

Abstract
Objective
To report 4 novel TUBB4A mutations leading to laryngeal and cervical dystonia with frequent generalization.
Methods
We screened 4 families including a total of 11 definitely affected members with a clinical picture resembling the original description.
Results
Four novel variants in the TUBB4A gene have been identified: D295N, R46M, Q424H, and R121W. In silico modeling showed that all variants have characteristics similar to R2G. The variants segregate with the disease in 3 of the families with evidence of incomplete penetrance in 2 of them. All 4 variants would be classified as likely pathogenic. The clinical picture particularly included laryngeal dystonia (often the site of onset), associated with cervical and upper limb dystonia and frequent generalization. Laryngeal dystonia was extremely prevalent (>90%) both in the original cases and in this case series. The hobby horse gait was evident in only 1 patient in this case series.
Conclusions
Our interpretation is that laryngeal involvement is a hallmark feature of DYT-TUBB4A. Nevertheless, TUBB4A mutations remain an exceedingly rare cause of laryngeal or other isolated dystonia.
DYT-TUBB4A,1 formerly known as DYT4 or whispering dysphonia, was first described in 1985 by Parker2 in a large family of UK origin who immigrated to Australia. Onset of dystonia was usually in the third decade or earlier. Patients commonly presented with spasmodic dysphonia (SD) and/or a cranio-cervical dystonia progressing to generalized dystonia. A peculiar “hobby horse” gait (described in 5 of 9 Australian cases) and extrusional tongue movements have been emphasized.3 In a subsequent description of 8 patients from this family, 6 had dysphonia and 6 had cervical dystonia (CD).4 In 2013, 2 independent groups of researchers almost simultaneously identified a heterozygous missense mutation, c.4C>G;p.R2G, in exon 1 of the TUBB4A gene as causative in members of the original family.4,5 TUBB4A, located on chromosome 19p, codes for the protein beta-tubulin 4A, a neuronally expressed tubulin5 that is an essential component of microtubules that form the cytoskeleton and serve diverse cellular functions.6 Sequencing of TUBB4A in close to 400 unrelated patients with dystonia (among whom 124 had SD) revealed another missense variant, p.A271T, in a possibly familial case of segmental dystonia with SD.5
In 2013, TUBB4A mutations were also shown to result in hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC).7 This disorder, first described in 2002,8 derives its name from characteristic abnormalities demonstrated on brain MRI, whereas brain MRI is normal in DYT-TUBB4A. The clinical spectrum of this severe illness, which expresses during infancy and causes a hypomyelinating leukodystrophy, comprises not only dystonia but also developmental delay, choreoathetosis, rigidity, opisthotonus, oculogyric crises, progressive spastic tetraplegia, ataxia, and, more rarely, seizures.8 Originally, a single missense mutation in TUBB4A, p.D249N, was reported in 11 unrelated patients.7 However, many other mutations in TUBB4A associated with H-ABC have since been described in this gene.9 Noteworthy, mutations in the TUBB4A gene more commonly cause H-ABC than DYT-TUBB4A.1 It has been suggested that these disorders represent different ends of a spectrum, with H-ABC at the severe end and DYT-TUBB4A at the milder end, with movement disorders being the common feature.9 Supporting this concept is the report of 4 unrelated patients with TUBB4A mutations with imaging findings consistent with H-ABC syndrome who had severe generalized dystonia and complete aphonia, reminiscent of DYT-TUBB4A phenomenology, but also with a variety of other neurologic features.10
On the other hand, TUBB4A mutations are an extremely rare cause of isolated dystonia in the absence of other neurologic signs. In a study of >700 patients with isolated dystonia, the authors were unable to identify any possibly pathogenic sequence alteration,11 while another group, screening close to 500 patients with isolated dystonia, found 1 rare, potentially pathogenic, in-frame deletion in an Italian patient with CD.12 Thus, routine genetic testing of TUBB4A in individuals with isolated dystonia is currently not recommended.11,12
Here, we report a total of 11 patients from 4 families with 4 novel TUBB4A variants manifesting isolated dystonia. In silico modeling showed that all variants have characteristics similar to R2G and A271T.
Methods
Standard Protocol Approvals, Registrations, and Patient Consents
Brazilian families were screened as part of a research project on the genetics of dystonia, approved by the institutional review board of each participating center. The Canadian and American families were screened after the probands sought medical attention at our movement disorders centers. All participants provided written informed consent.
Mutation Screening and In Silico Analysis
Probands were screened by next-generation sequencing with a dystonia panel (TruSeq Custom Amplicon assay 1.5, Illumina for families 1 and 2) or whole-exome sequencing (family 3). Detailed sequencing methods are provided as supplemental material (supplemental Methods, available on Dryad, doi.org/10.5061/dryad.34tmpg4gt). All variants were validated by Sanger sequencing. Family 4 was tested with a commercial next-generation sequencing gene panel (Invitae Dystonia Comprehensive Panel13). Allele frequencies were assessed in population databases (Genome Aggregation Database [gnomAD],14 AbraOM,15 and BIPMed-WES-db16). In silico predictions of pathogenicity were performed with combined annotation-dependent depletion (CADD)17; other functional prediction programs used included SIFT, PolyPhen, M-CAP, Meta-SNP, and Mutation Taster.18,19 Amino acid conservation was assessed through Clustal Omega.20
TUBB4A: Homology Modeling
To verify the effect of variants on the 3D structure and function of beta-tubulin-4A (NP 006078) wild-type and variant models, we performed homology modeling by in silico analysis using Phyre2 server21 and University of California San Francisco Chimera 1.13 software.22 Residues not aligned were modeled by ab initio approach.21 For the ligand binding sites prediction, 3DLigandSite server was used.23,24
We analyzed structural and functional effects caused by the variants identified in our families with dystonia and by variants previously reported in patients with DYT-TUBB4A (p.R2G and p.A271T)5 and p.D295H, a rare variant described in GnomAD.
Data Availability
All data and supplemental data are available on Dryad, doi.org/10.5061/dryad.34tmpg4gt.
Results
We have identified heterozygous novel TUBB4A (NM_006087) single nucleotide variants in 4 families with isolated dystonia, including a singleton case, 2 sib pairs, and a multigeneration family (family 3). All variants change highly conserved amino acids and are predicted to be deleterious by in silico analysis (CADD scores 24.6, 31, 25.2, and 26.5 for families 1, 2, 3, and 4, respectively). None of them was found in population databases. Clinical summaries and mutation data are presented in table 1 and pedigrees in figure 1.
Table 1.
Clinical Features and Variants Found in the 3 Reported Families

Figure 1. Pedigree Trees.

Pedigree trees for families 1 (top left), 2 (top right), 3 (bottom left), and 4 (bottom right) with the definitely affected members in red and unaffected ones in white. Question marks indicate questionably affected members in families 2 and 3; case 3.III.4 from family 3 was clinically definitely affected but not genetically tested and hence has a question mark. A question mark in family 1 indicates cases without information. Whenever a case was genetically tested (whether clinically affected or not), the variant in TUBB4A gene (D295N, R46M, Q424H, and R121W for families 1, 2, 3, and 4, respectively) or wild-type is indicated. Exome sequencing was performed on cases 3.II.1 and 3.III.5.
Next-Generation Sequencing
Probands from 3 of the families were screened for mutations using a dystonia gene panel. The only potentially pathogenic variants identified in TUBB4A included a singleton case harboring p.D295N and 2 sib pairs segregating p.R46M and p.R121W. Exome sequencing was performed on individuals 3.II.1 and 3.III.5 (figure 1, bottom left) from family 3. Examination for shared rare heterozygous variants (minor allele frequency <0.0001) revealed 17 single nucleotide variants (doi.org/10.5061/dryad.34tmpg4gt). After filtering of these variants for CADD score, family segregation, disease phenotype, and variants not observed in our internal dystonia exomes (n > 500) or GnomAD, 2 variants remained: p.A180V in C19ORF47 and p.Q424H in TUBB4A (supplemental table 1). C19ORF47 has an unknown function, is conserved only in rodents, and has a low level of expression in the brain (Genotype-Tissue Expression portal25). Thus, the variant p.Q424H in TUBB4A is the most likely candidate variant to cause the phenotype in this family.
Protein Modeling
To assess the pathogenicity of the mutations, protein models were created for wild-type and variant primary sequences. The best template structure selected was C2p4nB (beta-tubulin-chain 2B, TUBB2B_Q6B856) according to the coverage (96% of the sequence) and confidence (100%) of the model. The predicted structure was a core of beta-sheets surrounded by alpha-helices as illustrated in figure 2.
Figure 2. TUBB4A (NP 006078) Wild-Type Protein Model.

Created with Phyre2 server based on C2p4nB (TUBB2B) structure with colored domains: nucleotide binding site (E-site, GTP/GDP) (yellow), alpha/beta domain interface (green), beta/alpha domain interface (magenta), and Taxol binding site (blue).
Table 2 shows the predicted structural modifications of each variant found in our families, as well as in the previously described mutation R2G5 and in the rare variant D295H described in gnomAD through TUBB4A homology modeling. Figure 3 presents the structural and interaction modifications in variants R46M, D295N, Q424H, and R121W compared to the wild-type TUBB4A model. Supplemental figure S1 (doi.org/10.5061/dryad.34tmpg4gt) shows the wild-type TUBB4A model (left) and the loss of hydrogen bonds in the R46M, D295N, and R2G TUBB4A models (right; see also first row of table 2). Supplemental figure S2 shows the modification of the M-loop region in the R46M and R121W models.
Table 2.
Predicted Structural Modifications of Each Variant Found in our Families, in the Previously Described Mutation R2G, and in the Rare Variant D295H Described in GnomAD Through TUBB4A Homology Modeling

Figure 3. Structural and Interaction Modifications in Variants R46M, D295N, Q424H, and R121W Compared to Wild-Type TUBB4A Model.
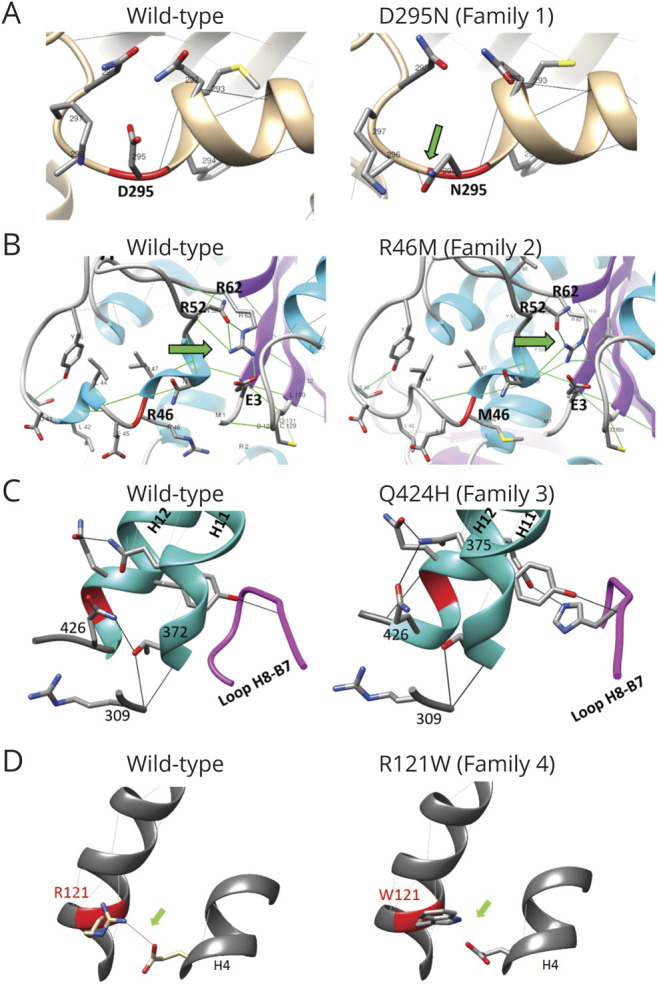
Positions of altered residues are indicated in red; hydrogen bonds are shown by lines between side chains; and green arrows point to the main changes. (A) Variant D295N (family 1) acquired a hydrogen bond between residues 295 and 296. (B) Variant R46M (family 2) modified the interactions among arginine 52 (R52), arginine 62 (R62), and glutamic acid 3 (E3) and changed the alpha-helix structure. (C) Variant Q424H (family 3) modified its interactions with residues 372 and 375 and extended the alpha-helix structure to residue 426; it also acquired a novel hydrogen bond between H12 alpha-helix and loop H8-B7. (D) Variant R121W (family 4) lost interaction between 121 and 158 residues (H3 and H8 alpha-helices).
The 3DLigandSite server predicted clusters of ligand binding sites with heterogens (GTP, magnesium, or zinc) for each model on the basis of the alignment with known 3D structure domains (table 3). The functions of the predicted sites were annotated according to the Universal Protein Resource (UniProtKB) P04350, Conserved Domain Database (National Center for Biotechnology Information) cd02187, and previous studies.26,27 Variants R46M, D295N, Q424H, R121W, and A271T were predicted to lose the binding site in residue N99 (alpha/beta domain interface), and specifically in the R121W model, this residue lost all interactions with other residues (supplemental figure S5, doi.org/10.5061/dryad.34tmpg4gt). Except for R121W, the variants were predicted to acquire a zinc-binding site in residue 280. Variants R46M, D295N, R2G, and A271T were predicted to acquire a GTP-binding site in residue 72. The 3D surface models are shown in supplemental figure S3.
Table 3.
Changes in the Ligand Binding Sites Predicted by 3Dligandsite for Variant Models24

Discussion
Here, we describe 4 novel variants in the TUBB4A gene in 3 unrelated families with dystonia, 1 from Brazil (Portuguese and African descent), 1 from Canada (French descent), and 1 from the United States (Czech and Norwegian descent), as well as in a singleton affected case also from Brazil. All variants change highly conserved protein regions, and in silico and protein modeling analyses suggest that they are deleterious. These are the first 3 families with dystonia segregating TUBB4A mutations with disease since the original family was described,4,5 thus confirming this dystonia locus.
In total, we describe 11 definitely affected cases (10 mutation positive, 1 mutation status unknown, see table 1 and figure 1). The dystonia varied between mild and very severe (in 1 case, leading to death at a very young age). The mean age at onset was 17 (range 2–30) years (median 21 years). The site of onset was most often the larynx (n = 6), arm (n = 5), or neck (n = 4), with the distribution extending from segmental to generalized. Final sites involved included the larynx (n = 8, 1 unknown) with both adductor (predominant) and possibly abductor (in case 1.II.1) types of SD, neck (n = 10), and upper limb (n = 10, 1 unknown), with isolated writer's cramp in the most mildly affected cases (n = 2) and more severe arm dystonia in the others. Generalized dystonia28 was found in 8 of 11 cases. The famous hobby horse gait described in the original Australian family was seen in only 1 of our cases (case 4.II.2); rather, SD associated with CD was the most consistent features of DYT-TUBB4A: 6 of 8 cases in the Australian family and 7 of 10 (plus one unknown, case 3.III.4, who was the most severe of all and therefore likely presented with SD and CD) cases in this series.
There was a marked variability of the progression of this disorder, especially emphasized by the early death in case 3.III.4 and the late striking progression in case 4.II.2, contrasting with the more benign course in some others.
Overall, the most consistent feature was laryngeal involvement, present in >90% of the reported case (all 9 cases of the original family3 and in 8 of 10 [plus 1 unknown] of our cases). Compared with other genetic causes of isolated dystonia, laryngeal involvement is found in only 14% of DYT-TOR1A (DYT1),29 in 44% of DYT-THAP1 (DYT6),30 in 53% of DYT-KMT2B (DYT28),31 and occasionally in DYT-GNAL (DYT25).32 Rarely is laryngeal involvement the presenting feature in these patients, in contrast to DYT-TUBB4A. Thus, laryngeal involvement, highlighted in the descriptions of the original family and present in the isolated case with the p.A271T variant,5 is a hallmark feature of DYT-TUBB4A (DYT4).
Although most dystonia genes show reduced penetrance, because families with DYT-TUBB4A are rare, overall penetrance estimations might not be reliable. The p.R2G TUBB4A mutation appears to be highly penetrant in the original family with DYT4.4,5 This effect could be specific to this variant, while other variants might be under the influence of modifiers. There are only 2 other previously described cases with DYT-TUBB4A dystonia, but neither had DNA available from relatives for segregation analysis, so penetrance could not be assessed.5,12
In the singleton case and in family 4, variants p.D295N and p.R121W, respectively, were also present in unaffected members; this could either be noncausal or due to reduced penetrance, which is a common feature for most dystonia genes.33 Because almost no unaffected family members were tested in families 2 and 3, no determination about penetrance can be made; however, examining the pedigree structure, we found almost equal numbers of affected and unaffected individuals as well as affected individuals in all generations, consistent with high penetrance of a dominant disorder, similar to what was described in the original family.5,33
Considering the clinical characteristics, particularly the marked laryngeal dystonia, as well as the in silico data including pathogenicity predictors (CADD scores >20), the high conservation of the amino acids across species, and our protein structure modeling analysis (see below), coupled with the fact that all of the variants are novel, the 4 variants we identified in TUBB4A would be classified as likely pathogenic according to the American College of Medical Genetics and Genomics guidelines for the interpretation of sequence variants.34 In the case of p.D295N and R121W, there are 2 very rare variants described in gnomAD14 at the same codon (p.D295H, allele frequency 8.8 ×10−6; and p.R121Q, allele frequency 3.9 ×10−6). Both in silico analysis and various functional prediction programs (SIFT, PolyPhen, M-CAP, Meta-SNP Mutation Taster, and CADD18,19) suggest that these variants are pathogenic (supplemental table 2, doi.org/10.5061/dryad.34tmpg4gt). No clinical information is provided in gnomAD, but given the age at onset and phenotypic expression associated with mutations in this gene, it is unlikely that the p.D295H and R121Q carriers expressed dystonia, thus supporting reduced penetrance of mutations at these residues. However, in vitro assays similar to recently published studies35–37 will be necessary to definitively define the pathogenicity of the variants described here. It should be noted that in family 3, a variant in C19ORF47 also segregates with the disease, and although nothing is known about the function of this gene and no missense variants have been reported to cause disease in ClinVar,38 it is possible this variant could, by itself or in combination with the TUBB4A variant, influence the phenotype.
Tubulins (alpha and beta) have 3 domains: N-terminal domain (GTP binding), intermediary domain (with Taxol-binding site, involved in microtubule stabilization), and C-terminal domain with 2 alpha-helices (H11 and H12), which form a crest on the outside surface of the microtubule protofilament 6.
The correct interaction between residues of the protein core is key to the monomer stability and efficiency of the nucleotide-binding domain. The interaction between residues that participate in the rotation of the N-terminal and second domains, which occurs after microtubule depolymerization and nucleotide hydrolysis, is essential for the function of beta-tubulin.26 The loss or modification of hydrogen bonds (as observed in the R46M, R121W, D295N, and Q424H models) may contribute to the destabilization of beta-tubulin after activation or oligomerization in a head-to-tail arrangement between alpha- and beta-tubulin.
Loop regions, T3, T5, T7, and M loops are important for the interaction between beta-tubulin and other structures (supplemental figure S4, doi.org/10.5061/dryad.34tmpg4gt).39,40 The loop H8-B7 is in the region of the beta/alpha interface domain. Variant Q424H modified the interaction of H12 with H11 and loop H8-B7 (figure 3C). H12 and H11 alpha-helices are in the C-terminal of beta-tubulin and are important for the assembly of M loops between protofilaments, as well as for the interaction with motor proteins.26 In the R121W variant model, interactions between H3 and these alpha-helices were lost (supplemental figure S5).
In silico modeling indicates an alpha-helix structure in the region of residues 277 to 279, but this was not confirmed by crystallographic studies.41 However, this region mediates lateral interactions between protofilaments; therefore, variant R46M may have functional consequences. The helix H3 in the N-terminal GTPase domain of a beta-tubulin subunit (containing R46M and R121W) contacts the M loop of the intermediary domain of an adjacent beta subunit. The M loop also contains a zinc-binding site, involved in the assembly of beta-beta subunits. The Taxol domain stabilizes the M loop, reducing dynamic instability of beta-tubulin26,39,40 (figure 2 and figure S2, doi.org/10.5061/dryad.34tmpg4gt).
In relation to the ligand binding sites, the R46M, R121W, D295N, and Q424H models are predicted to lose sites in the N-terminal region that are involved in GTP binding and polymerization (doi.org/10.5061/dryad.34tmpg4gt). In the R121W model, the loss of the N99 residue interactions with active sites of alpha/beta interface and GTP binding can impair the assembly of the tubulin dimer and conformation of the E site (supplemental figure S5).
Although functional studies of TUBB4A variants, including R2G and A271T, showed variable results,36,37,42 they all point to disorganization of microtubule dynamics, with findings including lack of typical radial tubulin networks, diminished interaction between beta- and alpha-tubulin, neuronal or oligodendrocyte morphological changes, abnormalities in motor protein binding to microtubules, and disrupted mitochondrial transport. In silico modeling reveals that variants R46M, R121W, D295N, and Q424H have characteristics similar to R2G and A271T, consistent with the idea that they also have functional consequences on microtubule stability.
This study describes 11 affected members in 4 different families with isolated dystonia with 4 distinct TUBB4A mutations that are likely to be pathogenic. The clinical picture particularly included laryngeal dystonia (often the site of onset), associated with cervical and upper limb dystonia and frequent generalization. Despite the very high prevalence of laryngeal dystonia in DYT-TUBB4A, mutations in TUBB4A remain an exceedingly rare cause of laryngeal dystonia or other isolated dystonia; only a single family and 2 isolated cases have been described before the present case series.4,5,11,12
Acknowledgment
The authors acknowledge Sao Paulo Research Foundation grants 2016/17211-2 (C.O.d.S.) and 2014/17128-2 (P.d.C.A.), NIH NS087997 (L.J.O.). The authors would like to thank the gnomAD and the groups who provided exome and genome variant data to this resource. A full list of contributing groups can be found at gnomad.broadinstitute.org/about. The authors would also like to thank Dr. Conor Fearon for editing the videos.
Glossary
- CADD
combined annotation-dependent depletion
- CD
cervical dystonia
- gnomAD
Genome Aggregation Database
- H-ABC
hypomyelination with atrophy of the basal ganglia and cerebellum
- SD
spasmodic dysphonia
Appendix. Authors
Study Funding
No targeted funding reported.
Disclosure
J.F. Bally reports travel grant for a congress paid by Abbvie and Zambon. S. Camargos reports honoraria from Roche and Teva Pharmaceuticals. C. Oliveira dos Santos reports no conflicts. D.S. Kern has served as an advisor for Colorado Clinical and Translational Sciences Institute Data Safety Monitoring Board and AbbVie Pharmaceutics; received honorarium from AbbVie Pharmaceutics and Boston Scientific; and received grants from the NIH. T. Lee, F. Pereira da Silva-Junior, R.D. Puga, F. Cardoso, E.R. Barbosa, and R. Yadav report no conflicts. L.J. Ozelius reports NIH grants and patent royalties from Athena Diagnostics, Inc. P. de Carvalho Aguiar received research grants from São Paulo Research Foundation (FAPESP)2017/24022-4 and is employed at the Hospital Israelita Albert Einstein. A.E. Lang received support for advisory work from Biogen, Janssen, Lundbeck, Merck, Roche, Sun Pharma, Theravance, and Corticobasal Degeneration Solutions; honoraria from Sun Pharma and AbbVie; and grants from Brain Canada, Canadian Institutes of Health Research, Corticobasal Degeneration Solutions, Edmond J. Safra Philanthropic Foundation, Michael J. Fox Foundation, Ontario Brain Institute, Parkinson Foundation, Parkinson Society Canada, and W. Garfield Weston Foundation. Go to Neurology.org/N for full disclosures.
References
- 1.Marras C, Lang A, van de Warrenburg BP, et al. Nomenclature of genetic movement disorders: recommendations of the International Parkinson and Movement Disorder Society Task Force. Mov Disord 2016;31:436–457. [DOI] [PubMed] [Google Scholar]
- 2.Parker N. Hereditary whispering dysphonia. J Neurol Neurosurg Psychiatry 1985;48:218–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wilcox RA, Winkler S, Lohmann K, Klein C. Whispering dysphonia in an Australian family (DYT4): a clinical and genetic reappraisal. Mov Disord 2011;26:2404–2408. [DOI] [PubMed] [Google Scholar]
- 4.Hersheson J, Mencacci NE, Davis M, et al. Mutations in the autoregulatory domain of β-tubulin 4a cause hereditary dystonia. Ann Neurol 2013;73:546–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lohmann K, Wilcox RA, Winkler S, et al. Whispering dysphonia (DYT4 dystonia) is caused by a mutation in the TUBB4 gene. Ann Neurol 2013;73:537–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lohmann K, Klein C. Update on the genetics of dystonia. Curr Neurol Neurosci Rep 2017;17:26. [DOI] [PubMed] [Google Scholar]
- 7.Simons C, Wolf NI, McNeil N, et al. A de novo mutation in the β-tubulin gene TUBB4A results in the leukoencephalopathy hypomyelination with atrophy of the basal ganglia and cerebellum. Am J Hum Genet 2013;92:767–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van der Knaap MS, Naidu S, Pouwels PJW, et al. New syndrome characterized by hypomyelination with atrophy of the basal ganglia and cerebellum. AJNR Am J Neuroradiol 2002;23:1466–1474. [PMC free article] [PubMed] [Google Scholar]
- 9.Hamilton EM, Polder E, Vanderver A, et al. Hypomyelination with atrophy of the basal ganglia and cerebellum: further delineation of the phenotype and genotype-phenotype correlation. Brain 2014;137:1921–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Erro R, Hersheson J, Ganos C, et al. H-ABC syndrome and DYT4: variable expressivity or pleiotropy of TUBB4 mutations? Mov Disord 2015;30:828–833. [DOI] [PubMed] [Google Scholar]
- 11.Zech M, Boesch S, Jochim A, et al. Large-scale TUBB4A mutational screening in isolated dystonia and controls. Parkinsonism Relat Disord 2015;21:1278–1281. [DOI] [PubMed] [Google Scholar]
- 12.Vulinovic F, Schaake S, Domingo A, et al. Screening study of TUBB4A in isolated dystonia. Parkinsonism Relat Disord 2017;41:118–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Invitae Dystonia Comprehensive Panel [online]. Available at: www.invitae.com/en/physician/tests/03351/. Accessed November 20th, 2020.
- 14.Genome Aggregation Database [online]. Available at: gnomad.broadinstitute.org. Accessed November 20th, 2020.
- 15.Naslavsky MS, Yamamoto GL, de Almeida TF, et al. Exomic variants of an elderly cohort of Brazilians in the ABraOM database. Hum Mutat 2017;38:751–763. [DOI] [PubMed] [Google Scholar]
- 16.Brazilian Initiative on Precision Medicine (BIPMed) [online]. Available at: bipmed.org. Accessed November 20th, 2020.
- 17.Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 2019;47:D886–D894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu X, Wu C, Li C, Boerwinkle E. dbNSFP v3.0: a one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum Mutat 2016;37:235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jagadeesh KA, Wenger AM, Berger MJ, et al. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat Genet 2016;48:1581–1586. [DOI] [PubMed] [Google Scholar]
- 20.Conway Institute UCD Dublin. Clustal Omega [online]. Available at: clustal.org/omega/. Accessed November 20th, 2020.
- 21.Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 2015;10:845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.UCSF Chimera. Available at: cgl.ucsf.edu/chimera/download.html. Accessed November 20th, 2020. [Google Scholar]
- 23.Wass MN, Kelley LA, Sternberg MJE. 3DLigandSite: predicting ligand-binding sites using similar structures. Nucleic Acids Res 2010;38(Web Server issue):W469–W473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Structural Bioinformatics Group. 3DLigandSite: Ligand binding site prediction server [online]. 3DLigandSite. Available at: www.sbg.bio.ic.ac.uk/3dligandsite/. Accessed November 20th, 2020. [Google Scholar]
- 25.Broad Institute of MIT and Harvard. GTEx Portal [online]. Available at: gtexportal.org/home/. Accessed November 20th, 2020.
- 26.Löwe J, Li H, Downing KH, Nogales E. Refined structure of alpha beta-tubulin at 3.5 A resolution. J Mol Biol 2001;313:1045–1057. [DOI] [PubMed] [Google Scholar]
- 27.Alushin GM, Lander GC, Kellogg EH, Zhang R, Baker D, Nogales E. High-resolution microtubule structures reveal the structural transitions in αβ-tubulin upon GTP hydrolysis. Cell 2014;157:1117–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Albanese A, Bhatia K, Bressman SB, et al. Phenomenology and classification of dystonia: a consensus update. Movement Disord 2013;28:863–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fasano A, Nardocci N, Elia AE, Zorzi G, Bentivoglio AR, Albanese A. Non-DYT1 early-onset primary torsion dystonia: comparison with DYT1 phenotype and review of the literature. Mov Disord 2006;21:1411–1418. [DOI] [PubMed] [Google Scholar]
- 30.LeDoux MS, Xiao J, Rudzińska M, et al. Genotype-phenotype correlations in THAP1 dystonia: molecular foundations and description of new cases. Parkinsonism Relat Disord 2012;18:414–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gorman KM, Meyer E, Kurian MA. Review of the phenotype of early-onset generalised progressive dystonia due to mutations in KMT2B. Eur J Paediatr Neurol 2018;22:245–256. [DOI] [PubMed] [Google Scholar]
- 32.Blitzer A, Brin MF, Simonyan K, Ozelius LJ, Frucht SJ. Phenomenology, genetics, and CNS network abnormalities in laryngeal dystonia: a 30-year experience. Laryngoscope 2018;128:S1–S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lohmann K, Klein C. Genetics of dystonia: what's known? What's new? What's next? Mov Disord 2013;28:899–905. [DOI] [PubMed] [Google Scholar]
- 34.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet medicine : official J Am Coll Med Genet 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Curiel J, Bey GR, Takanohashi A, et al. TUBB4A mutations result in specific neuronal and oligodendrocytic defects that closely match clinically distinct phenotypes. Hum Mol Genet 2017;26:4506–4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vulinovic F, Krajka V, Hausrat TJ, et al. Motor protein binding and mitochondrial transport are altered by pathogenic TUBB4A variants. Hum Mutat 2018;39:1901–1915. [DOI] [PubMed] [Google Scholar]
- 37.Watanabe N, Itakaoka M, Seki Y, et al. Dystonia-4 (DYT4)-associated TUBB4A mutants exhibit disorganized microtubule networks and inhibit neuronal process growth. Biochem Biophys Res Commun 2018;495:346–352. [DOI] [PubMed] [Google Scholar]
- 38.National Center for Biotechnology Information. ClinVar [online]. Available at: ncbi.nlm.nih.gov/clinvar/.
- 39.Amos LA. Focusing-in on microtubules. Curr Opin Struct Biol 2000;10:236–241. [DOI] [PubMed] [Google Scholar]
- 40.McKean PG, Vaughan S, Gull K. The extended tubulin superfamily. J Cel Sci 2001;114:2723–2733.. [DOI] [PubMed] [Google Scholar]
- 41.Nogales E, Wolf SG, Downing KH. Structure of the alpha beta tubulin dimer by electron crystallography. Nature 1998;391:199–203. [DOI] [PubMed] [Google Scholar]
- 42.Curiel J, Rodríguez Bey G, Takanohashi A, et al. TUBB4A mutations result in specific neuronal and oligodendrocytic defects that closely match clinically distinct phenotypes. Hum Mol Genet 2017;26:4506–4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data and supplemental data are available on Dryad, doi.org/10.5061/dryad.34tmpg4gt.