

Abstract
Mitochondria are organelles central to myriad cellular processes. To maintain mitochondrial health, various processes co-operate at both the molecular and organelle level. At the molecular level, mitochondria can sense imbalances in their homeostasis and adapt to these by signaling to the nucleus. This mito-nuclear communication leads to the expression of nuclear stress response genes. Upon external stimuli, mitochondria can also alter their morphology accordingly, by inducing fission or fusion. In an extreme situation, mitochondria are degraded by mitophagy. Adequate function and regulation of these mitochondrial quality control pathways are crucial for cellular homeostasis. As we discuss, alterations in these processes have been linked to several pathologies including neurodegenerative diseases and cancer.
Keywords: Mitochondrial dysfunction, UPRmt, ISR, Mitochondrial fission, Mitochondrial fusion, Mitophagy, PINK1, Parkin, Mitochondrial diseases
Introduction
Mitochondria are implicated in an expanding array of biological processes including redox balance, calcium homeostasis, energy production, metabolism and cell death [1, 2]. To maintain function, cells have evolved various processes that sense and respond to defective mitochondrial activity. These mechanisms react differently depending on the nature or intensity of the stress that mitochondria face. For instance, mitochondria can sense internal stresses such as misfolded proteins, mitochondrial DNA (mtDNA) mutations, metabolic or oxidative stress [3]. Mitochondria can adapt to these by retrograde signaling to the nucleus leading to the transcriptional upregulation of stress response proteins [4, 5]. Besides these stresses, mitochondria also face external ones such as mechanical stress, infection and environmental stress (e.g., hypoxia) [3]. Hence, when cells are challenged, mitochondria can maintain function through a second line of defense, by altering their mitochondrial dynamics. However, in the face of persistent damage, another homeostatic mechanism is to remove damaged mitochondria through a process called mitophagy [3, 6, 7].
Alterations in mitochondrial quality control responses have been described in several mitochondrial diseases such as neurodegenerative diseases (Parkinson’s, Alzheimer’s and Huntington’s disease) [8–11], cardiomyopathies [12], ocular diseases [13, 14] and cancer [15–17], highlighting the importance of an adequate balance of these pathways for maintaining homeostasis.
In this review, we will discuss key mechanisms of mitochondrial quality control pathways. Due to the complexity and diversity of these pathways we have divided them into two broad areas: regulation at the molecular level (focusing on mitochondrial-to-nuclear communication) and regulation at the organelle level (including mitochondrial dynamics and mitophagy). Our discussion is not intended to be exhaustive, for instance, for in-depth discussion of the cellular response to ROS, NAD + (nicotinamide adenine dinucleotide) and calcium signaling, the reader is referred to recent, comprehensive reviews [18–20].
Regulation of mitochondrial quality control at the molecular level
Mitochondria are organelles of bacterial ancestry. Thus, mitochondria have their own genome (mtDNA) in the matrix, which is surrounded by the inner (IMM) and outer mitochondrial membrane (OMM). While mitochondria have their own genome, the vast majority of mitochondrial proteins are encoded in the nucleus [21] and therefore require import to maintain mitochondrial function [22]. Additionally, mitochondria also need to cope with oxidative stress. Therefore, to ensure homeostasis, mitochondria have developed several mechanisms to sense and respond to stress via nuclear communication. This mito-nuclear communication—also termed mitochondrial retrograde signaling or mitohormesis—is an evolutionary conserved process. It can be triggered by several stressors such as misfolded proteins, inhibition of the ETC, mitochondrial depolarization, nutrient deprivation and/or redox imbalances [23–27]. Hence, different pathways are involved in regulating mito-nuclear communication (Fig. 1).
Fig. 1.

Mitochondrial retrograde signaling. The mitochondrial integrated stress response (ISRmt) is regulated by phosphorylation of the elongation transcription factor eIF2α, which enhances the translation of ATF4. The kinases HRI and GCN2 phosphorylate eIF2α (P-eIF2α) following a mitochondrial stress. In addition, the ISR is also part of the mitochondrial unfolded protein response (UPRmt), activated by misfolded mitochondrial proteins. Moreover, other mechanisms of mito-nuclear communication (mTORC, SIRT1, GPS2) are activated following changes in mitochondrial ROS, NAD, ATP/ADP ratio, calcium (Ca2+) or membrane potential (Δψ). Once the transcription factors ATF4, ATF5, ATF2 and/or GPS2 are in the nucleus, they regulate the expression of nuclear stress-response genes
Mitochondrial retrograde response
One of the first descriptions of mitochondrial retrograde signaling was made in the yeast Saccromyces cerevisiae, where the genes RTG1 and RTG2 were found to regulate the expression of the nuclear gene citrate synthase (CIT2) in response to alteration of mitochondrial function [28]. Stemming from this finding, Jia et al. described that the transcription factor involved in the regulation of CIT2 expression in yeast is a heterodimeric complex of Rtg1p and Rtg3p proteins (Retrograde regulation protein 1 and 3) [29]. Recently, it has been shown that inhibition of protein import into mitochondria and consequently cytoplasmic accumulation of misfolded mitochondrial proteins caused global changes at the transcriptome and proteome levels [30]. This led to an increased expression of proteasome subunit and cytosolic chaperones and a decrease in oxidative phosphorylation (OXPHOS) proteins. However, these changes were regulated by different transcription factors; Hsf1 and Rpn4 were involved in the upregulation of the proteasome and chaperones proteins; whilst the inactivation of the HAP complex led to a downregulation of OXPHOS proteins [30]. This study shows that there are multiple pathways that regulate mito-nuclear communication. Indeed, several mitochondrial signaling pathways that respond to inhibition of protein import have been recently described in yeast such as the unfolded protein response activated by mistargeting of proteins (UPRam) [31], mitochondrial precursor over-accumulation stress (mPOS) [32], mitochondrial compromised protein import response (MitoCPR) [33] and mitochondrial protein translocation-associated degradation (mitoTAD) [34], reviewed in [35].
Mitochondrial retrograde signaling has also been described in the nematode Caenorhabditis elegans, where mitochondria can sense and respond to unfolded proteins by activating the mitochondrial unfolded protein response (UPRmt), and thereby regulating the expression of mitochondrial chaperone genes, amongst others [36]. Mechanistically, Haynes and colleagues found that upon a proteotoxic stress, the mitochondrial matrix protease CLPP and the homeodomain-containing transcription factor DVE-1 in complex with the small ubiquitin-like protein UBL-5, are required for UPRmt [37]. Additionally, they found that both CLPP and the mitochondrial peptide transporter, HAF-1, are required for activation of the transcription factor ZC376.7, which binds to and activates the expression of nuclear genes involved in the UPRmt [38]. The transcription factor ZC376.7 (mammalian homolog of ATF5), was later re-named as ATFS-1 (Activating Transcription Factor associated with Stress-1). Thus, Nargund et al. showed that in C. elegans, ATFS-1 is imported into mitochondria and degraded under basal conditions, but following a mitochondrial stress, ATFS-1 accumulates in the cytosol, promoting its nuclear transport [39]. In the nucleus, ATFS-1 regulates the expression of several nuclear genes involved in immunity, mitochondrial chaperones and metabolism, amongst others [40, 41].
Besides these, other complementary mitochondrial retrograde signaling pathways have been observed in worms in response to mitochondrial dysfunction and oxidative stress (ROS). For instance, the translation initiator factor eIF2α (Eukaryotic Translation Initiation Factor 2A) is phosphorylated by the kinase CGN2 (general control non-derepressible-2) and modulates cytosolic protein synthesis [42]. On the other hand, the mitochondrial protein monooxygenase CLK-1 can translocate to the nucleus where it regulates ROS metabolism and proteostasis, by suppressing a subset of UPRmt genes [43]. Additionally, Herholz et al. found that Krüppel-like factor 1 (KLF-1) is the key transcription factor that regulates the expression of genes involved in xenobiotic detoxification processes [44]. This argues that distinct mechanisms are also involved in mito-nuclear communication in nematodes.
Furthermore, mitochondrial retrograde signaling has also been described in the fruit fly Drosophila melanogaster [45, 46] and in mammals. In mammals, there is evidence that treatments that alter mitochondria health, such as loss of membrane potential, unfolded proteins, inhibition of protein translation or mutations in mitochondrial DNA, signal to the nucleus in order to regulate the expression of nuclear genes and adapt towards adverse conditions [23–27]. Thus, indicating—as a generalized process—retrograde signaling is evolutionary conserved.
One of the mitochondrial retrograde signaling pathways in mammals is the UPRmt, which, as in other species, is activated upon the accumulation of misfolded mitochondrial proteins. A similar mechanism has been previously documented in other mammalian organelles, notably the endoplasmic reticulum (ER), where three different pathways (ATF6, PERK, IRE1α) are known to regulate the endoplasmic reticulum unfolded protein response (UPRer) [47]. Upon the discovery of UPRmt in other organisms, there has been intensive research trying to identify transcription factors involved in the mammalian UPRmt. One possibility is the protein ATF5 (Activated Transcription Factor 5), that appears to be activated similarly to ATFS-1 [48], meanwhile others have reported ATF4 (Activated Transcription Factor 4) as the transcription factor activated upon mitochondrial stress [5]. In addition, there is also evidence in both nematodes and mammalian cells, that NAD + can activate the UPRmt through activation of the Sirtuin pathway (specifically SIRT1) [49].
Another mechanism involved in the mitochondrial retrograde signaling is the integrated stress response (ISR). This pathway is activated upon several stresses such as proteostasis defects, nutrient deprivation, redox imbalances and viral infection, amongst others. However, all of them converge in the phosphorylation of the translation initiator factor eIF2α [50]. Various kinases that phosphorylate eIF2α have been identified: the double-stranded RNA-activated protein kinase (PKR) [51, 52], the general control non-derepressible-2 (GCN2) [53], the endoplasmic reticulum (ER) resident kinase (PERK) [54] and heme-regulated inhibitor (HRI) [55]. Upon phosphorylation of eIF2α (P-eIF2α), the activity of eIF2B (eIF2′s guanine nucleotide exchange factor) is blocked, therefore inhibiting protein synthesis [56]. Nevertheless, P-eIF2α can enhance the translation of the transcription factor ATF4 [57]. ATF4 was first described as a transcription factor involved in the UPRer upon activation by PERK [57]. Nevertheless, different studies have shown that eIF2α and ATF4 can be activated not only upon ER stress but also upon mitochondrial stress engaged by other signaling pathways [5, 42]. Two recent papers have described a mechanism of activation of ATF4 following a mitochondrial stress [58, 59]. Employing a genome-wide CRISPR screen, they found that OMA1 (a mitochondrial stress protease located on the inner mitochondrial membrane) is activated upon mitochondrial stress and cleaves DELE1 (another inner mitochondrial membrane protein). Cleavage of DELE1 leads to its accumulation in the cytosol and therefore activation of HRI, one of the kinases that phosphorylates eIF2α [58, 59].
Interestingly, the expression of not only ATF4 but also ATF5 is enhanced by eIF2α [60], thereby indicating that activation of the ISR is required for activation of UPRmt in mammals. In addition, in muscle, mTORC1 has been shown to regulate the one carbon metabolism and metabolic cytokines (FGF21) via ATF4 signaling [61], indicating a role for mTORC1 also in the ISR. Furthermore, Cardamone and colleagues described that the protein GPS2, involved in inflammation and lipid homeostasis, regulate the transcription of several ISR regulated genes [62]. They found that this protein resides in the mitochondria and, upon mitochondrial depolarization induced by FCCP treatment (a mitochondrial uncoupler of oxidative phosphorylation that disrupts ATP synthesis); GPS2 translocates to the nucleus, upregulating the expression of nuclear genes involved in the tricarboxylic acid (TCA) cycle, the electron transport chain (ETC), lipid synthesis and interleukin signaling pathways [62].
Although less well-described, apart from these, other mechanisms of mitochondrial retrograde signaling have also been reported in mammals. For instance, in myocytes, different mitochondrial stressors (decrease in mitochondrial membrane potential, inhibition of electron transport chain, reduced mtDNA or increased intracellular calcium levels) caused alterations in the expression of nuclear genes. These nuclear genes were regulated by the transcription factors NFAT (cytosolic counterpart of activated T-cell-specific nuclear factor) and ATF2 (Activated transcription factor 2) [27].
In vivo studies have confirmed the existence of several mitochondrial stress pathways in mammals [63–65]. Thus, Gomes et al. showed that when nicotinamide mononucleotide adenylyltransferase (NMNAT1) was decreased in the nucleus, it caused a decrease in expression of mitochondrial OXPHOS genes, mtDNA and ATP levels, by activating SIRT1 [65]. Moreover, mice that lack the expression of the mitochondrial aspartyl-tRNA synthetase, DARS2 (DARS2-deficient mice) display deregulated mitochondrial protein synthesis and thereby activation of the UPRmt [64]. However, although these mice demonstrated alterations in the respiratory chain, activation of the mitochondrial stress response was independent of the respiratory defects observed [64]. Nevertheless, deletion of CLPP in DARS2-deficient mice decreased the observed respiratory defects but did not rescue the UPRmt signaling, indicating a role of CLPP in OXPHOS regulation [63]. In addition, unlike in the nematode C. elegans where CLPP has a central role in regulating UPRmt [37], this study showed no role for CLPP in UPRmt regulation in mammals [63]. Hence, these data argue against different mitochondrial stress pathways involved in mito-nuclear communication in vivo that act cooperatively to maintain homeostasis.
Mitochondrial retrograde signaling in health and disease
Since the decline of mitochondria function is a hallmark of aging, neurogenerative disease and cancer [66], understanding how those pathways work is important for human health. In fact, several studies have demonstrated the importance of the UPRmt for development and longevity [42, 67]. Thus, mutations or deletions in genes involved in the ETC [68] and increased levels of NAD + [49], activated the UPRmt and extended lifespan. How mito-communication regulates lifespan remains unclear. Gomes et al. demonstrated that SIRT1/HIF1α pathway contributes to aging-associated mitochondrial dysfunction in mice and that this phenotype could be reversed by increasing NAD + levels [65]. This indicates that increasing the levels of NAD + could be beneficial and a possible treatment option for patients with mitochondrial diseases. Similarly, another strategy for treating these patients may be using PARP-1 inhibitors as PARP-1 (poly(ADP-ribose) polymerase-1) is a NAD + consuming enzyme. Indeed, treatment with PARP-1 inhibitors has been demonstrated to restore mitochondrial function [69]. On the other hand, others have suggested that activation of UPRmt extends longevity due to the induction of changes in chromatin structure through histone modifications [70]. In addition, a role for ROS in longevity has also been proposed [43, 46, 71], although others have shown that in mice, accumulation of mtDNA mutations that are associated with an aging phenotype and reduced lifespan, did not affect ROS levels [72]. Recently, a study has found that KLF-1 regulates longevity independently of the UPRmt but dependently on redox signaling [44]. Importantly, that extended longevity was due to the expression of xenobiotic detoxification genes such as cytochrome P450 oxidases (CYPs) [44], proposing CYPs as the key effectors of lifespan extension. However, whether or not the association between mitochondrial dysfunction and longevity is due to ROS, epigenetics or any other mechanisms remains unclear.
Alteration of mitochondrial stress-signaling pathways has also been dysregulated in cancer. For instance, in gastric cancer, cisplatin resistance was found to be due to upregulation of the cystine/glutamate antiporter (xCT) via ROS/GCN2-eIF2α-ATF4 pathway [73]. In addition, other studies have shown that tumours upregulate the ISR as a means of chemoresistance. BRAF-mutated melanoma cells, acquire resistance to vemurafenib by GCN2/ATF4 activation [74] and overexpression of ATF5 conferred radioresistance in lung cancer cells [75] and in malignant glioma cells, ATF5 promoted survival through transcription of anti-apoptotic MCL-1 [76]. Although the involvement of mitochondria was not shown directly in these studies, they offer proof of principle that ISR can induce drug resistance. Recently, ATF4 has also been shown to be oncogenic; in Drosophila, activation of ATF4 induced a Warburg-like phenotype and consequently tumorigenic growth [77] and in MYC-driven tumours, deletion of ATF4 prolonged survival [17], indicating that ATF4 is necessary for tumorigenesis. Collectively, although several advances have been made over the recent years in understanding the mitochondrial retrograde response in several organisms, much remains to be elucidated.
Regulation of mitochondrial quality control at the organelle level
As dynamic organelles, mitochondria can also alter their morphology by undergoing mitochondrial fission or fusion and thereby adapt toward stresses and fulfill the cellular needs. However, if the stress is severe or prolonged, cells can eliminate mitochondria through a process called mitophagy. Both mitochondrial dynamics and mitophagy, have demonstrated their importance for homeostasis as alterations in genes involved in these processes have been found in several diseases [10, 13, 14, 78].
Mitochondrial dynamics
Mitochondria are dynamic organelles that adapt their network according to cellular requirements. These changes are determined by rates of fission and fusion, the regulation of which varies with regards to the severity of the stresses that they face. Upon a high or prolonged stress, mitochondria undergo fission, meanwhile, upon a mild or low stress, mitochondria favour fusion [79]. Furthermore, studies have shown that mitochondria can also alter their morphology depending on the nature of stress; starvation induces fusion, meanwhile, low glucose and ETC inhibition can induce fission [80–83] (Fig. 2).
Fig. 2.
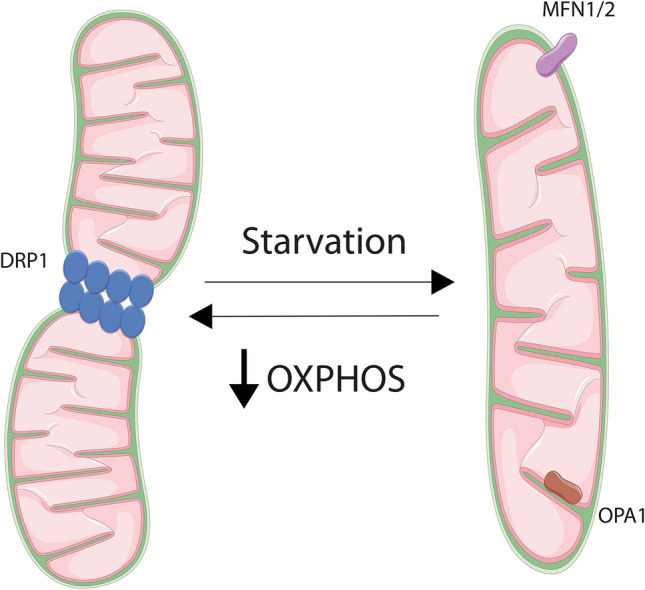
Mitochondrial dynamics. Mitochondria are dynamic organelles that adapt their network according to cellular requirements. For instance, a decrease of respiration leads to mitochondrial fission and starvation induces fusion. Both processes are regulated by different proteins; dynamin-related protein 1 (DRP1) is involved in fission, meanwhile Mitofusin 1 and Mitofusin 2 (MFN1/2) and optic atrophy 1 (OPA1) allows fusion. OXPHOS = oxidative phosphorylation
An appropriate balance in mitochondrial dynamics is important for mitochondrial health as each of them is implicated in different processes. Fusion is involved in the maintenance of mtDNA integrity, mitochondrial respiration, mitochondrial membrane potential, apoptosis and calcium signalling [84–89]. Fission has been shown to be important for preventing oxidative damage and to enable degradation of damaged mitochondria [6, 87, 90]. Additionally, mitochondrial dynamics have also been implicated in life span regulation [91].
Mitochondrial fusion
Mitochondrial fusion is regulated at both mitochondrial membranes; the outer membrane (OMM) and the inner membrane (IMM). Mitofusin 1 (MFN1) and Mitofusin 2 (MFN2) are GTPases required to allow fusion of the OM [92, 93] by forming homodimers or heterodimers [94]. This process is essential for development because combined knockout of MFN1 and MFN2 leads to embryonic lethality. Single knockouts of MFN1 or MFN2 are viable, however, deletion of each one alone promotes mitochondrial fragmentation, demonstrating their role in mitochondrial fusion [94]. In addition, mutations in MFN2 have been found in axonal Charcot-Marie-Tooth disease type 2A [9] and mice with the mutation R95Q in MFN2 developed this pathology [78]. MFN2 deficient mice showed cerebellar defects; dysfunctional ETC and loss of mtDNA nucleoids in Purkinje cells due to dysfunctional mitochondria (fusion-deficient cells) [95]. In another study, MFN2 mutant mice demonstrated a decrease in locomotive activity and a lack of MFN2 in dopaminergic neurons displayed fragmented mitochondria and decreased mitochondrial motility [96]. These data emphasise the importance of this protein and mitochondrial fusion for neurological development.
The protein optic atrophy 1 (OPA1) mediates fusion of the mitochondrial inner membrane [97, 98]. OPA1 has multiple isoforms due to splicing or variants generated via proteolytic cleavage. The long form of OPA1 (L-OPA1) is anchored in the inner membrane and it promotes mitochondrial fusion, whereas the short form of OPA1 (S-OPA1) is soluble. Mitochondrial fusion depends on the presence of both isoforms [99], by forming oligomeric complexes that maintain the cristae structure [100, 101]. Cleavage of L-OPA1 into S-OPA1 is mediated by the AAA-proteases OMA1 [102, 103] and YME1L [99, 104, 105]. Stresses that activate OMA1 or YME1L, for instance loss of mitochondrial potential or OXPHOS activity, lead to the proteolytic process of L-OPA1 into S-OPA1, liberating this protein from the IMM and therefore promoting fission [99, 103, 106, 107]. Thus, regulation of OPA1 proteolysis is a key process for mitochondrial network maintenance and function. Accordingly, mutations in this gene cause dominant optic atrophy (ADOA) [13, 14] and heterozygous mutations have been also associated with Behr syndrome [108].
Mitochondrial fission
The main proteins involved in mitochondrial fission are DRP1 (dynamin-related protein 1) [109], mitochondrial fission factor (MFF) [110] and mitochondrial fission protein 1 (FIS1) [111], amongst others [112]. DRP1 is a GTPase that resides in the cytosol. Upon specific stimulus DRP1 is recruited onto the OMM by receptors such as MFF and FIS1 [110]. However, there are differences between the yeast FIS1 and the human Fis1 (hFis1) in their activity. A recent study has shown that hFis1 induces fission independently of DRP1, instead, hFis1 negatively regulates fusion by binding to OPA1, MFN1 and MFN2 and blocking its activity [113]. Nevertheless, once DRP1 gets onto the OMM it multimerizes and forms rings on the mitochondrial outer membrane [109]. Once there, by hydrolysing GTP, the complex changes conformation and allows the constriction of the membrane inducing its division [114]. DRP1 activity is not only regulated by its translocation, but also by post-translational modifications including phosphorylation, ubiquitylation, and SUMOylation. Phosphorylation of S616 allows DRP1 activity [115], meanwhile, phosphorylation of S637 and S656 inhibits it [116, 117]. However, several kinases have been involved in the phosphorylation of its residues. For instance, ERK1/2 [15, 118] and mitotic kinase cyclin B-CDK1 (cyclin-dependent kinase 1 [115] have been described to phosphorylate DRP1 in S616 [119]. On the other hand, S656 is phosphorylated by cyclic AMP‐dependent protein kinase, PKA, and dephosphorylated by calcineurin [117].
DRP1 is essential for development; whole-body deletion of DRP1 is embryonic lethal [120, 121], showing the relevance of this protein for mitochondrial homeostasis. However, unlike MFN1/2 or OPA1, no autosomal diseases have been linked to mutations in DRP1, although alterations in mitochondrial fission have been related to neurodegenerative diseases [8]. Moreover, in different tumour models, such as pancreas [16], melanoma [118], lung cancer [122] and ovarian cancer [123], DRP1 and mitochondrial fission have shown to be important for tumour growth [15]. In addition, tissues from Alzheimer’s disease (AD) [124] and Huntington's disease (HD) [125] patients demonstrated increased expression of DRP1 and FIS1, and decreased expression of MFN1, MFN2 and OPA1, indicating that an impairment on mitochondrial dynamics are associated with AD and HD. Thus, an adequate function of mitochondrial fusion and fission is important for homeostasis and alterations in these processes are associated with several pathologies, such as cancer, neurodegenerative, neuroinflammatory and autoimmune disease [126].
Mitophagy
Autophagy is a regulated process through which cells can degrade unnecessary or non-functional cytosolic components through their engulfment in a double membrane vesicle, called autophagosome, and their delivery into the lysosomes. There are different types of autophagy such as macroautophagy, microautophagy, and chaperone-mediated autophagy (Reviewed in [127, 128]). The selective removal of mitochondria by autophagy, namely mitophagy, helps maintain adequate cellular homeostasis through the elimination of damaged mitochondria [7, 129] (Fig. 3). How was mitophagy discovered? Dysfunctional mitochondria were first observed inside vacuoles in yeast [130]. Later, the term mitophagy was coined by Lemaster’s group after finding mitochondria inside of lysosomes in rat hepatocytes upon nutrient deprivation [131]. These observations raised several questions—how do cells sense damaged mitochondria and how is mitophagy regulated? There are many mechanisms of mitophagy described [132–136]. For instance, the protein NIX has been shown to be involved in the removal of non-damaged mitochondria during erythropoiesis [137, 138]. Here we will focus on the PINK1/Parkin pathway, as the mechanism is better described and this pathway is involved in regulating mitochondrial stress response.
Fig. 3.
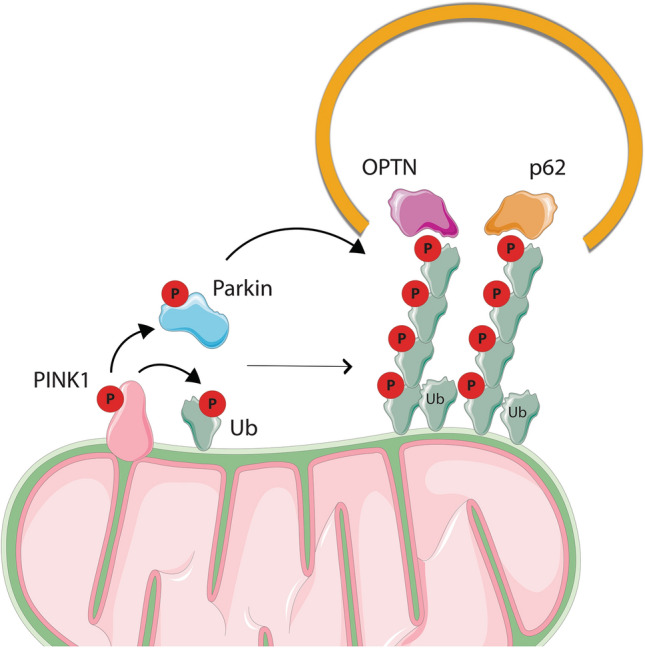
Mitophagy. Mitophagy allows the removal of damaged mitochondria by the PINK/Parkin pathway. When mitochondria are damaged, PINK1 is accumulated on the outer mitochondrial membrane (OMM) where it gets activated and phosphorylates and therefore activates Ubiquitin (Ub) and Parkin. Parkin ubiquitinates different OMM proteins in a feed-forward mechanism, leading to a general mitochondrial ubiquitination. That ubiquitination recruits autophagy receptor proteins (OPTN, p62), and, in turn, autophagosomes into the mitochondria, leading to the induction of autophagy
PINK1/Parkin pathway
The importance of PINK1 (PTEN-induced putative kinase 1) and Parkin for mitochondrial homeostasis is supported by the accumulation of dysfunctional mitochondria in Parkinson’s disease (PD) models and patients [10, 139]. In fact, both of these proteins have been found mutated in this disease; PINK1 mutation causes Hereditary Early-Onset Parkinson [10] meanwhile mutations in Parkin occurs in Autosomal recessive juvenile Parkinsonism (AR-JP) [11]. In Drosophila, studies have shown that this pathway is needed for maintaining adequate mitochondria function in muscle and neurons, and loss of function mutations of both proteins mimic the characteristic of PD [140]. Furthermore, in mice, deletion of Parkin leads to loss of dopaminergic neurons (DA), which is a hallmark of PD [141].
Under normal conditions, PINK1 is partially imported into mitochondria, where it is cleaved by the IMM protease PARL. This cleavage enables PINK1 ubiquitination and proteasome-dependent degradation in the cytosol [142]. Therefore, in healthy mitochondria, the levels of PINK1 are low in cells. Upon stresses such as mitochondrial depolarization [143], impairment of protein import [144], and others [145–147], PINK1 import into mitochondria is blocked and as a result, it accumulates on the OMM [148]. Once there, it binds the mitochondrial TOM complex [149, 150] and it is activated by auto-phosphorylation. Its activation leads to a signaling cascade by recruiting and activating other proteins like Parkin [143, 151] and Ubiquitin (Ub) [152]. Parkin is an E3 ubiquitin ligase that is activated upon PINK1 phosphorylation in its UBL domain [153, 154] and binding of phospho-Ub [152, 155–158]. Once activated, Parkin ubiquitinates different OMM proteins in a feed-forward mechanism, leading to a general mitochondrial ubiquitination. Then, receptor proteins on the mitochondria recruit the autophagic machinery which results in the engulfment of mitochondria by autophagosomes. Different autophagy receptors have been shown to play a role in mitophagy such as NDP52, optineurin (OPTN) and p62 [159, 160].
In vivo, the role of PINK1 and Parkin in mitochondrial homeostasis (mitophagy) is unclear. On the one hand, using a mouse model that lacks the proofreading function of the mitochondrial DNA polymerase g (POLG) (called Mutator mice), Pickrell and colleagues demonstrated that Parkin protects dopaminergic neurons from mitochondrial dysfunction. They show that Parkin loss in Mutator mice led to DA neuron loss and decrease in OXPHOS activity [161], thereby suggesting that Parkin has a role in maintaining mitochondrial homeostasis in vivo, or at least in DA neurons. On the other hand, mice that lack Parkin present normal cardiac function and their mitochondrial activity was unaltered. However, when those mice are challenged with a mitochondrial stress such as myocardial infarction or hypoxia, Parkin played a critical role in removing damaged mitochondria [162]. In line with these findings, another study has corroborated that in heart, the expression of Parkin is dispensable for mitophagy under basal conditions [163]. However, it contributes to mitochondrial removal in cardiomyopathies that present defects in mitochondrial fission [163], thus indicating a role of Parkin in regulating stress-induced mitophagy. In fact, studies have also shown that mice with PINK1 loss display normal basal mitophagy [135] and research in Drosophila has corroborated that under basal mitophagy, PINK1 and Parkin are dispensable [164]. Therefore, these studies together will indicate that the PINK1/Parkin pathway may not be implicated in regulating mitophagy under physiological conditions and/or that there are other additional pathways that regulate mitophagy [135]. In addition, although Parkin deficient mice demonstrated no role of Parkin for basal mitophagy, the mitochondria of those mice, although functional, were smaller [162], which may indicate that Parkin has additional roles independently of mitophagy. In fact, Parkin has also been involved in regulating bioenergetics [129], necroptosis [165], mitochondrial protein import [166], mitochondrial biogenesis [167] and inflammation [168].
Another question that remains controversial is whether PD is due to PINK1 or Parkin deficiency or due to a general mitochondrial dysfunction. The aforementioned study demonstrates a role of Parkin and PD [161]. However, pathogenic mutations of PD (T415N and G430D) that abolish the E3 activity of Parkin, altered its mitochondrial localization, whereas other pathological mutations (D280N or G328E) that do not alter its E3 activity did not impact Parkin recruitment to mitochondria [169]. In addition, mutations in PINK1 and Parkin are not exclusive for this pathology as other alterations such as mtDNA mutations [170, 171] and LRRK2 mutations [172, 173] have been described. Moreover, in some PD patients, impairment of MIRO1 degradation, an OMM protein that anchors mitochondria to microtubule motors, has also been observed [174]. Finally, another challenge of studying mitophagy is finding a physiological method of triggering it. Recently, Kovalchuke and colleagues have demonstrated that other more physiological oxidative stressors like L-DOPA, lead to Parkin degradation. Although the exact mechanism of how Parkin is degraded following L-DOPA remains unclear, they show that PINK1 and phospho-Ub are involved in this pathway [175], demonstrating a similar mechanism to other mitochondrial stressors such as CCCP (mitochondrial uncoupler) [156]. This study suggests that oxidative stress or phospho-Ub may contribute to the loss of Parkin in PD. Moreover, that finding opens a new strategy to treat those patients by blocking the association of Parkin with phospho-Ub. Another possible approach for PD treatment comes from the finding that PINK1 is cleaved by OMA1 in depolarised mitochondria. This indicates that inhibition of OMA1 could be used to increase the levels of mitophagy in PD patients [150].
To conclude, mitochondrial damage and defects in mitophagy have been observed in several neurodegenerative diseases, such as Parkinson’s (PD) [176] Alzheimer’s (AD) and Huntington’s disease (HD) [177]. Moreover, mitophagy impairment has also been associated with myopathies, metabolic disorders, inflammation and cancer [178]. Therefore, it is important to understand the mechanisms behind it and its role in the diseases, as it will help to develop new approaches for improved treatments.
Conclusion
Although the UPRmt is mainly caused by protein misfolding [26], other stresses have also been found to activate the UPRmt [179]. In addition, several conditions that were found to activate the ISR such as alteration of mitochondrial membrane potential, mitochondrial respiration, block in mitochondrial translation or in mitochondrial protein import [5], also alter mitochondrial dynamics [80, 180] and/or activated mitophagy [143, 144]. It is likely that activation of those pathways depends on the severity or duration of the stress. However, even though a threshold may exist, a connection between UPRmt and mitophagy has been reported [144]. Similarly, studies have also demonstrated a relation between mitochondrial dynamics and PINK or mitophagy [181, 182]. Therefore, these studies, amongst others, suggest a connection between the mitochondrial quality pathways, which requires further investigation.
As discussed, imbalances in quality control pathways have been associated with multiple pathologies. For instance, aberrations in mitochondrial dynamics have been found in cancer patients [15, 16], neurodegenerative diseases [8, 9] and ocular diseases [13, 14]. In addition, mutations in mitophagy-related genes have also been reported in neurodegenerative pathological conditions [10, 11]. All these patients have alterations in mitochondrial morphology and function. However, these patients also present mutations in other genes [183], thereby is still unclear whether the development of the diseases is due to alterations in mitophagy itself or rather a general mitochondrial dysfunction. Alterations in the expression of proteins involved in mitochondrial retrograde signaling (ISR) have also been linked to resistance and tumour growth [17, 73, 74, 77]. Hence, even though in the recent years new findings have given some clarity in understanding the mitochondrial quality control pathways, some mechanisms underlying mitochondrial homeostasis remain still unclear. Therefore, investigation of those pathways should provide knowledge for understanding, and potentially treating, mitochondrial pathologies.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Alba Roca-Portoles, Email: alba.roca@cbm.csic.es.
Stephen W. G. Tait, Email: stephen.tait@glasgow.ac.uk
References
- 1.Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148(6):1145–1159. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bock FJ, Tait SWG. Mitochondria as multifaceted regulators of cell death. Nat Rev Mole Cell Biol. 2019;21(2):85–100. doi: 10.1038/s41580-019-0173-8. [DOI] [PubMed] [Google Scholar]
- 3.Eisner V, Picard M, Hajnoczky G. Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat Cell Biol. 2018;20(7):755–765. doi: 10.1038/s41556-018-0133-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chandel NS. Mitochondria as signaling organelles. BMC Biol. 2014;12:34–34. doi: 10.1186/1741-7007-12-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Quirós PM, Prado MA, Zamboni N, Damico D, Williams RW, Finley D, Gygi SP, Auwerx J. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J Cell Biol. 2017;216(7):2027–2045. doi: 10.1083/jcb.201702058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Youle RJ, Van Der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337(6098):1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ashrafi G, Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013;20(1):31–42. doi: 10.1038/cdd.2012.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Costa V, Giacomello M, Hudec R, Lopreiato R, Ermak G, Lim D, Malorni W, Davies KJ, Carafoli E, Scorrano L. Mitochondrial fission and cristae disruption increase the response of cell models of Huntington’s disease to apoptotic stimuli. EMBO Mole Med. 2010;2(12):490–503. doi: 10.1002/emmm.201000102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bienfait HME, Baas F, Koelman JHTM, de Haan RJ, van Engelen BGM, Gabreëls-Festen AAWM, Ongerboer de Visser BW, Meggouh F, Weterman MAJ, De Jonghe P, Timmerman V, de Visser M. Phenotype of charcot–marie–tooth disease type 2. Neurology. 2007;68(20):1658–1667. doi: 10.1212/01.wnl.0000263479.97552.94. [DOI] [PubMed] [Google Scholar]
- 10.Valente EM, Abou-Sleiman PM, Caputo V, Muqit MMK, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, Gonzalez-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, Wood NW. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science (New York, N Y ) 2004;304(5674):1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- 11.Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392(6676):605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 12.Song M, Mihara K, Chen Y, Scorrano L, Dorn GW., 2nd Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab. 2015;21(2):273–286. doi: 10.1016/j.cmet.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alexander C, Votruba M, Pesch UE, Thiselton DL, Mayer S, Moore A, Rodriguez M, Kellner U, Leo-Kottler B, Auburger G. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet. 2000;26(2):211. doi: 10.1038/79944. [DOI] [PubMed] [Google Scholar]
- 14.Delettre C, Lenaers G, Griffoin J-M, Gigarel N, Lorenzo C, Belenguer P, Pelloquin L, Grosgeorge J, Turc-Carel C, Perret E. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet. 2000;26(2):207. doi: 10.1038/79936. [DOI] [PubMed] [Google Scholar]
- 15.Kashatus JA, Nascimento A, Myers LJ, Sher A, Byrne FL, Hoehn KL, Counter CM, Kashatus DF. Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol Cell. 2015;57(3):537–551. doi: 10.1016/j.molcel.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagdas S, Kashatus JA, Nascimento A, Hussain SS, Trainor RE, Pollock SR, Adair SJ, Michaels AD, Sesaki H, Stelow EB. Drp1 promotes KRas-driven metabolic changes to drive pancreatic tumor growth. Cell Rep. 2019;28(7):1845–1859. e1845. doi: 10.1016/j.celrep.2019.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tameire F, Verginadis II, Leli NM, Polte C, Conn CS, Ojha R, Salas Salinas C, Chinga F, Monroy AM, Fu W, Wang P, Kossenkov A, Ye J, Amaravadi RK, Ignatova Z, Fuchs SY, Diehl JA, Ruggero D, Koumenis C. ATF4 couples MYC-dependent translational activity to bioenergetic demands during tumour progression. Nat Cell Biol. 2019;21(7):889–899. doi: 10.1038/s41556-019-0347-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shadel GS, Horvath TL. Mitochondrial ROS signaling in organismal homeostasis. Cell. 2015;163(3):560–569. doi: 10.1016/j.cell.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boyman L, Karbowski M, Lederer WJ. Regulation of mitochondrial ATP production: Ca2+ signaling and quality control. Trends Mol Med. 2020;26(1):21–39. doi: 10.1016/j.molmed.2019.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Canto C, Menzies KJ, Auwerx J. NAD+ metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab. 2015;22(1):31–53. doi: 10.1016/j.cmet.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adams KL, Palmer JD. Evolution of mitochondrial gene content: gene loss and transfer to the nucleus. Mol Phylogenet Evol. 2003;29(3):380–395. doi: 10.1016/s1055-7903(03)00194-5. [DOI] [PubMed] [Google Scholar]
- 22.Chacinska A, Koehler CM, Milenkovic D, Lithgow T, Pfanner N. Importing mitochondrial proteins: machineries and mechanisms. Cell. 2009;138(4):628–644. doi: 10.1016/j.cell.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Michel S, Canonne M, Arnould T, Renard P. Inhibition of mitochondrial genome expression triggers the activation of CHOP-10 by a cell signaling dependent on the integrated stress response but not the mitochondrial unfolded protein response. Mitochondrion. 2015;21:58–68. doi: 10.1016/j.mito.2015.01.005. [DOI] [PubMed] [Google Scholar]
- 24.Moisoi N, Klupsch K, Fedele V, East P, Sharma S, Renton A, Plun-Favreau H, Edwards RE, Teismann P, Esposti MD, Morrison AD, Wood NW, Downward J, Martins LM. Mitochondrial dysfunction triggered by loss of HtrA2 results in the activation of a brain-specific transcriptional stress response. Cell Death Differ. 2009;16(3):449–464. doi: 10.1038/cdd.2008.166. [DOI] [PubMed] [Google Scholar]
- 25.Martinus RD, Garth GP, Webster TL, Cartwright P, Naylor DJ, Hoj PB, Hoogenraad NJ. Selective induction of mitochondrial chaperones in response to loss of the mitochondrial genome. Eur J Biochem. 1996;240(1):98–103. doi: 10.1111/j.1432-1033.1996.0098h.x. [DOI] [PubMed] [Google Scholar]
- 26.Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002;21(17):4411–4419. doi: 10.1093/emboj/cdf445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Biswas G, Adebanjo OA, Freedman BD, Anandatheerthavarada HK, Vijayasarathy C, Zaidi M, Kotlikoff M, Avadhani NG. Retrograde Ca2+ signaling in C2C12 skeletal myocytes in response to mitochondrial genetic and metabolic stress: a novel mode of inter-organelle crosstalk. EMBO J. 1999;18(3):522–533. doi: 10.1093/emboj/18.3.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liao X, Butow RA. RTG1 and RTG2: two yeast genes required for a novel path of communication from mitochondria to the nucleus. Cell. 1993;72(1):61–71. doi: 10.1016/0092-8674(93)90050-z. [DOI] [PubMed] [Google Scholar]
- 29.Jia Y, Rothermel B, Thornton J, Butow RA. A basic helix-loop-helix-leucine zipper transcription complex in yeast functions in a signaling pathway from mitochondria to the nucleus. Mol Cell Biol. 1997;17(3):1110–1117. doi: 10.1128/mcb.17.3.1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boos F, Kramer L, Groh C, Jung F, Haberkant P, Stein F, Wollweber F, Gackstatter A, Zoller E, van der Laan M, Savitski MM, Benes V, Herrmann JM. Mitochondrial protein-induced stress triggers a global adaptive transcriptional programme. Nat Cell Biol. 2019;21(4):442–451. doi: 10.1038/s41556-019-0294-5. [DOI] [PubMed] [Google Scholar]
- 31.Wrobel L, Topf U, Bragoszewski P, Wiese S, Sztolsztener ME, Oeljeklaus S, Varabyova A, Lirski M, Chroscicki P, Mroczek S. Mistargeted mitochondrial proteins activate a proteostatic response in the cytosol. Nature. 2015;524(7566):485–488. doi: 10.1038/nature14951. [DOI] [PubMed] [Google Scholar]
- 32.Wang X, Chen XJ. A cytosolic network suppressing mitochondria-mediated proteostatic stress and cell death. Nature. 2015;524(7566):481–484. doi: 10.1038/nature14859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weidberg H, Amon A. MitoCPR—A surveillance pathway that protects mitochondria in response to protein import stress. Science. 2018;360(6385):10–19. doi: 10.1126/science.aan4146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mårtensson CU, Priesnitz C, Song J, Ellenrieder L, Doan KN, Boos F, Floerchinger A, Zufall N, Oeljeklaus S, Warscheid B. Mitochondrial protein translocation-associated degradation. Nature. 2019;569(7758):679–683. doi: 10.1038/s41586-019-1227-y. [DOI] [PubMed] [Google Scholar]
- 35.Boos F, Labbadia J, Herrmann JM. How the mitoprotein-induced stress response safeguards the cytosol: a unified view. Trends Cell Biol. 2020;30(3):241–254. doi: 10.1016/j.tcb.2019.12.003. [DOI] [PubMed] [Google Scholar]
- 36.Yoneda T, Benedetti C, Urano F, Clark SG, Harding HP, Ron D. Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J Cell Sci. 2004;117(Pt 18):4055–4066. doi: 10.1242/jcs.01275. [DOI] [PubMed] [Google Scholar]
- 37.Haynes CM, Petrova K, Benedetti C, Yang Y, Ron D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev Cell. 2007;13(4):467–480. doi: 10.1016/j.devcel.2007.07.016. [DOI] [PubMed] [Google Scholar]
- 38.Haynes CM, Yang Y, Blais SP, Neubert TA, Ron D. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mole Cell. 2010;37(4):529–540. doi: 10.1016/j.molcel.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science. 2012;337(6094):587–590. doi: 10.1126/science.1223560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nargund AM, Fiorese CJ, Pellegrino MW, Deng P, Haynes CM. Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPR(mt) Mol Cell. 2015;58(1):123–133. doi: 10.1016/j.molcel.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pellegrino MW, Nargund AM, Kirienko NV, Gillis R, Fiorese CJ, Haynes CM. Mitochondrial UPR-regulated innate immunity provides resistance to pathogen infection. Nature. 2014;516(7531):414–417. doi: 10.1038/nature13818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baker BM, Nargund AM, Sun T, Haynes CM. Protective coupling of mitochondrial function and protein synthesis via the eIF2alpha kinase GCN-2. PLoS Genet. 2012;8(6):e1002760. doi: 10.1371/journal.pgen.1002760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Monaghan RM, Barnes RG, Fisher K, Andreou T, Rooney N, Poulin GB, Whitmarsh AJ. A nuclear role for the respiratory enzyme CLK-1 in regulating mitochondrial stress responses and longevity. Nat Cell Biol. 2015;17(6):782–792. doi: 10.1038/ncb3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Herholz M, Cepeda E, Baumann L, Kukat A, Hermeling J, Maciej S, Szczepanowska K, Pavlenko V, Frommolt P, Trifunovic A. KLF-1 orchestrates a xenobiotic detoxification program essential for longevity of mitochondrial mutants. Nat Commun. 2019;10(1):3323. doi: 10.1038/s41467-019-11275-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Owusu-Ansah E, Yavari A, Mandal S, Banerjee U. Distinct mitochondrial retrograde signals control the G1-S cell cycle checkpoint. Nat Genet. 2008;40(3):356–361. doi: 10.1038/ng.2007.50. [DOI] [PubMed] [Google Scholar]
- 46.Owusu-Ansah E, Song W, Perrimon N. Muscle mitohormesis promotes longevity via systemic repression of insulin signaling. Cell. 2013;155(3):699–712. doi: 10.1016/j.cell.2013.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8(7):519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 48.Fiorese CJ, Schulz AM, Lin YF, Rosin N, Pellegrino MW, Haynes CM. The transcription factor ATF5 mediates a mammalian mitochondrial UPR. Curr Biol. 2016;26(15):2037–2043. doi: 10.1016/j.cub.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mouchiroud L, Houtkooper RH, Moullan N, Katsyuba E, Ryu D, Cantó C, Mottis A, Jo Y-S, Viswanathan M, Schoonjans K. The NAD+/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell. 2013;154(2):430–441. doi: 10.1016/j.cell.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. The integrated stress response. EMBO Rep. 2016;17(10):1374–1395. doi: 10.15252/embr.201642195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rath E, Berger E, Messlik A, Nunes T, Liu B, Kim SC, Hoogenraad N, Sans M, Sartor RB, Haller D. Induction of dsRNA-activated protein kinase links mitochondrial unfolded protein response to the pathogenesis of intestinal inflammation. Gut. 2012;61(9):1269–1278. doi: 10.1136/gutjnl-2011-300767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Taylor SS, Haste NM, Ghosh G. PKR and eIF2alpha: integration of kinase dimerization, activation, and substrate docking. Cell. 2005;122(6):823–825. doi: 10.1016/j.cell.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 53.Dever TE, Feng L, Wek RC, Cigan AM, Donahue TF, Hinnebusch AG. Phosphorylation of initiation factor 2α by protein kinase GCN2 mediates gene-specific translational control of GCN4 in yeast. Cell. 1992;68(3):585–596. doi: 10.1016/0092-8674(92)90193-g. [DOI] [PubMed] [Google Scholar]
- 54.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397(6716):271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- 55.Zhan K, Vattem KM, Bauer BN, Dever TE, Chen J-J, Wek RC. Phosphorylation of eukaryotic initiation factor 2 by heme-regulated inhibitor kinase-related protein kinases in Schizosaccharomyces pombe is important for resistance to environmental stresses. Mol Cell Biol. 2002;22(20):7134–7146. doi: 10.1128/MCB.22.20.7134-7146.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Costa-Mattioli M, Walter P. The integrated stress response: From mechanism to disease. Science. 2020;368(6489):10–23. doi: 10.1126/science.aat5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6(5):1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- 58.Fessler E, Eckl EM, Schmitt S, Mancilla IA, Meyer-Bender MF, Hanf M, Philippou-Massier J, Krebs S, Zischka H, Jae LT. A pathway coordinated by DELE1 relays mitochondrial stress to the cytosol. Nature. 2020;579(7799):433–437. doi: 10.1038/s41586-020-2076-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Guo X, Aviles G, Liu Y, Tian R, Unger BA, Lin YT, Wiita AP, Xu K, Correia MA, Kampmann M. Mitochondrial stress is relayed to the cytosol by an OMA1-DELE1-HRI pathway. Nature. 2020;579(7799):427–432. doi: 10.1038/s41586-020-2078-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhou D, Palam LR, Jiang L, Narasimhan J, Staschke KA, Wek RC. Phosphorylation of eIF2 directs ATF5 translational control in response to diverse stress conditions. J Biol Chem. 2008;283(11):7064–7073. doi: 10.1074/jbc.M708530200. [DOI] [PubMed] [Google Scholar]
- 61.Khan NA, Nikkanen J, Yatsuga S, Jackson C, Wang L, Pradhan S, Kivela R, Pessia A, Velagapudi V, Suomalainen A. mTORC1 regulates mitochondrial integrated stress response and mitochondrial myopathy progression. Cell Metab. 2017;26(2):419–428. e415. doi: 10.1016/j.cmet.2017.07.007. [DOI] [PubMed] [Google Scholar]
- 62.Cardamone MD, Tanasa B, Cederquist CT, Huang J, Mahdaviani K, Li W, Rosenfeld MG, Liesa M, Perissi V. Mitochondrial retrograde signaling in mammals is mediated by the transcriptional cofactor GPS2 via direct mitochondria-to-nucleus translocation. Mol Cell. 2018;69(5):757–772.e757. doi: 10.1016/j.molcel.2018.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Seiferling D, Szczepanowska K, Becker C, Senft K, Hermans S, Maiti P, König T, Kukat A, Trifunovic A. Loss of CLPP alleviates mitochondrial cardiomyopathy without affecting the mammalian UPR mt. EMBO Rep. 2016;17(7):953–964. doi: 10.15252/embr.201642077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dogan SA, Pujol C, Maiti P, Kukat A, Wang S, Hermans S, Senft K, Wibom R, Rugarli EI, Trifunovic A. Tissue-specific loss of DARS2 activates stress responses independently of respiratory chain deficiency in the heart. Cell Metab. 2014;19(3):458–469. doi: 10.1016/j.cmet.2014.02.004. [DOI] [PubMed] [Google Scholar]
- 65.Gomes AP, Price NL, Ling AJ, Moslehi JJ, Montgomery MK, Rajman L, White JP, Teodoro JS, Wrann CD, Hubbard BP, Mercken EM, Palmeira CM, de Cabo R, Rolo AP, Turner N, Bell EL, Sinclair DA. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013;155(7):1624–1638. doi: 10.1016/j.cell.2013.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Haas RH. Mitochondrial dysfunction in aging and diseases of aging. Basel: Multidisciplinary Digital Publishing Institute; 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Durieux J, Wolff S, Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 2011;144(1):79–91. doi: 10.1016/j.cell.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang Y, Hekimi S. Mitochondrial dysfunction and longevity in animals: untangling the knot. Science. 2015;350(6265):1204–1207. doi: 10.1126/science.aac4357. [DOI] [PubMed] [Google Scholar]
- 69.Pirinen E, Canto C, Jo YS, Morato L, Zhang H, Menzies KJ, Williams EG, Mouchiroud L, Moullan N, Hagberg C, Li W, Timmers S, Imhof R, Verbeek J, Pujol A, van Loon B, Viscomi C, Zeviani M, Schrauwen P, Sauve AA, Schoonjans K, Auwerx J. Pharmacological Inhibition of poly(ADP-ribose) polymerases improves fitness and mitochondrial function in skeletal muscle. Cell Metab. 2014;19(6):1034–1041. doi: 10.1016/j.cmet.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tian Y, Garcia G, Bian Q, Steffen KK, Joe L, Wolff S, Meyer BJ, Dillin A. Mitochondrial stress induces chromatin reorganization to promote longevity and UPR(mt) Cell. 2016;165(5):1197–1208. doi: 10.1016/j.cell.2016.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee SJ, Hwang AB, Kenyon C. Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr Biol. 2010;20(23):2131–2136. doi: 10.1016/j.cub.2010.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Trifunovic A, Hansson A, Wredenberg A, Rovio AT, Dufour E, Khvorostov I, Spelbrink JN, Wibom R, Jacobs HT, Larsson NG. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc Natl Acad Sci USA. 2005;102(50):17993–17998. doi: 10.1073/pnas.0508886102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang SF, Chen MS, Chou YC, Ueng YF, Yin PH, Yeh TS, Lee HC. Mitochondrial dysfunction enhances cisplatin resistance in human gastric cancer cells via the ROS-activated GCN2-eIF2alpha-ATF4-xCT pathway. Oncotarget. 2016;7(45):74132–74151. doi: 10.18632/oncotarget.12356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nagasawa I, Kunimasa K, Tsukahara S, Tomida A. BRAF-mutated cells activate GCN2-mediated integrated stress response as a cytoprotective mechanism in response to vemurafenib. Biochem Biophys Res Commun. 2017;482(4):1491–1497. doi: 10.1016/j.bbrc.2016.12.062. [DOI] [PubMed] [Google Scholar]
- 75.Ishihara S, Yasuda M, Ishizu A, Ishikawa M, Shirato H, Haga H. Activating transcription factor 5 enhances radioresistance and malignancy in cancer cells. Oncotarget. 2015;6(7):4602–4614. doi: 10.18632/oncotarget.2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sheng Z, Li L, Zhu LJ, Smith TW, Demers A, Ross AH, Moser RP, Green MR. A genome-wide RNA interference screen reveals an essential CREB3L2-ATF5-MCL1 survival pathway in malignant glioma with therapeutic implications. Nat Med. 2010;16(6):671–677. doi: 10.1038/nm.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sorge S, Theelke J, Yildirim K, Hertenstein H, McMullen E, Muller S, Altburger C, Schirmeier S, Lohmann I. ATF4-induced warburg metabolism drives over-proliferation in drosophila. Cell Rep. 2020;31(7):107659. doi: 10.1016/j.celrep.2020.107659. [DOI] [PubMed] [Google Scholar]
- 78.Cartoni R, Arnaud E, Médard J-J, Poirot O, Courvoisier DS, Chrast R, Martinou J-C. Expression of mitofusin 2R94Q in a transgenic mouse leads to Charcot–Marie–Tooth neuropathy type 2A. Brain. 2010;133(5):1460–1469. doi: 10.1093/brain/awq082. [DOI] [PubMed] [Google Scholar]
- 79.van der Bliek AM. Fussy mitochondria fuse in response to stress. EMBO J. 2009;28(11):1533–1534. doi: 10.1038/emboj.2009.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Toyama EQ, Herzig S, Courchet J, Lewis TL, Losón OC, Hellberg K, Young NP, Chen H, Polleux F, Chan DC. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science. 2016;351(6270):275–281. doi: 10.1126/science.aab4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci USA. 2011;108(25):10190–10195. doi: 10.1073/pnas.1107402108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lee J-Y, Kapur M, Li M, Choi M-C, Choi S, Kim H-J, Kim I, Lee E, Taylor JP, Yao T-P. MFN1 deacetylation activates adaptive mitochondrial fusion and protects metabolically challenged mitochondria. J Cell Sci. 2014;127(22):4954–4963. doi: 10.1242/jcs.157321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. 2011;13(5):589–598. doi: 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chen Y, Csordás G, Jowdy C, Schneider TG, Csordás N, Wang W, Liu Y, Kohlhaas M, Meiser M, Bergem S. Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca2+ crosstalk. Circ Res. 2012;111(7):863–875. doi: 10.1161/CIRCRESAHA.112.266585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, Chan DC. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell. 2010;141(2):280–289. doi: 10.1016/j.cell.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M, Cipolat S, Costa V, Casarin A, Gomes LC. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell. 2013;155(1):160–171. doi: 10.1016/j.cell.2013.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27(2):433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Olichon A, Baricault L, Gas N, Guillou E, Valette A, Belenguer P, Lenaers G. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem. 2003;278(10):7743–7746. doi: 10.1074/jbc.C200677200. [DOI] [PubMed] [Google Scholar]
- 89.Weaver D, Eisner V, Liu X, Várnai P, Hunyady L, Gross A, Hajnóczky G. Distribution and apoptotic function of outer membrane proteins depend on mitochondrial fusion. Mol Cell. 2014;54(5):870–878. doi: 10.1016/j.molcel.2014.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kageyama Y, Zhang Z, Roda R, Fukaya M, Wakabayashi J, Wakabayashi N, Kensler TW, Reddy PH, Iijima M, Sesaki H. Mitochondrial division ensures the survival of postmitotic neurons by suppressing oxidative damage. J Cell Biol. 2012;197(4):535–551. doi: 10.1083/jcb.201110034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tang S, Le PK, Tse S, Wallace DC, Huang T. Heterozygous mutation of Opa1 in drosophila shortens lifespan mediated through increased reactive oxygen species production. PLoS ONE. 2009;4(2):e4492. doi: 10.1371/journal.pone.0004492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Santel A, Frank S, Gaume B, Herrler M, Youle RJ, Fuller MT. Mitofusin-1 protein is a generally expressed mediator of mitochondrial fusion in mammalian cells. J Cell Sci. 2003;116(Pt 13):2763–2774. doi: 10.1242/jcs.00479. [DOI] [PubMed] [Google Scholar]
- 93.Santel A, Fuller MT. Control of mitochondrial morphology by a human mitofusin. J Cell Sci. 2001;114(Pt 5):867–874. doi: 10.1242/jcs.114.5.867. [DOI] [PubMed] [Google Scholar]
- 94.Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell biol. 2003;160(2):189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chen H, McCaffery JM, Chan DC. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell. 2007;130(3):548–562. doi: 10.1016/j.cell.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 96.Pham AH, Meng S, Chu QN, Chan DC. Loss of Mfn2 results in progressive, retrograde degeneration of dopaminergic neurons in the nigrostriatal circuit. Hum Mol Genet. 2012;21(22):4817–4826. doi: 10.1093/hmg/dds311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Griparic L, van der Wel NN, Orozco IJ, Peters PJ, van der Bliek AM. Loss of the intermembrane space protein Mgm1/OPA1 induces swelling and localized constrictions along the lengths of mitochondria. J Biol Chem. 2004;279(18):18792–18798. doi: 10.1074/jbc.M400920200. [DOI] [PubMed] [Google Scholar]
- 98.Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci USA. 2004;101(45):15927–15932. doi: 10.1073/pnas.0407043101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Song Z, Chen H, Fiket M, Alexander C, Chan DC. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J Cell Biol. 2007;178(5):749–755. doi: 10.1083/jcb.200704110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, Bartoli D, Polishuck RS, Danial NN, De Strooper B, Scorrano L. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126(1):177–189. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 101.Yamaguchi R, Lartigue L, Perkins G, Scott RT, Dixit A, Kushnareva Y, Kuwana T, Ellisman MH, Newmeyer DD. Opa1-mediated cristae opening is Bax/Bak and BH3 dependent, required for apoptosis, and independent of Bak oligomerization. Mol Cell. 2008;31(4):557–569. doi: 10.1016/j.molcel.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ehses S, Raschke I, Mancuso G, Bernacchia A, Geimer S, Tondera D, Martinou J-C, Westermann B, Rugarli EI, Langer T. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J Cell Biol. 2009;187(7):1023–1036. doi: 10.1083/jcb.200906084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Head B, Griparic L, Amiri M, Gandre-Babbe S, van der Bliek AM. Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J Cell Biol. 2009;187(7):959–966. doi: 10.1083/jcb.200906083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Anand R, Wai T, Baker MJ, Kladt N, Schauss AC, Rugarli E, Langer T. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J Cell Biol. 2014;204(6):919–929. doi: 10.1083/jcb.201308006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Griparic L, Kanazawa T, van der Bliek AM. Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J Cell Biol. 2007;178(5):757–764. doi: 10.1083/jcb.200704112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mishra P, Carelli V, Manfredi G, Chan DC. Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. 2014;19(4):630–641. doi: 10.1016/j.cmet.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ishihara N, Fujita Y, Oka T, Mihara K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 2006;25(13):2966–2977. doi: 10.1038/sj.emboj.7601184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Marelli C, Amati-Bonneau P, Reynier P, Layet V, Layet A, Stevanin G, Brissaud E, Bonneau D, Durr A, Brice A. Heterozygous OPA1 mutations in Behr syndrome. Brain. 2011;134(4):e169. doi: 10.1093/brain/awq306. [DOI] [PubMed] [Google Scholar]
- 109.Smirnova E, Griparic L, Shurland D-L, Van Der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell. 2001;12(8):2245–2256. doi: 10.1091/mbc.12.8.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, Mihara K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol. 2010;191(6):1141–1158. doi: 10.1083/jcb.201007152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yu T, Fox RJ, Burwell LS, Yoon Y. Regulation of mitochondrial fission and apoptosis by the mitochondrial outer membrane protein hFis1. J Cell Sci. 2005;118(18):4141–4151. doi: 10.1242/jcs.02537. [DOI] [PubMed] [Google Scholar]
- 112.Losón OC, Song Z, Chen H, Chan DC. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell. 2013;24(5):659–667. doi: 10.1091/mbc.E12-10-0721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yu R, Jin SB, Lendahl U, Nistér M, Zhao J. Human Fis1 regulates mitochondrial dynamics through inhibition of the fusion machinery. EMBO J. 2019;38(8):12–23. doi: 10.15252/embj.201899748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ferguson SM, De Camilli P. Dynamin, a membrane-remodelling GTPase. Nat Rev Mol Cell Biol. 2012;13(2):75–88. doi: 10.1038/nrm3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem. 2007;282(15):11521–11529. doi: 10.1074/jbc.M607279200. [DOI] [PubMed] [Google Scholar]
- 116.Chang C-R, Blackstone C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J Biol Chem. 2007;282(30):21583–21587. doi: 10.1074/jbc.C700083200. [DOI] [PubMed] [Google Scholar]
- 117.Cribbs JT, Strack S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007;8(10):939–944. doi: 10.1038/sj.embor.7401062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Serasinghe MN, Wieder SY, Renault TT, Elkholi R, Asciolla JJ, Yao JL, Jabado O, Hoehn K, Kageyama Y, Sesaki H, Chipuk JE. Mitochondrial division is requisite to RAS-induced transformation and targeted by oncogenic MAPK pathway inhibitors. Mol Cell. 2015;57(3):521–536. doi: 10.1016/j.molcel.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kashatus DF, Lim K-H, Brady DC, Pershing NL, Cox AD, Counter CM. RALA and RALBP1 regulate mitochondrial fission at mitosis. Nat Cell Biol. 2011;13(9):1108. doi: 10.1038/ncb2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, Otera H, Nakanishi Y, Nonaka I, Goto Y-I, Taguchi N, Morinaga H, Maeda M, Takayanagi R, Yokota S, Mihara K. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol. 2009;11(8):958–966. doi: 10.1038/ncb1907. [DOI] [PubMed] [Google Scholar]
- 121.Wakabayashi J, Zhang Z, Wakabayashi N, Tamura Y, Fukaya M, Kensler TW, Iijima M, Sesaki H. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J Cell Biol. 2009;186(6):805–816. doi: 10.1083/jcb.200903065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rehman J, Zhang HJ, Toth PT, Zhang Y, Marsboom G, Hong Z, Salgia R, Husain AN, Wietholt C, Archer SL. Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. Faseb J. 2012;26(5):2175–2186. doi: 10.1096/fj.11-196543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tanwar DK, Parker DJ, Gupta P, Spurlock B, Alvarez RD, Basu MK, Mitra K. Crosstalk between the mitochondrial fission protein, Drp1, and the cell cycle is identified across various cancer types and can impact survival of epithelial ovarian cancer patients. Oncotarget. 2016;7(37):60021–60037. doi: 10.18632/oncotarget.11047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Manczak M, Calkins MJ, Reddy PH. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: implications for neuronal damage. Hum Mol Genet. 2011;20(13):2495–2509. doi: 10.1093/hmg/ddr139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Shirendeb U, Reddy AP, Manczak M, Calkins MJ, Mao P, Tagle DA, Reddy PH. Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington’s disease: implications for selective neuronal damage. Hum Mol Genet. 2011;20(7):1438–1455. doi: 10.1093/hmg/ddr024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Corrado M, Scorrano L. Campello S (2012) Mitochondrial dynamics in cancer and neurodegenerative and neuroinflammatory diseases. Internat J Cell Biol. 2012;10(123):140–156. doi: 10.1155/2012/729290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cuervo AM. Autophagy: in sickness and in health. Trends Cell Biol. 2004;14(2):70–77. doi: 10.1016/j.tcb.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 128.Boya P, Reggiori F, Codogno P. Emerging regulation and functions of autophagy. Nat Cell Biol. 2013;15(7):713–720. doi: 10.1038/ncb2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Gong G, Song M, Csordas G, Kelly DP, Matkovich SJ, Dorn GW., 2nd Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science (New York, NY) 2015;350(6265):2459. doi: 10.1126/science.aad2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Takeshige K, Baba M, Tsuboi S, Noda T, Ohsumi Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J Cell Biol. 1992;119(2):301–311. doi: 10.1083/jcb.119.2.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Rodriguez-Enriquez S, Kim I, Currin RT, Lemasters JJ. Tracker dyes to probe mitochondrial autophagy (mitophagy) in rat hepatocytes. Autophagy. 2006;2(1):39–46. doi: 10.4161/auto.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhang J, Randall MS, Loyd MR, Dorsey FC, Kundu M, Cleveland JL, Ney PA. Mitochondrial clearance is regulated by Atg7-dependent and-independent mechanisms during reticulocyte maturation. Blood. 2009;114(1):157–164. doi: 10.1182/blood-2008-04-151639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kundu M, Lindsten T, Yang C-Y, Wu J, Zhao F, Zhang J, Selak MA, Ney PA, Thompson CB. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood. 2008;112(4):1493–1502. doi: 10.1182/blood-2008-02-137398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14(2):177. doi: 10.1038/ncb2422. [DOI] [PubMed] [Google Scholar]
- 135.McWilliams TG, Prescott AR, Montava-Garriga L, Ball G, Singh F, Barini E, Muqit MMK, Brooks SP, Ganley IG. Basal mitophagy occurs independently of PINK1 in mouse tissues of high metabolic demand. Cell Metab. 2018;27(2):439–449.e435. doi: 10.1016/j.cmet.2017.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Glick D, Zhang W, Beaton M, Marsboom G, Gruber M, Simon MC, Hart J, Dorn GW, 2nd, Brady MJ, Macleod KF. BNip3 regulates mitochondrial function and lipid metabolism in the liver. Mol Cell Biol. 2012;32(13):2570–2584. doi: 10.1128/MCB.00167-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, Kundu M, Opferman JT, Cleveland JL, Miller JL, Ney PA. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci USA. 2007;104(49):19500–19505. doi: 10.1073/pnas.0708818104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 2008;454(7201):232–235. doi: 10.1038/nature07006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, Wacker M, Klose J, Shen J. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem. 2004;279(18):18614–18622. doi: 10.1074/jbc.M401135200. [DOI] [PubMed] [Google Scholar]
- 140.Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim J-M, Chung J. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441(7097):1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- 141.Shin J-H, Ko HS, Kang H, Lee Y, Lee Y-I, Pletinkova O, Troconso JC, Dawson VL, Dawson TM. PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson’s disease. Cell. 2011;144(5):689–702. doi: 10.1016/j.cell.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yamano K, Youle RJ. PINK1 is degraded through the N-end rule pathway. Autophagy. 2013;9(11):1758–1769. doi: 10.4161/auto.24633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Narendra D, Tanaka A, Suen D-F, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183(5):795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Jin SM, Youle RJ. The accumulation of misfolded proteins in the mitochondrial matrix is sensed by PINK1 to induce PARK2/Parkin-mediated mitophagy of polarized mitochondria. Autophagy. 2013;9(11):1750–1757. doi: 10.4161/auto.26122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Campanella M, Seraphim A, Abeti R, Casswell E, Echave P, Duchen MR. IF1, the endogenous regulator of the F1Fo-ATPsynthase, defines mitochondrial volume fraction in HeLa cells by regulating autophagy. Biochimica et Biophysica Acta (BBA)-Bioenergetics. 2009;1787(5):393–401. doi: 10.1016/j.bbabio.2009.02.023. [DOI] [PubMed] [Google Scholar]
- 146.Kim I, Lemasters JJ. Mitophagy selectively degrades individual damaged mitochondria after photoirradiation. Antioxid Redox Signal. 2011;14(10):1919–1928. doi: 10.1089/ars.2010.3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Suen D-F, Narendra DP, Tanaka A, Manfredi G, Youle RJ. Parkin overexpression selects against a deleterious mtDNA mutation in heteroplasmic cybrid cells. Proc Natl Acad Sci USA. 2010;107(26):11835–11840. doi: 10.1073/pnas.0914569107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015;85(2):257–273. doi: 10.1016/j.neuron.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Lazarou M, Jin SM, Kane LA, Youle RJ. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell. 2012;22(2):320–333. doi: 10.1016/j.devcel.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Sekine S, Wang C, Sideris DP, Bunker E, Zhang Z, Youle RJ. Reciprocal roles of Tom7 and OMA1 during mitochondrial import and activation of PINK1. Mol Cell. 2019;73(5):1028–1043.e1025. doi: 10.1016/j.molcel.2019.01.002. [DOI] [PubMed] [Google Scholar]
- 151.Narendra DP, Jin SM, Tanaka A, Suen D-F, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8(1):e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol. 2014;205(2):143–153. doi: 10.1083/jcb.201402104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kondapalli C, Kazlauskaite A, Zhang N, Woodroof HI, Campbell DG, Gourlay R, Burchell L, Walden H, Macartney TJ, Deak M. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012;2(5):120080. doi: 10.1098/rsob.120080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Shiba-Fukushima K, Imai Y, Yoshida S, Ishihama Y, Kanao T, Sato S, Hattori N. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Scient Rep. 2012;2:1002. doi: 10.1038/srep01002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kazlauskaite A, Kondapalli C, Gourlay R, Campbell DG, Ritorto MS, Hofmann K, Alessi DR, Knebel A, Trost M, Muqit MMK. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem J. 2014;460(1):127–139. doi: 10.1042/BJ20140334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, Endo T, Fon EA, Trempe J-F, Saeki Y, Tanaka K, Matsuda N. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014;510(7503):162–166. doi: 10.1038/nature13392. [DOI] [PubMed] [Google Scholar]
- 157.Schubert AF, Gladkova C, Pardon E, Wagstaff JL, Freund SM, Steyaert J, Maslen SL, Komander D. Structure of PINK1 in complex with its substrate ubiquitin. Nature. 2017;552(7683):51. doi: 10.1038/nature24645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Rose CM, Isasa M, Ordureau A, Prado MA, Beausoleil SA, Jedrychowski MP, Finley DJ, Harper JW, Gygi SP. Highly multiplexed quantitative mass spectrometry analysis of ubiquitylomes. Cell Syst. 2016;3(4):395–403.e394. doi: 10.1016/j.cels.2016.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12(2):119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- 160.Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524(7565):309–314. doi: 10.1038/nature14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Pickrell AM, Huang C-H, Kennedy SR, Ordureau A, Sideris DP, Hoekstra JG, Harper JW, Youle RJ. Endogenous parkin preserves dopaminergic substantia nigral neurons following mitochondrial DNA mutagenic stress. Neuron. 2015;87(2):371–381. doi: 10.1016/j.neuron.2015.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kubli DA, Zhang X, Lee Y, Hanna RA, Quinsay MN, Nguyen CK, Jimenez R, Petrosyan S, Murphy AN, Gustafsson AB. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J Biol Chem. 2013;288(2):915–926. doi: 10.1074/jbc.M112.411363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Song M, Gong G, Burelle Y, Gustafsson AB, Kitsis RN, Matkovich SJ, Dorn GW., 2nd Interdependence of Parkin-mediated mitophagy and mitochondrial fission in adult mouse hearts. Circ Res. 2015;117(4):346–351. doi: 10.1161/CIRCRESAHA.117.306859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Lee JJ, Sanchez-Martinez A, Zarate AM, Benincá C, Mayor U, Clague MJ, Whitworth AJ. Basal mitophagy is widespread in Drosophila but minimally affected by loss of Pink1 or parkin. J Cell Biol. 2018;217(5):1613–1622. doi: 10.1083/jcb.201801044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Lee SB, Kim JJ, Han S-A, Fan Y, Guo L-S, Aziz K, Nowsheen S, Kim SS, Park S-Y, Luo Q, Chung JO, Choi SI, Aziz A, Yin P, Tong S-Y, Fiesel FC, Springer W, Zhang J-S, Lou Z. The AMPK-Parkin axis negatively regulates necroptosis and tumorigenesis by inhibiting the necrosome. Nat Cell Biol. 2019;21(8):940–951. doi: 10.1038/s41556-019-0356-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Jacoupy M, Hamon-Keromen E, Ordureau A, Erpapazoglou Z, Coge F, Corvol JC, Nosjean O, Mannoury la Cour C, Millan MJ, Boutin JA, Harper JW, Brice A, Guedin D, Gautier CA, Corti O. The PINK1 kinase-driven ubiquitin ligase Parkin promotes mitochondrial protein import through the presequence pathway in living cells. Scient Rep. 2019;9(1):11829. doi: 10.1038/s41598-019-47352-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kuroda Y, Mitsui T, Kunishige M, Shono M, Akaike M, Azuma H, Matsumoto T. Parkin enhances mitochondrial biogenesis in proliferating cells. Hum Mol Genet. 2006;15(6):883–895. doi: 10.1093/hmg/ddl006. [DOI] [PubMed] [Google Scholar]
- 168.Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, Burman JL, Li Y, Zhang Z, Narendra DP, Cai H, Borsche M, Klein C, Youle RJ. Parkin and PINK1 mitigate STING-induced inflammation. Nature. 2018;561(7722):258–262. doi: 10.1038/s41586-018-0448-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou Y-S, Saiki S, Kawajiri S, Sato F, Kimura M, Komatsu M, Hattori N, Tanaka K. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. 2010;189(2):211–221. doi: 10.1083/jcb.200910140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet. 2006;38(5):515. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- 171.Kraytsberg Y, Kudryavtseva E, McKee AC, Geula C, Kowall NW, Khrapko K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet. 2006;38(5):518. doi: 10.1038/ng1778. [DOI] [PubMed] [Google Scholar]
- 172.Walter J, Bolognin S, Antony PM, Nickels SL, Poovathingal SK, Salamanca L, Magni S, Perfeito R, Hoel F, Qing X. Neural stem cells of Parkinson’s disease patients exhibit aberrant mitochondrial morphology and functionality. Stem Cell Rep. 2019;12(5):878–889. doi: 10.1016/j.stemcr.2019.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44(4):601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 174.Hsieh C-H, Shaltouki A, Gonzalez AE, Bettencourt da Cruz A, Burbulla LF, St Lawrence E, Schule B, Krainc D, Palmer TD, Wang X. Functional impairment in miro degradation and mitophagy is a shared feature in familial and sporadic Parkinson’s disease. Cell Stem Cell. 2016;19(6):709–724. doi: 10.1016/j.stem.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Kovalchuke L, Mosharov EV, Levy OA, Greene LA. Stress-induced phospho-ubiquitin formation causes parkin degradation. Scientific reports. 2019;9(1):11682. doi: 10.1038/s41598-019-47952-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Schapira AHV. Mitochondrial pathology in Parkinson’s disease. Mt Sinai J Med. 2011;78(6):872–881. doi: 10.1002/msj.20303. [DOI] [PubMed] [Google Scholar]
- 177.Batlevi Y, La Spada AR. Mitochondrial autophagy in neural function, neurodegenerative disease, neuron cell death, and aging. Neurobiol Dis. 2011;43(1):46–51. doi: 10.1016/j.nbd.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Hayat M. Autophagy: cancer, other pathologies, inflammation, immunity, infection, and aging. London: Academic Press; 2017. [Google Scholar]
- 179.Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, Williams RW, Auwerx J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. 2013;497(7450):451–457. doi: 10.1038/nature12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Ishihara N, Jofuku A, Eura Y, Mihara K. Regulation of mitochondrial morphology by membrane potential, and DRP1-dependent division and FZO1-dependent fusion reaction in mammalian cells. Biochem Biophys Res Commun. 2003;301(4):891–898. doi: 10.1016/s0006-291x(03)00050-0. [DOI] [PubMed] [Google Scholar]
- 181.Burman JL, Pickles S, Wang C, Sekine S, Vargas JNS, Zhang Z, Youle AM, Nezich CL, Wu X, Hammer JA, Youle RJ. Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J Cell Biol. 2017;216(10):3231–3247. doi: 10.1083/jcb.201612106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Han H, Tan J, Wang R, Wan H, He Y, Yan X, Guo J, Gao Q, Li J, Shang S, Chen F, Tian R, Liu W, Liao L, Tang B, Zhang Z. PINK1 phosphorylates Drp 1(S616) to regulate mitophagy-independent mitochondrial dynamics. EMBO Rep. 2020;10:e48686. doi: 10.15252/embr.201948686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Guerreiro R, Bras J, Hardy J. SnapShot: genetics of ALS and FTD. Cell. 2015;160(4):798–798.e791. doi: 10.1016/j.cell.2015.01.052. [DOI] [PubMed] [Google Scholar]