

Abstract
Background
Exacerbations of chronic obstructive pulmonary disease (COPD) are a major cause of hospital admissions, disease‐related morbidity and mortality. COPD is a heterogeneous disease with distinct inflammatory phenotypes, including eosinophilia, which may drive acute exacerbations in a subgroup of patients. Monoclonal antibodies targeting interleukin 5 (IL‐5) or its receptor (IL‐5R) have a role in the care of people with severe eosinophilic asthma, and may similarly provide therapeutic benefit for people with COPD of eosinophilic phenotype.
Objectives
To assess the efficacy and safety of monoclonal antibody therapies targeting IL‐5 signalling (anti‐IL‐5 or anti‐IL‐5Rα) compared with placebo in the treatment of adults with COPD.
Search methods
We searched the Cochrane Airways Trials Register, CENTRAL, MEDLINE, Embase, clinical trials registries, manufacturers' websites, and reference lists of included studies. Our most recent search was 23 September 2020.
Selection criteria
We included randomised controlled trials comparing anti‐IL‐5 therapy with placebo in adults with COPD.
Data collection and analysis
Two review authors independently extracted data and analysed outcomes using a random‐effects model.The primary outcomes were exacerbations requiring antibiotics or oral steroids, hospitalisations due to exacerbation of COPD, serious adverse events, and quality of life. We used standard methods expected by Cochrane. We used the GRADE approach to assess the certainty of the evidence.
Main results
Six studies involving a total of 5542 participants met our inclusion criteria. Three studies used mepolizumab (1530 participants), and three used benralizumab (4012 participants). The studies were on people with COPD, which was similarly defined with a documented history of COPD for at least one year. We deemed the risk of bias to be generally low, with all studies contributing data of robust methodology.
Mepolizumab 100 mg reduces the rate of moderate or severe exacerbations by 19% in those with an eosinophil count of at least 150/μL (rate ratio (RR) 0.81, 95% confidence interval (CI) 0.71 to 0.93; participants = 911; studies = 2, high‐certainty evidence). When participants with lower eosinophils are included, mepolizumab 100 mg probably reduces the exacerbation rate by 8% (RR 0.92, 95% CI 0.82 to 1.03; participants = 1285; studies = 2, moderate‐certainty evidence). Mepolizumab 300 mg probably reduces the rate of exacerbations by 14% in participants all of whom had raised eosinophils (RR 0.86, 95% CI 0.70 to 1.06; participants = 451; studies = 1, moderate‐certainty evidence); the evidence was uncertain for a single small study of mepolizumab 750 mg. In participants with high eosinophils, mepolizumab probably reduces the rate of hospitalisation by 10% (100 mg, RR 0.90, 95% CI 0.65 to 1.24; participants = 911; studies = 2, moderate‐certainty evidence) and 17% (300 mg, RR 0.83, 95% CI 0.51 to 1.35; participants = 451; studies = 1, moderate‐certainty evidence). Mepolizumab 100 mg increases the time to first moderate or severe exacerbation compared to the placebo group, in people with the eosinophilic phenotype (hazard ratio (HR) 0.78, 95% CI 0.66 to 0.92; participants = 981; studies 2, high‐certainty evidence). When participants with lower eosinophils were included this difference was smaller and less certain (HR 0.87, 95% CI 0.75 to 1.0; participants = 1285; studies 2, moderate‐certainty evidence). Mepolizumab 300 mg probably increases the time to first moderate or severe exacerbation in participants who all had eosinophilic phenotype (HR 0.77, 95% CI 0.60 to 0.99; participants = 451; studies = 1, moderate‐certainty evidence).
Benralizumab 100 mg reduces the rate of severe exacerbations requiring hospitalisation in those with an eosinophil count of at least 220/μL (RR 0.63, 95% CI 0.49 to 0.81; participants = 1512; studies = 2, high‐certainty evidence). Benralizumab 10 mg probably reduces the rate of severe exacerbations requiring hospitalisation in those with an eosinophil count of at least 220/μL (RR 0.68, 95% CI 0.49 to 0.94; participants = 765; studies = 1, moderate‐certainty evidence).
There was probably little or no difference between the intervention and placebo for quality of life measures. Where there were differences the mean difference fell below the pre‐specified minimum clinically significant difference.
Treatment with mepolizumab and benralizumab appeared to be safe. All pooled analyses showed that there was probably little or no difference in serious adverse events, adverse events, or side effects between the use of a monoclonal antibody therapy compared to placebo.
Authors' conclusions
We found that mepolizumab and benralizumab probably reduce the rate of moderate and severe exacerbations in the highly selected group of people who have both COPD and higher levels of blood eosinophils. This highlights the importance of disease phenotyping in COPD, and may play a role in the personalised treatment strategy in disease management.
Further research is needed to elucidate the role of monoclonal antibodies in the management of COPD in clinical practice. In particular, it is not clear whether there is a threshold blood eosinophil level above which these drugs may be effective. Studies including cost effectiveness analysis may be beneficial given the high cost of these therapies, to support use if appropriate.
Plain language summary
Mepolizumab or benralizumab for people with chronic obstructive pulmonary disease (COPD)
Background to the question
Chronic obstructive pulmonary disease (COPD) is a lung condition in which people can experience severe difficulties with breathing and an associated reduction in their quality of life.
For people with COPD, episodes in which the condition of patients seriously worsens are a major concern. We examined the findings of clinical trials to see whether mepolizumab or benralizumab, two new drugs, are better than placebo (dummy treatment) for people with COPD, and whether they reduce the number of episodes when the condition of patients seriously worsens.
Study characteristics
Six clinical studies compared either mepolizumab or benralizumab to placebo in a total of 5542 people with COPD. We examined the findings of the studies in terms of episodes when patients' conditions flared up requiring additional treatment, patient quality of life, patient performance in breathing tests, and side effects of the medication.
Main results
Three studies used mepolizumab, and the other three studies used benralizumab.
Mepolizumab 100 mg reduced the rate of flare‐ups in a group of people with both COPD and higher levels of blood eosinophils (a type of white blood cells involved in inflammatory and allergic reactions). When mepolizumab is given in a higher dose (300 mg or 750 mg) the rate of flare‐ups is probably reduced.
Benralizumab at a dose of 100mg resulted in a clear reduction in the number of episodes requiring admission to hospital, and when given at a lower dose (10mg) probably reduces flare‐ups requiring hospitalisation. This is in people with COPD and higher levels of blood eosinophils.
Further studies comparing mepolizumab or benralizumab to a placebo may provide more clarity on the role of these drugs for COPD.
Quality of the evidence
The included studies were for the most part very well‐designed and robust, and the evidence was generally of high quality.
Summary of findings
Summary of findings 1. Mepolizumab 100 mg compared with placebo for chronic obstructive pulmonary disease.
| Mepolizumab 100 mg compared with placebo for chronic obstructive pulmonary disease | ||||||
|
Patient or population: individuals with COPD Settings: outpatient Intervention: mepolizumab 100 mg Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Mepolizumab 100 mg | |||||
| Rate of moderate or severe exacerbations Eosinophilic phenotype |
1.60 moderate or severe exacerbations per year | 1.30 (1.14 to 1.49) moderate or severe exacerbations per year | Rate ratio 0.81 (0.71 to 0.93) | 911 (2 RCTs) | ⊕⊕⊕⊕ High |
|
| Rate of moderate or severe exacerbations All participants |
1.51 moderate or severe exacerbations per year | 1.39 (1.24 to 1.56) moderate or severe exacerbations per year | Rate ratio 0.92 (0.82 to 1.03) | 1285 (2 RCTs) | ⊕⊕⊕⊖ Moderatea |
|
| Time to first moderate or severe exacerbation Eosinophilic phenotype |
‐ | ‐ | Hazard ratio 0.78 (0.66 to 0.92) | 981 (2 RCTs) | ⊕⊕⊕⊕ High |
|
| Time to first moderate or severe exacerbation All participants |
‐ | ‐ | Hazard ratio 0.87 (0.75 to 1.0) | 1285 (2 RCTs) | ⊕⊕⊕⊖ Moderatea |
|
| Rate of exacerbations with ED visit or hospitalisations Eosinophilic phenotype |
0.27 exacerbations per year with ED visit | 0.24 (0.18 to 0.33) exacerbations per year with ED visit | Rate ratio 0.90 (0.65 to 1.24) | 911 (2 RCTs) | ⊕⊕⊕⊖ Moderatea |
|
| Rate of exacerbations with ED visit or hospitalisations All participants |
0.27 exacerbations per year with ED visit | 0.25 (0.19 to 0.33) exacerbations per year with ED visit | Rate ratio 0.94 (0.72 to 1.22) | 1285 (2 RCTs) | ⊕⊕⊕⊖ Moderatea |
|
| Serious adverse events | 199 serious adverse events out of 645 participants | 172 serious adverse events out of 640 participants | Odds ratio 0.82 (0.65 to 1.05) | 1285 (2 RCTs) | ⊕⊕⊕⊖ Moderateb |
|
| Health‐related quality of life: change in SGRQ total score Scale: 0 to 100 (lower is better) Eosinophilic phenotype |
‐ | The MD was −0.90 lower (−2.91 to 1.10). | ‐ | 911 (2 RCTs) | ⊕⊕⊕⊖ Moderateb |
A change of ≥ 4 is considered the minimum clinically significant difference. |
| Health‐related quality of life: change in SGRQ total score Scale: 0 to 100 (lower is better) All participants |
‐ | The MD was −0.30 lower (−2.00 to 1.41). | ‐ | 1285 (2 RCTs) | ⊕⊕⊕⊖ Moderateb |
A change of ≥ 4 is considered the minimum clinically significant difference. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; COPD: chronic obstructive pulmonary disease; ED: emergency department; MD: mean difference; RCT: randomised controlled trial; SGRQ: St George's Respiratory Questionnaire | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: We are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: Our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: We have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded once due to imprecision. There is high heterogeneity between the two studies. bDowngraded once due to imprecision. The confidence intervals include the possibility of a small or no effect and important benefit or harm.
Summary of findings 2. Mepolizumab 300 mg compared with placebo for chronic obstructive pulmonary disease.
| Mepolizumab 300 mg compared with placebo for chronic obstructive pulmonary disease | ||||||
|
Patient or population: individuals with COPD Settings: outpatient Intervention: mepolizumab 300 mg Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Mepolizumab 300 mg | |||||
| Rate of moderate or severe exacerbations Eosinophilic phenotype |
1.49 moderate or severe exacerbations per year | 1.28 (1.04 to 1.58) moderate or severe exacerbations per year | Rate ratio 0.86 (0.70 to 1.06) | 451 (1 RCT) | ⊕⊕⊕⊖ Moderatea |
|
| Time to first moderate or severe exacerbation Eosinophilic phenotype |
‐ | ‐ | Hazard ratio 0.77 (0.60 to 0.99) | 451 (1 RCT) | ⊕⊕⊕⊖ Moderatea |
|
| Rate of exacerbations with ED visit or hospitalisations Eosinophilic phenotype |
0.28 exacerbations with ED visit per year | 0.23 (0.14 to 0.38) exacerbations with ED visit per year | Rate ratio 0.83 (0.51 to 1.35) | 451 (1 RCT) | ⊕⊕⊕⊖ Moderatea |
|
| Serious adverse events | 68 serious adverse events out of 226 participants | 60 serious adverse events out of 225 participants | Odds ratio 0.84 (0.56 to 1.27) | 451 (1 RCT) | ⊕⊕⊕⊖ Moderatea |
|
| Health‐related quality of life: change in SGRQ total score Scale: 0 to 100 (lower is better) Eosinophilic phenotype |
‐ | The MD was −0.10 lower (−2.80 to 2.60). | ‐ | 451 (1 RCT) | ⊕⊕⊕⊖ Moderatea |
A change of ≥ 4 is considered the minimum clinically significant difference. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; COPD: chronic obstructive pulmonary disease; ED: emergency department; MD: mean difference; RCT: randomised controlled trial; SGRQ: St George's Respiratory Questionnaire | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: We are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: Our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: We have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded once as this is a single study. The true effect is likely to be close to the estimate of the effect, but further studies could be substantially different.
Summary of findings 3. Mepolizumab 750 mg compared with placebo for chronic obstructive pulmonary disease.
| Mepolizumab 750 mg compared with placebo for chronic obstructive pulmonary disease | ||||||
|
Patient or population: individuals with COPD Settings: outpatient Intervention: mepolizumab 750 mg Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Mepolizumab 750 mg | |||||
| Number of participants experiencing an exacerbation within 6 months | 7 out 10 | 4 out of 8 | Odds ratio 0.43 (0.06 to 2.97) | 18 (1 RCT) | ⊕⊕⊖⊖ Lowa |
|
| Number of participants experiencing an exacerbation in 4‐month follow‐up period | 1 out of 10 | 4 out of 8 | Odds ratio 9.00 (0.75 to 108.31) | 18 (1 RCT) | ⊕⊕⊖⊖ Lowa |
|
| Serious adverse events | 1 participant out of 10 | 2 participants out of 8 | Odds ratio 3.00 (0.22 to 40.93) | 18 (1 RCT) | ⊕⊕⊖⊖ Lowa |
|
| Heath‐related quality of life (CRQ at 6 months) | Mean 102.11 (SD 15.55) | MD 1.14 higher (−17.28 to 19.56) | ‐ | 18 (1 RCT) | ⊕⊕⊖⊖ Lowa |
A higher score indicates better health‐related quality of life. |
| Lung function (FEV₁) (litres post‐bronchodilator) at 6 months | Mean 1.33 (SD 0.71) | MD 0.25 higher (−0.36 to 0.86) | ‐ | 18 (1 RCT) | ⊕⊕⊖⊖ Lowa |
|
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; COPD: chronic obstructive pulmonary disease; CRQ: Chronic Respiratory Disease Questionnaire; FEV₁: forced expiratory volume in 1 second; MD: mean difference; RCT: randomised controlled trial; SD: standard deviation | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: We are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: Our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: We have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded twice due to imprecision. The study is very small with few participants, and consequently wide confidence intervals.
Summary of findings 4. Benralizumab 10 mg compared with placebo for chronic obstructive pulmonary disease.
| Benralizumab 10 mg compared with placebo for chronic obstructive pulmonary disease | ||||||
|
Patient or population: individuals with COPD Settings: outpatient Intervention: benralizumab 10 mg Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Benralizumab 10 mg | |||||
| Rate of moderate or severe exacerbations Eosinophils ≥ 220/μL | 1.17 moderate or severe exacerbations per year | 0.99 (0.83 to 1.19) moderate or severe exacerbations per year | Rate ratio 0.85 (0.71 to 1.02) | 765 (1 RCT) | ⊕⊕⊕⊖ Moderatea |
|
| Rate of moderate or severe exacerbations Eosinophils < 220/μL | 1.18 moderate or severe exacerbations per year | 1.23 (0.97 to 1.56) moderate or severe exacerbations per year | Rate ratio 1.04 (0.82 to 1.32) | 365 (1 RCT) | ⊕⊕⊕⊖ Moderatea |
|
| Rate of severe exacerbations requiring hospitalisation Eosinophils ≥ 220/μL |
0.32 severe exacerbations requiring hospitalisation per year | 0.22 (0.16 to 0.30) severe exacerbations requiring hospitalisation per year | Rate ratio 0.68 (0.49 to 0.94) | 765 (1 RCT) | ⊕⊕⊕⊖ Moderatea |
|
| Serious adverse events | 158 serious adverse events out of 568 participants | 144 serious adverse events out of 561 participants | Odds ratio 0.90 (0.69 to 1.17) | 1129 (1 RCT) | ⊕⊕⊕⊖ Moderatea |
|
| Heath‐related quality of life, change in SGRQ total score Scale: 0 to 100 (lower is better) Eosinophils ≥ 220/μL |
‐ | The MD was −0.87 lower (−3.23 to 1.49). | ‐ | 680 (1 RCT) | ⊕⊕⊕⊖ Moderatea |
A change of ≥ 4 is considered the minimum clinically significant difference. |
| Lung function (FEV₁) Eosinophils ≥ 220/μL |
‐ | The MD was 0.01 higher (−0.04 to 0.05). | ‐ | 669 (1 RCT) | ⊕⊕⊕⊖ Moderatea |
|
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; COPD: chronic obstructive pulmonary disease; FEV₁: forced expiratory volume in 1 second; MD: mean difference; RCT: randomised controlled trial; SGRQ: St George's Respiratory Questionnaire | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: We are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: Our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: We have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded once as this is a single study. The true effect is likely to be close to the estimate of the effect, but further studies could be substantially different.
Summary of findings 5. Benralizumab 30 mg compared with placebo for chronic obstructive pulmonary disease.
| Benralizumab 30 mg compared with placebo for chronic obstructive pulmonary disease | ||||||
|
Patient or population: individuals with COPD Settings: outpatient Intervention: benralizumab 30 mg Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Benralizumab 30 mg | |||||
| Rate of moderate or severe exacerbations Eosinophils ≥ 220/μL | 1.20 moderate or severe exacerbations per year | 1.20 (1.07 to 1.37) moderate or severe exacerbations per year | Rate ratio 1.00 (0.89 to 1.13) | 1523 (2 RCTs) | ⊕⊕⊕⊖ Moderatea |
|
| Rate of moderate or severe exacerbations Eosinophils < 220/μL | 1.24 moderate or severe exacerbations per year | 1.33 (1.13 to 1.57) moderate or severe exacerbations per year | Rate ratio 1.07 (0.91 to 1.27) | 711 (2 RCTs) | ⊕⊕⊕⊖ Moderatea |
|
| Rate of severe exacerbations requiring hospitalisation Eosinophils ≥ 220/μL |
0.29 severe exacerbations requiring hospitalisation per year | 0.28 (0.22 to 0.35) severe exacerbations requiring hospitalisation per year | Rate ratio 0.96 (0.75 to 1.22) | 1523 (2 RCTs) | ⊕⊕⊕⊖ Moderatea |
|
| Serious adverse events | 334 serious adverse events out of 1118 participants | 328 serious adverse events out of 1117 participants | Odds ratio 0.98 (0.81 to 1.17) | 2235 (2 RCTs) | ⊕⊕⊕⊖ Moderatea |
|
| Heath‐related quality of life, change in SGRQ total score Scale: 0 to 100 (lower is better) Eosinophils ≥ 220/μL |
‐ | The MD was −1.42 lower (−3.13 to 0.29). | ‐ | 1333 (2 RCTs) | ⊕⊕⊕⊖ Moderatea |
A change of ≥ 4 is considered the minimum clinically significant difference. |
| Lung function (FEV₁) Eosinophils ≥ 220/μL |
‐ | There was no MD −0.00 (−0.03 to 0.03). | ‐ | 1312 (2 RCTs) | ⊕⊕⊕⊖ Moderatea |
|
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; COPD: chronic obstructive pulmonary disease; FEV₁: forced expiratory volume in 1 second; MD: mean difference; RCT: randomised controlled trial; SGRQ: St George's Respiratory Questionnaire | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: We are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: Our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: We have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded once due to imprecision. The confidence intervals include the possibility of a small or no effect and important benefit or harm.
Summary of findings 6. Benralizumab 100 mg compared with placebo for chronic obstructive pulmonary disease.
| Benralizumab 100 mg compared with placebo for chronic obstructive pulmonary disease | ||||||
|
Patient or population: individuals with COPD Settings: outpatient Intervention: benralizumab 100 mg Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Benralizumab 100 mg | |||||
| Rate of moderate or severe exacerbations | 1.18 moderate or severe exacerbations per year | 1.11 (1.00 to 1.22) moderate or severe exacerbations per year | Rate ratio 0.94 (0.85 to 1.03) | 2314 (3 RCTs) | ⊕⊕⊕⊖ Moderatea |
|
| Rate of moderate or severe exacerbations Eosinophils ≥ 220/μL | 1.20 moderate or severe exacerbations per year | 1.06 (0.94 to 1.2) moderate or severe exacerbations per year | Rate ratio 0.88 (0.78 to 1.00) | 1512 (2 RCTs) | ⊕⊕⊕⊖ Moderatea |
|
| Rate of moderate or severe exacerbations Eosinophils < 220/μL | 1.24 moderate or severe exacerbations per year | 1.26 (1.08 to 1.49) moderate or severe exacerbations per year | Rate ratio 1.02 (0.87 to 1.20) | 720 (2 RCTs) | ⊕⊕⊕⊖ Moderatea |
|
| Rate of severe exacerbations requiring hospitalisation Eosinophils ≥ 220/μL |
0.29 severe exacerbations requiring hospitalisation per year | 0.18 (0.14 to 0.23) severe exacerbations requiring hospitalisation per year | Rate ratio 0.63 (0.49 to 0.81) | 1512 (2 RCTs) | ⊕⊕⊕⊕ High |
|
| Serious adverse events | 343 serious adverse events out of 1168 participants | 318 serious adverse events out of 1165 participants | Odds ratio 0.90 (0.75 to 1.08) | 2333 (3 RCTs) | ⊕⊕⊕⊖ Moderatea |
|
| Heath‐related quality of life, change in SGRQ total score Scale: 0 to 100 (lower is better) |
‐ | The MD was −1.45 lower (−2.84 to −0.07). | ‐ | 1433 (3 RCTs) | ⊕⊕⊕⊕ High |
A change of ≥ 4 is considered the minimum clinically significant difference. |
| Heath‐related quality of life, change in SGRQ total score Scale: 0 to 100 (lower is better) Eosinophils ≥ 220/μL |
‐ | The MD was −1.47 lower (−2.89 to −0.05). | ‐ | 1351 (2 RCTs) | ⊕⊕⊕⊕ High |
A change of ≥ 4 is considered the minimum clinically significant difference. |
| Lung function (FEV₁) | ‐ | The MD was 0.03 higher (−0.00 to 0.06). | ‐ | 1425 (3 RCTs) | ⊕⊕⊕⊕ High |
|
| Lung function (FEV₁) Eosinophils ≥ 220/μL |
‐ | The mean difference was 0.02 higher (−0.01 to 0.05). | ‐ | 1334 (2 RCTs) | ⊕⊕⊕⊖ Moderatea |
|
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; COPD: chronic obstructive pulmonary disease; FEV₁: forced expiratory volume in 1 second; MD: mean difference; RCT: randomised controlled trial; SGRQ: St George's Respiratory Questionnaire | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: We are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: Our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: We have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded once due to imprecision. The confidence intervals include the possibility of a small or no effect and important benefit or harm.
Background
Description of the condition
Chronic obstructive pulmonary disease (COPD) is a prevalent respiratory disease, affecting 251 million people worldwide and accounting for 5% of all deaths globally. It is projected to become the third‐leading cause of death by 2030 (WHO 2019a; WHO 2019b). In the UK, 4.5% of the population aged over 40 have a diagnosis of COPD, which poses a substantial socio‐economic burden (BLF 2019). COPD is a progressive disease that involves a spectrum of clinical features including dyspnoea, wheeze, cough and/or sputum production. Chronic exposure to noxious particles or gases, most commonly tobacco smoke, drives COPD development. Hallmarks of the disease are airway inflammation (bronchitis), airflow limitation, and lung parenchymal destruction (emphysema) (GOLD 2019; GOLD 2020). Unlike in asthma, the other common chronic airways disease, airflow obstruction in COPD is not fully reversible.
Exacerbations of COPD, characterised by acute worsening of symptoms beyond usual day‐to‐day variability, often require changes in treatment and are a major cause of hospitalisation and disease‐related morbidity and mortality.
COPD is a heterogeneous disease with distinct inflammatory phenotypes. Whilst neutrophils, macrophages, and B lymphocytes are the predominant inflammatory cell types in some patients (Hogg 2004), a significant proportion of patients demonstrate airway eosinophilia (Singh 2014). Phenotypic clusters have also been identified during acute exacerbations, with up to 40% showing an eosinophil‐predominant T helper type 2 (Th2) inflammatory profile (Shironjit 2006). Inflammatory phenotypes of COPD have clinical and therapeutic implications. Not only is blood eosinophilia significantly associated with increased severe exacerbation rates (Couillard 2017), the use of inhaled and systemic corticosteroids have demonstrated increased efficacies in preventing and treating COPD exacerbations in those with eosinophilia (Bafadhel 2012; Bafadhel 2014; Pascoe 2015). This may suggest that eosinophilia plays a role in the pathogenesis of COPD and may drive acute exacerbations in a subgroup of patients.
Description of the intervention
Corticosteroids suppress inflammation non‐specifically and are effective in many individuals with asthma or COPD; a notable proportion, however, are poorly responsive. Moreover, frequent or continuous systemic corticosteroid use carries the risk of added morbidity, such as adrenal suppression, hyperglycaemia, osteoporosis, and skin thinning.
In the search for more targeted treatments, monoclonal antibody (MAb) technology has been employed, with anti‐interleukin 5 (anti‐IL‐5) a commonly used MAb. The appeal of this approach is that MAbs can offer high affinity and specificity for targets not amenable to small‐molecule drugs. They have revolutionised the management of other conditions, particularly certain connective tissue diseases, inflammatory bowel disease, and cancers (Adegbola 2018; Bittner 2018). In all cases, biomarkers are needed which can predict therapeutic responses, for example eosinophils, which infiltrate the airways. MAbs can then be directed against immune pathways which may contribute to the presence of eosinophils, such as IL‐5.
Th2 cells and eosinophils are implicated in both COPD and asthma. Mediators including IL‐3, IL‐5, and IL‐13 are prominent in Th2‐type inflammation, where they promote eosinophil maturation. IL‐5 is particularly key for the differentiation, proliferation, and activation of eosinophils. Th2 cells can also drive airway inflammation via an immunoglobulin E (IgE) and mast cell mechanism. Several biologic drugs targeting Th2‐type inflammation have demonstrated efficacy as an adjunct to corticosteroids in the management of severe eosinophilic or atopic asthma, with acceptable side effect profiles (Farne 2017; Normansell 2014). Consequently, a number of these drugs have been approved for use in this context, namely omalizumab (anti‐IgE), mepolizumab (anti‐IL‐5), reslizumab (anti‐IL‐5), and benralizumab (anti‐IL‐5 receptor).
There may also be useful drug targets outside the Th2‐eosinophil pathway, although to date these have not shown such efficacy in airway diseases (Durham 2016; Nixon 2017).
How the intervention might work
Eosinophilic inflammation has been implicated in a proportion of individuals with COPD, most prominently during exacerbations (Singh 2014; Siva 2007; Vedel‐Krogh 2016). This process has been effectively targeted in severe eosinophilic asthma, therefore it is reasonable to expect that MAbs directed against similar targets in COPD patients with eosinophilic phenotypes may provide therapeutic benefit.
Why it is important to do this review
Whilst COPD is an irreversible disease, management of the condition is directed at slowing or halting the decline in lung function, preventing and aborting exacerbations, and optimising quality of life. Monoclonal antibody therapies have proven to be a useful tool for asthma. A recent Cochrane Review supports the use of anti‐IL‐5 treatments as an adjunct to standard treatment in people with severe eosinophilic asthma, with treatments roughly halving asthma exacerbations (Farne 2017). Given the number of pathological similarities between asthma and COPD, it may be that anti‐IL‐5 treatments can also benefit at least a subset of COPD patients. Anti‐IL‐5 treatments have not been approved for use in COPD, and they are not mentioned in guidelines, but as there is an emerging literature in this field, it is important to establish whether or not they have a role to play (Tan 2018). COPD is such a common condition that any additional treatments have the potential to benefit a large number of individuals. Exacerbations are a major determinant of both quality of life and healthcare usage. These drugs reduce exacerbations of asthma (Farne 2017); if they also reduced exacerbations of COPD in those with eosinophils it would be advantageous for both patients and healthcare systems.
Objectives
To assess the efficacy and safety of monoclonal antibody therapies targeting IL‐5 signalling (anti‐IL‐5 or anti‐IL‐5Rα) compared with placebo in the treatment of adults with COPD.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised controlled trials (RCTs). We included studies reported in full text, those published as an abstract only, and unpublished data.
Types of participants
We included adults (≥ 40 years old) with a diagnosis of COPD as defined by GOLD 2020. We recorded study authors' definitions of the severity of COPD. We did not exclude participants with comorbidities. Where possible, we excluded participants with a substantial asthma component to their disease, either with a label of 'asthma COPD overlap syndrome' (Pavord 2015), or excessive variation in lung function, defined by a variation of more than 12% and 200 mL in forced expiratory volume in one second (FEV₁), either between tests or with a bronchodilator at trial entry (GINA 2019).
Types of interventions
We included studies comparing anti‐IL‐5 therapy with placebo. Specifically, we considered anti‐IL‐5 therapies developed for use in other airway diseases such as those directed against various IL‐5 targets. We included studies that allowed participants to continue using their inhaled therapies including inhaled corticosteroids (ICS), long‐acting beta₂‐agonist (LABA), and long‐acting muscarinic antagonist (LAMA) or combination inhalers, as long as these co‐interventions were not part of the randomised treatment.
Types of outcome measures
Primary outcomes
All exacerbations
Hospitalisations due to COPD exacerbation
Serious adverse events
Quality of life (as measured on a validated scale, e.g. St George's Respiratory Questionnaire (SGRQ) or Chronic Respiratory Disease Questionnaire (CRQ))
Secondary outcomes
Measures of pulmonary function such as FEV₁, and forced vital capacity (FVC)
Exercise performance: six‐minute walk test and other measures
-
Self‐rated symptom score/symptoms of breathlessness such as:
inhaled rescue medication used during the treatment period and concomitant medication usage, including antibiotics and steroids;
number of days (or nights) participant experienced symptoms;
COPD Assessment Test (CAT) score; or
COPD Control Questionnaire (CCQ) score.
Mortality
Adverse events/side effects
Reporting one or more of the outcomes listed here in the study was not an inclusion criterion for the review.
Search methods for identification of studies
Electronic searches
We identified studies from searches of the following databases and trial registries:
Cochrane Airways Trials Register, via the Cochrane Register of Studies, all years to 23 September 2020 (Cochrane Airways 2019);
Cochrane Central Register of Controlled Trials (CENTRAL), via the Cochrane Register of Studies, all years to 23 September 2020;
MEDLINE Ovid SP 1946 to 23 September 2020;
Embase Ovid SP 1974 to 23 September 2020;
US National Institutes of Health Ongoing Trials Register ClinicalTrials.gov (www.clinicaltrials.gov)
World Health Organization International Clinical Trials Registry Platform (apps.who.int/trialsearch)
The database search strategies are listed in Appendix 1. The search strategy was developed in MEDLINE by the Cochrane Airways Information Specialist, in collaboration with the review authors, and then adapted for use in the other databases.
The Cochrane Airways Information Specialist searched all databases and trials registries from their inception to September 2020, using no restriction on language or type of publication. We identified handsearched conference abstracts and grey literature through the Cochrane Airways Trials Register and the CENTRAL database in the Cochrane Library.
Searching other resources
We checked the reference lists of all primary studies and review articles for additional references. We searched relevant manufacturers' websites for study information.
We searched on PubMed for errata or retractions from included studies published in full text on 19 June 2020.
Data collection and analysis
Selection of studies
We used Cochrane’s Screen4Me workflow to help assess the search results. Screen4Me comprises three components: known assessments – a service that matches records in the search results to records that have already been screened in Cochrane Crowd and been labelled as an RCT or as Not an RCT; the RCT classifier – a machine learning model that distinguishes RCTs from non‐RCTs; and if appropriate, Cochrane Crowd (crowd.cochrane.org), Cochrane’s citizen science platform where the Crowd help to identify and describe health evidence. More detailed information about the Screen4Me components can be found in the following publications: Marshall 2018, McDonald 2017, Noel‐Storr 2018, Thomas 2017.
Following this initial assessment, two review authors (RW and TD) independently screened the titles and abstracts identified by the search results and coded them as 'retrieve' (eligible or potentially eligible/unclear) or 'do not retrieve'. We retrieved the full‐text study reports of all potentially eligible studies, and two review authors (IC and PB) independently screened them for inclusion and recorded the reasons for exclusion of ineligible studies. Any disagreements were resolved through discussion or by consulting a third person/review author (RW, TD, or SM) if required. We identified and excluded duplicates and collated multiple reports of the same study so that each study, rather than each report, was the unit of interest in the review. We recorded the selection process in sufficient detail to complete a PRISMA flow diagram and Characteristics of excluded studies table (Moher 2009).
Data extraction and management
We used a data collection form for study characteristics and outcome data that had been piloted on at least one study in the review. Two review authors (SM and TD) extracted the following study characteristics from the included studies.
Methods: study design, total duration of study, details of any 'run‐in' period, number of study centres and location, study setting, withdrawals, and date of study.
Participants: N, mean age, age range, gender, severity of condition, diagnostic criteria, baseline lung function, smoking history, inclusion criteria and exclusion criteria.
Interventions: intervention, comparison, concomitant medications, and excluded medications.
Outcomes: primary and secondary outcomes specified and collected, and time points reported.
Notes: funding for studies and notable conflicts of interest of trial authors.
Two review authors (SM and EB) independently extracted outcome data from the included studies. We noted in the Characteristics of included studies table if outcome data were not reported in a useable way. Any disagreements were resolved by consensus or by involving a third person/review author (RW, TD, or SM). Two review authors (SM and EB) transferred data into the Review Manager 5 file (Review Manager 2020). We double‐checked that data were entered correctly by comparing the data presented in the systematic review with the study reports. A second review author (TD) spot‐checked study characteristics for accuracy against the study report.
Assessment of risk of bias in included studies
Two review authors (SM and TD) independently assessed risk of bias for each study using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). Any disagreements were resolved by discussion or by involving another review author (IC, PB, or RW). We assessed the risk of bias according to the following domains.
Random sequence generation
Allocation concealment
Blinding of participants and personnel
Blinding of outcome assessment
Incomplete outcome data
Selective outcome reporting
Other bias
We judged each potential source of bias as high, low, or unclear and provided a quote from the study report together with a justification for our judgement in the 'Risk of bias' table in Characteristics of included studies. We summarised the 'Risk of bias' judgements across different studies for each of the domains listed. We considered blinding separately for different key outcomes where necessary (e.g. for unblinded outcome assessment, risk of bias for all‐cause mortality may be very different than for a patient‐reported pain scale).
When considering treatment effects, we took into account the risk of bias for the studies that contributed to that outcome.
Measures of treatment effect
We analysed dichotomous data as odds ratios (OR) and continuous data as the inverse variance for rate ratios (RR) and hazard ratios (HR), mean difference (MD), or standardised mean difference (SMD). Where we combined data from rating scales in a meta‐analysis, we ensured that they were entered with a consistent direction of effect (e.g. lower scores always indicate improvement).
We undertook meta‐analyses only where this was meaningful, that is if the treatments, participants, and the underlying clinical question were similar enough for pooling to make sense.
We planned to describe reported skewed data narratively (e.g. as medians and interquartile ranges for each group); however, this was not an issue with the reported data.
Where multiple trial arms were reported in a single study, we included only the relevant arms. If we combined two comparisons (e.g. drug A versus placebo and drug B versus placebo) in the same meta‐analysis, we either combined the active arms or halved the control group to avoid double‐counting.
If adjusted analyses were available (analysis of variance (ANOVA) or analysis of covariance (ANCOVA)), we would use these as a preference in our meta‐analyses. If both change‐from‐baseline scores and endpoint scores were available for continuous data, we used change‐from‐baseline scores. If a study reported outcomes at multiple time points, we preferentially used 12‐month data but reported other time points where appropriate.
We used intention‐to‐treat (ITT), or 'full analysis set' analyses where they were reported (i.e. where data have been imputed for participants who were randomly assigned but did not complete the study) instead of completer or per‐protocol analyses.
Unit of analysis issues
For dichotomous outcomes we used participants, rather than events, as the unit of analysis (i.e. number of people admitted to hospital, rather than number of admissions per person). Where RRs were reported in a study, we analysed them on this basis. We planned to only meta‐analyse data from cluster‐RCTs if the available data had been adjusted to account for the clustering; however, the need to do so did not arise as no cluster‐RCTs were included in the review.
Dealing with missing data
We contacted investigators or study sponsors in order to verify key study characteristics and to obtain missing numerical outcome data where possible (e.g. when a study was identified as an abstract only). Where this was not possible, and we considered the missing data to introduce serious bias, we took this into consideration in the GRADE rating for the affected outcomes.
Assessment of heterogeneity
We used the I² statistic to measure heterogeneity amongst the studies in each analysis. Where we identified substantial heterogeneity we reported it and explored the possible causes by prespecified subgroup analysis.
Assessment of reporting biases
We were not able to pool more than 10 studies, therefore we did not create and examine a funnel plot to explore possible small‐study and publication biases.
Data synthesis
We used a random‐effects model, reported with 95% CIs, and performed a sensitivity analysis with a fixed‐effect model. We synthesised and reported dichotomous and continuous data separately for each outcome (e.g. hospitalisation/no hospitalisation or duration of hospitalisation). We also analysed ORs and reported them separately. For a given outcome measure, we combined effect estimates, such as differences at endpoint and change from baseline. We planned to combine outcomes measured using different scales (e.g. health‐related quality of life) by employing SMDs in the analyses; however, this was not necessary with the available data.
Subgroup analysis and investigation of heterogeneity
We planned to carry out the following subgroup analyses.
Baseline serum eosinophil counts (> 0.3 versus ≤ 0.3 × 10⁹ per litre of blood)
Baseline COPD severity using GOLD 2020 classification
We planned to use our primary outcomes in the subgroup analyses.
We would have used the formal test for subgroup interactions in Review Manager 5 (Review Manager 2020); however, due to the limited number of included studies, no subgroup analyses were carried out.
Sensitivity analysis
We planned to carry out the following sensitivity analyses, removing the following from the primary outcome analyses.
A comparison of available‐case analysis to true ITT analyses, where the ITT analyses are imputed.
A comparison based on the 'Risk of bias' assessment, where trials are judged to be at high risk of bias for any of the six 'Risk of bias' domains.
However, due to the limited number of included studies, no sensitivity analyses were carried out.
Summary of findings and assessment of the certainty of the evidence
We created a 'Summary of findings' table using the following outcomes: all exacerbations, hospitalisations due to COPD, serious adverse events, lung function (FEV₁), and quality of life. We used the five GRADE considerations (risk of bias, consistency of effect, imprecision, indirectness, and publication bias) to assess the quality of a body of evidence as it relates to the studies that contribute data for the prespecified outcomes. We used the methods and recommendations described in Section 8.5 and Chapter 12 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011), employing GRADEpro GDT software (GRADEpro GDT). We justified all decisions to downgrade the quality of studies using footnotes and made comments to aid the reader's understanding of the review where necessary.
Results
Description of studies
Results of the search
We identified 717 records in our literature searches (Figure 1), the last search being conducted in September 2020. We screened the 481 records that remained after removal of duplicates for eligibility. We excluded 183 records on the basis of title and abstract screening, 242 records were excluded by Cochrane RCT Classifier, and 16 records were excluded by Cochrane Crowd Known Assessments.
1.
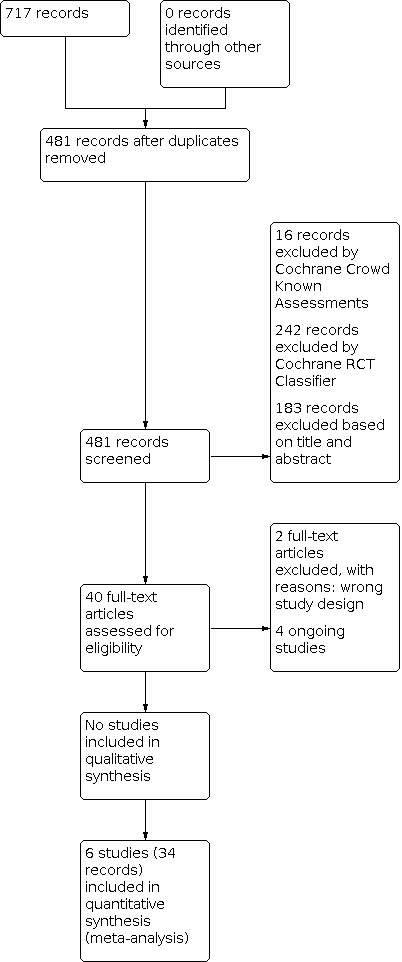
Study flow diagram.
Six studies met our inclusion criteria (Characteristics of included studies), and four others were included as ongoing studies (Characteristics of ongoing studies). The six included studies included had 34 records, as follows.
The three included studies comparing mepolizumab versus placebo had 16 records: four for Dasgupta 2016, six for NCT02105948 (METREX), and six for NCT02105961 (METREO).
The three included studies comparing benralizumab versus placebo had 18 records: nine for Brightling 2014, five for NCT02138916 (GALATHEA), and four for NCT02155660 (TERRANOVA).
There are four ongoing studies (see Characteristics of ongoing studies), two comparing benralizumab versus placebo NCT04053634 and NCT04098718 (the ABRA study), and two comparing mepolizumab versus placebo NCT04075331 and NCT04133909 (the MATINEE study).
We found no studies looking at reslizumab.
The definition used for exacerbation of COPD varied slightly amongst the included studies. GOLD 2020 defines three levels of severity of exacerbation depending on the treatment required: mild exacerbations no more than short‐acting bronchodilators, moderate exacerbations need antibiotics or oral steroids, or both, and severe exacerbations result in hospital attendance.
In NCT02138916 (GALATHEA) and NCT02155660 (TERRANOVA), exacerbations were defined as "a symptomatic worsening ... resulting in the use of systemic glucocorticoids, the use of antibiotics, or hospitali[s]ation or COPD‐related death". This definition of (any) exacerbation maps to moderate and severe exacerbations using the GOLD 2020 definitions. Brightling 2014, NCT02105948 (METREX), and NCT02105961 (METREO) used definitions that approximate the GOLD 2020 definition, but only recorded moderate and severe exacerbations. Dasgupta 2016 does not define exacerbation.
In this review, we have used the terms 'moderate and severe exacerbations' and 'severe exacerbation' where we believe the working definition used is sufficiently close to the GOLD 2020 definition to be practically equivalent.
Included studies
Mepolizumab
We included three studies comparing mepolizumab versus placebo (see Characteristics of included studies table), involving a total of 1530 participants distributed as follows: Dasgupta 2016 n = 19; NCT02105948 (METREX) n = 837; and NCT02105961 (METREO) n = 674. Mepolizumab was administered intravenously (IV) in Dasgupta 2016 (at a dose of 750 mg). In NCT02105948 (METREX) administration was subcutaneous (SC) (at a dose of 100 mg), and in NCT02105961 (METREO) administration was SC (at a dose of 100 mg or 300 mg). In NCT02105948 (METREX) and NCT02105961 (METREO), administration was every 4 weeks for up to 52 weeks, whilst in Dasgupta 2016 it was once a month.
The three studies only included participants with frequent exacerbations of COPD, with at least one "major" exacerbation in the previous year (Dasgupta 2016), or two moderate exacerbations (NCT02105948 (METREX); NCT02105961 (METREO)). Diagnosis in all three studies was in accordance with the American Thoracic Society/European Respiratory Society (ATS/ERS) definition, with a documented history of COPD for at least one year. Dasgupta 2016 specified FEV₁/FVC < 70% and FEV₁ < 60% of predicted normal values calculated using National Health and Nutrition Examination Survey (NHANES) III reference equations at screening visit. In addition, NCT02105948 (METREX) and NCT02105961 (METREO) specified a measured post‐salbutamol FEV₁ > 20% and ≤ 80% of predicted normal values calculated using NHANES III reference equations. Participants in Dasgupta 2016 were current or former smokers, whereas in NCT02105948 (METREX) and NCT02105961 (METREO) participation was independent of smoking status and smoking history.
Dasgupta 2016 would have allowed > 12% FEV₁ reversibility with prednisone as a surrogate for sputum eosinophilia. All participants were meant to have less than 12% FEV₁ reversibility with a bronchodilator. In the event, all participants had more than 3% sputum eosinophilia, and the prednisone surrogate was not used (Milan 2020 [pers comm]), so we considered this study as meeting our inclusion criteria. It appears that some individuals were included in this study despite not meeting the bronchodilator reversibility criteria.
Benralizumab
We included three studies comparing benralizumab versus placebo (see Characteristics of included studies table), involving a total of 4012 participants distributed as follows: Brightling 2014 n = 101; NCT02138916 (GALATHEA) n = 1656; and NCT02155660 (TERRANOVA) n = 2255. Benralizumab was administered SC in Brightling 2014 (at a dose of 100 mg), SC in NCT02138916 (GALATHEA) (at a dose of 30 mg or 100 mg), and SC in NCT02155660 (TERRANOVA) (at a dose of 10 mg, 30 mg, or 100 mg). Administration was every four weeks for the first three doses and then every eight weeks for the next five doses in Brightling 2014. In NCT02138916 (GALATHEA) and NCT02155660 (TERRANOVA), administration was every four weeks for the first three doses and every eight weeks thereafter, with the last dose administered at week 48.
The studies included participants with a diagnosis of COPD and a documented history of one or more annualised incidence rate of moderate or severe acute exacerbations of chronic obstructive pulmonary disease (Brightling 2014), or two or more moderate or one or more severe exacerbations in the previous year (NCT02138916 (GALATHEA); NCT02155660 (TERRANOVA)). NCT02138916 (GALATHEA) and NCT02155660 (TERRANOVA) specified a post‐bronchodilator FEV₁ > 20% and ≤ 65%. All participants were current or former smokers with ≥ 10 pack‐year exposure.
Excluded studies
Of the full‐text studies assessed for eligibility, three were ongoing studies and two were excluded with reasons (one was not a randomised trial, and the other was an aggregation of two studies investigating modulation of blood inflammatory markers) (see Characteristics of excluded studies).
Risk of bias in included studies
We assessed the risk of bias using the 'Risk of bias' tool described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011).
Allocation
We determined one study to be at low risk of selection bias across two domains (Brightling 2014). Four studies provided details on random sequence generation, and although it is highly likely that the allocation concealment was adequate, we were unable to find any details on this in the trial reports (NCT02105948 (METREX); NCT02105961 (METREO); NCT02138916 (GALATHEA); NCT02155660 (TERRANOVA)). One study presented no details on either random sequence generation or allocation concealment and was judged to be at unclear risk of bias for both domains (Figure 2) (Dasgupta 2016).
2.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Blinding
We assessed all six studies as at low risk of performance and detection bias.
Incomplete outcome data
We assessed five studies as at low risk of attrition bias. One study provided no information about incomplete outcome data (Dasgupta 2016).
Selective reporting
We assessed five studies as at low risk of reporting bias. Information for one study was insufficient to permit a judgement (Dasgupta 2016).
Other potential sources of bias
It is likely that Dasgupta 2016 included people with asthma. The inclusion criteria for this study required < 12% FEV₁ reversibility to a bronchodilator, but it appears that a number of participants were included (particularly in the placebo arm) despite having greater than 12% reversibility.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5; Table 6
Mepolizumab 100 mg versus placebo
Primary outcomes
All exacerbations
The two trials NCT02105961 (METREO) and NCT02105948 (METREX) providing data to this comparison reported their results for participants with higher levels of blood eosinophils (defined as ≥ 150 cells per mm³ at screening or ≥ 300 cells per mm³ in the year before trial entry) separately and their total sample. To reflect the information in these studies we have followed the same rationale here.
With regard to rate of moderate or severe exacerbations, we found evidence that participants with higher blood eosinophils had a lower rate of exacerbations when receiving mepolizumab 100 mg compared to those receiving placebo (rate ratio (RR) 0.81, 95% confidence interval (CI) 0.71 to 0.93; participants = 911; studies = 2, Analysis 1.1, high‐certainty evidence). There was probably a smaller reduction in the exacerbation rate with the inclusion of participants with lower blood eosinophils (RR 0.92, 95% CI 0.82 to 1.03; participants = 1285; studies = 2, Analysis 1.1, moderate‐certainty evidence).
1.1. Analysis.

Comparison 1: Mepolizumab 100 mg versus placebo, Outcome 1: Rate of moderate or severe exacerbations
Data were also available for the time to first moderate or severe exacerbation. The eosinophilic‐phenotype participants receiving mepolizumab 100 mg experienced a longer duration to first moderate or severe exacerbation than those in the placebo group (hazard ratio (HR) 0.78, 95% CI 0.66 to 0.92; participants = 981; studies = 2, Analysis 1.2, high‐certainty evidence). Evidence for a reduction within the total sample was less certain (HR 0.87, 95% CI 0.75 to 1.00; participants = 1285; studies = 2, Analysis 1.2, moderate‐certainty evidence) (Table 1).
1.2. Analysis.

Comparison 1: Mepolizumab 100 mg versus placebo, Outcome 2: Time to first moderate or severe exacerbation
Hospitalisations due to COPD exacerbation
Mepolizumab 100 mg probably reduces the rate of exacerbations leading to an emergency department visit or hospitalisation for the higher eosinophil participants (RR 0.90, 95% CI 0.65 to 1.24; participants = 911; studies = 2, Analysis 1.3, moderate‐certainty evidence), and there was a similar result within the total sample (RR 0.94, 95% CI 0.72 to 1.22; Analysis 1.3, moderate‐certainty evidence). We are not confident about these results due to high statistical heterogeneity between the two studies (Table 1).
1.3. Analysis.

Comparison 1: Mepolizumab 100 mg versus placebo, Outcome 3: Rate of exacerbations with ED visit or hospitalisation
Serious adverse events
There was probably a reduction in serious adverse events between mepolizumab 100 mg and placebo groups (odds ratio (OR) 0.82, 95% CI 0.65 to 1.05; participants = 1285; studies = 2, Analysis 1.4, moderate‐certainty evidence). Although both studies were large with a robust methodology, the confidence intervals include the possibility of benefit or harm (Table 1).
1.4. Analysis.

Comparison 1: Mepolizumab 100 mg versus placebo, Outcome 4: Serious adverse events
Quality of life
In the higher blood eosinophil participants, there was probably only a small difference in St George's Respiratory Questionnaire (SGRQ) total scores between mepolizumab 100 mg and placebo groups (mean difference (MD) −0.90, 95% CI −2.91 to 1.10; participants = 911; studies = 2, Analysis 1.5, moderate‐certainty evidence). The minimal important difference on this scale is a change of four units. We found similar results for the total sample of participants (MD −0.30, 95% CI −2.00 to 1.41; participants = 1285; studies = 2, Analysis 1.5, moderate‐certainty evidence). Although both studies were large with a robust methodology, the confidence intervals include the possibility of benefit or harm (Table 1).
1.5. Analysis.

Comparison 1: Mepolizumab 100 mg versus placebo, Outcome 5: Health‐related quality of life: change in SGRQ total score
Secondary outcomes
Measures of pulmonary function
NCT02105961 (METREO) and NCT02105948 (METREX) did not include these specific measures of pulmonary function as an outcome measure.
Exercise performance
NCT02105961 (METREO) and NCT02105948 (METREX) did not include these specific measures of exercise performance as an outcome measure.
Self‐rated symptom score/symptoms of breathlessness
NCT02105961 (METREO) and NCT02105948 (METREX) did not include any self‐rated symptom score/symptoms of breathlessness or number of days (or nights) that participants experienced symptoms as outcome measures.
COPD Assessment Test score
The COPD Assessment Test scores revealed evidence of a small difference between mepolizumab 100 mg and placebo groups for participants with higher blood eosinophils (MD −0.95, 95% CI −1.80 to −0.10; participants = 911; studies = 2, Analysis 1.6) indicating a benefit in favour of mepolizumab 100 mg. A similar benefit was observed for the total sample of participants (MD −0.78, 95% CI −1.50 to −0.06; participants = 1285; studies = 2, Analysis 1.6). The minimum important difference on this scale was a change of two units.
1.6. Analysis.

Comparison 1: Mepolizumab 100 mg versus placebo, Outcome 6: COPD Assessment Test: change in CAT score
COPD Control Questionnaire score
NCT02105961 (METREO) and NCT02105948 (METREX) did not include COPD Control Questionnaire (CCQ) score as an outcome measure.
Mortality
There was uncertainty between mepolizumab 100 mg and placebo groups with regard to mortality (OR 0.77, 95% CI 0.42 to 1.39; participants = 1285; studies = 2; I2 = 15%, Analysis 1.7).
1.7. Analysis.

Comparison 1: Mepolizumab 100 mg versus placebo, Outcome 7: Mortality
Adverse events/side effects
We are uncertain if there is a difference between mepolizumab 100 mg and placebo groups with regard to adverse events/side effects (OR 0.97, 95% CI 0.77 to 1.21; participants = 1285; studies = 2; I² = 63%, Analysis 1.8); the statistical heterogeneity for this outcome was high.
1.8. Analysis.

Comparison 1: Mepolizumab 100 mg versus placebo, Outcome 8: Adverse events
Mepolizumab 300 mg versus placebo
Primary outcomes
All exacerbations
Only one study contributed data for this outcome (NCT02105961 (METREO)). All participants in this study had higher levels of blood eosinophils using the same definition of higher eosinophils as the mepolizumab 100 mg versus placebo comparison above.
There was probably a reduction in the rate of moderate or severe exacerbations for mepolizumab 300 mg versus placebo for the study participants (RR 0.86, 95% CI 0.70 to 1.06; participants = 451; studies = 1, Analysis 2.1, moderate‐certainty evidence).
2.1. Analysis.

Comparison 2: Mepolizumab 300 mg versus placebo, Outcome 1: Rate of moderate or severe exacerbations
There was probably a difference favouring mepolizumab 300 mg in terms of the time to first moderate or severe exacerbation (HR 0.77, 95% CI 0.60 to 0.99; participants = 451; studies = 1, Analysis 2.2, moderate‐certainty evidence). The analysis included only one study, which has a robust methodology (Table 2).
2.2. Analysis.

Comparison 2: Mepolizumab 300 mg versus placebo, Outcome 2: Time to first moderate or severe exacerbation
Hospitalisations due to COPD exacerbation
Mepolizumab 300 mg probably reduces the rate of hospitalisations due to COPD exacerbation when compared with placebo (RR 0.83, 95% CI 0.51 to 1.35; participants = 451; studies = 1, Analysis 2.3, moderate‐certainty evidence) (Table 2).
2.3. Analysis.

Comparison 2: Mepolizumab 300 mg versus placebo, Outcome 3: Rate of exacerbations with ED visit or hospitalisation
Serious adverse events
There was probably a difference between mepolizumab 300 mg and placebo groups in the number of participants experiencing serious adverse events (OR 0.84, 95% CI 0.56 to 1.27; participants = 451; studies = 1, Analysis 2.4, moderate‐certainty evidence), with fewer serious adverse events in the intervention group (Table 2).
2.4. Analysis.

Comparison 2: Mepolizumab 300 mg versus placebo, Outcome 4: Serious adverse events
Quality of life
There was probably little or no difference in SGRQ total scores between mepolizumab 300 mg and placebo groups (MD −0.10, 95% CI −2.80 to 2.60; participants = 451; studies = 1; Analysis 2.5, moderate‐certainty evidence) (Table 2).
2.5. Analysis.

Comparison 2: Mepolizumab 300 mg versus placebo, Outcome 5: Health‐related quality of life: change in SGRQ total score
Secondary outcomes
Measures of pulmonary function
NCT02105961 (METREO) did not include these specific measures of pulmonary function as an outcome measure.
Exercise performance
NCT02105961 (METREO) did not include these specific measures of exercise performance as an outcome measure.
Self‐rated symptom score/symptoms of breathlessness
NCT02105961 (METREO) did not include any self‐rated symptom score/symptoms of breathlessness or number of days (or nights) participants experienced symptoms as outcome measures.
COPD Assessment Test score
There was probably little or no difference between mepolizumab 300 mg and placebo groups in CAT scores (MD −0.40, 95% CI −1.50 to 0.70; participants = 451; studies = 1, Analysis 2.6). The minimum important difference on this scale was a change of two units.
2.6. Analysis.

Comparison 2: Mepolizumab 300 mg versus placebo, Outcome 6: COPD Assessment Test: change in CAT score
COPD Control Questionnaire score
NCT02105961 (METREO) did not include the CCQ score as an outcome measure.
Mortality
There was considerable uncertainty between mepolizumab 300 mg and placebo groups with regard to mortality (OR 0.89, 95% CI 0.34 to 2.35; participants = 451; studies = 1, Analysis 2.7).
2.7. Analysis.

Comparison 2: Mepolizumab 300 mg versus placebo, Outcome 7: Mortality
Adverse events/side effects
We are uncertain if there is a difference between mepolizumab 300 mg and placebo groups in adverse events/side effects (OR 1.01, 95% CI 0.69 to 1.48; participants = 451; studies = 1, Analysis 2.8).
2.8. Analysis.

Comparison 2: Mepolizumab 300 mg versus placebo, Outcome 8: Adverse events
Mepolizumab 750 mg versus placebo
Primary outcomes
All exacerbations
Only one trial involving 19 participants compared mepolizumab 750 mg versus placebo (Dasgupta 2016). There is great uncertainty between mepolizumab 750 mg and placebo groups in the number of participants experiencing an exacerbation within six months (OR 0.43, 95% CI 0.06 to 2.97; participants = 18; studies = 1, Analysis 3.1, low‐certainty evidence). Similarly, there is great uncertainty between mepolizumab 750 mg and placebo groups in the number of participants experiencing an exacerbation in the four‐month follow‐up period (OR 9.00, 95% CI 0.75 to 108.31; participants = 18; studies = 1, Analysis 3.2, low‐certainty evidence) (Table 3).
3.1. Analysis.

Comparison 3: Mepolizumab 750 mg versus placebo, Outcome 1: Number of participants experiencing an exacerbation within 6 months
3.2. Analysis.

Comparison 3: Mepolizumab 750 mg versus placebo, Outcome 2: Number of participants experiencing an exacerbation in 4‐month follow‐up period
Hospitalisations due to COPD exacerbation
Dasgupta 2016 did not include data relating to hospitalisations as a specific outcome measure.
Serious adverse events
Data for this outcome were obtained through correspondence with the study authors. We are uncertain if there is a difference between the two study arms with regard to serious adverse events (OR 3.00, 95% CI 0.22 to 40.93; participants = 18; studies = 1, Analysis 3.3, low‐certainty evidence) (Table 3).
3.3. Analysis.

Comparison 3: Mepolizumab 750 mg versus placebo, Outcome 3: Serious adverse events
Quality of life
We are uncertain if there is a difference between mepolizumab 750 mg and placebo groups with regard to health‐related quality of life (HRQoL) (measured with the Chronic Respiratory Disease Questionnaire (CRQ)) at three months (MD 6.92, 95% CI −11.28 to 25.12; participants = 18; studies = 1, Analysis 3.4, low‐certainty evidence). There is also great uncertainty between groups in HRQoL at six months (MD 1.14, 95% CI −17.28 to 19.56; participants = 18; studies = 1, Analysis 3.5, low‐certainty evidence) (Table 3).
3.4. Analysis.

Comparison 3: Mepolizumab 750 mg versus placebo, Outcome 4: Health‐related quality of life (CRQ at 3 months)
3.5. Analysis.

Comparison 3: Mepolizumab 750 mg versus placebo, Outcome 5: Health‐related quality of life (CRQ at 6 months)
Secondary outcomes
Measures of pulmonary function
Post‐bronchodilator FEV₁ was assessed at three and six months. We are uncertain if there is a difference between mepolizumab 750 mg and placebo groups at three months (MD 0.26, 95% CI −0.35 to 0.87; participants = 18; studies = 1, Analysis 3.6) or six months (MD 0.25, 95% CI −0.36 to 0.86; participants = 18; studies = 1, Analysis 3.7).
3.6. Analysis.

Comparison 3: Mepolizumab 750 mg versus placebo, Outcome 6: FEV₁ (litres post‐bronchodilator) at 3 months
3.7. Analysis.

Comparison 3: Mepolizumab 750 mg versus placebo, Outcome 7: FEV₁ (litres post‐bronchodilator) at 6 months
FVC was similarly assessed at three and six months. In both cases, we are uncertain if there is a difference between the two groups: the FVC % post‐bronchodilator at three months for mepolizumab 750 mg was median 82.50 (interquartile range (IQR) 43 to 90) versus placebo median 64.50 (IQR 31 to 94). At six months, the authors observed median 75.50 (IQR 46 to 87) for mepolizumab 750 mg versus median 66.50 (IQR 31 to −84) for placebo.
Exercise performance
Dasgupta 2016 did not include these specific measures of exercise performance as an outcome measure.
Self‐rated symptom score/symptoms of breathlessness
Dasgupta 2016 did not include any self‐rated symptom score/symptoms of breathlessness or number of days (or nights) participants experienced symptoms as an outcome measure.
COPD Assessment Test score
The CAT was measured at three and six months. The scores were mepolizumab 750 mg median 13 (IQR 6 to 23) versus placebo median 22 (IQR 0 to 27) at three months, and mepolizumab 750 mg median 14 (IQR 3 to 29) versus placebo median 23 (IQR 4 to 39) at six months. The minimum important difference on this scale was a change of two units. These results are uncertain, as the quality of evidence is low due to the limited number of participants (Table 3).
COPD Control Questionnaire score
Dasgupta 2016 did not include the CCQ questionnaire as an outcome measure.
Mortality
Dasgupta 2016 did not include mortality as an outcome measure.
Adverse events/side effects
Data for this outcome were obtained through correspondence with the study authors. We are uncertain if there is a difference between the two study arms for adverse events (OR 0.78, 95% CI 0.04 to 14.75; participants = 18; studies = 1, Analysis 3.8, low‐certainty evidence) (Table 3).
3.8. Analysis.

Comparison 3: Mepolizumab 750 mg versus placebo, Outcome 8: Adverse events
Benralizumab 10 mg versus placebo
Primary outcomes
All exacerbations
Data for this comparison were available only from NCT02155660 (TERRANOVA). The data in this trial were reported separately for participants with eosinophils ≥ 220/μL and for those with eosinophils < 220/μL. To remain consistent with the reporting of that trial, we followed the same strategy. Regarding moderate of severe exacerbations for participants with eosinophils ≥ 220/μL, benralizumab 10 mg probably reduces the exacerbation rate (RR 0.85, 95% CI 0.71 to 1.02; participants = 765; studies = 1, Analysis 4.1, moderate‐certainty evidence); however, there was probably little or no difference between groups for those with eosinophils < 220/μL (RR 1.04, 95% CI 0.82 to 1.32; participants = 365; studies = 1, Analysis 4.1, moderate‐certainty evidence).
4.1. Analysis.

Comparison 4: Benralizumab 10 mg versus placebo, Outcome 1: Rate of moderate or severe exacerbations
The annual EXAcerbations of Chronic pulmonary disease Tool (EXACT‐PRO) exacerbation rate was also reported for participants with eosinophils ≥ 220/μL in NCT02155660 (TERRANOVA) (RR 0.98, 95% CI 0.81 to 1.19; participants = 765; studies = 1, Analysis 4.2, moderate‐certainty evidence), indicating there was probably little or no difference between benralizumab 10 mg and placebo for this outcome.
4.2. Analysis.

Comparison 4: Benralizumab 10 mg versus placebo, Outcome 2: Annual EXACT‐PRO exacerbation rate
Hospitalisations due to COPD exacerbation
However, regarding the rate of severe exacerbations requiring hospitalisation, there was probably a difference favouring benralizumab 10 mg versus placebo (RR 0.68, 95% CI 0.49 to 0.94; participants = 765; studies = 1, Analysis 4.3, moderate‐certainty evidence) (Table 4).
4.3. Analysis.

Comparison 4: Benralizumab 10 mg versus placebo, Outcome 3: Rate of severe exacerbations requiring hospitalisation
Serious adverse events
Serious adverse events were reported for the complete sample in NCT02155660 (TERRANOVA), and there was probably little or no difference between benralizumab 10 mg and placebo groups for this outcome (OR 0.90, 95% CI 0.69 to 1.17; participants = 1129; studies = 1, Analysis 4.4, moderate‐certainty evidence) (Table 4).
4.4. Analysis.

Comparison 4: Benralizumab 10 mg versus placebo, Outcome 4: Serious adverse events
Quality of life
The SGRQ total score for participants with baseline ≥ 220/μL was reported in NCT02155660 (TERRANOVA). The data revealed little or no difference between benralizumab and placebo for this outcome (MD −0.87, 95% CI −3.23 to 1.49; participants = 680; studies = 1, Analysis 4.5, moderate‐certainty evidence) (Table 4).
4.5. Analysis.

Comparison 4: Benralizumab 10 mg versus placebo, Outcome 5: SGRQ total score
Secondary outcomes
Measures of pulmonary function
Data were reported for FEV₁ for participants with baseline ≥ 220/μL in NCT02155660 (TERRANOVA). There was probably little or no difference between benralizumab and placebo for this outcome (MD 0.01, 95% CI −0.04 to 0.05; participants = 669; studies = 1, Analysis 4.6, moderate‐certainty evidence) (Table 4).
4.6. Analysis.

Comparison 4: Benralizumab 10 mg versus placebo, Outcome 6: FEV₁ (L)
Exercise performance
No separate data were available for this outcome.
Self‐rated symptom score/symptoms of breathlessness
Total rescue medication use for participants with baseline ≥ 220/μL was reported in NCT02155660 (TERRANOVA). There was probably a slight difference between groups favouring benralizumab for this outcome (MD −0.59 puffs per day, 95% CI −1.11 to −0.07; participants = 619; studies = 1, Analysis 4.7).
4.7. Analysis.

Comparison 4: Benralizumab 10 mg versus placebo, Outcome 7: Rescue medication use
Data for nights with awakenings for participants with baseline ≥ 220/μL were also available from NCT02155660 (TERRANOVA), and there was probably little or no difference between the two treatment arms for this outcome (MD −0.04, 95% CI −0.09 to 0.01; participants = 638; studies = 1, Analysis 4.8).
4.8. Analysis.

Comparison 4: Benralizumab 10 mg versus placebo, Outcome 8: Proportion of nights with awakenings
COPD Assessment Test score
There was probably little or no difference between benralizumab and placebo groups for participants with baseline ≥ 220/μL on the CAT score (MD 0.18, 95% CI −0.82 to 1.18; participants = 682; studies = 1, Analysis 4.9). The minimum important difference on this scale was a change of two units.
4.9. Analysis.

Comparison 4: Benralizumab 10 mg versus placebo, Outcome 9: COPD Assessment Test (CAT)
COPD Control Questionnaire score
No data were available for this outcome.
Mortality
Mortality data were reported for the complete sample in NCT02155660 (TERRANOVA), and there was probably little difference between benralizumab 10 mg and placebo groups for this outcome (OR 0.90, 95% CI 0.46 to 1.76; participants = 1129; studies = 1, Analysis 4.10).
4.10. Analysis.

Comparison 4: Benralizumab 10 mg versus placebo, Outcome 10: Mortality
Adverse events/side effects
Adverse events were reported for the complete sample in NCT02155660 (TERRANOVA), and there was probably little or no difference between benralizumab 10 mg and placebo groups for this outcome (OR 0.96, 95% CI 0.76 to 1.21; participants = 1129; studies = 1, Analysis 4.11).
4.11. Analysis.

Comparison 4: Benralizumab 10 mg versus placebo, Outcome 11: Adverse events
Benralizumab 30 mg versus placebo
Primary outcomes
All exacerbations
The rate of moderate or severe exacerbations reported in NCT02155660 (TERRANOVA) and NCT02138916 (GALATHEA) for participants in the eosinophils ≥ 220/μL category indicated that there was probably little or no difference between benralizumab 30 mg and placebo groups for this outcome (RR 1.00, 95% CI 0.89 to 1.13; participants = 1523; studies = 2 Analysis 5.1, moderate‐certainty evidence). Similarly, the data for participants in the eosinophils < 220/μL subgroup indicated that there was probably little or no difference between benralizumab 30 mg and placebo groups for this outcome (RR 1.07, 95% CI 0.91 to 1.27; participants = 711; studies = 2, Analysis 5.1, moderate‐certainty evidence). Data for the EXACT‐PRO exacerbation rate for participants in the eosinophils ≥ 220/μL category also indicated that there was probably little or no difference between benralizumab 30 mg and placebo groups (RR 1.03, 95% CI 0.90 to 1.17; participants = 1522; studies = 2, Analysis 5.2, moderate‐certainty evidence). Although the studies were large with a robust methodology, the confidence intervals include the possibility of a small or no effect and some benefit or harm (Table 5).
5.1. Analysis.

Comparison 5: Benralizumab 30 mg versus placebo, Outcome 1: Rate of moderate or severe exacerbations
5.2. Analysis.

Comparison 5: Benralizumab 30 mg versus placebo, Outcome 2: Annual EXACT‐PRO exacerbation rate
Hospitalisations due to COPD exacerbation
There was similarly probably little or no difference between benralizumab 30 mg and placebo in rate of severe exacerbations for participants in the eosinophils ≥ 220/μL category (RR 1.01, 95% CI 0.77 to 1.33; participants = 1523; studies = 2, Analysis 5.3, moderate‐certainty evidence). This was also the case with regard to rate of severe exacerbations requiring hospitalisation for participants in the eosinophils ≥ 220/μL subgroup (RR 0.96, 95% CI 0.75 to 1.22; participants = 1523; studies = 2, Analysis 5.4, moderate‐certainty evidence) (Table 5).
5.3. Analysis.

Comparison 5: Benralizumab 30 mg versus placebo, Outcome 3: Rate of severe exacerbations
5.4. Analysis.

Comparison 5: Benralizumab 30 mg versus placebo, Outcome 4: Rate of severe exacerbations requiring hospitalisation
Serious adverse events
There was similarly probably little or no difference between benralizumab 30 mg and placebo in serious adverse events (OR 0.98, 95% CI 0.81 to 1.17; participants = 2235; studies = 2; I² = 79%, Analysis 5.5, moderate‐certainty evidence). There was high statistical heterogeneity between the two studies, although they both have a robust methodology (Table 5).
5.5. Analysis.

Comparison 5: Benralizumab 30 mg versus placebo, Outcome 5: Serious adverse events
Quality of life
Mean change from baseline in SGRQ total score for participants with baseline ≥ 220/μL was reported in both NCT02155660 (TERRANOVA) and NCT02138916 (GALATHEA). There was probably only a small difference between benralizumab 30 mg and placebo for this outcome (MD −1.42, 95% CI −3.13 to 0.29; participants = 1333; studies = 2, Analysis 5.6, moderate‐certainty evidence). The minimal important difference on this scale was a change of four units. Although both studies were large with a robust methodology, the confidence intervals include the possibility of a small or no effect and some benefit or harm (Table 5).
5.6. Analysis.

Comparison 5: Benralizumab 30 mg versus placebo, Outcome 6: SGRQ total score
Secondary outcomes
Measures of pulmonary function
Data on FEV₁ performance were available from both NCT02155660 (TERRANOVA) and NCT02138916 (GALATHEA) relating to participants with baseline ≥ 220/μL. There was probably no difference between benralizumab 30 mg and placebo groups for this outcome (MD −0.00, 95% CI −0.03 to 0.03; participants = 1312; studies = 2, Analysis 5.7, moderate‐certainty evidence).
5.7. Analysis.

Comparison 5: Benralizumab 30 mg versus placebo, Outcome 7: FEV₁ (L)
Exercise performance
No separate data were available for this outcome.
Self‐rated symptom score/symptoms of breathlessness
Both NCT02155660 (TERRANOVA) and NCT02138916 (GALATHEA) provided data on inhaled rescue medication used during the treatment period; however, this was reported only for participants with baseline ≥ 220/μL. A small difference between benralizumab 30 mg versus placebo was indicated, favouring benralizumab (MD −0.40 puffs per day, 95% CI −0.77 to −0.03; participants = 1216; studies = 2; I² = 0%, Analysis 5.8).
5.8. Analysis.

Comparison 5: Benralizumab 30 mg versus placebo, Outcome 8: Rescue medication use
The number of nights participants experienced symptoms was reported in both trials as a measure of nights with awakenings for participants with baseline ≥ 220/μL. A small difference favouring benralizumab was indicated (MD −0.06, 95% CI −0.09 to −0.02; participants = 1242; studies = 2; I² = 11%, Analysis 5.9).
5.9. Analysis.

Comparison 5: Benralizumab 30 mg versus placebo, Outcome 9: Proportion of nights with awakenings
COPD Assessment Test score
The CAT score was reported in both trials for participants with baseline eosinophils ≥ 220/μL. No evidence of a difference between benralizumab 30 mg and placebo was observed for this outcome (MD −0.18, 95% CI −0.90 to 0.55; participants = 1338; studies = 2; I² = 0%, Analysis 5.10). The minimum important difference on this scale was a change of two units.
5.10. Analysis.

Comparison 5: Benralizumab 30 mg versus placebo, Outcome 10: COPD Assessment Test (CAT)
COPD Control Questionnaire score
No data were available relating to CCQ score.
Mortality
We are uncertain if there is a difference in mortality between benralizumab 30 mg and placebo groups (OR 1.13, 95% CI 0.70 to 1.84; participants = 2235; studies = 2; I² = 0%, Analysis 5.11).
5.11. Analysis.

Comparison 5: Benralizumab 30 mg versus placebo, Outcome 11: Mortality
Adverse events/side effects
There was probably little or no difference between benralizumab 30 mg and placebo groups in adverse events (OR 0.94, 95% CI 0.80 to 1.12; participants = 2235; studies = 2; I² = 51%, Analysis 5.12).
5.12. Analysis.

Comparison 5: Benralizumab 30 mg versus placebo, Outcome 12: Adverse events
Benralizumab 100 mg versus placebo
Primary outcomes
All exacerbations
The rate of moderate or severe exacerbations was reported in Brightling 2014, NCT02155660 (TERRANOVA), and NCT02138916 (GALATHEA), with no certain difference observed between benralizumab 100 mg and placebo groups (RR 0.94, 95% CI 0.85 to 1.03; participants = 2314; studies = 3, Analysis 6.1, moderate‐certainty evidence).
6.1. Analysis.

Comparison 6: Benralizumab 100 mg versus placebo, Outcome 1: Rate of moderate or severe exacerbations
The annual EXACT‐PRO exacerbation rate was also reported for participants in the eosinophils ≥ 220/μL category in NCT02155660 (TERRANOVA) and NCT02138916 (GALATHEA), with no certain difference indicated between benralizumab 100 mg and placebo groups (RR 0.95, 95% CI 0.82 to 1.09; participants = 1509; studies = 2, Analysis 6.2, moderate‐certainty evidence). Although both studies were large with a robust methodology, the confidence intervals include the possibility of a small or no effect and important benefit or harm (Table 6).
6.2. Analysis.

Comparison 6: Benralizumab 100 mg versus placebo, Outcome 2: Annual EXACT‐PRO exacerbation rate
Hospitalisations due to COPD exacerbation
There was probably a small difference favouring benralizumab 100 mg versus placebo in absolute number of participants experiencing exacerbations (OR 0.39, 95% CI 0.07 to 2.13; participants = 82; studies = 1; Analysis 6.3, moderate certainty evidence) (Brightling 2014). However, with regard to rate of exacerbations for participants in the eosinophils ≥ 220/μL category in NCT02155660 (TERRANOVA) and NCT02138916 (GALATHEA), there was a clear advantage with benralizumab 100 mg versus placebo (RR 0.63, 95% CI 0.49 to 0.81; participants = 1512; studies = 2, Analysis 6.4, high‐certainty evidence) (Table 6).
6.3. Analysis.

Comparison 6: Benralizumab 100 mg versus placebo, Outcome 3: Hospitalisations due to COPD exacerbation
6.4. Analysis.

Comparison 6: Benralizumab 100 mg versus placebo, Outcome 4: Rate of exacerbations requiring hospitalisation
Serious adverse events
Three studies reported this outcome (Brightling 2014; NCT02138916 (GALATHEA); NCT02155660 (TERRANOVA)). There was probably little or no difference between benralizumab 100 mg and placebo groups (OR 0.90, 95% CI 0.75 to 1.08; participants = 2333; studies = 3, Analysis 6.5, moderate‐certainty evidence) (Table 6).
6.5. Analysis.

Comparison 6: Benralizumab 100 mg versus placebo, Outcome 5: Serious adverse events
Quality of life
There may be little or no difference between benralizumab 100 mg and placebo in change in SGRQ total score in Brightling 2014 (MD −1.08, 95% CI −7.34 to 5.18; participants = 82; studies = 1, Analysis 6.6). However, for participants in the eosinophils ≥ 220/μL category in NCT02155660 (TERRANOVA) and NCT02138916 (GALATHEA), there was a small difference in change in SGRQ total score favouring benralizumab 100 mg versus placebo (MD −1.47, 95% CI −2.89 to −0.05; participants = 1351; studies = 2, Analysis 6.6, high‐certainty evidence), although this difference was not greater than the minimum clinically significant difference of four units (Table 6).
6.6. Analysis.

Comparison 6: Benralizumab 100 mg versus placebo, Outcome 6: Quality of life: change in SGRQ total score
Secondary outcomes
Measures of pulmonary function such as FEV₁, and FVC
NCT02155660 (TERRANOVA) and NCT02138916 (GALATHEA) reported FEV₁ data only for participants with baseline eosinophils ≥ 220/μL. There was no certain difference between benralizumab 100 mg and placebo groups for this outcome (MD 0.02, 95% CI −0.01 to 0.05; participants = 1334; studies = 2; I² = 0%, Analysis 6.7).
6.7. Analysis.

Comparison 6: Benralizumab 100 mg versus placebo, Outcome 7: FEV₁ (L)
However, in Brightling 2014, FEV₁ data were reported for the complete sample. A difference was observed favouring the benralizumab arm (MD 0.19, 95% CI 0.05 to 0.33; participants = 91; studies = 1, Analysis 6.7).
Exercise performance
No separate data were available for this outcome.
Self‐rated symptom score/symptoms of breathlessness
Data on inhaled rescue medication used during the treatment period were reported for participants with baseline eosinophils ≥ 220/μL in both NCT02155660 (TERRANOVA) and NCT02138916 (GALATHEA). A small difference favouring benralizumab 100 mg versus placebo was indicated (MD −0.49, 95% CI −0.83 to −0.15; participants = 1237; studies = 2; I² = 0%, Analysis 6.8).
6.8. Analysis.

Comparison 6: Benralizumab 100 mg versus placebo, Outcome 8: Rescue medication use
Data relating to the proportion of nights participants were awake were also provided by NCT02155660 (TERRANOVA) and NCT02138916 (GALATHEA) for those with baseline eosinophils ≥ 220/μL. There was probably a small difference favouring benralizumab for this outcome (MD −0.03, 95% CI −0.07 to 0.00; participants = 1263; studies = 2; I² = 0%, Analysis 6.9). The minimum important difference on this scale was a change of two units.
6.9. Analysis.

Comparison 6: Benralizumab 100 mg versus placebo, Outcome 9: Proportion of nights with awakenings
COPD Assessment Test score
Data were also available for participants with baseline eosinophils ≥ 220/μL in NCT02155660 (TERRANOVA) and NCT02138916 (GALATHEA), indicating probably a small difference favouring benralizumab on this measure (MD −0.60, 95% CI −1.29 to 0.10; participants = 1358; studies = 2; I² = 66%, Analysis 6.10).
6.10. Analysis.

Comparison 6: Benralizumab 100 mg versus placebo, Outcome 10: COPD Assessment Test (CAT)
COPD Control Questionnaire score
No data were available for this outcome.
Mortality
Three studies reported this outcome (Brightling 2014; NCT02138916 (GALATHEA); NCT02155660 (TERRANOVA)), with probably no difference observed between benralizumab 100 mg and placebo groups (OR 0.94, 95% CI 0.57 to 1.55; participants = 2333; studies = 3; I² = 0%, Analysis 6.11).
6.11. Analysis.

Comparison 6: Benralizumab 100 mg versus placebo, Outcome 11: Mortality
Adverse events/side effects
Data from three studies revealed that there was probably no difference between study arms for this outcome (OR 1.06, 95% CI 0.90 to 1.26; participants = 2333; studies = 3; I² = 64%, Analysis 6.12) (Brightling 2014; NCT02138916 (GALATHEA); NCT02155660 (TERRANOVA)).
6.12. Analysis.

Comparison 6: Benralizumab 100 mg versus placebo, Outcome 12: Adverse events
Discussion
Summary of main results
Six studies met the inclusion criteria for this review (Brightling 2014; Dasgupta 2016; NCT02105948 (METREX); NCT02105961 (METREO); NCT02138916 (GALATHEA); NCT02155660 (TERRANOVA)). Three studies compared mepolizumab to placebo (Dasgupta 2016; NCT02105948 (METREX); NCT02105961 (METREO)), and three compared benralizumab to placebo (Brightling 2014; NCT02138916 (GALATHEA); NCT02155660 (TERRANOVA)). No head‐to‐head trials were identified. All studies included only participants with frequent exacerbations of COPD.
For our primary outcome, rate of moderate or severe exacerbations, mepolizumab 100 mg reduces exacerbations by 19% in those with an eosinophil count of at least 150/μL, based on high‐certainty evidence. With the inclusion of participants with lower eosinophils, mepolizumab 100 mg probably reduces the exacerbation rate by 8%, based on moderate‐certainty evidence. Mepolizumab 300 mg also probably reduces the rate of exacerbations by 14% in participants all of whom had raised eosinophils. The evidence in a single small study of mepolizumab 750 mg was very uncertain.
Participants receiving mepolizumab 100 mg experienced a longer duration to first moderate or severe exacerbation than those in the placebo group, but only those with the eosinophilic phenotype; within the total sample this difference was smaller and less certain. The certainty of the evidence for the eosinophilic group was high. There was also a small increase in time to first moderate or severe exacerbation in all participants receiving mepolizumab 300 mg, based on moderate‐certainty evidence.
The COPD Assessment Test (CAT score), a questionnaire designed to measure the impact of COPD on a person's life, revealed a modest benefit in favour of the mepolizumab 100 mg group for all participants, including the eosinophilic phenotype participants. There was probably little or no difference between mepolizumab 300 mg and placebo groups in CAT scores. This difference may be due to measurement imprecision, with the mepolizumab 100 mg data based on two studies with high heterogeneity (NCT02105948 (METREX); NCT02105961 (METREO)), and the mepolizumab 300 mg data having wide confidence intervals (NCT02105961 (METREO)).
For all other outcomes where mepolizumab was compared to placebo, there were no certain differences between the intervention and placebo.
Benralizumab 100 mg reduces the rate of severe exacerbation requiring hospitalisation in participants with an eosinophil count of at least 220/μL, based on high‐certainty evidence. Benralizumab 10 mg probably reduces the rate of severe exacerbations in those with an eosinophil count of at least 220/μL, based on moderate‐certainty evidence.
For participants in the eosinophils ≥ 220/μL category in NCT02155660 (TERRANOVA) and NCT02138916 (GALATHEA), there was an improvement in the St George's Respiratory Questionnaire (SGRQ) total score in favour of benralizumab 100 mg; however, the difference fell below the minimum clinically significant difference.
Self‐rated symptom score/symptoms of breathlessness, such as inhaled rescue medication used during the treatment period and concomitant medication usage, including antibiotics and steroids, showed a treatment advantage for benralizumab SC 30 mg and 100 mg. The number of nights participants experienced symptoms, as a measure of nights with awakenings, was also reported in NCT02155660 (TERRANOVA) and NCT02138916 (GALATHEA), with a difference favouring benralizumab 30 mg, and a probable benefit of benralizumab 100 mg.
No clinically meaningful changes in lung function were seen. The included trials were conducted in populations selected for fixed airflow obstruction, so it would be surprising if large changes in pulmonary physiological measurements were observed.
Treatment with mepolizumab SC and benralizumab SC appeared to be safe. All pooled analyses showed that there was probably little or no difference in serious adverse events, adverse events, or side effects between the use of a monoclonal antibody therapy compared to placebo.
Anti‐IL‐5 therapies appear to be safe in individuals with COPD, and have demonstrated some modest efficacy in the reduction of exacerbation rates and disease‐related symptoms. Nevertheless, these efficacies were more certain in those participants with higher blood eosinophil levels. (Note that the definition of higher blood eosinophils differed between the mepolizumab and the benralizumab trials, but in both cases included participants with blood eosinophils at the higher end of the normal range as well as those with true blood eosinophilia).
Overall completeness and applicability of evidence
The participant demographics are representative of individuals with COPD, with mean age between 63 to 67 years. The aim of this review was to assess the efficacy and safety of monoclonal antibody therapies targeting IL‐5 signalling (anti‐IL‐5 or anti‐IL‐5Rα) compared with placebo in the treatment of adults with COPD. Whilst exacerbation rate, hospitalisation, health‐related quality of life, and adverse events were consistently reported in all studies, other outcome measures such as exercise tolerance, self‐rated symptoms, and lung function were not.
We identified four studies that are ongoing and have yet to be completed.
There are a number of monoclonal antibody therapies approved for use in the context of eosinophilic or atopic asthma. We did not find evidence in COPD for monoclonal antibodies other than mepolizumab (anti‐IL‐5) and benralizumab (anti‐IL‐5 receptor). The information does not lend itself to a direct dose response interpretation as there is no evidence of greater effects at higher doses.
Certainty of the evidence
We applied the GRADE system and judged the certainty of the evidence for most comparisons to be at least moderate. Although we are more certain about the benefits for participants with higher levels of blood eosinophils in reducing the rate of severe exacerbations, we cannot deduce that the intervention does not work in those with lower levels of blood eosinophils; we are less certain about this group due to the wider confidence intervals which include the possibility of a small or no effect and important benefit or harm. The limitations in some of the included studies are noted in the Results, Figure 2, and Characteristics of included studies. A funnel plot was not feasible due to the small number of included studies, therefore a formal assessment of publication bias using such methods was not possible. Nevertheless, our search strategy was comprehensive and robust, and included searching conference abstracts and ongoing studies to find unpublished studies.
Potential biases in the review process
Our review adhered as closely as possible to our published protocol (Donovan 2019). In order to align with the GOLD 2020 definition, we have used different labels for two of our primary outcomes compared with our protocol (Donovan 2019): "severe exacerbation" is functionally identical to "hospitalisations due to COPD exacerbation", and the switch from "any exacerbation" to "moderate or severe exacerbation" allows meaningful comparisons between the two drugs.
As with most systematic reviews, there remains the possibility that we may have failed to identify unpublished trials contributing positive or negative results, and we are aware of the potential for publication bias. Six trials meeting our inclusion criteria were identified through comprehensive and systematic database searches, and two review authors independently evaluated all the identified studies to endeavour to address any study selection bias or errors.
Agreements and disagreements with other studies or reviews
We are not aware of any other systematic reviews addressing this question.
The results of NCT02105948 (METREX) and NCT02105961 (METREO) were published together, along with both pre‐planned and post hoc analyses of their combined data (Pavord 2017). These analyses found a 23% reduction in the rate of moderate or severe exacerbations in participants with baseline blood eosinophil counts greater than 300 cells per microlitre treated with mepolizumab compared with placebo. This reduction was possibly confined to exacerbations that required treatment with corticosteroids, rather than those requiring antibiotics alone (Pavord 2018). This reduction in exacerbation rate is consistent with our review (19% reduction in exacerbation rate in those with an eosinophilic phenotype treated with mepolizumab 100 mg), although we did not specifically examine the treatments chosen for exacerbations.
A Cochrane Review found that both mepolizumab and benralizumab reduced exacerbation rates in severe asthma by around 50% (Farne 2017). Whilst we did find some evidence of a reduction in exacerbations in COPD with a eosinophilic phenotype with these drugs, the decrease was far more modest, not consistent across drug doses, and in the case of benralizumab, confined to exacerbations severe enough to require hospitalisation.
Authors' conclusions
Implications for practice.
Mepolizumab and benralizumab may have a small role as add‐on therapies in a highly selected group of chronic obstructive pulmonary disease (COPD) patients who have both higher levels of blood eosinophils and frequent moderate to severe exacerbations. In this group, these treatments appear to modestly reduce the rate of severe exacerbations (and for mepolizumab, possibly moderate exacerbations). The included studies did not compare frequent with infrequent exacerbators, and our conclusions here relate only to the former reflecting the samples in the included studies; we are not in a position to comment on infrequent exacerbators. Importantly, there were no safety concerns or an excess of serious adverse events. Lung function and health‐related quality of life were not improved.
Implications for research.
Based on the available evidence, it seems unlikely that interleukin 5 (IL‐5) or its receptor (IL‐5R) therapies will be of use for the majority of people with COPD. Given the mechanisms of action of these drugs, they would only be expected to be of benefit in those with type 2 inflammation. The cut‐off points used to define higher levels of blood eosinophils were relatively low (150 to 220/μL), so even the high‐eosinophil groups may have included substantial numbers of participants with only modest degrees of type 2 inflammation. Future trials of anti‐IL‐5 therapies in COPD should target those with true peripheral blood eosinophilia.
Without direct comparisons between benralizumab versus mepolizumab from head‐to‐head trials there is considerable uncertainty to guide practice. Whilst a network meta‐analysis could potentially illuminate this issue, in the absence of such studies, the differing definitions of eosinophilia in the included studies are a major barrier to this.
History
Protocol first published: Issue 9, 2019 Review first published: Issue 12, 2020
Acknowledgements
SM and TD acknowledge the support of Lancaster University and the University of Cumbria for providing the time to develop and complete this review.
The Background and Methods sections of this review are based on a standard template used by Cochrane Airways.
The review authors and Airways Editorial team are grateful to the following peer and consumer reviewers for their time and comments:
Nicholas Hopkinson (UK);
Huib AM Kerstjens (the Netherlands);
Stella O'Brien (UK); and
Cho Naing (Malaysia).
The review authors are especially grateful to Chris Cates for additional guidance on the statistical analysis of this review, and to the rest of the staff at the editorial base (Emma Dennett, Liz Stovold, Emma Jackson, and Rebecca Fortescue) for their excellent support throughout the development of the review. We would particularly like to thank Dr Parameswaran Nair for kindly providing additional data for his study.
This project was supported by the National Institute for Health Research (NIHR), via Cochrane Infrastructure funding to Cochrane Airways. The views and opinions expressed herein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS, or the Department of Health.
Appendices
Appendix 1. Database search strategies
Cochrane Airways Register via the Cochrane Register of Studies
| 1 | MeSH DESCRIPTOR Pulmonary Disease, Chronic Obstructive Explode All AND INSEGMENT |
| 2 | MeSH DESCRIPTOR Bronchitis, Chronic AND INSEGMENT |
| 3 | (obstruct*) near3 (pulmonary or lung* or airway* or airflow* or bronch* or respirat*) AND INSEGMENT |
| 4 | COPD:MISC1 AND INSEGMENT |
| 5 | (COPD OR AECOPD OR AECB):TI,AB,KW AND INSEGMENT |
| 6 | #1 OR #2 OR #3 OR #5 OR #4 AND INSEGMENT |
| 7 | MESH DESCRIPTOR Antibodies, Monoclonal AND INSEGMENT |
| 8 | MESH DESCRIPTOR Antibodies, Monoclonal, Humanized AND INSEGMENT |
| 9 | mepolizumab AND INSEGMENT |
| 10 | SB24056 or SB‐24056 AND INSEGMENT |
| 11 | Bosatria or Nucala AND INSEGMENT |
| 12 | benralizumab* AND INSEGMENT |
| 13 | MEDI‐563 AND INSEGMENT |
| 14 | Reslizumab* AND INSEGMENT |
| 15 | Cinquil or Cinqair AND INSEGMENT |
| 16 | CEP‐38072 AND INSEGMENT |
| 17 | anti‐interleukin 5 AND INSEGMENT |
| 18 | anti‐IL5 AND INSEGMENT |
| 19 | anti‐IL‐5 AND INSEGMENT |
| 20 | MESH DESCRIPTOR Interleukin‐5 EXPLODE ALL AND INSEGMENT |
| 21 | MESH DESCRIPTOR Receptors, Interleukin‐5 EXPLODE ALL AND INSEGMENT |
| 22 | #7 OR #8 OR #9 OR #10 OR #11 OR #12 OR #13 OR #14 OR #15 OR #16 OR #17 OR #18 OR #19 OR #20 OR #21 AND INSEGMENT |
| 23 | #22 AND #6 AND INSEGMENT |
| 24 | INREGISTER |
| 25 | #23 AND #24 |
CENTRAL via the Cochrane Register of Studies
| 1 | MeSH DESCRIPTOR Pulmonary Disease, Chronic Obstructive Explode All AND CENTRAL:TARGET |
| 2 | MeSH DESCRIPTOR Bronchitis, Chronic AND CENTRAL:TARGET |
| 3 | (obstruct*) near3 (pulmonary or lung* or airway* or airflow* or bronch* or respirat*) AND CENTRAL:TARGET |
| 4 | COPD:MISC1 AND CENTRAL:TARGET |
| 5 | (COPD OR AECOPD OR AECB):TI,AB,KW AND CENTRAL:TARGET |
| 6 | #1 OR #2 OR #3 OR #5 OR #4 AND CENTRAL:TARGET |
| 7 | MESH DESCRIPTOR Antibodies, Monoclonal AND CENTRAL:TARGET |
| 8 | MESH DESCRIPTOR Antibodies, Monoclonal, Humanized AND CENTRAL:TARGET |
| 9 | mepolizumab AND CENTRAL:TARGET |
| 10 | SB24056 or SB‐24056 AND CENTRAL:TARGET |
| 11 | Bosatria or Nucala AND CENTRAL:TARGET |
| 12 | benralizumab* AND CENTRAL:TARGET |
| 13 | MEDI‐563 AND CENTRAL:TARGET |
| 14 | Reslizumab* AND CENTRAL:TARGET |
| 15 | Cinquil or Cinqair AND CENTRAL:TARGET |
| 16 | CEP‐38072 AND CENTRAL:TARGET |
| 17 | anti‐interleukin 5 AND CENTRAL:TARGET |
| 18 | anti‐IL5 AND CENTRAL:TARGET |
| 19 | anti‐IL‐5 AND CENTRAL:TARGET |
| 20 | MESH DESCRIPTOR Interleukin‐5 EXPLODE ALL AND CENTRAL:TARGET |
| 21 | MESH DESCRIPTOR Receptors, Interleukin‐5 EXPLODE ALL AND CENTRAL:TARGET |
| 22 | #7 OR #8 OR #9 OR #10 OR #11 OR #12 OR #13 OR #14 OR #15 OR #16 OR #17 OR #18 OR #19 OR #20 OR #21 AND CENTRAL:TARGET |
| 23 | #22 AND #6 AND CENTRAL:TARGET |
MEDLINE (Ovid) ALL
| 1 | Lung Diseases, Obstructive/ |
| 2 | exp Pulmonary Disease, Chronic Obstructive/ |
| 3 | emphysema$.tw. |
| 4 | (chronic$ adj3 bronchiti$).tw. |
| 5 | (obstruct$ adj3 (pulmonary or lung$ or airway$ or airflow$ or bronch$ or respirat$)).tw. |
| 6 | (COPD or AECOPD or AECB).ti,ab. |
| 7 | or/1‐6 |
| 8 | exp Antibodies, Monoclonal, Humanized/ |
| 9 | Antibodies, Monoclonal/ |
| 10 | mepolizumab.tw. |
| 11 | (SB24056 or SB‐24056).tw. |
| 12 | (Bosatria or Nucala).tw. |
| 13 | benralizumab.tw. |
| 14 | MEDI‐563.tw. |
| 15 | Reslizumab.tw. |
| 16 | (Cinquil or Cinqair).tw. |
| 17 | CEP‐38072.tw. |
| 18 | anti‐interleukin$ 5.tw. |
| 19 | anti‐IL5.tw. |
| 20 | anti‐IL‐5.tw. |
| 21 | Interleukin‐5/ |
| 22 | exp Receptors, Interleukin‐5/ |
| 23 | or/8‐22 |
| 24 | 7 and 23 |
| 25 | (controlled clinical trial or randomised controlled trial).pt. |
| 26 | (randomised or randomised).ab,ti. |
| 27 | placebo.ab,ti. |
| 28 | dt.fs. |
| 29 | randomly.ab,ti. |
| 30 | trial.ab,ti. |
| 31 | groups.ab,ti. |
| 32 | or/25‐31 |
| 33 | Animals/ |
| 34 | Humans/ |
| 35 | 33 not (33 and 34) |
| 36 | 32 not 35 |
| 37 | 24 and 36 |
Embase (Ovid)
| 1 | exp chronic obstructive lung disease/ |
| 2 | obstructive airway disease/ |
| 3 | (obstruct$ adj3 (pulmonary or lung$ or airway$ or airflow$ or bronch$ or respirat$)).tw. |
| 4 | (chronic$ adj3 bronchiti$).tw. |
| 5 | emphysema$.tw. |
| 6 | (COPD or AECOPD or AECB).ti,ab. |
| 7 | or/1‐6 |
| 8 | mepolizumab/ |
| 9 | mepolizumab.tw. |
| 10 | (SB24056 or SB‐24056).tw. |
| 11 | (Bosatria or Nucala).tw. |
| 12 | benralizumab/ |
| 13 | benralizumab.tw. |
| 14 | MEDI‐563.tw. |
| 15 | reslizumab/ |
| 16 | Reslizumab.tw. |
| 17 | (Cinquil or Cinqair).tw. |
| 18 | CEP‐38072.tw. |
| 19 | anti‐interleukin$ 5.tw. |
| 20 | anti‐IL5.tw. |
| 21 | anti‐IL‐5.tw. |
| 22 | interleukin 5/ |
| 23 | interleukin 5 receptor/ |
| 24 | or/8‐22 |
| 25 | 7 and 24 |
| 26 | Randomized Controlled Trial/ |
| 27 | randomisation/ |
| 28 | controlled clinical trial/ |
| 29 | Double Blind Procedure/ |
| 30 | Single Blind Procedure/ |
| 31 | Crossover Procedure/ |
| 32 | (clinica$ adj3 trial$).tw. |
| 33 | ((singl$ or doubl$ or trebl$ or tripl$) adj3 (mask$ or blind$ or method$)).tw. |
| 34 | exp Placebo/ |
| 35 | placebo$.ti,ab. |
| 36 | random$.ti,ab. |
| 37 | ((control$ or prospectiv$) adj3 (trial$ or method$ or stud$)).tw. |
| 38 | (crossover$ or cross‐over$).ti,ab. |
| 39 | or/26‐38 |
| 40 | exp animals/ or exp invertebrate/ or animal experiment/ or animal model/ or animal tissue/ or animal cell/ or nonhuman/ |
| 41 | human/ or normal human/ or human cell/ |
| 42 | 40 and 41 |
| 43 | 40 not 42 |
| 44 | 39 not 43 |
| 45 | 25 and 44 |
ClinicalTrials.gov
| Search field | Search terms |
| Study type | Interventional |
| Condition | COPD |
| Intervention | mepolizumab OR benralizumab OR Reslizumab |
WHO ICTRP
| Search field | Search terms |
| Condition | COPD |
| Intervention | mepolizumab OR benralizumab OR Reslizumab |
Data and analyses
Comparison 1. Mepolizumab 100 mg versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 Rate of moderate or severe exacerbations | 2 | Rate Ratio (IV, Fixed, 95% CI) | Subtotals only | |
| 1.1.1 Eosinophilic phenotype | 2 | 911 | Rate Ratio (IV, Fixed, 95% CI) | 0.81 [0.71, 0.93] |
| 1.1.2 All participants | 2 | 1285 | Rate Ratio (IV, Fixed, 95% CI) | 0.92 [0.82, 1.03] |
| 1.2 Time to first moderate or severe exacerbation | 2 | Hazard Ratio (IV, Fixed, 95% CI) | Subtotals only | |
| 1.2.1 Eosinophilic phenotype | 2 | 981 | Hazard Ratio (IV, Fixed, 95% CI) | 0.78 [0.66, 0.92] |
| 1.2.2 All participants | 2 | 1285 | Hazard Ratio (IV, Fixed, 95% CI) | 0.87 [0.75, 1.00] |
| 1.3 Rate of exacerbations with ED visit or hospitalisation | 2 | Rate Ratio (IV, Fixed, 95% CI) | Subtotals only | |
| 1.3.1 Eosinophilic phenotype | 2 | 911 | Rate Ratio (IV, Fixed, 95% CI) | 0.90 [0.65, 1.24] |
| 1.3.2 All participants | 2 | 1285 | Rate Ratio (IV, Fixed, 95% CI) | 0.94 [0.72, 1.22] |
| 1.4 Serious adverse events | 2 | 1285 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.82 [0.65, 1.05] |
| 1.5 Health‐related quality of life: change in SGRQ total score | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.5.1 Eosinophilic phenotype | 2 | 911 | Mean Difference (IV, Fixed, 95% CI) | ‐0.90 [‐2.91, 1.10] |
| 1.5.2 All participants | 2 | 1285 | Mean Difference (IV, Fixed, 95% CI) | ‐0.30 [‐2.00, 1.41] |
| 1.6 COPD Assessment Test: change in CAT score | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.6.1 Eosinophilic phenotype | 2 | 911 | Mean Difference (IV, Fixed, 95% CI) | ‐0.95 [‐1.80, ‐0.10] |
| 1.6.2 All participants | 2 | 1285 | Mean Difference (IV, Fixed, 95% CI) | ‐0.78 [‐1.50, ‐0.06] |
| 1.7 Mortality | 2 | 1285 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.77 [0.42, 1.39] |
| 1.8 Adverse events | 2 | 1285 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.77, 1.21] |
Comparison 2. Mepolizumab 300 mg versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 2.1 Rate of moderate or severe exacerbations | 1 | Rate Ratio (IV, Fixed, 95% CI) | Subtotals only | |
| 2.1.1 Eosinophilic phenotype | 1 | 451 | Rate Ratio (IV, Fixed, 95% CI) | 0.86 [0.70, 1.06] |
| 2.2 Time to first moderate or severe exacerbation | 1 | Hazard Ratio (IV, Fixed, 95% CI) | Subtotals only | |
| 2.2.1 Eosinophilic phenotype | 1 | 451 | Hazard Ratio (IV, Fixed, 95% CI) | 0.77 [0.60, 0.99] |
| 2.3 Rate of exacerbations with ED visit or hospitalisation | 1 | Rate Ratio (IV, Fixed, 95% CI) | Subtotals only | |
| 2.3.1 Eosinophilic phenotype | 1 | 451 | Rate Ratio (IV, Fixed, 95% CI) | 0.83 [0.51, 1.35] |
| 2.4 Serious adverse events | 1 | 451 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.84 [0.56, 1.27] |
| 2.5 Health‐related quality of life: change in SGRQ total score | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2.5.1 Eosinophilic phenotype | 1 | 451 | Mean Difference (IV, Fixed, 95% CI) | ‐0.10 [‐2.80, 2.60] |
| 2.6 COPD Assessment Test: change in CAT score | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2.6.1 Eosinophilic phenotype | 1 | 451 | Mean Difference (IV, Fixed, 95% CI) | ‐0.40 [‐1.50, 0.70] |
| 2.7 Mortality | 1 | 451 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.34, 2.35] |
| 2.8 Adverse events | 1 | 451 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.01 [0.69, 1.48] |
Comparison 3. Mepolizumab 750 mg versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 3.1 Number of participants experiencing an exacerbation within 6 months | 1 | 18 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.43 [0.06, 2.97] |
| 3.2 Number of participants experiencing an exacerbation in 4‐month follow‐up period | 1 | 18 | Odds Ratio (M‐H, Fixed, 95% CI) | 9.00 [0.75, 108.31] |
| 3.3 Serious adverse events | 1 | 18 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.00 [0.22, 40.93] |
| 3.4 Health‐related quality of life (CRQ at 3 months) | 1 | 18 | Mean Difference (IV, Fixed, 95% CI) | 6.92 [‐11.28, 25.12] |
| 3.5 Health‐related quality of life (CRQ at 6 months) | 1 | 18 | Mean Difference (IV, Fixed, 95% CI) | 1.14 [‐17.28, 19.56] |
| 3.6 FEV₁ (litres post‐bronchodilator) at 3 months | 1 | 18 | Mean Difference (IV, Fixed, 95% CI) | 0.26 [‐0.35, 0.87] |
| 3.7 FEV₁ (litres post‐bronchodilator) at 6 months | 1 | 18 | Mean Difference (IV, Fixed, 95% CI) | 0.25 [‐0.36, 0.86] |
| 3.8 Adverse events | 1 | 18 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.78 [0.04, 14.75] |
Comparison 4. Benralizumab 10 mg versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 4.1 Rate of moderate or severe exacerbations | 1 | 1130 | Rate Ratio (IV, Fixed, 95% CI) | 0.91 [0.79, 1.06] |
| 4.1.1 Eosinophils ≧ 220/μL | 1 | 765 | Rate Ratio (IV, Fixed, 95% CI) | 0.85 [0.71, 1.02] |
| 4.1.2 Eosinophils < 220/μL | 1 | 365 | Rate Ratio (IV, Fixed, 95% CI) | 1.04 [0.82, 1.32] |
| 4.2 Annual EXACT‐PRO exacerbation rate | 1 | 765 | Rate Ratio (IV, Fixed, 95% CI) | 0.98 [0.81, 1.19] |
| 4.2.1 Eosinophils ≧ 220/μL | 1 | 765 | Rate Ratio (IV, Fixed, 95% CI) | 0.98 [0.81, 1.19] |
| 4.3 Rate of severe exacerbations requiring hospitalisation | 1 | 765 | Rate Ratio (IV, Fixed, 95% CI) | 0.68 [0.49, 0.94] |
| 4.3.1 Eosinophils ≧ 220/μL | 1 | 765 | Rate Ratio (IV, Fixed, 95% CI) | 0.68 [0.49, 0.94] |
| 4.4 Serious adverse events | 1 | 1129 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.90 [0.69, 1.17] |
| 4.5 SGRQ total score | 1 | 680 | Mean Difference (IV, Fixed, 95% CI) | ‐0.87 [‐3.23, 1.49] |
| 4.5.1 Eosinophils ≧ 220/μL | 1 | 680 | Mean Difference (IV, Fixed, 95% CI) | ‐0.87 [‐3.23, 1.49] |
| 4.6 FEV₁ (L) | 1 | 669 | Mean Difference (IV, Fixed, 95% CI) | 0.01 [‐0.04, 0.05] |
| 4.6.1 Eosinophils ≧ 220/μL | 1 | 669 | Mean Difference (IV, Fixed, 95% CI) | 0.01 [‐0.04, 0.05] |
| 4.7 Rescue medication use | 1 | 619 | Mean Difference (IV, Fixed, 95% CI) | ‐0.59 [‐1.11, ‐0.07] |
| 4.7.1 Eosinophils ≧ 220/μL | 1 | 619 | Mean Difference (IV, Fixed, 95% CI) | ‐0.59 [‐1.11, ‐0.07] |
| 4.8 Proportion of nights with awakenings | 1 | 638 | Mean Difference (IV, Fixed, 95% CI) | ‐0.04 [‐0.09, 0.01] |
| 4.8.1 Eosinophils ≧ 220/μL | 1 | 638 | Mean Difference (IV, Fixed, 95% CI) | ‐0.04 [‐0.09, 0.01] |
| 4.9 COPD Assessment Test (CAT) | 1 | 682 | Mean Difference (IV, Fixed, 95% CI) | 0.18 [‐0.82, 1.18] |
| 4.9.1 Eosinophils ≧ 220/μL | 1 | 682 | Mean Difference (IV, Fixed, 95% CI) | 0.18 [‐0.82, 1.18] |
| 4.10 Mortality | 1 | 1129 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.90 [0.46, 1.76] |
| 4.11 Adverse events | 1 | 1129 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.96 [0.76, 1.21] |
Comparison 5. Benralizumab 30 mg versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 5.1 Rate of moderate or severe exacerbations | 2 | 2234 | Rate Ratio (IV, Fixed, 95% CI) | 1.03 [0.93, 1.13] |
| 5.1.1 Eosinophils ≧ 220/μL | 2 | 1523 | Rate Ratio (IV, Fixed, 95% CI) | 1.00 [0.89, 1.13] |
| 5.1.2 Eosinophils < 220/μL | 2 | 711 | Rate Ratio (IV, Fixed, 95% CI) | 1.07 [0.91, 1.27] |
| 5.2 Annual EXACT‐PRO exacerbation rate | 2 | 1522 | Rate Ratio (IV, Fixed, 95% CI) | 1.03 [0.90, 1.17] |
| 5.2.1 Eosinophils ≧ 220/μL | 2 | 1522 | Rate Ratio (IV, Fixed, 95% CI) | 1.03 [0.90, 1.17] |
| 5.3 Rate of severe exacerbations | 2 | 1523 | Rate Ratio (IV, Fixed, 95% CI) | 1.01 [0.77, 1.33] |
| 5.3.1 Eosinophils ≧ 220/μL | 2 | 1523 | Rate Ratio (IV, Fixed, 95% CI) | 1.01 [0.77, 1.33] |
| 5.4 Rate of severe exacerbations requiring hospitalisation | 2 | 1523 | Rate Ratio (IV, Fixed, 95% CI) | 0.96 [0.75, 1.22] |
| 5.4.1 Eosinophils ≧ 220/μL | 2 | 1523 | Rate Ratio (IV, Fixed, 95% CI) | 0.96 [0.75, 1.22] |
| 5.5 Serious adverse events | 2 | 2235 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.98 [0.81, 1.17] |
| 5.6 SGRQ total score | 2 | 1333 | Mean Difference (IV, Fixed, 95% CI) | ‐1.42 [‐3.13, 0.29] |
| 5.6.1 Eosinophils ≧ 220/μL | 2 | 1333 | Mean Difference (IV, Fixed, 95% CI) | ‐1.42 [‐3.13, 0.29] |
| 5.7 FEV₁ (L) | 2 | 1312 | Mean Difference (IV, Fixed, 95% CI) | ‐0.00 [‐0.03, 0.03] |
| 5.7.1 Eosinophils ≧ 220/μL | 2 | 1312 | Mean Difference (IV, Fixed, 95% CI) | ‐0.00 [‐0.03, 0.03] |
| 5.8 Rescue medication use | 2 | 1216 | Mean Difference (IV, Fixed, 95% CI) | ‐0.40 [‐0.77, ‐0.03] |
| 5.8.1 Eosinophils ≧ 220/μL | 2 | 1216 | Mean Difference (IV, Fixed, 95% CI) | ‐0.40 [‐0.77, ‐0.03] |
| 5.9 Proportion of nights with awakenings | 2 | 1242 | Mean Difference (IV, Fixed, 95% CI) | ‐0.06 [‐0.09, ‐0.02] |
| 5.9.1 Eosinophils ≧ 220/μL | 2 | 1242 | Mean Difference (IV, Fixed, 95% CI) | ‐0.06 [‐0.09, ‐0.02] |
| 5.10 COPD Assessment Test (CAT) | 2 | 1338 | Mean Difference (IV, Fixed, 95% CI) | ‐0.18 [‐0.90, 0.55] |
| 5.10.1 Eosinophils ≧ 220/μL | 2 | 1338 | Mean Difference (IV, Fixed, 95% CI) | ‐0.18 [‐0.90, 0.55] |
| 5.11 Mortality | 2 | 2235 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.13 [0.70, 1.84] |
| 5.12 Adverse events | 2 | 2235 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.80, 1.12] |
Comparison 6. Benralizumab 100 mg versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 6.1 Rate of moderate or severe exacerbations | 3 | 2314 | Rate Ratio (IV, Fixed, 95% CI) | 0.94 [0.85, 1.03] |
| 6.1.1 All | 1 | 82 | Rate Ratio (IV, Fixed, 95% CI) | 1.03 [0.67, 1.58] |
| 6.1.2 Eosinophils ≧ 220/μL | 2 | 1512 | Rate Ratio (IV, Fixed, 95% CI) | 0.88 [0.78, 1.00] |
| 6.1.3 Eosinophils < 220/μL | 2 | 720 | Rate Ratio (IV, Fixed, 95% CI) | 1.02 [0.87, 1.20] |
| 6.2 Annual EXACT‐PRO exacerbation rate | 2 | 1509 | Rate Ratio (IV, Fixed, 95% CI) | 0.95 [0.82, 1.09] |
| 6.2.1 Eosinophils ≧ 220/μL | 2 | 1509 | Rate Ratio (IV, Fixed, 95% CI) | 0.95 [0.82, 1.09] |
| 6.3 Hospitalisations due to COPD exacerbation | 1 | 82 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.39 [0.07, 2.13] |
| 6.4 Rate of exacerbations requiring hospitalisation | 2 | 1512 | Rate Ratio (IV, Fixed, 95% CI) | 0.63 [0.49, 0.81] |
| 6.4.1 Eosinophils ≧ 220/μL | 2 | 1512 | Rate Ratio (IV, Fixed, 95% CI) | 0.63 [0.49, 0.81] |
| 6.5 Serious adverse events | 3 | 2333 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.90 [0.75, 1.08] |
| 6.6 Quality of life: change in SGRQ total score | 3 | 1433 | Mean Difference (IV, Fixed, 95% CI) | ‐1.45 [‐2.84, ‐0.07] |
| 6.6.1 All participants | 1 | 82 | Mean Difference (IV, Fixed, 95% CI) | ‐1.08 [‐7.34, 5.18] |
| 6.6.2 Eosinophils ≧ 220/μL | 2 | 1351 | Mean Difference (IV, Fixed, 95% CI) | ‐1.47 [‐2.89, ‐0.05] |
| 6.7 FEV₁ (L) | 3 | 1425 | Mean Difference (IV, Fixed, 95% CI) | 0.03 [‐0.00, 0.06] |
| 6.7.1 Eosinophils ≧ 220/μL | 2 | 1334 | Mean Difference (IV, Fixed, 95% CI) | 0.02 [‐0.01, 0.05] |
| 6.7.2 All participants | 1 | 91 | Mean Difference (IV, Fixed, 95% CI) | 0.19 [0.05, 0.33] |
| 6.8 Rescue medication use | 2 | 1237 | Mean Difference (IV, Fixed, 95% CI) | ‐0.49 [‐0.83, ‐0.15] |
| 6.8.1 Eosinophils ≧ 220/μL | 2 | 1237 | Mean Difference (IV, Fixed, 95% CI) | ‐0.49 [‐0.83, ‐0.15] |
| 6.9 Proportion of nights with awakenings | 2 | 1263 | Mean Difference (IV, Fixed, 95% CI) | ‐0.03 [‐0.07, 0.00] |
| 6.9.1 Eosinophils ≧ 220/μL | 2 | 1263 | Mean Difference (IV, Fixed, 95% CI) | ‐0.03 [‐0.07, 0.00] |
| 6.10 COPD Assessment Test (CAT) | 2 | 1358 | Mean Difference (IV, Fixed, 95% CI) | ‐0.60 [‐1.29, 0.10] |
| 6.10.1 Eosinophils ≧ 220/μL | 2 | 1358 | Mean Difference (IV, Fixed, 95% CI) | ‐0.60 [‐1.29, 0.10] |
| 6.11 Mortality | 3 | 2333 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.57, 1.55] |
| 6.12 Adverse events | 3 | 2333 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.07 [0.91, 1.26] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Brightling 2014.
| Study characteristics | ||
| Methods | Multicentre, phase 2a, randomised, double‐blind, placebo‐controlled study Study locations: Canada, Denmark, Germany, Poland, Spain, the United Kingdom, the United States |
|
| Participants | 421 participants were screened, and 101 participants with a diagnosis of COPD were randomised into the following 2 study arms. Benralizumab 100 mg: 51 participants, mean age 62.9 years (SD 8.2); females 16 (31.4%) Placebo: 50 participants, mean age 64.6 years (SD 7.5); females 21 (42%) Inclusion criteria:
Exclusion criteria:
|
|
| Interventions | Benralizumab (MEDI‐563) 100 mg versus placebo matched to benralizumab (MEDI‐563). In both intervention and control arms of the study: injection subcutaneously every 4 weeks for the first 3 doses and then every 8 weeks for the next 5 doses (Day 1, 29, 57, 113, 169, 225, 281, and 337). |
|
| Outcomes | Primary outcome measures:
Secondary outcome measures:
|
|
| Notes | Principal Investigator: Rene van der Merwe, MBChB. MedImmune LLC Sponsor: MedImmune LLC. Collaborator: AstraZeneca |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomly assigned (1:1), via computer‐generated permuted block randomisation (block size of 4) with a central telephone and web‐based system, to receive 100 mg benralizumab or matched placebo, subcutaneously. |
| Allocation concealment (selection bias) | Low risk | All other study site personnel, participants, and sponsors, including data analysts, were masked to treatment allocation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants and personnel were blinded (clearly stated in clinicaltrials.gov/ct2/show/nct01227278). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Investigators were blinded (clearly stated in clinicaltrials.gov/ct2/show/nct01227278). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Incomplete outcome data were comprehensively reported. |
| Selective reporting (reporting bias) | Low risk | No apparent indication of selective outcome reporting. |
| Other bias | Low risk | No other apparent sources of bias. |
Dasgupta 2016.
| Study characteristics | ||
| Methods | Phase 3, randomised, double‐blind, placebo‐controlled study Study location: Firestone Institute of Respiratory Health, St Joseph's Hospital, Hamilton, Ontario, Canada, L8N 4A6 |
|
| Participants | 19 participants aged 40 to 80 years with a diagnosis of COPD with eosinophilic bronchitis were randomised into 2 study arms. 1 participant (from placebo group) left the study just after randomisation because of severe exacerbation requiring hospitalisation, therefore the study was conducted in 18 participants. Mepolizumab 750 mg: 8 participants, mean age 65.1 years (SD 6.3); females 4 (50%). Placebo: 10 participants, mean age 66.9 years (SD 5.9); females 1 (10%). Inclusion criteria:
Exclusion criteria:
|
|
| Interventions | Mepolizumab 750 mg versus placebo. Mepolizumab (an anti‐IL‐5, given once a month intravenously at a dose of 750 mg). The placebo consisted of 100 mL normal saline solution (0.9%, 154 mmol/L sodium chloride). |
|
| Outcomes | Primary outcome measures:
Secondary outcome measures:
|
|
| Notes | Principal Investigator: Parameswaran Nair, MD, PhD, FRCP. Associate Professor of Medicine, Division of Respiratory, McMaster University Sponsor: McMaster University |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information in the trial report to permit a judgement. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information in the trial report to permit a judgement. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This is clearly stated in clinicaltrials.gov/show/NCT01463644. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This is clearly stated in clinicaltrials.gov/show/NCT01463644. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Insufficient information in the trial report to permit a judgement. |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information in the trial report to permit a judgement. |
| Other bias | High risk | The inclusion criteria for this study required < 12% FEV₁ reversibility to a bronchodilator. It appears that some participants were entered despite not meeting this criterion, therefore it is likely that some people in this study had current asthma. |
NCT02105948 (METREX).
| Study characteristics | ||
| Methods | Multicentre, phase 3, randomised, placebo‐controlled, double‐blind, parallel‐group study. Study locations: Australia, Belgium, Canada, the Czech Republic, Estonia, France, Greece, Italy, Mexico, Norway, Peru, Poland, the Russian Federation, Spain, Sweden, the United States Study duration: 52 weeks A 4‐arm study: mepolizumab 100 mg ‐ high stratum versus placebo ‐ high stratum; and mepolizumab 100 mg ‐ low stratum versus placebo ‐ low stratum. |
|
| Participants | 837 participants aged at least 40 years with frequent exacerbations of COPD. Unselected participants in the mITT population with an eosinophilic phenotype were stratified according to blood eosinophil count (≥ 150 per cubic millimetre at screening or ≥ 300 per cubic millimetre during the previous year). Mepolizumab 100 mg ‐ high stratum: 233 participants, mean age 65.2 years (SD 8.36); females 84 (36.1%) Placebo ‐ high stratum: 229 participants, mean age 65.3 years (SD 8.53); females 79 (34.5%) Mepolizumab 100 mg ‐ low stratum: 184 participants, mean age 66.1 years (SD 9.14); females 76 (41.3%) Placebo ‐ low stratum: 190 participants, mean age 65.2 years (SD 8.62); females 77 (40.5%) Inclusion criteria:
Exclusion criteria:
|
|
| Interventions | Mepolizumab 100 mg versus placebo. Each participant received 100 mg mepolizumab SC injection or placebo every 4 weeks (13 administrations during 52‐week treatment period) along with optimised standard of care background therapy. Placebo: sterile 0.9% sodium chloride solution Salbutamol MDI was issued for use as rescue medication throughout the study. |
|
| Outcomes | Primary outcome measures:
Secondary outcome measures
|
|
| Notes | Principal Investigator: GSK Clinical Trials Sponsor: GlaxoSmithKline |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was performed using a centralised, computer‐generated, permuted‐block design with fixed block size of 6; separate schedules were generated for each country. |
| Allocation concealment (selection bias) | Unclear risk | It is highly likely that the allocation concealment was adequate, but no details provided in the trial report. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participant, investigator, outcomes assessor masked (confirmed in clinicaltrials.gov/ct2/show/results/NCT02105948). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Participant, investigator, outcomes assessor masked (confirmed in clinicaltrials.gov/ct2/show/results/NCT02105948). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing data sensitivity analyses conducted indicating robustness of primary efficacy results. |
| Selective reporting (reporting bias) | Low risk | No apparent indication of selective outcome reporting |
| Other bias | Low risk | No apparent indication of other sources of bias. |
NCT02105961 (METREO).
| Study characteristics | ||
| Methods | Multicentre, phase 3, randomised, placebo‐controlled, double‐blind, parallel‐group study Study locations: Argentina, Australia, Canada, Chile, Denmark, Germany, Japan, Republic of Korea, the Netherlands, Romania, Slovakia, Taiwan, Ukraine, the United Kingdom, the United States Study duration: 52 weeks |
|
| Participants | 674 participants aged at least 40 years with COPD. All participants had a blood eosinophil count of at least 150 per cubic millimetre at screening or at least 300 per cubic millimetre during the previous year. Mepolizumab 300 mg: 225 participants, mean age 64.8 years (SD 8.96); females 67 (29.8%) Mepolizumab 100 mg: 223 participants, mean age 64.8 (SD 9.06); females 91 (40.8%) Placebo: 226 participants, mean age 65.8 years (SD 8.64); females 70 (31.0%) Inclusion criteria:
Exclusion criteria:
|
|
| Interventions | 3 arm trial: mepolizumab 100 mg versus mepolizumab 300 mg versus placebo. Each participant received 100 mg or 300 mg mepolizumab SC injection or placebo every 4 weeks (13 administrations during 52‐week treatment period) along with their baseline standard of care COPD medication. Placebo: sterile 0.9% sodium chloride solution Salbutamol MDI was issued for use as rescue medication throughout the study. |
|
| Outcomes | Primary outcome measures:
Secondary outcome measures:
|
|
| Notes | Principal Investigator: GSK Clinical Trials Sponsor: GlaxoSmithKline |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was performed using a centralised, computer‐generated, permuted‐block design with fixed block size of 6; separate schedules were generated for each country. |
| Allocation concealment (selection bias) | Unclear risk | It is highly likely that the allocation concealment was adequate, but no details provided in the trial report. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participant, investigator, and outcomes assessor were masked (confirmed in clinicaltrials.gov/ct2/show/results/NCT02105961). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Participant, investigator, and outcomes assessor were masked (confirmed in clinicaltrials.gov/ct2/show/results/NCT02105961). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing data sensitivity analyses conducted indicating robustness of primary efficacy results. |
| Selective reporting (reporting bias) | Low risk | No apparent indication of selective outcome reporting. |
| Other bias | Low risk | No apparent indication of other sources of bias. |
NCT02138916 (GALATHEA).
| Study characteristics | ||
| Methods | Multicentre, phase 3, randomised, placebo‐controlled, double‐blind, parallel‐group study A 3‐arm study: benralizumab 30 mg versus benralizumab 100 mg versus placebo. Study locations: Austria, Canada, the Czech Republic, Germany, Hungary, Italy, Japan, Republic of Korea, the Netherlands, Poland, Romania, the Russian Federation, South Africa, Spain, Switzerland, the United Kingdom, the United States Duration of study: 56 weeks |
|
| Participants | 1656 participants aged at least 40 to 85 years with moderate to very severe COPD. Benralizumab 30 mg: 554 participants, mean age 65.9 years (SD 7.77); females 172 (31.0%) Benralizumab 100 mg: 552 participants, mean age 65.3 years (SD 8.05); females 180 (32.6%) Placebo: 550 participants, mean age 65.2 years (SD 8.22); females 175 (31.8%) Inclusion criteria:
Exclusion criteria:
|
|
| Interventions | 3‐arm trial: benralizumab 30 mg versus benralizumab 100 mg versus placebo Each participant received 30 mg or 100 mg benralizumab or placebo subcutaneously on study week 0 until study week 48 inclusive. Participants were randomised to receive benralizumab 30 mg or 100 mg or placebo by SC injection every 8 weeks throughout the 56‐week study. |
|
| Outcomes | Primary outcome measures:
Secondary outcome measures:
|
|
| Notes | Principal Investigator: Gerard Criner, MD. Temple University School of Medicine, 3401 North Broad Street, Suite 745 PP, Philadelphia, PA 19140 Sponsor: AstraZeneca. Collaborator: MedImmune LLC |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Detailed account of stratification of eligible participants. |
| Allocation concealment (selection bias) | Unclear risk | Unable to find confirmation on this point in the trial reports. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Blinding of participants and personnel is explicit in clinicaltrials.gov/ct2/show/NCT02138916. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | As detailed in clinicaltrials.gov/ct2/show/NCT02138916. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | As detailed in clinicaltrials.gov/ct2/show/NCT02138916. |
| Selective reporting (reporting bias) | Low risk | No indication of selective outcome reporting. |
| Other bias | Low risk | No indication of other sources of bias. |
NCT02155660 (TERRANOVA).
| Study characteristics | ||
| Methods | Multicentre, phase 3, randomised, placebo‐controlled, double‐blind, parallel‐group study A 4‐arm study: benralizumab 10 mg versus benralizumab 30 mg versus benralizumab 100 mg versus placebo. Study locations: Argentina, Australia, Belgium, Brazil, Bulgaria, Chile, Colombia, Croatia, Denmark, France, Israel, Mexico, New Zealand, Norway, Peru, the Philippines, Poland, Serbia, Slovenia, Sweden, Taiwan, Thailand, Turkey, Ukraine, the United States, Vietnam Study duration: 56 weeks |
|
| Participants | 2255 participants aged at least 40 to 85 years with moderate to very severe COPD Benralizumab 10 mg: 562 participants, mean age 64.7 years (SD 8.47); females 196 (34.9%) Benralizumab 30 mg: 562 participants, mean age 65.6 years (SD 8.61); females 194 (34.5%) Benralizumab 100 mg: 562 participants, mean age 65.0 years (SD 8.23); females 207 (36.8%) Placebo: 568 participants, mean age 65.3 years (SD 8.44); females 209 (36.8%) Inclusion criteria:
Exclusion criteria:
|
|
| Interventions | 4‐arm trial: benralizumab 10 mg versus benralizumab 30 mg versus benralizumab 100 mg versus placebo Each participant received 10 mg, 30 mg, or 100 mg benralizumab or placebo subcutaneously on study week 0 until study week 48 inclusive. Participants were randomised to receive benralizumab 10 mg, benralizumab 30 mg, benralizumab 100 mg, or placebo by SC injection every 8 weeks throughout the 56‐week study. |
|
| Outcomes | Primary outcome measures:
Secondary outcome measures:
|
|
| Notes | Principal Investigator: Bartolome R Celli, MD. Brigham and Women's Hospital, Pulmonary Division, 75 Francis Street, PBB Clinics 3, Boston, MA 02115 Sponsor: AstraZeneca. Collaborator: MedImmune LLC |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Detailed account of stratification of eligible participants. |
| Allocation concealment (selection bias) | Unclear risk | Unable to find confirmation on this point in the trial reports. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | As indicated in clinical trial information (clinicaltrials.gov/ct2/show/NCT02155660). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | As indicated in clinical trial information (clinicaltrials.gov/ct2/show/NCT02155660). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | As indicated in clinical trial information (clinicaltrials.gov/ct2/show/NCT02155660). |
| Selective reporting (reporting bias) | Low risk | All endpoints reported. |
| Other bias | Low risk | No apparent indication of other sources of bias. |
AECOPD: acute exacerbation of chronic obstructive pulmonary disease; anti‐IL‐5: anti‐interleukin 5; ATS: American Thoracic Society; BODE: body mass index obstruction dyspnoea exercise capacity; CAT: COPD Assessment Test; COPD: chronic obstructive pulmonary disease; CRQ‐SAS: Chronic Respiratory Questionnaire Self‐Administered Standardized Format; ECG: electrocardiogram; ECOPD: exacerbation of chronic obstructive pulmonary disease; ED: emergency department; EGPA: eosinophilic granulomatosis with polyangiitis; EOS: elevated blood eosinophils; E‐RS:COPD: Evaluating Respiratory Symptoms in Chronic Obstructive Pulmonary Disease; ERS: European Respiratory Society; EXACT‐PRO: EXAcerbations of Chronic pulmonary disease Tool; FEV₁: forced expiratory volume in 1 second; FVC: forced vital capacity; GINA: Global Initiative for Asthma; GOLD: Global Initiative for Chronic Obstructive Lung Disease; ICS; inhaled corticosteroid; IM: intramuscular; IP: investigational product; IV: intravenous; LABA: long‐acting beta₂‐agonist; LAMA: long‐acting muscarinic antagonist; MDI: metered dose inhaler; mMRC: modified Medical Research Council Scale; mITT: modified intention‐to‐treat; NHANES: National Health and Nutrition Examination Survey; NYHA: New York Heart Association; PEFR: peak expiratory flow rate; RCT: randomised controlled trial; SABA: short‐acting beta₂‐agonist; SAMA: short‐acting muscarinic antagonist; SC: subcutaneous; SD: standard deviation; SGRQ: St George's Respiratory Questionnaire; SGRQ‐C: St George's Respiratory Questionnaire for COPD patients; TB: active tuberculosis; TEAE: treatment‐emergent adverse events; TESAE: treatment‐emergent serious adverse events
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Condreay 2019 | Investigation of genetic associations with frequency of moderate or severe COPD exacerbations (or both) in COPD participants receiving mepolizumab. Not a randomised trial |
| Sridhar 2019 | Aggregation of 2 studies investigating modulation of blood inflammatory markers |
COPD: chronic obstructive pulmonary disease
Characteristics of ongoing studies [ordered by study ID]
NCT04053634.
| Study name | Efficacy and safety of benralizumab in moderate to very severe chronic obstructive pulmonary disease (COPD) with a history of frequent exacerbations (RESOLUTE) |
| Methods | Multicentre, randomised, double‐blind, chronic‐dosing, parallel‐group, placebo‐controlled phase 3 study Study locations: Argentina, Austria, Brazil, Canada, Chile, Colombia, the Czech Republic, Denmark, Germany, Hungary, Italy, Japan, Republic of Korea, the Philippines, Poland, Spain, Sweden, Turkey, the United Kingdom, the United States |
| Participants | Patients with a history of ≥ 2 moderate and/or severe COPD exacerbations in the previous year. Estimated enrolment: 868 participants Inclusion criteria:
Exclusion criteria:
|
| Interventions | Benralizumab versus placebo Benralizumab solution for injection in accessorised prefilled syringe or matching placebo will be administered SC every 4 weeks for the first 3 doses ‐ Weeks 0, 4, and 8, and then every 8 weeks until the end of treatment. |
| Outcomes | Primary outcome measure:
Secondary outcome measures:
Additional predefined outcome measures:
|
| Starting date | August 2019 |
| Contact information | AstraZeneca Clinical Study Information Center |
| Notes | Sponsor: AstraZeneca |
NCT04075331.
| Study name | Mepolizumab for COPD Hospital Eosinophilic Admissions Pragmatic Trial (COPD‐HELP) |
| Methods | Single‐centre, double‐blinded, randomised, placebo‐controlled trial Study location: Leicestershire, UK |
| Participants | Patients admitted to hospital for a severe exacerbation of eosinophilic COPD. Estimated enrolment: 238 participants Inclusion criteria:
Exclusion criteria:
|
| Interventions | Mepolizumab 100 mg versus placebo Both mepolizumab and placebo are delivered SC. The placebo is saline solution for subcutaneous injection. |
| Outcomes | Primary outcome measure:
Secondary outcome measures:
|
| Starting date | February 2020 |
| Contact information | Neil Greening +44 (0)116 258 3474; neil.greening@leicester.ac.uk |
| Notes | Sponsor: University of Leicester. Collaborators: Leicester Clinical Trials Unit and GlaxoSmithKline |
NCT04098718.
| Study name | Acute Exacerbations Treated With BenRAlizumab (The ABRA Study) (ABRA) |
| Methods | Randomised, double‐blinded, randomised, placebo‐controlled, phase 2 study Study location: UK |
| Participants | Patients with a diagnosis of COPD or asthma (or both COPD and asthma) Estimated enrolment: 158 participants Inclusion criteria:
Exclusion criteria:
Additional exclusion criteria on day of exacerbation (Visit 2):
|
| Interventions | 4‐arm study: standard care prednisolone 30 mg given daily for 5 days to treat an exacerbation versus benralizumab as a single 100 mg SC injection and oral placebo tablet daily for 5 days versus benralizumab as a single 100 mg SC injection and oral prednisolone 30 mg daily for 5 days versus placebo SC injection and oral prednisolone 30 mg daily for 5 days |
| Outcomes | Primary outcome measures:
Secondary outcome measures:
Additional predefined outcome measures:
|
| Starting date | October 2019 |
| Contact information | Mona Bafadhel, PhD, MBChB. Nuffield Department of Medicine, University of Oxford, UK |
| Notes | Sponsor: University of Oxford |
NCT04133909.
| Study name | Mepolizumab as Add‐on Treatment IN Participants With COPD Characterized by Frequent Exacerbations and Eosinophil Level (MATINEE) |
| Methods | Multicentre, randomised, double‐blind, parallel‐group, placebo‐controlled phase 3 study Study locations: Argentina, Australia, Belgium, Canada, Denmark, France, Germany, Hungary, Israel, Republic of Korea, Mexico, the Netherlands, New Zealand, Poland, Spain, the United Kingdom, the United States |
| Participants | Inclusion criteria:
Exclusion criteria:
|
| Interventions | This study is designed to confirm the benefits of mepolizumab treatment on moderate or severe exacerbations in COPD participants given as an add on to their optimised maintenance COPD therapy. The maximum duration of participation is approximately 57 weeks, consisting of 2 screening visits, run‐in period, and a 52‐week intervention period. 800 to 1000 participants will be randomised in 1:1 ratio to receive mepolizumab 100 mg or placebo every 4 weeks for a total of 13 doses. |
| Outcomes | Primary outcome measure:
Secondary outcome measures:
|
| Starting date | October 2019 |
| Contact information | GSKClinicalSupportHD@gsk.com |
| Notes | Sponsor: GlaxoSmithKline |
ACQ‐6: Asthma Control Questionnaire; ACT: Asthma Control Test; ADA: anti‐drug antibodies; AE: acute exacerbation;AECOPD: acute exacerbation of chronic obstructive pulmonary disease; ALT: alanine aminotransferase; APFS: accessorised prefilled syringe; AST: aspartate transaminase; AQLQ: Asthma Quality of Life Questionnaire; ATS: American Thoracic Society; βHCG: beta human chorionic gonadotropin; BiPAP: bi‐level positive airway pressure; BODE: body mass index obstruction dyspnoea exercise capacity; bpm: beats per minute; CAT: COPD Assessment Test; COPD: chronic obstructive pulmonary disease; CPAP: continuous positive airway pressure; CRQ‐SAS: Chronic Respiratory Questionnaire Self‐Administered Standardized Format; ECG: electrocardiogram; ED: emergency department; EGPA: eosinophilic granulomatosis with polyangiitis; eMCR: extended Medical Research Council; EOS: elevated blood eosinophils; E‐RS:COPD: Evaluating Respiratory Symptoms in Chronic Obstructive Pulmonary Disease; ERS: European Respiratory Society; EXACT‐PRO: EXAcerbations of Chronic pulmonary disease Tool; FENO: fractional exhaled nitric oxide; FEV₁: forced expiratory volume in 1 second; FVC: forced vital capacity; GINA: Global Initiative for Asthma; HBsAg: hepatitis B surface antigen; ICS; inhaled corticosteroid; ICU: intensive care unit; IM: intramuscular; IMP: progestogen‐only implant; IP: investigational product; IV: intravenous; LABA: long‐acting beta₂‐agonist; LAMA: long‐acting muscarinic antagonist; LCADL: London Chest Activities of Daily Living Questionnaire; mMRC: modified Medical Research Council Scale; mITT: modified intention‐to‐treat; NHANES: National Health and Nutrition Examination Survey; NIPPV: non‐invasive positive pressure ventilation device; NYHA: New York Heart Association; Pc20: The provocative concentration of methacholine that results in a 20% drop in FEV1; PEFR: peak expiratory flow rate; PDE4: Phosphodiesterase‐4; PPB: parts‐per‐billion; PPD: purified protein derivative; RCT: randomised controlled trial; SABA: short‐acting beta₂‐agonist; SAE: serious acute exacerbation; SAMA: short‐acting muscarinic antagonist; SC: subcutaneous; SD: standard deviation; SGRQ: St George's Respiratory Questionnaire; SGRQ‐C: St George's Respiratory Questionnaire for COPD patients; SPPB: short physical performance battery; TB: active tuberculosis; TEAE: treatment‐emergent adverse events; TESAE: treatment‐emergent serious adverse events; WEMWBS: Warwick‐Edinburgh Mental Wellbeing Scale; WOCBP: women of childbearing potential
Differences between protocol and review
We used Cochrane’s Screen4Me workflow to help assess the search results, which was not in the published protocol.
Contributions of authors
The review authors contributed to the following sections.
TD: Methods, Data collection, and Analysis sections SM: Background, Methods, Data collection, Analysis, and Methods sections RW: Methods, Data collection, and Analysis sections EB: Analysis and Methods sections PB: Background and Methods sections IC: Methods section
Contributions of the editorial team
Chris Cates (Co‐ordinating Editor): checked the methods and the data; edited the review; advised on methodology; approved the review prior to publication. Han Ni (Contact Editor): edited the review and advised on content. Emma Dennett (Managing Editor): co‐ordinated the editorial process; advised on content; edited the review. Emma Jackson (Assistant Managing Editor): conducted peer review; edited the references and other sections of the review. Elizabeth Stovold (Information Specialist): designed the search strategy and conducted the searches.
Sources of support
Internal sources
-
Tim Donovan, UK
University of Cumbria provided Tim Donovan's salary
-
Steve Milan, UK
Lancaster University provided Steve Milan's salary
External sources
The authors declare that no such funding was received for this systematic review, UK
Declarations of interest
TD: none known. SM: none known. RW: I work in a clinically relevant speciality (respiratory medicine). EB: none known. PB: I work in a clinically relevant speciality (respiratory medicine). IC: I work in a clinically relevant speciality (respiratory medicine). I have been involved as a local investigator for a GSK‐sponsored drug trial of inhaled nemiralisib for COPD, but did not directly receive funding for this.
New
References
References to studies included in this review
Brightling 2014 {published data only}
- Brightling C, Bafadhel M, Bleecker E, Panettieri R, She D, Ward CK, et al. A phase 2a study of benralizumab in adults with COPD [Abstract]. European Respiratory Journal 2014;44(Suppl 58):P2818. [Google Scholar]
- Brightling C, Bafadhel M, Bleecker E, Panettieri R, She D, Ward CK, et al. Late-breaking abstract: a phase 2a study of benralizumab in adults with COPD. European Respiratory Journal 2014;44:P2818. [Google Scholar]
- Brightling CE, Van Der Merwe R, She D, Birrell C. A phase 2a study of benralizumab, an anti-interleukin-5 receptor α monoclonal antibody, in adults with chronic obstructive pulmonary disease and sputum eosinophilia [Abstract]. American Journal of Respiratory and Critical Care Medicine 2014;189:A3771. [Google Scholar]
- Brightling CE, Bleecker ER, Panettieri RA Jr, Bafadhel M, She D, Ward CK, et al. Benralizumab for chronic obstructive pulmonary disease and sputum eosinophilia: a randomised, double-blind, placebo-controlled, phase 2a study. Lancet Respiratory Medicine 2014;2(11):891-901. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damera G, Brightling C, Bleecker E, She D, Van Der Merwe R, Ward C. Pharmacodynamic effect of benralizumab on blood basophils and serum biomarkers in adults with COPD with sputum eosinophilia. European Respiratory Journal 2015;46:PA2118. [DOI: 10.1183/13993003.congress2015.PA2118] [DOI] [Google Scholar]
- Damera G, Brightling CE, Bleecker ER, She D, Van Der Merwe R, Ward CK. Pharmacodynamic effect of benralizumab on blood basophils and serum biomarkers in adults with chronic obstructive pulmonary disease and sputum eosinophilia. American Journal of Respiratory and Critical Care Medicine 2015;191:PA2118. [Google Scholar]
- EUCTR2010-020127-52-GB. A Phase 2a, double blind, placebo controlled study to evaluate the efficacy of MEDI-563 in subjects with moderate to severe COPD and sputum eosinophilia. who.int/trialsearch/Trial2.aspx?TrialID=EUCTR2010-020127-52-GB (first received 3 August 2010).
- NCT01227278. A study to evaluate the effectiveness of MEDI-563 in subjects with chronic obstructive pulmonary disease (COPD). clinicaltrials.gov/show/nct01227278 (first received 25 October 2010).
- Yan L, Roskos L, Ward CK, She D, Merwe R, Wang B. Pharmacokinetics and pharmacodynamics of benralizumab in subjects with moderate-to-severe chronic obstructive pulmonary disease. Clinical Pharmacology and Therapeutics 2015;97:S95. [Google Scholar]
Dasgupta 2016 {published and unpublished data}
- Dasgupta A, Kjarsgaard M, Capaldi D, Radford K, Aleman F, Boylan C, et al. A pilot randomised clinical trial of mepolizumab in COPD with eosinophilic bronchitis. European Respiratory Journal 2017;49(3):1602486. [DOI: 10.1183/13993003.02486-2016] [DOI] [PubMed] [Google Scholar]
- Dasgupta A, Kjarsgaard M, Capaldi D, Radford K, Aleman F, Parraga G, et al. In: Mepolizumab in COPD with eosinophilic bronchitis: a randomized clinical trial. European Respiratory Society 26th Annual Congress; 2016 Sep 3-7; London. Vol. 48. 2016:PA305. [DOI: 10.1183/13993003.congress-2016.PA305] [DOI] [PubMed]
- Nair PK, Dasgupta A, Kjarsgaard M, Capaldi D, Radford K, Aleman FP, et al. Mepolizumab in COPD with eosinophilic bronchitis: a randomized clinical trial. Journal of Allergy and Clinical Immunology 2016;137(2 Suppl 1):AB392. [Google Scholar]
- NCT01463644. Mepolizumab in chronic obstructive pulmonary diseases (COPD) with eosinophilic bronchitis. clinicaltrials.gov/show/NCT01463644 (first received 2 November 2011).
NCT02105948 (METREX) {published data only}
- EUCTR2013-004298-28-SE. Study evaluating safety and efficacy of mepolizumab in the treatment of COPD patients with frequent exacerbations. www.who.int/trialsearch/Trial2.aspx?TrialID=EUCTR2013-004298-28-SE (first received 15 April 2014).
- Jose A. Mepolizumab for eosinophilic chronic obstructive pulmonary disease. Journal of Clinical Outcomes Management 2018;25:1. [Google Scholar]
- NCT02105948. Study to evaluate efficacy and safety of mepolizumab for frequently exacerbating chronic obstructive pulmonary disease (COPD) patients. clinicaltrials.gov/show/nct02105948 (first received 7 April 2014).
- PER-013-14. Study 117106: Mepolizumab vs. placebo as add-on treatment for frequently exacerbating COPD patients. www.who.int/trialsearch/Trial2.aspx?TrialID=PER-013-14 (first received 12 June 2014).
- Sciurba F, Chanez P, Martinot J, Lugogo N, Yancey SW, Harris SS, et al. Mepolizumab reduces exacerbations in eosinophilic COPD. European Respiratory Journal 2017;50(Suppl 61):OA3194. [Google Scholar]
- Sciurba F, Chanez P, Martinot J-B, Lugogo N, Yancey SW, Harris SS, et al. Late breaking abstract - mepolizumab reduces exacerbations in eosinophilic COPD. European Respiratory Journal 2017;50(Suppl 61):OA3194. [DOI: 10.1183/1393003.congress-2017.OA3194] [DOI] [Google Scholar]
NCT02105961 (METREO) {published data only}
- DRKS00007259. Study MEA117113: mepolizumab vs. placebo as add-on treatment for frequently exacerbating COPD patients characterized by eosinophil level. www.drks.de/DRKS00007259 (first received 4 March 2016).
- EUCTR2013-004297-98-NL. English study evaluating safety and efficacy of mepolizumab in the treatment of COPD patients with frequent exacerbations. https://www.clinicaltrialsregister.eu/ctr-search/trial/2013-004297-98/results (first received 1 September 2014).
- NCT02105961. Efficacy and safety of mepolizumab as an add-on treatment in chronic obstructive pulmonary disease (COPD). clinicaltrials.gov/show/nct02105961 (first received 7 April 2014).
- Pavord ID, Chanez P, Criner GJ, Kerstjens HAM, Korn S, Lugogo N, et al. Mepolizumab for eosinophilic chronic obstructive pulmonary disease. New England Journal of Medicine 2017;377(17):1613-29. [DOI: 10.1056/NEJMoa1708208] [DOI] [PubMed] [Google Scholar]
- Pavord ID, Kerstjens HAM, Korn S, Criner GJ, Sagara H, Rubin DB, et al. Dose-ranging study of mepolizumab in eosinophilic COPD. European Respiratory Journal 2017;50:PA1366. [Google Scholar]
- Pavord ID, Kerstjens HAM, Korn S, Criner GJ, Sagara H, Rubin DB, et al. Late breaking abstract - dose-ranging study of mepolizumab in eosinophilic COPD. European Respiratory Journal 2017;50(Suppl 61):PA1366. [DOI: 10.1183/1393003.congress-2017.PA1366] [DOI] [Google Scholar]
NCT02138916 (GALATHEA) {published data only}
- Criner GJ, Celli BR, Brightling CE, Agusti A, Papi A, Singh D, et al. Benralizumab for the prevention of COPD exacerbations. New England Journal of Medicine 2019;381(11):1023-34. [DOI: 10.1056/NEJMoa1905248] [DOI] [PubMed] [Google Scholar]
- EUCTR2013-004590-27-AT. Efficacy and safety of 2 benralizumab doses in moderate to very severe COPD patients with prior exacerbation history. www.who.int/trialsearch/Trial2.aspx?TrialID=EUCTR2013-004590-27-AT (first received 27 June 2014).
- NCT02138916. Benralizumab efficacy in moderate to very severe chronic obstructive pulmonary disease (COPD) with exacerbation history. clinicaltrials.gov/show/NCT02138916 (first received 15 May 2014).
NCT02155660 (TERRANOVA) {published data only}
- NCT02155660. Efficacy and safety of benralizumab in moderate to very severe chronic obstructive pulmonary disease (COPD) with exacerbation history. clinicaltrials.gov/show/NCT02155660 (first received 4 June 2014).
- PER-040-14. A randomised, double-blind, double dummy, chronic dosing (56 week) placebo-controlled, parallel group, multicentre, phase iii study to evaluate the efficacy and safety of 3 doses of benralizumab (medi-563) in patients with moderate to very severe chronic obstructive pulmonary disease (COPD) with a history of COPD exacerbations (Terranova). www.who.int/trialsearch/Trial2.aspx?TrialID=PER-040-14 (first received 13 November 2014).
References to studies excluded from this review
Condreay 2019 {published data only}
- Condreay LD, Gao C, Bradford E, Yancey SW, Ghosh S. No genetic associations with mepolizumab efficacy in COPD with peripheral blood eosinophilia. Respiratory Medicine 2019;155:26-8. [DOI: 10.1016/j.rmed.2019.07.004] [DOI] [PubMed] [Google Scholar]
Sridhar 2019 {published data only}
- Sridhar S, Liu H, Pham TH, Damera G, Newbold P. Modulation of blood inflammatory markers by benralizumab in patients with eosinophilic airway diseases. Respiratory Research 2019;20(1):14. [DOI: 10.1186/s12931-018-0968-8] [DOI] [PMC free article] [PubMed] [Google Scholar]
References to ongoing studies
NCT04053634 {published data only}
- NCT04053634. Efficacy and safety of benralizumab in moderate to very severe chronic obstructive pulmonary disease (COPD) with a history of frequent exacerbations. clinicaltrials.gov/show/nct04053634 (first received 12 August 2019).
NCT04075331 {published data only}
- NCT04075331. Mepolizumab for COPD hospital eosinophilic admissions pragmatic trial (COPD-HELP). clinicaltrials.gov/show/NCT04075331 (first received 30 August 2019).
NCT04098718 {published data only}
- NCT04098718. Acute exacerbations treated with benralizumab (The ABRA Study) (ABRA). clinicaltrials.gov/show/nct04098718 (first received 23 September 2019).
NCT04133909 {published data only}
- NCT04133909. Mepolizumab as Add-on Treatment IN Participants With COPD Characterized by Frequent Exacerbations and Eosinophil Level (MATINEE). clinicaltrials.gov/ct2/show/NCT04133909 (first received 21 October 2019).
Additional references
Adegbola 2018
- Adegbola SO, Sahnan K, Warusavitarne J, Hart A, Tozer P. Anti-TNF therapy in Crohn's disease. International Journal of Molecular Sciences 2018;19(8):2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bafadhel 2012
- Bafadhel M, McKenna S, Terry S, Mistry V, Reid C, Haldar P, et al. Acute exacerbations of chronic obstructive pulmonary disease: identification of biologic clusters and their biomarkers. American Journal of Respiratory and Critical Care Medicine 2012;184(6):662-71. [DOI] [PubMed] [Google Scholar]
Bafadhel 2014
- Bafadhel M, Davies L, Calverley PMA, Aaron SD, Brightling CE, Pavord ID. Blood eosinophil guided prednisolone therapy for exacerbations of COPD: a further analysis. European Respiratory Journal 2014;44(3):789-91. [DOI] [PubMed] [Google Scholar]
Bittner 2018
- Bittner B, Richter W, Schmidt J. Subcutaneous administration of biotherapeutics: an overview of current challenges and opportunities. BioDrugs 2018;32(5):425-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
BLF 2019
- British Lung Foundation. Chronic obstructive pulmonary disease (COPD) statistics. statistics.blf.org.uk/copd (accessed prior to 7 June 2019).
Cochrane Airways 2019
- Cochrane Airways Trials Register. airways.cochrane.org/trials-register (accessed 10 June 2019).
Couillard 2017
- Couillard S, Larivée P, Courteau J, Vanasse A. Eosinophils in COPD exacerbations are associated with increased readmissions. Chest 2017;151(2):366-73. [DOI] [PubMed] [Google Scholar]
Donovan 2019
- Donovan T, Crossingham I, Milan SJ, Wang R, Bradley P. Anti-IL5 therapies for chronic obstructive pulmonary disease. Cochrane Database of Systematic Reviews 2019, Issue 9. Art. No: CD013432. [DOI: 10.1002/14651858.CD013432] [DOI] [PMC free article] [PubMed] [Google Scholar]
Durham 2016
- Durham AL, Caramori G, Chung KF, Adcock IM. Targeted anti-inflammatory therapeutics in asthma and chronic obstructive lung disease. Translational Research 2016;167(1):192-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
Farne 2017
- Farne H, Wilson A, Powell C, Bax L, Milan S. Anti-IL5 therapies for asthma. Cochrane Database of Systematic Reviews 2017, Issue 9. Art. No: CD010834. [DOI: 10.1002/14651858.CD010834.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
GINA 2019
- Global Initiative for Asthma. 2019 GINA Report, Global Strategy for Asthma Management and Prevention. ginasthma.org/wp-content/uploads/2019/06/GINA-2019-main-report-June-2019-wms.pdf (accessed 5 July 2019).
GOLD 2019
- Singh D, Agusti A, Anzueto A, Barnes PJ, Bourbeau J, Celli BR, et al. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Lung Disease: the GOLD science committee report 2019. European Respiratory Journal 2019;53(5):1900164. [DOI: 10.1183/13993003.00164-2019] [DOI] [PubMed] [Google Scholar]
GOLD 2020
- 2020 Global Strategy for Prevention, Diagnosis and Management of COPD. goldcopd.org/wp-content/uploads/2019/12/GOLD-2020-FINAL-ver1.2-03Dec19_WMV.pdf.
GRADEpro GDT [Computer program]
- GRADEpro GDT. Version accessed prior to 7 June 2019. Hamilton (ON): McMaster University (developed by Evidence Prime), 2019. Available at gradepro.org.
Higgins 2011
- Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Hogg 2004
- Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. New England Journal of Medicine 2004;350(26):2645-53. [DOI] [PubMed] [Google Scholar]
Marshall 2018
- Marshall IJ, Noel-Storr AH, Kuiper J, Thomas J, Wallace BC. Machine learning for identifying randomized controlled trials: an evaluation and practitioner’s guide. Research Synthesis Methods 2018;9(4):602-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
McDonald 2017
- McDonald S, Noel-Storr AH, Thomas J. Harnessing the efficiencies of machine learning and Cochrane Crowd to identify randomised trials for individual Cochrane reviews. In: Global Evidence Summit; 2017 September 13-16; Cape Town, South Africa. 2017.
Milan 2020 [pers comm]
- Milan S. Prednisone surrogate in the trial. Email to: P Nair 15 September 2020.
Moher 2009
- Moher D, Liberati A, Tetzlaff J, Altman D. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLOS Medicine 2009;6(7):e1000097. [DOI: 10.1371/journal.pmed.1000097] [DOI] [PMC free article] [PubMed] [Google Scholar]
Nixon 2017
- Nixon J, Newbold P, Mustelin T, Anderson GP, Kolbeck R. Monoclonal antibody therapy for the treatment of asthma and chronic obstructive pulmonary disease with eosinophilic inflammation. Pharmacology and Therapeutics 2017;169:57-77. [DOI] [PubMed] [Google Scholar]
Noel‐Storr 2018
- Noel-Storr AH, Project Transform team. Cochrane Crowd: new ways of working together to produce health evidence. In: Evidence Live; 2018 June 18-20; Oxford, UK. 2018.
Normansell 2014
- Normansell R, Walker S, Milan SJ, Walters EH, Nair P. Omalizumab for asthma in adults and children. Cochrane Database of Systematic Reviews 2014, Issue 1. Art. No: CD003559. [DOI: 10.1002/14651858.CD003559.pub4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Pascoe 2015
- Pascoe S, Locantore N, Dransfield MT, Barnes NC, Pavord ID. Blood eosinophil counts, exacerbations, and response to the addition of inhaled fluticasone furoate to vilanterol in patients with chronic obstructive pulmonary disease: a secondary analysis of data from two parallel randomised controlled trials. Lancet Respiratory Medicine 2015;3(6):435-42. [DOI] [PubMed] [Google Scholar]
Pavord 2015
- Pavord I, Bush A. Two lovely black eyes; oh, what a surprise! Thorax 2015;70(7):609. [DOI] [PubMed] [Google Scholar]
Pavord 2017
- Pavord ID, Chanez P, Criner GJ, Kerstjens HAM, Korn S, Lugogo N, et al. Mepolizumab for eosinophilic chronic obstructive pulmonary disease. New England Journal of Medicine 2017;377(17):1613-29. [DOI] [PubMed] [Google Scholar]
Pavord 2018
- Pavord ID. Biologics and chronic obstructive pulmonary disease. Journal of Allergy and Clinical Immunology 2018;141(6):1983-91. [DOI] [PubMed] [Google Scholar]
Review Manager 2020 [Computer program]
- Review Manager 5 (RevMan 5). Version 5.4. The Cochrane Collaboration, 2020.
Shironjit 2006
- Shironjit S, Brightling CE. Eosinophilic airway inflammation in COPD. International Journal of Chronic Obstructive Pulmonary Disease 2006;1(1):39-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
Singh 2014
- Singh D, Kolsum U, Brightling CE, Locantore N, Agusti A, Tal-Singer R, et al. Eosinophilic inflammation in COPD: prevalence and clinical characteristics. European Respiratory Journal 2014;44(6):1697-700. [DOI] [PubMed] [Google Scholar]
Siva 2007
- Siva R, Green RH, Brightling CE, Shelley M, Hargadon B, McKenna S, et al. Eosinophilic airway inflammation and exacerbations of COPD: a randomised controlled trial. European Respiratory Journal 2017;29(5):906-13. [DOI] [PubMed] [Google Scholar]
Tan 2018
- Tan IR, Villalobos R, Jaen A, Wang A. Anti-IL-5 agents in the treatment of eosinophilic COPD: a systematic review and meta-analysis. Chest 2018;154(4):759A. [DOI: 10.1016/j.chest.2018.08.684] [DOI] [Google Scholar]
Thomas 2017
- Thomas J, Noel-Storr AH, Marshall I, Wallace B, McDonald S, Mavergames C, et al. Living systematic review network. Living systematic reviews: 2. Combining human and machine effort. Journal of Clinical Epidemiology 2017;91:31-7. [DOI] [PubMed] [Google Scholar]
Vedel‐Krogh 2016
- Vedel-Krogh S, Nielsen SF, Lange P, Vestbo J, Nordestgaard BG. Blood eosinophils and exacerbations in chronic obstructive pulmonary disease. The Copenhagen General Population Study. American Journal of Respiratory and Critical Care Medicine 2016;193(9):965-74. [DOI] [PubMed] [Google Scholar]
WHO 2019a
- World Health Organization. Chronic obstructive pulmonary disease (COPD). www.who.int/news-room/fact-sheets/detail/chronic-obstructive-pulmonary-disease-(copd) (accessed prior to 7 June 2019).
WHO 2019b
- World Health Organization. Burden of COPD. www.who.int/respiratory/copd/burden/en/ (accessed prior to 7 June 2019).