

Abstract
Background.
Transplant glomerulopathy (TG) is a pathologic feature of chronic active antibody-mediated rejection (cAMR) and is associated with renal allograft failure. The specific role of B cells in the pathogenesis of TG is unclear.
Methods.
We utilized a minor mismatched rat kidney transplant model with B cell deficient recipients, generated by CRISPR/Cas9 technology, to investigate the impact of B cell depletion on the pathogenesis of TG. We hypothesized B cell deficiency would prevent TG in the rat kidney transplant model of cAMR. Treatment groups included syngeneic, allogeneic, sensitized allogeneic, and B cell deficient allogeneic transplant recipients.
Results.
B cell deficient recipients demonstrated reduced TG lesions, decreased microvascular inflammation, reduced allograft infiltrating macrophages, and reduced IFNγ transcripts within the allograft. Allograft transcript levels of IFNγ, MCP, and IL-1β correlated with numbers of intragraft macrophages. B cell deficient recipients lacked circulating donor-specific antibodies and had an increased splenic T regulatory cell population.
Conclusions.
In this model of cAMR, B cell depletion attenuated the development of TG with effects on T cell and innate immunity.
INTRODUCTION
Over recent decades, the field of kidney transplantation has advanced in the diagnosis and treatment of acute rejection. However, improvements in maintaining long-term allograft survival have been of substantially smaller magnitude and remain a major challenge in clinical practice. Approximately half of transplanted kidneys fail within 10 years of transplantation.1 The predominant cause of late allograft failure is chronic active antibody-mediated rejection (cAMR).2,3 Transplant glomerulopathy (TG) is a diagnostic feature of cAMR and is a common finding in allografts failing due to cAMR.4,5 TG is defined as the duplication of the glomerular basement membrane, confirmed by visualization with electron or light microscopy.4 The cumulative incidence of TG increases over time; approximately 20% of allograft biopsies demonstrate TG within 5 years after transplantation.6 The incidence of TG is greater in high-risk populations and is found in up to 55% of sensitized recipients.7 Once TG develops, it is associated with poor clinical outcomes including allograft failure and patient mortality.5–10 Therefore, TG represents a common problem encountered in transplant recipients and carries a large impact on allograft and patient survival.
During AMR, donor-specific antibodies (DSA) injure the allograft endothelium, resulting in remodeling of the vessel walls and ultimately generating glomerular pathology in the form of TG.11 Eventually the nephron ceases to function and kidney failure ensues.11 To date, no therapeutics have shown efficacy to reverse the trajectory to allograft failure from TG, constituting a major unmet clinical need.12 The role of B cell-directed therapy in cAMR is unclear as clinical trials of B cell targeted therapies show conflicting results. Pretransplant B cell-directed therapy with anti-CD20 therapy (rituximab) as part of induction may prevent cAMR.13,14 However, other studies found B cell-directed therapy with induction did not reduce the incidence of acute rejection.15–17 Similarly, rituximab as treatment for active AMR improved allograft survival in some studies,18–21 but not in others.22–24 Reasons for these discrepant findings may, in part, be due to the limited ability of rituximab to completely eliminate DSA.25 The ability of B cell depletion to prevent allograft failure due to cAMR and TG remains unclear.
The transplant animal model is an important tool to investigate the pathogenesis of cAMR and allows us to study the role of B cells in more detail than in clinical studies. Along these lines, the Fischer-344 to Lewis rat kidney transplant model is a well-established model for the study of chronic rejection.26–28 A major advantage of this model is that it replicates the clinical pathology of cAMR seen in kidney transplant patients, including TG.29 Whether it is possible to prevent formation of the chronic pathologic lesions due to cAMR, such as TG, via B cell depletion remains unknown. In order to investigate the impact of B cell depletion to prevent the development of TG, we utilized a minor mismatched rat kidney transplant model with genetically B cell deficient recipients. The genetically B cell deficient recipients were generated by CRISPR/Cas9 technology with a targeted deletion in the gene encoding IgM, which results in an early truncation of IgM.30 Since membrane immunoglobulin expression is mandatory for B cell maturation, this genetic modification produces a very early block of B cell production. We hypothesized B cell deficiency would prevent TG lesions in a rat kidney transplant model of cAMR.
MATERIALS AND METHODS
Study design
All procedures were performed in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals, the Public Health Service Policy on Humane Care and Use of Laboratory Animals, and a University of Wisconsin-Madison Institutional Animal Care and Use Committee protocol. Fischer and Lewis rats (Envigo) were housed in a pathogen-free animal care facility. Four experimental groups were included: 1) syngeneic, 2) allogeneic, 3) sensitized allogeneic, and 4) B cell deficient (B−/−) allogeneic kidney transplant recipients (Figure 1). Syngeneic kidney transplants were performed between Lewis donor to Lewis recipient. Minor mismatch allogeneic kidney transplants were performed between Fischer donor (MHC haplotype RT1lv1) to Lewis recipient (MHC haplotype RT1l), as previously described.31 Sensitized allogeneic Lewis recipients received an intravenous blood transfusion from a Fischer donor 21 days prior to kidney transplant. Allogeneic B−/− Lewis recipients received a kidney transplant from a Fischer donor. B−/− Lewis rats were generated via clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 technology as described previously.30 Sample size was determined from prior published work.32,33 To avoid graft loss from acute T cell mediated rejection, all recipients were given cyclosporine (1.5 mg/kg/day) via intraperitoneal injection for 10 days. For the remainder of the experimental duration, recipients were free of immunosuppression. See Supplemental Methods for complete details on transplant microsurgery.
FIGURE 1.
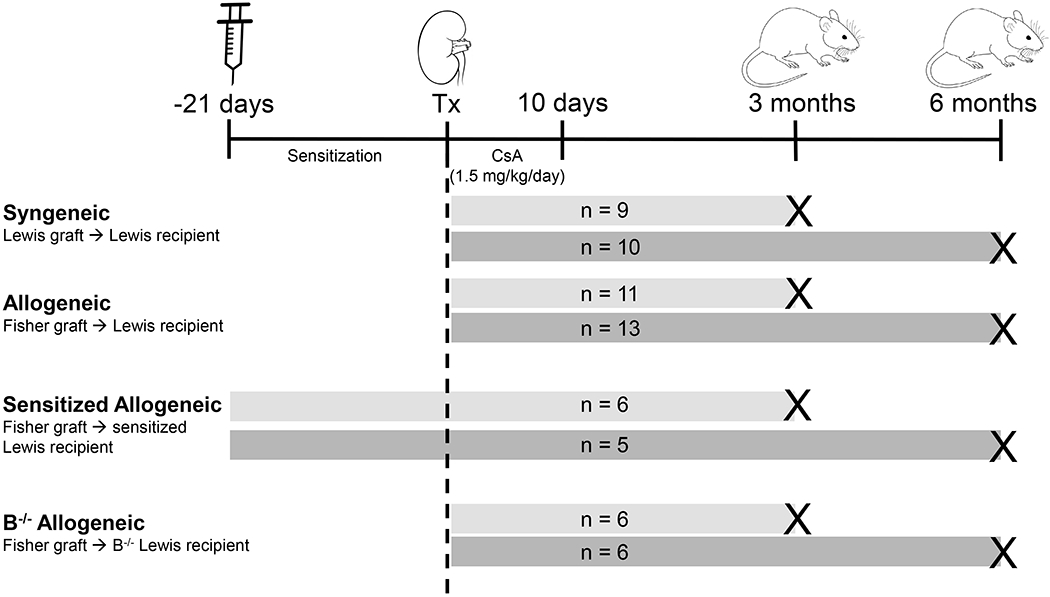
Study design included 4 experimental groups: syngeneic, allogeneic, sensitized allogeneic, and B cell deficient (B−/−) allogeneic kidney transplant recipients. Syngeneic transplants were performed between a Lewis donor and Lewis recipient. Allogeneic transplants were performed between a Fischer donor and Lewis recipient. Sensitized Lewis recipients received a blood transfusion from a Fischer donor 21 days prior to kidney transplantation from a Fischer donor. B−/− Lewis recipients received a kidney transplant from a Fischer donor. All recipients received cyclosporine for the first 10 days posttransplant and underwent native nephrectomy 10 days after transplant. Samples were collected at 3-month (light gray bars) or 6-month (dark gray bars) time points after transplantation. Group designations and sample sizes are indicated in the figure. CsA, cyclosporine; Tx, transplant.
Donor-specific antibody assay
Recipient DSA measurement was performed as described previously.30 Donor splenocytes were incubated with recipient plasma, staining was measured by flow cytometry (BD LSR II), and analyzed using FlowJo (TreeStar, Inc., Ashland, OR). Antibodies used were directed against: IgG (112-096-003, Jackson ImmunoResearch), IgG1 (clone RG11/39.4, BD Biosciences, San Diego, CA), IgG2a (clone RG7/1.30, BD Biosciences), IgG2b (clone RG7/11.1, BD Biosciences), IgG2c (biotinylated clone A92-1, BD Biosciences), IgM (clone G53-238, BD Biosciences), and CD3 (clone 1F4, BioLegend; or clone G4.18, BD Biosciences).
Flow cytometry
Single cell suspension of splenocytes were stained for B cells, T cells, and macrophages and analyzed by flow cytometry (BD LSR II) using FlowJo software (TreeStar, version 10.5.3), as previously described.32 B cell subsets were identified as transitional B cells (CD3−IgD+CD45R+CD38+CD24++) or naïve B cells (CD3−IgD+CD45R+CD27−). T cell subsets were identified as T helper cells (CD3+CD4+), cytotoxic T cells (CD3+CD8+), T regulatory cells (CD3+CD4+CD25+FoxP3+), or T follicular helper cells (CD3+CD4+CD278+CXCR5+). Antibodies for flow staining included: anti-CD3 (clone 1F4, BioLegend), anti-IgD (close MARD3, BioRad), anti-CD45R (B220) (clone HIS24 eBioscience), anti-CD38 (clone 14.27 BioLegend), anti-CD24 (clone ML5, BD Horizon), anti-CD27 (clone LG.3A10, BD Horizon), anti-CD4 (clone W3/25, Biolegend), anti-CD8 (cloneOX-8, BioLegend), anti-CD278 (clone C398.4A, BioLegend), anti-CXCR5 (clone EPR8837, Abcam), anti-CD25 (clone OX-39, BioLegend), and anti-FoxP3 (clone 150D, BioLegend). Ghost Dye Red 780 Viability Dye was used in all experiments.
Histology and electron microscopy
Kidney tissue was fixed in formalin, embedded in paraffin, cut into 5 µm sections, and stained with hematoxylin and eosin, periodic acid-Schiff, or Jones’ Methenamine Silver stain. Light microscopy images were obtained on an Olympus BX51 microscope with an Olympus KP70 camera (Olympus America Inc.). Kidney pathology was scored according to Banff 2017 criteria4 by a transplant pathologist blinded to study groups.
Kidney tissue for electron microscopy was fixed in 2% glutaraldehyde/2% paraformaldehyde with 0.1 M cacodylic acid and 3% sucrose, embedded in resin, and cut into ultrathin sections. The total length of glomerular basement membrane (GBM) and duplicated GBM was measured on electron photomicrographs (William S. Middleton Memorial Veterans Hospital Electron Microscopy Facility) using Image J software (National Institute of Health). The proportion of duplicated GBM was calculated by dividing the length of duplicated GBM segment by the total length of GBM (Figure S1).
ELISA, immunohistochemistry, immunofluorescence microscopy, and immunoblotting
Serum levels of IgM and IgG were determined by ELISA according to manufacturer’s instructions (Affymetrix/Invitrogen, Inc.). Immunoperoxidase, immunofluorescence, and Western blot of kidney tissues were performed as previously described.30 Images for quantitative analysis were obtained on an Olympus BX51 microscope with an Olympus KP70 camera (Olympus America Inc.) and quantified using Image J (National Institute of Health). A transplant pathologist, blinded to study groups, counted the number of CD68+ cells within 20 glomeruli per allograft and determined the mean number of CD68+ cells per glomerulus. Immunoblot was performed on kidney lysates and reported as relative protein levels normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Antibodies directed at the following were used: CD68 (clone ED1, Biorad), GAPDH (clone 6C5, Abcam), C3 (55463, MP Biomedicals), C4d (12-5000, American Research Products), CD3 (ab5690, Abcam).
Reverse transcriptase polymerase chain reaction analysis
Total RNA was extracted from frozen kidney tissue with Trizol (Life Technologies) and purified using the RNAeasy kit (Qiagen). cDNA was synthesized using the SuperScript IV first-strand synthesis system (Invitrogen). TaqMan gene expression assays (Applied Biosystems) were run on a 7500 Reverse Transcriptase polymerase chain reaction (PCR) System (Applied Biosystems). Mean cycle threshold (CT), standard deviation and ∆∆CT normalized to syngeneic animals were determined for IL-1β (Rn00580432_m1), IL-6 (Rn01410330_m1), IFNγ (Rn00594078_m1), MCP (Rn00580555_m1), and GAPDH (Rn01775763_g1).
Mixed lymphocyte reaction, ELISpot assay, and cellular proliferation
Recipient (Lewis [MHC haplotype RT1l]) lymph node cells were isolated from mesenteric lymph nodes and labeled with CellTrace Violet (Invitrogen). Donor (Fischer [MHC haplotype RT1lv1]) splenocytes were isolated and irradiated with a dose of 20 Gy. Cells were cocultured for 3 days. IFNγ production of Lewis lymph node cells in response to stimulation with donor splenocytes, anti-CD3 (BioLegend, 201401), and anti-CD28 (BioLegend, 200902). IFNγ production was determined by IFNγ ELISpot kit (R&D Systems) and measured on an automated ELISpot reader. Proliferation of Lewis lymph node cells was measured by CellTrace Violet Cell Proliferation kit (Invitrogen) and T cell populations were identified by flow cytometry. Data were analyzed using FlowJo software (TreeStar, version 10.5.3) and proliferation was calculated using the proliferation module of FlowJo.
Statistical analysis
Data were analyzed using Prism statistical software (GraphPad Software, Version 6.07). Continuous variables are presented as means ± standard deviation. Ordinal data, such as Banff scores, are presented as counts and proportions of cases with a defined score. One-way ANOVA followed by a post hoc Tukey’s multiple comparisons test was used to compare multiple independent groups. Pearson correlation was used to assess the associations between allograft transcript and macrophage levels. A P value less than 0.05 was considered significant.
RESULTS
B cell deficient recipients lacked donor-specific antibodies, B cells, and immunoglobulins
Recipient DSA levels were determined by flow cytometry (Figure 2A). IgM and IgG DSA were not present in B−/− allogeneic recipients at 3 or 6 months (B−/− allogeneic compared to allogeneic and sensitized allogeneic groups, 1-way ANOVA P<0.0001). IgG DSA subclasses (IgG1, IgG2a, IgG2b, and IgG2c) were not present in B−/− allogeneic recipients. Allogeneic recipients generated higher IgG DSA levels compared to syngeneic recipients (P<0.001 at both 3 and 6 months). Sensitized allogeneic recipients developed greater IgM and IgG DSA levels compared to allogeneic recipients (P<0.001 for both 3 and 6 months).
FIGURE 2.
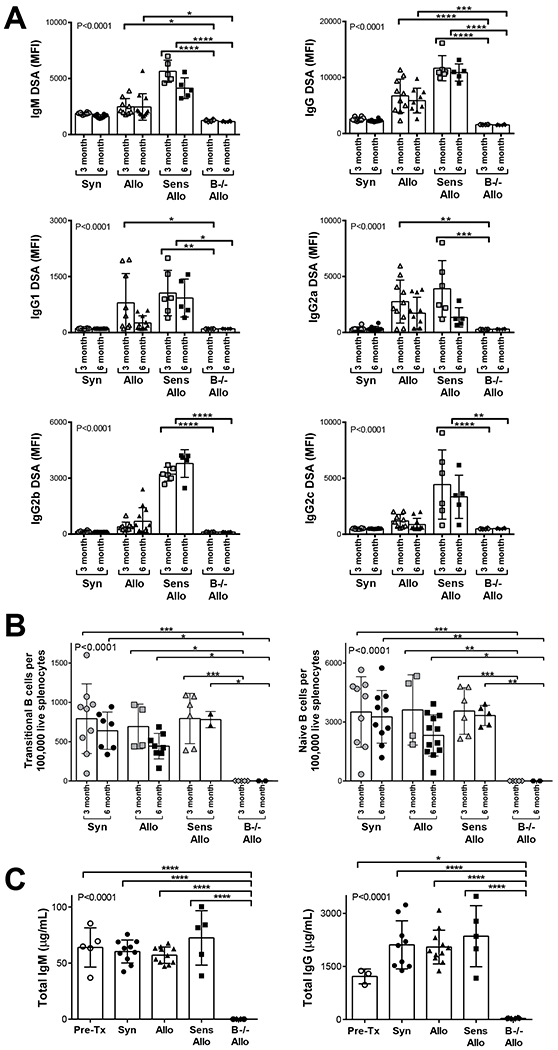
B−/− allogeneic recipients did not generate DSA, B cells, and immunoglobulins. A, B−/− allogeneic recipients did not develop IgM or IgG DSA compared to allogeneic or sensitized allogeneic recipients at 3 or 6 months (P<0.0001 in 1-way ANOVA). IgM and IgG DSA were increased in sensitized allogeneic compared to syngeneic recipients at both 3 and 6 months compared to all other groups. B−/− allogeneic recipients did not generate any IgG DSA subtypes (IgG1, IgG2a, IgG2b, and IgG2c). B, B−/− allogeneic recipients lacked naïve and transitional splenic B cells compared to all other groups at 3 and 6 months (P<0.0001 in 1-way ANOVA). C, B−/− allogeneic recipients demonstrated absence of circulating total IgM and IgG at 6 months after transplant compared to pretransplant wild-type Lewis rats, syngeneic, allogeneic, and sensitized allogeneic recipients (P<0.0001 in 1-way ANOVA). Data presented as mean ± standard deviation. Data points on graph represent individual animals. Syn, syngeneic; Allo, allogeneic; Sens Allo, sensitized allogeneic; B−/− Allo, B cell deficient allogeneic; MFI, mean fluorescence intensity; Pre-Tx, pretransplant wild-type Lewis rats. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001
Flow cytometry was performed to enumerate splenic B cells. Figure 2B demonstrates transitional B cells (CD3−IgD+CD45R+CD38+CD24++) and naïve B cells (CD3−IgD+CD45R+CD27−) were absent in B−/− allogeneic compared to syngeneic, allogeneic, and sensitized allogeneic recipients (1-way ANOVA, P< 0.0001).
Circulating total IgM and IgG levels were determined by ELISA on serum collected from recipients 6 months after transplant (Figure 2C). IgM and IgG were not present in B−/− allogeneic recipients (B−/− allogeneic compared to pretransplant wild-type Lewis rats, syngeneic, allogeneic, and sensitized allogeneic groups, 1-way ANOVA P< 0.0001).
B cell deficiency reduced microvascular inflammation within the allograft
Banff scoring was performed on all kidney allografts by a renal pathologist. Figure 3 shows the distribution of Banff scores for glomerulitis, peritubular capillaritis, and microvascular inflammation by treatment group. Glomerulitis was less frequent in B−/− allogeneic compared to allogeneic allografts (3 months: 67% of B−/− allogeneic versus 100% of allogeneic allografts had a g score ≥ 1, P<0.0001; 6 months: 50% of B−/− allogeneic versus 100% of allogeneic allografts had a g score ≥ 1, P<0.0001). Peritubular capillaritis was less frequent in B−/− allogeneic compared to allogeneic allografts (3 months: 17% of B−/− allogeneic versus 73% of allogeneic allografts had a ptc score ≥ 1, P<0.0001; 6 months: 0% of B−/− allogeneic versus 70% of allogeneic allografts had a ptc score ≥ 1, P<0.0001). The proportion of allografts with microvascular inflammation was less in B−/− allogeneic compared to allogeneic allografts (3 months: 17% of B−/− allogeneic versus 82% of allogeneic allografts had an mvi score ≥ 2, P<0.0001; 6 months: 0% of B−/− allogeneic versus 62% allografts had an mvi score ≥ 2, P<0.0001).
FIGURE 3.
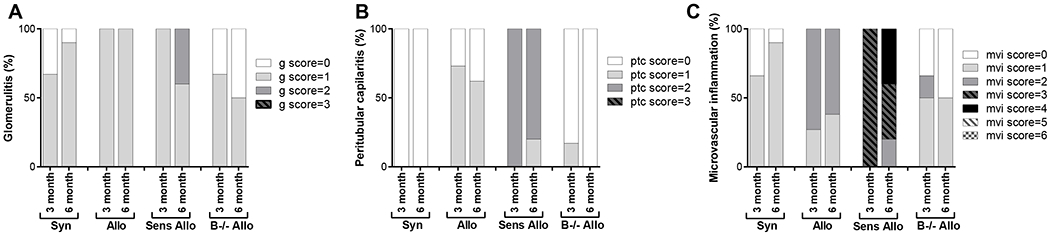
Banff scoring was performed on kidney allografts. The frequency of Banff scores for glomerulitis, peritubular capillaritis, and microvascular inflammation are shown by treatment group. g, glomerulitis; ptc, peritubular capillaritis; mvi, microvascular inflammation
B cell deficiency attenuated transplant glomerulopathy
The majority of chronic glomerulopathy (cg) lesions were at the Banff 1a score level, i.e. visible by electron microscopy only. Since the cg score is based on the extent of glomerular basement membrane (GBM) double contours, we performed quantitative measurements of the length of GBM with duplication visualized by electron microscopy for each transplant recipient (Figure 4). GBM duplication was reduced in B−/− allogeneic recipients compared allogeneic recipients at 6 months (P<0.0001), and compared to sensitized allogeneic recipients at 6 months (P<0.0001).
FIGURE 4.
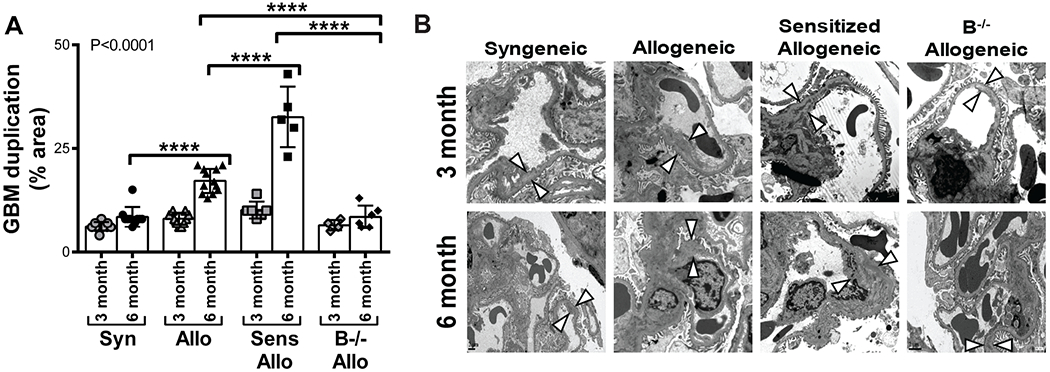
B cell deficiency attenuated the double contour lesions of transplant glomerulopathy. A, The mean proportion of length of GBM with double contours was quantified for each recipient. GBM double contours were reduced in B−/− allogeneic recipients compared to allogeneic recipients at 6 months and compared to sensitized allogeneic recipients at 3 and 6 months. Allogeneic recipients had a higher proportion of GBM with double contours compared to syngeneic recipients at 6 months. Sensitized allogeneic recipients had a greater percentage of GBM with double contours compared to allogeneic recipients at 6 months. Data presented as mean ± standard deviation. Data points on graph represent individual animals. P<0.0001 by 1-way ANOVA, ****P<0.0001. B, Representative images of GBM duplication (arrowheads) as determined by electron microscopy are shown (3 month: magnification × 15,000. 6 month: magnification × 8,000). Syn, syngeneic; Allo, allogeneic; Sens Allo, sensitized allogeneic; B−/− Allo, B cell deficient allogeneic; GBM, glomerular basement membrane.
Allogeneic recipients developed a higher proportion of duplicated GBM compared to syngeneic recipients at 6 months (P<0.0001). Sensitized allogeneic recipients had a greater amount of GBM duplication compared to allogeneic recipients at 6 months (P<0.0001). Collectively, these data highlight an attenuation of GBM duplication in B−/− allogeneic allografts compared to the more extensive GBM duplication in allogeneic and sensitized allogeneic allografts.
C4d deposition was observed in the allografts of sensitized allogeneic recipients
To examine the nature of complement deposition within the allograft, kidney sections were stained for C4d and C3 (Figure 5). Levels of C4d deposition were greater in allogeneic allografts than in syngeneic allografts at 6 months after transplant (P<0.05). Sensitized recipients had the highest levels of C4d within the allograft (P<0.0001 for 3 and 6 month sensitized allogeneic versus allogeneic recipients). C4d deposition was reduced in B−/− recipients compared to sensitized recipients (P<0.0001 at 3 and 6 months). Deposition of C3 was primarily located in the glomeruli. Sensitized recipients had the highest levels of C3 staining (P<0.05 for 6 month sensitized versus allogeneic recipients).
FIGURE 5.
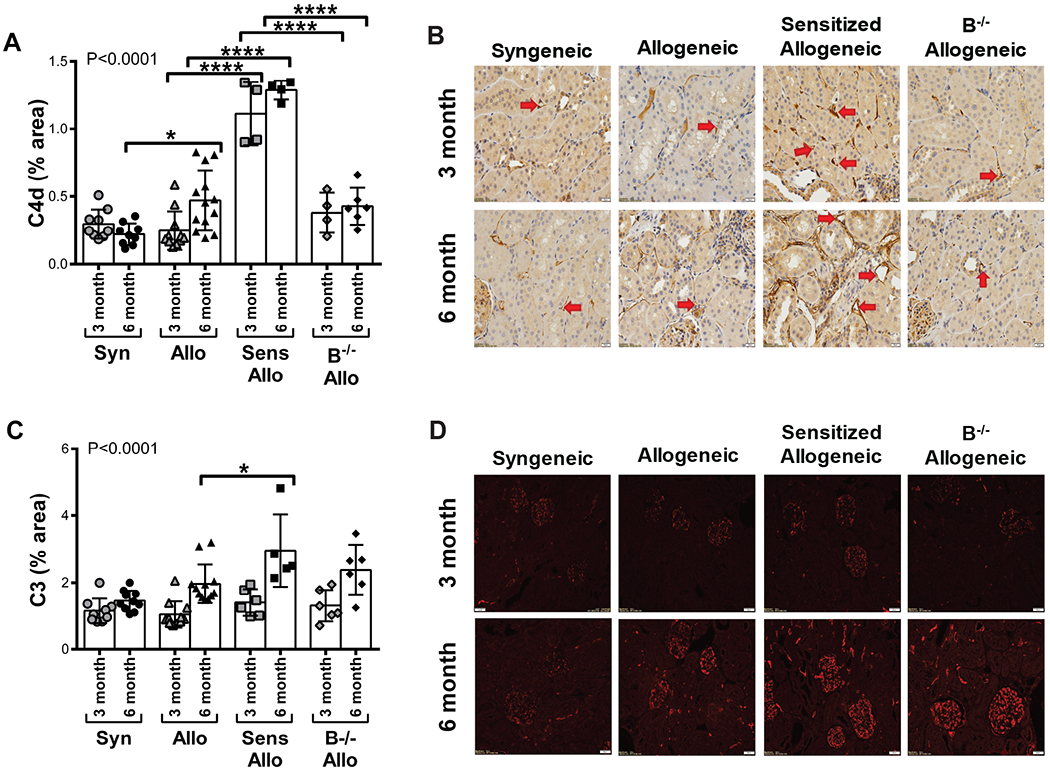
Intragraft deposition of complement proteins C4d and C3. A, At 6 months, C4d deposition was increased in allogeneic recipients compared to syngeneic recipients. Sensitized allogeneic recipients had greater deposition of C4d compared to allogeneic recipients at both time points. C4d deposition was reduced in B−/− allogeneic recipients compared to sensitized recipients at 3 and 6 months. P<0.0001 by 1-way ANOVA. B, Representative histology of C4d staining (brown stain) within the allograft is shown. Red arrows indicate peritubular capillaries with C4d deposition (magnification × 400). C, At 6 months, sensitized allogeneic recipients had greater C3 deposition compared to allogeneic recipients. P<0.0001 by 1-way ANOVA. D, Representative immunofluorescence of C3 deposition (red) within the allograft is shown (magnification × 200). Data presented as mean ± standard deviation. Data points on graph represent individual animals. *P<0.05, ****P<0.0001. Syn, syngeneic; Allo, allogeneic; Sens Allo, sensitized allogeneic; B−/− Allo, B cell deficient allogeneic
B cell deficiency reduced intragraft macrophages
Allograft infiltrating macrophages were assessed by quantitative immunohistochemistry, immunoblot of CD68 protein within allograft lysates, and quantitative analysis of glomerulitis due to macrophages (Figure 6). Immunohistochemistry demonstrated allogeneic recipients had a greater amount of intragraft macrophages than syngeneic recipients (P<0.05 at both 3 and 6 months). Sensitized allogeneic recipients had the highest levels of intragraft macrophages (P<0.05 for 3 and 6 month sensitized allogeneic compared to allogeneic recipients). B−/− allogeneic recipients had reduced intragraft macrophages compared to sensitized allogeneic recipients at 3 (P<0.05) and 6 months (P<0.01, Figure 6A–B). Western blot of CD68 protein from kidney lysates demonstrated allogeneic recipients had greater levels of CD68 protein than syngeneic recipients (P<0.05 at both 3 and 6 months, Figure 6C). The highest levels of CD68 were seen in allografts from sensitized allogeneic recipients (P<0.05 for 3 and 6 month sensitized compared to allogeneic recipients). B−/− allogeneic recipients had reduced levels of CD68 protein compared to allogeneic recipients at 6 months (P<0.05) and compared to sensitized recipients at 3 (P<0.01) and 6 months (P<0.05). To evaluate the contribution of glomerulitis to the development of TG, a transplant pathologist counted the number of CD68+ cells within 20 glomeruli per allograft and then determined the mean number of CD68+ cells per glomerulus (Figure 6D). Fewer CD68+ cells per glomerulus were seen in B−/− allografts at 3 and 6 months compared to sensitized recipients (P<0.05 for both). Thus, a reduction in intragraft macrophages, glomerular macrophages, and glomerulitis in B−/− animals may contribute to the reduced prevalence of TG in B−/− recipients.
FIGURE 6.
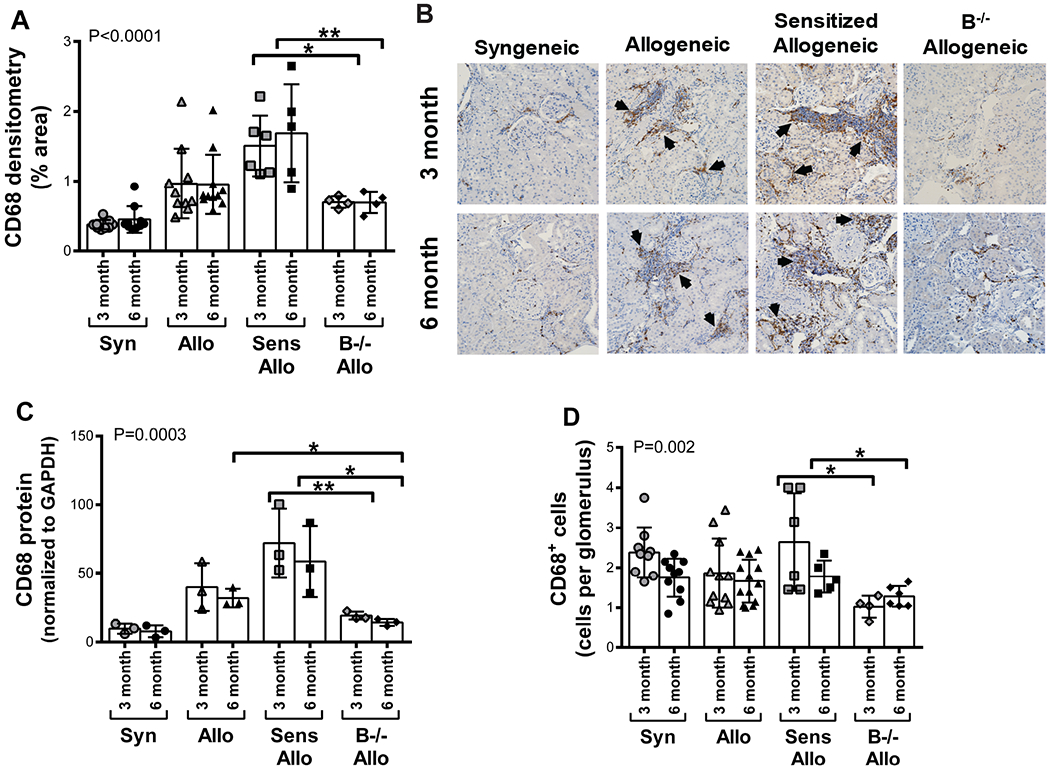
B cell deficiency reduced intragraft macrophages. A, Quantitative immunohistochemistry demonstrated increased levels of CD68 within allogeneic allografts compared to syngeneic allografts. Sensitized recipients had greater levels of CD68 within the allograft compared to allogeneic recipients. B−/− allogeneic recipients had reduced levels of CD68 compared to sensitized recipients. P<0.0001 by 1-way ANOVA. B, Representative histology of CD68 deposition (brown stain, indicated by arrows) within the allograft is shown (magnification x200). C, CD68 protein was measured by immunoblot of kidney lysates and reported as relative protein level normalized to GAPDH. CD68 protein was increased in allogeneic compared to syngeneic allografts. Similarly, CD68 protein levels were greater in sensitized compared to allogeneic allografts. CD68 protein levels were decreased in allografts from B−/− allogeneic recipients compared to allogeneic recipients at 6 months and compared to sensitized recipients at 3 and 6 months. P=0.0003 by 1-way ANOVA. D, The mean number of CD68+ cells per glomerulus were determined. There were fewer CD68+ cells per glomerulus in B−/− allogeneic recipients compared to sensitized allogeneic recipients. P=0.002 by 1-way ANOVA. Data presented as mean ± standard deviation. Data points on graph represent individual animals. *P<0.05, **P<0.01 Syn, syngeneic; Allo, allogeneic; Sens Allo, sensitized allogeneic; B−/− Allo, B cell deficient allogeneic
B cell deficiency reduced IFNγ intragraft transcript levels
Quantitative reverse transcription polymerase chain reaction (RT-qPCR) was used to assess transcript levels of cytokines and chemokines within the allograft (Figure 7). Transcripts were normalized to GAPDH and the fold-change was determined relative to syngeneic allograft transcript levels. IFNγ transcript levels in B−/− allogeneic recipients were reduced compared to allogeneic recipients at 3 months and compared to sensitized allogeneic recipients at 3 and 6 months (P<0.05 for all). IL-1β transcript levels were decreased in B−/− allogeneic recipients compared to sensitized allogeneic recipients at 6 months (P<0.05). There were no significant differences in IL-6 or MCP transcript levels across treatment groups at either time point.
FIGURE 7.
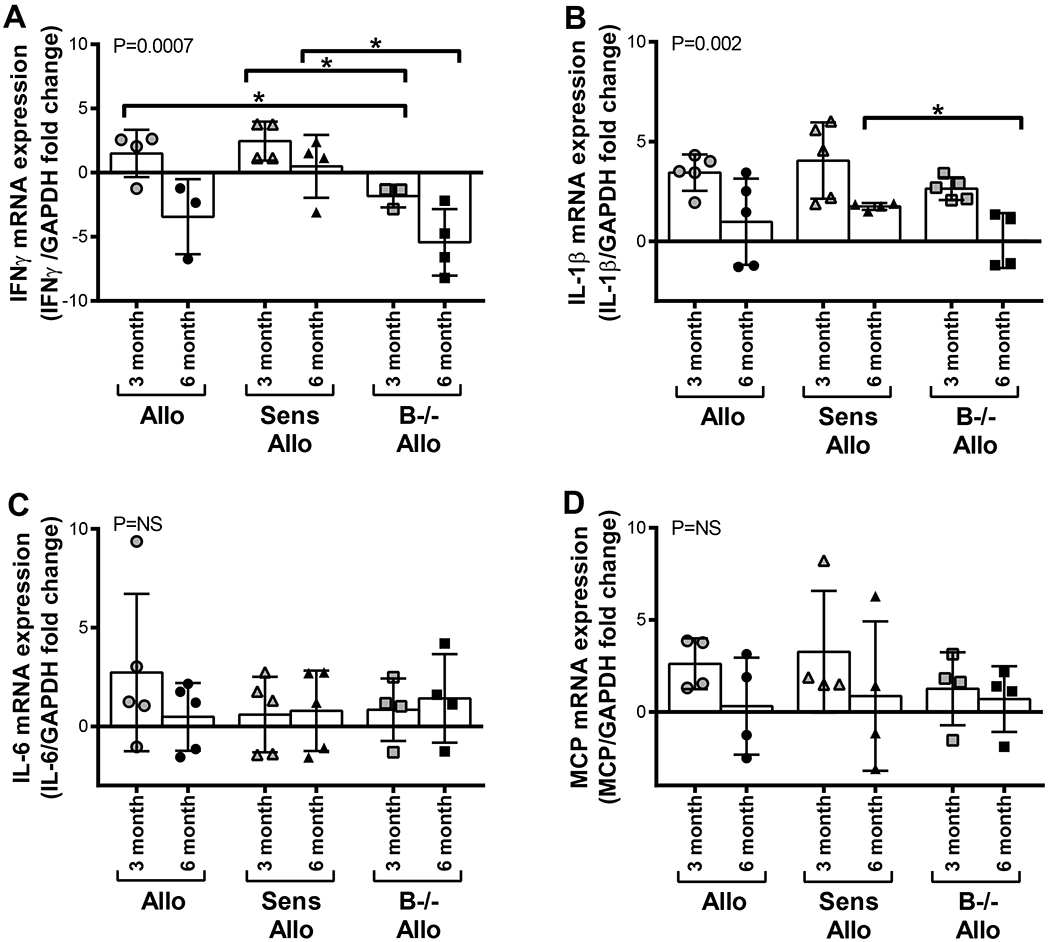
B cell deficiency reduced intragraft transcript levels of IFNγ and IL-1β. A, IFNγ transcript levels were reduced in allografts of B−/− allogeneic recipients compared to allogeneic recipients at 3 months and compared to sensitized allogeneic recipients at 3 and 6 months. P=0.0007 by 1-way ANOVA. B, IL-1β transcript levels were decreased in allografts from B−/− recipients compared to sensitized recipients at 6 months. P=0.002 by 1-way ANOVA. C and D, There were no significant differences in IL-6 or MCP transcript levels among treatment groups. Data presented as mean ± standard deviation. Data points on graph represent individual animals. *P<0.05. Syn, syngeneic; Allo, allogeneic; Sens Allo, sensitized allogeneic; B−/− Allo, B cell deficient allogeneic; NS, not significant
Intragraft transcript levels of IL-1β, IFNγ, and MCP correlated with levels of intragraft macrophages
The levels of CD68 protein, as determined by immunoblot of whole kidney lysate, correlated with allograft transcript levels of IL-1β (P<0.001, R2=0.61), IFNγ (P=0.005, R2=0.33), and MCP (P=0.001, R2=0.40) (Figure 8). A similar trend was observed between CD68, determined by quantitative immunohistochemistry, and allograft transcript levels. Levels of CD68 from quantitative immunohistochemistry correlated with allograft transcript levels of IL-1β (P<0.001, R2=0.37), IFNγ (P=0.04, R2=0.14), and MCP (P=0.002, R2=0.31). The differences in P and R2 values between immunoblot and immunohistochemistry correlations are likely reflective of immunoblot data generated from whole kidney lysate and immunohistochemistry performed on renal cortical areas only. There was no correlation between IL-6 transcript levels and CD68 determined by either immunoblot or immunohistochemistry. No correlations were observed between numbers of glomerular CD68+ cells and intragraft cytokine transcript levels.
FIGURE 8.
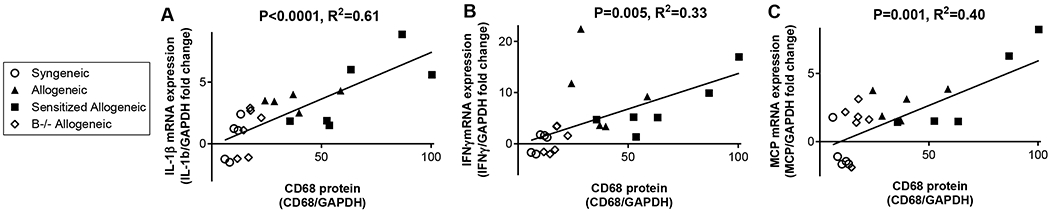
Increased transcript levels of IL-1β, IFNγ, and MCP correlated with increased levels of intragraft macrophages, as determined by immunoblot of CD68 protein, from whole kidney lysates. Data points on graph represent individual animals. Pearson correlation values are given.
B cell deficient recipients had better allograft function following transplant
Blood was collected at 3 and 6-month time points for analysis of serum creatinine and BUN levels (Table 1). B−/− allogeneic recipients had lower serum creatinine than allogeneic recipients at 3 months (P=0.02). B−/− allogeneic recipients had lower BUN levels than either allogeneic (P=0.01) or sensitized allogeneic recipients (P=0.04) at 6 months.
TABLE 1.
Kidney function by treatment group
| Syngeneic | Allogeneic | Sensitized Allogeneic | B−/− Allogeneic | P value | |
|---|---|---|---|---|---|
| Creatinine (mg/dL) | 0.007 | ||||
| 3 months | 0.48 ± 0.13 | 0.61 ± 0.09a | 0.53 ± 0.05 | 0.48 ± 0.10b | |
| 6 months | 0.42 ± 0.12 | 0.51 ± 0.09 | 0.60 ± 0.17 | 0.44 ± 0.09 | |
| BUN (mg/dL) | <0.0001 | ||||
| 3 months | 23.4 ± 2.9 | 26.3 ± 2.6a | 27.2 ± 1.5 | 26.0 ± 2.9 | |
| 6 months | 19.8 ± 3.0 | 24.1 ± 3.3a | 24.4 ± 4.0 | 23.4 ± 1.3b,c | |
Data presented as mean ± standard deviation. P value determined by 1-way ANOVA with multiple comparisons.
P<0.05 for syngeneic versus allogeneic
P<0.05 for allogeneic versus B−/− allogeneic
P<0.05 for sensitized allogeneic versus B−/− allogeneic
B cell deficiency increased splenic T regulatory cell populations
To investigate the effect of B cell depletion on T cell populations, splenocytes were analyzed by flow cytometry (Figure 9A). We observed an increased population of splenic T regulatory cells (CD3+CD4+CD25+FoxP3+) in B−/− allogeneic recipients at 6 months posttransplant compared to other groups (P<0.05 for all). T follicular helper cells (CD3+CD4+CD278+CXCR5+) were elevated in sensitized allogeneic recipients compared to allogeneic recipients at 6 months posttransplant (P=0.01). To determine if splenic cell populations were broadly affected due to the deficit of B cells within the spleen, we examined the additional leukocyte population of macrophages. B cell deficiency did not impact numbers of splenic macrophages (Figure 9A). Quantitative histologic analysis of kidney sections revealed no differences in total CD3+ T cells in the allograft between treatment groups.
FIGURE 9.
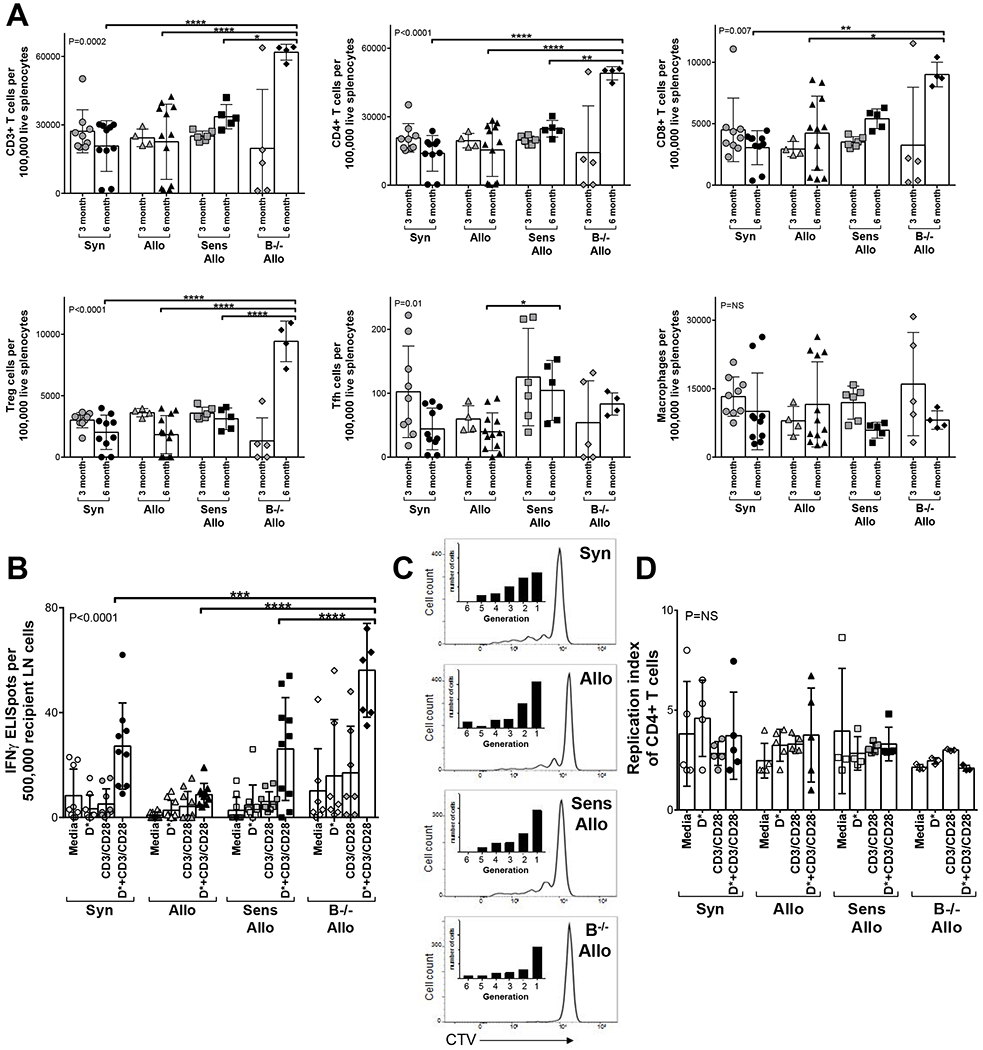
A, Ex vivo splenic T cell populations were analyzed by flow cytometry. There was an increase in the populations of splenic T regulatory cells in B−/− allogeneic recipients compared to all other groups at 6 months, P<0.001 by 1-way ANOVA). T follicular helper cells were not elevated in B−/− allogeneic recipients compared to any other group. In contrast to the changes in T cell populations, splenic macrophages had no differences across all groups (P=NS by 1-way ANOVA). B, T cell function was assessed in vitro by determination of IFNγ production of recipient lymph node cells in a mixed lymphocyte reaction and ELISpot assay. Recipient (Lewis) lymph node cells were cultured with media, irradiated donor (Fischer) cells, T cell stimulation (anti-CD3 and anti-CD28), or irradiated donor cells with anti-CD3/CD28. B cell deficient recipients demonstrated donor-specific T cell response by increased IFNγ production compared to all other groups (P<0.0001 by 1-way ANOVA). C and D, In vitro, the proliferation of T cells was assessed by mixed lymphocyte reaction and cell division was tracked by flow cytometry. Recipient lymph node cells were labeled with Cell Trace Violet and were cultured with media, irradiated donor (Fischer) cells, T cell stimulation (anti-CD3 and anti-CD28), or irradiated donor cells with anti-CD3/CD28. C, A representative experiment from each experimental group is shown for all proliferating cells in culture (Cell Trace Violet positive cells) treated with anti-CD3/CD28. The histograms show the proliferation distribution for all Cell Trace Violet positive cells. The inset bar graphs show the distribution of cells by number of generations of division (generations 1 through 6), which demonstrated adequate ability to resolve generations. D, The replication index (or fold expansion of responding cells) was determined for recipient CD4+ T cells in culture. There were no differences in the in vitro replication index of CD4+ T cells across groups (P=NS by 1-way ANOVA). Data presented as mean ± standard deviation. Data points on graph represent individual animals. Syn, syngeneic; Allo, allogeneic; Sens Allo, sensitized allogeneic; B−/− Allo, B cell deficient allogeneic; NS, not significant; LN, lymph node; D*, irradiated donor (Fischer) cells; CTV, cell trace violet. *P<0.05, **P<0.01, ****P<0.0001
B cell deficiency impact on T cell function and proliferation in vitro
To evaluate the impact of B cell deficiency on donor-specific T cell reactivity, we examined IFNγ production of recipient lymphoid cells by ELISpot assay in a mixed lymphocyte reaction of recipient (Lewis) lymph node cells cocultured with irradiated donor (Fischer) splenocytes (Figure 9B). Recipient cell cultures were treated with media, donor antigen (irradiated donor cells), T cell stimulation (anti-CD3 and anti-CD28), or a combination of donor antigen with T cell stimulation (irradiated donor cells with anti-CD3/CD28). In response to T cell stimulation with donor antigen, B cell deficient lymphoid cells increased IFNγ production compared to all other groups (P<0.0001 by 1-way ANOVA). This demonstrates the T cells from B cell deficient recipients have the ability to respond to donor antigen in vitro.
To assess the effect of B cell deficiency on T cell proliferation, we performed a mixed lymphocyte reaction and tracked cell division by flow cytometry. Recipient lymph node cells were labeled with Cell Trace Violet and cultured with media, irradiated donor (Fischer) cells, T cell stimulation (anti-CD3 and anti-CD28), or irradiated donor cells with anti-CD3/CD28. In Figure 9C, representative histograms of the proliferation distribution for all dividing cells in culture (Cell Trace Violet positive) in culture with anti-CD3/CD28 are shown. The inset black bar graphs show the distribution of cells by number of generations of division (generations 1 through 6). Typically, the maximum number of resolvable generations is 8, as this is the point at which fluorescence due to the tracer approaches 1%.34 The distribution of cells in generations 1 through 6 demonstrate adequate staining and ability to resolve generations. The replication index (or fold expansion of responding cells) was determined for recipient CD4+ T cells in culture (Figure 9D). There were no differences in the in vitro replication index of CD4+ T cells across groups (P=NS by 1-way ANOVA). Analysis of CD8+ T cells also demonstrated no differences in replication index across groups (P=NS by 1-way ANOVA). Thus, T cells from B cell deficient recipients demonstrated similar proliferative function in response to donor antigen as in other experimental groups.
DISCUSSION
In this study, we demonstrated B cell deficiency attenuated development of TG in a rat model of cAMR. In addition to the impact on glomerular basement membrane pathology, B cell deficiency reduced microvascular inflammation, macrophage infiltration, and IFNγ transcripts within the allograft compared to allogeneic recipients. Increased transcript levels of IFNγ, IL-1β, and MCP correlated with increased macrophage numbers within the allograft. B cell deficiency also corresponded to a reduction in circulating DSA levels and increased T regulatory cell populations within lymphoid tissues. In vitro, T cells from B cell deficient recipients demonstrated the functional ability to respond to donor-specific antigen with increased production of IFNγ.
The role of B cell depletion in the treatment of clinical cAMR remains an area of debate. Some retrospective clinical studies demonstrated B cell depletion as a viable therapeutic strategy for AMR,18,19 while several clinical trials have not.24,35 Thus, a better understanding of the role of B cells in cAMR and TG is needed.36 Animal models are informative in this regard. Our study demonstrated complete B cell deficiency throughout the transplant course lessened the extent of TG. The attenuated TG lesions may be the result of a less inflammatory microenvironment within the allograft, as we observed B cell depletion reduced intragraft IFNγ transcript levels, microvascular inflammation, and infiltrating macrophages. A recent study in a mouse model of cAMR demonstrated prophylactic depletion of B cells by anti-CD20 antibody improved allograft survival.37 However, if anti-CD20 was administered once high DSA titers had developed, B cell therapy did not prevent allograft failure due to cAMR.37 Taken together, these findings support a role of B cell therapeutics in cAMR and suggest the timing and duration of B cell depletion impacts allograft inflammation.
In this study, complete B cell deficiency attenuated, but did not completely eliminate, TG. Low levels of glomerulitis were detected by Banff score and immunohistochemistry for CD68+ cells within the glomerulus in the B cell deficient group. Nonhuman primate studies of the natural history of AMR demonstrate the progression from glomerulitis to subsequent TG, and eventually to overt renal dysfunction (proteinuria and increased creatinine).11 Clinical studies demonstrate 95% of patients who developed TG had a preceding biopsy with glomerulitis.38 Additionally, glomerulitis due to increased intraglomerular monocytes was associated with glomerular capillary wall remodeling, elevated IL-1β and IL-6 production by peripheral mononuclear cells, and a decline in renal function in kidney transplant patients.39 It is likely that some degree of ongoing glomerulitis contributed to the mild TG lesions seen in the B cell deficient group of our model.
We found the highest amount of allograft C4d deposition in the sensitized allogeneic recipient group. C4d deposition in the B−/− allogeneic recipients was not significantly different compared to the allogeneic recipients; this lack of difference in C4d between these 2 groups could be due to the transient nature of C4d staining in the chronic setting.40,41 Additionally, while the classical pathway of complement is viewed as the primary means of complement activation during AMR, it is interesting to consider other pathways that result in C4d beyond the classical pathway. This is a consideration, as generation of C4d occurs with activation of the lectin pathway of complement. Recent studies demonstrate the lectin pathway can be activated by binding ligands on injured, ischemic, or dying cells.42,43 Thus, there is potential for cell injury to activate the lectin pathway through this mechanism and result in C4d deposition. Along these lines, lectin pathway activation was identified during T cell mediated rejection, AMR, and delayed graft function, and mannose-binding lectin levels were associated with severity of rejection and allograft failure.44–47 Thus, the potential exists for multiple pathways of the complement system to contribute to allograft injury.
We observed allografts from B cell deficient recipients had lower levels of intragraft macrophages. These findings are consistent with previous work, by ourselves and others, in which B cell depletion reduced intragraft macrophages in acute rejection in rodent transplant models.30,48 Recent clinical studies highlight the importance of intragraft macrophages; intragraft macrophages correlated with severity of rejection, development of interstitial fibrosis, and lower allograft and patient survival.49–52 Macrophages are recruited to the allograft by monocyte chemotactic protein (MCP, also known as C-C motif chemokine ligand 2 (CCL2)). In the current study, MCP transcript levels correlated with increased intragraft macrophages. Once macrophages have infiltrated into the allograft, they demonstrate a high degree of plasticity in response to their microenvironment and can generate inflammatory, fibrotic, or reparative responses. An environment rich in IFNγ drives macrophages towards a proinflammatory phenotype.53 Proinflammatory macrophages generate potent cytokines (such as IL-1 and IL-6), which can promote tissue injury and have been associated with development of transplant glomerulopathy in transplant patients.54 We observed an association between increased intragraft macrophages and allograft IFNγ and IL-1β transcript levels. B cell deficient recipients had a significant reduction in allograft IFNγ transcripts, likely reflective of a less inflammatory microenvironment within the allograft in the setting of B cell deficiency.
Clinical studies of gene transcripts identified IFNγ, chemokine, and macrophage-associated transcripts among the top profiles in the setting of TG or cAMR.55–58 Among patients with TG, increased IFNγ transcripts were associated with allograft failure compared to those without allograft failure.59,60 In agreement with clinical cohorts and mouse models of rejection in which increased IFNγ transcripts were present in rejection,61–63 we found higher levels of IFNγ transcripts in the allogeneic and sensitized allogeneic groups compared to the B cell deficient group in our rat model of cAMR. IFNγ enhances MHC expression on renal microvascular endothelial cells,64,65 which increases antigen availability and, thus, contributes to the pathogenesis of cAMR (microvascular inflammation and TG).
In this model, the changes in histology were more extensive than the differences in renal function. This observation of allograft pathology in the setting of preserved or mild renal dysfunction is analogous to subclinical AMR as observed in human studies.66 Clinical studies utilizing protocol biopsies demonstrate subclinical AMR (evidence of glomerulitis and/or capillaritis along with DSA) with preserved renal function.67 Studies demonstrated that at 1-year follow-up, patients with subclinical AMR are at risk for interstitial fibrosis/tubular atrophy, reduced GFR, and are at high risk for development of TG (43% for those with prior subclinical AMR compared to 0% for those without).68 Additionally, subclinical AMR at 1 year after transplant was independently associated with an increased risk of allograft failure.69 We suspect had we conducted a longer experiment we would observe further progression of allograft pathology with subsequent development of overt renal dysfunction.
We observed a shift towards T regulatory cell populations in spleens of B cell deficient recipients at 6 months posttransplant. This is consistent with our previous study, which we demonstrated increased splenic T regulatory cells in acute rejection in the B cell deficient rat strain.30 A recent study utilizing a mouse kidney transplant model given inhibitors directed at B Cell Activating Factor of the Tumor Necrosis Factor Family (BAFF) or dual blockade with BAFF and A Proliferation Inducing Ligand (APRIL) also found increased splenic T regulatory cells.70,71 Similar to our observations in animal models, numerous studies report an increase in T regulatory cells following B cell depletion with rituximab in human patients with lupus nephritis, nephrotic syndrome, and kidney transplant recipients with AMR.72–75 However, not all clinical studies observed this. A recent study that included a cohort of patients treated with rituximab for AMR, desensitization, native glomerular disease, or autoimmune disease saw a nonsignificant trend of increased T regulatory cells in both rituximab treated and untreated patients.76 These discrepancies may be related to the mixed cohort, variations in rituximab dosing due to underlying diagnoses, and concurrent immunosuppressive medications.
In our study, we did not find enhanced T cell numbers in the allografts of B cell deficient recipients. In our in vitro studies, we demonstrated T cells from B cell deficient recipients retained the functional ability to produce IFNγ and proliferate in response to donor-specific stimuli. As we observed donor-specific T cell function in vitro, we suspect the lack of T cell infiltration into the allograft was due to a reduction in T cell homing to the allograft. This could be the result of lack of professional antigen presenting cells (B cells) to appropriately prime T cells, lack of DSA, and lack of sufficient allograft injury in B cell deficiency to stimulate T cell migration.
Our study has several limitations. We have limited sample sizes. Nevertheless, we did find significant differences in the setting of B cell deficiency with the current sample sizes. We suspect additional numbers would further strengthen these findings. Some cases of off-target effects of the CRISPR/Cas9 system have been reported.77 However, our rat colony generated by CRISPR/Cas9 technology has not demonstrated any adverse phenotypic effects after many generations.
In summary, we provide evidence that complete B cell deficiency during transplantation attenuated the development of TG in a model of chronic active AMR. As TG was attenuated but not eliminated, this demonstrates B cells are critical to the development of TG but not solely sufficient for its development. Contributions from additional arms of the immune system, such as innate immunity, likely contribute to microvascular injury and subsequent development of TG. Questions remain if TG can be halted or reversed at an early stage, such as when double contours first appear or when endothelial cell injury is identified by electron microscopy. Future studies of therapeutics to prevent and treat TG are needed involving a comprehensive immunologic assessment.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Kristy Meyer, Joan Sempf, and the William S. Middleton Memorial Veterans Hospital Electron Microscopy Facility for their assistance and expertise. The authors thank Dana Clark, MA, for her editorial assistance with this manuscript.
Financial Disclosure
This project was supported by the NIH grant K23 DK122136 (SEP), the American Society of Nephrology John Merrill Grant in Transplantation (SEP), and the UW Transplant Research Training Grant T32 AI1256231 (KRD). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
ABBREVIATIONS
- Allo
allogeneic
- B−/−
B cell deficient
- BUN
blood urea nitrogen
- C3
complement component 3
- C4d
complement component 4d
- cAMR
chronic active antibody-mediated rejection
- CD
cluster of differentiation
- cg
chronic glomerulopathy
- CRISPR
clustered regularly interspaced short palindromic repeats
- DSA
donor-specific antibody
- ELISA
enzyme-linked immunosorbent assay
- ELISpot
enzyme-linked immunosorbent spot assay
- g
glomerulitis
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GBM
glomerular basement membrane
- IgG
immunoglobulin G
- IgM
immunoglobulin M
- IL
interleukin
- INFγ
interferon gamma
- MCP
monocyte chemoattractant protein-1
- MFI
mean fluorescence intensity
- MHC
major histocompatibility complex
- mvi
microvascular inflammation
- ptc
peritubular capillaritis
- RNA
ribonucleic acid
- RT-qPCR
quantitative reverse transcription polymerase chain reaction
- Sens
sensitized
- Syn
syngeneic
- TG
transplant glomerulopathy
Footnotes
Disclaimer
The authors declare no conflicts of interest.
REFERENCES
- 1.USRDS. 2018 USRDS Annual Data Report: Epidemiology of Kidney Disease in the United States. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; 2018. [Google Scholar]
- 2.Sellares J, de Freitas DG, Mengel M, et al. Understanding the causes of kidney transplant failure: the dominant role of antibody-mediated rejection and nonadherence. Am J Transplant. 2012;12(2):388–399. [DOI] [PubMed] [Google Scholar]
- 3.Einecke G, Sis B, Reeve J, et al. Antibody-mediated microcirculation injury is the major cause of late kidney transplant failure. Am J Transplant. 2009;9(11):2520–2531. [DOI] [PubMed] [Google Scholar]
- 4.Haas M, Loupy A, Lefaucheur C, et al. The Banff 2017 Kidney Meeting Report: Revised diagnostic criteria for chronic active T cell-mediated rejection, antibody-mediated rejection, and prospects for integrative endpoints for next-generation clinical trials. Am J Transplant. 2018;18(2):293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.El-Zoghby ZM, Stegall MD, Lager DJ, et al. Identifying specific causes of kidney allograft loss. Am J Transplant. 2009;9(3):527–535. [DOI] [PubMed] [Google Scholar]
- 6.Gloor JM, Sethi S, Stegall MD, et al. Transplant glomerulopathy: subclinical incidence and association with alloantibody. Am J Transplant. 2007;7(9):2124–2132. [DOI] [PubMed] [Google Scholar]
- 7.Bentall A, Cornell LD, Gloor JM, et al. Five-year outcomes in living donor kidney transplants with a positive crossmatch. Am J Transplant. 2013;13(1):76–85. [DOI] [PubMed] [Google Scholar]
- 8.Maryniak RK, First MR, Weiss MA. Transplant glomerulopathy: evolution of morphologically distinct changes. Kidney Int. 1985;27(5):799–806. [DOI] [PubMed] [Google Scholar]
- 9.Panzer SE, Joachim E, Parajuli S, et al. Glomerular C3 deposition is an independent risk factor for allograft failure in kidney transplant recipients with transplant glomerulopathy. Kidney Int Rep. 2019;4(4):582–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Issa N, Cosio FG, Gloor JM, et al. Transplant glomerulopathy: risk and prognosis related to anti-human leukocyte antigen class II antibody levels. Transplantation. 2008;86(5):681–685. [DOI] [PubMed] [Google Scholar]
- 11.Smith RN, Kawai T, Boskovic S, et al. Four stages and lack of stable accommodation in chronic alloantibody-mediated renal allograft rejection in Cynomolgus monkeys. Am J Transplant. 2008;8(8):1662–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abreu R, Carvalho F, Viana H, et al. Morphologic patterns and treatment of transplant glomerulopathy: A retrospective analysis. Clin Transplant. 2017;31(4). [DOI] [PubMed] [Google Scholar]
- 13.Loupy A, Suberbielle-Boissel C, Zuber J, et al. Combined posttransplant prophylactic IVIg/anti-CD 20/plasmapheresis in kidney recipients with preformed donor-specific antibodies: a pilot study. Transplantation. 2010;89(11):1403–1410. [DOI] [PubMed] [Google Scholar]
- 14.Kohei N, Hirai T, Omoto K, et al. Chronic antibody-mediated rejection is reduced by targeting B-cell immunity during an introductory period. Am J Transplant. 2012;12(2):469–476. [DOI] [PubMed] [Google Scholar]
- 15.Clatworthy MR, Watson CJ, Plotnek G, et al. B-cell-depleting induction therapy and acute cellular rejection. N Engl J Med. 2009;360(25):2683–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tyden G, Genberg H, Tollemar J, et al. A randomized, doubleblind, placebo-controlled, study of single-dose rituximab as induction in renal transplantation. Transplantation. 2009;87(9):1325–1329. [DOI] [PubMed] [Google Scholar]
- 17.van den Hoogen MW, Kamburova EG, Baas MC, et al. Rituximab as induction therapy after renal transplantation: a randomized, double-blind, placebo-controlled study of efficacy and safety. Am J Transplant. 2015;15(2):407–416. [DOI] [PubMed] [Google Scholar]
- 18.Redfield RR, Ellis TM, Zhong W, et al. Current outcomes of chronic active antibody mediated rejection - A large single center retrospective review using the updated BANFF 2013 criteria. Hum Immunol. 2016;77(4):346–352. [DOI] [PubMed] [Google Scholar]
- 19.Kaposztas Z, Podder H, Mauiyyedi S, et al. Impact of rituximab therapy for treatment of acute humoral rejection. Clin Transplant. 2009;23(1):63–73. [DOI] [PubMed] [Google Scholar]
- 20.Lefaucheur C, Loupy A, Vernerey D, et al. Antibody-mediated vascular rejection of kidney allografts: a population-based study. Lancet. 2013;381(9863):313–319. [DOI] [PubMed] [Google Scholar]
- 21.Parajuli S, Mandelbrot DA, Muth B, et al. Rituximab and monitoring strategies for late antibody-mediated rejection after kidney transplantation. Transplant Direct. 2017;3(12):e227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sautenet B, Blancho G, Büchler M, et al. One-year results of the effects of rituximab on acute antibody-mediated rejection in renal transplantation: RITUX ERAH, a multicenter double-blind randomized placebo-controlled trial. Transplantation. 2016;100(2):391–399. [DOI] [PubMed] [Google Scholar]
- 23.Zarkhin V, Li L, Kambham N, et al. A randomized, prospective trial of rituximab for acute rejection in pediatric renal transplantation. Am J Transplant. 2008;8(12):2607–2617. [DOI] [PubMed] [Google Scholar]
- 24.Moreso F, Crespo M, Ruiz JC, et al. Treatment of chronic antibody mediated rejection with intravenous immunoglobulins and rituximab: A multicenter, prospective, randomized, double-blind clinical trial. Am J Transplant. 2018;18(4):927–935. [DOI] [PubMed] [Google Scholar]
- 25.Jackson AM, Kraus ES, Orandi BJ, et al. A closer look at rituximab induction on HLA antibody rebound following HLA-incompatible kidney transplantation. Kidney Int. 2015;87(2):409–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.White E, Hildemann WH. Allografts in genetically defined rats: Difference in survival between kidney and skin. Science. 1968;162(3859):1293–1295. [DOI] [PubMed] [Google Scholar]
- 27.Joosten SA, van Dixhoorn MG, Borrias MC, et al. Antibody response against perlecan and collagen types IV and VI in chronic renal allograft rejection in the rat. Am J Pathol. 2002;160(4):1301–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hanaoka K, Kawato Y, Kubo K, et al. A chronic renal rejection model with a fully MHC-mismatched rat strain combination under immunosuppressive therapy. Transpl Immunol. 2016;38:19–26. [DOI] [PubMed] [Google Scholar]
- 29.Grau V, Zeuschner P, Immenschuh S, et al. Immune complex-type deposits in the Fischer-344 to Lewis rat model of renal transplantation and a subset of human transplant glomerulopathy. Transplantation. 2016;100(5):1004–1014. [DOI] [PubMed] [Google Scholar]
- 30.Panzer SE, Wilson NA, Verhoven BM, et al. Complete B cell deficiency reduces allograft inflammation and intragraft macrophages in a rat kidney transplant model. Transplantation. 2018;102(3):396–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.White E, Hildemann WH, Mullen Y. Chronic kidney allograft reactions in rats. Transplantation. 1969;8(5):602–617. [DOI] [PubMed] [Google Scholar]
- 32.Huang G, Wilson NA, Reese SR, et al. Characterization of transfusion-elicited acute antibody-mediated rejection in a rat model of kidney transplantation. Am J Transplant. 2014;14(5):1061–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reese SR, Wilson NA, Huang G, et al. Calcineurin inhibitor minimization with ixazomib, an investigational proteasome inhibitor, for the prevention of antibody mediated rejection in a preclinical model. Transplantation. 2015;99(9):1785–1795. [DOI] [PubMed] [Google Scholar]
- 34.Roederer M Interpretation of cellular proliferation data: avoid the panglossian. Cytometry A. 2011;79(2):95–101. [DOI] [PubMed] [Google Scholar]
- 35.Wan SS, Ying TD, Wyburn K, et al. The treatment of antibody-mediated rejection in kidney transplantation: An updated systematic review and meta-analysis. Transplantation. 2018;102(4):557–568. [DOI] [PubMed] [Google Scholar]
- 36.Schinstock CA, Mannon RB, Budde K, et al. Recommended treatment for antibody-mediated rejection after kidney transplantation: The 2019 Expert Consensus from the Transplantion Society Working Group. Transplantation. 2020;104(5):911–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abe T, Ishii D, Gorbacheva V, et al. Anti-huCD20 antibody therapy for antibody-mediated rejection of renal allografts in a mouse model. Am J Transplant. 2015;15(5):1192–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bagnasco SM, Zachary AA, Racusen LC, et al. Time course of pathologic changes in kidney allografts of positive crossmatch HLA-incompatible transplant recipients. Transplantation. 2014;97(4):440–445. [DOI] [PubMed] [Google Scholar]
- 39.Batal I, De Serres SA, Mfarrej BG, et al. Glomerular inflammation correlates with endothelial injury and with IL-6 and IL-1beta secretion in the peripheral blood. Transplantation. 2014;97(10):1034–1042. [DOI] [PubMed] [Google Scholar]
- 40.Nickeleit V, Zeiler M, Gudat F, et al. Detection of the complement degradation product C4d in renal allografts: diagnostic and therapeutic implications. J Am Soc Nephrol. 2002;13(1):242–251. [DOI] [PubMed] [Google Scholar]
- 41.Regele H, Bohmig GA, Habicht A, et al. Capillary deposition of complement split product C4d in renal allografts is associated with basement membrane injury in peritubular and glomerular capillaries: a contribution of humoral immunity to chronic allograft rejection. J Am Soc Nephrol. 2002;13(9):2371–2380. [DOI] [PubMed] [Google Scholar]
- 42.Farrar CA, Tran D, Li K, et al. Collectin-11 detects stress-induced L-fucose pattern to trigger renal epithelial injury. J Clin Invest. 2016;126(5):1911–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nauta AJ, Raaschou-Jensen N, Roos A, et al. Mannose-binding lectin engagement with late apoptotic and necrotic cells. Eur J Immunol. 2003;33(10):2853–2863. [DOI] [PubMed] [Google Scholar]
- 44.Berger SP, Roos A, Mallat MJ, et al. Association between mannose-binding lectin levels and graft survival in kidney transplantation. Am J Transplant. 2005;5(6):1361–1366. [DOI] [PubMed] [Google Scholar]
- 45.Imai N, Nishi S, Alchi B, et al. Immunohistochemical evidence of activated lectin pathway in kidney allografts with peritubular capillary C4d deposition. Nephrol Dial Transplant. 2006;21(9): 2589–2595. [DOI] [PubMed] [Google Scholar]
- 46.Golshayan D, Wojtowicz A, Bibert S, et al. Polymorphisms in the lectin pathway of complement activation influence the incidence of acute rejection and graft outcome after kidney transplantation. Kidney Int. 2016;89(4):927–938. [DOI] [PubMed] [Google Scholar]
- 47.Bobka S, Ebert N, Koertvely E, et al. Is early complement activation in renal transplantation associated with later graft outcome? Kidney Blood Press Res. 2018;43(5):1488–1504. [DOI] [PubMed] [Google Scholar]
- 48.Gorbacheva V, Fan R, Beavers A, et al. Anti-donor MHC class II alloantibody induces glomerular injury in mouse renal allografts subjected to prolonged cold ischemia. J Am Soc Nephrol. 2019;30(12):2413–2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Girlanda R, Kleiner DE, Duan Z, et al. Monocyte infiltration and kidney allograft dysfunction during acute rejection. Am J Transplant. 2008;8(3):600–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bergler T, Jung B, Bourier F, et al. Infiltration of macrophages correlates with severity of allograft rejection and outcome in human kidney transplantation. PLoS One. 2016;11(6):e0156900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brasen JH, Khalifa A, Schmitz J, et al. Macrophage density in early surveillance biopsies predicts future renal transplant function. Kidney Int. 2017;92(2):479–489. [DOI] [PubMed] [Google Scholar]
- 52.Toki D, Zhang W, Hor KL, et al. The role of macrophages in the development of human renal allograft fibrosis in the first year after transplantation. Am J Transplant. 2014;14(9):2126–2136. [DOI] [PubMed] [Google Scholar]
- 53.Mannon RB. Macrophages: contributors to allograft dysfunction, repair, or innocent bystanders? Curr Opin Organ Transplant. 2012;17(1):20–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.De Serres SA, Vadivel N, Mfarrej BG, et al. Monocyte-secreted inflammatory cytokines are associated with transplant glomerulopathy in renal allograft recipients. Transplantation. 2011;91(5):552–559. [DOI] [PubMed] [Google Scholar]
- 55.Hayde N, Bao Y, Pullman J, et al. The clinical and genomic significance of donor-specific antibody-positive/C4d-negative and donor-specific antibody-negative/C4d-negative transplant glomerulopathy. Clin J Am Soc Nephrol. 2013;8(12):2141–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Loupy A, Lefaucheur C, Vernerey D, et al. Molecular microscope strategy to improve risk stratification in early antibody-mediated kidney allograft rejection. J Am Soc Nephrol. 2014;25(10):2267–2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Venner JM, Hidalgo LG, Famulski KS, et al. The molecular landscape of antibody-mediated kidney transplant rejection: evidence for NK involvement through CD16a Fc receptors. Am J Transplant. 2015;15(5):1336–1348. [DOI] [PubMed] [Google Scholar]
- 58.Parkes MD, Halloran PF, Hidalgo LG. Mechanistic sharing between NK cells in ABMR and effector T cells in TCMR. Am J Transplant. 2018;18(1):63–73. [DOI] [PubMed] [Google Scholar]
- 59.Kamal L, Broin PO, Bao Y, et al. Clinical, histological, and molecular markers associated with allograft loss in transplant glomerulopathy patients. Transplantation. 2015;99(9):1912–1918. [DOI] [PubMed] [Google Scholar]
- 60.Lubetzky M, Hayde N, Ó Broin P, et al. Molecular signatures and clinical outcomes of transplant glomerulopathy stratified by microvascular inflammation and donor-specific antibody. Clin Transplant. 2019;33(3):e13469. [DOI] [PubMed] [Google Scholar]
- 61.Halloran PF, Venner JM, Famulski KS. Comprehensive analysis of transcript changes associated with allograft rejection: Combining universal and selective features. Am J Transplant. 2017;17(7):1754–1769. [DOI] [PubMed] [Google Scholar]
- 62.Reeve J, Einecke G, Mengel M, et al. Diagnosing rejection in renal transplants: a comparison of molecular- and histopathology-based approaches. Am J Transplant. 2009;9(8):1802–1810. [DOI] [PubMed] [Google Scholar]
- 63.Famulski KS, Einecke G, Reeve J, et al. Changes in the transcriptome in allograft rejection: IFN-gamma-induced transcripts in mouse kidney allografts. Am J Transplant. 2006;6(6):1342–1354. [DOI] [PubMed] [Google Scholar]
- 64.Muczynski KA, Ekle DM, Coder DM, et al. Normal human kidney HLA-DR-expressing renal microvascular endothelial cells: characterization, isolation, and regulation of MHC class II expression. J Am Soc Nephrol. 2003;14(5):1336–1348. [DOI] [PubMed] [Google Scholar]
- 65.Wilson NA, Dylewski J, Degner KR, et al. An in vitro model of antibody-mediated injury to glomerular endothelial cells: Upregulation of MHC class II and adhesion molecules. Transpl Immunol. 2019:101261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Arias M, Seron D, Herrero I, et al. Subclinical antibody-mediated rejection. Transplantation. 2017;101(6S Suppl 1):S1–S18. [DOI] [PubMed] [Google Scholar]
- 67.Parajuli S, Joachim E, Alagusundaramoorthy S, et al. Subclinical antibody-mediated rejection after kidney transplantation: Treatment outcomes. Transplantation. 2019;103(8):1722–1729. [DOI] [PubMed] [Google Scholar]
- 68.Loupy A, Suberbielle-Boissel C, Hill GS, et al. Outcome of subclinical antibody-mediated rejection in kidney transplant recipients with preformed donor-specific antibodies. Am J Transplant. 2009;9(11):2561–2570. [DOI] [PubMed] [Google Scholar]
- 69.Loupy A, Vernerey D, Tinel C, et al. Subclinical rejection phenotypes at 1 year post-transplant and outcome of kidney allografts. J Am Soc Nephrol. 2015;26(7):1721–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bath NM, Ding X, Wilson NA, et al. Desensitization and treatment with APRIL/BLyS blockade in rodent kidney transplant model. PLoS One. 2019;14(2):e0211865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wilson NA, Bath NM, Verhoven BM, et al. APRIL/BLyS blockade reduces donor-specific antibodies in allosensitized mice. Transplantation. 2019;103(7):1372–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vigna-Perez M, Hernandez-Castro B, Paredes-Saharopulos O, et al. Clinical and immunological effects of Rituximab in patients with lupus nephritis refractory to conventional therapy: a pilot study. Arthritis Res Ther. 2006;8(3):R83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vallerskog T, Gunnarsson I, Widhe M, et al. Treatment with rituximab affects both the cellular and the humoral arm of the immune system in patients with SLE. Clin Immunol. 2007;122(1):62–74. [DOI] [PubMed] [Google Scholar]
- 74.Sfikakis PP, Souliotis VL, Fragiadaki KG, et al. Increased expression of the FoxP3 functional marker of regulatory T cells following B cell depletion with rituximab in patients with lupus nephritis. Clin Immunol. 2007;123(1):66–73. [DOI] [PubMed] [Google Scholar]
- 75.Degner KR, Wilson NA, Reese S, et al. Short-term immunopathological changes associated with pulse steroids/IVIG/rituximab therapy in late kidney allograft antibody mediated rejection. Kidney360. 2020;1(5):389–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sentis A, Diekmann F, Llobell A, et al. Kinetic analysis of changes in T- and B-lymphocytes after anti-CD20 treatment in renal pathology. Immunobiology. 2017;222(4):620–630. [DOI] [PubMed] [Google Scholar]
- 77.Pattanayak V, Lin S, Guilinger JP, et al. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol. 2013;31(9): 839–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.