

Staphylococcus epidermidis is a pathogen emerging worldwide as a leading cause of health care-associated infections. A standardized high-resolution typing method to document transmission and dissemination of multidrug-resistant S. epidermidis strains is needed.
KEYWORDS: Staphylococcus epidermidis, cgMLST, typing
ABSTRACT
Staphylococcus epidermidis is a pathogen emerging worldwide as a leading cause of health care-associated infections. A standardized high-resolution typing method to document transmission and dissemination of multidrug-resistant S. epidermidis strains is needed. Our aim was to provide a core genome multilocus sequence typing (cgMLST) scheme for S. epidermidis to improve the international surveillance of S. epidermidis. We defined a cgMLST scheme based on 699 core genes and used it to investigate the population structure of the species and the genetic relatedness of isolates recovered from infants hospitalized in several wards of a French hospital. Our results show the long-lasting endemic persistence of S. epidermidis clones within and across wards of hospitals and demonstrate the ability of our cgMLST approach to identify and track these clones. We made the scheme publicly available through the Institut Pasteur BIGSdb server (http://bigsdb.pasteur.fr/epidermidis/). This tool should enable international harmonization of the epidemiological surveillance of multidrug-resistant S. epidermidis clones. By comparing gene distribution among infection and commensal isolates, we also confirmed the association of the mecA locus with infection isolates and of the fdh gene with commensal isolates. (This study has been registered at ClinicalTrials.gov under registration no. NCT03374371.)
INTRODUCTION
Staphylococcus epidermidis, a member of coagulase-negative staphylococci, is an abundant and ubiquitous resident of the human skin and mucosal microbiota (1). The most frequently isolated microorganisms in intensive care unit (ICU)-acquired bloodstream infections are coagulase-negative staphylococci, among which S. epidermidis is prominent (2). S. epidermidis infections are almost exclusively associated with health care and occur primarily in patients with implanted medical devices, such as intravascular devices or orthopedic and cardiac implants. In addition, neonates and immunocompromised patients represent a population at particular risk for nosocomial infections (3). The ability of S. epidermidis to form biofilms contributes to the chronicity and difficulties to treat these infections (3, 4).
The proportion of methicillin-resistant S. epidermidis (MRSE) among clinical isolates is greater than 70% (5), leading to the probabilistic prescription of vancomycin in patients with suspected S. epidermidis infections. Resistance to linezolid is rare but emerging (5, 6). The emergence and global spread of multidrug-resistant S. epidermidis clones causing invasive diseases have been largely documented (7, 8). In addition, the transmission of S. epidermidis strains among patients and between patients and medical staff, mostly in neonatal intensive care units, is possible (3, 9–11). However, the significance of S. epidermidis transmission is likely to be underestimated, and the identification of subtypes that could represent higher risks for transmission or pathogenesis remains difficult using available subtyping methods.
The classification of S. epidermidis isolates has so far largely relied on multilocus sequence typing (MLST) based on the sequencing of 7 conserved loci (12). This typing method allows the assignment of a sequence type (ST) to each unique allelic profile. STs can be grouped into clonal complexes (CCs) with the eBURST algorithm (13). While an extensive strain level diversity has been observed between healthy individuals (1), only a few health care-associated clones are found in hospital environments worldwide (4, 14). Indeed, more than 50% of the clinical strains and less than 10% of the community strains belong to a clonal complex encompassing ST2 and ST5 (4, 5, 14). The global dissemination of these two genotypes suggests an adaptation to the hospital environment. ST2 strains are almost all resistant to methicillin (15, 16). Similar to the closely related Staphylococcus aureus species, the mecA gene confers resistance to methicillin and is found within the staphylococcal cassette chromosome mec (SCCmec) genomic island (17, 18).
The high-resolution whole-genome sequencing (WGS) approach has been used to study S. epidermidis linezolid-resistant isolate epidemiology (6, 19). A major insight from WGS-based epidemiological studies is the description of long-lasting endemic persistence of S. epidermidis clones across wards of hospitals (20, 21).
The population structure of S. epidermidis is divided into three main phylogenetic lineages (17, 22), which encompass six previously defined clonal groups (23, 24). These subsets of S. epidermidis show distinct associations with clinical infections and carriage. In addition, they have distinctive genetic and phenotypic features, including association with biofilm synthesis genes, SSCmec, and antimicrobial resistance. However, both asymptomatic carriage and invasive isolates are found in all major phylogenetic groups (22), and the epidemiological relationships of carriage and invasive isolates need to be deciphered at microevolutionary timescales.
Genotyping of bacterial isolates is best performed with WGS for molecular epidemiological studies (25–28). Core genome MLST (cgMLST) provides the high resolution of genome-wide methods while maintaining the benefits of standardization, reproducibility, and portability of classical 7-gene MLST (28, 29). While infections with multidrug-resistant S. epidermidis strains are an increasingly important issue in hospitals worldwide, an effective tool for international strain comparison is still lacking.
The main aims of this study were to (i) define a cgMLST scheme for S. epidermidis genotyping, (ii) provide a publicly accessible international database for S. epidermidis genomic typing, and (iii) investigate the genetic relatedness of isolates recovered from infants hospitalized in several wards of a French hospital, including a neonatal intensive care unit (NICU). We also explored the population structure of S. epidermidis and the association of previously reported marker genes and lineages.
MATERIALS AND METHODS
Routine procedures for bacterial isolation from clinical samples.
The 25 isolates sequenced here were recovered from one healthy adult and from cultures performed routinely on clinical samples in the clinical microbiology laboratory at Necker-Enfants Malades Hospital (Paris, France). The strains were grown on Columbia agar supplemented with 5% horse blood and incubated 24 to 48 h at 37°C (Incucell incubator; MMM group, Germany). Coagulase-negative staphylococcus isolates were identified at the species level by matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS) (Bruker Daltonics, Bremen, Germany).
cgMLST scheme definition.
For the determination of the cgMLST scheme, 5 closed S. epidermidis genomes and 299 draft genome assemblies available at NCBI GenBank (on 22 December 2015) were downloaded. We also downloaded 83 draft genome assemblies deposited by Meric et al. (17) in the Dryad repository. All used assemblies are publicly available from the Institut Pasteur instance of the BIGSdb at https://bigsdb.pasteur.fr/epidermidis/.
Low-quality assemblies were excluded based on the following criteria: assembled genome size of >150% of the reference genome, organism identified as non-S. epidermidis as determined using Kraken v0.10.5 (30), and average nucleotide identity of <95% compared to the ATCC 12228 strain reference (RefSeq accession number GCF_000007645) (31). This resulted in the selection of 371 genomes for core gene definition. Table S1 in the supplemental material provides a detailed description of the strain collection used for the establishment of the cgMLST scheme (e.g., date of isolation, geographic origin, and source).
From this set, we inferred the species core genome using the CoreGeneBuilder pipeline default settings (26) and the S. epidermidis ATCC 12228 strain (RefSeq accession number GCF_000007645) as a reference. The pipeline’s first step relies on the eCAMBer software (32), which consists of a de novo annotation of the genomes (except the reference) using Prodigal (33) and the harmonization of the positions of the stop and start codons. In the next step, the core genome is inferred with a bidirectional best hits (BBH) approach as previously described by Touchon et al. (34). We used CoreGeneBuilder default settings. A gene was considered as part of the core genome if it was found in at least 95% of the genomes under study. This resulted in an initial core genome containing 1,623 genes. We then filtered out some genes based on the following criteria. (i) First, we removed 14 potential paralogous loci. The rationale behind this is that the presence of paralogs inside a typing scheme can lead to ambiguities, as a candidate gene might be attributable to two different core gene loci. To detect those potential paralogs, we compared each allele of each locus against all of the alleles of all of the other loci using the software BLAT (35). If a single hit was found between two different loci (more than 70% protein identity between two alleles), we removed both loci. (ii) Second, we removed 840 loci whose length varies among alleles, which is useful in reducing ambiguities during the genotyping process. (iii) We also removed 10 loci for which alleles contained ambiguous characters. (iv) Last, we removed 582 loci with variation within the start and stop codons; the rationale is that allele calling relies on blastn, which may not produce full-length alignments when the start or stop codons differ, resulting in incomplete alleles. Note that these sets of filtered loci can overlap. After applying these four steps, 699 core genes were retained and constitute our cgMLST scheme.
cgMLST allele definitions and tagging of publicly available genomes using BIGSdb.
The BIGSdb server is designed to host separately a database of allele sequence definitions and a database of genomic assemblies with attached source information (36). Newly imported assemblies were scanned using BIGSdb to designate alleles of each locus. A locus was tagged when its sequence had >90% nucleotide sequence identity over at least 90% of the alignment length with a reference allele. In the case of an exact match with an allele found in the database, the locus was designated by the allele number. In case of partial match, if the locus corresponded to a protein-coding sequence (beginning with a start codon and ending with a stop codon), the allele was added to the allele database. Then, a subsequent scan allowed tagging isolates with novel alleles. A locus was considered missing when there was no match with alleles in the database.
To evaluate the conservation of the chosen genes in an independent sample of the S. epidermidis population, we analyzed 289 additional genome assemblies from NCBI GenBank and from Harris et al. (4) that were not yet available at the time of scheme definition. The validation set is described in Table S1.
All assemblies, allele sequence designations, and profiles are available at http://bigsdb.pasteur.fr/epidermidis/.
Assessment of the population structure of S. epidermidis.
The population structure of S. epidermidis was assessed based on 703 available genomes. To obtain the alignment required for the phylogenetic analysis, we extracted, for each genome assembly, the allelic sequences of the 699 loci. For each locus, the allele sequences were translated to proteins, aligned using MAFFT (37) and the G-INS-i algorithm, and back-translated to nucleotide sequences, thus resulting in codon alignments. We finally concatenated the 699 alignments into one supermatrix, containing 703 sequences (isolates) and 539,151 sites (the nucleotide positions of missing alleles were replaced by gaps in the concatenation of alignments of individual loci). A phylogenetic tree was obtained with the maximum likelihood (ML) criterion after purging recombination using Gubbins v2.2.0 (38) with default parameters resulting in an alignment of 31,385 sites. RAxML v8.2.8 (39) was used for the phylogenetic inference under the GTRCAT model (final tree optimized under the GTRGAMMA model). The tree was drawn using iTOL (40).
The same methodology was applied to compare clusters obtained based on cgMLST allelic profiles with those observed by phylogenetic analysis of the concatenated allele sequences of the 699 core loci.
Patients and isolates.
To evaluate the ability of cgMLST to identify potential transmission events within hospital settings, we selected 25 isolates including 24 clinical isolates from patients hospitalized in several wards between November 2014 and August 2016.
The 25 isolates were recovered from 21 hospitalized individuals (16 hospitalized neonates and 5 hospitalized children) and one nonhospitalized healthy adult. All neonates have stayed in a NICU, 3 children have stayed in a gastroenterology ward, and 2 children have stayed in an orthopedic ward. One isolate was sequenced per patient except for 3 patients who had 2 isolates sequenced (patients 3, 7, and 21). Information on patients and sources of isolates is summarized in Table 1.
TABLE 1.
S. epidermidis isolates sequenced in this study
| Isolate (BIGSdb ID) | Isolation date (yr/mo/day) | Patient information (patient no.) | ST | Source | Methicillin susceptibility | Ward |
|---|---|---|---|---|---|---|
| NCK_SE0001 (510) | 15/12/24 | Healthy adult | Skin | MSSEa | Community | |
| NCK_SE0002 (54) | 14/12/17 | Hospitalized child (1) | 2 | Blood | MRSE | Gastroenterology |
| NCK_SE0004 (59) | 14/12/10 | Hospitalized child (2) | 5 | Blood | MRSE | Gastroenterology |
| NCK_SE0005 (511) | 15/7/10 | Hospitalized neonate (3) | 2 | Blood | MRSE | NICU |
| NCK_SE0006 (520) | 15/7/17 | Hospitalized neonate (3) | 2 | Blood | MRSE | NICU |
| NCK_SE0008 (521) | 15/10/16 | Hospitalized neonate (4) | 2 | Blood | MRSE | NICU |
| NCK_SE0009 (522) | 15/12/1 | Hospitalized neonate (5) | 2 | Blood | MRSE | NICU |
| NCK_SE0010 (512) | 16/4/18 | Hospitalized neonate (6) | 2 | Blood | MRSE | NICU |
| NCK_SE0011 (523) | 15/10/24 | Hospitalized neonate (7) | 5 | Blood | MRSE | NICU |
| NCK_SE0012 (524) | 15/11/13 | Hospitalized neonate (7) | 5 | Central catheter | MRSE | NICU |
| NCK_SE0013 (513) | 16/6/17 | Hospitalized neonate (8) | 2 | Blood | MRSE | NICU |
| NCK_SE0014 (525) | 15/11/24 | Hospitalized neonate (9) | 2 | Blood | MRSE | NICU |
| NCK_SE0015 (526) | 15/12/18 | Hospitalized neonate (10) | 22 | Blood | MRSE | NICU |
| NCK_SE0016 (527) | 15/12/19 | Hospitalized neonate (11) | 5 | Blood | MRSE | NICU |
| NCK_SE0017 (514) | 16/7/25 | Hospitalized neonate (12) | 2 | Blood | MRSE | NICU |
| NCK_SE0019 (515) | 16/7/12 | Hospitalized neonate (13) | 2 | Blood | MRSE | NICU |
| NCK_SE0020 (528) | 16/6/27 | Hospitalized neonate (14) | 22 | Blood | MRSE | NICU |
| NCK_SE0021 (529) | 15/12/15 | Hospitalized children (15) | 5 | Bone biopsy | MRSE | Orthopedic |
| NCK_SE0022 (530) | 16/5/19 | Hospitalized neonate (16) | 2 | Trachea | MRSE | NICU |
| NCK_SE0023 (516) | 16/1/19 | Hospitalized neonate (17) | 5 | Postoperative wound | MRSE | NICU |
| NCK_SE0027 (518) | 16/6/26 | Hospitalized neonate (18) | 2 | Blood | MRSE | NICU |
| NCK_SE0028 (519) | 16/4/15 | Hospitalized neonate (19) | 22 | Blood | MRSE | NICU |
| NCK_SE0029 (75) | 14/12/8 | Hospitalized child (20) | 59 | Blood | MSSE | Gastroenterology |
| NCK_SE0030 (82) | 15/4/10 | Hospitalized child (21) | 5 | Wound biopsy | MRSE | Orthopedic |
| NCK_SE0031 (99) | 15/5/18 | Hospitalized child (21) | 5 | Bone biopsy | MRSE | Orthopedic |
MSSE, methicillin susceptible S. epidermidis.
The study of S. epidermidis clinical isolates collected from the Necker-Enfants Malades Hospital was approved by the ethics review board of our institution and registered with ClinicalTrials.gov under registration number NCT03374371. All experiments were performed in accordance with the guidelines and regulations described by the Declaration of Helsinki and the Huriet-Serusclat law on human research ethics, and informed consent was obtained for all participating subjects.
Whole-genome sequencing and assembly.
DNA extractions were performed on pure culture using DNeasy blood and tissue kit (Qiagen) following the manufacturer’s instructions with addition of lysostaphin (Sigma) to the Gram-positive lysis buffer (final concentration, 20 μg/ml).
Whole-genome shotgun libraries sequenced on an Illumina MiSeq sequencing platform (2 × 150-bp paired-end reads) were prepared with the Nextera XT kit (Illumina). Whole-genome shotgun libraries sequenced on an Illumina HiSeq 2500 sequencing platform (2 × 130-bp paired-end reads) were prepared with the Ovation Ultralow V2 kit (NuGEN).
Reads were trimmed using Trimmomatic v0.36 (41) and assembled using SPAdes v3.9.0 (42). De novo assembly quality was assessed with QUAST (43). In silico MLST was performed on the de novo assemblies with MLST v2.6 (https://github.com/tseemann/mlst). De novo assemblies were imported and scanned in BIGSdb.
Cluster analysis of clinical isolates using BIGSdb.
To validate the ability of cgMLST to cluster related isolates, we first used sequence data from a recently published study of linezolid-resistant isolates (6). Raw Illumina reads (accession number SRP039360) were downloaded from NCBI Sequence Read Archive (SRA) and processed as newly sequenced genomes. Assemblies were imported and scanned in BIGSdb except isolate UCLA-Sa-LR-01, which was discarded due to the poor quality of its de novo assembly. We also tested our cgMLST scheme using the 25 newly sequenced clinical isolates (Table 1). The PHYLOViZ tool (integrated as a plugin of BIGSdb) was used to generate minimum spanning trees based on the cgMLST allelic profiles (44). Here, clusters were defined as groups of genotypes showing ≤21 cgMLST allele mismatches with at least one other member of the group.
Data availability.
This whole-genome shotgun project has been deposited in GenBank under BioProject number PRJNA608162. All assemblies, sequences, and their allelic designations and cgMLST profiles are available from the Institut Pasteur BIGSdb access Web page at https://bigsdb.pasteur.fr/epidermidis/.
RESULTS
Definition and evaluation of a cgMLST scheme for S. epidermidis using public genomes.
Using 371 publicly available genomes, we developed a cgMLST scheme consisting of 699 core genes, which includes 30% of the 2,348 coding DNA sequences (CDS) of the S. epidermidis ATCC 12228 reference genome. As shown in Fig. S1 in the supplemental material, the distribution along the S. epidermidis genome of the 699 cgMLST genes was even.
To evaluate the conservation of gene loci of the newly defined cgMLST scheme, a validation set of 289 additional publicly available genome assemblies was analyzed (NCBI genomes, last accessed on 27 September 2017). These additional genomes were made public after the definition of our cgMLST scheme and thus represent a nonoverlapping strain collection. These genomes had 97.5% tagged loci on average, showing that the cgMLST scheme loci are highly conserved and can, therefore, be expected to be mostly present in future sequenced genomes. Figure S2 in the supplemental material shows the distribution of the number of missing loci among the 699 genes of the scheme.
The cgMLST genotyping system is publicly available through the Institut Pasteur BIGSdb server (https://bigsdb.pasteur.fr/epidermidis/).
Population structure of S. epidermidis genomes.
We next used 703 genomes to assess the phylogenetic structure of S. epidermidis based on cgMLST gene sequences (see Fig. S1), which confirmed that the population is divided into 3 main clades (A to C) as previously reported (17). Most of the isolates belong to clade A, which encompasses clonal groups 5 and 6 associated with the two major nosocomial clones ST2 and ST5, respectively (23, 24).
Figure S3 in the supplemental material shows the concordance with clades A, B, and C as defined previously by Meric and colleagues (17), with the clonal groups defined previously (23, 24) and with classical 7-gene MLST.
In addition to the core genes, we scanned the assemblies for the presence of accessory genes associated with the SCCmec locus (mecA, ccrA, and ccrB) and the ica genes associated with biofilm formation capacity through polymeric N-acetylglucosamine (PNAG) synthesis. In addition, we searched for the putative commensal marker fdh encoding a formate dehydrogenase (14).
The distribution of SCCmec-associated genes among infection and commensal isolates showed a positive association between infection strains and the SCCmec locus (Fisher, <1e−3) (Fig. 1). The search of the cooccurrence of the icaAD genes, commonly used as a proxy to evaluate the biofilm formation potential of isolates (45), yielded no significant association (Fig. 1). In contrast, a positive association between commensal isolates compared to infection isolates was observed for the putative commensal marker gene fdh (Fisher, <0.005) (Fig. 1).
FIG 1.
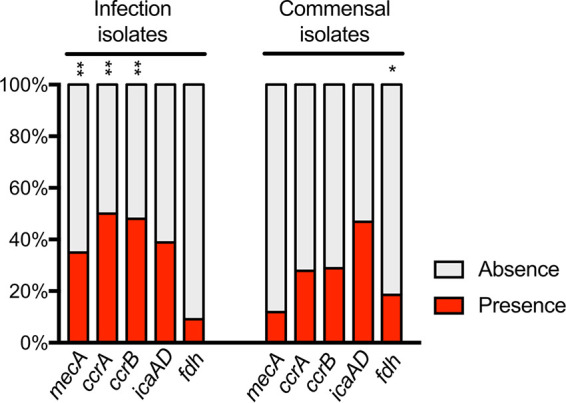
Distribution of gene presence and absence for mecA, ccrAB, ica, and fdh loci among 252 infection isolates and 194 commensal isolates from the BIGSdb. mecA encodes penicillin-binding protein 2 (PBP2); ccrA and ccrB encode cassette chromosome recombinase A and B, respectively; icaA and icaD genes are involved in polysaccharide intercellular adhesin (PIA) production; fdh encodes a formate dehydrogenase. There was a positive association between infection isolates and mecA, ccrA, and ccrB loci compared to commensal isolates (**, Fisher < 0.001), and there was a positive association between commensal isolates and fdh locus compared to infection isolates (*, Fisher < 0.005).
Application of the S. epidermidis cgMLST scheme to hospital linezolid-resistant isolates.
To evaluate the ability of cgMLST to cluster S. epidermidis isolates, we analyzed the relationships among isolates from a previous study of linezolid-resistant isolates (6).
All genomes had >93% of tagged loci (95.5% on average). Cluster analysis based on cgMLST allelic profiles showed 4 clusters and 1 outgroup isolate (Fig. 2). The same clusters were found after phylogenetic analysis of the concatenated sequences of core genes (Fig. 2). These results were concordant with the ones reported by Tewhey et al. based on the alignments of 2,093 core genes (6). However, there was no description of potential epidemiological links between the isolates of this study, which were compared only because of their common phenotypic linezolid resistance.
FIG 2.
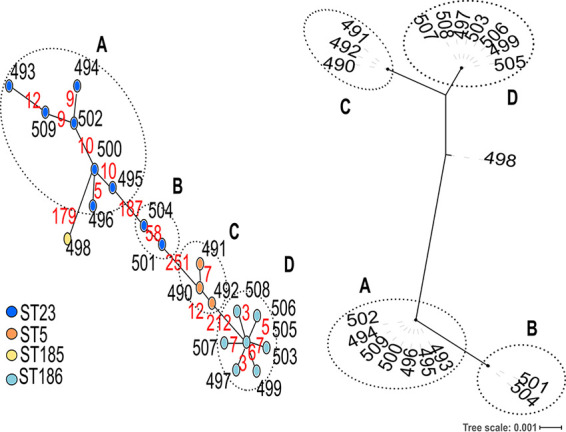
Comparison of cluster analysis based on cgMLST allelic profiles with phylogenetic analysis based on core gene alignments for 20 linezolid-resistant isolates. The minimum spanning tree based on cgMLST allelic profiles is shown on the left side. The tree was generated from allelic profiles of 699 core genes and visualized with PHYLOViZ. Each node represents an isolate and is colored according to the 7-locus ST. In silico MLST performed on the de novo assemblies revealed that isolates UCLA-Sa-LR-14 (497) and UCLA-Sa-LR-21 (503) had been erroneously assigned to ST24 and ST186-like, respectively, in the original publication (6). We found that in fact both strains belonged to ST186. Black numbers next to the nodes indicate the BIGSdb identifiers of each isolate. Red numbers on the connecting lines indicate the number of core gene allele differences between adjacent nodes. Link lengths are loosely related to the number of allelic differences. The tree based on 699 core gene sequence alignments is shown on the right side. The tip labels indicate the BIGSdb identifiers of each isolate. Clusters reported by Tewhey et al. based on the alignments of 2,093 core genes (6) are indicated by circles and letters (A, B, C, and D).
Application to S. epidermidis epidemiology in a French hospital.
To assess the ability of cgMLST to cluster S. epidermidis isolates with defined epidemiological sources, we next sequenced 24 clinical isolates recovered from several wards of a pediatric French hospital and an isolate from a healthy adult (total of 25 sequenced isolates). The mean number of contigs was 211 (range, 137 to 410) (see Table S2 in the supplemental material). After cgMLST genotyping of the genomic assemblies, the mean percentage of tagged loci was 97.6% (range, 96.1% to 100%) (Table S2). As expected, most isolates recovered from hospitalized patients (20 out of 24) belonged to ST2 (n = 12) and ST5 (n = 8) (Table 1).
Nineteen isolates were grouped into 6 clusters (≤21 allele differences), whereas there were 6 ungrouped isolates (Fig. 3). Five clusters were observed among isolates from the NICU during 1 year (span time of clusters ranged from 49 to 354 days), whereas another cluster contained isolates from the orthopedic and the gastroenterology wards. The maximum number of differing alleles among isolates of the same cluster was 21, whereas the minimum number of differing alleles between isolates of distinct clusters was 39 (Fig. 3). For the 3 pairs of isolates recovered from 3 individual patients, collected between 7 and 38 days apart, we observed a maximum of 7 differences. The timeline shown in Fig. 4 illustrates that isolates belonging to the same cluster were sometimes found in patients hospitalized in distinct wards (i.e., cluster IV in patients hospitalized in gastroenterology and orthopedic units) and in patients hospitalized up to 1 year apart (i.e., cluster I for patients 3, 16, and 18).
FIG 3.

Comparison of clustering analyses based on cgMLST allelic profiles and on core gene sequences alignment for 25 S. epidermidis isolates sequenced in this study. The minimum spanning tree based on cgMLST allelic profiles is shown on the left side. The tree was generated from allelic profiles of 699 core genes. Each node represents an allelic profile and corresponds to a single isolate except the node corresponding to cluster VI where 2 isolates have the same profile. Nodes are colored according to the ward where patients were hospitalized. Black numbers next to the nodes indicate the BIGSdb identifiers of each isolate. Red numbers on the connecting lines indicate the number of core gene allele differences between adjacent nodes. Link lengths are not proportional to the number of allelic differences. Clusters of closely related genotypes (≤21 allele differences) are surrounded by circles and are numbered (I to VI). The tree based on 699 core gene alignment is shown on the right side. The tip labels indicate the BIGSdb identifiers and names of each isolate. Clusters are indicated by circles and are numbered (I to VI) as in the minimum spanning tree.
FIG 4.

Timeline of the 21 hospitalized patients from which 24 S. epidermidis isolates were sequenced. The patient numbers indicated in the colored boxes and the patient isolate numbers indicated between brackets correspond to those in Table 1. The clusters to which isolates belong correspond to those in Fig. 3. The isolate sequence type (ST) is indicated on the right side of the graph.
DISCUSSION
S. epidermidis is a leading cause of bacteremia and device-related infections and can cause severe infections in immunocompromised patients and neonates. S. epidermidis infections are mostly caused by strains belonging to multidrug-resistant nosocomial clones ST2 and ST5 (5, 10, 19, 46). Our results corroborate the predominance of ST2 and ST5 among hospitalized patients. Although 7-gene MLST has played an important role in recognizing the worldwide emergence and nosocomial endemic persistence of multidrug-resistant clones, there is a need for higher-resolution genotyping, allowing strain comparison within endemic clones. Moreover, standardization is paramount for worldwide tracking of strain dissemination and recognizing subtypes with medically important properties. We, therefore, explored a core genome MLST (cgMLST) approach, which provides a high resolution and has the potential for a universal typing method both for epidemiological investigations and for population biology studies. Standardized cgMLST schemes are available for many pathogens including Klebsiella pneumoniae, Campylobacter jejuni, Campylobacter coli, Staphylococcus aureus, and Listeria monocytogenes (28, 47–49). However, a cgMLST scheme has been lacking until now for S. epidermidis.
The potential of the developed S. epidermidis cgMLST scheme to delineate closely related isolates was investigated by comparing genomes obtained from children hospitalized in several wards of the same hospital. Our analysis showed that the cgMLST scheme of 699 genes had an adequate discriminatory power to delineate distinct clusters of very closely related isolates within the same ST. The maximum allelic distance among epidemiologically related isolates (2 isolates recovered from 2 patients hospitalized several months apart in the orthopedic ward) was 21, and we chose this cutoff to define clusters. This relatively high number of differences is likely to reflect the fact that clones are able to persist several years in hospitals where they diversify and are likely transferred to patients on multiple occasions (7, 20, 21). For instance, Dortet et al. investigated a long-lasting outbreak spanning 2 years caused by linezolid-resistant S. epidermidis clones and report 22 ST2 isolates with 13 single nucleotide polymorphisms (SNPs) along their core genome (19). Our findings also show that several clones can coexist and persist over months in a NICU. This is concordant with previous observations showing that patients acquire genetically related strains belonging to multidrug-resistant endemic clones that persist in clinical settings (10, 50).
In addition, we leveraged the ability of the BIGSdb tool to analyze the presence of genes of medical interest to compare the distribution of several putative marker genes associated with infection or carriage. We confirmed a positive association between infection strains and the SCCmec locus (22) and a positive association between commensal isolates and the fdh gene (14). The cooccurrence of icaAD genes has been proposed as a marker for biofilm formation ability (45) linked with isolate virulence. However, the presence of the ica locus does not always correlate with biofilm production in vitro (45). For instance, in a study of 200 S. epidermidis isolates from hospitalized patients in Germany, among 124 icaAD-positive isolates, only 49% were able to produce biofilm in vitro, whereas only 50% of in vitro biofilm-producing isolates were ica positive (51). The weak correlation between ica presence and biofilm formation ability is corroborated by Du et al. who reported biofilm production in less than 60% of their icaA-positive isolates (52).
In another study, when nosocomial isolates associated with infection or contamination were compared, only mecA, but neither icaA nor fdh, was differentially found among infection and contamination isolates (24).
In conclusion, we developed a cgMLST scheme for WGS-based typing of S. epidermidis, integrated it in a publicly available BIGSdb Web application platform, and illustrated its application to the molecular epidemiology of S. epidermidis in a French university hospital. We hope that the novel genotyping tool will contribute to allowing reproducible comparisons among clinical laboratories for the epidemiological surveillance and biological studies of S. epidermidis clones causing infections.
Supplementary Material
ACKNOWLEDGMENTS
We thank C. Bréchot for having stimulated the collaboration between the Necker Hospital and Institut Pasteur in the frame of the Pasteur International biobanking network (PIBnet) program. We thank Bryan Brancotte for maintenance of the Institut Pasteur Web application.
We declare that there are no conflicts of interest.
This work was funded by Contrat de Recherche Clinique, French Ministry of Health (CRC17040); the sponsor was Assistance Publique des Hôpitaux de Paris (APHP) (Département de la Recherche Clinique et du Dévelopement).
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Oh J, Byrd AL, Deming C, Conlan S, Program NCS, Kong HH, Segre JA, NISC Comparative Sequencing Program. 2014. Biogeography and individuality shape function in the human skin metagenome. Nature 514:59–64. doi: 10.1038/nature13786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wisplinghoff H, Bischoff T, Tallent SM, Seifert H, Wenzel RP, Edmond MB. 2004. Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin Infect Dis 39:309–317. doi: 10.1086/421946. [DOI] [PubMed] [Google Scholar]
- 3.Becker K, Heilmann C, Peters G. 2014. Coagulase-negative staphylococci. Clin Microbiol Rev 27:870–926. doi: 10.1128/CMR.00109-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harris LG, Murray S, Pascoe B, Bray J, Meric G, Mageiros L, Wilkinson TS, Jeeves R, Rohde H, Schwarz S, de Lencastre H, Miragaia M, Rolo J, Bowden R, Jolley KA, Maiden MCJ, Mack D, Sheppard SK. 2016. Biofilm morphotypes and population structure among Staphylococcus epidermidis from commensal and clinical samples. PLoS One 11:e0151240. doi: 10.1371/journal.pone.0151240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mendes RE, Deshpande LM, Costello AJ, Farrell DJ. 2012. Molecular epidemiology of Staphylococcus epidermidis clinical isolates from U.S. hospitals. Antimicrob Agents Chemother 56:4656–4661. doi: 10.1128/AAC.00279-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tewhey R, Gu B, Kelesidis T, Charlton C, Bobenchik A, Hindler J, Schork NJ, Humphries RM. 2014. Mechanisms of linezolid resistance among coagulase-negative staphylococci determined by whole-genome sequencing. mBio 5:e00894-14. doi: 10.1128/mBio.00894-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee JYH, Monk IR, Goncalves da Silva A, Seemann T, Chua KYL, Kearns A, Hill R, Woodford N, Bartels MD, Strommenger B, Laurent F, Dodemont M, Deplano A, Patel R, Larsen AR, Korman TM, Stinear TP, Howden BP. 2018. Global spread of three multidrug-resistant lineages of Staphylococcus epidermidis. Nat Microbiol 3:1175–1185. doi: 10.1038/s41564-018-0230-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li X, Arias CA, Aitken SL, Galloway Pena J, Panesso D, Chang M, Diaz L, Rios R, Numan Y, Ghaoui S, DebRoy S, Bhatti MM, Simmons DE, Raad I, Hachem R, Folan SA, Sahasarabhojane P, Kalia A, Shelburne SA. 2018. Clonal emergence of invasive multidrug-resistant Staphylococcus epidermidis deconvoluted via a combination of whole-genome sequencing and microbiome analyses. Clin Infect Dis 67:398–406. doi: 10.1093/cid/ciy089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roach DJ, Burton JN, Lee C, Stackhouse B, Butler-Wu SM, Cookson BT, Shendure J, Salipante SJ. 2015. A year of infection in the intensive care unit: prospective whole genome sequencing of bacterial clinical isolates reveals cryptic transmissions and novel microbiota. PLoS Genet 11:e1005413. doi: 10.1371/journal.pgen.1005413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Soeorg H, Huik K, Parm U, Ilmoja ML, Metsvaht T, Lutsar I. 2017. Molecular epidemiology of Staphylococcus epidermidis in neonatal intensive care units. APMIS 125:63–73. doi: 10.1111/apm.12637. [DOI] [PubMed] [Google Scholar]
- 11.Milisavljevic V, Wu F, Cimmotti J, Haas J, Della-Latta P, Larson E, Saiman L. 2005. Genetic relatedness of Staphylococcus epidermidis from infected infants and staff in the neonatal intensive care unit. Am J Infect Control 33:341–347. doi: 10.1016/j.ajic.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 12.Thomas JC, Vargas MR, Miragaia M, Peacock SJ, Archer GL, Enright MC. 2007. Improved multilocus sequence typing scheme for Staphylococcus epidermidis. J Clin Microbiol 45:616–619. doi: 10.1128/JCM.01934-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feil EJ, Li BC, Aanensen DM, Hanage WP, Spratt BG. 2004. eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J Bacteriol 186:1518–1530. doi: 10.1128/jb.186.5.1518-1530.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Conlan S, Mijares LA, Program NCS, Becker J, Blakesley RW, Bouffard GG, Brooks S, Coleman H, Gupta J, Gurson N, Park M, Schmidt B, Thomas PJ, Otto M, Kong HH, Murray PR, Segre JA, NISC Comparative Sequencing Program. 2012. Staphylococcus epidermidis pan-genome sequence analysis reveals diversity of skin commensal and hospital infection-associated isolates. Genome Biol 13:R64. doi: 10.1186/gb-2012-13-7-r64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Argudin MA, Vanderhaeghen W, Vandendriessche S, Vandecandelaere I, Denis O, Coenye T, Butaye P. 2015. Biofilm formation of ica operon-positive Staphylococcus epidermidis from different sources. APMIS 123:1081–1089. doi: 10.1111/apm.12472. [DOI] [PubMed] [Google Scholar]
- 16.Lee JY, Monk IR, Pidot SJ, Singh S, Chua KY, Seemann T, Stinear TP, Howden BP. 2016. Functional analysis of the first complete genome sequence of a multidrug resistant sequence type 2 Staphylococcus epidermidis. Microb Genom 2:e000077. doi: 10.1099/mgen.0.000077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meric G, Miragaia M, de Been M, Yahara K, Pascoe B, Mageiros L, Mikhail J, Harris LG, Wilkinson TS, Rolo J, Lamble S, Bray JE, Jolley KA, Hanage WP, Bowden R, Maiden MC, Mack D, de Lencastre H, Feil EJ, Corander J, Sheppard SK. 2015. Ecological overlap and horizontal gene transfer in Staphylococcus aureus and Staphylococcus epidermidis. Genome Biol Evol 7:1313–1328. doi: 10.1093/gbe/evv066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gill SR, Fouts DE, Archer GL, Mongodin EF, Deboy RT, Ravel J, Paulsen IT, Kolonay JF, Brinkac L, Beanan M, Dodson RJ, Daugherty SC, Madupu R, Angiuoli SV, Durkin AS, Haft DH, Vamathevan J, Khouri H, Utterback T, Lee C, Dimitrov G, Jiang L, Qin H, Weidman J, Tran K, Kang K, Hance IR, Nelson KE, Fraser CM. 2005. Insights on evolution of virulence and resistance from the complete genome analysis of an early methicillin-resistant Staphylococcus aureus strain and a biofilm-producing methicillin-resistant Staphylococcus epidermidis strain. J Bacteriol 187:2426–2438. doi: 10.1128/JB.187.7.2426-2438.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dortet L, Glaser P, Kassis-Chikhani N, Girlich D, Ichai P, Boudon M, Samuel D, Creton E, Imanci D, Bonnin R, Fortineau N, Naas T. 2018. Long-lasting successful dissemination of resistance to oxazolidinones in MDR Staphylococcus epidermidis clinical isolates in a tertiary care hospital in France. J Antimicrob Chemother 73:41–51. doi: 10.1093/jac/dkx370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chong J, Quach C, Blanchard AC, Poliquin PG, Golding GR, Laferriere C, Levesque S. 2016. Molecular epidemiology of a vancomycin-intermediate heteroresistant Staphylococcus epidermidis outbreak in a neonatal intensive care unit. Antimicrob Agents Chemother 60:5673–5681. doi: 10.1128/AAC.00726-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rodriguez-Lucas C, Rodicio MR, Camara J, Dominguez MA, Alaguero M, Fernandez J. 2019. Long-term endemic situation caused by a linezolid- and meticillin-resistant clone of Staphylococcus epidermidis in a tertiary hospital. J Hosp Infect 105:64–69. doi: 10.1016/j.jhin.2019.10.013. [DOI] [PubMed] [Google Scholar]
- 22.Meric G, Mageiros L, Pensar J, Laabei M, Yahara K, Pascoe B, Kittiwan N, Tadee P, Post V, Lamble S, Bowden R, Bray JE, Morgenstern M, Jolley KA, Maiden MCJ, Feil EJ, Didelot X, Miragaia M, de Lencastre H, Moriarty TF, Rohde H, Massey R, Mack D, Corander J, Sheppard SK. 2018. Disease-associated genotypes of the commensal skin bacterium Staphylococcus epidermidis. Nat Commun 9:5034. doi: 10.1038/s41467-018-07368-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thomas JC, Zhang L, Robinson DA. 2014. Differing lifestyles of Staphylococcus epidermidis as revealed through Bayesian clustering of multilocus sequence types. Infect Genet Evol 22:257–264. doi: 10.1016/j.meegid.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tolo I, Thomas JC, Fischer RSB, Brown EL, Gray BM, Robinson DA. 2016. Do Staphylococcus epidermidis genetic clusters predict isolation sources? J Clin Microbiol 54:1711–1719. doi: 10.1128/JCM.03345-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salipante SJ, SenGupta DJ, Cummings LA, Land TA, Hoogestraat DR, Cookson BT. 2015. Application of whole-genome sequencing for bacterial strain typing in molecular epidemiology. J Clin Microbiol 53:1072–1079. doi: 10.1128/JCM.03385-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guglielmini J, Bourhy P, Schiettekatte O, Zinini F, Brisse S, Picardeau M. 2019. Genus-wide Leptospira core genome multilocus sequence typing for strain taxonomy and global surveillance. PLoS Negl Trop Dis 13:e0007374. doi: 10.1371/journal.pntd.0007374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruppitsch W, Pietzka A, Prior K, Bletz S, Fernandez HL, Allerberger F, Harmsen D, Mellmann A. 2015. Defining and evaluating a core genome multilocus sequence typing scheme for whole-genome sequence-based typing of Listeria monocytogenes. J Clin Microbiol 53:2869–2876. doi: 10.1128/JCM.01193-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cody AJ, Bray JE, Jolley KA, McCarthy ND, Maiden MCJ. 2017. Core genome multilocus sequence typing scheme for stable, comparative analyses of Campylobacter jejuni and C. coli human disease isolates. J Clin Microbiol 55:2086–2097. doi: 10.1128/JCM.00080-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maiden MC, Jansen van Rensburg MJ, Bray JE, Earle SG, Ford SA, Jolley KA, McCarthy ND. 2013. MLST revisited: the gene-by-gene approach to bacterial genomics. Nat Rev Microbiol 11:728–736. doi: 10.1038/nrmicro3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wood DE, Salzberg SL. 2014. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol 15:R46. doi: 10.1186/gb-2014-15-3-r46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haubold B, Klotzl F, Pfaffelhuber P. 2015. andi: Fast and accurate estimation of evolutionary distances between closely related genomes. Bioinformatics 31:1169–1175. doi: 10.1093/bioinformatics/btu815. [DOI] [PubMed] [Google Scholar]
- 32.Wozniak M, Wong L, Tiuryn J. 2014. eCAMBer: efficient support for large-scale comparative analysis of multiple bacterial strains. BMC Bioinformatics 15:65. doi: 10.1186/1471-2105-15-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hyatt D, Chen G-L, LoCascio PF, Land ML, Larimer FW, Hauser LJ. 2010. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Touchon M, Hoede C, Tenaillon O, Barbe V, Baeriswyl S, Bidet P, Bingen E, Bonacorsi S, Bouchier C, Bouvet O, Calteau A, Chiapello H, Clermont O, Cruveiller S, Danchin A, Diard M, Dossat C, Karoui ME, Frapy E, Garry L, Ghigo JM, Gilles AM, Johnson J, Le Bouguénec C, Lescat M, Mangenot S, Martinez-Jéhanne V, Matic I, Nassif X, Oztas S, Petit MA, Pichon C, Rouy Z, Ruf CS, Schneider D, Tourret J, Vacherie B, Vallenet D, Médigue C, Rocha EPC, Denamur E. 2009. Organised genome dynamics in the Escherichia coli species results in highly diverse adaptive paths. PLoS Genet 5:e1000344. doi: 10.1371/journal.pgen.1000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kent WJ. 2002. BLAT—The BLAST-like alignment tool. Genome Res 12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jolley KA, Maiden MC. 2010. BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 11:595. doi: 10.1186/1471-2105-11-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Croucher NJ, Page AJ, Connor TR, Delaney AJ, Keane JA, Bentley SD, Parkhill J, Harris SR. 2015. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res 43:e15. doi: 10.1093/nar/gku1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Letunic I, Bork P. 2019. Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res 47:W256–W259. doi: 10.1093/nar/gkz239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gurevich A, Saveliev V, Vyahhi N, Tesler G. 2013. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29:1072–1075. doi: 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nascimento M, Sousa A, Ramirez M, Francisco AP, Carrico JA, Vaz C. 2017. PHYLOViZ 2.0: providing scalable data integration and visualization for multiple phylogenetic inference methods. Bioinformatics 33:128–129. doi: 10.1093/bioinformatics/btw582. [DOI] [PubMed] [Google Scholar]
- 45.Cafiso V, Bertuccio T, Santagati M, Campanile F, Amicosante G, Perilli MG, Selan L, Artini M, Nicoletti G, Stefani S. 2004. Presence of the ica operon in clinical isolates of Staphylococcus epidermidis and its role in biofilm production. Clin Microbiol Infect 10:1081–1088. doi: 10.1111/j.1469-0691.2004.01024.x. [DOI] [PubMed] [Google Scholar]
- 46.Shelburne SA, Dib RW, Endres BT, Reitzel R, Li X, Kalia A, Sahasrabhojane P, Chaftari AM, Hachem R, Vargas-Cruz NS, Jiang Y, Garey K, Fowler VG, Jr, Holland TL, Gu J, Miller W, Sakurai A, Arias CA, Aitken SL, Greenberg DE, Kim J, Flores AR, Raad I. 2020. Whole-genome sequencing of Staphylococcus epidermidis bloodstream isolates from a prospective clinical trial reveals that complicated bacteremia is caused by a limited number of closely related sequence types. Clin Microbiol Infect 26:646.e1–646.e8. doi: 10.1016/j.cmi.2019.10.008. [DOI] [PubMed] [Google Scholar]
- 47.Bialek-Davenet S, Criscuolo A, Ailloud F, Passet V, Jones L, Delannoy-Vieillard AS, Garin B, Le Hello S, Arlet G, Nicolas-Chanoine MH, Decre D, Brisse S. 2014. Genomic definition of hypervirulent and multidrug-resistant Klebsiella pneumoniae clonal groups. Emerg Infect Dis 20:1812–1820. doi: 10.3201/eid2011.140206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moura A, Tourdjman M, Leclercq A, Hamelin E, Laurent E, Fredriksen N, Van Cauteren D, Bracq-Dieye H, Thouvenot P, Vales G, Tessaud-Rita N, Maury MM, Alexandru A, Criscuolo A, Quevillon E, Donguy MP, Enouf V, de Valk H, Brisse S, Lecuit M. 2017. Real-time whole-genome sequencing for surveillance of Listeria monocytogenes. Emerg Infect Dis 23:1462–1470. doi: 10.3201/eid2309.170336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leopold SR, Goering RV, Witten A, Harmsen D, Mellmann A. 2014. Bacterial whole-genome sequencing revisited: portable, scalable, and standardized analysis for typing and detection of virulence and antibiotic resistance genes. J Clin Microbiol 52:2365–2370. doi: 10.1128/JCM.00262-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Krediet TG, Mascini EM, van Rooij E, Vlooswijk J, Paauw A, Gerards LJ, Fleer A. 2004. Molecular epidemiology of coagulase-negative staphylococci causing sepsis in a neonatal intensive care unit over an 11-year period. J Clin Microbiol 42:992–995. doi: 10.1128/jcm.42.3.992-995.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mertens A, Ghebremedhin B. 2013. Genetic determinants and biofilm formation of clinical Staphylococcus epidermidis isolates from blood cultures and indwelling devises. Eur J Microbiol Immunol 3:111–119. doi: 10.1556/EuJMI.3.2013.2.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Du X, Zhu Y, Song Y, Li T, Luo T, Sun G, Yang C, Cao C, Lu Y, Li M. 2013. Molecular analysis of Staphylococcus epidermidis strains isolated from community and hospital environments in China. PLoS One 8:e62742. doi: 10.1371/journal.pone.0062742. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This whole-genome shotgun project has been deposited in GenBank under BioProject number PRJNA608162. All assemblies, sequences, and their allelic designations and cgMLST profiles are available from the Institut Pasteur BIGSdb access Web page at https://bigsdb.pasteur.fr/epidermidis/.