

Abstract
We report the results of a phase 1 dose-escalation study of belinostat and bortezomib in adult patients with acute leukemia or MDS or CML with blast crisis. Thirty-eight patients received IV belinostat days 1-5 and 8-12 with IV bortezomib days 1, 4, 8, and 11 every 21 days. QTc prolongation was the only identified DLT. The RP2Ds were 1.3 mg/m2 bortezomib and 1000 mg/m2 belinostat. One patient with highly refractory MLL-ENL rearranged biphenotypic AML with multiple karyotypic aberrations had a complete pathologic and karyotypic response. One patient with post-MPN AML remained on study with stable disease (SD) for 32 cycles. Whole-exome sequencing revealed no aberrations in the first patient and a hyper-mutator genotype in the second. Eighteen patients had a best response of SD. We conclude that this treatment strategy is feasible but has limited activity in this population. Nevertheless, the factors that predict exceptional responses to this strategy warrant further investigation.
Keywords: Acute leukemia, belinostat, bortezomib, myelodysplastic syndrome, phase 1 clinical trial
Introduction
Outcomes for patients with relapsed and refractory acute leukemia remain poor. In newly diagnosed acute myeloid leukemia (AML) patients less than age 60, approximately 35% are cured [1, 2]. Outcomes are worse for certain subgroups including older patients, patients with therapy-related AML, and patients with antecedent hematologic disorders such as myelodysplastic syndrome (MDS) or myeloproliferative neoplasms (MPN). Clearly, novel therapies and treatment paradigms are urgently needed.
Bortezomib, a reversible inhibitor of the 26S proteasome, has been approved for the treatment of multiple myeloma and mantle cell lymphoma [3]. In clinical studies, bortezomib has shown minimal single-agent activity in acute leukemia [4, 5]. However, in vivo administration of bortezomib has been shown to attenuate leukemic cell proteasome and NF-κB activities [4, 5]. Other possible mechanisms of proteasome inhibitor (PI)-induced toxicity include disruption of stromal cell actions [6], anti-angiogenic actions [7], interference with DNA repair [8], and induction of oxidative injury [9]. Such pre-clinical findings provide a basis for employing bortezomib in combination with other anti-leukemic agents, particularly those whose activity might be enhanced by proteasome or NF-κB inhibition. In this context, the reliance of leukemia stem cells on NF-κB for survival [10] has prompted investigation of bortezomib with standard chemotherapy (e.g., cytarabine and daunorubicin) in patients with AML [11].
HDAC inhibitors (HDACIs) constitute a chemically diverse group of epigenetic agents that modify chromatin structure and gene expression, selectively promoting neoplastic cell death, including AML cells. HDACIs also perturb all 3 arms of the DNA-damage response—DNA repair, cell cycles arrest, and apoptosis [12]. They also activate NF-κB in leukemia cells, and prevention of this process e.g., IKK (IκB kinase) inhibitors significantly increases lethality [13]. Notably, the lethality of HDACIs has been shown to be synergistically enhanced by proteasome inhibitors such as bortezomib in leukemic [14] and other malignant hematopoietic cell types [15].
Belinostat, a pan-HDACI approved for relapsed/refractory peripheral T-cell lymphoma, has no significant single-agent activity in AML [16]. Nevertheless, in pre-clinical models of AML and ALL, bortezomib was shown to interact synergistically with belinostat at extremely low concentrations (e.g., nM) to induce cell death [17]. This was associated with evidence of interruption of both canonical and non-canonical NF-κB signaling pathways as well as down-regulation of NF-κB-dependent apoptotic proteins accompanied by up-regulation of the pro-apoptotic protein Bim. Similar findings were extended to primary patient-derived acute leukemia cells. The central goal of this study was to conduct a phase 1 trial of belinostat and bortezomib in patients with acute leukemia or MDS, document the safety of the combination, and identify a recommended phase 2 dose (RP2D). Secondary goals were to observe preliminary signals of activity and determine the feasibility of pharmacodynamic correlative studies.
Materials and Methods
Drug supply
Belinostat was supplied by TopoTarget A/S – Spectrum Pharmaceuticals, Inc (now Acrotech Biopharma). Bortezomib (VELCADE) was supplied by Millennium Pharmaceuticals, Inc (now Takeda).
Eligibility criteria
Eligible patients were 18 years of age and older with a diagnosis of relapsed or refractory acute leukemia (AML other than acute promyelocytic leukemia; ALL), acute leukemia that evolved from a prior MDS (no requirement for prior therapy), acute leukemia in patients at least 60 years of age (no requirement for prior therapy), MDS (International Prognostic Scoring System intermediate-2 or greater), or CML with blast crisis.
Additional inclusion criteria included Eastern Cooperative Oncology Group performance status 0 to 2, creatinine less than 1.5 × the upper limit of normal (ULN) or creatinine clearance greater than 45 mL/min, aspartate aminotransferase (AST) and alanine aminotransferase (ALT) less than 2.5 × the ULN, and serum total bilirubin less than 1.5 × the ULN. Patients with prior allogeneic stem cell transplantation were eligible if the interval from transplant was at least 12 months and there was no graft-versus-host disease or current immunosuppressive therapy.
Patients were excluded from the study if they had a white blood cell count greater than 50 × 109/L; known CNS leukemia; prior allergic reaction to components of the investigational regimen; grade 1 with pain or grade 2 or greater peripheral neuropathy or paresthesia by NCI Common Terminology Criteria for Adverse Events (CTCAE) criteria (v4.0) within 14 days before enrollment; significant cardiovascular disease; known risk factors for torsades de pointes or medications associated with prolonged QT interval; persistent blood pressure greater than 160/95; treatment with strong or moderate CYP3A4 inhibitors; or steroids for cancer control within one week prior to start of study treatment. All patients provided written informed consent. The study was approved and conducted in accordance with the policies of the Institutional Review Boards of Virginia Commonwealth University and MD Anderson Cancer Center. This clinical trial is registered at Clinicaltrials.gov, NCT01075425.
Study design
This was a phase 1 traditional 3+3 dose-escalation study designed to determine the RP2D for the combination of belinostat and bortezomib. Belinostat was administered by a 30-minute intravenous (IV) infusion on days 1 through 5 and days 8 through 12 of each cycle. Bortezomib was administered by a bolus IV infusion preceding belinostat on days 1, 4, 8, and 11 of each cycle. The treatments were repeated on 21-day cycles until disease progression or unacceptable toxicity. Initially, there were 6 planned dose levels of the combination; however, an additional 3 dose levels were added to permit increased belinostat dosing. The starting dose for bortezomib was 1.0 mg/m2; the starting dose for belinostat was 500 mg/m2.
All adverse events were characterized in terms of severity and relatedness to study treatment and were reported according to NCI CTCAE version 4.0. Dose-limiting toxicity (DLT) was defined as any of the following occurring during the first cycle of treatment and determined to be possibly, probably, or definitely related to study treatment: (a) grade 4 infection; (b) any other grade 4 non-hematologic toxicity not directly related to a hematologic toxicity; (c) any grade 3 non-hematologic toxicity that persisted for more than 7 days; or (d) grade 4 thrombocytopenia or neutropenia that persisted more than 6 weeks in the absence of leukemia. For DLT evaluation, patients received greater than 70% of prescribed study drug doses in cycle 1. Patients who were not DLT evaluable were replaced.
To assess response, a bone marrow aspiration was performed during the rest week of cycle 2 and again as clinically indicated. Response was evaluated using the revised response criteria developed by an international working group for AML [18]. Complete response (CR) required a normal bone marrow aspirate with greater than 20% cellularity and less than 5% blast cells as well as count recovery with absolute neutrophil count (ANC) of 1000/μL or higher, platelet count of 100,000/μL or higher, absence of leukemic blasts in peripheral blood as well as red blood cell (RBC) transfusion independence. Complete response with incomplete blood count recovery (CRi) was the same as CR but with platelet count less than 100,000/μL or ANC less than 1000/μL. Partial response (PR) was defined as the presence of trilineage hematopoiesis in the marrow but with 5-25% marrow blasts. Progressive disease (PD) was defined as a greater than 50% increase in bone marrow or peripheral blood blasts from baseline or the development of extramedullary leukemia. Stable disease (SD) was assigned to patients who did not have CR, CRi, PR, or PD. Patients could receive up to 12 cycles of study therapy unless they derived clinical benefit and tolerated study therapy without significant toxicity.
Pharmacodynamic analysis
Research samples were collected from consenting patients with ≥ 65% leukemic blasts in blood or bone marrow aspirate. Both prior to and 24 hours (± 5 hours) following treatment, triplicate peripheral blood samples or a single bone marrow aspirate were obtained. Whole blood (5-6 mL) was collected into tubes containing either sodium heparin or EDTA. Bone marrow aspirate (10 mL) was collected in a heparinized syringe. Peripheral blood mononuclear cells were isolated using Accuspin System-Histopaque-1077 tubes (Sigma) according to the manufacturer’s instructions, and the cells were subsequently stored at −80°C.
Protein extraction and Western blot analysis was done as previously described [19]. Antibodies to the following proteins were used: Bcl-XL (Cell Signaling), Bim (Calbiochem), NF-kB p65/RelA (Chemicon), and XIAP (BD Biosciences). Western blot analysis of p65/RelA was performed on a nuclear fraction isolated according to Suzuki et al. [20]. An Odyssey Imager (LI-COR Biosciences) was used to quantify binding of IRDye 680LT-conjugated secondary antibody (LI-COR Biosciences).
Genomic analysis
Tumors from 2 patients, unusual responders, and corresponding paired normal tissue were de-identified, shipped to Duke University, and processed in accordance with a protocol approved by the Institutional Review Board at Duke University. Genomic DNA was extracted from the tissue using column-based methods [21]. See Supplementary Methods for details .
Results
Patient characteristics
A total of 41 patients were enrolled and 38 patients were treated on study. Three patients enrolled but were not treated due to disease-related events that rendered initiation of study treatment inappropriate. The characteristics of the treated patients are reported in Table 1. The median age was 62 (range, 27-83) and 50% (19 of 38) were women. The majority of patients (28 of 38) had AML and all but one were previously treated. The median number of prior regimens was 2 (range, 0-5). Only one patient had a prior allogeneic stem cell transplant. Twenty patients received at least one prior hypomethylating agent.
Table 1.
Patient characteristics (38 treated patients)
| Gender | No. of patients |
| Female | 19 |
| Male | 19 |
| Race | No. of patients |
| Asian | 1 |
| Black or African-American | 9 |
| White | 27 |
| More than one race | 2 |
| Ethnicity | No. of patients |
| Hispanic or Latino | 2 |
| Non-Hispanic | 36 |
| Age | Years |
| Median | 62 |
| Range | 27-83 |
| Performance Status | No. of patients |
| 0 | 1 |
| 1 | 28 |
| 2 | 9 |
| Diagnosis | No. of patients |
| AML* | 28 |
| ALL | 2 |
| MDS | 6 |
| CML-BC | 2 |
| Prior Treatment | No. of regimens |
| Median | 2 |
| Range | 0-5 |
| Prior allogeneic stem cell transplant | 1 |
Includes 1 patient with previously untreated AML
Safety and tolerability
Patients were enrolled to 7 of 9 planned dose levels. Details of patient enrollment and cycle administration are provided in Table 2. The summary of drug-related toxicity is provided in Table 3. Patients received a median of 2 cycles of study treatment with a range of 1 to 32 cycles (up to 126 weeks). Eleven patients were not evaluable for DLT due to early discontinuation of study treatment. Eight of the 11 had disease progression in cycle 1 leading to insufficient drug exposure. One patient electively withdrew from the study; one tolerated drug poorly and had insufficient drug exposure; one had insufficient drug exposure in cycle 1 due to febrile neutropenia but was able to start cycle 2. The most common non-hematologic treatment-related adverse events were nausea, vomiting, diarrhea, and fatigue; the majority of the gastrointestinal toxicities were grade 1/2. The most common grade 3/4 treatment-related toxicity was QTc prolongation, which was identified in 5 patients. QTc prolongation was also the only DLT identified during study treatment. The DLT grade 3 QTc prolongation, occurred in 4 patients at dose levels 6 and 7 but readily resolved without sequelae permitting patients to continue study treatment. One patient at dose level 5 had grade 3 QTc prolongation in cycle 6 leading to discontinuation of study treatment. The RP2D of the combination was dose level 5, bortezomib 1.3 mg/m2 and belinostat 1000 mg/m2.
Table 2.
Dose levels and dose-limiting toxicities
| Dose Level | Bortezomib (mg/m2) D1,4,8,11 IV Push | Belinostat (mg/m2) D1-5, 8-12 IV | Number of Patients Treated/Number of Patients DLT Evaluable | DLT Event | Median Number of Cycles (Range) |
|---|---|---|---|---|---|
| 1 | 1.0 | 500 | 6/3 | 1.5 (1-4) | |
| 2 | 1.3 | 500 | 6/4 | 1.5 (1-4) | |
| 3 | 1.3 | 650 | 4/3 | 2 (1-2) | |
| 4 | 1.3 | 850 | 3/3 | 6 (2-10) | |
| 5* | 1.3 | 1000 | 9/6 | 3 (1-14) | |
| 6 | 1.3 | 1200 | 7/6 | Gr3 QTc prolongation (2 patients) | 2 (1-32) |
| 7 | 1.3 | 1400 | 3/2 | Gr3 QTc prolongation (2 patients) | 2 (1-8) |
Maximum tolerated dose (MTD).
Table 3.
Toxicity summary for treatment-related toxicities*
| Event | All Grades N (%) | Grades 3-4 N (%) | |
|---|---|---|---|
| Cardiac | Cardiac disorders - other, specify | 10 (26) | 1 (3) |
| Prolonged QTc | 20 (5) | 5 (13) | |
| Gastrointestinal | Anorexia | 12 (32) | 0 (0) |
| Diarrhea | 15 (39) | 3 (8) | |
| Nausea | 26 (68) | 1 (3) | |
| Vomiting | 18 (47) | 0 (0) | |
| General | Fatigue/malaise | 23 (61) | 3 (8) |
| Infusion-related reaction | 9 (24) | 0 (0) | |
| Hematologic | Platelet count decreased | 9 (24) | 9 (24) |
| White blood cell decreased | 11 (29) | 9 (24) | |
| Laboratory | Increased AST/ALT | 9 (24) | 2 (5) |
| Alkaline phosphatase increased | 13 (34) | 1 (3) | |
| Neurologic | Dysgeusia | 12 (32) | 0 (0) |
| Paresthesia | 10 (26) | 0 (0) | |
| Peripheral sensory neuropathy | 11 (29) | 2 (5) | |
Includes toxicities with incidence greater than 20%
Responses
One patient with AML had a CR after 2 cycles and proceeded to stem cell transplant after an additional 2 cycles of study treatment. Eighteen patients had a best response of SD (AML = 12, ALL = 1, MDS = 4, CML-BC = 1). The median duration of SD was 3.8 months. One patient with post-MPN AML remained on study with SD for 32 cycles of study treatment.
Correlative Studies
Pre- and post-treatment peripheral blood samples for pharmacodynamic analysis were only able to be obtained from 2 patients due to specimen requirements or lack of consent. While correlative studies were integrated, patients were required to have greater than 65% blasts to be eligible for sample collection. Only one sample from a consenting patient contained a sufficient percentage of blasts in pre- and post-treatment testing to be analyzed by Western blotting. Relative to the pre-treatment samples, the expression levels of Bim (Fig. 1A) and Bcl-XL (Fig. 1A) increased in the post-treatment samples, and the expression levels of XIAP (Fig. 1A) and NF-κB (Fig. 1B) decreased in the post-treatment samples. These limited findings are concordant with pre-clinical observations [17].
Figure 1.
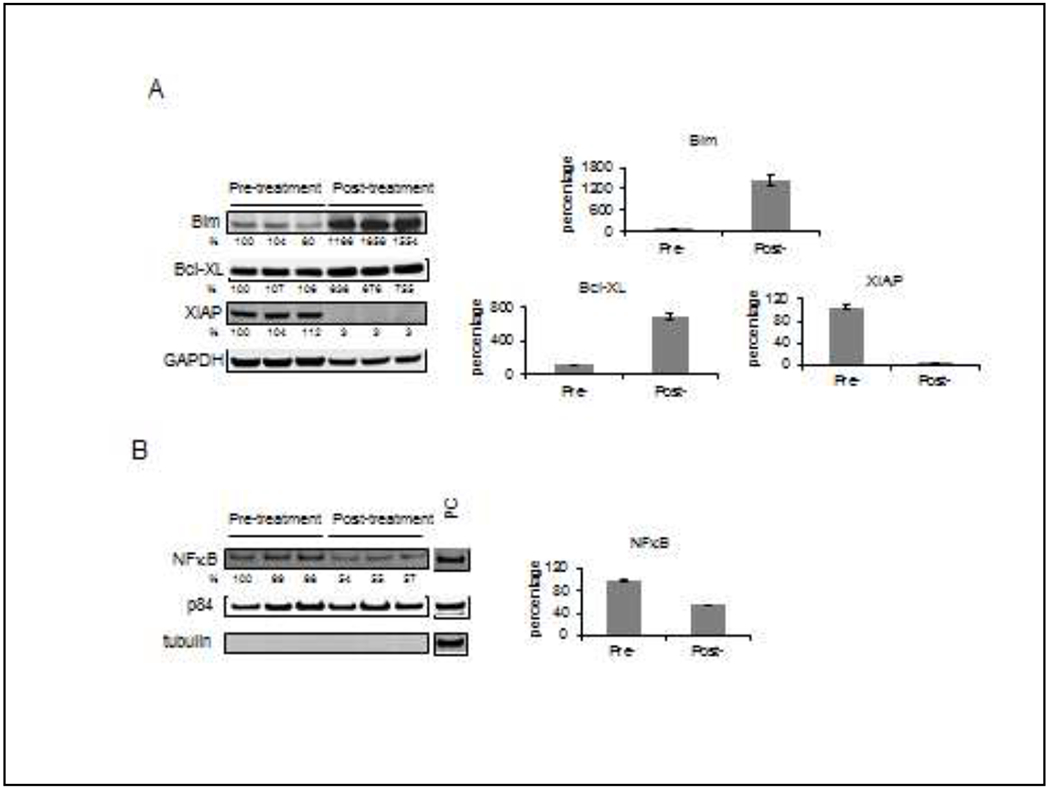
Western blot analysis of pre- and post-treatment patient peripheral blood samples PBMCs were isolated at baseline (pre-) and 24 hours after (post-) treatment. A, triplicate whole cell lysate samples were run and analyzed for their level of expression of Bim, Bcl-XL, and XIAP. The numbers below the figures represent the relative level of the assayed proteins normalized to the GAPDH level, where the normalized level of first pre-treatment sample is defined as 100%. The graphs show the relative level of assayed proteins in the samples (average ± standard error). B, nuclear extracts from the samples were analyzed for NF-κB. The numbers below the NF-κB figure represent the relative level of the assayed protein normalized to the p84 level, where the normalized level of the first pre-treatment sample is defined as 100%. To show the purity of nuclear fraction, α-tubulin was run as a Marker of cytoplasmic contamination. PC: positive control sample. The graph shows the relative level of assayed proteins in the samples (average ± standard error).
Discussion
Pre-clinical studies demonstrating anti-tumor synergism between HDAC and proteasome inhibitors [15] led to the evaluation of this strategy in multiple myeloma, in which addition of the pan-HDACI panobinostat to bortezomib and dexamethasone led to approval of this regimen in patients who had failed multiple prior therapies [22]. To the best of our knowledge, the phase 1 trial of bortezomib and belinostat represents the first to test the HDACI/proteasome inhibitor strategy in relapsed/refractory acute leukemia and MDS patients, although based upon pre-clinical findings [23], a phase I/II trial of vorinostat and bortezomib in patients with infant ALL has been launched (NCT02553460). Clearly, the limited clinical activity of the belinostat/bortezomib regimen in AML/MDS argues against further exploration in this setting. This may reflect several factors, including the very modest (belinostat) [16] or absent (bortezomib) [24] single-agent activity of these agents in acute leukemia. Nevertheless, despite the minimal activity of this regimen, 2 patients had highly unusual responses that merit further consideration.
To identify potential genetic mechanisms of sensitivity to the belinostat and bortezomib combination, whole-exome sequencing of pre-treatment tumor and germline DNA from the 2 unusual responders was performed. The first patient, a 59-year old male, had mixed phenotype acute leukemia (MPAL), T/myeloid. At initial diagnosis, he had complex cytogenetics including MLL (11q23). His disease was refractory to initial induction with cytarabine and daunorubicin and then re-induction with fludarabine, cytarabine, and idarubicin. At the time of study enrollment, the marrow aspirate blast count was 86%. He achieved a pathologic and karyotypic CR after 2 cycles with normal FISH and cytogenetics. He received an additional 2 cycles of study treatment before proceeding to allogeneic stem cell transplant. Contrary to expectations, whole-exome sequencing identified no discernible events. This is in contrast to findings in other exceptional responders, in which aberrations in relevant pathways were identified as a potential contributor to response [25]. In the present case, it is possible that alternative aberrations other than mutations e.g., involving miRNA or other epigenetic abnormalities may underlie the dramatic response of this patient to this regimen.
The second patient, a 49-year old man, was diagnosed with JAK2 mutated primary myelofibrosis 3 years prior to AML diagnosis. He was treated with 11 cycles of azacitidine until disease progression to AML. At the time of study enrollment, the marrow aspirate had 38% blasts with complex cytogenetics. After 2 cycles, the aspirate blasts could not be reported but peripheral blasts were unchanged at 31%. His best response to study treatment was SD, but marrow fibrosis complicated monitoring of blasts on multiple subsequent marrow aspirates. During this period, his peripheral blast count ranged from 18 to 59%, but he remained stable and appeared to be deriving clinical benefit. Evaluation of his bone marrow aspirate after cycle 32 showed 47% blasts and evaluation of peripheral blood showed 66% blasts, after which he stopped study treatment. Whole-exome sequencing showed 244 individual events. Among the somatically mutated genes, events were observed in the DNA-repair genes MYH1 and MYH8; the signaling genes ALK, FLT1, NOTCH3, VEGFA, and VEGFB; and the chromatin modifiers EP400 and INO80B. It is impossible to define the effects of any one of these mutations, although their large number suggests a hypermutated phenotype.
In the seminal work by The Cancer Genome Atlas Network, AML genomes from adult patients with de novo AML had an average of only 13 mutations [26]. Of those, only an average of 5 genes were recurrently mutated in AML. Thus, the 2 patients with notable responses represented the 2 extremes in mutational frequencies in AML. Despite the absence of an objective response in the latter patient, his clinical course was remarkable. Notably, while the average survival of patients with post-MPN AML is in the range of 3-6 months [27, 28, 29], this patient survived with SD and remained functional for 32 months on treatment. One speculative possibility is that the bortezomib/belinostat regimen may have targeted and reduced the number of leukemia stem cell-like cells, thereby preventing the rapid expansion of the total leukemia cell burden.
A major goal of this phase 1 study was to describe the toxicity of the novel regimen. In a combination regimen, it can be difficult to attribute toxicity to a single drug. The most common grade 3/4 treatment-related toxicity as well as the only identified DLT was QTc prolongation which was likely related to belinostat. As a class, HDACIs have been associated with QTc interval prolongation on the ECG. The clinical significance of this phenomenon is not completely clear. For belinostat, the incidence of QTc prolongation has been reported as 11% (all grades) with 4% grade 3/4 incidence [30]. At the time of the study administration, there was concern regarding risk of QTc prolongation with belinostat, which led to frequent ECG monitoring for study patients. Furthermore, the concurrent administration of drugs with a risk of torsades de pointes was prohibited. Drugs with a possible or conditional risk of torsades de pointes could be used at investigator discretion. It is notable that drugs routinely used in the care of patients with leukemia such as azoles, quinolones, and ondansetron are also associated with QTc prolongation. Of the 4 patients with QTc prolongation as a DLT, none had to discontinue treatment as ECG abnormalities readily resolved, which suggests supportive care medications such as ondansetron were likely confounding. One patient had QTc prolongation after the DLT evaluation period, which ultimately led to discontinuation of study treatment. The role of QTc monitoring on study as well as with concomitant medications remains to be fully resolved.
In summary, the results of this phase I study indicate that the limited overall efficacy of the belinostat/bortezomib regimen in patients with relapsed/refractory AML does not warrant further clinical evaluation. However, it is important to note that 2 extremely high-risk patients experienced remarkable clinical responses e.g., a CR after 2 cycles, and SD for 32 cycles. Interestingly, whole-exome sequencing revealed multiple mutations in the patient with SD, but no apparent mutations in the patient who achieved a CR. These disparate findings complicate our capacity to understand the genetic mechanism responsible for sensitivity to this regimen. However, as HDACI activity primarily affects the epigenome, simultaneous characterization of both epigenetic and genetic alterations may provide insights into the factors determining responses to such HDACI-based regimens. Furthermore, the specificity of HDACIs for individual HDACs may play a key role in determining responses in different hematologic malignancies. Regardless of these considerations, one potentially important implication of these findings is that combining 2 agents that individually have little or no anti-leukemic activity may be highly active in a sub-set of AML patients. Additionally, the possibility exists that HDACIs with demonstrated single-agent activity might be viable options for this combination strategy. For example, the HDACI pracinostat has shown single-agent activity in AML [31, 32], and trials combining this agent with hypomethylating agents are currently underway (NCT03151304, NCT03151408). Accordingly, the strategy of combining pracinostat with proteasome inhibitors in AML is currently being explored at the pre-clinical level. Similarly, a change in the proteasome inhibitor may improve synergism. Ixazomib, an oral long acting proteasome inhibitor, may confer benefit through a more constant therapeutic AUC.
Supplementary Material
Table 4.
Best response by diagnosis
| Best response | AML | ALL | MDS | CML-BC | Total |
|---|---|---|---|---|---|
| (number of patients) | |||||
| Complete remission (CR) | 1 | 0 | 0 | 0 | 1 |
| Stable disease (SD) | 12 | 1 | 4 | 1 | 18 |
| Progressive disease (PD) | 13 | 1 | 1 | 1 | 16 |
| Not assessed / Not evaluable‡ | 2 | 0 | 1 | 0 | 3 |
Study participation discontinued prior to disease assessment timepoint (1 withdrew for personal reasons; 2 discontinued study treatment for unrelated adverse events)
Grant Support:
The study was supported by grants from the National Institutes of Health (NCI RC2 CA148431, NCI P30 CA016059 [Cancer Center Support Grant to Massey Cancer Center; supported in part the Biostatistics Shared Resource], UL1TR002649 [CTSA award from the National Center for Advancing Translational Sciences to the VCU C. Kenneth and Dianne Wright Center for Clinical and Translational Research]), and through funding from Millennium-Spectrum.
Footnotes
Disclosure of Possible Conflict of Interests: No potential conflicts of interest were disclosed by any of the authors.
References
- 1.Buchner T, Berdel WE, Haferlach C, et al. Age-related risk profile and chemotherapy dose response in acute myeloid leukemia: a study by the German Acute Myeloid Leukemia Cooperative Group. J Clin Oncol. 2009. January 1;27(1):61–9. doi: 10.1200/JCO.2007.15.4245. [DOI] [PubMed] [Google Scholar]
- 2.Moore JO, George SL, Dodge RK, et al. Sequential multiagent chemotherapy is not superior to high-dose cytarabine alone as postremission intensification therapy for acute myeloid leukemia in adults under 60 years of age: Cancer and Leukemia Group B Study 9222. Blood. 2005. May 1;105(9):3420–7. doi: 10.1182/blood-2004-08-2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moorthy AK, Savinova OV, Ho JQ, et al. The 20S proteasome processes NF-kappaB1 p105 into p50 in a translation-independent manner. EMBO J. 2006. May 3;25(9):1945–56. doi: 10.1038/sj.emboj.7601081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Orlowski RZ, Voorhees PM, Garcia RA, et al. Phase 1 trial of the proteasome inhibitor bortezomib and pegylated liposomal doxorubicin in patients with advanced hematologic malignancies. Blood. 2005. April 15;105(8):3058–65. doi: 10.1182/blood-2004-07-2911. [DOI] [PubMed] [Google Scholar]
- 5.Cortes J, Thomas D, Koller C, et al. Phase I study of bortezomib in refractory or relapsed acute leukemias. Clin Cancer Res. 2004. May 15;10(10):3371–6. doi: 10.1158/1078-0432.CCR-03-0508. [DOI] [PubMed] [Google Scholar]
- 6.Hideshima T, Chauhan D, Hayashi T, et al. Proteasome inhibitor PS-341 abrogates IL-6 triggered signaling cascades via caspase-dependent downregulation of gp130 in multiple myeloma. Oncogene. 2003. November 20;22(52):8386–93. doi: 10.1038/sj.onc.1207170. [DOI] [PubMed] [Google Scholar]
- 7.Roccaro AM, Hideshima T, Raje N, et al. Bortezomib mediates antiangiogenesis in multiple myeloma via direct and indirect effects on endothelial cells. Cancer Res. 2006. January 1;66(1):184–91. doi: 10.1158/0008-5472.CAN-05-1195. [DOI] [PubMed] [Google Scholar]
- 8.Jacquemont C, Taniguchi T. Proteasome function is required for DNA damage response and fanconi anemia pathway activation. Cancer Res. 2007. August 1;67(15):7395–405. doi: 10.1158/0008-5472.CAN-07-1015. [DOI] [PubMed] [Google Scholar]
- 9.Yu C, Rahmani M, Dent P, et al. The hierarchical relationship between MAPK signaling and ROS generation in human leukemia cells undergoing apoptosis in response to the proteasome inhibitor Bortezomib. Exp Cell Res. 2004. May 1;295(2):555–66. doi: 10.1016/j.yexcr.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 10.Guzman ML, Swiderski CF, Howard DS, et al. Preferential induction of apoptosis for primary human leukemic stem cells. Proc Natl Acad Sci U S A. 2002. December 10;99(25):16220–5. doi: 10.1073/pnas.252462599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Attar EC, Johnson JL, Amrein PC, et al. Bortezomib added to daunorubicin and cytarabine during induction therapy and to intermediate-dose cytarabine for consolidation in patients with previously untreated acute myeloid leukemia age 60 to 75 years: CALGB (Alliance) study 10502. J Clin Oncol. 2013. March 1;31(7):923–9. doi: 10.1200/JCO.2012.45.2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Petruccelli LA, Pettersson F, Del Rincon SV, et al. Expression of leukemia-associated fusion proteins increases sensitivity to histone deacetylase inhibitor-induced DNA damage and apoptosis. Mol Cancer Ther. 2013. August;12(8):1591–604. doi: 10.1158/1535-7163.MCT-12-1039. [DOI] [PubMed] [Google Scholar]
- 13.Dai Y, Rahmani M, Dent P, et al. Blockade of histone deacetylase inhibitor-induced RelA/p65 acetylation and NF-kappaB activation potentiates apoptosis in leukemia cells through a process mediated by oxidative damage, XIAP downregulation, and c-Jun N-terminal kinase 1 activation. Mol Cell Biol. 2005. July;25(13):5429–44. doi: 10.1128/MCB.25.13.5429-5444.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu C, Rahmani M, Conrad D, et al. The proteasome inhibitor bortezomib interacts synergistically with histone deacetylase inhibitors to induce apoptosis in Bcr/Abl+ cells sensitive and resistant to STI571. Blood. 2003. November 15;102(10):3765–74. doi: 10.1182/blood-2003-03-0737. [DOI] [PubMed] [Google Scholar]
- 15.Pei XY, Dai Y, Grant S. Synergistic induction of oxidative injury and apoptosis in human multiple myeloma cells by the proteasome inhibitor bortezomib and histone deacetylase inhibitors. Clin Cancer Res. 2004. June 1;10(11):3839–52. doi: 10.1158/1078-0432.CCR-03-0561. [DOI] [PubMed] [Google Scholar]
- 16.Kirschbaum MH, Foon KA, Frankel P, et al. A phase 2 study of belinostat (PXD101) in patients with relapsed or refractory acute myeloid leukemia or patients over the age of 60 with newly diagnosed acute myeloid leukemia: a California Cancer Consortium Study. Leuk Lymphoma. 2014. October;55(10):2301–4. doi: 10.3109/10428194.2013.877134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dai Y, Chen S, Wang L, et al. Bortezomib interacts synergistically with belinostat in human acute myeloid leukaemia and acute lymphoblastic leukaemia cells in association with perturbations in NF-kappaB and Bim. British Journal of Haematology. 2011. April;153(2):222–35. doi: 10.1111/j.1365-2141.2011.08591.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheson BD, Bennett JM, Kopecky KJ, et al. Revised recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia. J Clin Oncol. 2003. December 15;21(24):4642–9. doi: 10.1200/JCO.2003.04.036. [DOI] [PubMed] [Google Scholar]
- 19.Holkova B, Perkins EB, Ramakrishnan V, et al. Phase I trial of bortezomib (PS-341; NSC 681239) and alvocidib (flavopiridol; NSC 649890) in patients with recurrent or refractory B-cell neoplasms. Clin Cancer Res. 2011. May 15;17(10):3388–97. doi: 10.1158/1078-0432.CCR-10-2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suzuki K, Bose P, Leong-Quong RY, et al. REAP: A two minute cell fractionation method. BMC Res Notes. 2010. November 10;3:294. doi: 10.1186/1756-0500-3-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jima DD, Zhang J, Jacobs C, et al. Deep sequencing of the small RNA transcriptome of normal and malignant human B cells identifies hundreds of Novel microRNAs. Blood. 2010. August 23;116(23):e118–27. doi: blood-2010-05-285403 [pii] 10.1182/blood-2010-05-285403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.San-Miguel JF, Hungria VT, Yoon SS, et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. The Lancet Oncology. 2014. October;15(11):1195–206. doi: 10.1016/S1470-2045(14)70440-1. [DOI] [PubMed] [Google Scholar]
- 23.Koss C, Nance S, Connelly M, et al. Targeted inhibition of the MLL transcriptional complex by proteosome inhibitors elicits a high response rate in relapsed/refractory MLL rearranged leukemia. Blood. 2014;124(21):972–972. doi: 10.1182/blood.V124.21.972.972. [DOI] [Google Scholar]
- 24.Horton TM, Pati D, Plon SE, et al. A phase 1 study of the proteasome inhibitor bortezomib in pediatric patients with refractory leukemia: a Children’s Oncology Group study. Clin Cancer Res. 2007. March 1;13(5):1516–22. doi: 10.1158/1078-0432.CCR-06-2173. [DOI] [PubMed] [Google Scholar]
- 25.Iyer G, Hanrahan AJ, Milowsky MI, et al. Genome sequencing identifies a basis for everolimus sensitivity. Science. 2012. October 12;338(6104):221. doi: 10.1126/science.1226344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cancer Genome Atlas Research N, Ley TJ, Miller C, et al. Genomic and epigenomic landscapes of adult de Novo acute myeloid leukemia. N Engl J Med. 2013. May 30;368(22):2059–74. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bjorkholm M, Derolf AR, Hultcrantz M, et al. Treatment-related risk factors for transformation to acute myeloid leukemia and myelodysplastic syndromes in myeloproliferative neoplasms. J Clin Oncol. 2011. June 10;29(17):2410–5. doi: 10.1200/JCO.2011.34.7542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khan M, Siddiqi R, Gangat N. Therapeutic options for leukemic transformation in patients with myeloproliferative neoplasms. Leukemia Research. 2017. December;63:78–84. doi: 10.1016/j.leukres.2017.10.009. [DOI] [PubMed] [Google Scholar]
- 29.Mesa RA, Li CY, Ketterling RP, et al. Leukemic transformation in myelofibrosis with myeloid metaplasia: a single-institution experience with 91 cases. Blood. 2005. February 1;105(3):973–7. doi: 10.1182/blood-2004-07-2864. [DOI] [PubMed] [Google Scholar]
- 30.Sawas A, Radeski D, O’Connor OA. Belinostat in patients with refractory or relapsed peripheral T-cell lymphoma: a perspective review. Ther Adv Hematol. 2015. August;6(4):202–8. doi: 10.1177/2040620715592567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abaza YM, Kadia TM, Jabbour EJ, et al. Phase 1 dose escalation multicenter trial of pracinostat alone and in combination with azacitidine in patients with advanced hematologic malignancies. Cancer. 2017. December 15;123(24):4851–4859. doi: 10.1002/cncr.30949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bose P, Grant S. Orphan drug designation for pracinostat, volasertib and alvocidib in AML. Leukemia Research. 2014. August;38(8):862–5. doi: 10.1016/j.leukres.2014.06.007. [DOI] [PubMed] [Google Scholar]
- 33.Meyer M, Kircher M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb Protoc. 2010. June;2010(6):pdb prot5448. doi: 10.1101/pdb.prot5448. [DOI] [PubMed] [Google Scholar]
- 34.Cock PJ, Fields CJ, Goto N, et al. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 2010. April;38(6):1767–71. doi: gkp1137 [pii] 10.1093/nar/gkp1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research. 2010. September;20(9):1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Love C, Sun Z, Jima D, et al. The genetic landscape of mutations in Burkitt lymphoma. Nat Genet. 2012. November 11. doi: 10.1038/ng.2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang J, Grubor V, Love CL, et al. Genetic heterogeneity of diffuse large B-cell lymphoma. Proc Natl Acad Sci U S A. 2013. January 22;110(4):1398–403. doi: 10.1073/pnas.1205299110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang J, Jima D, Moffitt AB, et al. The genomic landscape of mantle cell lymphoma is related to the epigenetically determined chromatin state of normal B cells. Blood. 2014. May 8;123(19):2988–96. doi: 10.1182/blood-2013-07-517177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010. March 1;26(5):589–95. doi: btp698 [pii] 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009. August 15;25(16):2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010. March 15;26(6):841–2. doi: btq033 [pii] 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li H A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011. November 1;27(21):2987–93. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cibulskis K, Lawrence MS, Carter SL, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nature Biotechnology. 2013. March;31(3):213–9. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang H, Wang K. Genomic variant annotation and prioritization with ANNOVAR and wANNovAR. Nat Protoc. 2015. October;10(10):1556–66. doi: 10.1038/nprot.2015.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010. September;38(16):e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Love C, Sun Z, Jima D, et al. The genetic landscape of mutations in Burkitt lymphoma. Nature Genetics. 2012. December;44(12):1321–5. doi: 10.1038/ng.2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.