

Abstract
The KCNQ1 voltage-gated potassium channel regulates the repolarization of cardiac cells and a plurality of point mutations in its voltage sensing domain (VSD) are associated with toxic gain or loss of pore function resulting in disease. As is the case with many disease-associated membrane proteins, there are hundreds of human variants of interest identified for KCNQ1, but a significant portion of these variants have not been characterized in relation to their functional and disease associations. Additionally, as the VSD consists of four transmembrane helices, studies into dynamic structural differences among KCNQ1 VSD variants are hindered by the current limitations and deficits in high resolution structure determination of membrane proteins. Here, we use native ion mobility – mass spectrometry and collision induced unfolding (CIU) to address the need for a high throughput compatible method for structural characterization of membrane protein variants of unknown significance using the KCNQ1 VSD as a model system. We perform CIU on wild-type and three mutant KCNQ1 VSD forms associated with toxic gain or loss of function, and show through automated feature detection and comprehensive difference analysis of the CIU datasets that the variants are clearly grouped by function and disease association. We also construct a classification scheme based on the CIU datasets which is able to differentiate the variant functional groups and robustly classify a recently characterized variant to its correct grouping. Further, we probe the stability of the KCNQ1 VSD variants when liberated from C12E8 micelles at pH 8.0 and find preliminary evidence that the R231C mutation associated with gain of pore function is destabilized relative to wild-type and loss of function variants.
Graphical Abstract
Membrane proteins maintain cellular homeostasis through regulating signaling, facilitating ion transport across biological membranes, and performing a wide array of enzymatic reactions.1 Disruption of any of these vital functions can cause cellular dyshomeostasis and disease.2 The point mutations can significantly alter membrane protein folding, trafficking, and activity, and thus many such sequence variants have been linked to disease.3 With recent advances in genome sequencing, more human membrane protein variants are being discovered, but the significance of these variants is not always readily apparent.4,5 Methods for identifying structural differences in these variants is complicated by their insolubility in aqueous solutions and difficulty in obtaining pure, high concentration samples.6–8 As there can be hundreds of variants of interest for a particular membrane protein,3,9,10 there is an active need for high-throughput compatible methods to obtain such structural information on variants of unknown significance (VUS) and classify them according to their functional and disease associations.
Mass spectrometry (MS) is readily capable of overcoming the complexity and challenges associated with studying membrane proteins structure.11 In particular, native MS has enabled insights into the ligand binding,12–14 local lipid environment,15 and protein-protein interactions of membrane proteins.16 Coupling ion mobility (IM) separation with mass spectrometry (IM-MS) produces information on the orientationally averaged size of ions which allows for the evaluation of structural changes within membrane proteins.17,18 The use of IM-MS also facilitates collision induced unfolding (CIU) experiments, in which protein ions are subjected to ramped collision energies within the instrument, causing unfolding.19 CIU experiments have been shown to be sensitive to lipid binding in membrane proteins,20,21 and classification schemes built on CIU experiments,22,23 which has differentiated disulfide bonding patterns24 and ligand binding modes.25 However, the use of CIU to differentiate classes of membrane protein variants associated with disease have yet to be completely explored.
Here, we report the first use of CIU to differentiate variants of the 18 kDa voltage sensor domain of the KCNQ1 voltage-gated potassium channel. KCNQ1 plays a key role in the tightly controlled process of action potential repolarization in cardiac cells26 and the pore opening is modulated by the tetraspan integral voltage sensor domain (VSD).27,28 Specific heritable mutations in the KCNQ1 VSD are associated with losses or gains of pore activity, where a loss of function (LOF) causes Long QT Syndrome (LQTS)29,30 and a gain of function (GOF) causes Short QT Syndrome (SQTS) or atrial fibrillation.31,32 Both conditions are associated with an increased risk for cardiac arrhythmias and sudden cardiac death.33,34 There are over 600 human variants of KCNQ1 associated with LQTS.10 However, the significance of many of these variants and the mechanism by which the mutations cause disregulation is not well understood.
In this work we use native IM-MS and CIU to differentiate KCNQ1 VSD variants, each having different disease and functional significance. We first show that wild-type (WT) KCNQ1 VSD can be liberated from detergent micelles in a manner that retains native like folding in the gas-phase. Then we explore the CIU of WT KCNQ1 alongside three mutants: R231C which is associated with SQTS and GOF,35,36 E115G which is associated with LQTS and LOF,37 and H126L which was originally a VUS associated with LQTS, and has recently been shown to exhibit LOF.38 Our feature detection and comprehensive difference analysis approach highlights changes in the unfolding pathway of WT, GOF, and LOF KCNQ1 VSD variants, where our data support the finding that H126L behaves most similar to the LOF type. We demonstrate the ability of CIU classification to differentiate WT, GOF, and LOF KCNQ1 VSD variants and show that the H126L fingerprints classify as LOF. Further, we quantify the relative stability of the KCNQ1 VSD variants through CIU-based stability measurements and find preliminary evidence that the R231C mutation is destabilized relative to WT, which we discuss in the context of cellular data showing increased expression and cellular trafficking. Lastly, we project the utility of CIU methods in screening large numbers of VUS and discuss the future of such technologies for advancing our understanding of membrane protein function.
Methods
Membrane Protein Sample Preparation.
WT KCNQ1 and the mutant variants H126L, E115G, and R231C were expressed in E. Coli and purified using protocols described elsewhere.38 Octaethylene Glycol Monododecyl Ether (C12E8) was purchased from Anatrace and ammonium acetates was purchased from Sigma Aldrich (St. Louis, MO). For mass spectrometry experiments, 150 μM KNQ1 in buffer of 50 mM 2-(N-morpholino)ethanesulfonic acid(MES), 0.5 mM ethylenediaminetetraacetic acid (EDTA), 2 mM tris(2-carboxyethyl)phosphine (TCEP), and 56 μM n-dodecyl-β-d-maltoside (DDM) at pH 5.5 was simultaneously buffer and detergent-exchanged to 200 mM ammonium acetate buffer, pH 8.0, containing 0.02% C12E8 (~4 × CMC) using 10 kDa Amicon Ultra-0.5 centrifugal filter units (MilliporeSigma, Burlington, MA). The final protein concentration used in mass spectrometry experiments was approximately 25 μM.
IM-MS and CIU Experiments.
All IM-MS and CIU data was collected using a Synapt G2 HDMS IM-Q-ToF mass spectrometer (Waters, Milford, MA), with a direct infusion nESI source set to positive ion mode. Our instrument settings were tuned to generate intact protein ions while completely dissociating detergent adducts prior to the IM separator, including appropriately tuned settings for the source temperature (30° C), source gas flow (50 mL/min), and the sampling cone (120 V). Trapping cell, helium cell and IM cell gas flow rates were 6 mL/min, 200 mL/min, and 60 mL/min, respectively. Trapping cell wave velocity and height were 116 m/s and 0.1 V. IMS wave velocity and height were 250 m/s and 15 V. Transfer cell wave velocity and height were 300 m/s and 10 V, with an accelerating potential of 70 V used to dissociate empty micelles and salt clusters. Collision cross section analysis was performed using IMSCal-19v4, a program written in C. The theoretical collision cross section of the KCNQ1 VSD was computed using PDB entry 6MIE and IMPACT.39–41 All CIU analyses were performed by increasing the trap collision voltage in a stepwise manner 5 – 50 V in 5 V increments, as severe signal loss was observed at voltages above 50 V for mutant KCNQ1. CIU data from the 8+ charge state of KCNQ1 was extracted into a text-based format using TWIMExtract42, then processed and analyzed using CIUSuite 2.1.24 Data processing included three rounds of 2D Savitzky-Golay smoothing with a window of five bins and interpolation of the collision voltage axis by a factor of four.
Protein stability values, which reflect the trap collision voltage at which 50% of the gas phase protein has transitioned to an unfolded state, were extracted as described previously and are referred to here as CIU50 values.21 For mutant KCNQ1, excess surfactant related noise was noted in the lower drift time region from 35 – 50 V. This noise was removed from the text-based files manually to eliminate any biased contributions to RMSD analysis, as shown in Figure S1. To ensure the noise removal did not alter the reported protein unfolding trajectory, CIU50 analysis was performed before and after noise removal, and reported CIU50 values were identical. Classification of these denoised data was performed using the “All Data” mode of CIUSuite 2.1, with automatic feature selection. The algorithms that enable our CIU classification analysis are described in detail elsewhere.21,22,24
Results and Discussion
IM-MS of KCNQ1 VSD.
Sequence variants associated with toxic LOF or GOF occur throughout the sequence of the KCNQ1 VSD, as shown in Figure 1A.38 For our assay, we chose mutations from disparate locations in the protein sequence, with E115G in soluble helix S0, H126L in membrane spanning helix S1, and R231C in membrane spanning helix S4. The KCNQ1 potassium channel functions as a tetrameric complex, where four pore domains interact to form a functional channel.27 The tetraspanning VSD of KCNQ1 is connected to the pore domain by a flexible linker between helices S4 and S5, as well as a 99 residue soluble domain before helix S1. The VSD is positioned at the periphery of the pore, and as such is not thought to be directly involved in pore oligomerization.27
Figure 1.
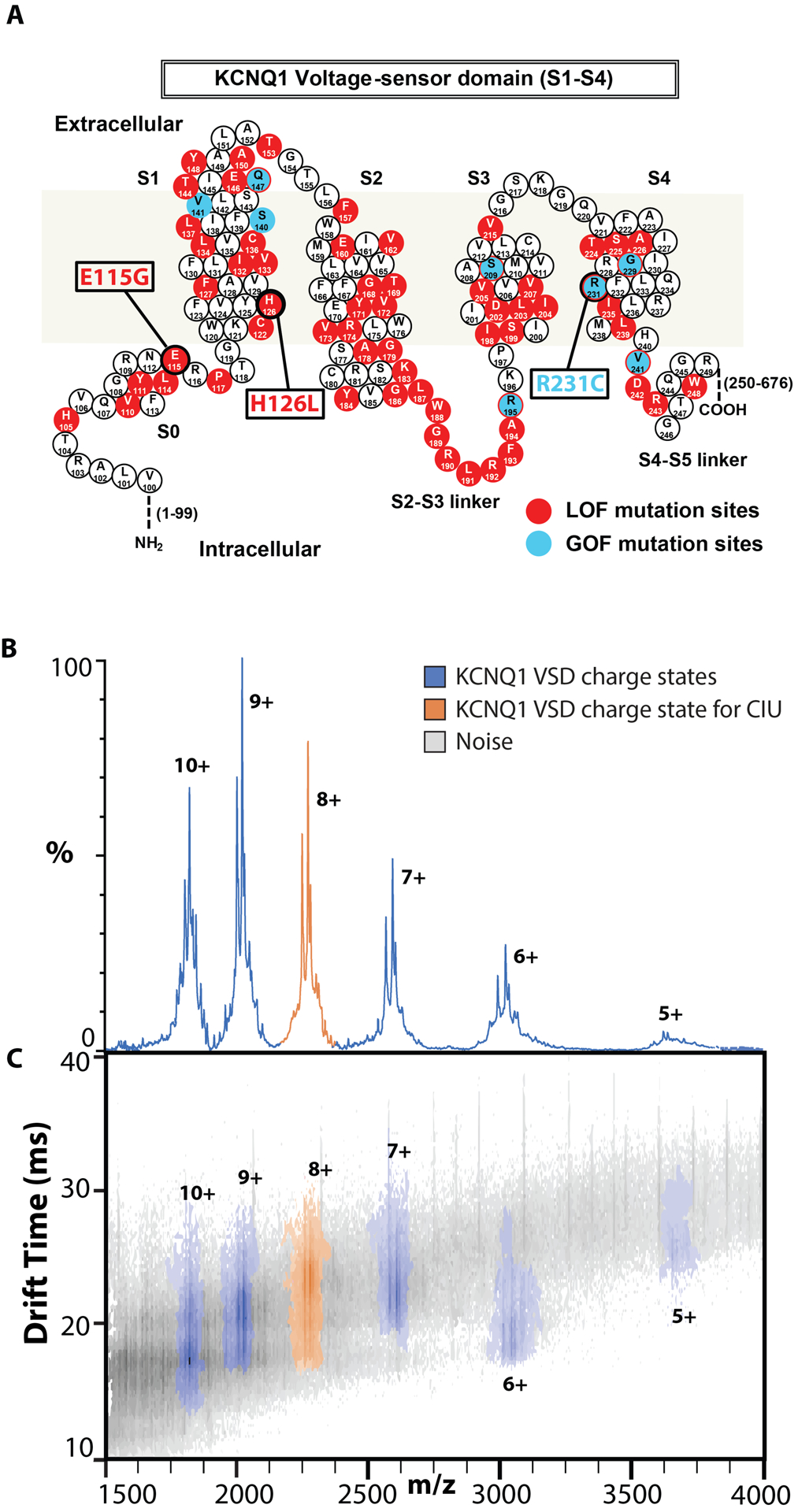
Native IM-MS of the KCNQ1 VSD. A. Sequence and structure of the KCNQ1 VSD with residues known to mutate and result in disease phenotypes associated with LOF (red) or GOF (blue). Variants E115G, H126L, and R231C are studied in this work. B,C. Representative mass spectra and IM-MS data for WT KCNQ1 at 80 V trap collision voltage. One distribution of charge states, 5–10+, is observed for all variants corresponding to KCNQ1 VSD monomers (Figure S2). The charge state envelope and CCS analysis indicate native like folding, and the 8+ charge state (orange) was chosen for further analysis.
Figure 1B shows an example native IM-MS dataset for WT KCNQ1, where a range of signals can be detected after successful dissociation of the detergent from the associated membrane protein of interest. MS analysis and charge state assignment of these signals reveal an ion population with an intact mass of 18208 ± 44 Da, which agrees well with the expected sequence mass of a KCNQ1 VSD monomer, 18158 Da. The range of charge states we observe for KCNQ1, 5–10+ indicate native like structure for the gas phase protein (Figure 1C, S2). Collision cross section analysis of the 8+ monomeric WT KCNQ1 VSD indicated slightly larger gas phase structures relative to the theoretical cross section computed using a recently published structure for the KCNQ1 VSD intermediate state (2456Å2 and 2039Å2, respectively).41 Notably, for all four sequence variants studied here we observe monomodal IM distributions at low collision energies for the KCNQ1 VSD and find no evidence of oligomeric species (Figure S2). As has been documented for other IM-MS experiments of membrane proteins,21,43–46 we observe noise signals related to detergent and salt clusters that persist despite sample and instrumental optimization (Figure 1B). These noise signals were more intense in the IM-MS datasets recorded for the mutant protein forms, perhaps due to the lower protein concentrations used in these experiments in comparison to those involving WT (Figure S1). The 8+ charge state of the KCNQ1 VSD (orange) was chosen for subsequent CIU experiments due to its large S/N and relatively low charge state.40
Differences in CIU features correlate to KCNQ1 VSD variant function.
CIU analysis often focuses on defining regions of stability, or features, within the recorded data. CIU feature analysis has been used to characterize domain level unfolding47 and noncovalent interactions48 within proteins, and we employed such an approach to interrogate the CIU data collected for the four KCNQ1 VSD sequence variants discussed above. CIU fingerprints of the KCNQ1 VSD 8+ charge state were collected in triplicate using a collision voltage range of 5–50 V. While the interference from noise signals was minimal for WT KCNQ1 VSD, each variant dataset contained noise appearing primarily at larger collision voltage (CV) values and at drift times shorter than those occupied by the protein ion signals tracked in our experiments (Figure S1). These noise signals did not alter the CIU features detected in our fingerprints; however, to eliminate any bias in our workflow, detergent noise was uniformly removed as described in the methods section for all datasets interrogated in this study. Figure 2 shows the de-noised CIU fingerprints (left) and detected features (right) for each KCNQ1 variant. For WT KCNQ1 VSD fingerprints, three features are observed with median drift times of 16.4 ± 0.3 ms, 19.9 ± 0.0 ms, and 21.8 ± 0.0 ms. Similarly, the GOF variant R231C fingerprints also exhibit three features with median drift times comparable to those observed for WT at 17.0 ± 0.3 ms, 20.0 ± 0.3 ms, and 22.7 ± 0.2 ms respectively. The two LOF variants, E115G and H126L, show two features each at drift times of 17 ± 0.0 ms and 21.0 ± 0.0 ms, and 16.4 ± 0.3 ms and 20.4 ± 0.5 ms, respectively.
Figure 2.
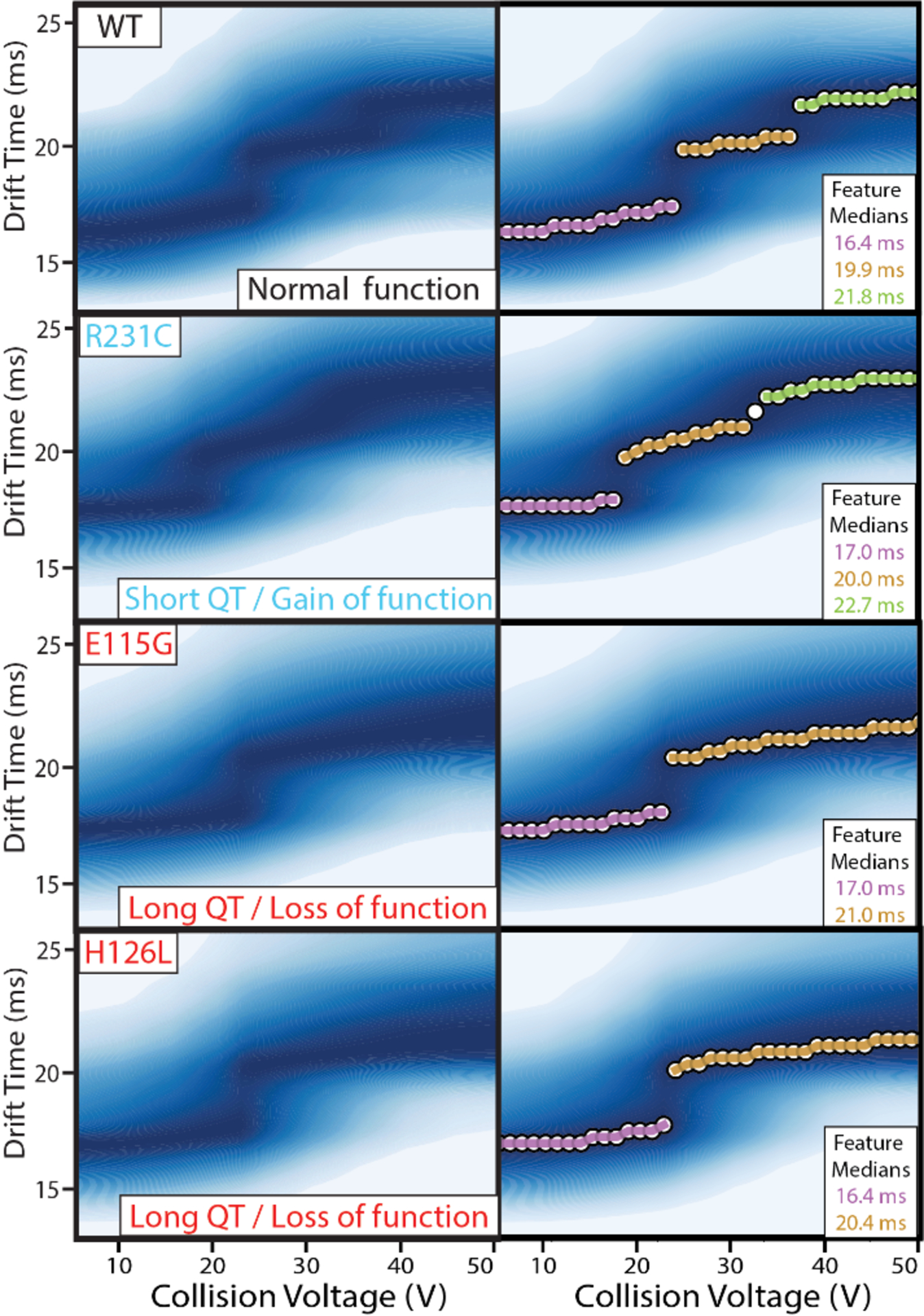
CIU fingerprints of KCNQ1 VSD variants. (Top) Fingerprints were collected for the 8+ charge state in triplicate for WT, R231C, E115G, and H126L variants, from 5–50 V, and then denoised and averaged to produce the images shown here. (Bottom) Automated feature detection of the fingerprints finds three discrete features between 5–50 V for WT and R231C, and two discrete features for E115G and H126L. The similarity in starting drift times of the four fingerprints indicate all forms begin at similar orientationally averaged sizes.
The most compact KCNQ1 CIU feature occurs at similar starting drift times for each variant studied here, which indicates that there are no significant differences in the orientationally averaged size of the gas phase structures at low collision energies. However, differences were detected in the overall number of features occurring between 5–50 V across the mutants we studied, where we specifically detect two CIU features over the voltage range for E115G and H126L, and three features for WT and R231C. The third feature for WT and R231C indicates that those gas-phase proteins experience an additional unfolded state relative to E115G and H126L across the 5 – 50 V range. The similarity observed in the CIU data recorded for E115G and H126L is further apparent in the similar range of voltages values over which their features persist, as seen in Figure 2. These results mimic previous cellular assays which classify these mutants together in terms of their LOF severity.38
Quantifying differences in CIU fingerprints of KCNQ1 VSD variants through comprehensive analysis (RMSD).
To further investigate the relative differences between KCNQ1 VSD variants, we performed comprehensive difference analyses using a root mean squared deviation (RMSD) approach. Such analyses have been used previously to quantify differences detected in CIU data recorded for antibodies based on glycosylation state.49 Figure 3A shows a difference plot for the averaged CIU fingerprints recorded for E115G and H126L KCNQ1 mutants. We find an overall RMSD across these CIU fingerprints to be 6.6%, which is similar to RSMD values encountered for comparisons between KCNQ1 technical replicates alone (a range from 6.4% to 9.5%). In contrast, Figure 3B shows a similar CIU difference plot generated for averaged H126L and R231C fingerprints. In this comparison, the regions of differences are more pronounced, with stronger R231C signal apparent at longer drift times relative to H126L. Unsurprisingly, we find a larger overall RMSD for this comparison, resulting a value almost three times that for the analysis shown in Figure 3A, at 18.6%.
Figure 3.
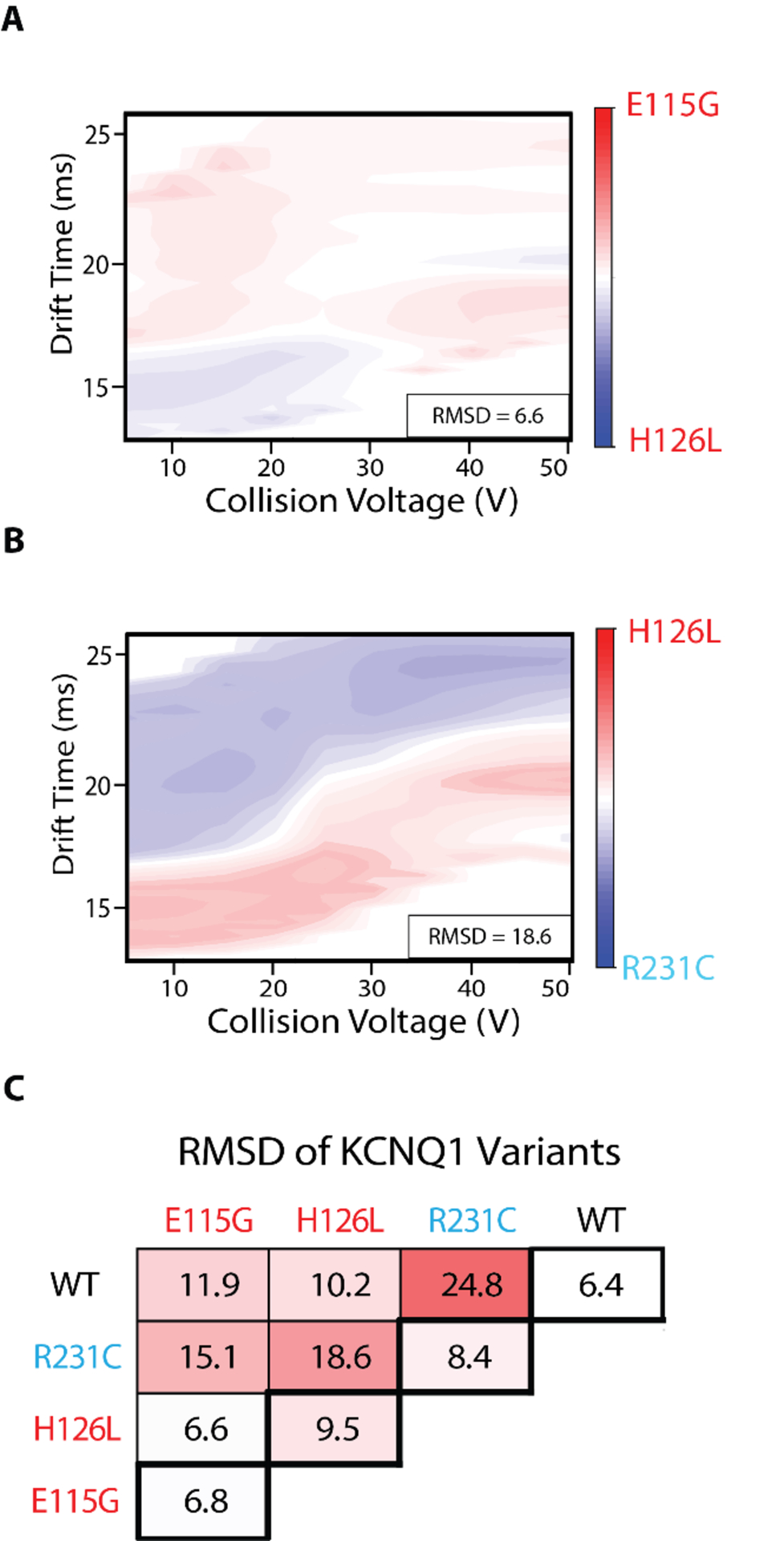
Comprehensive difference analysis of KCNQ1 VSD variants CIU fingerprints, N = 3 for each variant. A, B. Example difference plots of a low RMSD comparison (E115G and H126L, 6.6%) and high RMSD comparison (H126L and R231C, 18.6%). C. Pairwise comparisons of WT, E115G, H126L, and R231C. RMSD baseline values are shown in the thick bordered boxes. Differences of 2–3x above the baseline are considered significant. All difference plots are shown in Figure S3.
A comprehensive comparative analysis of our KCNQ1 variant CIU datasets reveals those mutants that are most and least similar to one another (Figure 3). In this cross-comparison, we see differences 2–3 times greater than the baseline RMSD values computed through the analysis of technical replicates of identical samples (thick bordered boxes, values range from 6.4% for WT to 9.5% for H126L), as indicated by the color scale in Figure 3C. Specifically, we observe no significant differences for the CIU data recorded for H126L and E115G, while the RMSD recorded for the comparisons of H126L and R231C CIU data is significant.
Interestingly, the datasets exhibiting the largest RMSD value in our cross-comparison CIU analyses are WT and R231C, producing a value of 24.8%, despite the similarities observed between these data in Figure 2 (also see Figure S3). A careful analysis of our comparative CIU data indicates that R231C is destabilized relative to WT and other KCNQ1 VSD variants. Overall, the comprehensive difference analysis shown in Figure 3C indicates significant differences between R231C and all other variants, as well as similarities between E115G and H126L. These results align with the fact that R231C is the lone GOF mutant studied here, as well as prior trafficking and cellular expression data recorded for this variant.38
CIU classification of phenotypic function.
As shown in Figure 1A, there are many possible disease-associated variants of the KCNQ1 VSD. Additionally, some former VUS forms of KCNQ1 were found to exhibit WT-like function, indicating they may not contribute to disease.38 To probe the ability of CIU to differentiate these phenotypes, we endeavored to build and test a three-way CIU classification scheme with the KCNQ1 variants studied here, grouped into the following classes: WT, gain of function (GOF), and loss of function (LOF). We started with at least three denoised replicates each of WT, E115G, and R231C KCNQ1 VSD used in the training dataset to represent each functional phenotype: WT, LOF, and GOF, respectively (Figure 4A). Our efforts identified the most differentiating region within the datasets as those between 10–20 V, and cross validation revealed that classifiers using only the 15 V datasets produced the greatest assignment accuracy (0.81 AUC-ROC, Figure 4A inset).
Figure 4.
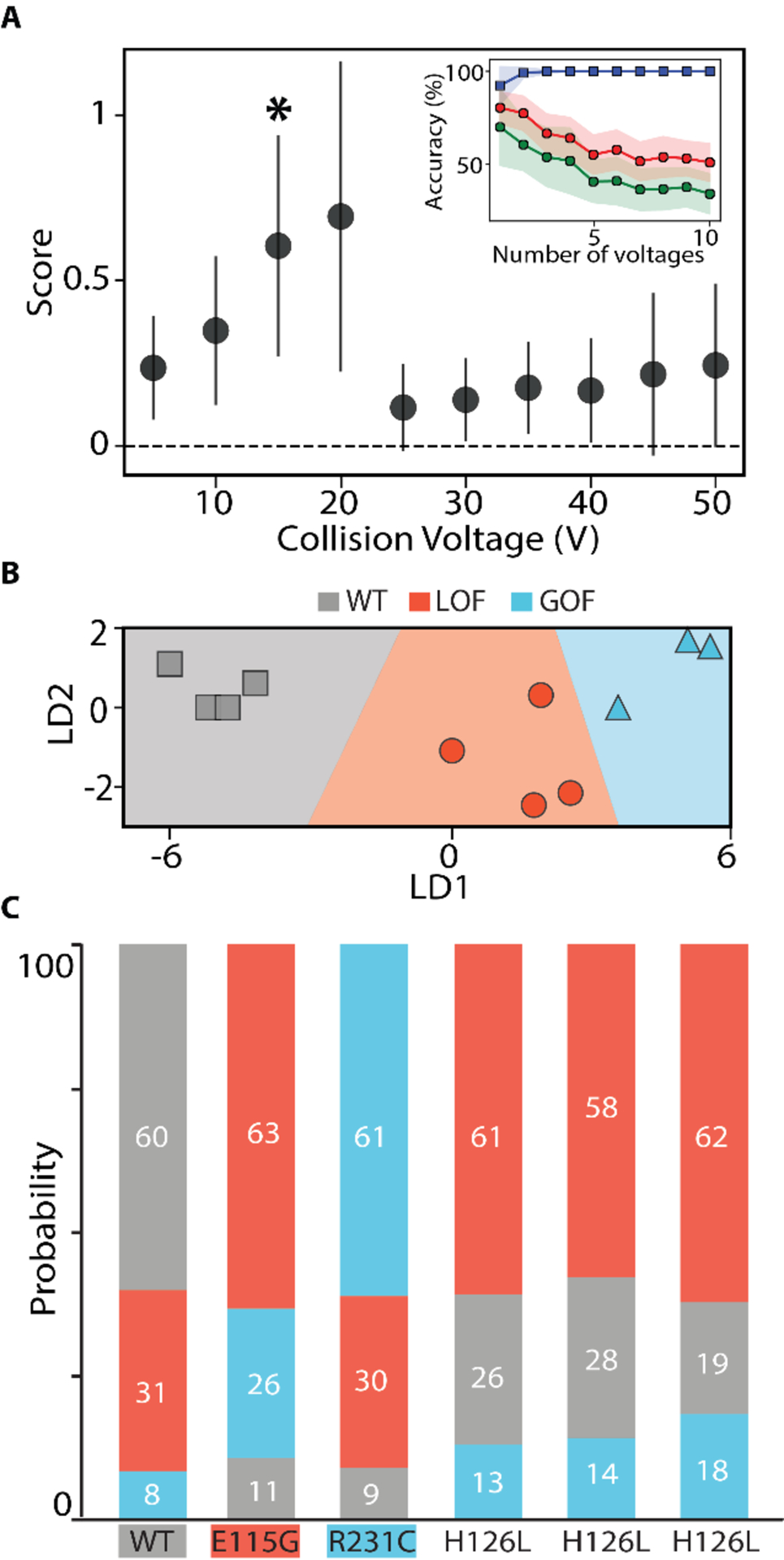
CIU based classification of KCNQ1 VSD functional variants A. Each voltage is scored on its ability to differentiate the three classes: WT, LOF, and GOF, using at least three WT, E115G, and R231C replicates. All voltages were used in a ‘leave one out’ cross-validation test, shown in the inset plot, where we tracked the accuracy achieved with the training data (blue),non-training data (green), and the area under the ROC curve (red) as a function of the number of voltages included in the classification scheme. These tests indicated one voltage (*) was best for classification B. Using the voltage indicated in A, the training data set is plotted in linear discriminant and shows clear separation of the data into the three classes. C. The probability of assignment for replicates not part of the training data set is displayed in a bar chart. Each replicate is correctly assigned, including replicates of the H126L variant for which no example was included in the training set.
A classification scheme was built using the chosen 15 V data and the results plotted linear discriminant space in Figure 4B, where gray data points indicate the WT phenotype, red indicate the LOF phenotype, and blue indicate the GOF phenotype. To test the performance of our classification scheme, we input CIU replicates to our classifier that were not part of the training dataset as unknowns. The classification scheme was then used to analyze these unknowns, and the resulting probabilities associated with assigning each of these datasets to one of the three KCNQ1 classes defined above is shown in Figure 4C. Data for WT, E115G, and R231C all classify as expected, producing an assignment probability of at least 60% for the correct class. In order to further test our classifier, we employed it to analyze CIU data collected for the H126L variant, none of which was in our training data. Our H126L CIU data classifies, as expected, as an LOF KCNQ1 variant,38 with a total class assignment probability of 60 ± 2%. As such, our results both support prior assignment of H126L as an LOF KCNQ1 variant, and demonstrate the ability of CIU fingerprints to classify KCNQ1 VSD mutants based on their associated phenotypic function.
CIU reveals evidence of mutant destabilization.
To further probe and quantify differences among KNCQ1 VSD variants relative to WT, we performed CIU50 analysis in order to quantify stability differences among the mutants studied here. Due to the different number of CIU features detected across KCNQ1 VSD variants, WT and R231C fingerprints were fit with two sigmoidal curves and E115G and H126L fingerprints were fit with one sigmoidal curve, as shown in Figure 5A, during our analysis. As such, all comparisons here will focus on the first CIU50 value recorded for WT and R231C variants.
Figure 5.
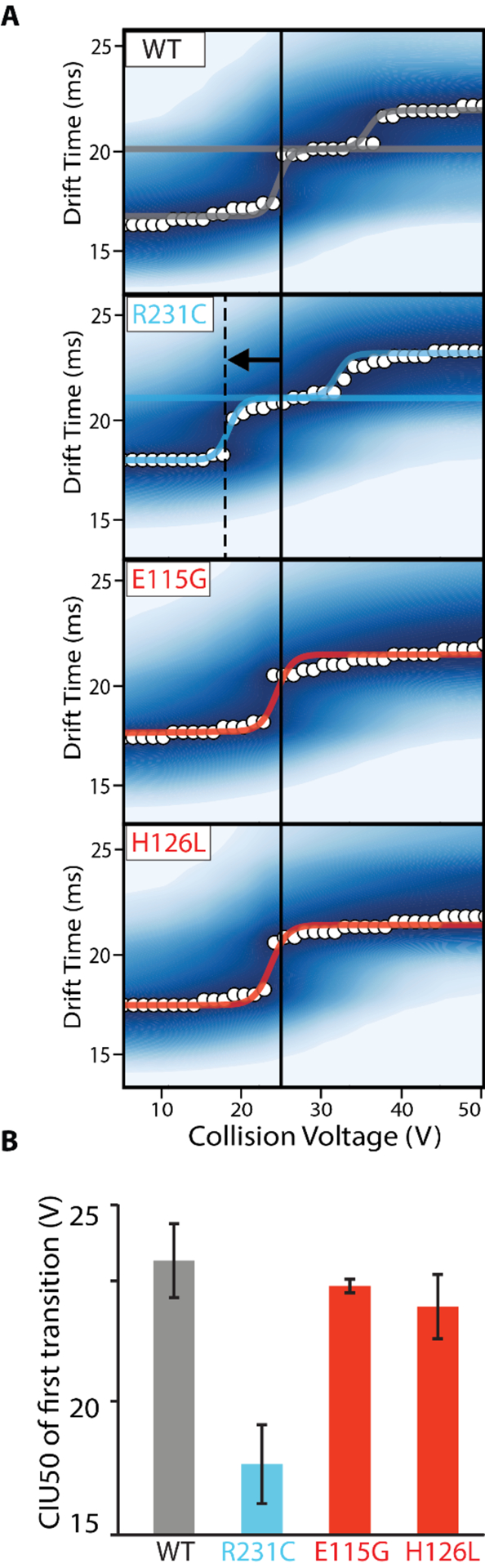
CIU50 stability analysis of KCNQ1 variants. A. Sigmoidal curves are fit between identified features to describe the transition, where WT and R231C have two transitions and E115G and H126L have one transition. B. A bar chart of average CIU50 values extracted from the inflection point determined for each first transition detected, N = 3. Gray is WT, blue is GOF, and red is LOF.
Note that this CIU50 value represents the transition between the first two features in all variants and these features possess drift time centroids within error of each other (Figure 2). We therefore consider this transition to be equivalent across all variants and suitable for a meaningful comparison. Figure 5B summarizes the average CIU50 values extracted for the first CIU transitions shown in Figure 5A. We record the stability of the WT as 25.8 ± 1.5 V, which is within error of the stability values we observe for the two LOF mutants E115G and H126L at 24.8 ± 0.3 V and 24.0 ± 1.3 V respectively. The GOF mutant, R231C, exhibits CIU50 values for the first transition that occur at significantly lower energies relative to the other three variants, producing a value of 17.7 ± 1.6 V. In addition, the CIU50 we record for the second CIU transition for R231C also occurs at lower average voltages than WT, but the experimental error we compute for these values renders the difference insignificant , with R231C producing a value of 30.0 ± 4.9 V and WT having a CIU50 of 37.0 ± 3.3 V.
Overall, we take these CIU50 data to support the observations made in Figures 2 and 3, in that the GOF mutant R231C produces CIU data that is significantly different from that observed from either WT or LOF KCNQ1 mutants, primarily driven by the destabilization of R231C. This data is interesting to consider in the context of cellular assays which demonstrate that full-length R231C KCNQ1 is a super-trafficker, and is highly-expressed relative to the other variants studied here.50 As protein mistrafficking has been linked to destablizaiton,3 our R231C results may appear initially surprising. However, it is important to note that the construct used in our MS assays only includes the VSD of KCNQ1, whereas cellular trafficking assays involve the entire KCNQ1 sequence, which is tetrameric in its native state. In addition, our in vivo data does not necessarily indicate that the overall folding stability of R231C is higher than that of WT. Alternative mechanisms that integrate both our CIU and in vivo data for R231C include the possibility that the intrinsic trafficking efficiency of the channel comprised of fully folded voltage sensors populating the inactive state is much higher than for a channel populating an intermediate state. In addition, the protein-protein interactions formed with cellular trafficking machinery may not depend on the VSD region of KCNQ1. Overall, the R231C-induced destabilization of the KCNQ1 may provide unexpected insights into the molecular mechanisms that underlie KCNQ1 trafficking. Future native IM-MS assays of full length KCNQ1 may further reveal how the destabilization of R231C KCNQ1 results in increased trafficking and GOF.
Conclusions
CIU is amenable to the characterization of membrane proteins and many past studies have focused mainly on its ability to characterize lipid binding behavior in such systems.21,48,51 Here we describe a workflow for using CIU to classify membrane protein variants according to their associated functional role within a given phenotype using the KCNQ1 VSD as a model system. We first performed comprehensive difference analysis on three types of KCNQ1 VSD variants: WT, GOF, and LOF, and found strong evidence to support clustering our CIU data along the known functional consequences associated with each variant analyzed. We then used an established machine learning approach to build a classification scheme using CIU data recorded for example WT, GOF, and LOF variants, and found that this approach was reliably able to group further CIU data into classes based on the above phenotypes. Lastly, we explored the stability differences associated with these variants using CIU50 analysis and found evidence that the GOF R231C mutant is destabilized relative to WT and LOF variants for the KCNQ1 VSD.
Native IM-MS is a method compatible with high throughput analyses, as is our classification workflow. Using the methods presented here, we estimate that the native IM-MS data collection needed to classify a VUS with our scheme could be performed in triplicate within 6 minutes. Clearly, our KCNQ1 classification scheme would be strengthened by the addition of more known variants for each class prior to testing such high throughput analyses, where we envision that only the most differentiating regions of CIU space need be collected for each unknown interrogated. We also project that this same type of CIU classification scheme could be deployed to detect the five LOF classes described previously for KCNQ1.38 Overall, we anticipate that the workflow presented here could be expanded to broadly classify KCNQ1 VUSs, or be expanded to accommodate a wide range of membrane protein systems possessing variants that lack functional annotation.
Supplementary Material
ACKNOWLEDGMENT
Membrane protein research in the Ruotolo lab is supported by the National Institute of Health under R01 GM105942. KCNQ1 research at the Sanders lab is supported by the National Institute of Health under grant R01 HL122010.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
The authors declare no competing financial interest.
REFERENCES
- (1).Almén MS; Nordström KJV; Fredriksson R; Schiöth HB Mapping the Human Membrane Proteome: A Majority of the Human Membrane Proteins Can Be Classified According to Function and Evolutionary Origin. BMC Biol. 2009, 7, 50. 10.1186/1741-7007-7-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Escribá PV; González-Ros JM; Goñi FM; Kinnunen PKJ; Vigh L; Sánchez-Magraner L; Fernández AM; Busquets X; Horváth I; Barceló-Coblijn G Membranes: A Meeting Point for Lipids, Proteins and Therapies: Translational Medicine. J. Cell. Mol. Med 2008, 12 (3), 829–875. 10.1111/j.1582-4934.2008.00281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Marinko JT; Huang H; Penn WD; Capra JA; Schlebach JP; Sanders CR Folding and Misfolding of Human Membrane Proteins in Health and Disease: From Single Molecules to Cellular Proteostasis. 2019. 10.1021/acs.chemrev.8b00532. [DOI] [PMC free article] [PubMed]
- (4).Ackerman MJ Genetic Purgatory and the Cardiac Channelopathies: Exposing the Variants of Uncertain/Unknown Significance Issue. Hear. Rhythm 2015, 12 (11), 2325–2331. 10.1016/j.hrthm.2015.07.002. [DOI] [PubMed] [Google Scholar]
- (5).Oulas A; Minadakis G; Zachariou M; Spyrou GM Selecting Variants of Unknown Significance through Network-Based Gene-Association Significantly Improves Risk Prediction for Disease-Control Cohorts. Sci. Rep 2019, 9 (1), 1–15. 10.1038/s41598-019-39796-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Moraes I; Evans G; Sanchez-Weatherby J; Newstead S; Stewart PDS Membrane Protein Structure Determination - The next Generation. Biochimica et Biophysica Acta - Biomembranes. Elsevier; January 1, 2014, pp 78–87. 10.1016/j.bbamem.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Weekes MP; Antrobus R; Lill JR; Duncan LM; Hör S; Lehner PJ Comparative Analysis of Techniques to Purify Plasma Membrane Proteins; 2010; Vol. 21. [PMC free article] [PubMed] [Google Scholar]
- (8).Lacapère JJ; Pebay-Peyroula E; Neumann JM; Etchebest C Determining Membrane Protein Structures: Still a Challenge! Trends Biochem. Sci 2007, 32 (6), 259–270. 10.1016/j.tibs.2007.04.001. [DOI] [PubMed] [Google Scholar]
- (9).Moeller HB; Rittig S; Fenton RA Nephrogenic Diabetes Insipidus: Essential Insights into the Molecular Background and Potential Therapies for Treatment. Endocrine Reviews. Oxford Academic April 1, 2013, pp 278–301. 10.1210/er.2012-1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Zhang T; Moss A; Cong P; Pan M; Chang B; Zheng L; Fang Q; Zareba W; Robinson J; Lin C; Li Z; Wei J; Zeng Q; Qi M LQTS Gene LOVD Database. Hum. Mutat 2010, 31 (11), E1801–E1810. 10.1002/humu.21341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Zhan LP; Liu CZ; Nie ZX Mass Spectrometry of Membrane Proteins. In Membrane Biophysics: New Insights and Methods; Springer; Singapore, 2017; pp 285–317. 10.1007/978-981-10-6823-2_10. [DOI] [Google Scholar]
- (12).Gault J; Liko I; Landreh M; Shutin D; Bolla JR; Jefferies D; Agasid M; Yen H-Y; Ladds MJGW; Lane DP; Khalid S; Mullen C; Remes P; Huguet R; McAlister G; Goodwin M; Viner R; Syka J; Robinson CV Combining ‘Native’ with ‘Omics’ Based Mass Spectrometry to Identify Endogenous Ligands Bound to Membrane Proteins. Nat. Methods 2020, 17, 505–508. 10.1038/s41592-020-0821-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Reddy Bolla J; Agasid MT; Mehmood S; Robinson C V; ARjatscls, R. Membrane Protein-Lipid Interactions Probed Using Mass Spectrometry. Annu. Rev. Biochem 2019, 88, 85–111. 10.1146/annurev-biochem-013118. [DOI] [PubMed] [Google Scholar]
- (14).Landreh M; Marty MT; Gault J; Robinson CV A Sliding Selectivity Scale for Lipid Binding to Membrane Proteins. Curr. Opin. Struct. Biol 2016, 39, 54–60. 10.1016/j.sbi.2016.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Marty MT; Hoi KK; Gault J; Robinson CV Probing the Lipid Annular Belt by Gas-Phase Dissociation of Membrane Proteins in Nanodiscs. Angew. Chemie - Int. Ed 2016, 55 (2), 550–554. 10.1002/anie.201508289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Keener JE; Zambrano DE; Zhang G; Zak CK; Reid DJ; Deodhar BS; Pemberton JE; Prell JS; Marty MT Chemical Additives Enable Native Mass Spectrometry Measurement of Membrane Protein Oligomeric State within Intact Nanodiscs. J. Am. Chem. Soc 2019, 141 (2), 1054–1061. 10.1021/jacs.8b11529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Konijnenberg A; Yilmaz D; Ingólfsson HI; Dimitrova A; Marrink SJ; Li Z; Vénien-Bryan C; Sobott F; Koçer A Global Structural Changes of an Ion Channel during Its Gating Are Followed by Ion Mobility Mass Spectrometry. Proc. Natl. Acad. Sci 2014, 111 (48), 17170–17175. 10.1073/pnas.1413118111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Henrich E; Peetz O; Hein C; Laguerre A; Hoffmann B; Hoffmann J; Dötsch V; Bernhard F; Morgner N Analyzing Native Membrane Protein Assembly in Nanodiscs by Combined Non-Covalent Mass Spectrometry and Synthetic Biology. Elife 2017, 6, 1–19. 10.7554/eLife.20954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Dixit SM; Polasky DA; Ruotolo BT Collision Induced Unfolding of Isolated Proteins in the Gas Phase: Past, Present, and Future. Current Opinion in Chemical Biology. Elsevier Ltd; February 1, 2018, pp 93–100. 10.1016/j.cbpa.2017.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Liu Y; Cong X; Liu W; Laganowsky A Characterization of Membrane Protein–Lipid Interactions by Mass Spectrometry Ion Mobility Mass Spectrometry. J. Am. Soc. Mass Spectrom 2017, 28 (4), 579–586. 10.1007/s13361-016-1555-1. [DOI] [PubMed] [Google Scholar]
- (21).Fantin SM; Parson KF; Niu S; Liu J; Polasky DA; Dixit SM; Ferguson-Miller SM; Ruotolo BT Collision Induced Unfolding Classifies Ligands Bound to the Integral Membrane Translocator Protein. Anal. Chem 2019, 91, 44. 10.1021/acs.analchem.9b03208. [DOI] [PubMed] [Google Scholar]
- (22).Polasky DA; Dixit SM; Fantin SM; Ruotolo BT CIUSuite 2 : Next-Generation Software for the Analysis of Gas-Phase Protein Unfolding Data CIUSuite 2 : Next-Generation Software for the Analysis of Gas-. Anal. Chem 2019, Just Accepted Manuscript. 10.1021/acs.analchem.8b05762. [DOI] [PubMed] [Google Scholar]
- (23).Polasky DA; Dixit SM; Vallejo DD; Kulju KD; Ruotolo BT An Algorithm for Building Multi-State Classifiers Based on Collision-Induced Unfolding Data. Anal. Chem 2019, 91 (16), 10407–10412. 10.1021/acs.analchem.9b02650. [DOI] [PubMed] [Google Scholar]
- (24).Vallejo DD; Polasky DA; Kurulugama RT; Eschweiler JD; Fjeldsted JC; Ruotolo BT A Modified Drift Tube Ion Mobility-Mass Spectrometer for Charge-Multiplexed Collision-Induced Unfolding. 2019. 10.1021/acs.analchem.9b00427. [DOI] [PubMed]
- (25).Rabuck-Gibbons JN; Keating JE; Ruotolo BT Collision Induced Unfolding and Dissociation Differentiates ATP-Competitive from Allosteric Protein Tyrosine Kinase Inhibitors. Int. J. Mass Spectrom 2018, 427, 151–156. 10.1016/j.ijms.2017.12.002. [DOI] [Google Scholar]
- (26).Abbott GW KCNE1 and KCNE3: The Yin and Yang of Voltage-Gated K+ Channel Regulation. Gene. Elsevier B.V. January 15, 2016, pp 1–13. 10.1016/j.gene.2015.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Cui J Voltage-Dependent Gating: Novel Insights from KCNQ1 Channels. Biophysical Journal. Biophysical Society; January 5, 2016, pp 14–25. 10.1016/j.bpj.2015.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Brewer KR; Kuenze G; Vanoye CG; George AL; Meiler J; Sanders CR Structures Illuminate Cardiac Ion Channel Functions in Health and in Long QT Syndrome. Frontiers in Pharmacology. Frontiers Media S.A. May 4, 2020, p 550. 10.3389/fphar.2020.00550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Wu J; Ding WG; Horie M Molecular Pathogenesis of Long QT Syndrome Type 1. Journal of Arrhythmia. Elsevier B.V. October 1, 2016, pp 381–388. 10.1016/j.joa.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Wu W; Sanguinetti MC Molecular Basis of Cardiac Delayed Rectifier Potassium Channel Function and Pharmacology. Cardiac Electrophysiology Clinics. W.B. Saunders; June 1, 2016, pp 275–284. 10.1016/j.ccep.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Campuzano O; Fernandez-Falgueras A; Lemus X; Sarquella-Brugada G; Cesar S; Coll M; Mates J; Arbelo E; Jordà P; Perez-Serra A; del Olmo B; Ferrer-Costa C; Iglesias A; Fiol V; Puigmulé M; Lopez L; Pico F; Brugada J; Brugada R Short QT Syndrome: A Comprehensive Genetic Interpretation and Clinical Translation of Rare Variants. J. Clin. Med 2019, 8 (7), 1035. 10.3390/jcm8071035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Zhang H; Kharche S; Holden AV; Hancox JC Repolarisation and Vulnerability to Re-Entry in the Human Heart with Short QT Syndrome Arising from KCNQ1 Mutation-A Simulation Study. Progress in Biophysics and Molecular Biology. Pergamon: January 1, 2008, pp 112–131. 10.1016/j.pbiomolbio.2007.07.020. [DOI] [PubMed] [Google Scholar]
- (33).Schwartz PJ; Crotti L; Insolia R Long-QT Syndrome. Circ. Arrhythmia Electrophysiol 2012, 5 (4), 868–877. 10.1161/CIRCEP.111.962019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Gussak I; Brugada P; Brugada J; Wright RS; Kopecky SL; Chaitman BR; Bjerregaard P Idiopathic Short QT Interval:A New Clinical Syndrome? Cardiology 2000, 94 (2), 99–102. 10.1159/000047299. [DOI] [PubMed] [Google Scholar]
- (35).Henrion U; Zumhagen S; Steinke K; Strutz-Seebohm N; Stallmeyer B; Lang F; Schulze-Bahr E; Seebohm G Overlapping Cardiac Phenotype Associated with a Familial Mutation in the Voltage Sensor of the KCNQ1 Channel. Cell. Physiol. Biochem 2012, 29 (5–6), 809–818. 10.1159/000178470. [DOI] [PubMed] [Google Scholar]
- (36).Bartos DC; Duchatelet S; Burgess DE; Klug D; Denjoy I; Peat R; Lupoglazoff JM; Fressart V; Berthet M; Ackerman MJ; January CT; Guicheney P; Delisle BP R231C Mutation in KCNQ1 Causes Long QT Syndrome Type 1 and Familial Atrial Fibrillation. Hear. Rhythm 2011, 8 (1), 48–55. 10.1016/j.hrthm.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Tester DJ; Will ML; Haglund CM; Ackerman MJ Compendium of Cardiac Channel Mutations in 541 Consecutive Unrelated Patients Referred for Long QT Syndrome Genetic Testing. Hear. Rhythm 2005, 2 (5), 507–517. 10.1016/j.hrthm.2005.01.020. [DOI] [PubMed] [Google Scholar]
- (38).Huang H; Kuenze G; Smith JA; Taylor KC; Duran AM; Hadziselimovic A; Meiler J; Vanoye CG; George AL; Sanders CR Mechanisms of KCNQ1 Channel Dysfunction in Long QT Syndrome Involving Voltage Sensor Domain Mutations. Sci. Adv 2018, 4 (3), eaar2631. 10.1126/sciadv.aar2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Marklund EG; Degiacomi MT; Baldwin AJ; Benesch JLP; Marklund EG; Degiacomi MT; Robinson CV; Baldwin AJ; Benesch JLP Collision Cross Sections for Structural Proteomics Resource Collision Cross Sections for Structural Proteomics. Struct. Des 2015, 23 (4), 791–799. 10.1016/j.str.2015.02.010. [DOI] [PubMed] [Google Scholar]
- (40).Ruotolo BT; Benesch JLP; Sandercock AM; Hyung SJ; Robinson CV Ion Mobility-Mass Spectrometry Analysis of Large Protein Complexes. Nat. Protoc 2008, 3 (7), 1139–1152. 10.1038/nprot.2008.78. [DOI] [PubMed] [Google Scholar]
- (41).Taylor KC; Kang PW; Hou P; N. Du Yang; Kuenze G; Smith JA; Shi J; Huang H; White KM; Peng D; George AL; Meiler J; McFeeters RL; Cui J; Sanders CR Structure and Physiological Function of the Human KCNQ1 Channel Voltage Sensor Intermediate State. Elife 2020, 9. 10.7554/eLife.53901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Haynes SE; Polasky DA; Dixit SM; Majmudar JD; Neeson K; Ruotolo BT; Martin BR Variable-Velocity Traveling-Wave Ion Mobility Separation Enhancing Peak Capacity for Data-Independent Acquisition Proteomics. Anal. Chem 2017, 89 (11), 5669–5672. 10.1021/acs.analchem.7b00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Calabrese AN; Watkinson TG; Henderson PJF; Radford SE; Ashcroft AE Amphipols Outperform Dodecylmaltoside Micelles in Stabilizing Membrane Protein Structure in the Gas Phase. Anal. Chem 2015, 87 (2), 1118–1126. 10.1021/ac5037022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Borysik AJ; Robinson CV The “sticky Business” of Cleaning Gas-Phase Membrane Proteins: A Detergent Oriented Perspective. Phys. Chem. Chem. Phys 2012, 14 (42), 14439–14449. 10.1039/c2cp41687e. [DOI] [PubMed] [Google Scholar]
- (45).Gupta K; Donlan JAC; Hopper JTS; Uzdavinys P; Landreh M; Struwe WB; Drew D; Baldwin AJ; Stansfeld PJ; Robinson CV The Role of Interfacial Lipids in Stabilizing Membrane Protein Oligomers. Nature 2017, 541 (7637), 421–424. 10.1038/nature20820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Borysik AJ; Hewitt DJ; Robinson CV Detergent Release Prolongs the Lifetime of Native-like Membrane Protein Conformations in the Gas-Phase. J. Am. Chem. Soc 2013, 135 (16), 6078–6083. 10.1021/ja401736v. [DOI] [PubMed] [Google Scholar]
- (47).Zhong Y; Han L; Ruotolo BT Collisional and Coulombic Unfolding of Gas-Phase Proteins: High Correlation to Their Domain Structures in Solution. Angew. Chemie Int. Ed 2014, 53 (35), 9209–9212. 10.1002/anie.201403784. [DOI] [PubMed] [Google Scholar]
- (48).Landreh M; Marklund EG; Uzdavinys P; Degiacomi MT; Coincon M; Gault J; Gupta K; Liko I; Benesch JLP; Drew D; Robinson CV Integrating Mass Spectrometry with MD Simulations Reveals the Role of Lipids in Na+/H+ Antiporters. Nat. Commun 2017, 8, 1–9. 10.1038/ncomms13993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Tian Y; Han L; Buckner AC; Ruotolo BT Collision Induced Unfolding of Intact Antibodies: Rapid Characterization of Disulfide Bonding Patterns, Glycosylation, and Structures. Anal. Chem 2015, 87 (22), 11509–11515. 10.1021/acs.analchem.5b03291. [DOI] [PubMed] [Google Scholar]
- (50).Laganowsky A; Reading E; Allison TM; Ulmschneider MB; Degiacomi MT; Baldwin AJ; Robinson CV Membrane Proteins Bind Lipids Selectively to Modulate Their Structure and Function. Nature 2014, 510 (7503), 172–175. 10.1038/nature13419. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.