

Abstract
Activation of ERK1/2 signaling in cardiovascular regulatory regions of the brain contributes to sympathetic excitation in myocardial infarction (MI)-induced heart failure (HF) by increasing brain renin-angiotensin system (RAS) activity, neuroinflammation, and endoplasmic reticulum (ER) stress. The mechanisms eliciting brain ERK1/2 signaling in HF are still poorly understood. We tested the involvement of the epidermal growth factor receptor (EGFR) which, upon activation, stimulates ERK1/2 activity. Adult male Sprague-Dawley rats received bilateral microinjections of a lentiviral vector encoding a small interfering RNA (siRNA) for EGFR, or a scrambled siRNA, into the hypothalamic paraventricular nucleus (PVN), a recognized source of sympathetic overactivity in HF. One week later, coronary artery ligation was performed to induce HF. Four weeks later, the EGFR siRNA-treated HF rats, compared with the scrambled siRNA-treated HF rats, had lower mRNA and protein levels of EGFR, lower levels of phosphorylated (p-) EGFR and p-ERK1/2 and lower mRNA levels of the inflammatory mediators TNF-α, IL-1β and cyclooxygenase-2, the RAS components angiotensin-converting enzyme and angiotensin II type 1a receptor and the ER stress markers BIP and ATF4 in the PVN. They also had lower plasma and urinary norepinephrine levels and improved peripheral manifestations of HF. Additional studies revealed that p-EGFR was increased in the PVN of HF rats, compared with sham-operated control rats. These results suggest that activation of EGFR in the PVN triggers ERK1/2 signaling, along with ER stress, neuroinflammation and RAS activity, in MI-induced HF. Brain EGFR may be a novel target for therapeutic intervention in MI-induced HF.
Keywords: brain, extracellular signal-regulated protein kinases 1 and 2, p44/42 mitogen-activated protein kinase, neuroinflammation, renin-angiotensin system, endoplasmic reticulum stress
INTRODUCTION
In rats with myocardial infarction (MI)-induced heart failure (HF), the mitogen-activated protein kinase (MAPK) signaling pathway leading to phosphorylation of extracellular signal-regulated protein kinases 1 and 2 (ERK1/2), also known as p44/42 MAPK, is activated in cardiovascular regions of the brain (Wei et al., 2016b; Wei et al., 2008b; Yu et al., 2016). ERK1/2 signaling contributes to sympathetic excitation by increasing the expression of pro-inflammatory cytokines and renin-angiotensin system (RAS) components and inducing endoplasmic reticulum (ER) stress in these brain regions (Wei et al., 2016b; Yu et al., 2016). Multiple excitatory agonists that increase in the brain in HF, including angiotensin II (Ang II), aldosterone and pro-inflammatory cytokines (PICs), are known to activate brain ERK1/2 signaling (Wei et al., 2009; Wei et al., 2008a; Wei et al., 2014; Zhang et al., 2012), but the precise mechanisms by which they do so are not completely understood.
One well-recognized pathway to ERK1/2 signaling is activation of the epidermal growth factor receptor (EGFR) (Wee and Wang, 2017). While epidermal growth factor and transforming growth factor-alpha are its primary endogenous ligands (Voldborg et al., 1997), EGFR may also be activated - or transactivated - by Ang II and PICs (Jamroz-Wisniewska et al., 2008;Konishi and Berk, 2003;Pastore et al., 2008;Takeyama et al., 2000;Taniguchi et al., 2013;Touyz et al., 2002). Upon activation and dimerization of EGFR, autophosphorylation of tyrosine kinase residues initiates a series of intracellular molecular events that activate the Ras/Raf/MEK signaling pathway leading to the phosphorylation (activation) of ERK1/2 (Hunter, 1998).
EGFR plays a major role in the differentiation of neurons and glial cells in early brain development (Ferrer et al., 1996). In that setting, activated (phosphorylated) EGFR is highly expressed in neuron and astrocytes (Tavassoly et al., 2020). In the adult brain under normal conditions, neurons continue to express EGFR at lower levels, but expression is minimal to absent in astrocytes (Tavassoly et al., 2020). However, in pathological states, including neurodegenerative diseases and ischemic or traumatic brain injury, the expression EGFR and activated (phosphorylated) EGFR is upregulated in neurons and astrocytes (Tavassoly et al., 2020). Oxidative stress has also been identified as a potential contributor to activation of neuronal EGFR (Wakatsuki et al., 2015).
We hypothesized that EGFR might contribute to increased ERK1/2 signaling and its downstream effects to augment sympathetic excitation in HF, a setting in which RAS activity, inflammation, and oxidative stress all increase within cardiovascular regions of the brain. We tested this hypothesis by downregulating EGFR expression in the hypothalamic paraventricular nucleus (PVN), a forebrain center in which RAS activity, inflammation and oxidative stress are known to drive the sympathetic overactivity that plays a major role in the pathophysiology of HF (Ferguson et al., 2008). The results suggest that PVN EGFR activity is a major contributor to sympathetic excitation in HF and a potential novel target for therapeutic intervention.
EXPERIMENTAL PROCEDURES
Animals
Adult male Sprague-Dawley rats (250–300 g) were purchased from Envigo/Harlan (Indianapolis, IN). Animals were housed in a room with controlled temperature (23 ± 2°C) and 12:12 h light-dark cycle, and standard rat chow and water were given ad libitum. Experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All procedures were approved by the Institutional Animal Care and Use Committee of the University of Iowa. All efforts were made to minimize the number of animals used and their suffering.
Experimental protocols
Three rats were transcardially perfused with 4% paraformaldehyde under deep anesthesia for immunofluorescent studies to examine EGFR protein expression in the PVN.
Sixteen rats were studied to determine the effect of HF on activation (phosphorylation, p-) of EGFR in the PVN. These rats underwent coronary artery ligation (CL) to induce HF (n = 10), or an identical sham operative procedure (Sham, n = 6). The severity of HF was determined by echocardiography within 24 hours after CL and by repeat echocardiography and anatomical assessments of cardiac remodeling and pulmonary congestion at the conclusion of the 4-week protocol (see below). Three HF rats that died before the end of the protocol and one HF rat with a small myocardial infarction (MI) on initial echocardiogram (ischemic zone ≤ 30%, as defined below) were excluded from the study.
Fifty-four rats received bilateral PVN microinjections of an EGFR siRNA (0.3 μl of 109 IU/ml, n = 27 rats) or a scrambled siRNA (n = 27 rats) lentiviral vector, each encoding green fluorescent protein (GFP). Among these, 18 rats (9 rats/group) were euthanized one week later to collect brain tissue for immunofluorescent studies to verify the transfection potential of the siRNAs (3 rats/group) or for real-time PCR to determine the knockdown efficiency of the EGFR siRNA (6 rats/group). Age-matched normal rats (n = 9 rats) served as control.
The remaining thirty-six rats underwent CL to induce HF. Echocardiography was performed within 24 hours of CL to evaluate the extent of myocardial injury and left ventricular function. Three rats with a small MI, 1 treated with EGFR siRNA and 2 treated with scrambled siRNA, were excluded from further study. Ten rats did not survive the study protocol: 7 (4 treated with EGFR siRNA, 3 treated with scrambled siRNA) died within 24 hours of CL, and 3 more (1 treated with EGFR siRNA, 2 treated with scrambled siRNA) died between 24 hours and the end of the protocol.
Thus, the final study groups from which data were acquired regarding the effects of EGFR knockdown were: HF + EGFR siRNA (n=12) and HF + scrambled siRNA (n=11). A second echocardiogram was performed on these rats at 4 weeks to evaluate treatment effects. Finally, these animals were anesthetized for invasive measurements of left ventricular hemodynamics and then euthanized to collect brain and urine for molecular studies. The heart and lungs were also harvested and weighed to evaluate effects of treatment on peripheral indicators of HF.
Experimental methods
PVN microinjections of siRNA.
Bilateral PVN microinjections were performed as previously described (Yu et al., 2016). Briefly, animals were anesthetized (ketamine 100 mg/kg + xylazine 10 mg/kg IP) and positioned in a stereotaxic apparatus (Kopf Instruments; Tujunga, CA). A longitudinal skin incision was made to expose the skull, and two small holes were drilled at 1.8 mm posterior to bregma and 0.4 mm from midline. A 29-gauge guide cannula was inserted to a position 7.6 mm ventral to dura. A 35-gauge stainless injection cannula connected via calibrated polyethylene tubing to a 1-μl Hamilton microsyringe was inserted into the guide cannula and extended 0.5 mm beyond the tip of the guide cannula. Bilateral microinjections were made in a volume of 0.3 μl over 30 sec for each side.
The EGFR siRNA and scrambled siRNA were obtained from Applied Biological Materials Inc (Richmond, BC, Canada).
Induction of HF.
HF was induced by CL as described before (Yu et al., 2012). Briefly, rats were anesthetized (ketamine 100 mg/kg + xylazine 10 mg/kg IP), intubated and placed on a ventilator. Under sterile conditions, a left thoracotomy was performed and the left anterior descending coronary artery was tied off between the pulmonary artery outflow tract and the left atrium with a polyester suture. After CL, the lungs were re-inflated and the chest was quickly closed. Buprenorphine (0.03 mg/kg, SC) was administered immediately postoperatively and then every 12 hours for 48 hours to minimize postsurgical pain.
Echocardiography.
Rats were anesthetized with ketamine (60 mg/kg IP) for echocardiographic assessment of ischemic zone as a percent of the left ventricular (LV) circumference (%IZ), LV ejection fraction (LVEF), and LV end-diastolic volume (LVEDV), as described before (Yu et al., 2016; Yu et al., 2012). Echocardiography was performed using a Vevo 2100 Imaging System (Visualsonics, Inc., Toronto).
Hemodynamic assessment.
Rats were anesthetized with urethane (1.5 g/kg, IP). A Millar catheter was inserted via the right carotid artery into the LV as described previously (Yu et al., 2016;Yu et al., 2012), and systolic blood pressure (SBP), diastolic blood pressure (DBP), LV end-diastolic pressure (LVEDP), LV peak systolic pressure (LVPSP), and maximum rate of rise of LV pressure (LV dP/dtmax) were assessed. HR (beats/min) was derived from the frequency of arterial pressure pulses.
Anatomic measurements.
Wet lung weight, LV weight and right ventricular (RV) weight were measured and corrected by body weight (BW) to evaluate two anatomical indicators of the severity of HF: pulmonary congestion and cardiac remodeling.
Immunofluorescent studies.
To assess EGFR protein expression in the PVN, rats were deeply anesthetized with urethane (1.5 g/kg, IP) and transcardially perfused with 4% paraformaldehyde. Brains were removed, post-fixed in 4% paraformaldehyde for 24 h at 4°C and then cryoprotected in 30% sucrose for 48 h at 4°C. Brains were frozen in OCT compound on dry ice and cut into 20 μm coronal sections using a cryostat. EGFR immunoreactivity was detected using a rabbit anti-rat primary antibody to EGFR (1:250, Catalog No: ab52894, Abcam, Cambridge, MA). Neurons were identified using a mouse anti-rat primary antibody to NeuN (1:200, Catalog No: MAB377, Millipore, Billerica, MA). Immunoreactivity for EGFR and NeuN was visualized using secondary antibodies Alexa fluor 488 goat anti-rabbit IgG (1:200, Catalog No: ab150077, Abcam, Cambridge, MA) and Alexa fluor 568 goat anti-mouse IgG (1:200, Catalog No: ab 175702, Abcam, Cambridge, MA), respectively.
To verify the transfection potential of the siRNAs, as indicated by visualization of GFP in the PVN, rats were sacrificed without perfusion and brains were removed quickly. Brains were immediately frozen in liquid nitrogen and then cut into 20 μm coronal sections using a cryostat.
Immunofluorescence for EGFR and NeuN and GFP fluorescence were visualized using a confocal laser-scanning microscope (Zeiss LSM 710, Carl Zeiss, Inc, Oberkochen, Germany).
Real-time PCR.
Gene expression was used to assess the effect of EGFR knockdown on representative components of the major neuroexcitatory systems that are activated in the PVN in HF. The PVN region, including small amounts of surrounding tissues, was punched using a 15-gauge needle stub (inner diameter: 1.5 mm), and total RNA was extracted with RNeasy® Plus Mini Kit (QIAGEN, Germantown, MD). mRNA levels for EGFR, ER stress markers binding immunoglobulin protein (BIP), activating transcription factor (ATF)4 and ATF6, inflammatory mediators tumor necrosis factor (TNF)-α, interleukin (IL)-1β and cyclooxygenase (COX)-2, and RAS components angiotensin-converting enzyme (ACE) and angiotensin II type 1a receptor (AT1aR) in the PVN were analyzed with SYBR Green real-time PCR following reverse transcription of total RNA. The sequences for the primers used are summarized in Table 1. Real-time PCR was performed using the ABI prism 7000 Sequence Detection System (Applied Biosystems, Carlsbad, CA). The values were corrected by β-actin and presented as fold changes relative to control or HF + scrambled siRNA group.
Table 1.
Sequences for primers
| Gene | Primers | Sequences |
|---|---|---|
| BIP | Forward primer: Reverse primer: |
5’-AAGGTGAACGACCCCTAACAAA-3’ 5’-GTCACTCGGAGAATACCATTAACATCT-3’ |
| ATF4 | Forward primer: Reverse primer: |
5’-CTACTAGGTACCGCCAGAAG-3’ 5’-GCCTTACGGACCTCTTCTAT-3’ |
| ATF6 | Forward primer: Reverse primer: |
5’-GATTTGATGCCTTGGGAGTC-3’ 5’-GGACCGAGGAGAAGAGACAG-3’ |
| TNF-α | Forward primer: Reverse primer: |
5’-CCTTATCTACTCCCAGGTTCTC-3’ 5’-TTTCTCCTGGTATGAATGGC-3’ |
| IL-1β | Forward primer: Reverse primer: |
5’-CGACAGAATCTAGTTGTCC-3’ 5’-TCATAAACACTCTCATCCACAC-3’ |
| COX-2 | Forward primer: Reverse primer: |
5’-GGCACAAATATGATGTTCGCA-3’ 5’-CCTCGCTTCTGATCTGTCTTGA-3’ |
| ACE | Forward primer: Reverse primer: |
5’-GTGTTGTGGAACGAATACGC-3’ 5’-CCTTCTTTATGATCCGCTTGA-3’ |
| AT1aR | Forward primer: Reverse primer: |
5’-ACTCACAGCAACCCTCCAAG-3’ 5'-ATCACCACCAAGCTGTTTCC-3' |
| β-actin | Forward primer: Reverse primer: |
5’-CCGCGAGTACAACCTTCT-3’ 5'-CGTCATCCATGGCGAACT-3' |
BIP: binding immunoglobulin protein; ATF4: activating transcription factor 4; ATF6: activating transcription factor 6; TNF-α: tumor necrosis factor-α; IL-1β: interleukin-1β; COX-2: cyclooxygenase-2; ACE: angiotensin converting enzyme; AT1aR: angiotensin II type 1a receptor.
Western blot analysis.
Protein was extracted from PVN punches using N-PER Neuronal Protein Extraction Reagent (Thermo Fisher Scientific, Rockford, IL). Protein levels for total EGFR and phosphorylated (p-) EGFR, total ERK1/2 and p-ERK1/2, and β-actin were determined by Western blot analysis, as previously described (Yu et al., 2016) using primary antibodies to EGFR (1:1000, Catalog No: ab52894, Abcam, Cambridge, MA) and p-EGFR (1:500, Catalog No: 09-310, Millipore, Temecula, CA), ERK1/2 (p44/42) and p-ERK1/2 (p-p44/42), and β-actin (1:1000, Catalog No: 4695 for ERK1/2; 1:500, Catalog No: 4377 for p-ERK1/2; 1:1000, Catalog No: 4970 for β-actin, Cell Signaling Technology, Danvers, MA). The density of the bands was quantified using Image Lab analysis software (Bio-Rad, Hercules, CA).
Biochemical assays.
Immediately after animals were sacrificed, trunk blood was collected for plasma samples and urine samples were collected from bladder using a 27 Ga needle attached to a 1 mL syringe. Plasma and urinary norepinephrine levels were measured with a commercial ELISA kit (NE; Labor Diagnostika Nord, Nordhorn, Germany) according to the manufacturers’ instruction.
Statistical analysis
All data are expressed as mean ± SEM. Statistical analyses were performed with GraphPad Prism 8.0 (GraphPad Software, Inc., San Diego, CA). The significance of differences in mean values was analyzed using unpaired t-test, one-way or two-way ANOVA followed by Tukey’s multiple comparison tests. P<0.05 was considered statistically significant.
RESULTS
EGFR expression in the PVN
Immunohistochemical studies in normal rats confirmed the presence of EGFR immunoreactivity in PVN neurons (Figure 1).
Figure 1.

Representative confocal images of the PVN region of a normal rat, showing immunoreactivity for EGFR (green) and the neuronal marker NeuN (red). Upper panels: low power (x10) magnification, 3rd ventricle to the right. Lower panels: higher power (x40) images taken from the area indicated by the yellow boxes in the upper panels. Merged image shows localization of EGFR in PVN neurons (yellow)
Western blots revealed no differences in levels of total EGFR in the PVN of sham-operated and HF rats, but the levels of p-EGFR in the PVN were significantly increased in the HF rats compared with the sham-operated rats (Figure 2).
Figure 2.
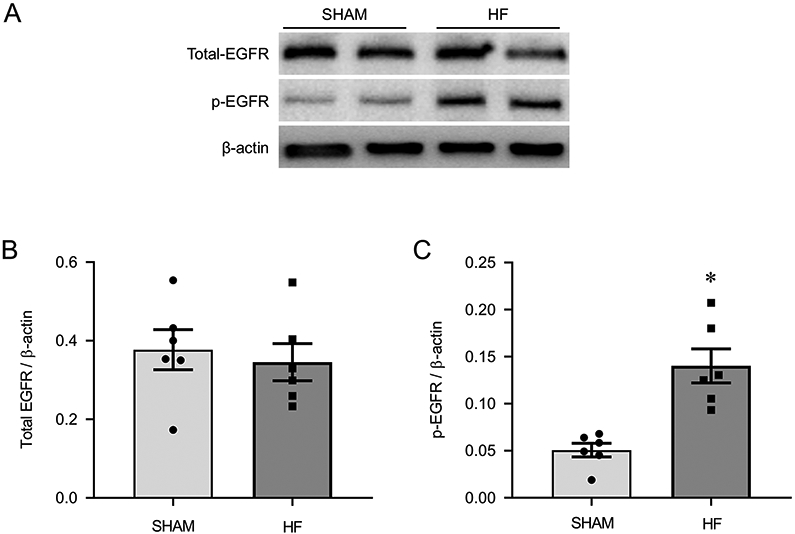
Representative Western blots (A) and protein levels of total EGFR (B) and phosphorylated (p-) EGFR (C) in the PVN of heart failure (HF) rats 4 weeks after coronary artery ligation and age-matched sham-operated (Sham) control rats. Values are mean ± SEM (n = 6 for each group). *p<0.05, vs Sham.
Validation of EGFR knockdown
One week after bilateral PVN microinjections of the lentiviral vector carrying EGFR siRNA or a scrambled siRNA, GFP fluorescence demonstrated effective viral transfection of the PVN (Figure 3A). Real-time PCR revealed significantly lower EGFR mRNA levels in the PVN of animals that had received the EGFR siRNA (Figure 3B), but not in animals that had received the scrambled siRNA, compared with the abundant EGFR mRNA expression in normal control rats (average Ct value: 25 ± 0.2). These observations verified the effectiveness of EGFR gene knockdown in the PVN.
Figure 3.

Validation of PVN transfection and EGFR knockdown. A lentiviral vector encoding a scrambled (Scr) siRNA + green fluorescent protein (GFP) or EGFR siRNA + GFP was microinjected bilaterally in PVN. One week later, GFP was distributed diffusely throughout the PVN (A). A normal control rat is shown for comparison. Rats treated with the EGFR siRNA had significantly lower levels of EGFR mRNA, compared with normal control rats and rats treated with the Scr siRNA (B). Values in (B) are mean ± SEM (n = 6 for each group) and are expressed as a fold change compared to Control. *p<0.05, vs Control or Scr siRNA.
EGFR knockdown in HF rats
The effects of EGFR knockdown on central mediators of sympathetic excitation in HF were studied 5 weeks after PVN microinjection of EGFR siRNA or scrambled siRNA lentiviral particles and 4 weeks after CL, a time point at which HF is well established in this rat model (Francis et al., 2001b). On initial assignment to the EGFR siRNA and scrambled siRNA treatment groups, HF rats were well-matched with regard to the echocardiographic indices of LV function measured within 24 hours of CL (Table 3).
HF rats that received the EGFR siRNA, compared with HF rats that received the scrambled siRNA, had significantly lower levels of EGFR mRNA (Figure 4A) and lower levels of total EGFR (Figure 4B and 4C) and p-EGFR (Figure 4B and 4D) protein in the PVN. These lower levels of EGFR were accompanied by a reduced ratio of phosphorylated to total ERK1/2 protein (Figure 5A and 5B), indicating reduced ERK1/2 activity. There was no statistically significant difference in the level of total ERK1/2 between two experimental groups (Figure 5A and 5C).
Figure 4.
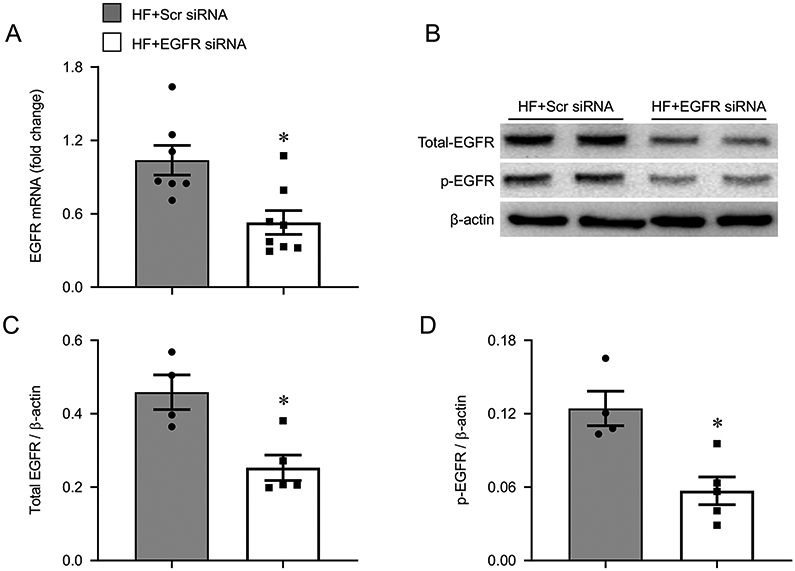
mRNA expression of EGFR (A), representative Western blots (B) and protein levels of total EGFR (C) and phosphorylated (p-) EGFR (D) in the hypothalamic paraventricular nucleus (PVN) of heart failure (HF) rats 4 weeks after coronary artery ligation and 5 weeks after bilateral PVN microinjection of a lentiviral vector encoding scrambled (Scr) siRNA or EGFR siRNA. mRNA data are expressed as a fold change compared to HF+Scr siRNA. Values are mean ± SEM (n = 4-8 for each group). *p<0.05, vs HF+Scr siRNA.
Figure 5.
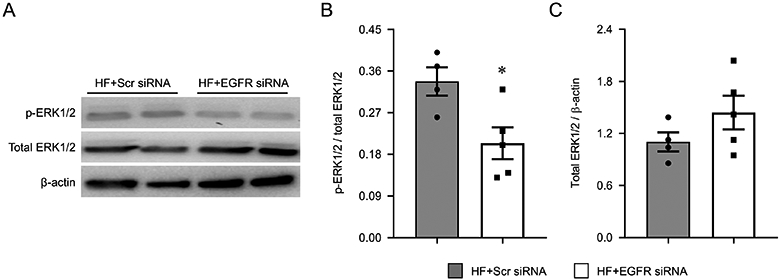
Representative Western blots (A), ratio of phosphorylated (p-) ERK1/2 to total ERK1/2 protein (B) and total ERK1/2 protein levels (C) in the hypothalamic paraventricular nucleus (PVN) of heart failure (HF) rats 4 weeks after coronary artery ligation and 5 weeks after bilateral PVN microinjection of a lentiviral vector encoding a scrambled (Scr) siRNA or EGFR siRNA. Levels of total ERK1/2 were corrected by β-actin. Values are mean ± SEM (n = 4-5 for each group). *p<0.05, vs HF+Scr siRNA.
Effect of EGFR knockdown on excitatory mediators in the PVN
RAS components, inflammatory mediators, and ER stress are all upregulated in the PVN of HF rats and are known to contribute to sympathetic excitation in this model (Wei et al., 2016b;Yu et al., 2016). The present study examined the effect of EGFR knockdown in PVN on mRNA expression of representative components of these three major neuroexcitatory systems. Compared with HF rats that received scrambled siRNA, HF rats that received EGFR siRNA had significantly lower mRNA levels of the ER stress markers BIP (Figure 6A) and ATF4 (Figure 6B), the inflammatory mediators TNF-α (Figure 6D), IL-1β (Figure 6E), and COX-2 (Figure 6F), and the RAS components ACE (Figure 6G) and AT1aR (Figure 6H) in the PVN. There was no difference in mRNA expression of ATF6 (Figure 6C) in the PVN between the 2 groups.
Figure 6.
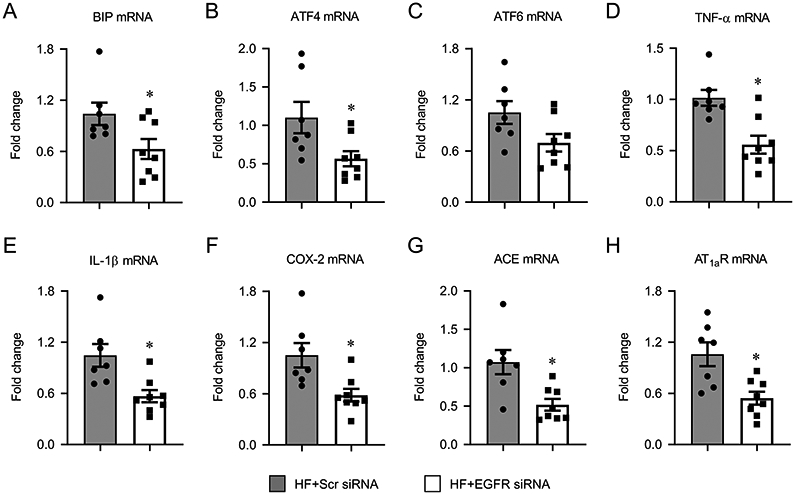
mRNA expression of excitatory mediators, including ER stress markers BIP (A), ATF4 (B) and ATF6 (C), inflammatory mediators tumor necrosis factor (TNF)-α (D), interleukin (IL)-1β (E) and cyclooxygenase (COX)-2 (F), and RAS components angiotensin-converting enzyme (ACE, G) and angiotensin II type 1a receptor (AT1aR, H), in the hypothalamic paraventricular nucleus (PVN) of heart failure (HF) rats 4 weeks after coronary artery ligation and 5 weeks after PVN microinjection of a lentiviral vector encoding a scrambled (Scr) siRNA or EGFR siRNA. Values are mean ± SEM (n = 7-8 for each group) and are expressed as a fold change compared to HF+Scr siRNA. *p<0.05, vs HF+Scr siRNA.
Effects of EGFR knockdown in PVN on peripheral indicators of HF
Echocardiography.
Echocardiography performed within 24 hours of CL and 4 weeks after CL revealed that LVEDV was significantly increased and LVEF was markedly decreased in HF rats, compared with sham-operated rats. In the HF group, LVEDV had increased further at 4 weeks compared with 24 hours. The %IZ was similar at the two time points (Table 2).
Table 2.
Echocardiographic and anatomical measurements:
HF vs sham-operated rats
| Sham | HF | |
|---|---|---|
|
Echocardiographic variables ~24 h |
||
| LVEDV (ml) | 0.38 ± 0.04 | 0.60 ± 0.03* |
| LVEF | 0.85 ± 0.03 | 0.37 ± 0.02* |
| %IZ | - | 40 ± 2* |
|
Echocardiographic variables at 4 weeks |
||
| LVEDV (ml) | 0.39 ± 0.07 | 1.14 ± 0.08*§ |
| LVEF | 0.83 ± 0.03 | 0.36 ± 0.03* |
| %IZ | - | 37 ± 3* |
|
Anatomic variables at 4 weeks |
||
| BW (g) | 362 ± 5 | 355 ± 5 |
| LV/BW (mg/g) | 2.38 ± 0.04 | 2.35 ± 0.13 |
| RV/BW (mg/g) | 0.53 ± 0.03 | 0.94 ± 0.11* |
| Lung/BW (mg/g) | 3.41 ± 0.09 | 8.97 ± 1.38* |
LVEDV: left ventricular end-diastolic volume; LVEF: left ventricular ejection fraction; %IZ: ischemic zone as a percent of left ventricular circumference; BW: body weight; LV: left ventricular; RV: right ventricular; Values are expressed as mean ± SEM (n = 6 for each group).
p<0.05, versus Sham
p<0.05, versus baseline at 24 hours.
In HF rats treated with EGFR siRNA or scrambled siRNA, echocardiography within 24 hours revealed comparable degrees of cardiac injury and similar values of LVEDV and LVEF. At 4 weeks after CL, LVEDV had increased further in both treatment groups but %IZ and LVEF were unchanged. There were no significant differences in LVEDV, %IZ, or LVEF between the two treatment groups (Table 3).
Table 3.
Echocardiographic measurements:
HF + EGFR siRNA and HF + Scr siRNA rats
| HF + Scr siRNA | HF + EGFR siRNA | |
|---|---|---|
| Variables ~24 h after MI | ||
| LVEDV (ml) | 0.71 ± 0.07 | 0.76 ± 0.04 |
| LVEF | 0.33 ± 0.03 | 0.31 ± 0.02 |
| %IZ | 45 ± 3 | 43 ± 2 |
| Variables 4 weeks after MI | ||
| LVEDV (ml) | 1.16 ± 0.05* | 1.05 ± 0.07* |
| LVEF | 0.30 ± 0.02 | 0.32 ± 0.02 |
| %IZ | 46 ± 2 | 44 ± 1 |
HF + Scr siRNA, HF rats treated with scrambled siRNA; HF + EGFR siRNA, HF rats treated with EGFR siRNA. LVEDV: left ventricular end-diastolic volume; LVEF: left ventricular ejection fraction; %IZ: ischemic zone as a percent of left ventricular circumference. Values are expressed as mean ± SEM (n = 11-12 for each group).
p<0.05, versus baseline at 24 hours.
Hemodynamics.
At 4 weeks after CL, HF rats that had received EGFR siRNA had significantly lower LVEDP and higher LV dP/dtmax than HF rats that received scrambled siRNA (Table 4). There no differences in HR, SBP, DBP and LVPSP between the two treatment groups.
Table 4.
Anatomical and hemodynamic measurements:
HF + EGFR siRNA vs HF + Scr siRNA
| Variables | HF + Scr siRNA | HF + EGFR siRNA |
|---|---|---|
| BW (g) | 360 ± 6 | 363 ± 5 |
| LV/BW (mg/g) | 2.24 ± 0.08 | 2.19 ± 0.07 |
| RV/BW (mg/g) | 0.98 ± 0.04 | 0.83 ± 0.05* |
| Lung/BW (mg/g) | 9.06 ± 0.90 | 6.65 ± 0.67* |
| HR (beats/min) | 365 ± 12 | 369 ± 14 |
| SBP (mmHg) | 103 ± 4 | 107 ± 5 |
| DBP (mmHg) | 80 ± 2 | 78 ± 3 |
| LVPSP (mmHg) | 100 ± 4 | 109 ± 5 |
| LVEDP (mmHg) | 14 ± 2 | 10 ± 1* |
| dP/dt (mmHg/s) | 6199 ± 416 | 7537 ± 459* |
HF + Scr siRNA, HF rats treated with scrambled siRNA; HF + EGFR siRNA, HF rats treated with EGFR siRNA. BW: body weight; LV: left ventricular; RV: right ventricular; HR: heart rate; SBP: systolic blood pressure; DBP: diastolic blood pressure; LVPSP: LV peak systolic pressure; LVEDP: LV end-diastolic pressure. dP/dt: maximum rate of rise of LV pressure. Values are expressed as mean ± SEM (n = 11-12 for each group).
p<0.05, versus HF+ Scr siRNA.
Anatomy.
Body weight (BW) and LV/BW were comparable between the sham-operated and HF rats 4 weeks after CL. However, RV/ BW and wet lung/BW ratios were markedly higher in HF rats than sham-operated rats (Table 2).
In HF rats treated with EGFR siRNA or scrambled siRNA, BW and LV/BW ratio were comparable between the two treatment groups (Table 4). However, compared with HF rats that had received scrambled siRNA, HF rats that had received EGFR siRNA had less pulmonary congestion as evidenced by a reduced wet lung/BW ratio, and less RV hypertrophy as evidenced by a reduced RV/BW ratio.
Norepinephrine levels.
Plasma and urinary NE levels, indicators of sympathetic excitation in HF (Wafi et al., 2019;Zheng et al., 2016), were significantly lower in the HF rats that had received EGFR siRNA, compared with HF rats that had received scrambled siRNA (Figure 7).
Figure 7.
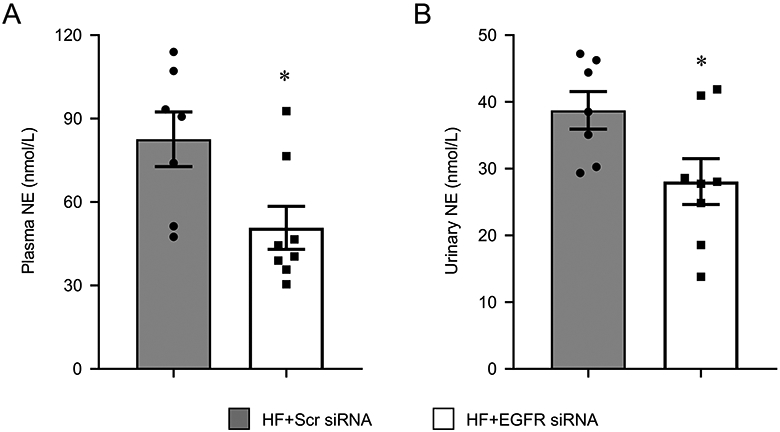
Plasma (A) and urinary (B) norepinephrine levels in heart failure (HF) rats 4 weeks after coronary artery ligation and 5 weeks after bilateral hypothalamic paraventricular nucleus (PVN) microinjection of a lentiviral vector encoding a scrambled (Scr) siRNA or EGFR siRNA. Values are mean ± SEM (n = 7-8 for each group). *p<0.05, HF+EGFR siRNA vs HF+Scr siRNA.
DISCUSSION
The present study addressed a potential role for EGFR as a gateway to ERK1/2 MAPK signaling in the PVN of HF rats. We found that EGFR is abundantly expressed in PVN neurons of normal rats, that EGFR is activated (phosphorylated) in the PVN in HF rats, and that knockdown of EGFR mRNA in the PVN significantly reduces not only total and phosphorylated EGFR but also phosphorylated ERK1/2 in HF rats. These changes were accompanied by reductions in PVN markers of ER stress, inflammation and RAS activity, which we have previously shown are dependent at least in part on ERK1/2 signaling (Wei et al., 2016b;Wei et al., 2008b). Prior work has shown that reducing the activity of these excitatory mediators in cardiovascular regions of the brain ameliorates sympathetic excitation and the peripheral manifestations of HF (Francis et al., 2004; Huang et al., 2009; Kang et al., 2010; Kang et al., 2008b; Wei et al., 2016a; Yu et al., 2017; Yu et al., 2018; Yu et al., 2007), as occurred in this study. Accordingly, we propose that activation of EGFR in the PVN is one mechanism that stimulates the ERK1/2 signaling that we have shown sustains sympathetic activation in HF (Wei et al., 2008a; Yu et al., 2016).
EGFR is a transmembrane protein belonging to the ErbB family of receptor tyrosine kinases that are activated by multiple stimuli and are critical signaling mechanisms in many pathological states such as cancer, diabetes and cardiovascular dysfunction (Akhtar and Benter, 2013; Khan et al., 2019; Makki et al., 2013). The altered neurochemical milieu of the PVN in HF, characterized by increased RAS activity and inflammation, is highly conducive to activation of EGFR. EGFR may be transactivated by Ang II (Pastore et al., 2008; Touyz et al., 2002) and PICs (Pastore et al., 2008), and directly activated by the oxidative stress induced by these major excitatory systems. Yet, to our knowledge, the present study is the first to demonstrate that EGFR is activated in a cardiovascular region of the brain in HF. Preliminary data (unpublished results) from our laboratory suggest that TGFα, an endogenous ligand for EGFR, is also upregulated in the PVN of HF rats (Yu et al., 2020). The TNF-α converting enzyme (TACE), also known as a disintegrin and a metalloproteinase (ADAM)17, is upregulated in the PVN of HF rats (Yu et al., 2019a), and one of its effects is to induce membrane shedding of soluble TGFα (Khan et al., 2019). Thus, several mechanisms that promote phosphorylation of EGFR are active in the PVN in HF.
The ERK1/2 MAPK signaling pathway is one of several key molecular pathways mediating the downstream effects of EGFR (Hsieh and Conti, 2005; Tomas et al., 2014). Activation of EGFR induces receptor dimerization, leading to autophosphorylation and tyrosine kinase activity (Bae and Schlessinger, 2010) that eventuates in activation of the Ras/Raf/Mek pathway leading to phosphorylation of ERK1/2 (Boutros et al., 2008; Hunter, 1998; Roberts and Der, 2007). There are at least two potential mechanisms by which ERK1/2 activity might contribute to sympathetic excitation in HF rats. In cardiovascular regions of the brain, phosphorylation of ERK1/2 leads to increased gene expression of RAS components, inflammatory mediators and markers of ER stress (Wei et al., 2016b; Yu et al., 2016), all of which contribute to sympathetic excitation. By this mechanism, ERK1/2 signaling may perpetuate the sympatho-excitatory neurochemical milieu. ERK1/2 signaling also has a direct channel effect. ERK1/2 phosphorylates and inactivates the potassium channel subunit Kv4.2 (Adams et al., 2000), and by that mechanism may reduce A-current and thereby alter the excitability of presympathetic PVN neurons (Sonner and Stern, 2007). Further studies are needed to address the relative contribution of these two mechanisms to the sympatho-excitatory influences of ERK1/2.
Our studies in normal and sham-operated rats revealed abundant expression of EGFR mRNA and protein in the PVN, with confocal images showing EGFR expression in PVN neurons. Under normal conditions, one might anticipate minimal expression of p-EGFR in PVN neurons and minimal or no expression of EGFR in glial elements (Tavassoly et al., 2020). In HF, as in other neuroinflammatory conditions (Tavassoly et al., 2020), one might anticipate upregulation of EGFR and p-EGFR and a functional role in astrocytes and microglial cells as well as in neurons. For example, transactivation of EGFR in astrocytes has been implicated in the release of gliotransmitters and astrocyte-to-neuron signaling (Sharif and Prevot, 2010). In microglial cultures, inhibiting phosphorylation of EGFR prevents microglia activation, EGFR-induced MAPK signaling, and MAPK-mediated production of IL-1β and TNF-α (Qu et al., 2012). Notably, in that study, inhibition of ERK1/2 signaling was most effective in preventing EGFR-mediated cytokine production. Similar findings were observed in vivo. In a spinal cord injury model, inhibition of EGFR signaling substantially reduced the activation of microglia and astrocytes (Qu et al., 2012), MAPK signaling and cytokine production. The present study found that p-EGFR was dramatically upregulated in the PVN in our HF model. The cell types mediating the effects of EGFR knockdown on the neurochemistry of the PVN and sympathetic activation were not determined, but reductions in EGFR-mediated glial and neuronal mechanisms likely contributed.
In the present study, we assessed the effect of a reduction in EGFR signaling on a representative sampling of neuroexcitatory mediators whose expression has been shown to be dependent upon ERK1/2 signaling. Consistent with our previous studies in which ERK1/2 activity in the PVN of HF rats was reduced by selective inhibition (Wei et al., 2016b) or by siRNA knockdown (Yu et al., 2016), the present study found that the reduction in ERK1/2 activity in the PVN of HF rats treated with EGFR siRNA was accompanied by lower mRNA levels of the ER stress indicators BIP and ATF4, the inflammatory mediators TNF-α, IL-1β and COX-2, and the RAS components ACE and AT1aR in the PVN. Similarly, like HF rats in which ERK1/2 activity has been reduced, HF rats treated with EGFR siRNA had lower plasma NE levels and improved peripheral manifestations of HF. LV end-diastolic pressure was reduced and LV dP/dtmax was increased, and indicators of cardiac remodeling (RV/BW) and pulmonary congestion (lung/BW) were improved. These hemodynamic and anatomic indices of improved cardiac function occurred in the absence of any significant change in echocardiographic indices of cardiac function, which is typical of this HF model (Wei et al., 2016a; Wei et al., 2016b; Wei et al., 2008b; Yu et al., 2017; Yu et al., 2018; Yu et al., 2016; Yu et al., 2012) in which the ischemic injury to the heart muscle is permanent.
The purpose of this study was to determine whether and how activation of EGFR in the PVN contributes to sympathetic excitation and the progression of HF. Since previous studies have established that ERK1/2 signaling, RAS activity and inflammation are all upregulated in the PVN in this rat model of HF (Tan et al., 2004; Wei et al., 2008b; Yu et al., 2016; Yu et al., 2012), the use of sham-operated controls to demonstrate that point was considered unjustified in this study. An effect of EGFR knockdown in sham-operated rats was also considered irrelevant to the issue being addressed – i.e., the contribution of EGFR to mechanisms driving the augmented sympathetic nerve activity in HF. To further minimize animal use, mRNA measurements were used as indicators of the levels of RAS activity, inflammation and ER stress in the PVN. In multiple previous studies in which we have measured both mRNA and protein levels at various time points from 1-6 weeks after the induction HF (Yu et al., 2019b; Yu et al., 2008; Yu et al., 2007; Yu et al., 2010; Yu et al., 2012), the protein levels have consistently paralleled the mRNA levels as might be expected in the HF setting, in which gene expression is chronically upregulated. However, potential limitations of the study are the selection of only a few representative components of the major central excitatory systems known to contribute to sympathetic excitation in HF and the reliance on mRNA rather than protein values.
The interactions between EGFR, ERK1/2, brain RAS, neuroinflammation and ER stress in the PVN are complex. Both angiotensin II and TNF-α transactivate EGFR (Jamroz-Wisniewska et al., 2008; Konishi and Berk, 2003; Pastore et al., 2008; Takeyama et al., 2000; Taniguchi et al., 2013; Touyz et al., 2002), initiate ERK1/2 signaling (Wei et al., 2021; Wei et al., 2009), and induce ER stress (Duvigneau et al., 2019; Menikdiwela et al., 2019; Takayanagi et al., 2015; Xue et al., 2005; Yap et al., 2020; Young et al., 2012). Recent work in our laboratory revealed that TNF-α-induced phosphorylation of ERK1/2 is largely dependent on activation of EGFR (Wei et al., 2021). In HF, in which both RAS activity and PICs are increased in the PVN, ERK1/2 signaling contributes to the PVN production of RAS components and inflammatory mediators (Yu et al., 2016) and the induction of ER stress (Wei et al., 2016b), while chronic central inhibition of ER stress reduces ERK1/2 signaling in the PVN, along with the expression of inflammatory mediators (Wei et al., 2016a). Thus, it appears that ERK1/2 signaling contributes to the production of the same mediators that activate it. These convoluted and apparent feed-forward interactions may provide a molecular substrate for the sustained sympathetic excitation in HF that leads to increased preload (i.e., volume accumulation), afterload (i.e., arterial vasoconstriction) and cardiac remodeling, and precipitates serious ventricular arrhythmias. Notably, interventions that selectively interrupt forebrain RAS activity (Francis et al., 2004; Huang et al., 2009; Yu et al., 2018), neuroinflammation (Kang et al., 2010; Kang et al., 2008b; Yu et al., 2017; Yu et al., 2007) or ER stress (Wei et al., 2016a) have all been effective in reducing sympathetic excitation with beneficial effects on cardiovascular function in HF. Interventions within the PVN EGFR-ERK1/2 pathway have the potential to reduce the influences of all three of these systems on the sympathetic overactivity that promotes adverse outcomes in HF. In the present study, a reduction in EGFR activity in the PVN that reduced sympathetic activity improved cardiac function, as evidenced by a reduction in LVEDP, an increase in LV dP/dtmax and reduced pulmonary congestion and right ventricular hypertrophy. Echocardiographic indices of left ventricular function were unchanged, as is commonly observed in this HF model in which the ischemia-induced myocardial injury is permanent (Francis et al., 2001a; Francis et al., 2004; Guggilam et al., 2008; Kang et al., 2008a, Kang et al., 2008b).
In conclusion, the present study demonstrates that EGFR signaling in the PVN contributes to sympathetic excitation and the progression of HF. While not directly tested in this study, we speculate that the beneficial effects of EGFR knockdown were mediated by the resulting reduction in ERK1/2 activity and the associated, and likely subsequent, reduction in ER stress, neuroinflammation and brain RAS activity. Notably, alternative pathways to activation of ERK1/2 (Roskoski, 2012) were unable to compensate for the effects of EGFR knockdown, emphasizing the importance of EGFR as a gateway to ERK1/2 signaling in HF. The findings suggest that brain EGFR activity may be a novel target for therapeutic intervention in the MI-induced HF syndrome.
Highlights.
The hypothalamic paraventricular nucleus (PVN) is a source of augmented sympathetic nerve activity in heart failure (HF).
Epidermal growth factor receptor (EGFR) activates extracellular signal-related kinase 1 and 2 (ERK1/2) in PVN of HF rats.
ERK1/2 upregulates PVN expression of excitatory mediators that drive sympathetic activity in HF rats.
siRNA knockdown of EGFR in the PVN reduces sympathetic activity and improves the peripheral manifestations of HF.
Brain EGFR is a potential target for therapeutic intervention in the augmented sympathetic activity that promotes HF progression.
ACKNOWLEDGEMENTS
We acknowledge Kathy Zimmerman, RDCS, for diligent and expert assistance in the performance of the echocardiograms.
DECLARATION OF INTERESTS
This material is based upon work supported in part by the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development, and by the National Heart, Lung, and Blood Institute of the National Institutes of Health under award numbers R01 HL136149 (to RB Felder), R01 HL-139521 (to S-G Wei) and S10 OD019941 (to RM Weiss). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations
- ACE
angiotensin converting enzyme
- ADAM17
a disintegrin and a metalloproteinase 17
- AT1aR
angiotensin II type 1a receptors
- ATF4
activating transcription factor 4
- ATF6
activating transcription factor 6
- Ang II
angiotensin II
- BIP
binding immunoglobulin protein
- BW
body weight
- CL
coronary artery ligation
- CON
control
- COX-2
cyclooxygenase-2
- DBP
diastolic blood pressure
- dP/dtmax
maximum rate of rise of LV pressure
- EGFR
epidermal growth factor receptor
- ELISA
enzyme-linked immunosorbent assay
- ER
endoplasmic reticulum
- ERK1/2
extracellular signal-regulated kinases 1 and 2
- GFAP
glial fibrillary acidic protein
- GFP
green fluorescent protein
- HF
heart failure
- HR
heart rate
- IL-1β
interleukin-1 beta
- %IZ
ischemic zone as a percent of left ventricular circumference
- LV
left ventricle
- LVEDP
left ventricular end diastolic pressure
- LVEDV
left ventricular end diastolic volume
- LVEF
left ventricular ejection fraction
- LVPSP
left ventricular peak systolic pressure
- MAPK
mitogen-activated protein kinase
- MI
myocardial infarction
- NE
norepinephrine
- p-
phosphorylated
- PICs
proinflammatory cytokines
- PVN
hypothalamic paraventricular nucleus
- RAS
renin-angiotensin system
- RV
right ventricle
- SBP
systolic blood pressure
- SFO
subfornical organ
- siRNA
small interfering RNA
- TACE
tumor necrosis factor – alpha converting enzyme
Footnotes
The authors have no other interests to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Adams JP, Anderson AE, Varga AW, Dineley KT, Cook RG, Pfaffinger PJ, Sweatt JD (2000) The A-type potassium channel Kv4.2 is a substrate for the mitogen-activated protein kinase ERK. J Neurochem 75:2277–2287. [DOI] [PubMed] [Google Scholar]
- Akhtar S, Benter IF (2013) The role of epidermal growth factor receptor in diabetes-induced cardiac dysfunction. Bioimpacts 3:5–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae JH, Schlessinger J (2010) Asymmetric tyrosine kinase arrangements in activation or autophosphorylation of receptor tyrosine kinases. Mol Cells 29:443–448. [DOI] [PubMed] [Google Scholar]
- Boutros T, Chevet E, Metrakos P (2008) Mitogen-activated protein (MAP) kinase/MAP kinase phosphatase regulation: roles in cell growth, death, and cancer. Pharmacol Rev 60:261–310. [DOI] [PubMed] [Google Scholar]
- Duvigneau JC, Luis A, Gorman AM, Samali A, Kaltenecker D, Moriggl R, Kozlov AV (2019) Crosstalk between inflammatory mediators and endoplasmic reticulum stress in liver diseases. Cytokine 124:154577. [DOI] [PubMed] [Google Scholar]
- Ferguson AV, Latchford KJ, Samson WK (2008) The paraventricular nucleus of the hypothalamus - a potential target for integrative treatment of autonomic dysfunction. Expert Opin Ther Targets 12:717–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Alcantara S, Ballabriga J, Olive M, Blanco R, Rivera R, Carmona M, Berruezo M, et al. (1996) Transforming growth factor-alpha (TGF-alpha) and epidermal growth factor-receptor (EGF-R) immunoreactivity in normal and pathologic brain. Prog Neurobiol 49:99–123. [DOI] [PubMed] [Google Scholar]
- Francis J, Wei SG, Weiss RM, Felder RB (2004) Brain angiotensin-converting enzyme activity and autonomic regulation in heart failure. Am J Physiol Heart Circ Physiol 287:H2138–H2146. [DOI] [PubMed] [Google Scholar]
- Francis J, Weiss RM, Wei SG, Johnson AK, Beltz TG, Zimmerman K, Felder RB (2001a) Central mineralocorticoid receptor blockade improves volume regulation and reduces sympathetic drive in heart failure. Am J Physiol Heart Circ Physiol 281:H2241–H2251. [DOI] [PubMed] [Google Scholar]
- Francis J, Weiss RM, Wei SG, Johnson AK, Felder RB (2001b) Progression of heart failure after myocardial infarction in the rat. Am J Physiol Regul Integr Comp Physiol 281:R1734–1745. [DOI] [PubMed] [Google Scholar]
- Guggilam A, Patel KP, Haque M, Ebenezer PJ, Kapusta DR, Francis J (2008) Cytokine blockade attenuates sympathoexcitation in heart failure: cross-talk between nNOS, AT-1R and cytokines in the hypothalamic paraventricular nucleus. Eur J Heart Fail 10:625–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh M, Conti M (2005) G-protein-coupled receptor signaling and the EGF network in endocrine systems. Trends Endocrinol Metab 16:320–326. [DOI] [PubMed] [Google Scholar]
- Huang BS, Ahmad M, Tan J, Leenen FH (2009) Chronic central versus systemic blockade of AT(1) receptors and cardiac dysfunction in rats post-myocardial infarction. Am J Physiol Heart Circ Physiol 297:H968–975. [DOI] [PubMed] [Google Scholar]
- Hunter T (1998) The Croonian Lecture 1997. The phosphorylation of proteins on tyrosine: its role in cell growth and disease. Philos Trans R Soc Lond B Biol Sci 353:583–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamroz-Wisniewska A, Wojcicka G, Lowicka E, Ksiazek M, Beltowski J (2008) Transactivation of epidermal growth factor receptor in vascular and renal systems in rats with experimental hyperleptinemia: role in leptin-induced hypertension. Biochem Pharmacol 75:1623–1638. [DOI] [PubMed] [Google Scholar]
- Kang YM, Ma Y, Elks C, Zheng JP, Yang ZM, Francis J (2008a) Cross-talk between cytokines and renin-angiotensin in hypothalamic paraventricular nucleus in heart failure: role of nuclear factor-kappaB. Cardiovasc Res 79:671–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang YM, Wang Y, Yang LM, Elks C, Cardinale J, Yu XJ, Zhao XF, Zhang J, et al. (2010) TNF-alpha in hypothalamic paraventricular nucleus contributes to sympathoexcitation in heart failure by modulating AT1 receptor and neurotransmitters. Tohoku J Exp Med 222:251–263. [DOI] [PubMed] [Google Scholar]
- Kang YM, Zhang ZH, Xue B, Weiss RM, Felder RB (2008b) Inhibition of brain proinflammatory cytokine synthesis reduces hypothalamic excitation in rats with ischemia-induced heart failure. Am J Physiol Heart Circ Physiol 295:H227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan K, Valeri N, Dearman C, Rao S, Watkins D, Starling N, Chau I, Cunningham D (2019) Targeting EGFR pathway in metastatic colorectal cancer- tumour heterogeniety and convergent evolution. Crit Rev Oncol Hematol 143:153–163. [DOI] [PubMed] [Google Scholar]
- Konishi A, Berk BC (2003) Epidermal growth factor receptor transactivation is regulated by glucose in vascular smooth muscle cells. J Biol Chem 278:35049–35056. [DOI] [PubMed] [Google Scholar]
- Makki N, Thiel KW, Miller FJ Jr. (2013) The epidermal growth factor receptor and its ligands in cardiovascular disease. Int J Mol Sci 14:20597–20613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menikdiwela KR, Ramalingam L, Allen L, Scoggin S, Kalupahana NS, Moustaid-Moussa N (2019) Angiotensin II Increases Endoplasmic Reticulum Stress in Adipose Tissue and Adipocytes. Sci Rep 9:8481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastore S, Mascia F, Mariani V, Girolomoni G (2008) The epidermal growth factor receptor system in skin repair and inflammation. J Invest Dermatol 128:1365–1374. [DOI] [PubMed] [Google Scholar]
- Qu WS, Tian DS, Guo ZB, Fang J, Zhang Q, Yu ZY, Xie MJ, Zhang HQ, et al. (2012) Inhibition of EGFR/MAPK signaling reduces microglial inflammatory response and the associated secondary damage in rats after spinal cord injury. J Neuroinflammation 9:178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts PJ, Der CJ (2007) Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 26:3291–3310. [DOI] [PubMed] [Google Scholar]
- Roskoski R Jr. (2012) ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol Res 66:105–143. [DOI] [PubMed] [Google Scholar]
- Sharif A, Prevot V (2010) ErbB receptor signaling in astrocytes: a mediator of neuron-glia communication in the mature central nervous system. Neurochem Int 57:344–358. [DOI] [PubMed] [Google Scholar]
- Sonner PM, Stern JE (2007) Functional role of A-type potassium currents in rat presympathetic PVN neurones. J Physiol 582:1219–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayanagi T, Kawai T, Forrester SJ, Obama T, Tsuji T, Fukuda Y, Elliott KJ, Tilley DG, et al. (2015) Role of epidermal growth factor receptor and endoplasmic reticulum stress in vascular remodeling induced by angiotensin II. Hypertension 65:1349–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeyama K, Dabbagh K, Jeong Shim J, Dao-Pick T, Ueki IF, Nadel JA (2000) Oxidative stress causes mucin synthesis via transactivation of epidermal growth factor receptor: role of neutrophils. J Immunol 164:1546–1552. [DOI] [PubMed] [Google Scholar]
- Tan J, Wang H, Leenen FH (2004) Increases in brain and cardiac AT1 receptor and ACE densities after myocardial infarct in rats. Am J Physiol Heart Circ Physiol 286:H1665–1671. [DOI] [PubMed] [Google Scholar]
- Taniguchi K, Xia L, Goldberg HJ, Lee KW, Shah A, Stavar L, Masson EA, Momen A, et al. (2013) Inhibition of Src kinase blocks high glucose-induced EGFR transactivation and collagen synthesis in mesangial cells and prevents diabetic nephropathy in mice. Diabetes 62:3874–3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavassoly O, Sato T, Tavassoly I (2020) Inhibition of Brain Epidermal Growth Factor Receptor Activation: A Novel Target in Neurodegenerative Diseases and Brain Injuries. Mol Pharmacol 98:13–22. [DOI] [PubMed] [Google Scholar]
- Tomas A, Futter CE, Eden ER (2014) EGF receptor trafficking: consequences for signaling and cancer. Trends Cell Biol 24:26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touyz RM, Wu XH, He G, Salomon S, Schiffrin EL (2002) Increased angiotensin II-mediated Src signaling via epidermal growth factor receptor transactivation is associated with decreased C-terminal Src kinase activity in vascular smooth muscle cells from spontaneously hypertensive rats. Hypertension 39:479–485. [DOI] [PubMed] [Google Scholar]
- Voldborg BR, Damstrup L, Spang-Thomsen M, Poulsen HS (1997) Epidermal growth factor receptor (EGFR) and EGFR mutations, function and possible role in clinical trials. Ann Oncol 8:1197–1206. [DOI] [PubMed] [Google Scholar]
- Wafi AM, Yu L, Gao L, Zucker IH (2019) Exercise training upregulates Nrf2 protein in the rostral ventrolateral medulla of mice with heart failure. J Appl Physiol 127:1349–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakatsuki S, Furuno A, Ohshima M, Araki T (2015) Oxidative stress-dependent phosphorylation activates ZNRF1 to induce neuronal/axonal degeneration. J Cell Biol 211:881–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wee P, Wang Z (2017) Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 9:doi: 10.3390/cancers9050052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei SG, Yu Y, Felder RB (2021) TNF-α-induced sympathetic excitation requires EGFR and ERK1/2 signaling in cardiovascular regulatory regions of the forebrain. Am J Physiol Heart Circ Physiol 320:H772–H786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei SG, Yu Y, Weiss RM, Felder RB (2016a) Endoplasmic reticulum stress increases brain MAPK signaling, inflammation and renin-angiotensin system activity and sympathetic nerve activity in heart failure. Am J Physiol Heart Circ Physiol 311:H871–H880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei SG, Yu Y, Weiss RM, Felder RB (2016b) Inhibition of brain mitogen-activated protein kinase signaling reduces central endoplasmic reticulum stress and inflammation and sympathetic nerve activity in heart failure rats. Hypertension 67:229–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei SG, Yu Y, Zhang ZH, Felder RB (2009) Angiotensin II upregulates hypothalamic AT1 receptor expression in rats via the mitogen-activated protein kinase pathway. Am J Physiol Heart Circ Physiol 296:H1425–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei SG, Yu Y, Zhang ZH, Weiss RM, Felder RB (2008a) Angiotensin II-triggered p44/42 mitogen-activated protein kinase mediates sympathetic excitation in heart failure rats. Hypertension 52:342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei SG, Yu Y, Zhang ZH, Weiss RM, Felder RB (2008b) Mitogen-activated protein kinases mediate upregulation of hypothalamic angiotensin II type 1 receptors in heart failure rats. Hypertension 52:679–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei SG, Zhang ZH, Yu Y, Felder RB (2014) Central SDF-1/CXCL12 expression and its cardiovascular and sympathetic effects: the role of angiotensin II, TNF-alpha, and MAP kinase signaling. Am J Physiol Heart Circ Physiol 307:H1643–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue X, Piao JH, Nakajima A, Sakon-Komazawa S, Kojima Y, Mori K, Yagita H, Okumura K, et al. (2005) Tumor necrosis factor alpha (TNFalpha) induces the unfolded protein response (UPR) in a reactive oxygen species (ROS)-dependent fashion, and the UPR counteracts ROS accumulation by TNFalpha. J Biol Chem 280:33917–33925. [DOI] [PubMed] [Google Scholar]
- Yap J, Chen X, Delmotte P, Sieck GC (2020) TNFalpha selectively activates the IRE1alpha/XBP1 endoplasmic reticulum stress pathway in human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 318:L483–L493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young CN, Cao X, Guruju MR, Pierce JP, Morgan DA, Wang G, Iadecola C, Mark AL, et al. (2012) ER stress in the brain subfornical organ mediates angiotensin-dependent hypertension. J Clin Invest 122:3960–3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Cao Y, Bell B, Chen X, Weiss RM, Felder RB, Wei SG (2019a) Brain TACE (Tumor Necrosis Factor-alpha-Converting Enzyme) contributes to sympathetic excitation in heart failure rats. Hypertension 74:63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Wei SG, Weiss RM, Felder RB (2017) TNF-alpha receptor 1 knockdown in the subfornical organ ameliorates sympathetic excitation and cardiac hemodynamics in heart failure rats. Am J Physiol Heart Circ Physiol 313:H744–H756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Wei SG, Weiss RM, Felder RB (2018) Angiotensin II Type 1a Receptors in the Subfornical Organ Modulate Neuroinflammation in the Hypothalamic Paraventricular Nucleus in Heart Failure Rats. Neuroscience 381:46–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Wei SG, Weiss RM, Felder RB (2019b) Sex differences in the central and peripheral manifestations of ischemia-induced heart failure in rats. Am J Physiol Heart Circ Physiol 316:H70–H79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Wei SG, Weiss RM, Felder RB (2020) Transforming Growth Factor-α Acts via Epidermal Growth Factor Receptor to Increase p44/42 Mitogen-Activated Protein Kinase Signaling and Expression of Excitatory Mediators in the Hypothalamic Paraventricular Nucleus in Rats. FASEB J 34 (S1): 10.1096/fasebj.2020.34.s1.03644 [DOI] [Google Scholar]
- Yu Y, Wei SG, Zhang ZH, Gomez-Sanchez E, Weiss RM, Felder RB (2008) Does aldosterone upregulate the brain renin-angiotensin system in rats with heart failure? Hypertension 51:727–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Wei SG, Zhang ZH, Weiss RM, Felder RB (2016) ERK1/2 MAPK signaling in hypothalamic paraventricular nucleus contributes to sympathetic excitation in rats with heart failure after myocardial infarction. Am J Physiol Heart Circ Physiol 310:H732–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Zhang ZH, Wei SG, Chu Y, Weiss RM, Heistad DD, Felder RB (2007) Central gene transfer of interleukin-10 reduces hypothalamic inflammation and evidence of heart failure in rats after myocardial infarction. Circ Res 101:304–312. [DOI] [PubMed] [Google Scholar]
- Yu Y, Zhang ZH, Wei SG, Serrats J, Weiss RM, Felder RB (2010) Brain perivascular macrophages and the sympathetic response to inflammation in rats after myocardial infarction. Hypertension 55:652–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Zhang ZH, Wei SG, Weiss RM, Felder RB (2012) Peroxisome proliferator-activated receptor-gamma regulates inflammation and renin-angiotensin system activity in the hypothalamic paraventricular nucleus and ameliorates peripheral manifestations of heart failure. Hypertension 59:477–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZH, Yu Y, Wei SG, Felder RB (2012) Aldosterone-induced brain MAPK signaling and sympathetic excitation are angiotensin II type-1 receptor dependent. Am J Physiol Heart Circ Physiol 302:H742–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Liu X, Sharma NM, Patel KP (2016) Renal denervation improves cardiac function in rats with chronic heart failure: Effects on expression of beta-adrenoceptors. Am J Physiol Heart Circ Physiol 311:H337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]