assessment of QMG, MG-ADL, and MGC score responder rate (≥3.0-point improvement); incidence of adverse events (AEs; categorized as preferred terms, according to the Medical Dictionary for Regulatory Activities v.21.0); change in total serum IgG concentration; and change in MG-specific autoantibody concentrations.
Statistical Methods
The trial aimed to demonstrate that the reduction in QMG score between baseline and day 29 would be greater in the rozanolixizumab 7 mg/kg group compared with placebo. The sample size calculation used a one-sided 5% significance level, an estimate of the SD for change in QMG of 3.4, and an anticipated treatment effect of 3.4 (as observed previously in patients with severe disease).9 Based on these assumptions, a sample size of 40 would be required to detect a statistically significant treatment difference between rozanolixizumab and placebo in mean change from baseline in QMG at day 29 with a power >90%.
All randomized patients who received ≥1 dose of rozanolixizumab were included in the safety set (SS); efficacy analyses included all randomized patients who had received ≥1 dose of rozanolixizumab and had a baseline and ≥1 postbaseline QMG measurement during dosing period 1 (full analysis set; FAS). A subset of the FAS, the pharmacodynamic per protocol set (PD-PPS), was used for analysis of serum total IgG concentrations; these patients had no important protocol deviation affecting serum concentrations.
The primary analysis of mean change from baseline in QMG was based on a mixed-model repeated-measures analysis including terms for treatment group, baseline QMG score, and the interaction between treatment group and week. The model defined patient as a random effect and utilized an unstructured covariance pattern. We provide a 1-sided p value and upper confidence limits (UCLs).
Three interim analyses were performed, none of which had an impact on the α level. The first assessed futility (based on QMG, MG-ADL, and MGC scores) and took place once 20 patients had reached day 29; evidence of futility would have resulted in halting or amending the trial. The second interim analysis, conducted by the data monitoring committee, considered the safety of rozanolixizumab and took place once 20 patients had reached day 57. The third interim analysis was performed once all patients had reached day 29, when the data for the primary endpoint (mean change from baseline to day 29 in QMG score) and secondary efficacy variables (mean change from baseline to day 29 in MG-ADL and MGC scores) were generated.
Data Availability
Underlying data from this article may be requested by qualified researchers 6 months after product approval in the United States or Europe, or global development is discontinued; and 18 months after trial completion. Investigators may request access to anonymized individual patient data and redacted trial documents, which may include raw datasets, analysis-ready datasets, trial protocol, blank case report form, annotated case report form, statistical analysis plan, dataset specifications, and clinical trial report. Prior to use of the data, proposals need to be approved by an independent review panel at clinicalstudydatarequest.com and a signed data sharing agreement will need to be executed. All documents are available in English only, for a prespecified time, typically 12 months, on a password-protected portal.
Results
The first patient was enrolled on May 15, 2017, and the last patient completed on August 6, 2018. Sixty-nine patients were screened across 21 sites, of whom 43 (from 17 sites) initiated rozanolixizumab treatment (FAS and SS; figure 1).
Figure 1. CONSORT (Consolidated Standards of Reporting Trials) Diagram.

aDid not meet inclusion criteria. bDiscontinued treatment due to a severe adverse event (AE) (headache) but continued in the observation period.
Patient demographics (table 1) were broadly similar between treatment groups at baseline. The combination of the reported baseline MG-ADL and MGC scores, as well as the protocol-defined entry criteria of QMG score ≥11 and eligibility for IVIg/PLEX, indicated a population with moderate to severe MG.
Table 1.
Baseline Characteristics and Demographics of the Full Analysis Set

Improvements from baseline to day 29 in QMG score for rozanolixizumab compared with placebo were not statistically significant (least squares [LS] mean: −1.8 vs −1.2; difference: −0.7; 95% UCL: 0.8; p = 0.221; figure 2A). Improved day-to-day function, as measured by change from baseline to day 29 in MG-ADL, was observed following treatment with rozanolixizumab compared with placebo (LS mean: −1.8 vs −0.4; difference: −1.4; 95% UCL: −0.4; figure 2B). MGC score also showed changes from baseline to day 29 with rozanolixizumab compared with placebo (LS mean: −3.1 vs −1.2; difference: −1.8; 95% UCL 0.4; figure 2C). Following treatment with rozanolixizumab, LS mean reductions from baseline in QMG and MGC scores reached their nadir 1 week after the final period 1 infusion (day 22; −2.4 and −3.3, respectively) with increases towards baseline values in scores observed between day 22 and day 29, before initiation of period 2.
Figure 2. Change in Measures of Clinical Effect from Baseline to Day 29 (Period 1).
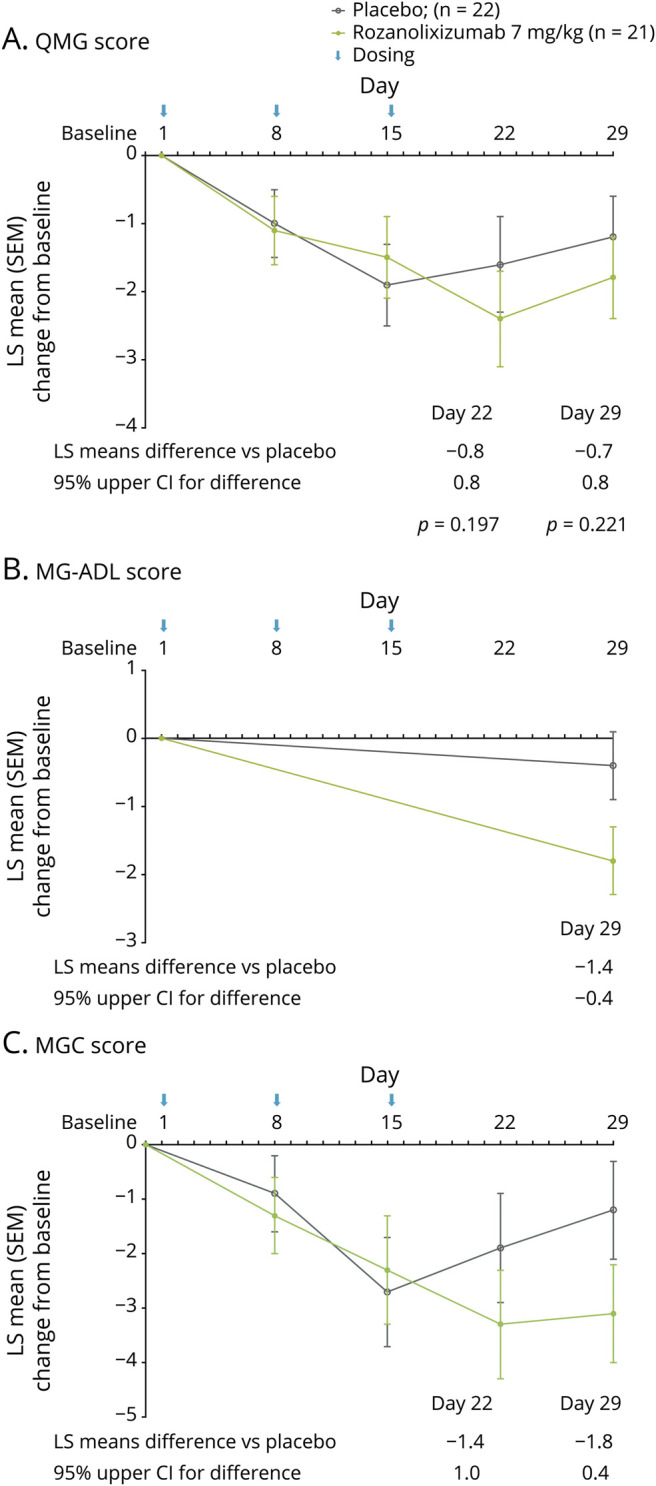
Change from baseline to day 29 in (A) Quantitative Myasthenia Gravis (QMG) score, (B) Myasthenia Gravis–Activities of Daily Living (MG-ADL) score, and (C) Myasthenia Gravis–Composite (MGC) score. Data are least squares (LS) mean change from baseline of the full analysis set; error bars show SEM. CI = confidence interval; SEM = standard error of the mean.
Day 29 responder rates (≥3.0-point improvement from baseline) were higher in patients receiving rozanolixizumab 7 mg/kg vs placebo for QMG (38% vs 23%), MG-ADL (48% vs 14%), and MGC (48% vs 27%) scores (figure 3, A–C). Compared with day 29, greater responder rates with rozanolixizumab were observed on day 22 (1 week after the final dose), when 52% of patients receiving rozanolixizumab 7 mg/kg achieved a clinically meaningful (3.0-point) improvement in QMG score and 55% of patients receiving rozanolixizumab 7 mg/kg achieved a clinically meaningful (3.0-point) improvement in MGC score. MG-ADL was not assessed at day 22, hence responder rates could not be determined.
Figure 3. Responder Rate Analysis (Full Analysis Set) at Day 29 for Measures of Clinical Effect.
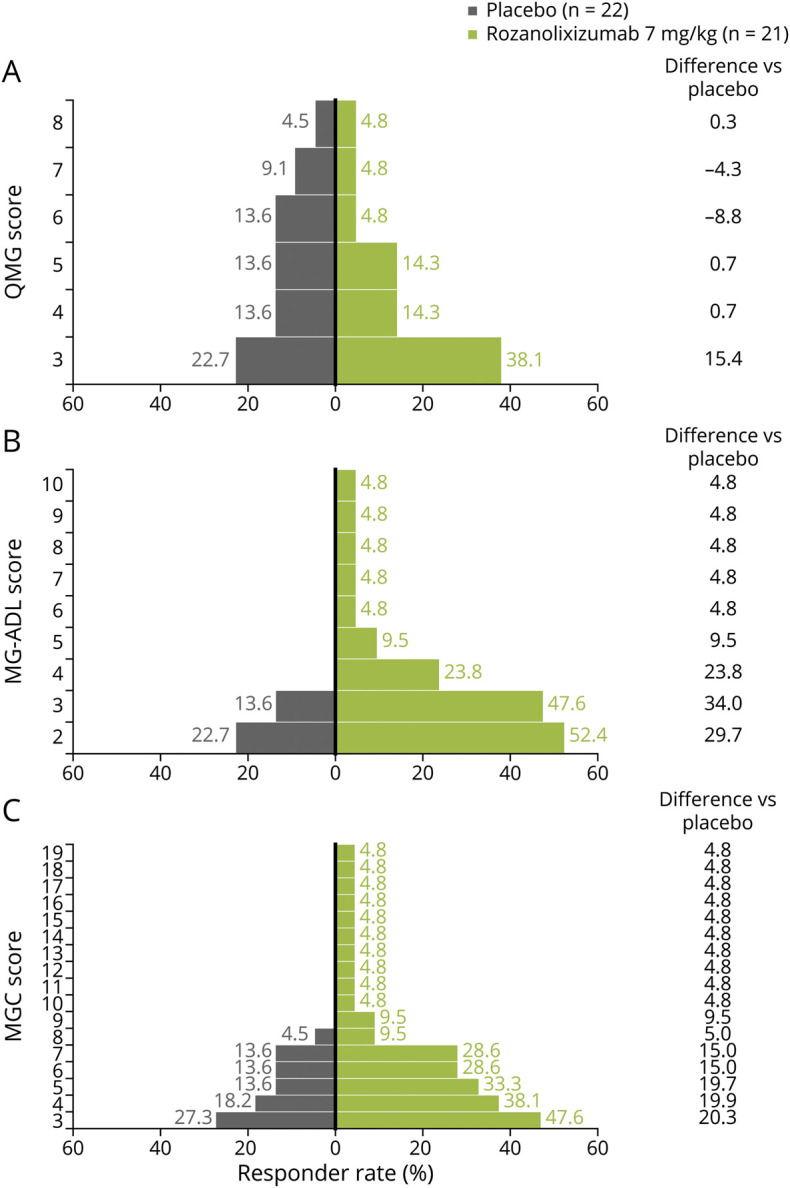
Day 29 responder rates (≥3.0-point improvement from baseline) for (A) Quantitative Myasthenia Gravis (QMG) score, (B) Myasthenia Gravis–Activities of Daily Living (MG-ADL) score, and (C) Myasthenia Gravis–Composite (MGC) score.
Continuation of rozanolixizumab 7 mg/kg treatment in period 2 resulted in further improvements in QMG, MG-ADL, and MGC scores. QMG and MG-ADL scores reached a maximum reduction 21 days after re-initiation of rozanolixizumab (7 mg/kg) treatment (day 50; figure 4, A and B). MGC score nadir was achieved 14 days after re-initiation of rozanolixizumab (7 mg/kg) treatment (day 43; figure 4C). Following cessation of period 2 treatment, QMG, MG-ADL, and MGC scores all increased towards the baseline values, to the end of the observation period (day 99; figure 4, A–C).
Figure 4. Change From Baseline in Measures of Clinical Effect (Full Analysis Set) According to Periods 1 and 2.

Change from baseline for (A) Quantitative Myasthenia Gravis (QMG) score, (B) Myasthenia Gravis–Activities of Daily Living (MG-ADL) score, and (C) Myasthenia Gravis–Composite (MGC) score. SEM = standard error of the mean.
Patients re-randomized from rozanolixizumab 7 mg/kg to rozanolixizumab 4 mg/kg in period 2 maintained modest reductions for QMG, MG-ADL, and MGC scores from day 29 (figure 4, A–C). Nadir was observed for all 3 measures 21 days after the first rozanolixizumab (4 mg/kg) dose was administered (day 50). The reduction in QMG score was maintained to day 78 (figure 4A).
Both the placebo/rozanolixizumab 4 mg/kg and placebo/rozanolixizumab 7 mg/kg groups showed improvements in QMG, MG-ADL, and MGC scores (figure 4, A–C), which appeared dose-related.
Exploratory analyses (PD-PPS) showed a reduction in total serum IgG concentration for all dose groups after administration of rozanolixizumab (figure 5A). In period 1, treatment with rozanolixizumab resulted in a rapid reduction of IgG concentrations vs placebo (52% vs 4%, respectively; day 29); the nadir in IgG concentration following treatment with rozanolixizumab occurred by day 22 (61%). Following period 2 re-randomization, continuation of rozanolixizumab 7 mg/kg further reduced IgG concentration (maximum decrease 68% by day 50). The rozanolixizumab 7 mg/kg/4 mg/kg dose group maintained a reduction in IgG concentration with the nadir observed on day 50 (59%). The placebo/rozanolixizumab 7 mg/kg and placebo/rozanolixizumab 4 mg/kg dose groups both showed dose-related reductions in IgG concentrations.
Figure 5. Mean Percent Change From Baseline in Serum Immunoglobulin G (IgG) (Pharmacodynamic per Protocol Analysis Set) and Anti–acetylcholine Receptor (AChR) (Full Analysis Set [FAS]) Following Rozanolixizumab Treatment.

(A) Serum IgG concentration. (B) Serum AChR autoantibody concentration. Data are from FAS group members who were AChR-positive at baseline; up to 40 patients in period 1 and up to 39 patients in period 2 are included in the analysis. For both graphs, data are presented as mean percent change from baseline, error bars show standard error of the mean (SEM).
Reductions in serum anti-AChR autoantibody titres (FAS) were observed following treatment with rozanolixizumab (figure 5B). In period 1, mean reductions were greater following rozanolixizumab treatment compared with placebo (44% reduction from baseline vs 6% increase from baseline, respectively), and showed a slight delay when compared with reductions in total IgG. The period 2 profile showed a similar pattern to that observed with the reduction in IgG concentration; rozanolixizumab 7 mg/kg / 7 mg/kg continued to reduce IgG to a nadir of 68% (day 36) and the reduction was maintained to day 50. Rozanolixizumab 7 mg/kg / 4 mg/kg maintained the reduction observed in period 1. The placebo/rozanolixizumab 7 mg/kg and placebo/rozanolixizumab 4 mg/kg dose groups showed comparable reductions in anti-AChR autoantibody concentrations in period 2. Anti-MuSK autoantibody concentrations are not described due to low patient numbers (n = 1).
During dosing period 1, 16/21 (76%) patients receiving rozanolixizumab and 16/22 (73%) patients receiving placebo reported at least 1 AE (table 2). None of the 21 patients receiving rozanolixizumab 7 mg/kg and 2/22 (9%) patients receiving placebo reported serious AEs (SAEs) during period 1. By the end of the observation period (day 99), 36/43 (84%) patients reported 1 or more AE, and 5/43 (12%) reported 1 or more SAE. Overall, 4 rozanolixizumab-treated patients withdrew from the trial, 1 due to MG crisis (SAE) and 3 due to headache (2 due to the prespecified protocol withdrawal criteria of severe headache [1 SAE]; 1 moderate headache). No deaths occurred during the trial.
Table 2.
Adverse Events (AEs), n (%)a

Most commonly reported AEs during period 1 (≥5% of all patients, SS) are summarized in table 2. In period 1, higher frequency of headaches was reported in patients receiving 7 mg/kg (57%) compared with placebo (14%). Most headaches were mild to moderate. All instances of headache resolved with standard therapies and without sequelae.
Period 1 AEs deemed by the investigator to be related to treatment were reported in 10/21 patients receiving rozanolixizumab 7 mg/kg and 5/22 patients receiving placebo (table 2). Headache was the most common treatment-related AE (8/21 [38%] patients receiving rozanolixizumab 7 mg/kg and 2/22 [9%] patients receiving placebo). Infusion site reactions occurred in 1 patient in each of the following groups: placebo (4.5%, period 1), placebo/rozanolixizumab 4 mg/kg (9.1%, period 2), rozanolixizumab 7 mg/kg/7 mg/kg (10%, period 2).
The incidence of infections between rozanolixizumab and placebo groups was similar (table 2). During period 1, the most common infections were nasopharyngitis and upper respiratory tract infection. During period 2, 1 patient in the rozanolixizumab 7 mg/kg/7 mg/kg group and 2 patients in each of the other arms reported infections and infestations. No serious or opportunistic infections were reported during the trial.
Discussion
Rozanolixizumab is an SC-infused FcRn inhibitor in development for the treatment of IgG autoantibody-mediated diseases, including MG. This is the first trial to assess the therapeutic potential of rozanolixizumab in patients with gMG and our results across efficacy measures, together with data reported by Howard et al.,2 offer important insights into the viability of inhibition of FcRn as a therapeutic approach in gMG. Although the primary endpoint (change from baseline in QMG score to day 29) was not statistically significant, overall, considering a range of prespecified clinical efficacy measures (QMG, MG-ADL, and MGC), the data suggest rozanolixizumab has potential to provide clinical benefit in patients with moderate to severe gMG. Furthermore, rozanolixizumab was associated with reductions in anti-AChR autoantibodies and was well-tolerated across 2 dose levels with no new safety findings compared to previously reported data.8,10 The combination of favorable clinical and proof-of-concept PD effects warrants further evaluation of rozanolixizumab in larger, adequately powered phase 3 trials.
This trial has provided important insights into the appropriate dose and optimal timing of rozanolixizumab infusion for measurement of efficacy; these insights have informed the design of the ongoing phase 3 trial. Of the 2 rozanolixizumab doses studied, 7 mg/kg yielded the greatest reductions from baseline for all efficacy and PD measures (e.g., the outcomes of the rozanolixizumab 7 mg/kg / 7 mg/kg dose group), making it a proposed dose for future studies. Analyses of efficacy and PD variables showed weekly infusions to be the optimal treatment regimen, since better responses were observed 1 week after last dose (day 22) rather than at the primary endpoint assessment (day 29, 2 weeks after last dose). Extension of the initial 3-week dosing period (period 1) with additional doses of rozanolixizumab in period 2 led to further reductions in QMG, MG-ADL, and MGC scores compared with period 1, suggesting that a dosing period of 6 weeks (Q1W) might yield a better clinical response. This is also consistent with data showing that the maximum response to PLEX generally occurs 6 weeks after treatment.11
Across studies, PD assessment of rozanolixizumab has consistently demonstrated rapid and dose-dependent reductions in serum IgG concentration, with maximum mean decreases of 43%–51% observed following rozanolixizumab SC 7 mg/kg.8,10 In the current trial, a maximum mean decrease in serum IgG concentration of 68% was observed for the rozanolixizumab 7 mg/kg / 7 mg/kg dose group. This proof of concept supports preclinical findings and adds clinical evidence that rozanolixizumab reduces circulating pathogenic IgG by disrupting the natural IgG recycling mechanism.7 Other treatments, such as PLEX, have demonstrated IgG reductions of up to 73%.11
Anti-AChR autoantibody reduction following rozanolixizumab treatment was similar to levels observed after PLEX11 (68% vs 71%, respectively). Despite treatment with rozanolixizumab resulting in up to 68% reduction in anti-AChR autoantibodies, no consistent correlation with clinical effects was observed. These data are consistent with previous studies reporting no associations between anti-AChR antibodies and clinical improvements in MG.12–14
In addition to issues around timing of efficacy assessments, it should also be considered whether there is a need for more robust MG endpoints. It is known that the clinical significance of a change in QMG score can be affected by baseline disease severity and that subtleties exist when assessing treatment response with QMG (percentage change or point change from baseline).15,16 Furthermore, reported changes in QMG score vary widely between clinical trials, even those using consistent treatment regimens.9,17,18 Consideration of these points in conjunction with the findings of this study suggests that further investigation to explore the sensitivity of clinical scales when assessing treatment response in MG studies could be beneficial.
Observations from this trial warrant further investigation as part of a larger phase 3 trial. For example, when the group of patients receiving 7 mg/kg rozanolixizumab in period 1 was analyzed by treatment allocation in period 2, clinical response appeared greater during period 1 in the subgroup that initially received 7 mg/kg and continued on 7 mg/kg compared with patients who initially received 7 mg/kg and continued on 4 mg/kg in period 2. These differences could be accounted for by an imbalance in baseline characteristics between the 2 groups. For example, the group that received 7 mg/kg throughout the trial had a lower mean age, milder Myasthenia Gravis Foundation of America disease classification scores, and lower QMG scores at baseline compared with patients who switched from 7 mg/kg to 4 mg/kg in period 2. Understanding the effect of patient age and disease severity on the clinical effects of rozanolixizumab in MG will be important considerations in future trials.
The safety results reported in this trial are consistent with previous studies of rozanolixizumab. Assessment of safety in 2 prior studies (a first-in-human, placebo-controlled, single-dose, dose-escalating trial [NCT02220153] and a phase 2 trial involving patients with primary immune thrombocytopenia [NCT02718716]) showed an acceptable safety profile after 7 mg/kg SC infusions of rozanolixizumab.8,10 Both studies reported headache as the most common AE, which is consistent with the current trial. The reason headaches appear to be associated with FcRn inhibitors remains unclear, but will continue to be explored as more data become available.
IVIg and PLEX are among the current treatment options for patients with worsening gMG despite their uncertain modes of action3,5,6 and limited evidence from randomized clinical trials.19–21 While effective at relieving the clinical symptoms for patients with MG, both IVIg and PLEX are associated with a high treatment burden and can be associated with considerable AEs that often require comedications.22,23 Both treatments also require specialist IV preparations and long administration times, resulting in a significant burden on the health care system.22–24 Exploratory results using novel anti-FcRn therapies, including rozanolixizumab reported here, and the recently reported phase 2 study of efgartigimod,2 suggest clinically meaningful improvements in patients with gMG. These therapeutic approaches have the potential to reduce treatment burden for a range of patients living with MG, as well as other IgG-driven autoimmune conditions.
Limitations of this trial are that a small number of patients were enrolled, and the testing of the primary efficacy hypothesis was single-sided; therefore, the level of interpretation/extrapolation that can be applied to these results may be limited. Data on the duration of MG in this trial population were not collected; consequently, no assessment of whether MG duration influences rozanolixizumab efficacy could be performed. As noted, the short treatment duration of 3 weeks and the measure of clinical efficacy 2 weeks after the final dose may have been insufficient, and a longer treatment period may be required to exert efficacy in the overall population.
In conclusion, the primary endpoint did not show significant improvement in QMG score (from baseline to day 29), but when all prespecified efficacy measures (QMG, MG-ADL, and MGC) are considered, the data overall suggest rozanolixizumab, a humanized monoclonal antibody specifically targeting FcRn, may provide clinical benefit for patients with moderate to severe gMG. Proof-of-concept PD effects were also seen with rozanolixizumab. These data support further evaluation in the ongoing phase 3 study (NCT03971422) to assess the efficacy of rozanolixizumab in the treatment of MG, with potential to offer a targeted approach to treatment.
Acknowledgment
The authors thank Linda Feighery, PhD, CMPP, and Veronica Porkess, PhD, CMPP, of UCB Pharma for publication and editorial support; the MG0002 trial investigators (appendix 2, links.lww.com/WNL/B262); and the patients, their caregivers, and the investigators and their teams who contributed to this trial.
Glossary
- AChR
acetylcholine receptor
- AE
adverse event
- FAS
full analysis set
- FcRn
neonatal Fc receptor
- gMG
generalized myasthenia gravis
- IgG
immunoglobulin G
- IVIg
IV immunoglobulin
- LS
least squares
- MG
myasthenia gravis
- MG-ADL
Myasthenia Gravis–Activities of Daily Living
- MGC
Myasthenia Gravis–Composite
- MuSK
muscle-specific kinase
- PD
pharmacodynamic
- PD-PPS
pharmacodynamic per protocol analysis set
- PLEX
plasma exchange
- Q1W
once-weekly
- QMG
Quantitative Myasthenia Gravis
- SAE
serious adverse event
- SC
subcutaneously
- SS
safety set
- UCL
upper confidence limit
- ULN
upper limit of normal
Appendix 1. Authors
Appendix 2. Coinvestigators

Footnotes
Class of Evidence: NPub.org/coe
Study Funding
The trial (NCT03052751) was funded by UCB Pharma, the manufacturer of rozanolixizumab. Medical writing support was provided by iMed Communications (an Ashfield Company, part of UDG Healthcare plc), Macclesfield, UK, and funded by UCB Pharma, in accordance with Good Publications Practice (GPP3) guidelines (ismpp.org/gpp3).
Disclosures
Dr. Bril received support for grants and consulting from UCB, CSL Behring, Octapharma, Grifols, Biogen, ArgenX, and Takeda (Shire); consulting fees from Powell Mansfield Inc., Akcea, Alnylam, Alexion, and Pfizer; and grants from Baxalta. Dr. Benatar received trial support and consulting fees from UCB and Ra Pharma. Dr. Andersen received trial support from UCB. Dr. Vissing received trial support and speaker honorarium from UCB. Dr. Brock is an employee of UCB Pharma and may hold stock or stock options. Dr. Greve is an employee of UCB Pharma and may hold stock or stock options. Dr. Kiessling is an employee of UCB Pharma and may hold stock or stock options. F. Woltering is an employee of UCB Pharma and may hold stock or stock options. Dr. Griffin is an employee of iMed Communications; iMed Communications received funds for medical writing support. Dr. Van den Bergh reports no disclosures relevant to the article. Go to Neurology.org/N for full disclosures.
References
- 1.Phillips WD, Vincent A. Pathogenesis of myasthenia gravis: update on disease types, models, and mechanisms. F1000Res 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Howard JF Jr, Bril V, Burns TM, et al. Randomized phase 2 study of FcRn antagonist efgartigimod in generalized myasthenia gravis. Neurology 2019;92:e2661–e2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol 2007;7:715–725. [DOI] [PubMed] [Google Scholar]
- 4.Sand KM, Bern M, Nilsen J, Noordzij HT, Sandlie I, Andersen JT. Unraveling the interaction between FcRn and albumin: opportunities for design of albumin-based therapeutics. Front Immunol 2014;5:682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chaigne B, Mouthon L. Mechanisms of action of intravenous immunoglobulin. Transfus Apher Sci 2017;56:45–49. [DOI] [PubMed] [Google Scholar]
- 6.Filomena CA, Filomena AP, Hudock J, Ballas SK. Evaluation of serum immunoglobulins by protein electrophoresis and rate nephelometry before and after therapeutic plasma exchange. Am J Clin Pathol 1992;98:243–248. [DOI] [PubMed] [Google Scholar]
- 7.Smith B, Kiessling A, Lledo-Garcia R, et al. Generation and characterization of a high affinity anti-human FcRn antibody, rozanolixizumab, and the effects of different molecular formats on the reduction of plasma IgG concentration. mAbs 2018;10:1111–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kiessling P, Lledo-Garcia R, Watanabe S, et al. The FcRn inhibitor rozanolixizumab reduces human serum IgG concentration: a randomized phase 1 study. Sci Transl Med 2017;9. [DOI] [PubMed] [Google Scholar]
- 9.Zinman L, Ng E, Bril V. IV immunoglobulin in patients with myasthenia gravis: a randomized controlled trial. Neurology 2007;68:837–841. [DOI] [PubMed] [Google Scholar]
- 10.Robak T, Jarque I, Musteata V, et al. Phase II, multiple-dose study of anti-FcRn antibody, rozanolixizumab (UCB7665), in patients with primary immune thrombocytopenia: interim analysis. Blood 2017;130:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guptill J, Juel V, Massey J, Chopra M, Howard J. Effects of therapeutic plasma exchange on myasthenic outcome measures. 68th annual meeting of the AAN; Vancouver, Canada; 2016.
- 12.Raja SM, Howard JF Jr., Juel VC, Massey JM, Chopra M, Guptill JT. Clinical outcome measures following plasma exchange for MG exacerbation. Ann Clin Transl Neurol 2019;6:2114–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olanow CW, Wechsler AS, Roses AD. A prospective study of thymectomy and serum acetylcholine receptor antibodies in myasthenia gravis. Ann Surg 1982;196:113–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanders DB, Burns TM, Cutter GR, Massey JM, Juel VC, Hobson-Webb L. Does change in acetylcholine receptor antibody level correlate with clinical change in myasthenia gravis? Muscle Nerve 2014;49:483–486. [DOI] [PubMed] [Google Scholar]
- 15.Benatar M, Howard JF Jr, Barohn R, Wolfe GI, Cutter G. Learning from the past: reflections on recently completed myasthenia gravis trials. Ann NY Acad Sci 2018;1412:5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.MGFA. Resources for Professionals [online]. Available at: myasthenia.org/For-Professionals/Resources-for-Professionals. Accessed October 7, 2019. [Google Scholar]
- 17.Wolfe GI, Barohn RJ, Foster BM, et al. Randomized, controlled trial of intravenous immunoglobulin in myasthenia gravis. Muscle Nerve 2002;26:549–552. [DOI] [PubMed] [Google Scholar]
- 18.Howard JF Jr, Barohn RJ, Cutter GR, et al. A randomized, double-blind, placebo-controlled phase II study of eculizumab in patients with refractory generalized myasthenia gravis. Muscle Nerve 2013;48:76–84. [DOI] [PubMed] [Google Scholar]
- 19.Gajdos P, Chevret S, Toyka KV. Intravenous immunoglobulin for myasthenia gravis. Cochrane Database Syst Rev 2012;12:Cd002277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bril V, Barnett-Tapia C, Barth D, Katzberg HD. IVIG and PLEX in the treatment of myasthenia gravis. Ann NY Acad Sci 2012;1275:1–6. [DOI] [PubMed] [Google Scholar]
- 21.Gajdos P, Chevret S, Toyka K. Plasma exchange for myasthenia gravis. Cochrane Database Syst Rev 2002:Cd002275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cherin P, Marie I, Michallet M, et al. Management of adverse events in the treatment of patients with immunoglobulin therapy: a review of evidence. Autoimmun Rev 2016;15:71–81. [DOI] [PubMed] [Google Scholar]
- 23.Szczeklik W, Wawrzycka K, Wludarczyk A, et al. Complications in patients treated with plasmapheresis in the intensive care unit. Anaesthesiol Intens Ther 2013;45:7–13. [DOI] [PubMed] [Google Scholar]
- 24.Heatwole C, Johnson N, Holloway R, Noyes K. Plasma exchange versus intravenous immunoglobulin for myasthenia gravis crisis: an acute hospital cost comparison study. J Clin Neuromuscul Dis 2011;13:85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Underlying data from this article may be requested by qualified researchers 6 months after product approval in the United States or Europe, or global development is discontinued; and 18 months after trial completion. Investigators may request access to anonymized individual patient data and redacted trial documents, which may include raw datasets, analysis-ready datasets, trial protocol, blank case report form, annotated case report form, statistical analysis plan, dataset specifications, and clinical trial report. Prior to use of the data, proposals need to be approved by an independent review panel at clinicalstudydatarequest.com and a signed data sharing agreement will need to be executed. All documents are available in English only, for a prespecified time, typically 12 months, on a password-protected portal.