

SUMMARY
The β-barrel assembly machine (BAM) integrates β-barrel proteins into the outer membrane (OM) of Gram-negative bacteria. An essential BAM subunit (BamA) catalyzes integration by promoting the formation of a hybrid-barrel intermediate-state between its own β-barrel domain and that of its client proteins. Here we show that in addition to catalyzing the integration of β-barrel proteins, BamA functions as a polypeptide export channel. In vivo structural mapping via intermolecular disulfide-crosslinking showed that the extracellular ‘passenger’ domain of a member of the ‘autotransporter’ superfamily of virulence factors traverses the OM through the BamA β-barrel lumen. Furthermore, we demonstrate that a highly conserved residue within autotransporter β-barrels is required to position the passenger inside BamA to initiate translocation, and that during translocation the passenger stabilizes the hybrid-barrel state. Our results not only establish a new function for BamA, but also unify the divergent functions of BamA and other ‘Omp85’ superfamily transporters.
Keywords: Autotransporter, BamA, BAM, Omp85 superfamily, outer membrane protein, protein folding, secretion
eTOC BLURB
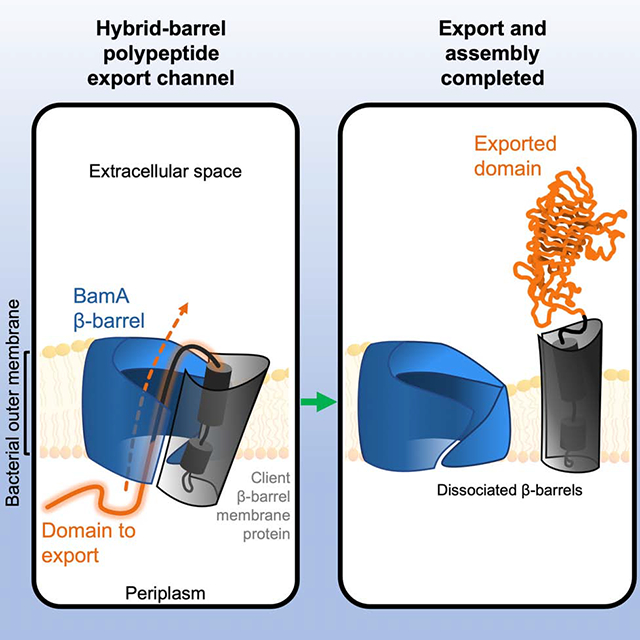
The mechanism by which the large soluble domains of outer membrane proteins such as autotransporters are secreted into the extracellular environment has been a long-standing mystery. Doyle and Bernstein show that the essential outer membrane protein BamA creates a unique channel that fulfils this export function.
Graphical Abstract
INTRODUCTION
The vast majority of integral outer membrane proteins (OMPs) produced by Gram-negative bacteria are anchored to the membrane by an amphipathic β-sheet that folds into a closed cylindrical ‘β-barrel’ structure (Konovalova et al., 2017; Schulz, 2000). In addition to containing a C-terminal β-barrel domain, a widespread superfamily of OMPs called ‘autotransporters’ also contain an N-terminal extracellular ‘passenger’ domain that varies in size (20–300 kDa) and that often possess a virulence function in pathogenic bacteria (Meuskens et al., 2019). Passenger domains are predominantly β-helical and are connected to a 12-stranded β-barrel by an α-helix that is embedded inside the β-barrel domain (Celik et al., 2012; Kajava and Steven, 2006). Due to their structural arrangement, autotransporters have proven to be valuable model proteins to study OMP assembly (Doyle and Bernstein, 2019; Ieva et al., 2011; Pavlova et al., 2013; Roman-Hernandez et al., 2014). Like other bacterial β-barrel proteins, autotransporter β-barrels are assembled (folded) and integrated into the outer membrane (OM) by the β-barrel assembly machinery (BAM) (Jain and Goldberg, 2007; Rossiter et al., 2011). In E. coli, the BAM consists of the membrane protein subunit BamA and the accessory lipoproteins BamBCDE (Wu et al., 2005). BamA is a member of the ‘Omp85’ superfamily of proteins that is found in both bacteria and eukaryotic organelles of bacterial origin (Heinz and Lithgow, 2014; Heinz et al., 2015). BamA is conserved in all Gram-negative species and, along with its homologs in organelles, is essential for cell viability (Kozjak et al., 2003; Patel et al., 2008; Voulhoux et al., 2003). Solved structures show that BamA contains N-terminal periplasmic domains that provide a scaffold for the lipoproteins and a C-terminal 16-stranded β-barrel (Gu et al., 2016; Han et al., 2016; Iadanza et al., 2016). Although the function of the BAM is poorly understood, multiple studies show that β-strands β1 and β16 of BamA must open laterally to allow an OMP β-barrel to assemble (Doerner and Sousa, 2017; Doyle and Bernstein, 2019; Lundquist et al., 2018; Noinaj et al., 2014; Tomasek et al., 2020). Recent crosslinking and structural investigations show the existence of an intermediate folding step in which the final β-strand of the client β-barrel is bound to BamAβ1 to form a unique ‘asymmetric hybrid-barrel’ structure (Doyle and Bernstein, 2019; Tomasek et al., 2020). Current models suggest that this structure is energetically favorable to allow for the closure and release of the new β-barrel into the OM (Doyle and Bernstein, 2019; Hohr et al., 2018; Tomasek et al., 2020). Interestingly, some Omp85 proteins (e.g., bacterial ‘two-partner secretion’ (TPS) transporters and the chloroplast Toc75 protein) catalyze protein translocation rather than membrane protein integration reactions (Baud et al., 2014; Ganesan and Theg, 2019).
While significant progress has been made to understand the assembly of the autotransporter β-barrel, the nature of the passenger translocation channel remains a mystery. After autotransporters are transported across the inner membrane through the SecYEG complex, the passenger domain is thought to remain in a largely unfolded conformation that is protected by periplasmic chaperones (Drobnak et al., 2015; Junker et al., 2009; Ruiz-Perez et al., 2009). A large body of evidence suggests that passenger domains are transported across the OM in a C- to N-terminal direction in a process initiated by the formation of a C-terminal hairpin, and that the folding of C-terminal segments on the cell surface is a major driving force that pulls the rest of the passenger across the membrane (Junker et al., 2006, 2009; Peterson et al., 2010; Yuan et al., 2018). Initial models proposed that after the autotransporter β-barrel is integrated into the OM, its lumen is used as a channel to secrete the passenger across the outer membrane (whence the name “autotransporter”) (Jose et al., 1995; Pohlner et al., 1987). However, when the first structures of autotransporter β-barrels where solved, it became clear that the luminal space is only wide enough to accommodate the α-helix or an unfolded hairpin (Barnard et al., 2007; Khalid and Sansom, 2006; Oomen et al., 2004). Subsequent in vivo proteolysis and biotinylation experiments indicated that the incorporation of an α-helical structure into the β-barrel occurs at an early stage before β-barrel folding is fully completed and thereby challenged the idea that passenger translocation is catalyzed by the β-barrel alone (Ieva et al., 2008). Moreover, molecular dynamics simulations and disulfide-loop experiments suggested that the folded β-barrel is too stable and narrow to allow passenger translocation (Khalid and Sansom, 2006; Sauri et al., 2012), yet translocation of passengers with at least small secondary structural elements has been observed (Leyton et al., 2011; Skillman et al., 2005). As a potential solution to this paradox, several observations suggest that BamA plays a role in passenger domain secretion. In vivo photo-crosslinking experiments showed that the passenger is in proximity to BamA during secretion (Ieva and Bernstein, 2009). Recent disulfide-bond formation experiments have also shown that the autotransporter β-barrel associates with the BamA β-barrel during assembly to form a hybrid-barrel structure in which both β-barrels are in an open conformation after passenger translocation had been initiated (Doyle and Bernstein, 2019). However, it is unclear from these results whether the lumen of the BamA β-barrel plays a direct role in passenger translocation, or if BamA merely maintains the autotransporter β-barrel in an expanded state to allow passenger translocation to occur.
Here, we sought to resolve the apparent paradox of autotransporter passenger domain secretion. To this end we utilized a derivative of EspP, a model autotransporter produced by E. coli O157:H7, that is trapped at the OM during the assembly and secretion process (Doyle and Bernstein, 2019). Using intermolecular disulfide-crosslinking to obtain a precise interaction map in vivo we found that when the autotransporter and BamA β-barrels are associated to form a hybrid-barrel structure, the passenger traverses the OM through the BamA β-barrel lumen and the α-helix is in close proximity to the BamA β-barrel. Interestingly, we found that the initiation of passenger translocation through this novel channel depends on a highly conserved residue within the autotransporter β-barrel that is required for the specific alignment of the α-helix and passenger in the channel. Finally, we obtained evidence that an incompletely secreted passenger domain inhibits the premature release of the autotransporter β-barrel from BamA and thereby couples passenger secretion to β-barrel assembly by the BAM. Overall, our results show that BamA functions as a polypeptide translocase – a finding that helps to explain the evolution of the Omp85 superfamily and illustrates the fundamental ability of these proteins to promote both membrane integration and translocation reactions.
RESULTS
The autotransporter passenger is secreted through the BamA β-barrel lumen
To locate the passenger secretion channel, we deployed a system that we recently developed to create an autotransporter assembly intermediate that remains associated with BamA in vivo (Doyle and Bernstein, 2019). In this system, we replaced most of the EspP passenger with maltose binding protein (MBP) to generate a fusion called MBP−76EspP (Figure 1A). When MBP−76EspP is expressed in E. coli, MBP folds rapidly in the periplasm and thereby creates a molecular ‘knot’ that prevents completion of passenger translocation (Figure 1B, right). The region between MBP and the β-barrel contains a short segment of native passenger residues (EspP948–984) that are trapped in the process of traversing the OM, a sequence of passenger residues that are already translocated and exposed on the cell surface, and an α-helix (EspP999–1031) that is likely embedded inside the β-barrel (Figures 1A and 1B). For some autotransporters such as EspP, once passenger translocation is completed and the β-barrel dissociates from BamA, the passenger is released from the cell surface by an intra-barrel auto-cleavage reaction that occurs within the α-helix (Figures 1A and 1B, left) and leaves a ‘mature’ β-barrel in the OM (Dautin et al., 2007). Because passenger translocation must be completed before autotransporters dissociate from BamA (Ieva and Bernstein, 2009; Ieva et al., 2011), MBP−76EspP forms a stable assembly intermediate in which its β-barrel remains associated with the BamA β-barrel as part of a hybrid-barrel (Figure 1B, right). The hybridization occurs by the formation of a rigid interface between BamA β-strand 1 (β1) and EspP β-strand 12 (β12) (Doyle and Bernstein, 2019).
Figure 1: Residue positions for autotransporter secretion channel mapping.

(A) Structure of MBP−76EspP. SS: signal sequence; TS: TwinStrepII-tag; MBP: maltose binding protein (tan); P: passenger domain (orange); α: a small portion of the passenger that is translocated plus the α-helix (black); green triangle: surface-exposed loop that is digestible by exogenous PK; white triangle: autoproteolytic cleavage site between N1023/N1024. Passenger residues substituted with cysteine for secretion channel mapping are indicated (numbering based on native EspP sequence). Binding sites for antibodies used in this study are shown. (B) Left: model of autotransporter assembly. The passenger is secreted in a C- to N-terminal direction while the β-barrel is joined with BamA in a hybrid-barrel state. For EspP, autoproteolytic cleavage separates the two domains after the passenger is fully secreted and the β-barrel is fully folded. Right: because the folded MBP moiety blocks the completion of passenger translocation, the assembly of MBP−76EspP arrests while it is associated with BamA. (C) Structure of BamA (from apo-state BAM, PDB: 5ljo). Red: β1; black: β2; green: β3; yellow: β16. Inset, zoom of β-barrel main-chain showing residues substituted with cysteine for secretion channel mapping. N427, G443, and G457 are exposed to the β-barrel lumen, V444 faces the lipid bilayer, I806 interfaces with β1 near N427, and N805 is oriented in an opposite direction to I806. See also Figures S1–S4.
To test the hypothesis that the lumen of the BamA β-barrel is directly involved in passenger domain translocation, we transformed E. coli with two plasmids. One plasmid expresses a derivative of MBP−76EspP containing a single cysteine substitution at one of 12 positions in the passenger that we predicted to be trapped inside the translocation (Figure 1A). The second plasmid expresses the BAM containing a His-tagged copy of BamA with an N427C substitution in β1 (Figure 1C). We chose N427C because we have previously shown that BamAβ1 forms a stable interface with MBP−76EspPβ12 during the hybrid-barrel intermediate stage and that BamAN427C forms a disulfide bond with MBP−76EspPR1297C. The data in that study, together with solved structures of the BAM in the apo state and folded EspP (Barnard et al., 2007, 2012; Gu et al., 2016; Han et al., 2016; Iadanza et al., 2016, 2020), demonstrated that both residues are oriented towards the lumen of the hybrid-barrel. Therefore, the placement of a cysteine at BamAN427 would allow us to determine if the traversing passenger is located close to BamAβ1 inside the hybrid-barrel using disulfide-crosslinking. After the MBP−76EspP and BamA cysteine mutants were expressed, bacteria were mock-treated or treated with the thiol-specific oxidizer 4,4’-dipyridyl disulfide (4-DPS). Disulfide-bonded MBP−76EspP•BamA species were then identified by immunoblots probed simultaneously with anti-StrepII (detects the N-terminus of MBP−76EspP) and anti-BamAC (detects a C-terminal epitope of BamA). Consistent with our hypothesis, upon chemical oxidation we detected disulfide-bond formation between BamAN427C and multiple cysteine positions in MBP−76EspP indicating that the passenger traverses the OM through a channel located near the luminal side of BamAβ1 (Figure 2A).
Figure 2: The autotransporter passenger secretion channel is located within the BamA lumen.

(A) E. coli BL21(DE3) expressing HisBamAN427CBCDE and MBP−76EspP with cysteine substitutions at the indicated residues were mock treated (−) or treated with 4-DPS (+). Intermolecular disulfide-bonds (•) between cysteine pairs were detected by double-immunoblotting with antibodies against the N-terminus of MBP−76EspP (αStrepII, top) or the C-terminus of BamA (αBamAC, bottom). Right, example of detected HisBamAN427C•MBP−76EspPT984C magnified from boxed region of immunoblot (and αStrepII (cyan) and αBamAC (red) signals overlaid). Asterisk: non-specific side-reactions. (B) Disulfide-bond levels (mean ± SEM) between MBP−76EspP and HisBamA for all cysteine substitutions defined in Figure 1. Experiments (N=3) conducted as in A except that immunoblots were probed with αStrepII only. Statistical tests are in Table S1. (C) Model of the autotransporter secretion channel based on disulfide-mapping. ‘1’ and ‘2’ portray the dynamicity of the passenger (orange) as it traverses the OM through the luminal space within the BamA-EspP hybrid-barrel. Some passenger residue positions used in A and B are depicted (orange/blue circles). EspP α-helix residue N1026 (purple circle) is also shown. BamA β1 (red), β2 (black), and β3 (green) are shown with luminal residues N427 (red circle), G443 (gray circle), and G457 (green circle), respectively. MBP prevents MBP−76EspP residues close to S953 from entry into the channel. (D) Surface-view cross-section of C. BamA β16 residues N805 (tan circle), I806 (yellow circle), and T809 (white circle), and EspP β1 (cyan square) are shown.
To expand on these initial findings, we sought to generate a detailed quantitative map of the secretion channel. To this end we constructed additional strains expressing each of the 12 MBP−76EspP passenger cysteine substitution mutants alongside BAM containing a BamA cysteine substitution at G443, V444, G457, N805, I806, or T809. Based on the solved structures, substitutions at G443 and G457 (located in β2 and β3, respectively) are expected to orient towards the β-barrel lumen (at a similar membrane depth as N427), whereas V444 points out into the lipid bilayer (Figure 1C). BamA I806 in β16 is adjacent to N427, whereas N805 is oriented in the opposite direction and is somewhat obstructed by BamA loop 6 (L6) (Figure 1C). The location of T809 is unresolved in current structures due to the dynamicity generated by a ‘kink’ at G807 (Lundquist et al., 2018). All 84 cysteine-pair expressing strains were 4-DPS treated and the levels of disulfide-bonding were quantified from immunoblots probed with anti-StrepII (Figure 2B, red circles; Table S1). Note that MBP−76EspP•BamA crosslinks were initially identified by simultaneously probing blots with antibodies against the N- or C-termini of both MBP−76EspP and BamA (Figures S1 and S2).
4-DPS treatment resulted in high levels of disulfide-bonding between the passenger and BamAN427C, BamAG443C, and BamAG457C (Figure 2B, red circles). In contrast, only very low levels of disulfide-bonding with BamAV444C were observed, demonstrating that the passenger traverses a channel that is on the luminal side of BamAβ1/β2/β3 (Figures 2B–D). Overall disulfide-bond levels between BamAβ1/β2/β3 and the passenger were lowest for BamAG457C and highest for BamAN427C (Figure 2B, red circles; Figure S3), showing that the passenger is located closest to the luminal side of BamAβ1 but can also be observed deeper inside the BamA β-barrel lumen during the earliest stages of translocation (Figures 2C and 2D). The passenger also formed high levels of disulfide-bonds with BamAI806C and BamAT809C (similar to the gross levels observed with BamAN427C) but formed significantly lower levels of disulfide-bonds with BamAN805C (oriented to the cell surface and obstructed by L6), especially when the cysteines were positioned near the N-terminus of the passenger (predicted to be close to, or within, the periplasm). These results show that the passenger traverses a space closely bordered by C-terminal residues of BamAβ16. Furthermore, while the overall level of disulfide-crosslinking between the passenger and BamAN427C or BamAI806C was similar, we observed an intriguing difference in the crosslinking pattern at the level of specific cysteine-pairs (Figure 2B, red circles, e.g.: MBP−76EspPK956C•BamAN427C vs. MBP−76EspPK956C•BamAI806C). The data suggest that during passenger translocation, BamA residues N427 and I806 are not as close to each other as they are in the apo-state (Figure 1C). This notion is not only consistent with previous observations that strands β1 and β16 must open laterally for BamA to catalyze OMP assembly through a hybrid-barrel state (Doyle and Bernstein, 2019; Noinaj et al., 2014; Tomasek et al., 2020), but also with a new model that explains how a space is created that can facilitate the translocation of extended polypeptides such as an autotransporter passenger (Figures 2C and 2D).
The extensive disulfide-bonding that we observed across locations in the passenger indicates that MBP−76EspP existed in multiple intermediate states that likely represent different stages of translocation at the moment of crosslinking. The significantly higher level of disulfide-bonding closer to the C-terminus of the passenger, regardless of the location of the cysteine in BamA (Figure 2B, red circles), indicates that the assembly of the majority of the MBP−76EspP molecules was arrested during the earliest phase of passenger translocation. It is also striking that at certain passenger positions there were discrete jumps in the efficiency of crosslinking. This multimodal pattern was particularly apparent for crosslinking with BamAG457C and BamAT809C but was also observed for BamAN427C and BamAG443C. The data are consistent with evidence that the sequential folding of C-terminal passenger β-strands at the cell surface (Figure 2C, transition between states ‘1’ to ‘2’) provides the energy to drive passenger translocation across the OM (Drobnak et al., 2015; Junker et al., 2006, 2009; Peterson et al., 2010; Yuan et al., 2018).
An unexpected finding in these experiments was that high levels of MBP−76EspP•BamA disulfide-bonds formed without the aid of a chemical oxidizer. This was observed for cysteines positioned near the middle of the MBP−76EspP passenger with BamAN427C (highest), BamAG443C, BamAI806C, and BamAT809C (Figure 2B, blue squares). Because the crosslinking was too site-specific to be a result of spontaneous oxidation, we wondered whether DsbA, the ~21kDa periplasmic enzyme that catalyzes disulfide-oxidation (Landeta et al., 2018), might be responsible. To test this hypothesis, we repeated the disulfide-bonding experiments with the cysteine-pairs MBP−76EspPS973C/BamAN427C or MBP−76EspPS966C/BamAT809C expressed in isogenic wild-type or dsbA− strains. Although significant levels of MBP−76EspPS973C•BamAN427C and MBP−76EspPS966C•BamAT809C crosslinks were observed even without 4-DPS in the wild-type strain, disulfide-bonds did not form without 4-DPS treatment in the dsbA− strain expressing MBP−76EspPS973C/BamAN427C or MBP−76EspPS966C/BamAT809C, (Figure S4A). This shows that DsbA is required for the formation of these disulfide-bonds in the absence of 4-DPS. While DsbA function is not the focus of this study, given the known steps of disulfide formation by DsbA (Kadokura and Beckwith, 2009), we propose that DsbA can form mixed-disulfide intermediates with accessible parts of the unfolded passenger (e.g., S973C) in the periplasm (Figure S4B, left). Upon movement of the passenger into the channel, the disulfide is subsequently transferred to form a disulfide bond with BamA (Figure S4B, right). This model is consistent with 4-DPS-dependent crosslinking data (Figure 2B) that suggest that C-terminal portions of the passenger (i.e. T984C) are embedded inside BamA early when MBP−76EspP assembly is arrested and are therefore inaccessible to DsbA. Furthermore, recently solved structures of the ~14kDa lipoprotein RcsF bound deep inside the BamA β-barrel (Rodriguez-Alonso et al., 2020) shows how small proteins (e.g. DsbA) can access the lumen of BamA from the periplasm during assembly reactions.
Because of its likely role in the initiation of passenger translocation, we also investigated the location of the autotransporter α-helix during the hybrid-barrel state. Based on evidence that the α-helix is placed within the autotransporter β-barrel at an early stage of assembly (Ieva et al., 2008), we predicted that it would be located within the hybrid-barrel in close proximity to the BamA lateral opening (Figure 2C, 2D). To test this idea, we constructed strains expressing BamAN427C, BamAG781C, BamAN805C, or BamAI806C alongside cysteine mutants MBP−76EspPN1026C (α-helix) or MBP−76EspPD1031C (C-terminal to α-helix) (Figures 3A and 3B). Based on previous structural analysis (Barnard et al., 2007, 2012), we predicted that N1026C and D1031C would be accessible for disulfide-bonding due to the open conformation of the hybrid-barrel state (Figure 2D). Upon 4-DPS treatment and doubly probing immunoblotted samples with anti-His (to detect the N-terminus of BamA) and an antiserum against a C-terminal epitope of EspP (anti-EspPβC), crosslinks were observed between all screened positions (Figure 3C, 3D). These results show that, during passenger translocation, the α-helix is embedded inside the hybrid-barrel in close proximity to BamA strands β1/15/16 (Figures 2C and 2D). While crosslinking between MBP−76EspPN1026C and BamAN805C or BamAI806C was similar (Figure 3C), MBP−76EspPD1031C formed disulfide-bonds less efficiently with BamAN805C than with BamAI806C (Figure 3D). This observation is consistent with the predicted location of EspPD1031 (close to the periplasm) and the orientation of BamAN805 (towards the cell surface and L6). Finally, the relatively low disulfide-bonding between MBP−76EspPN1026C or MBP−76EspPD1031C and BamAN427C is consistent with a model in which the passenger domain is secreted through the lumen of BamA near β1 and thereby obstructs interactions with the α-helix.
Figure 3: The autotransporter α-helix interacts with the BamA β-barrel during passenger translocation.

(A) Structure of BamA (as in Figure 1C) oriented to indicate residues investigated for proximity to the EspP α-helix and to show the interface of BamA β1 with β15/16. (B) Crystal structure of the EspP β-barrel pre-cleavage state (PDB: 3slj) is shown highlighting the α-helix (black), β1 (cyan), and β12 (green). Inset, zoom of membrane-spanning section of the β-barrel. Near side of the β-barrel is cut away to show the α-helix residue N1026 (purple) and α-helix flanking residue D1031 (blue) that were investigated for proximity to the BamA β-barrel. N1026 and D1031 are buried within the folded β-barrel and are oriented towards the opposite side of the lumen relative to β1. (C, D) E. coli BL21(DE3) expressing either MBP−76EspPN1026C (C) or MBP−76EspPD1031C (D), and either HisBamAN427CBCDE, HisBamAG781CBCDE, HisBamAN805CBCDE, or HisBamAI806CBCDE, were treated as in Figures 2A and 2B (N = 2). Intermolecular disulfide-bonds were detected by double-immunoblotting with antibodies against the N-terminus of HisBamA (αHis) (left) and the C-terminus of EspP (αEspPβC) (right). Asterisk: non-specific side-reactions.
A conserved tryptophan in autotransporter β-barrels primes passenger secretion
Recently, we serendipitously observed that the mutation of EspP residues G1040 or W1042 to cysteine abolishes passenger translocation (Doyle and Bernstein, 2019). Sequence analysis shows that G1040 is only weakly conserved, whereas W1042 (which faces the lumen of EspPβ1) is among the most conserved of all autotransporter β-barrel residues (Figures 4A and 4B). We sought to further investigate the importance of these residues in autotransporter assembly using our new insights into the nature of the passenger secretion channel. To that end, we introduced G1040A, W1042A, or W1042F substitutions into EspPΔ1, an EspP derivative that contains ~90 native passenger residues N-terminal to the α-helix (Szabady et al., 2005) (Figure 4C), and expressed these in E. coli. As mentioned, the EspP passenger is cleaved from the cell surface leaving a ‘mature’ β-barrel in the OM only when all other assembly steps are complete. By probing samples with anti-EspPβC, we found that both G1040A and W1042F substitutions had no effect on the accumulation of the mature β-barrel (Figure 4C, Table S1). In contrast, the W1042A mutation almost completely abolished the appearance of the mature β-barrel and therefore must interfere with a required stage of autotransporter assembly. Notably, constitutive expression of EspPΔ1W1042A led to a severe growth defect that might be due to the toxic accumulation of the mutant protein on BAM (Figure S5A). There was also no accumulation of the mature β-barrel when W1042A was introduced into full length EspP, and even the W1042F substitution caused a modest reduction in assembly (Figures S5B–S5D, Table S1). The results suggest that the conserved tryptophan plays an even more critical role in the assembly of large native passengers and that it might play a specific role in passenger translocation. Consistent with our hypothesis, we found that the W1042A substitution did not affect the assembly of EspPΔ5, a version of EspP that contains only the α-helix and β-barrel (Figure 4D).
Figure 4: A highly conserved tryptophan is critical for passenger translocation initiation.

(A) Hidden Markov model of autotransporter β-barrel sequence conservation in strand β1 and residues G1040 (circle) and W1042 (star) within EspPβ1 (cyan). (B) Cross-section of the EspP β-barrel. Magnified slice of the area in the dashed box on the EspP β-barrel (left) shows the location of residue W1042 (pink) and its importance in an array of potential interactions (dashed lines) including with α-helix residues R1028 (red) and M1029 (blue), and β4 residue Y1108 (teal). W1042 is also oriented towards G1066 (yellow) in β2 forming a ‘mortise-tenon joint’. Gray mesh; solvent-excluded protein surface. (C) Cell extracts from E. coli AD202 expressing wild-type (WT) or mutant versions of ‘EspPΔ1’ (EspP909–1300) were probed by immunoblotting with αEspPβC (left) and quantified (right) to monitor the conversion of proEspPΔ1 to a mature cleaved β-barrel (line at median, N = 3). Statistical tests are in Table S1, P < 0.0001 (****). (D) As in C except that strains expressed ‘EspPΔ5’ (EspP998–1300) or EspPΔ5W1042A. Two-tailed t-test: P = 0.0078 (**), N = 3. (E) E. coli BL21(DE3) expressing MBP−76EspP, MBP−76EspPW1042A, or MBP−76EspPW1042F and HisBamABCDE were mock treated (−) or treated with PK (+), and immunoblots were probed with αStrepII and αEspPβC to monitor surface exposure of the passenger loop (N = 3). (F) As in E except that strains expressed MBP−76EspP, MBP−76EspPR1028A, MBP−76EspPM1029A, or MBP−76EspPY1108F. In C-F, non-specific bands (~) and degradation products (#) are denoted. See also Figure S5.
To elucidate the function of W1042, we constructed strains expressing MBP−76EspP, MBP−76EspPW1042A, or MBP−76EspPW1042F, and bacteria were subsequently treated with proteinase K (PK) to assess the level of passenger surface exposure. Because a small loop of the passenger is translocated when the assembly of MBP−76EspP arrests (Figure 1A), the majority of the fusion was digested into N- and C-terminal fragments (Figure 4E) as observed previously (Doyle and Bernstein, 2019). Strikingly, however, MBP−76EspPW1042A remained resistant to PK digestion (and MBP−76EspPW1042F was only partially susceptible) indicating that the initiation of passenger translocation was severely disrupted. These results, along with clues from the folded structure of the EspP β-barrel that indicate W1042 may be required for secondary interactions with luminal β-barrel residues and the α-helix (Figure 4B), led us to hypothesize that W1042 acts as a folding nucleus to position the passenger and α-helix during the initiation of translocation. Accordingly, we predicted that the mutation of W1042-interacting residues such as R1028 or M1029 (α-helix), or its hydrogen-bond partner Y1108 (strand β4), may phenocopy the W1042A/F substitutions. Consistent with our hypothesis, MBP−76EspPR1028A (but not MBP−76EspPM1029A) was completely resistant to PK digestion (Figure 4F). Because MBP−76EspPY1108F was also only partially susceptible to PK digestion, folding assisted by hydrogen-bonding between W1042 and Y1108 is also likely to be required for the initiation of passenger translocation (Figure 4F). As expected, the introduction of the Y1108F mutation into EspPΔ5 had no effect on the accumulation of mature β-barrel (Figure S5E, Table S1). Although the R1028A mutation inhibited accumulation of the mature β-barrel as previously observed (Barnard et al., 2012; Dautin and Bernstein, 2011) (Figure S5E), all of the EspPΔ5 mutants that we tested (W1042A, R1028A, M1029A, and Y1108F) were resistant to denaturation during cold-SDS-PAGE (Figure S5F) and therefore folded into a native β-barrel structure in the absence of a passenger domain.
To determine if W1042 is required to correctly position the α-helix inside the hybrid-barrel, we introduced the W1042A mutation into MBP−76EspPN1026C. While the mutation did not change the level of disulfide-bonding with BamAG781C, it significantly reduced crosslinking between N1026C and BamAI806C (Figures 5A and 5B; Table S1) by presumably causing a change in conformation or location of the α-helix within the hybrid-barrel channel. We repeated this experiment with MBP−76EspPD1031C but saw no change in crosslinking due to the W1042A mutation, indicating that the effect is limited to the α-helix further embedded inside the β-barrel (Figure S5G). To test if W1042 is also required to correctly position the passenger in the channel, we introduced the W1042A substitution into MBP−76EspPS966C and MBP−76EspPT984C, two derivatives that form disulfide-bonds with BamAN427C at high levels and represent distal positions within the passenger. Subsequent assays revealed a loss or severe reduction of MBP−76EspPS966C/W1042A•BamAN427C and MBP−76EspPT984C/W1042A•BamAN427C disulfide-crosslinking (Figure 5C). To rule out the possibility that the W1042A substitution affects hybrid-barrel formation, we also introduced the mutation into MBP−76EspPS1299C, a derivative that crosslinks with BamAS425C at high levels when the hybrid-barrel forms during assembly (Doyle and Bernstein, 2019) (Figures 5D and 5E). Substantial disulfide-bonding was observed between MBP−76EspPS1299C and BamAS425C regardless of the presence of W1042A (Figure 5D) confirming that the mutation did not alter the interface between BamAβ1 and MBP−76EspPβ12. Overall, the data strongly suggest that the conserved tryptophan, through its interaction with residues within the β-barrel lumen and the α-helix, is required to position the α-helix and passenger C-terminus within the hybrid-barrel to prime secretion. In this model, W1042A disrupts the conformation of the α-helix which has compounding effects on the positioning of the passenger inside BamA and the initiation of translocation (Figure 5E, right).
Figure 5: The conserved tryptophan is required to position the autotransporter passenger inside the BamA β-barrel.

(A) Experiments as in Figure 2A except strains expressed either MBP−76EspPN1026C or MBP−76EspPN1026C/W1042A (cysteine positioned in the α-helix) and either HisBamAG781CBCDE or HisBamAI806CBCDE and disulfide-bonded proteins were detected using αHis (left) and αEspPβC (right). Asterisk: non-specific side-reactions. (B) Experiments conducted as in A except immunoblots probed with αStrepII and disulfide-bonded MBP−76EspP⦁HisBamA was quantified (line at median, N = 3). Statistical tests are in Table S1, P < 0.001 (***). (C) As in A except that strains expressed HisBamAN427CBCDE and MBP−76EspP passenger cysteine substitutions (S966C or T984C) with or without the W1042A substitution. Immunoblots were doubly probed with αBamAC (left) and αStrepII (right) (N = 2). (D) As in C except that strains expressed MBP−76EspPS1299C (with or without the W1042A substitution) and HisBamAS425C (cysteines are in close proximity during hybrid-barrel state). (E) Model showing the importance of the conserved β1 tryptophan in passenger secretion. W1042A disrupts the positioning/folding of the α-helix into the BamA-EspP hybrid-barrel channel (and, in turn, the positioning of the passenger) such that no detectible translocation occurs. Labelling as in Figure 2C.
Channel occupancy inhibits completion of autotransporter β-barrel folding
Having established that the passenger is secreted through a BamA-autotransporter hybrid-barrel, we hypothesized that the occupancy of the traversing passenger within the space between BamAβ1 and β16 may inhibit premature release of the autotransporter β-barrel to ensure completion of passenger translocation. To test this idea, we exploited the finding that the assembly of arrested MBP−76EspP can be restarted upon the addition of PK (Doyle and Bernstein, 2019). When PK digests the surface exposed loop of MBP−76EspP, an N-terminal periplasmic fragment is released along with a C-terminal fragment that can rapidly assemble into a mature β-barrel (Figure 6A, left). To enable controlled tethering of the passenger inside the channel (Figure 6A, right), we expressed a pair of cysteine mutants (MBP−76EspPT984C and BamAN427C) that form the highest levels of disulfide-bonds (Figure 2B). Cultures were divided and either mock or 4-DPS treated before incubation with PK for up to 15 min. To track fragments of MBP−76EspPT984C•BamAN427C, samples were double-immunoblotted using anti-StrepII and an antiserum generated against full-length BamA (anti-BamA) (Figure 6B). The same samples were also probed with anti-EspPβC to allow the tracking of levels of C-terminal EspP PK fragments and mature cleaved β-barrel (Figure 6C, 6D, Table S1).
Figure 6: Channel occupancy by a translocating passenger inhibits completion of β-barrel assembly.

(A) Summary of experimental design and results in B-D. Left, the assembly of MBP−76EspP is arrested due to inhibition of passenger translocation by MBP. Assembly can be restarted by addition of PK, which digests the exposed passenger loop and allows rapid secretion of the C-terminus of the passenger and autocatalytic release of a mature β-barrel. Right, the passenger was additionally tethered to the secretion channel via a disulfide-bond. PK treatment resulted in a pool of C-terminal EspP fragments and slowed conversion into a mature β-barrel. (B, C) E. coli BL21(DE3) expressing HisBamAN427CBCDE and MBP−76EspPT984C were treated as in Figure 2A and subsequently treated with PK for up to 15 min. Disulfide-bonded species, fragment formation, and EspP β-barrel maturation, were detected by double-immunoblot using antisera against full length BamA (αBamA, left) and StrepII (right) (B), or using only αEspPβC (C). Side-reactions (*), non-specific bands (~), endogenous BamA degradation fragments (#), and BamA PK fragments (<) are denoted. (D) Quantified levels (mean ± SEM) of C-terminal EspP PK fragments (right) or mature β-barrel (far right) from experiments in C (N = 3). Statistical tests are in Table S1, P <0.05 (*), P < 0.01 (**), P < 0.001 (***). See also Figure S6.
As expected, in mock treated samples nearly all MBP−76EspP was rapidly digested within 30 sec to release an N-terminal MBP-containing ~50 kDa fragment that is degraded by endogenous periplasmic proteases (Figure 6B, compare lanes 1 and 3, anti-StrepII) and a corresponding C-terminal ~30 kDa fragment that was rapidly converted to a mature cleaved β-barrel (Figure 6C, compare lanes 1 and 3). Both in the absence and presence of 4-DPS, free BamA was also digested by PK relatively slowly to generate an N-terminal ~70 kDa fragment (Figure 6B lanes 3–10, anti-BamA, Figure S6). The size of this fragment is consistent with previous observations that BamA L6 is susceptible to PK digestion (Doyle and Bernstein, 2019). In 4-DPS treated samples the majority of the MBP−76EspPT984C•BamAN427C disulfide-bonded species was digested within 30 sec to yield a band corresponding to the N-terminal MBP-containing fragment still tethered to BamAβ1 (Figure 6A, 6B compare lanes 2 and 7). A slow accumulation of a band corresponding to the crosslinked form of N-terminal PK fragments of both BamA and MBP−76EspP was also observed (Figure 6B lanes 7–10, Figure S6). Compared to mock-treated samples (Figure 6A left, 6C lanes 3–6), tethering of the passenger to BamAβ1 (Figure 6A right, 6C lanes 7–10) caused a persistence of C-terminal EspP PK fragments (Figure 6A) at significantly higher levels across all time points, as well as a decrease in the levels of mature β-barrel (Figure 6D). Given that only ~40% of MBP−76EspPT984C molecules were tethered to BamAN427C before addition of PK (Figure 2B) the difference in remaining C-terminal EspP PK fragment levels was striking (Figure 6D) and suggests that the tethered passenger strongly inhibits the assembly of the separated C-terminal fragment into the mature β-barrel form (Figure 6A, right).
DISCUSSION
Although autotransporters were identified more than 30 years ago (Pohlner et al., 1987), this study describes the first direct analysis of the location of the passenger secretion channel. We found that the BamA β-barrel lumen is the channel for autotransporter passenger translocation and that the α-helix is positioned within the hybrid-barrel where it interacts with the lateral opening of the BamA β-barrel. The results rule out the alternative possibility that the passenger is translocated through the lumen of the autotransporter β-barrel while it is held open by BamA. The traversing passenger is located relatively deep inside the BamA β-barrel lumen (close to BamAβ3) during early stages of translocation but closer to the interface between the two β-barrels (i.e., BamAβ1) at later stages. We also obtained evidence that the passenger segment that traverses the BamA β-barrel inhibits entry into the final stages of autotransporter β-barrel assembly. Perhaps the location of the passenger segment close to BamAβ1 prevents the premature collapse of the hybrid-barrel by physically obstructing the process of β-strand exchange (the proposed mechanism of release of new β-barrels from BamA (Doyle and Bernstein, 2019; Tomasek et al., 2020), thus ensuring that the passenger can be fully translocated.
Our results show that autotransporters can regulate the initiation of passenger translocation. We found that an extremely conserved tryptophan residue in EspPβ1 is required for the correct positioning of the α-helix and a C-terminal passenger segment inside BamA to initiate translocation, but not for the assembly of the β-barrel itself. Solved structures show that W1042 is located inside the fully folded EspP β-barrel and oriented towards G1066 in β2 (Barnard et al., 2012). This inter-strand interaction was previously described as a ‘mortise-tenon joint’ that provides thermal stability to the folded β-barrel (Leyton et al., 2014). Substitutions of G1066 in EspP (or the equivalent G1061 in the related autotransporter Pet), however, cause only moderate assembly defects that slightly delay the initiation of passenger translocation (Leyton et al., 2014; Pavlova et al., 2013). W1042 participates in numerous secondary interactions with inward-facing residues across the inner wall of the β-barrel including Y1108 in β4, and with residues in the α-helical segment closest to the periplasm including R1028. Interestingly, Y1108 also forms a hydrogen-bond with W1042 and is a conserved residue in β3 that forms another mortise-tenon joint with G1081. Here we show that R1028A and Y1108F mutations disrupt passenger translocation initiation in a similar manner to W1042A (and to W1042F, which was designed to disrupt hydrogen-bonding with Y1108). Coupled with the extremely high conservation of W1042, these interactions suggest that the residue has a significantly more important role in autotransporter biogenesis than simply stabilizing β1 with β2. We speculate that the conserved tryptophan acts as an early folding nucleus that both structurally stabilizes multiple strands at the N-terminus of the autotransporter β-barrel and affixes the C-terminal segment of the α-helix to the inner wall of the β-barrel in a conformation that promotes the initiation of passenger translocation. This scenario is supported by previous observations that the α-helix is positioned within the β-barrel at a very early stage of autotransporter assembly before the β-barrel is completely assembled into the OM, and that α-helical positioning is maintained even when large insertion mutations are introduced N- or C-terminal to residues such as R1028 that make contacts with W1042 (Ieva et al., 2008).
We also found that, in addition to catalyzing the assembly and integration of OM β-barrel proteins, BamA can function as a remarkable polypeptide-translocase. The traversal of the BamA β-barrel lumen is consistent with recent molecular dynamics experiments that show that much less force is required to pull a polypeptide through BamA than EspP (Ryoo et al., 2020). Furthermore, we recently found that crosslinking between cysteines positioned in BamAβ16 and EspPβ1 is unexpectedly low (Doyle and Bernstein, 2019) (e.g.: ~5% for BamAT809C-MBP−76EspPG1040C), but show here that crosslinking between BamAT809C and cysteines positioned in the passenger is exceptionally high (e.g.: ~50% for BamAT809C-MBP−76EspPT984C). These observations are well explained if the traversing passenger segment and α-helix occupies a space between BamAβ16 and EspPβ1 that physically obstructs their interaction. Interestingly, the residues of BamAβ16 C-terminal to G807 (including T809) are known to be extremely dynamic (Doerner and Sousa, 2017; Lundquist et al., 2018), yet the function of this dynamicity in β-barrel assembly remains unknown. Given the proximity of these residues to the traversing passenger, it is possible that autotransporters take advantage of the intrinsic dynamicity of BamAβ16 to promote the movement of polypeptide segments through the BamA β-barrel lumen.
Since we have shown that the lumen of BamA can promote the translocation of large autotransporter passengers, it seems reasonable that it plays a similar role in the translocation of hydrophilic surface loops and soluble domains present in other types of OMPs. Although the majority of loops are small and may traverse the OM without a significant energetic penalty, some OMPs have large and complex folded loops (Coppens et al., 2018; Lauber et al., 2018) that may require the BamA-translocase function that we have described here. Perhaps this export function also allows BamA to translocate lipoproteins (e.g. RcsF) when a hybrid-barrel state is formed upon engagement of client OMPs. Since BamA is ubiquitous among Gram-negative bacteria and is thought to represent the archetypical member of the Omp85-superfamily (Heinz and Lithgow, 2014), perhaps homologs such as mitochondrial Sam50 might possess similar polypeptide-translocase activities. This may explain how heterologously expressed autotransporters assemble properly into the mitochondrial OM and undergo passenger translocation (Muller et al., 2011; Ulrich et al., 2014). Our results also establish a functional link to other members of the Omp85-superfamily that promote protein translocation but not membrane integration reactions (e.g., TPS transporters, the chloroplast protein Toc75). Indeed, secreted proteins of the TPS pathway are like the passengers of autotransporters in that they are also largely β-helical (Guerin et al., 2017). It is intriguing to speculate that, instead of evolving a new function, specialized Omp85 family members involved in protein translocation simply preserved a pre-existing secretion function that was associated with a BamA-like ancestor. Finally, our findings have ramifications for the activity of a newly identified class of antibiotics that target BamA. These inhibitors are thought to stabilize the closed form of the BamA β-barrel (Imai et al., 2019), presumably preventing the formation of hybrid-barrel intermediates for OMP integration. Having shown that BamA serves to translocate autotransporter virulence factors, we predict that BamA inhibitors would also have a synergistic anti-virulence effect in addition to their demonstrated bactericidal activity.
Limitations
Although our placement of cysteines in the BamA β-barrel was sufficiently extensive to support the conclusion that the BamA lumen forms the passenger translocation channel, we cannot determine if the passenger infrequently occupies even deeper spaces within BamA. We also used only one model autotransporter fusion construct that we previously showed forms a stable assembly intermediate and therefore allows us to analyze secretion in vivo. While we expect our secretion model to be generalizable based on the conservation of BamA, future studies should focus on other autotransporters and species other than E. coli to confirm this assumption. Finally, we could not investigate the effect of increasing the length of the passenger because longer fusions are targeted by endogenous proteases.
STAR METHODS
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Harris D. Bernstein (harris_bernstein@nih.gov).
Materials Availability
Strains and plasmids generated in this study are available upon request to the Lead Contact.
Data and Code Availability
Original data for figures in the paper is available upon request to the Lead Contact or at Mendeley Data (http://dx.doi.org/10.17632/3cy3y7dnmk.1)
Experimental model and subject details
The E. coli B strain BL21(DE3) (Invitrogen catalog number C600003) was used for most experiments. E. coli K-12 strains AD202 (MC4100ompT::KmR) (Akiyama and Ito, 1990) and RI2 (AD202dsbA::CmR) (Ieva et al., 2008) were also used for experiments involving EspPΔ1 or EspPΔ5 (and mutants thereof), and experiments investigating the effect of DsbA on disulfide-bond formation during assembly of client proteins by the BAM. E. coli K-12 strain XL1-Blue (Agilent catalog number 200236) was routinely used for cloning and mutagenesis. For all experiments, strains were grown in Lysogeny broth (LB) (Miller formulation) supplemented with ampicillin (100 μg mL−1) and trimethoprim (50 μg mL−1) as necessary. For general strain propagation and cloning, cultures were grown at 37 °C. For experiments, cultures were grown as indicated in Method Details.
Method Details
Plasmid construction
The plasmids that were used in this study are listed in Table S2. Oligonucleotide primers and linear dsDNA fragments that were used in plasmid construction are listed in Table S3. To construct a plasmid that encodes EspPΔ1 under the tight control of an L-rhamnose inducible PrhaB promoter (pMTD405), pRha-MBP-EspPΔ1 was amplified by PCR with primers mtd96/mtd127 and the resulting fragment was mixed with mtd126 for Gibson assembly. MBP−76EspP was also expressed from a PrhaB controlled plasmid and HisBamABCDE was expressed form an IPTG inducible plasmid (Doyle and Bernstein, 2019). Substitution mutations were introduced into plasmids using the QuikChange II Site-directed Mutagenesis Kit (Agilent).
Disulfide-bond formation assay
To observe site-specific intermolecular protein-protein interactions, in vivo disulfide-bond formation assays were conducted essentially as described (Doyle and Bernstein, 2019). Strains doubly transformed with appropriate plasmids were grown from a single colony in 10 mL of LB at 25 °C with orbital shaking at 250 rpm overnight. Overnight cultures were pelleted (3000 × g. 5 min, 4 °C), washed with 10 mL LB, and resuspended with 10 mL LB before seeding 20 mL LB subcultures at OD600 = 0.05 in Erlenmeyer flasks with sealed lids. After subcultures were grown for 4 h (25 °C, 250 rpm) to OD600 ~ 0.4 – 0.6, a final concentration of 0.4 mM IPTG was added to induce expression of BAM for 1 h, and a final concentration of 0.2% L-rhamnose was then added to induce expression of MBP−76EspP for 45 min. Samples of the subcultures (1 mL) were aliquoted into 1.5 mL tubes on ice, pelleted (10,000 × g, 2 min, 4 °C), and resuspended in 1 mL ice-cold phosphate buffered saline (PBS). Bacteria were incubated with 4,4’-dipyridyl disulfide (4-DPS), a thiol-specific disulfide oxidizing catalyst, at a concentration of 0.2 mM (or an equivalent volume of ethanol for mock treatment controls), mixed, and incubated on ice for 30 min. Bacteria were then pelleted (10,000 × g, 2 min, 4 °C) and resuspended in 0.5 mL ice-cold PBS. Bacterial lysis and protein precipitation was conducted by incubating samples with final concentrations of 10% (v/v) trichloroacetic acid (TCA) and 4 mM phenylmethanesulfonyl fluoride (PMSF) for 10 min on ice. Precipitated proteins were pelleted (20,817 × g, 10 min, 4 °C), washed with 0.6 mL of ice-cold acetone, pelleted again, and air dried at 37 °C for 15 min.
Steady-state autotransporter assembly assay
To assess the ability of EspP derivatives to assemble and to release a mature cleaved β-barrel, strains transformed with appropriate plasmids were grown from a single colony in 10 mL LB at 25 °C with orbital shaking at 250 rpm overnight. Overnight cultures were pelleted (3000 × g. 5 min, 4 °C), washed with 10 mL LB, and resuspended with 10 mL LB before seeding 10 mL LB subcultures at OD600 = 0.05 in 50 mL tubes. Subcultures were grown for 4 h (25 °C, 250 rpm) to OD600 ~ 0.4 – 0.6 and EspP expression was induced for 30 min by the addition of 0.2% L-rhamnose (final concentration). 1 mL samples of the subcultures were then lysed, and proteins TCA precipitated, as described above.
Plate growth assay
Bacterial strains transformed with appropriate plasmids were streaked onto LB agar, or LB agar containing 0.2% L-rhamnose, and incubated at 37 °C for 16 h. Plates were photographed using a Gel Doc EZ imager (Bio-Rad).
Cell surface proteolysis
Bacteria expressing full length EspP or MBP−76EspP were grown as described in the steady-state autotransporter assembly assay or the disulfide-bond formation assay respectively. To monitor the level of cell surface exposure of passenger domains, Proteinase K (PK) digestions on live bacteria were conducted essentially as described (Doyle and Bernstein, 2019). 1 mL samples of induced bacterial subcultures were aliquoted into 1.5 mL tubes on ice. Bacteria were pelleted (10,000 × g, 2 min, 4 °C), resuspended in 0.5 mL ice-cold PBS, and incubated on ice for 30 min with 200 μg mL−1 (PK) or equivalent volume of PK buffer (5 mM CaCl2, 50 mM Tris-HCL pH 8) for mock-treated controls. The samples were then lysed, and proteins TCA precipitated, as described above.
Disulfide-bond formation and proteolytic arrest-release assay
To monitor the ability of MBP−76EspP to assemble after the introduction of an intermolecular disulfide-bond, site-specific disulfide-bonds were first generated as described as above. After incubation with 4-DPS (or ethanol), bacteria were pelleted (10,000 × g, 2 min, 4 °C), resuspended in 0.5 mL of pre-warmed PBS (25 °C). PK (200 μg mL−1) (or PK buffer for mock samples) was then added to the bacteria and incubated in a thermomixer at 25 °C with orbital shaking (350 rpm). After 0.5, 2, 5, or 15 min, bacteria were lysed and proteins were TCA precipitated as described above.
Heat-modifiable gel mobility-shift assay
To observe folded states of the EspP β-barrel, as described previously (Doyle and Bernstein, 2019), bacteria were grown as described in the steady-state autotransporter assembly assay. Bacteria were then resuspended in BugBuster Master Mix (EMD Millipore) containing EDTA-free SigmaFast protease inhibitor (Sigma-Aldrich) (volume, μL = OD600 × 100) and lysed on ice for 3 min. Aliquots of lysates were mixed with 2x SDS protein gel loading solution (Quality Biological) (3:1 ratio) to bring the final SDS concentration to 1%. Samples were either retained on ice or heated to 99 °C for 10 min. Proteins were then resolved by cold-SDS-PAGE (packing a gel tank in ice and running the gels at 4 °C) and transferred to nitrocellulose for immunoblotting as described below.
Western immunoblotting, imaging, and quantitation
Precipitated protein samples were resuspended in 2x SDS protein gel loading solution in a volume normalized to the final OD600 reading of the subculture from which the sample was taken (volume, μL = OD600 × 200) and heated at 99 °C for 15 min. Protein samples (5 μL) were separated by SDS-PAGE on 8–16% Tris-glycine gradient gels (Invitrogen) at room temperature for 1 h and 47 min at 150 V, and transferred to nitrocellulose membranes using iBlotII transfer devices (Life Technologies). For size markers, we routinely used Chameleon Duo Pre-stained Protein Ladder (Licor). Immunoblotting buffer [Odyssey Blocking Buffer (Licor) diluted at a 1:1 ratio with PBS and supplemented with 0.01% Tween-20] was used for all blocking and antibody/antiserum incubations, PBS supplemented with 0.01% Tween-20 (PBST) for initial washes, and PBS for final washes. Monoclonal mouse anti-StrepII and anti-His antibodies were obtained from Qiagen (catalog number 34850) and Genscript (catalog number A00186), respectively. Polyclonal rabbit antisera raised against peptides representing the N- or C-termini of the EspP β-barrel, or the C-terminus of BamA, and antisera against full length BamA, have been described previously (Doyle and Bernstein, 2019; Ieva and Bernstein, 2009; Pavlova et al., 2013; Szabady et al., 2005). Different combinations of antisera were chosen for double-blotting of disulfide-crosslinked samples to avoid probing of mutated epitopes. Infra-red Goat secondary antibodies that bind mouse Ig (anti-mouse 800CW IRDye, catalogue number 926–32210) or rabbit Ig (anti-rabbit 680LT IRDye, catalogue number 926–680210) were obtained from Licor. Membranes intended for quantitation of StrepII-tag detection were blocked overnight, incubated with anti-StrepII (1:5,000 dilution) for 1 h, washed (2 × 5min with PBST), incubated with goat anti-mouse 800CW (1:5,000 dilution) for 1 h, washed (3 × 5 min with PBST, 2 × 5 min with PBS), and air dried. Otherwise, membranes for single- or double-antibody immunoblots were incubated overnight with primary antibodies/antisera and 2 h with secondary antibodies. Dried membranes were imaged using an Amersham Typhoon 5 scanner (GE Healthcare) using maximum quality and resolution settings. All membranes for StrepII-tag detection and quantitation were scanned with a 785 nm laser, IRlong 825BP30 filter, and PMT set at 700V. Pixel intensities were measured using Fiji software (v2.0.0-rc-68/1.52g). Within-lane values were used to calculate percent disulfide-bond formation [(MBP−76EspP-HisBamA/(sum of MBP−76EspP signals)) × 100]. This approach to band intensity calculation was similarly applied to calculate the percentage of the protein that underwent proteolytic maturation.
Sequence and structure analysis
To generate a residue conservation sequence logo of the autotransporter β-barrel, sequence alignments from the SMART website (Letunic and Bork, 2018) for Hidden Markov model SM00869 were displayed using the Skylign webtool (Wheeler et al., 2014). Secondary interactions of EspP W1042 were analyzed with UCSF Chimera (Pettersen et al., 2004) (v1.13.1) using ‘FindHBond’ and ‘Find Clashes/Contacts’ tools with default settings. Protein Imager (Tomasello et al., 2020) was used to create a cartoon representation of the passenger for the graphical abstract.
Quantification and statistical analysis
All statistical analyses were conducted using GraphPad Prism (v8.4.0). Details of the number of experiments (N), definition of center, and variance measurements are noted in the figure legends. Details of statistical tests including (ANOVA and multiple comparisons) can be found in the Supplemental Information. Significant differences were defined as a P value <0.05.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Monoclonal mouse anti-StrepII | Qiagen | cat#34850 |
| Monoclonal mouse anti-His | Genescript | cat#A00186 |
| Polyclonal rabbit anti-EspP β-barrel (N-term peptide) | Doyle and Bernstein, 2019 | N/A |
| Polyclonal rabbit anti-EspP β-barrel (C-term peptide) | Szabady et al., 2005 | N/A |
| Polyclonal rabbit anti-BamA (C-term peptide) | Pavlova et al., 2013 | N/A |
| Polyclonal rabbit anti-BamA (full protein) | Ieva and Bernstein, 2009 | N/A |
| Goat anti-mouse Ig 800CW IRDye | Licor | cat#926-32210 |
| Goat anti-rabbit Ig 680LT IRDye | Licor | cat#926-680210 |
| Bacterial and Virus Strains | ||
| E. coli B strain BL21(DE3) | Invitrogen | cat#C600003 |
| E. coli K-12 strain AD202 | Akiyama and Ito, 1990 | N/A |
| E. coli K-12 strain RI2 | Ieva et al., 2008 | N/A |
| E. coli K-12 strain XL1-Blue | Agilent | cat#200236 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 4,4’-dipyridyl disulfide | Sigmaaldrich | cat#143057-5G |
| Proteinase K | Roche | cat#3115852001 |
| BugBuster Master Mix | EMD Milipore | cat#71456 |
| Deposited Data | ||
| Uncut immunoblots | This paper; Mendeley Data | http://dx.doi.org/10.17632/3cy3y7dnmk.1 |
| Experimental Models: Organisms/Strains | ||
| E. coli B strain BL21(DE3) | Invitrogen | cat#C600003 |
| E. coli K-12 strain AD202 | Akiyama and Ito, 1990 | N/A |
| E. coli K-12 strain RI2 | Ieva et al., 2008 | N/A |
| Oligonucleotides | ||
| See Table S8 in Supplementary Information | This paper | N/A |
| Recombinant DNA | ||
| See Table S7 in Supplementary Information | This paper | N/A |
| Software and Algorithms | ||
| UCSF Chimera | Pettersen et al., 2004 | https://www.cgl.ucsf.edu/chimera/ |
| Other | ||
| SMART website | Letunic and Bork, 2018 | http://smart.embl-heidelberg.de |
| Skylign webtool | Wheeler et al., 2014 | http://skylign.org |
HIGHLIGHTS.
The BamA β-barrel lumen can function as a polypeptide export channel.
Autotransporter passenger domains are secreted by BamA during a hybrid-barrel stage.
A conserved tryptophan in autotransporters primes passenger domain export.
Channel occupancy inhibits completion of autotransporter β-barrel folding.
ACKNOWLEDGEMENTS
We thank Thushani Nilaweera for providing helpful comments on the manuscript. This work was supported by the National Institute of Diabetes and Digestive and Kidney Diseases Intramural Research Program.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Akiyama Y, and Ito K (1990). SecY protein, a membrane-embedded secretion factor of E. coli, is cleaved by the ompT protease in vitro. Biochem Biophys Res Commun 167, 711–715. [DOI] [PubMed] [Google Scholar]
- Barnard TJ, Dautin N, Lukacik P, Bernstein HD, and Buchanan SK (2007). Autotransporter structure reveals intra-barrel cleavage followed by conformational changes. Nat Struct Mol Biol 14, 1214–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnard TJ, Gumbart J, Peterson JH, Noinaj N, Easley NC, Dautin N, Kuszak AJ, Tajkhorshid E, Bernstein HD, and Buchanan SK (2012). Molecular basis for the activation of a catalytic asparagine residue in a self-cleaving bacterial autotransporter. J Mol Biol 415, 128–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baud C, Guerin J, Petit E, Lesne E, Dupre E, Locht C, and Jacob-Dubuisson F (2014). Translocation path of a substrate protein through its Omp85 transporter. Nat Commun 5, 5271. [DOI] [PubMed] [Google Scholar]
- Celik N, Webb CT, Leyton DL, Holt KE, Heinz E, Gorrell R, Kwok T, Naderer T, Strugnell RA, Speed TP, et al. (2012). A bioinformatic strategy for the detection, classification and analysis of bacterial autotransporters. PLoS One 7, e43245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppens F, Castaldo G, Debraekeleer A, Subedi S, Moonens K, Lo A, and Remaut H (2018). Hop-family Helicobacter outer membrane adhesins form a novel class of Type 5-like secretion proteins with an interrupted beta-barrel domain. Mol Microbiol 110, 33–46. [DOI] [PubMed] [Google Scholar]
- Dautin N, Barnard TJ, Anderson DE, and Bernstein HD (2007). Cleavage of a bacterial autotransporter by an evolutionarily convergent autocatalytic mechanism. EMBO J 26, 1942–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dautin N, and Bernstein HD (2011). Residues in a conserved alpha-helical segment are required for cleavage but not secretion of an Escherichia coli serine protease autotransporter passenger domain. J Bacteriol 193, 3748–3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerner PA, and Sousa MC (2017). Extreme Dynamics in the BamA beta-Barrel Seam. Biochemistry 56, 3142–3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle MT, and Bernstein HD (2019). Bacterial outer membrane proteins assemble via asymmetric interactions with the BamA beta-barrel. Nat Commun 10, 3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drobnak I, Braselmann E, and Clark PL (2015). Multiple driving forces required for efficient secretion of autotransporter virulence proteins. J Biol Chem 290, 10104–10116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesan I, and Theg SM (2019). Structural considerations of folded protein import through the chloroplast TOC/TIC translocons. FEBS Lett 593, 565–572. [DOI] [PubMed] [Google Scholar]
- Gu Y, Li H, Dong H, Zeng Y, Zhang Z, Paterson NG, Stansfeld PJ, Wang Z, Zhang Y, Wang W, et al. (2016). Structural basis of outer membrane protein insertion by the BAM complex. Nature 531, 64–69. [DOI] [PubMed] [Google Scholar]
- Guerin J, Bigot S, Schneider R, Buchanan SK, and Jacob-Dubuisson F (2017). Two-Partner Secretion: Combining Efficiency and Simplicity in the Secretion of Large Proteins for Bacteria-Host and Bacteria-Bacteria Interactions. Front Cell Infect Microbiol 7, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L, Zheng J, Wang Y, Yang X, Liu Y, Sun C, Cao B, Zhou H, Ni D, Lou J, et al. (2016). Structure of the BAM complex and its implications for biogenesis of outer-membrane proteins. Nat Struct Mol Biol 23, 192–196. [DOI] [PubMed] [Google Scholar]
- Heinz E, and Lithgow T (2014). A comprehensive analysis of the Omp85/TpsB protein superfamily structural diversity, taxonomic occurrence, and evolution. Front Microbiol 5, 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz E, Selkrig J, Belousoff MJ, and Lithgow T (2015). Evolution of the Translocation and Assembly Module (TAM). Genome Biol Evol 7, 1628–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohr AIC, Lindau C, Wirth C, Qiu J, Stroud DA, Kutik S, Guiard B, Hunte C, Becker T, Pfanner N, et al. (2018). Membrane protein insertion through a mitochondrial beta-barrel gate. Science 359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadanza MG, Higgins AJ, Schiffrin B, Calabrese AN, Brockwell DJ, Ashcroft AE, Radford SE, and Ranson NA (2016). Lateral opening in the intact beta-barrel assembly machinery captured by cryo-EM. Nat Commun 7, 12865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadanza MG, Schiffrin B, White P, Watson MA, Horne JE, Higgins AJ, Calabrese AN, Brockwell DJ, Tuma R, Kalli AC, et al. (2020). Distortion of the bilayer and dynamics of the BAM complex in lipid nanodiscs. Commun Biol 3, 766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ieva R, and Bernstein HD (2009). Interaction of an autotransporter passenger domain with BamA during its translocation across the bacterial outer membrane. Proc Natl Acad Sci U S A 106, 19120–19125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ieva R, Skillman KM, and Bernstein HD (2008). Incorporation of a polypeptide segment into the beta-domain pore during the assembly of a bacterial autotransporter. Mol Microbiol 67, 188–201. [DOI] [PubMed] [Google Scholar]
- Ieva R, Tian P, Peterson JH, and Bernstein HD (2011). Sequential and spatially restricted interactions of assembly factors with an autotransporter beta domain. Proc Natl Acad Sci U S A 108, E383–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y, Meyer KJ, Iinishi A, Favre-Godal Q, Green R, Manuse S, Caboni M, Mori M, Niles S, Ghiglieri M, et al. (2019). A new antibiotic selectively kills Gram-negative pathogens. Nature 576, 459–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S, and Goldberg MB (2007). Requirement for YaeT in the outer membrane assembly of autotransporter proteins. J Bacteriol 189, 5393–5398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jose J, Jahnig F, and Meyer TF (1995). Common structural features of IgA1 protease-like outer membrane protein autotransporters. Mol Microbiol 18, 378–380. [DOI] [PubMed] [Google Scholar]
- Junker M, Besingi RN, and Clark PL (2009). Vectorial transport and folding of an autotransporter virulence protein during outer membrane secretion. Mol Microbiol 71, 1323–1332. [DOI] [PubMed] [Google Scholar]
- Junker M, Schuster CC, McDonnell AV, Sorg KA, Finn MC, Berger B, and Clark PL (2006). Pertactin beta-helix folding mechanism suggests common themes for the secretion and folding of autotransporter proteins. Proc Natl Acad Sci U S A 103, 4918–4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadokura H, and Beckwith J (2009). Detecting folding intermediates of a protein as it passes through the bacterial translocation channel. Cell 138, 1164–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajava AV, and Steven AC (2006). The turn of the screw: variations of the abundant beta-solenoid motif in passenger domains of Type V secretory proteins. J Struct Biol 155, 306–315. [DOI] [PubMed] [Google Scholar]
- Khalid S, and Sansom MS (2006). Molecular dynamics simulations of a bacterial autotransporter: NalP from Neisseria meningitidis. Mol Membr Biol 23, 499–508. [DOI] [PubMed] [Google Scholar]
- Konovalova A, Kahne DE, and Silhavy TJ (2017). Outer Membrane Biogenesis. Annu Rev Microbiol 71, 539–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozjak V, Wiedemann N, Milenkovic D, Lohaus C, Meyer HE, Guiard B, Meisinger C, and Pfanner N (2003). An essential role of Sam50 in the protein sorting and assembly machinery of the mitochondrial outer membrane. J Biol Chem 278, 48520–48523. [DOI] [PubMed] [Google Scholar]
- Landeta C, Boyd D, and Beckwith J (2018). Disulfide bond formation in prokaryotes. Nat Microbiol 3, 270–280. [DOI] [PubMed] [Google Scholar]
- Lauber F, Deme JC, Lea SM, and Berks BC (2018). Type 9 secretion system structures reveal a new protein transport mechanism. Nature 564, 77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I, and Bork P (2018). 20 years of the SMART protein domain annotation resource. Nucleic Acids Res 46, D493–D496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyton DL, Johnson MD, Thapa R, Huysmans GH, Dunstan RA, Celik N, Shen HH, Loo D, Belousoff MJ, Purcell AW, et al. (2014). A mortise-tenon joint in the transmembrane domain modulates autotransporter assembly into bacterial outer membranes. Nat Commun 5, 4239. [DOI] [PubMed] [Google Scholar]
- Leyton DL, Sevastsyanovich YR, Browning DF, Rossiter AE, Wells TJ, Fitzpatrick RE, Overduin M, Cunningham AF, and Henderson IR (2011). Size and conformation limits to secretion of disulfide-bonded loops in autotransporter proteins. J Biol Chem 286, 42283–42291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundquist K, Bakelar J, Noinaj N, and Gumbart JC (2018). C-terminal kink formation is required for lateral gating in BamA. Proc Natl Acad Sci U S A 115, E7942–E7949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuskens I, Saragliadis A, Leo JC, and Linke D (2019). Type V Secretion Systems: An Overview of Passenger Domain Functions. Front Microbiol 10, 1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller JE, Papic D, Ulrich T, Grin I, Schutz M, Oberhettinger P, Tommassen J, Linke D, Dimmer KS, Autenrieth IB, et al. (2011). Mitochondria can recognize and assemble fragments of a beta-barrel structure. Mol Biol Cell 22, 1638–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noinaj N, Kuszak AJ, Balusek C, Gumbart JC, and Buchanan SK (2014). Lateral opening and exit pore formation are required for BamA function. Structure 22, 1055–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oomen CJ, van Ulsen P, van Gelder P, Feijen M, Tommassen J, and Gros P (2004). Structure of the translocator domain of a bacterial autotransporter. EMBO J 23, 1257–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel R, Hsu SC, Bedard J, Inoue K, and Jarvis P (2008). The Omp85-related chloroplast outer envelope protein OEP80 is essential for viability in Arabidopsis. Plant Physiol 148, 235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova O, Peterson JH, Ieva R, and Bernstein HD (2013). Mechanistic link between beta barrel assembly and the initiation of autotransporter secretion. Proc Natl Acad Sci U S A 110, E938–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson JH, Tian P, Ieva R, Dautin N, and Bernstein HD (2010). Secretion of a bacterial virulence factor is driven by the folding of a C-terminal segment. Proc Natl Acad Sci U S A 107, 17739–17744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Pohlner J, Halter R, Beyreuther K, and Meyer TF (1987). Gene structure and extracellular secretion of Neisseria gonorrhoeae IgA protease. Nature 325, 458–462. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Alonso R, Letoquart J, Nguyen VS, Louis G, Calabrese AN, Iorga BI, Radford SE, Cho SH, Remaut H, and Collet JF (2020). Structural insight into the formation of lipoprotein-beta-barrel complexes. Nat Chem Biol 16, 1019–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman-Hernandez G, Peterson JH, and Bernstein HD (2014). Reconstitution of bacterial autotransporter assembly using purified components. Elife 3, e04234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossiter AE, Leyton DL, Tveen-Jensen K, Browning DF, Sevastsyanovich Y, Knowles TJ, Nichols KB, Cunningham AF, Overduin M, Schembri MA, et al. (2011). The essential beta-barrel assembly machinery complex components BamD and BamA are required for autotransporter biogenesis. J Bacteriol 193, 4250–4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Perez F, Henderson IR, Leyton DL, Rossiter AE, Zhang Y, and Nataro JP (2009). Roles of periplasmic chaperone proteins in the biogenesis of serine protease autotransporters of Enterobacteriaceae. J Bacteriol 191, 6571–6583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryoo D, Rydmark MO, Pang YT, Lundquist KP, Linke D, and Gumbart JC (2020). BamA is required for autotransporter secretion. Biochim Biophys Acta Gen Subj 1864, 129581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauri A, Ten Hagen-Jongman CM, van Ulsen P, and Luirink J (2012). Estimating the size of the active translocation pore of an autotransporter. J Mol Biol 416, 335–345. [DOI] [PubMed] [Google Scholar]
- Schulz GE (2000). beta-Barrel membrane proteins. Curr Opin Struct Biol 10, 443–447. [DOI] [PubMed] [Google Scholar]
- Skillman KM, Barnard TJ, Peterson JH, Ghirlando R, and Bernstein HD (2005). Efficient secretion of a folded protein domain by a monomeric bacterial autotransporter. Mol Microbiol 58, 945–958. [DOI] [PubMed] [Google Scholar]
- Szabady RL, Peterson JH, Skillman KM, and Bernstein HD (2005). An unusual signal peptide facilitates late steps in the biogenesis of a bacterial autotransporter. Proc Natl Acad Sci U S A 102, 221–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasek D, Rawson S, Lee J, Wzorek JS, Harrison SC, Li Z, and Kahne D (2020). Structure of a nascent membrane protein as it folds on the BAM complex. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasello G, Armenia I, and Molla G (2020). The Protein Imager: a full-featured online molecular viewer interface with server-side HQ-rendering capabilities. Bioinformatics 36, 2909–2911. [DOI] [PubMed] [Google Scholar]
- Ulrich T, Oberhettinger P, Schutz M, Holzer K, Ramms AS, Linke D, Autenrieth IB, and Rapaport D (2014). Evolutionary conservation in biogenesis of beta-barrel proteins allows mitochondria to assemble a functional bacterial trimeric autotransporter protein. J Biol Chem 289, 29457–29470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voulhoux R, Bos MP, Geurtsen J, Mols M, and Tommassen J (2003). Role of a highly conserved bacterial protein in outer membrane protein assembly. Science 299, 262–265. [DOI] [PubMed] [Google Scholar]
- Wheeler TJ, Clements J, and Finn RD (2014). Skylign: a tool for creating informative, interactive logos representing sequence alignments and profile hidden Markov models. BMC Bioinformatics 15, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T, Malinverni J, Ruiz N, Kim S, Silhavy TJ, and Kahne D (2005). Identification of a multicomponent complex required for outer membrane biogenesis in Escherichia coli. Cell 121, 235–245. [DOI] [PubMed] [Google Scholar]
- Yuan X, Johnson MD, Zhang J, Lo AW, Schembri MA, Wijeyewickrema LC, Pike RN, Huysmans GHM, Henderson IR, and Leyton DL (2018). Molecular basis for the folding of beta-helical autotransporter passenger domains. Nat Commun 9, 1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original data for figures in the paper is available upon request to the Lead Contact or at Mendeley Data (http://dx.doi.org/10.17632/3cy3y7dnmk.1)