

SUMMARY
Cellular inflammasome activation causes caspase-1 cleavage of the pore-forming protein gasdermin D (GSDMD) with subsequent pyroptotic cell death and cytokine release. Here, we clarify the ambiguous role of the related family member gasdermin E (GSDME) in this process. Inflammasome stimulation in GSDMD-deficient cells led to apoptotic caspase cleavage of GSDME. Endogenous GSDME activation permitted sublytic, continuous interleukin-1β (IL-1β) release and membrane leakage, even in GSDMD-sufficient cells, whereas ectopic expression led to pyroptosis with GSDME oligomerization and complete liberation of IL-1β akin to GSDMD pyroptosis. We find that NLRP3 and NLRP1 inflammasomes ultimately rely concurrently on both gasdermins for IL-1β processing and release separately from their ability to induce cell lysis. Our study thus identifies GSDME as a conduit for IL-1β release independent of its ability to cause cell death.
Graphical abstract
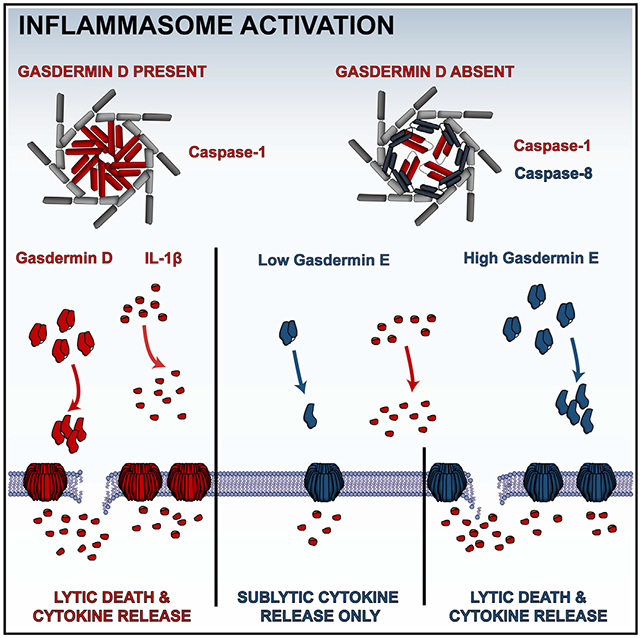
In brief
Gasdermin D is the immediate effector of pyroptosis after cellular inflammasome activation. Zhou and Abbott demonstrate that the homologous gasdermin E is activated secondarily to release of IL-1β in a sublytic phase that is independent of cell lysis, which requires higher gasdermin E presence and substantial pore formation.
INTRODUCTION
The innate immune system helps tailor an adaptive immune response to offending pathogens, and the induction of a pro-inflammatory cell death, termed pyroptosis, amplifies this response. Pyroptosis typically initiates after activation of a cellular inflammasome and most often results in activation of the pore-forming protein gasdermin D (GSDMD). Inflammasomes are supramolecular organizing centers that locally concentrate the inflammatory protease caspase-1 (CASP1) via its caspase activation and recruitment domain, or CARD, for proximity-induced auto-activation (Martinon et al., 2002). CASP1 drives inflammatory cell death through GSDMD activation and magnifies an immune response by cleavage of interleukin-1β (IL-1β) to allow its mature form to be released along with other cytokines (He et al., 2015; Kayagaki et al., 2015; Shi et al., 2015).
Each member of the gasdermin family contains an N-terminal pore-forming domain and a C-terminal autoinhibitory domain separated by a linker region. Of these family members, GSDMD is the best studied (Aglietti et al., 2016; Ding et al., 2016; Liu et al., 2016, 2019; Ruan et al., 2018). GSDMD can be cleaved by CASP1 after canonical inflammasome activation or by CASP4 after direct caspase detection of intracellular bacterial lipopolysaccharide (LPS) (Hagar et al., 2013; Kayagaki et al., 2015). More recently, a small-molecule inhibitor driving new putative NLRP1/CARD8 inflammasomes was described also to act through GSDMD (Johnson et al., 2018; Okondo et al., 2017). Proteolysis of GSDMD disinhibits association of the pore-forming domain with acidic membrane phospholipids, and membrane insertion promotes dimerization and further oligomerization of the pore-forming domain into heterogeneous pores ranging from 6 to 20 nm of inner diameter (Aglietti et al., 2016; Ding et al., 2016; Sborgi et al., 2016). These pores have been shown to allow exodus of smaller proteins, such as IL-1β, while the cell remains viable (Evavold et al., 2018; Heilig et al., 2018). During this interval, termed the sublytic phase, the pores also promote uncontrolled diffusion of ions and eventual full loss of membrane integrity, leading ultimately to cellular demise in the lytic phase. The underlying cellular mechanisms of GSDMD transition from the sublytic to lytic phase are unclear.
The next best-studied gasdermin family member is gasdermin E (GSDME). Initially described as a genetic cause of hearing loss (van Camp et al., 1995), GSDME was more recently characterized as a pyroptosis-inducing protein that is cleaved by apoptosis-associated CASP3 rather than inflammatory CASP1 or CASP4 (Rogers et al., 2017; Wang et al., 2017). The cleaved N-terminal fragment of GSDME was initially suggested to function nearly identically to the GSDMD pore-forming domain (Rogers et al., 2019; Yu et al., 2019; Zeng et al., 2019), yet conflicting reports (Chen et al., 2019; Heilig et al., 2020; Lee et al., 2018; Sarhan et al., 2018; Tixeira et al., 2018) were unable to ascribe pyroptotic activity to GSDME. Notably, there has been scant direct evidence of GSDME cleavage and oligomerization preceding lysis in a cell (Rogers et al., 2019). Finally, no study has actually directly shown GSDME to be responsible for GSDMD-independent IL-1β release after inflammasome activation, although this conclusion has been inferred in multiple studies (Aizawa et al., 2020; Monteleone et al., 2018; Sagulenko et al., 2018; Schneider et al., 2017).
In this work, we unify previous observations by showing within a single model system that higher GSDME concentration is sufficient to drive intracellular oligomerization and cell lysis after NLRP3 and NLRC4 inflammasome activation. We identify a previously undescribed GSDME sublytic phase in which GSDME pores allow substantial IL-1β release over time, akin to GSDMD. Finally, we observe a codependency for both gasdermins, although not their lytic capabilities, in Val-boroPro (VbP)-induced IL-1β processing and release. Our results thus reveal a parallel system of cytokine release by pore-forming gasdermins that is segregated from their ability to instigate lytic cell death.
RESULTS
Inflammasome activation triggers apoptotic cascade and GSDME cleavage in GSDMD-deficient macrophages
To study cellular consequences of inflammasome activation when classical pyroptotic executors are absent, we terminally differentiated wild-type and GSDMD−/− human THP-1 monocyte-like cells into macrophages with phorbol 12-myristate 13-acetate (PMA), then activated the NLRP3 inflammasome with nigericin. For wild-type cells, traditional hallmarks of GSDMD-associated lytic cell death were present: cell swelling, membrane permeability as measured by propidium iodide influx, and overt cell lysis as assessed with supernatant lactate dehydrogenase (LDH) activity (Figures 1A and 1B). Morphologically, we noted that classically pyroptotic cells also appeared speckled and translucent under light microscopy (Figure 1A). Genetic deletion of GSDMD prevented these sequelae at early times, yet these knockout cells eventually manifested cell death characteristics, as others have observed (He et al., 2015; Heilig et al., 2020; Kayagaki et al., 2015). A few classically pyroptotic cells were noted, but also present were apoptotic bodies and opaque, swollen cells. To rule out inflammasome-independent nigericin effects, we pre-incubated cells with NLRP3 inhibitor MCC950 and observed abrogation of cell lysis in GSDMD−/− cells (Figure 1B), consistent with this residual death being an inflammasome-driven event.
Figure 1. Inflammasome activation triggers apoptotic cascade and GSDME cleavage in GSDMD-deficient macrophages.
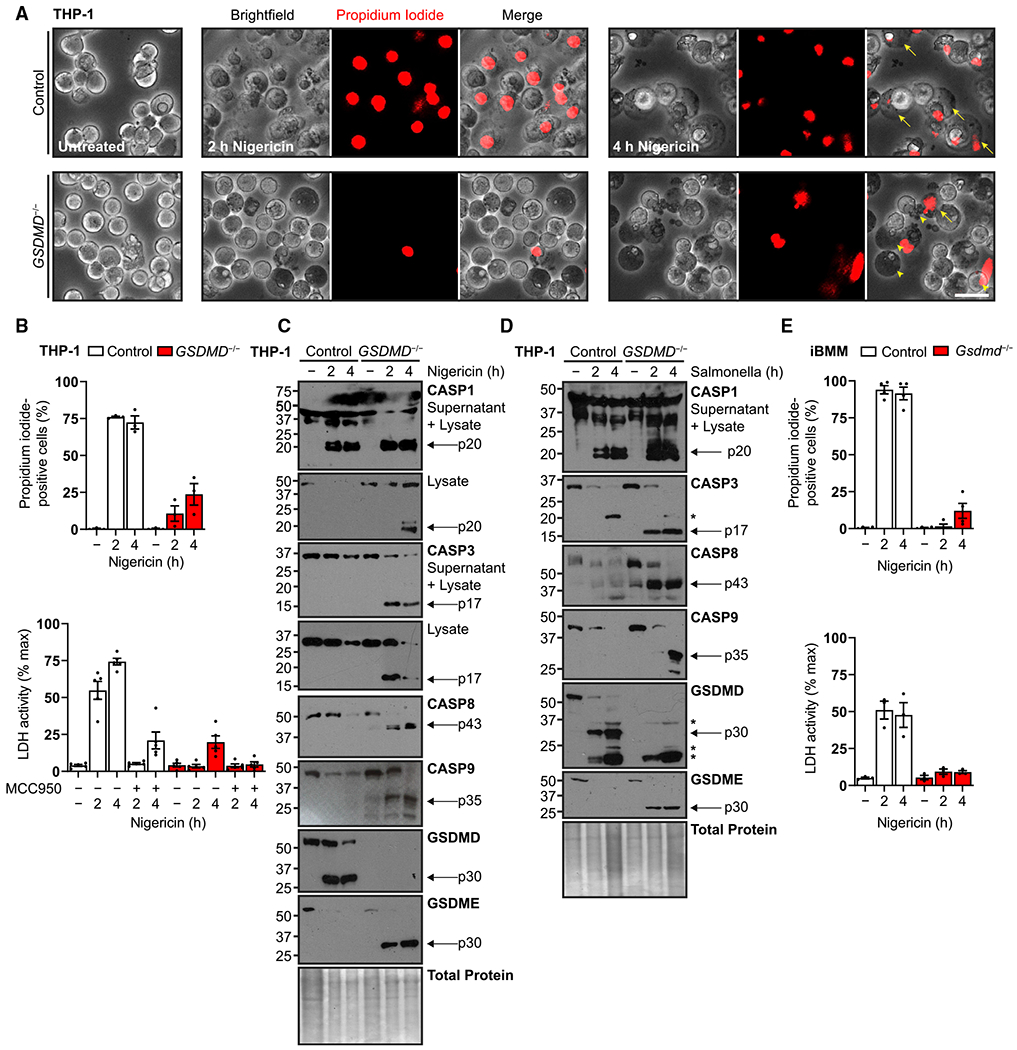
(A) Representative micrographs of human PMA-differentiated (100 ng/mL, 16 h) wild-type and GSDMD−/− THP-1 cells treated with nigericin (20 μM) and incubated with cell-impermeable propidium iodide dye to visualize membrane permeability upon inflammasome activation. Arrows indicate classically swollen pyroptotic cells that are transparent, speckled, and positive for propidium iodide uptake. Arrowheads indicate atypical opaque, impermeable cells and apoptotic bodies.
(B) Quantification of propidium iodide uptake and quantification of frank cell lysis by supernatant lactate dehydrogenase activity assay for similar experiments. For the latter, cells were also pre-treated with NLRP3 inhibitor MCC950 (10 μM, 0.5 h).
(C and D) Immunoblots of similar cells treated with nigericin (20 μM) to activate the NLRP3 inflammasome or S. typhimurium (MOI = 10) for the NLRC4 inflammasome. Combined supernatant and lysate were assayed to detect caspase-1 fragments expelled during pyroptosis.
(E) Quantification of propidium iodide uptake and quantification of cell lysis for wild-type and Gsdmd−/− murine immortalized bone marrow-derived macrophages (iBMMs) treated with S. typhimurium (MOI = 10) or primed with LPS (0.2 μg/mL, 4 h) before nigericin treatment (20 μM).
Scale bar, 50 μm. Graph bars represent mean ± standard error of biological replicates. Graph points represent pooled technical replicates per biological replicate. Immunoblots are representative of at least six independent experiments. Asterisks indicate cross-reactive S. typhimurium protein bands.
See also Figure S1.
To determine whether secondary activation of apoptotic caspases at the inflammasome (Heilig et al., 2020; Lee et al., 2018; Sagulenko et al., 2013, 2018) could be responsible for delayed cell death, we queried caspase activation after treatment with nigericin or the bacterium S. typhimurium, an NLRC4 inflammasome activator (Zhang et al., 2015). THP-1 macrophages treated with nigericin (Figure 1C) exhibited GSDMD cleavage and depletion of cellular protein over 4 h (Davis et al., 2019). In contrast, GSDMD−/− cells showed the presence of the residual CASP1 large catalytic p20 subunit (Boucher et al., 2018) and active subunits of apoptotic caspases, whereas wild-type cells expelled CASP1 p20 into the supernatant fraction. Also observed was cleavage of GSDME into its p30 pore-forming domain. Treatment with S. typhimurium showed comparable results (Figure 1D). Our results mirror those from other recent reports showing apoptotic caspase-driven GSDME cleavage downstream of inflammasome activation (Aizawa et al., 2020; Heilig et al., 2020; Tsuchiya et al., 2019; Zeng et al., 2019). To study an orthogonal system, we similarly treated immortalized murine bone marrow macrophages (iBMMs) described previously (Rathkey et al., 2018). Wild-type and Gsdmd−/− iBMMs showed a similar apoptotic caspase activation and morphological changes (Figures S1A–S1C), but the residual cell death upon Gsdmd deletion was blunted compared with THP-1 cells (Figure 1E). Notably, we did not detect GSDME in this lineage of iBMM. Taken together, these data initially suggested that macrophages undergoing inflammasome stimulation lose membrane integrity in the absence of GSDMD only when GSDME is present, and that this process depends on cellular expression and possibly species specificity.
Endogenous GSDME permits IL-1β release with limited secondary pyroptosis
Because Gsdmd−/− murine iBMMs lacked both GSDME and delayed cell death, we hypothesized that GSDME expression underlay species differences in secondary necrotic death post-inflammasome activation. To test this hypothesis, we generated GSDMD−/−GSDME−/− double knockouts in human THP-1 cells. Nigericin treatment revealed greatly reduced propidium iodide influx with a small drop in cell lysis as compared with GSDME-sufficient GSDMD−/− cells (Figure 2A). Thus, although some of the overt lysis in double-knockout cells was attributable to gasdermin-independent events, GSDME was responsible for almost all of the membrane leakage, with no clear morphological difference between treated single- and double-knockout cells (Figure 2B). Notably, GSDME single deletion did not alter cell death parameters in response to nigericin (Figure S2A), although somewhat increased GSDMD cleavage was noted (Figure S2B).
Figure 2. Endogenous GSDME permits IL-1β release with limited secondary pyroptosis.
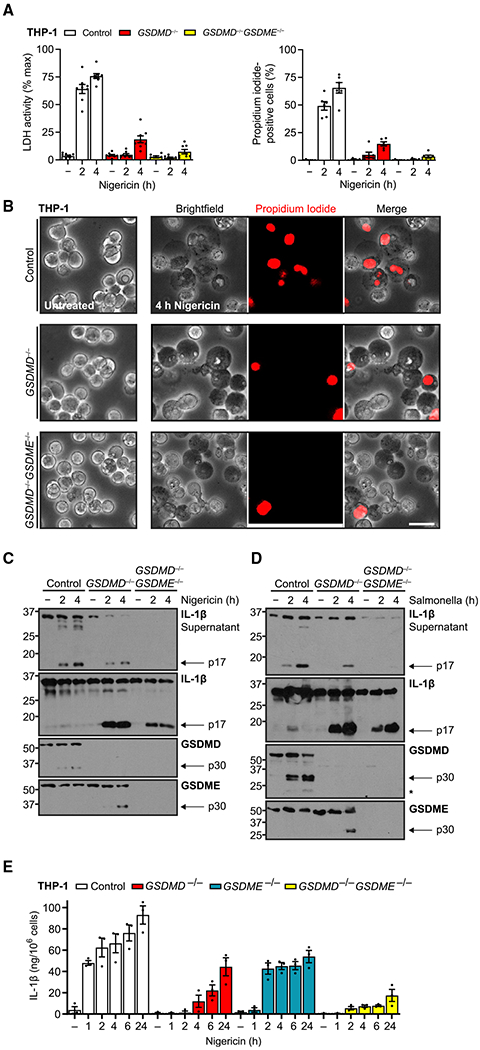
(A and B) PMA-differentiated (100 ng/mL, 16 h) wild-type, GSDMD−/−, and GSDMD−/−GSDME−/− THP-1 cells treated with nigericin (20 μM) assessed for propidium iodide uptake, cell lysis through supernatant lactate dehydrogenase activity assay (A), and cell morphology by microscopy (B).
(C and D) Immunoblots of these cells treated with nigericin or S. typhimurium (MOI = 10).
(E) Mature IL-1β ELISA for supernatants of wild-type and indicated gasdermin knockout differentiated THP-1 treated with nigericin (20 μM).
Immunoblots are representative of at least five independent experiments. Scale bar, 50 μm. Graph bars represent mean ± standard error of biological replicates. Graph points represent pooled technical replicates per biological replicate.
See also Figure S2.
This pattern of membrane leakiness without full lysis is most consistent with sublytic pores acting as a conduit for IL-1β as described for GSDMD (Evavold et al., 2018; Heilig et al., 2018). Therefore, we hypothesized that in our system, GSDME may be acting similarly, with overt lysis being prevented by multiple factors, such as membrane repair, insufficient pore number, or other undetermined mechanisms (Evavold et al., 2018; Heilig et al., 2018; Rühl et al., 2018). Thus, we next explored release of the cytokine IL-1β by THP-1 macrophages treated with nigericin or S. typhimurium (Figures 2C and 2D). Immunoblots showed that whereas wild-type cells readily released almost all intracellular, mature IL-1β to the cell-free supernatant, GSDMD−/− cells retained most mature IL-1β within the cell with modest release increasing over time and S. typhimurium treatment eliciting a slower effect. Double-gasdermin deletion further prevented release of both pro-IL-1β and mature p17 IL-1β into the supernatant, indicating dependency on GSDME, and this result was confirmed with two individual clones (Figure S3). We also noted baseline secretion of pro-IL-1β in untreated cells over the experimental time course, probably reflecting other mechanisms of GSDMD-dependent IL-1β secretion, such as the recently reported endosomal pathway (Bulek et al., 2020).
To quantify the dependency of IL-1β release on gasdermins, we conducted an extended time course of double and both individual gasdermin knockouts in THP-1 macrophages with nigericin treatment (Figure 2E). We found that control cells quickly released a large amount of IL-1β, which then increased over a day. GSDME-attributable IL-1β release in GSDMD−/− similarly rose over a day without the initial bolus. IL-1β release attributable to GSDMD alone in GSDME−/− cells, which exhibit similar lysis and propidium iodide uptake as control cells (Figure S2B), peaked at 2 h and remained level over a day, and double knockouts exhibited greatly diminished IL-1β release. Thus, the two gasdermins were almost fully responsible for all IL-1β release in a parallel yet seemingly additive manner with different kinetics. In support of this conclusion, wild-type THP-1 IL-1β release resembled the sum of that in the individual knockout cell lines. Thus, endogenous THP-1 GSDME is able to support movement of small ions and IL-1β across intact cell membrane with limited actual cellular lysis.
GSDME expression level determines secondary pyroptosis
For GSDMD, lack of lytic action is caused by too little cellular homeostatic disruption, purportedly because of insufficient spatial concentration of pores (Evavold et al., 2018). We reasoned that if GSDME followed a similar paradigm, simply increasing the steady-state concentration of full-length GSDME would confer lytic, secondary pyroptosis to cells. We thus virally overexpressed human GSDME and the non-cleavable GSDMED270A variant (altered at the CASP3 cleavage and activation site) into THP-1 double-knockout macrophages. Micrographs of cells reconstituted with wild-type GSDME and treated with nigericin showed the presence of swollen, pyroptotic cells with propidium iodide staining, whereas the negative controls, i.e., double-knockout and caspase-immune GSDME cells, did not (Figure 3A). Quantification of propidium iodide uptake and cell lysis further showed higher cell lysis per propidium iodide-positive cell (Figure 3B), in contrast with GSDMD−/− cells expressing endogenous, lower levels of GSDME (Figure 2A). Immunoblots of these cells additionally pretreated with NLRP3 inhibitor MCC950 or pan-caspase and calpain inhibitor z-VAD-fmk revealed, as expected, substantial mature IL-1β release only in wild-type, non-inhibited GSDME-competent cells, and the non-cleavable variant did not impair either CASP3 activation or generation of p17 IL-1β (Figure 3C).
Figure 3. GSDME expression level determines secondary pyroptosis.
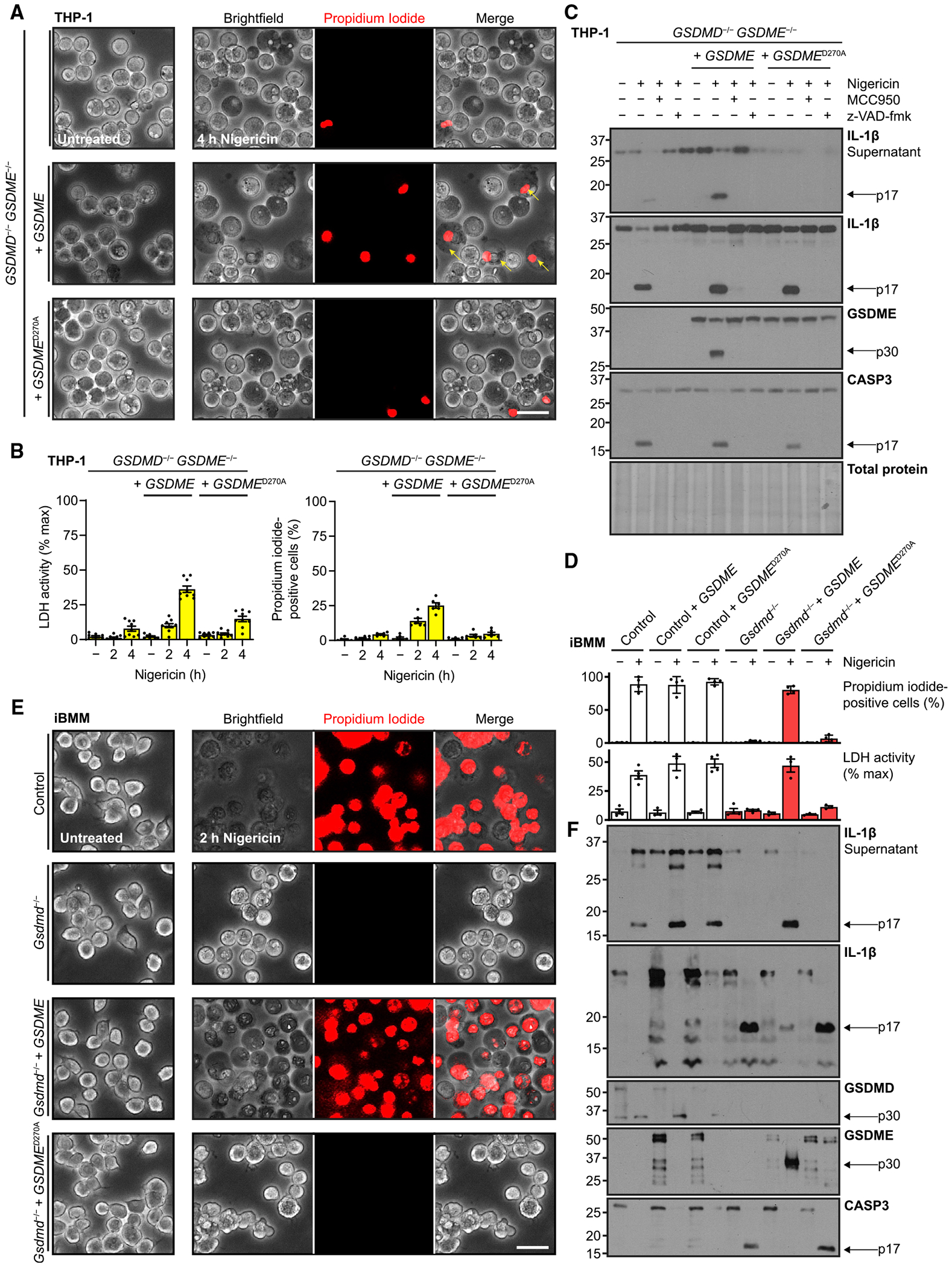
(A) Micrographs of GSDMD−/−GSDME−/− THP-1 cells rescued with constitutively expressed wild-type or caspase-immune D270A GSDME, PMA differentiated (100 ng/mL, 16 h), and treated with nigericin (20 μM).
(B) For similarly treated cells, quantification of propidium iodide uptake and quantification of cell lysis with supernatant lactate dehydrogenase activity assay.
(C) Immunoblots of indicated PMA-differentiated cells pre-treated with NLRP3 inhibitor MCC950 (10 μM, 0.5 h) or pan-caspase and calpain inhibitor z-VAD-fmk (20 μM, 0.5 h) before exposure to nigericin (20 μM).
(D–F) Identical analyses of murine control or Gsdmd−/− iBMMs with orthologous expression of human GSDME or caspase-immune variant D270A.
Immunoblots are representative of at least four independent experiments. Scale bar, 50 μm. Graph bars represent mean ± standard error of biological replicates. Graph points represent pooled technical replicates per biological replicate.
Next, we wanted to address the initial observations that murine Gsdmd−/− iBMMs did not exhibit meaningful amounts of delayed cell death (Figures S1A–S1C) using orthologous expression of human GSDME. Gsdmd-sufficient cell lines primed with bacterial LPS and then treated with nigericin exhibited similar, high measures of lysis and propidium iodide influx, with no meaningful effect of concurrent GSDME expression, while in Gsdmd−/− iBMMs, wild-type, but not non-cleavable, GSDME overexpression was able to recapitulate this phenotype (Figure 3D). Similarly, only wild-type GSDME was able to mirror the pyroptotic morphology conferred in primary pyroptosis by Gsdmd (Figure 3E). Immunoblots showed that in the presence of either active gasdermin, cleaved IL-1β was released almost in its entirety (Figure 3F). We again observed depletion of antibody-reactive protein within the 2 h of nigericin treatment, accounting for, for example, loss of CASP3 immunoreactivity. In summary, these results indicated that increasing the amount of GSDME in cells is sufficient to transform sublytic activation to bona fide lysis, and that this pathway is conserved between mouse and human.
Sublytic GSDME activity transitions to oligomer pore-dependent lysis
We next sought to confirm biochemically whether GSDME forms pores as GSDMD does to act as a plasma membrane channel for IL-1β and propidium iodide. One measure of pore formation is observation by immunoblot of higher-order gasdermin p30 forms. GSDMD first dimerizes, and the dimers subsequently oligomerize into higher-order structures, and this process is targetable by small-molecule inhibitors (Hu et al., 2020; Rathkey et al., 2018). In wild-type and GSDMD−/− THP-1 cells, treatment with nigericin induced GSDMD or GSDME cleavage, respectively (Figure 4A). When cell lysates were prepared under non-reducing conditions to maintain monomer association, we detected GSDMD dimers, but not appreciable GSDME self-association. Upon GSDME rescue in double-knockout cells, however, we did observe the GSDME p30 dimer (Figure 4B). In the murine iBMM system, we observed similar results, except GSDMD and GSDME dimers and an oligomer smear were detected (Figure 4C).
Figure 4. Sublytic GSDME activity transitions to pore-dependent lysis.
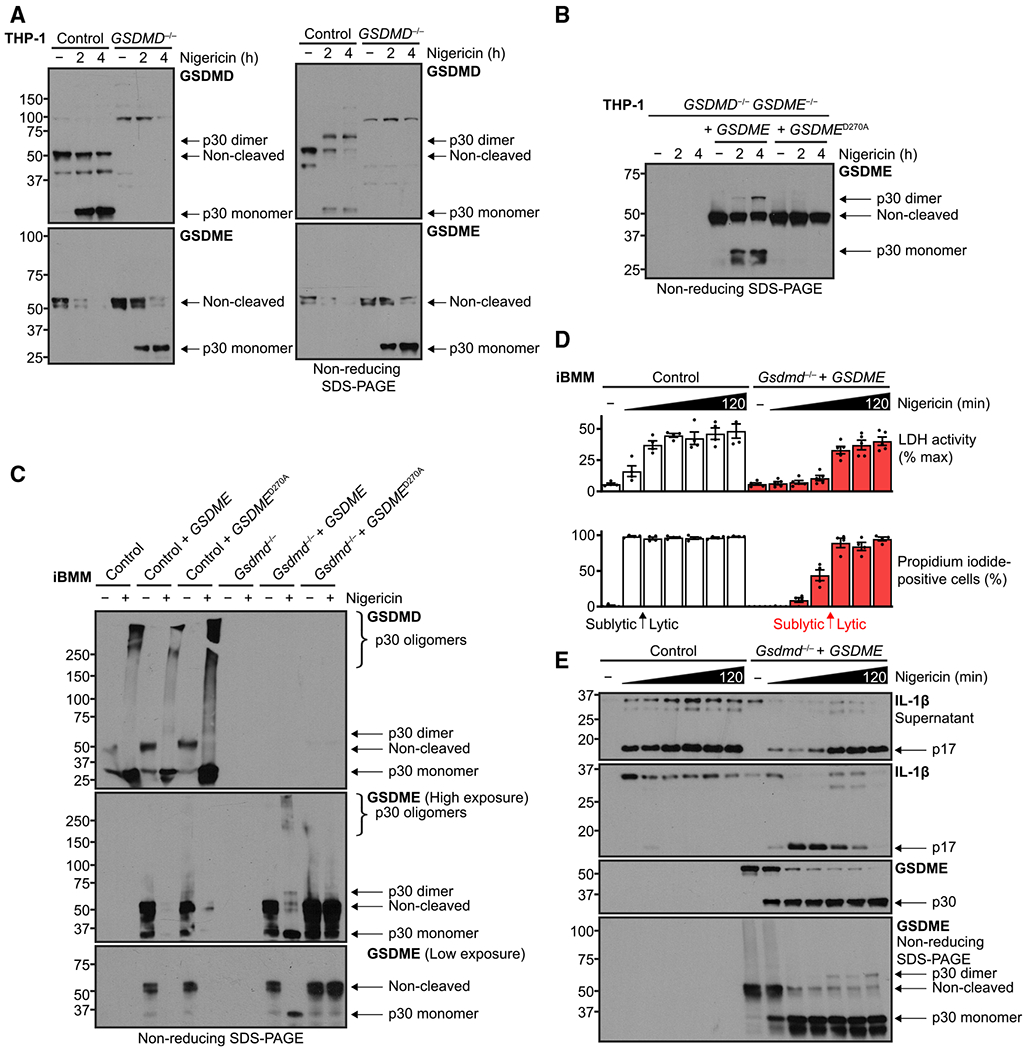
(A and B) Immunoblots of PMA-differentiated (100 ng/mL, 16h) human THP-1 cells with the indicated genotypes were treated with nigericin (20 μM). Where noted, lysates were processed under non-reducing conditions to assess higher-order self-association of GSDMD or GSDME p30 fragments as a readout for pore formation.
(C–E) Murine immortalized bone marrow-derived macrophages were LPS primed (0.2 μg/mL, 4 h) and treated with nigericin (20 μM) for up to 2 h and similarly prepared to assess gasdermin self-association.
(D) Quantification of propidium iodide uptake and quantification of frank cell lysis by supernatant lactate dehydrogenase activity assay for similar experiments. Arrows indicate transition from sublytic phase to full-blown cell lysis.
Immunoblots are representative of at least three independent experiments. Graph bars represent mean ± standard error of biological replicates. Graph points represent pooled technical replicates per biological replicate.
Finally, we asked whether the GSDME response was binary or a transition, i.e., if sublytic activation with concomitant IL-1β release and propidium iodide influx was present prior to secondary pyroptosis and cell lysis. To test this hypothesis, we performed a detailed time course over 2 h of nigericin treatment comparing wild-type to Gsdmd−/− with GSDME cells, in essence comparing the two gasdermins directly (Figures 4D and 4E). GSDMD activation caused maximum propidium iodide influx by 20 min, but not full lysis, measured by released LDH activity, until between 40 and 60 min. However, GSDME cleavage was apparent by 20 min and nearly complete by 40 min, yet a greater proportion of IL-1β p17 remained intracellular in the first hour. Compared with GSDMD, a similar transition point for GSDME between 60 and 80 min was observed, whereby increased IL-1β release coincided with both an increase of cell lysis (Figure 4D) and GSDME p30 dimer formation in non-reducing conditions (Figure 4E, bottom panel). This slower lysis coincides with the stoichiometrically lower presence of GSDME dimer versus the GSDMD dimer, even when overexpressed. In other words, for GSDME overexpression, we observed a sublytic phase similar to in THP-1 cells with endogenous GSDME, yet also an eventual transition to full lytic phase with greater IL-1β release and cellular demise.
Gasdermins segregate VbP pyroptosis from IL-1β release
Having established a role for GSDME in secondary IL-1β release from cells upon NLRP3 and NLRC4 inflammasome stimulation in the absence of GSDMD, we next asked whether this pathway extended to other inflammasomes. We treated our THP-1 cell lines with VbP, a small-molecule inhibitor of the two serine proteases dipeptidyl peptidase 8 and 9. VbP induces death with pyroptotic characteristics in a CASP1-dependent manner through either the NLRP1 inflammasome or another protein, CARD8, also containing a CARD, although exact mechanisms remain elusive (Gai et al., 2019; Johnson et al., 2018; Okondo et al., 2017). One of these studies (Johnson et al., 2018) noted that cell lines, including THP-1, treated with VbP exhibited LDH release and loss of viability but variable, sometimes even minimal, GSDMD cleavage. We hypothesized that GSDME instead underlies some of the cellular outcomes from VbP treatment.
We treated control or gasdermin-deficient THP-1 cells with VbP or with nigericin as a pyroptosis-positive control (Figure 5A). GSDME or GSDME deficiency resulted in a striking lack of secreted IL-1β in response to VbP, but as opposed to constrained release of processed cytokine in nigericin-treated GSDMD−/− cells, removal of either gasdermin resulted in less production of the cleaved IL-1β. The double gasdermin knockout further diminished cleaved IL-1β in both compartments, suggesting parallel instead of redundant pathways for IL-1β activation. These data indicate that more complicated signaling pathways, such as cooperativity or feed-forward loops, for GSDMD and GSDME in VbP treatment.
Figure 5. Gasdermins segregate Val-boroPro pyroptosis from IL-1β release.
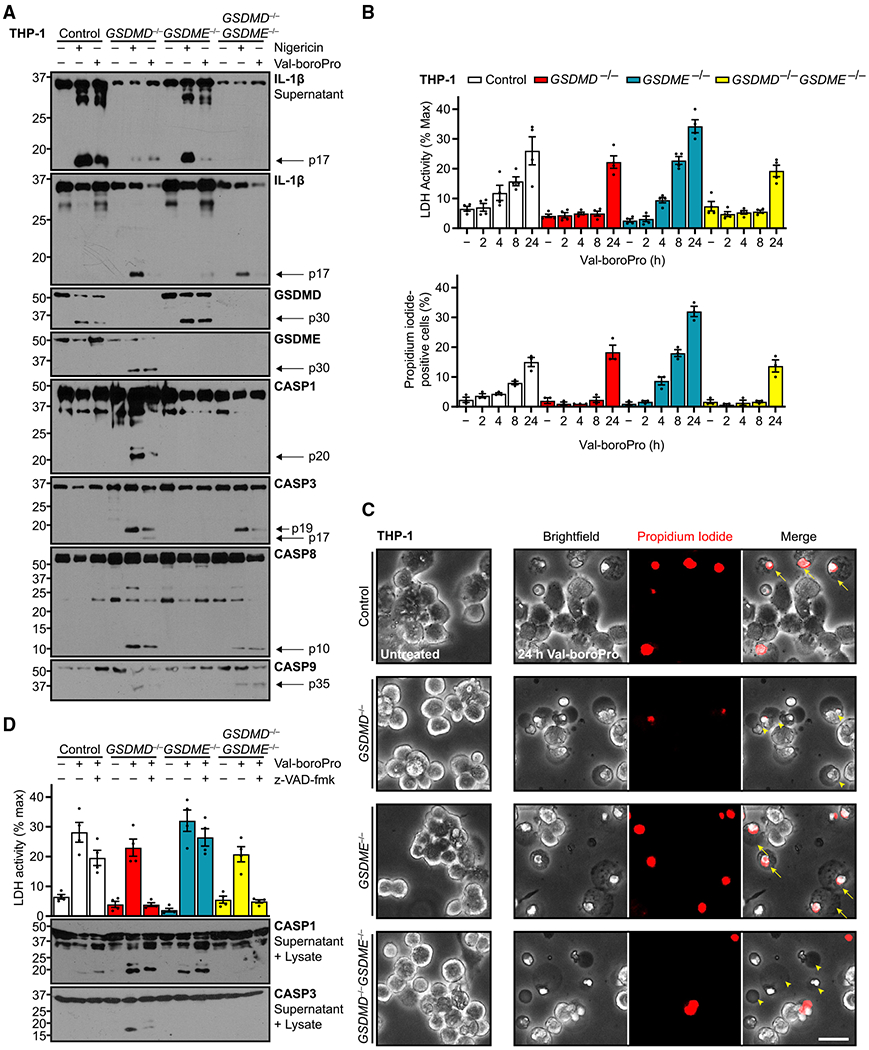
Indicated genotypes of PMA-differentiated (100 ng/mL, 16 h) human THP-1 cells were treated with NLRP1 inflammasome activator Val-boroPro (2 μM).
(A) Immunoblots of these cells also were treated with nigericin (20 μM, 3 h) as pyroptosis positive control.
(B) Quantification of propidium iodide uptake and cell lysis through supernatant lactate dehydrogenase activity assay.
(C) Micrographs of Val-boroPro treatment. Arrows indicate classically pyroptotic cells that are swollen, speckled, transparent, and permeable to propidium iodide. Arrowheads indicate atypical, opaque cells that are impermeable to propidium iodide and opaque.
(D) Quantification of cell lysis induced with Val-boroPro with additional pretreatment with calpain and pan-caspase inhibitor z-VAD-fmk (20 μM, 0.5 h).
Scale bar, 50 μm. Immunoblots are representative of at least three independent experiments. Graph bars represent mean ± standard error of biological replicates. Graph points represent pooled technical replicates per biological replicate.
IL-1β release was dissociated from cell death in these assays, however. Analysis of cell death parameters upon VbP treatment revealed modest cell death compared with nigericin-treated cells (25% versus 75% for nigericin in Figure 1B). This resulted regardless of gasdermin presence, because double-knockout cells exhibited the same amount as control cells (Figure 5B). However, GSDMD sufficiency in wild-type cells seemed to accelerate cell death toward this level. More surprising was that GSDME deletion exacerbated the GSDMD-dependent necrotic death, and cell morphology in these and control cells resembled classically pyroptotic cells (Figure 5C). In contrast with the NLRP3 and NLRC4 models, then, GSDME dissociated IL-1β processing from pyroptotic cell death.
Last, we tested the role of caspase activation in the VbP system by co-treating with the pan-caspase inhibitor z-VAD-fmk (Figure 5D). These experiments revealed that the baseline VbP-induced cell death in cells without GSDMD was completely blocked by caspase inhibition. However, the GSDMD-dependent death, in wild-type and GSDME−/− cells, was not. Further, although CASP3 activation was effectively neutralized by z-VAD-fmk, the levels of CASP1 p20 were decreased but only shifted to a higher molecular weight. This shift possibly reflected the blockage of terminal autoprocessing of CASP1 at the interdomain linker site (Boucher et al., 2018), and our findings indicate a z-VAD-fmk-resistant source of CASP1 activation in response to VbP treatment, a hypothesis consistent with the lower effectiveness of a CASP1 inhibitor in this system (Okondo et al., 2017). These data suggest that although both gasdermins can facilitate IL-1β processing and release, cell death mechanisms can be dissociated.
In summary, our experiments have shown that GSDME underlies mature IL-1β release in a multitude of inflammasome-dependent contexts. On one hand, when GSDMD is absent, NLRP3 or NLRC4 stimulation causes GSDME to be activated downstream of the apoptotic cascade. Alternatively, VbP induced pyroptosis-like cell death that utilized cell death-independent IL-1β processing that was dependent on the presence of gasdermins. Thus, gasdermin activation in general cannot be thought of as a straightforward cell death mechanism but depends heavily on cellular context ranging from activation method to protein abundance.
DISCUSSION
Similar domain architecture among gasdermin proteins initially led to the assumption of identical pyroptotic activity upon cleavage. Activation was assumed to take cues from cellular context to provide signal specificity for the all-or-nothing committal to concerted pyroptotic cell lysis and cytokine release. Thus, inflammatory caspases cleave GSDMD to cause pro-inflammatory pyroptosis, and GSDME-dependent lysis in an apoptotic environment would be immunologically silent. However, since the initial characterization of GSDMD (He et al., 2015; Kayagaki et al., 2015) and GSDME (Rogers et al., 2017; Wang et al., 2017), this simple model has been plagued with exceptions: GSDMD can instead be activated by apoptotic CASP8 (Orning et al., 2018; Sarhan et al., 2018); GSDMD can act as a channel for IL-1β in membrane-intact, living cells (Evavold et al., 2018; Heilig et al., 2018); and GSDME cleavage may not lead to necrotic death (Lee et al., 2018; Tixeira et al., 2018). Our study further challenges this initial dogma of gasdermin activation, showing that “apoptotically” activated GSDME possesses an immunological role. In stark contrast with the more commonly studied context of GSDME in cancer cells as an apoptosis-to-pyroptosis converter (Croes et al., 2019; Ding et al., 2020; Liang et al., 2020; Liu et al., 2020; Wang et al., 2017; Yu et al., 2019), we specifically establish a causal link between GSDME cleavage and mature IL-1β release into the extracellular space following inflammasome activation independent of cell death.
Further, our data address the discrepancies regarding the ability of cleaved GSDME to initiate pyroptosis, finding that a sufficient concentration of GSDME was necessary for dimerization and oligomerization, as suggested recently (Chen et al., 2020a). Human THP-1 macrophages do not possess sufficient GSDME for pore-induced cell death and detectable self-association (Figure 4A), but GSDME overexpression in both THP-1 and iBMM cells was sufficient to induce dimer and oligomer formation following nigericin treatment (Figures 4B and 4C). Importantly, we have directly demonstrated higher-order GSDME structures in cells, a principle that has been assumed but shown thus far only in a cell-free system (Rogers et al., 2019).
GSDME can function in either sublytic or lytic modes. In the former, low numbers of pores are postulated to act as a conduit for ions and small molecules, such as IL-1β. We did not explicitly observe sublytic pore formation, yet the combination of propidium iodide influx and IL-1β efflux with relatively low cell lysis, both attributable to endogenous GSDME, makes a strong case for this inference (Figures 2A, 2C–2E, and 4A). To engage in lytic activation, a critical threshold of pore number or local concentration need be surpassed, and membrane integrity is irreversibly compromised. Although the terminal mechanisms underlying this Anal step are still unknown (Heilig et al., 2020), it results in expulsion of larger molecules, a process commonly approximated by LDH activity (Figure 3B). We provide evidence that these two modes can be transitional, rather than mutually exclusive, for both gasdermins in the iBMM orthologous GSDME expression model, wherein GSDMD and GSDME both exhibited a sublytic-to-lytic transition period with different kinetics (Figure 4D). This result reconciles previous studies showing either ability (Rogers et al., 2017, 2019; Tsuchiya et al., 2019; Wang et al., 2017) or lack thereof (Chen et al., 2019; Heilig et al., 2020; Lee et al., 2018; Tixeira et al., 2018) by GSDME in induction of lytic cell death. In our experiments, lysis strictly coincided with presence of dimers and oligomers (Figures 5B and 5C), suggesting that any mechanisms opposing or delaying the lytic phase do so by removing or inhibiting higher-order GSDME structures.
More surprisingly, our findings also indicate that the immunological consequence of IL-1β release is independent of GSDME lytic death, because supernatant mature IL-1β increased gradually over time in both sublytic THP-1 and iBMM activation, but then en masse after lytic phase onset in murine iBMMs (Figures 2C–2E, 3C, and 4E). This mode of action for GSDME-dependent IL-1β release is consistent with some unexplained observations in recent studies. Monteleone et al. (2018) reported plasma membrane-localized murine IL-1β that was secreted in a fast, GSDMD-dependent manner, yet they also found a GSDMD-independent fraction that was continuously released in a slower, non-lytic manner over at least 5 h, as we similarly quantified with GSDME-dependent release (Figure 2E). Schneider et al. (2017) separately characterized IL-1β release with secondary lytic pyroptosis in primary Gsdmd−/− murine bone marrow-derived dendritic cells undergoing apoptotic caspase activation, although they were not able to identify the effector. Finally, the Broz group recently found that GSDME was not necessary for CASP1-initiated, GSDMD-independent secondary necrosis, yet they observed moderate amounts of IL-1β release and GSDME cleavage in Gsdmd-deficient murine bone marrow-derived macrophages (Heilig et al., 2020). Systematic comparison of GSDME expression and p30 self-association and among C57BL/6 primary and immortalized macrophages (Heilig et al., 2020; Monteleone et al., 2018), differentiated dendritic cells from crossed 129 and C57BL/6 mice (Schneider et al., 2017), and human cells would be necessary to further confirm our working model and rationalize species and cell-type differences.
Our central finding has implications for several lines of ongoing research. From the therapeutic standpoint, targeting GSDMD itself (Hu et al., 2020; Rathkey et al., 2018; Sollberger et al., 2018) may not be sufficient. In the case of IL-1β release, alternative release by GSDME over time may supersede GSDMD-dependent cellular lysis that terminates further IL-1β processing (Figure 2E). However, because our work was done in immortalized cellular models, whether physiologically relevant amounts of cytokine release in vivo are attained remains to be determined, although a recent preprint report noted a similar redundancy of GSDME for GSDMD in mice (Wang et al., 2021). In addition, our study, like most others, was performed under GSDMD deficiency or genetic ablation. Albeit a constructed condition in our work, cell types that are inflammasome competent and GSDMD deficient likely exist physiologically, and it is tempting to speculate that GSDME may act as the primary IL-1β conduit there. In contrast, when GSDMD is present, the kinetics of GSDME activation pathways likely differ. Follow-up studies using knockin GSDMD polymorphic variants, various expression levels as in physiology, or caspase-immune D276A versions may result in different conclusions, as use of the Casp1C284A mouse model versus knockouts has shown (Aizawa et al., 2020; Schneider et al., 2017).
Overall, results from our study and other similar recent reports coalesce in a redundant network of lytic cell death and cytokine release, a notion supported by discovery of mitochondrial crosstalk between GSDMD and GSDME (Rogers et al., 2019) and enhancement of inflammasome-dependent CASP3 processing through a mitochondrial axis (Heilig et al., 2020), hence blurring the lines of single gasdermin reliance in any cell death process. Along these lines, our results with VbP treatment of THP-1 cells indicated that even when both GSDMD and GSDME are present, they need not follow the paradigm of GSDMD primacy in either cell death or cytokine release. Here, both GSDMD and GSDME are important for IL-1β release and, surprisingly, processing, in a cooperative manner, as either single deletion disproportionately reduced IL-1β cleavage and output (Figure 5A). However, we found that GSDME single deletion was unexpectedly deleterious for cell viability. This finding further emphasizes the dissociation of IL-1β release through gasdermin conduits from lytic cell death.
Of note, non-differentiated THP-1 monocytes treated with VbP were previously shown to utilize the ASC-independent CARD8 inflammasome, with weak cytokine processing, as opposed to the fully effective NLRP1 inflammasome (Johnson et al., 2018). Although the relative contributions of these two inflammasomes in differentiated THP-1 macrophages may differ from monocytic conditions, it is also likely that in our system, activation of either gasdermin as a conduit may trigger downstream, feedforward NLRP3 activation, similarly to the non-canonical NLRP3 inflammasome or with pannexin-1 activity (Chen et al., 2020b; Kayagaki et al., 2013). Determination of the complex steps of intertwined activation and lytic pathways will be a strong focus in upcoming research as the mechanisms of individual pathways are resolved. Some investigators have recently suggested that the definition of pyroptosis be refined to necrotic gasdermin-dependent cell death with indifference to upstream activators (Broz et al., 2020). Our data indicate that this definition could be difficult to consolidate. As demonstrated, sublytic gasdermin activation permits a subset of activity, such as cytokine release, but downstream effectors, as in the case of VbP inflammasome activation, may be the ultimate arbiters of cell death.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Derek Abbott (dwa4@case.edu)
Materials availability
Plasmids and cell lines generated in this study will be available internationally upon request with appropriate materials transfer agreements.
Data and code availability
This study did not generate unique datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mammalian Cell Lines
Murine Nlrp3−/− immortalized bone marrow-derived macrophages overexpressing FLAG-NLRP3 and mCerulean-ASC (in summation referred to as iBMMs) were a gift from Eicke Latz and were characterized previously (Rathkey et al., 2018). DMEM with 10% SuperCalf serum (GeminiBio), 100 U mL−1 penicillin, and 100 μg mL−1 streptomycin was used as culture medium. Human THP-1 cells (TIB-202; American Type Culture Collection) were cultured in RPMI with 10% fetal bovine serum (Gemini Bio), 100 U mL−1 penicillin, and 100 μg mL−1 streptomycin. THP-1 and iBMMs were routinely tested for mycoplasma contamination by PCR.
METHOD DETAILS
Cell Line Modifications
Knockouts of iBMM Gsdmd were generated previously with stable lentiviral expression of FLAG-Cas9 and sgRNA targeting the protospacer adjacency motif at 77TGG (Russo et al., 2016) using pLentiCRISPRv2 (Addgene, a gift from Feng Zhang). THP-1 GSDMD knockouts were made with lentiviral targeting of 11CCT(−). These cell lines were selected with puromycin, 10 μg mL−1 for iBMM and 3 μg mL−1 for THP-1, for two weeks, then single cell clones were confirmed for knockout with immunoblotting and genomic DNA sequencing. THP-1 GSDME knockouts in control and GSDMD−/− cells, targeting 653CCT(−) and 170CCC(−) respectively, were generated with nucleoporation of Cas9 protein (Integrated DNA Technologies) coupled with Alt-R tracrRNA and Alt-R crRNA (Integrated DNA Technologies) according to manufacturer protocols. The 4D-Nucleofector, SG cell line kit, and default THP-1 program (Lonza) were used for transient Cas9 transfection and gene editing. Single cell clones per line were obtained by limiting dilution and verified with immunoblot and genomic sequencing.
For overexpression of GSDME and GSDMED270A, we used Gibson assembly to subclone open reading frames of the more common 142Pro human GSDME variant (NCBI reference sequence NM_004403.2) into a lentiviral expression plasmid with constitutive expression driven by the mammalian Elongation Factor-1α short promoter as described previously (Chirieleison et al., 2017). The expression cassette consisted of neomycin resistance coupled by a self-cleaving P2A peptide (Liu et al., 2017) to GSDME. This schema resulted in addition of a proline residue at the N terminus of GSDME. Transduced cells were selected with G418/geneticin (InvivoGen) at 0.5 μg mL−1 for three weeks, then total populations assayed for overexpression compared to knockouts with immunoblot.
Cell Culture Conditions
For experiments, iBMMS were seeded for overnight confluency. They were then primed with 0.2 μg mL−1 ultrapure S. minnesota lipopolysaccharide (InvivoGen) in fresh media for 4 hours. Cells were washed once in phosphate-buffered saline then incubated for treatment in live cell imaging solution (140 mM NaCl, 2.5 mM KCl, 1.8 mM CaCl2, 1.0 mM MgCl2, and 20 mM HEPES at pH 7.4 and 300 mOsm; ThermoFisher Scientific). THP-1 cells were differentiated terminally into adherent macrophages using 100 ng mL−1 PMA (MilliporeSigma) in fresh media for 16-18 hours. Fresh media plus PMA was used for THP-1 seeding at 4 × 106 cells in 4 mL for 60 mm plates or at 0.5 × 106 cells in 0.5 mL for 24-well dishes then replaced with fresh media for treatment. Treatment was staggered to achieve synchronous endpoints where possible.
Inflammasome activation and drug treatment
Cellular inflammasomes were activated with log phase S. typhimurium (14028; American Type Culture Collection) at a multiplicity of infection of 10, with 20 μM nigericin (MilliporeSigma), or with 2 μM Val-boroPro/Talabostat (R&D Systems) in 0.5 mL per well or 1.5 mL per plate. Propidium iodide at 1 μg mL−1 (ThermoFisher Scientific) was added concurrently for imaging experiments. The NLRP3 inhibitor MCC950 was used at 10 μM and pan-caspase and calpain inhibitor z-VAD-fmk at 20 μM for 30 minutes prior to inflammasome activation. Ethanol was used as a vehicle for 1000 × nigericin stock solution, DMSO for other 1000 × stock solutions.
Cell death assays
Propidium iodide influx was assayed directly through brightfield and epifluorescence images taken with 20 × objective magnification on a DM IL LED microscope (Leica Microsystems). Cell lysis was measured using the CyQUANT LDH Cytotoxicity Assay kit (ThermoFisher Scientific). Cellular supernatant was directly used, and maximum signal was obtained by lysing cells for 30 minutes in 1% Triton X-100. Absorbance of warmed media was used as baseline and subtracted from all other samples before dividing by the maximum signal for each cell genotype.
Immunoassays
For immunoblots and ELISA assays, cells were scraped with supernatant from plates and centrifuged at 20,000 × g for one minute. For cleaved human IL-1β ELISA (#437004, BioLegend), supernatant was then directly diluted and assayed. For immunoblots, cleared supernatant was incubated with 5 × SDS-PAGE sample buffer (5% sodium dodecyl sulfate, 0.25 M Tris pH 6.8, 50% glycerol, 5% β-mercaptoethanol) for 5 minutes at 98°C. Cell pellets were washed with PBS, re-centrifuged, disrupted with lysis buffer (1% SDS, 1 mM EDTA, 0.01 M Tris pH 8, and 150 mM NaCl) at 25°C, and then held for 5 minutes at 98°C. Sample buffer was added to 1 × and samples held for 5 minutes at 98°C. To assay gasdermin cleavage product dimerization and oligomerization, non-reducing lysis conditions were obtained by omitting β-mercaptoethanol from sample buffer.
Lysates and supernatant were separated using SDS-PAGE, and proteins were wet transferred onto 0.22 μm nitrocellulose membranes. Membranes were then washed with Tris-buffered saline with 0.01% Tween-20 (TBST) and incubated with primary antibody diluted in 5% bovine serum albumin in TBST for 16 hours at 4°C or 1-2 hours at 25°C. Membranes were washed thrice in TBST for 5 minutes then incubated with HRP-conjugated secondary antibody diluted in 5% milk in TBST for 1-2 hours at 25°C. Membranes were again washed thrice in TBST then developed with enhanced chemiluminescence.
Primary antibodies were used at 1:1000 dilution. Primary mouse-specific antibodies were CASP1 p20 (#AG-20B-0048-C100; AdipoGen Life Sciences), GSDMD (ab209845; Abcam), and IL-1β (AF-401-NA, R&D Systems). Human-specific antibodies were GSDMD (HPA044487, Atlas Antibodies) and IL-1β (AF-201-NA, R&D Systems). Dual specificity antibodies were GSDME (ab215191, Abcam), CASP1 p10 (ab179515, Abcam), CASP3 (#9662, Cell Signaling Technology [CST]), CASP8 (#4790, CST), cleaved CASP8 (#9429, CST), CASP9 (#9508, CST). Secondary antibodies were used at 1:10,000-1:5000 dilution: anti-goat (sc-2354, Santa Cruz Biotechnology), anti-mouse (#7076, CST), and anti-rabbit (#7074, CST).
QUANTIFICATION AND STATISTICAL ANALYSIS
Experiments were performed with at least three independent biological replicates. For propidium iodide quantification, at least 100 cells each in at least two random fields per replicate were manually counted in a non-blinded manner using the ImageJ cell counter module. Any visible trace of red channel pixels was considered a propidium iodide-positive cell. Data were compiled in Microsoft Excel and plotted using Graphpad Prism 8 as mean ± standard error of the mean. Exact values for statistical parameters are detailed in figure legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-Caspase-1 (p20, mouse) | AdipoGen Life Sciences | Cat# AG-20B-0042; RRID:AB_2490248 |
| Rabbit monoclonal anti-GSDMD (mouse) | Abcam | Cat# ab209845; RRID:AB_2783550 |
| Goat polyclonal anti-IL-1β (mouse) | R&D Systems | Cat# AF-401-NA; RRID:AB_416684 |
| Rabbit polyclonal anti-GSDMD (human) | Atlas Antibodies | Cat# HPA044487; RRID:AB_2678957 |
| Goat polyclonal anti-IL-1β (human) | R&D Systems | Cat# AF-201-NA; RRID:AB_354387 |
| Rabbit monoclonal anti-GSDME | Abcam | Cat# ab215191; RRID:AB_2737000 |
| Rabbit recombinant anti-Caspase-1 (p10 + p12) | Abcam | Cat# ab179515; RRID:AB_2884954 |
| Rabbit polyclonal anti-Caspase-3 | Cell Signaling Technology | Cat# 9662; RRID:AB_331439 |
| Rabbit monoclonal anti-Caspase-8 | Cell Signaling Technology | Cat# 4790; RRID:AB_10545768 |
| Rabbit polyclonal anti-Caspase-8 (cleaved) | Cell Signaling Technology | Cat# 9429; RRID:AB_2068300 |
| Mouse monoclonal anti-Caspase-9 | Cell Signaling Technology | Cat# 9508; RRID:AB_2068620 |
| Monoclonal anti-goat IgG-HRP | Santa Cruz Biotechnology | Cat# sc-2354; RRID:AB_628490 |
| Polyclonal anti-mouse IgG-HRP | Cell Signaling Technology | Cat# 7076; RRID:AB_330924 |
| Polyclonal anti-rabbit IgG-HRP | Cell Signaling Technology | Cat# 7074; RRID:AB_2099233 |
| Bacterial and virus strains | ||
| Salmonella enterica subsp. enterica serovar Typhimurium | American Type Culture Collection | Cat# 14028 |
| Chemicals, peptides, and recombinant proteins | ||
| Propidium iodide | ThermoFisher Scientific | Cat# P1304MP |
| Nigericin | MilliporeSigma | Cat# N7143 |
| Val-boroPro/Talabostat | R&D Systems | Cat# 3719 |
| Live cell imaging solution | ThermoFisher Scientific | Cat# A14291DJ |
| SuperCalf serum | GeminiBio | Cat# 100-510 |
| Fetal bovine serum | GeminiBio | Cat# 100-106 |
| Salmonella minnesota ultrapure lipopolysaccharide | InvivoGen | Cat# tlrl-smlps |
| Critical commercial assays | ||
| CyQUANT LDH Cytotoxicity Assay | ThermoFisher Scientific | Cat# C20301 |
| Human IL-1β ELISA MAX Deluxe Kit | BioLegend | Cat# 437004 |
| Experimental models: Cell lines | ||
| THP-1 human monocytic leukemia | American Type Culture Collection | Cat# TIB-202, RRID:CVCL_0006 |
| Immortalized murine bone marrow-derived macrophages overexpressing FLAG-NLRP3 and mCerulean-ASC (iBMMs) | Eicke Latz (De Nardo et al., 2018) | N/A |
| iBMM Gsdmd−/− | Rathkey et al., 2018 | N/A |
| Oligonucleotides | ||
| Human GSDMD crRNA targeting 11CCT(−): TCTCCGGACTACCCGCTCAA | This paper | N/A |
| Human GSDME crRNA targeting 653CCT(−): GTATAACTCAATGACACCGT | This paper | N/A |
| Human GSDME crRNA targeting 170CCC(−): TTCTATGAGTACATCGCCAA | This paper | N/A |
| Recombinant DNA | ||
| pLN.GSDME (third generation lentiviral vector based on pLentiCRISPR v2): EF-1α–PuroR–P2A–GSDME-WPRE | This paper | N/A |
| pLN.GSDMED270A | This paper | N/A |
Highlights.
Inflammasome activation in the absence of gasdermin D activates gasdermin E
Gasdermin E permits IL-1β release independent of cell death in a sublytic phase
Lysis depends on substantial gasdermin E oligomerization and pore formation
ACKNOWLEDGMENTS
We are grateful to Hannah Kondolf, Christopher Ryder, Tsan Xiao, and George Dubyak for insightful discussions and Jeffrey Tomalka for assistance in Cas9 ribonucleoprotein transfection. This work was supported by National Institutes of Health grants T32 GM007250 and T32 AI089474 (to B.Z.) and P01 AI 141350 and R01 GM086550 (to D.W.A.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.108998.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, Ciferri C, Dixit VM, and Dueber EC (2016). GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc. Natl. Acad. Sci. USA 113, 7858–7863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aizawa E, Karasawa T, Watanabe S, Komada T, Kimura H, Kamata R, Ito H, Hishida E, Yamada N, Kasahara T, et al. (2020). GSDME-dependent incomplete pyroptosis permits selective IL-1α release under caspase-1 inhibition. iScience 23, 101070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher D, Monteleone M, Coll RC, Chen KW, Ross CM, Teo JL, Gomez GA, Holley CL, Bierschenk D, Stacey KJ, et al. (2018). Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J. Exp. Med 215, 827–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz P, Pelegrín P, and Shao F (2020). The gasdermins, a protein family executing cell death and inflammation. Nat. Rev. Immunol 20, 143–157. [DOI] [PubMed] [Google Scholar]
- Bulek K, Zhao J, Liao Y, Rana N, Corridoni D, Antanaviciute A, Chen X, Wang H, Qian W, Miller-Little WA, et al. (2020). Epithelial-derived gasdermin D mediates nonlytic IL-1β release during experimental colitis. J. Clin. Invest 130, 4218–4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen KW, Demarco B, Heilig R, Shkarina K, Boettcher A, Farady CJ, Pelczar P, and Broz P (2019). Extrinsic and intrinsic apoptosis activate pannexin- 1 to drive NLRP 3 inflammasome assembly. EMBO J. 38, e101638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen KW, Demarco B, and Broz P (2020a). Beyond inflammasomes: emerging function of gasdermins during apoptosis and NETosis. EMBO J. 39, e103397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen KW, Demarco B, and Broz P (2020b). Pannexin-1 promotes NLRP3 activation during apoptosis but is dispensable for canonical or noncanonical inflammasome activation. Eur. J. Immunol 50, 170–177. [DOI] [PubMed] [Google Scholar]
- Chirieleison SM, Marsh RA, Kumar P, Rathkey JK, Dubyak GR, and Abbott DW (2017). Nucleotide-binding oligomerization domain (NOD) signaling defects and cell death susceptibility cannot be uncoupled in X-linked inhibitor of apoptosis (XIAP)-driven inflammatory disease. J. Biol. Chem 292, 9666–9679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croes L, Fransen E, Hylebos M, Buys K, Hermans C, Broeckx G, Peeters M, Pauwels P, Op de Beeck K, and Van Camp G (2019). Determination of the potential tumor-suppressive effects of Gsdme in a chemically induced and in a genetically modified intestinal cancer mouse model. Cancers (Basel) 11, 1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MA, Fairgrieve MR, Den Hartigh A, Yakovenko O, Duvvuri B, Lood C, Thomas WE, Fink SL, and Gale M Jr. (2019). Calpain drives pyroptotic vimentin cleavage, intermediate filament loss, and cell rupture that mediates immunostimulation. Proc. Natl. Acad. Sci. USA 116, 5061–5070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Nardo D, Kalvakolanu DV, and Latz E (2018). Immortalization of Murine Bone Marrow-Derived Macrophages. In Macrophages G Rousselet, ed. (Springer; New York: ), pp. 35–49. [DOI] [PubMed] [Google Scholar]
- Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, Sun H, Wang DC, and Shao F (2016). Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535, 111–116. [DOI] [PubMed] [Google Scholar]
- Ding Q, Zhang W, Cheng C, Mo F, Chen L, Peng G, Cai X, Wang J, Yang S, and Liu X (2020). Dioscin inhibits the growth of human osteosarcoma by inducing G2/M-phase arrest, apoptosis, and GSDME-dependent cell death in vitro and in vivo. J. Cell. Physiol 235, 2911–2924. [DOI] [PubMed] [Google Scholar]
- Evavold CL, Ruan J, Tan Y, Xia S, Wu H, and Kagan JC (2018). The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity 48, 35–44.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gai K, Okondo MC, Rao SD, Chui AJ, Ball DP, Johnson DC, and Bachovchin DA (2019). DPP8/9 inhibitors are universal activators of functional NLRP1 alleles. Cell Death Dis. 10, 587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagar JA, Powell DA, Aachoui Y, Ernst RK, and Miao EA (2013). Cytoplasmic LPS Activates Caspase-11: Implications in TLR4-Independent Endotoxic Shock. Science 341, 1250–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, Yang ZH, Zhong CQ, and Han J (2015). Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 25, 1285–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilig R, Dick MS, Sborgi L, Meunier E, Hiller S, and Broz P (2018). The Gasdermin-D pore acts as a conduit for IL-1β secretion in mice. Eur. J. Immunol 48, 584–592. [DOI] [PubMed] [Google Scholar]
- Heilig R, Dilucca M, Boucher D, Chen KW, Hancz D, Demarco B, Shkarina K, and Broz P (2020). Caspase-1 cleaves Bid to release mitochondrial SMAC and drive secondary necrosis in the absence of GSDMD. Life Sci. Alliance 3, e202000735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu JJ, Liu X, Xia S, Zhang Z, Zhang Y, Zhao J, Ruan J, Luo X, Lou X, Bai Y, et al. (2020). FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat. Immunol 21, 736–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DC, Taabazuing CY, Okondo MC, Chui AJ, Rao SD, Brown FC, Reed C, Peguero E, de Stanchina E, Kentsis A, and Bachovchin DA (2018). DPP8/DPP9 inhibitor-induced pyroptosis for treatment of acute myeloid leukemia. Nat. Med 24, 1151–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, Miyake K, Zhang J, Lee WP, Muszynski A, et al. (2013). Noncanonical Inflammasome Activation by Intracellular LPS Independent of TLR4. Science 341, 1246–1249. [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, et al. (2015). Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671. [DOI] [PubMed] [Google Scholar]
- Lee BL, Mirrashidi KM, Stowe IB, Kummerfeld SK, Watanabe C, Haley B, Cuellar TL, Reichelt M, and Kayagaki N (2018). ASC- and caspase-8-dependent apoptotic pathway diverges from the NLRC4 inflammasome in macrophages. Sci. Rep 8, 3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J, Zhou J, Xu Y, Huang X, Wang X, Huang W, and Li H (2020). Osthole inhibits ovarian carcinoma cells through LC3-mediated autophagy and GSDME-dependent pyroptosis except for apoptosis. Eur. J. Pharmacol 874, 172990. [DOI] [PubMed] [Google Scholar]
- Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, and Lieberman J (2016). Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Chen O, Wall JBJ, Zheng M, Zhou Y, Wang L, Vaseghi HR, Qian L, and Liu J (2017). Systematic comparison of 2A peptides for cloning multi-genes in a polycistronic vector. Sci. Rep 7, 2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Wang C, Yang J, Zhou B, Yang R, Ramachandran R, Abbott DW, and Xiao TS (2019). Crystal Structures of the Full-Length Murine and Human Gasdermin D Reveal Mechanisms of Autoinhibition, Lipid Binding, and Oligomerization. Immunity 51, 43–49.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Fang Y, Chen X, Wang Z, Liang X, Zhang T, Liu M, Zhou N, Lv J, Tang K, et al. (2020). Gasdermin E-mediated target cell pyroptosis by CAR T cells triggers cytokine release syndrome. Sci. Immunol 5, eaax7969. [DOI] [PubMed] [Google Scholar]
- Martinon F, Burns K, and Tschopp J (2002). The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10, 417–426. [DOI] [PubMed] [Google Scholar]
- Monteleone M, Stanley AC, Chen KW, Brown DL, Bezbradica JS, von Pein JB, Holley CL, Boucher D, Shakespear MR, Kapetanovic R, et al. (2018). Interleukin-1β Maturation Triggers Its Relocation to the Plasma Membrane for Gasdermin-D-Dependent and -Independent Secretion. Cell Rep. 24, 1425–1433. [DOI] [PubMed] [Google Scholar]
- Okondo MC, Johnson DC, Sridharan R, Go EB, Chui AJ, Wang MS, Poplawski SE, Wu W, Liu Y, Lai JH, et al. (2017). DPP8 and DPP9 inhibition induces pro-caspase-1-dependent monocyte and macrophage pyroptosis. Nat. Chem. Biol 13, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, Brooks A, Xia S, Wu H, Kelliher MA, et al. (2018). Pathogen blockade of TAK1 triggers caspase-8–dependent cleavage of gasdermin D and cell death. Science 362, 1064–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathkey JK, Zhao J, Liu Z, Chen Y, Yang J, Kondolf HC, Benson BL, Chirieleison SM, Huang AY, Dubyak GR, et al. (2018). Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci. Immunol 3, eaat2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, and Alnemri ES (2017). Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun 8, 14128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, and Alnemri ES (2019). Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat. Commun 10, 1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan J, Xia S, Liu X, Lieberman J, and Wu H (2018). Cryo-EM structure of the gasdermin A3 membrane pore. Nature 557, 62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rühl S, Shkarina K, Demarco B, Heilig R, Santos JC, and Broz P (2018). ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 362, 956–960. [DOI] [PubMed] [Google Scholar]
- Russo HM, Rathkey J, Boyd-Tressler A, Katsnelson MA, Abbott DW, and Dubyak GR (2016). Active Caspase-1 Induces Plasma Membrane Pores That Precede Pyroptotic Lysis and Are Blocked by Lanthanides. J. Immunol 197, 1353–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagulenko V, Thygesen SJ, Sester DP, Idris A, Cridland JA, Vajjhala PR, Roberts TL, Schroder K, Vince JE, Hill JM, et al. (2013). AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ. 20, 1149–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagulenko V, Vitak N, Vajjhala PR, Vince JE, and Stacey KJ (2018). Caspase-1 Is an Apical Caspase Leading to Caspase-3 Cleavage in the AIM2 Inflammasome Response, Independent of Caspase-8. J. Mol. Biol 430, 238–247. [DOI] [PubMed] [Google Scholar]
- Sarhan J, Liu BC, Muendlein HI, Li P, Nilson R, Tang AY, Rongvaux A, Bunnell SC, Shao F, Green DR, and Poltorak A (2018). Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc. Natl. Acad. Sci. USA 115, E10888–E10897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sborgi L, Rühl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, Farady CJ, Müller DJ, Broz P, and Hiller S (2016). GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 35, 1766–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider KS, Groß CJ, Dreier RF, Saller BS, Mishra R, Gorka O, Heilig R, Meunier E, Dick MS, Čikovič T, et al. (2017). The Inflammasome Drives GSDMD-Independent Secondary Pyroptosis and IL-1 Release in the Absence of Caspase-1 Protease Activity. Cell Rep. 21, 3846–3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, and Shao F (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. [DOI] [PubMed] [Google Scholar]
- Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, Kordes S, Menninger S, Eickhoff J, Nussbaumer P, Klebl B, et al. (2018). Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol 3, eaar6689. [DOI] [PubMed] [Google Scholar]
- Tixeira R, Shi B, Parkes MAF, Hodge AL, Caruso S, Hulett MD, Baxter AA, Phan TK, and Poon IKH (2018). Gasdermin E Does Not Limit Apoptotic Cell Disassembly by Promoting Early Onset of Secondary Necrosis in Jurkat T Cells and THP-1 Monocytes. Front. Immunol 9, 2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya K, Nakajima S, Hosojima S, Thi Nguyen D, Hattori T, Manh Le T, Hori O, Mahib MR, Yamaguchi Y, Miura M, et al. (2019). Caspase-1 initiates apoptosis in the absence of gasdermin D. Nat. Commun 10, 2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Camp G, Coucke P, Balemans W, van Velzen D, van de Bilt C, van Laer L, Smith RJ, Fukushima K, Padberg GW, Frants RR, et al. (1995). Localization of a gene for non-syndromic hearing loss (DFNA5) to chromosome 7p15. Hum. Mol. Genet 4, 2159–2163. [DOI] [PubMed] [Google Scholar]
- Wang Y, Gao W, Shi X, Ding J, Liu W, He H, Wang K, and Shao F (2017). Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547, 99–103. [DOI] [PubMed] [Google Scholar]
- Wang C, Yang T, Xiao J, Xu C, Alippe Y, Sun K, Kanneganti T-D, Monahan JB, Abu-Amer Y, Lieberman J, et al. (2021). Activation of GSDME compensates for GSDMD deficiency in a mouse model of NLRP3 inflammasomopathy. bioRxiv, 2021.01.06.425634. [Google Scholar]
- Yu J, Li S, Qi J, Chen Z, Wu Y, Guo J, Wang K, Sun X, and Zheng J (2019). Cleavage of GSDME by caspase-3 determines lobaplatin-induced pyroptosis in colon cancer cells. Cell Death Dis. 10, 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng CY, Li CG, Shu JX, Xu LH, Ouyang DY, Mai FY, Zeng QZ, Zhang CC, Li RM, and He XH (2019). ATP induces caspase-3/gasdermin E-mediated pyroptosis in NLRP3 pathway-blocked murine macrophages. Apoptosis 24, 703–717. [DOI] [PubMed] [Google Scholar]
- Zhang L, Chen S, Ruan J, Wu J, Tong AB, Yin Q, Li Y, David L, Lu A, Wang WL, et al. (2015). Cryo-EM structure of the activated NAIP2-NLRC4 inflammasome reveals nucleated polymerization. Science 350, 404–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate unique datasets or code.