

Abstract
Despite mounting evidence of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) engagement with immune cells, most express little, if any, of the canonical receptor of SARS-CoV-2, angiotensin-converting enzyme 2 (ACE2). Here, using a myeloid cell receptor-focused ectopic expression screen, we identified several C-type lectins (DC-SIGN, L-SIGN, LSECtin, ASGR1, and CLEC10A) and Tweety family member 2 (TTYH2) as glycan-dependent binding partners of the SARS-CoV-2 spike. Except for TTYH2, these molecules primarily interacted with spike via regions outside of the receptor-binding domain. Single-cell RNA sequencing analysis of pulmonary cells from individuals with coronavirus disease 2019 (COVID-19) indicated predominant expression of these molecules on myeloid cells. Although these receptors do not support active replication of SARS-CoV-2, their engagement with the virus induced robust proinflammatory responses in myeloid cells that correlated with COVID-19 severity. We also generated a bispecific anti-spike nanobody that not only blocked ACE2-mediated infection but also the myeloid receptor-mediated proinflammatory responses. Our findings suggest that SARS-CoV-2-myeloid receptor interactions promote immune hyperactivation, which represents potential targets for COVID-19 therapy.
Keywords: SARS-CoV-2, COVID-19, DC-SIGN, L-SIGN, LSECtin, ASGR1, CLEC10A, TTYH2, myeloid cells, proinflammatory responses
Graphical abstract
Most immune cells express little, if any, of the canonical SARS-CoV-2 receptor, ACE2. Lu et al. report that C-type lectins and TTYH2 act as SARS-CoV-2 myeloid cell-interacting partners that trigger immune hyperactivation but not infection. These findings raise the possibility that these virus-myeloid cell interactions are directly involved in COVID-19 immunopathogenesis and could be targeted for COVID-19 therapy.
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the etiological agent of the coronavirus disease 2019 (COVID-19) pandemic, has resulted in over 116 million confirmed cases and more than 2.5 million deaths as of early March 2021 (https://covid19.who.int/). The high morbidity and mortality of COVID-19 is associated with dysregulated immune responses (Zhou et al., 2020b). Excessive lung inflammation induced by SARS-CoV-2 infection is postulated to be a major driver of disease severity in individuals with COVID-19 (Mehta et al., 2020) and contributes to the clinical observations of acute respiratory distress syndrome, cytokine release syndrome, and lymphopenia (Bost et al., 2020; Diao et al., 2020; Feng et al., 2020; Giamarellos-Bourboulis et al., 2020; Gong et al., 2020; Liao et al., 2020; Moore and June, 2020; Vabret et al., 2020). Pharmacological interventions aimed at reducing SARS-CoV-2 replication have been relatively ineffective in improving clinical symptoms in individuals with severe COVID-19 (Grein et al., 2020), pointing to the hyperactive immunopathology as a driving force for late-stage diseases. However, little information is available about the mechanisms for immunopathogenesis, which is key for identification of therapeutic targets.
Angiotensin-converting enzyme 2 (ACE2) has been identified as the primary cellular receptor for SARS-CoV-2 entry into host epithelial cells (Hoffmann et al., 2020b; Wu et al., 2020a; Zhou et al., 2020a). Similar to SARS-CoV, SARS-CoV-2 spike (S) glycoprotein binds to ACE2 via the receptor-binding domain (RBD) (Lu et al., 2020; Zhou et al., 2020a). Following receptor engagement, several host serine proteases, including TMPRSS2, TMPRSS4, furin, and endosomal cathepsins, cleave the SARS-CoV-2 S protein at the junction between S1 and S2 fragments, enabling host and viral membrane fusion and delivery of the viral genome into the cytosol (Hoffmann et al., 2020a, 2020b; Zang et al., 2020). Although proteases such as TMPRSS2 and cathepsins are utilized by SARS-CoV-2 and SARS-CoV (Hoffmann et al., 2020b), S cleavage by furin is unique to SARS-CoV-2 because of the presence of a polybasic (RRAR) site at the S1-S2 junction (Xia et al., 2020). In addition to ACE2, a number of other host molecules reportedly support SARS-CoV-2 binding to cells and act as entry factors, including CD147 (Wang et al., 2020), neuropilin-1 (Cantuti-Castelvetri et al., 2020; Daly et al., 2020), sialic acid (Morniroli et al., 2020), and heparan sulfate (Clausen et al., 2020; Liu et al., 2020). However, the functional relevance of these attachment factors and receptors, especially to immune cells, is unclear. Nevertheless, these studies highlight that SARS-CoV-2 may engage with some target cells independent of ACE2 and that the current therapeutic approaches to block S protein RBD-ACE2 interaction (Brouwer et al., 2020; Cao et al., 2020; Chen et al., 2020; Ju et al., 2020; Wu et al., 2020b) may not be sufficient to disrupt such interactions.
Single-cell RNA sequencing (scRNA-seq) studies using bronchoalveolar lavage (BAL) fluid from individuals with COVID-19 detected SARS-CoV-2 RNA in pulmonary epithelial cells and immune cell populations, particularly in myeloid cells (Bost et al., 2020). Myeloid cells may have a central role in SARS-CoV-2 pathogenesis because inflammatory monocytes and expansion of monocyte-derived macrophages in infected pulmonary tissue (Liao et al., 2020; Wen et al., 2020) can serve as sources of proinflammatory cytokines and chemokines (Feng et al., 2020; Giamarellos-Bourboulis et al., 2020; Moore and June, 2020) and modulate T cell functionality (Zhou et al., 2020c). These hyperactive myeloid cells correlate positively with systemic levels of proinflammatory mediators, including interleukin-1 (IL-1), IL-6, IL-8, and C-X-C motif chemokine (CXCL) 10 (Diao et al., 2020; Feng et al., 2020; Giamarellos-Bourboulis et al., 2020; Gong et al., 2020; Liao et al., 2020; Moore and June, 2020; Vabret et al., 2020). Based on these results and the relatively low ACE2 expression or other putative SARS-CoV-2 receptors in myeloid cells, we sought to identify additional cellular binding partners of the S protein beyond ACE2 and define their roles in SARS-CoV-2-myeloid cell interactions.
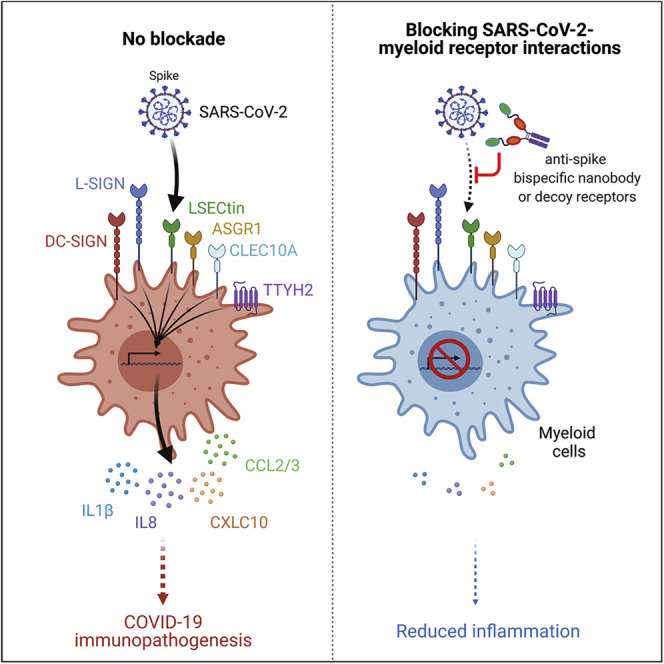
We proposed an immune asynchrony model to explain the possible mechanisms of COVID-19 pathogenesis that centers on how SARS-CoV-2 dysregulates myeloid cell responses (Zhou et al., 2020b). Here, using a high-throughput myeloid cell receptor-focused screening approach, we identified and characterized six host plasma membrane proteins that bound to the SARS-CoV-2 S protein and potentially mediated interactions with myeloid cells: the C-type lectin receptors DC-SIGN (encoded by CD209), L-SIGN (encoded by CLEC4M), LSECtin (encoded by CLEC4G), asialoglycoprotein receptor 1 (encoded by ASGR1), CLEC10A (encoded by CLEC10A), and Tweety family member 2 (encoded by TTYH2), a probable chloride channel with no known immunological functions. C-type lectins have been suggested as attachment factors or signaling receptors for several viruses, including HIV-1 and HIV-2 (Pöhlmann et al., 2001), dengue virus (Chen et al., 2008; Tassaneetrithep et al., 2003; Wu et al., 2013), Ebola virus (Brudner et al., 2013; Zhao et al., 2016), and respiratory viruses, including measles virus (de Witte et al., 2006), influenza A virus (Hillaire et al., 2013), and SARS-CoV (Jeffers et al., 2004; Marzi et al., 2004). However, the contribution of C-type lectins to immune dysregulation in the context of COVID-19 outcomes is unclear. Our findings suggest that these receptors on myeloid cells may have an ACE2-independent role in SARS-CoV-2 pathogenesis and immune hyperactivation.
Results
C-type lectins and TTYH2 interact with the SARS-CoV-2 S protein
To identify myeloid cell-associated receptors for SARS-CoV-2, we built a myeloid cell receptor array that comprised a human cDNA library of ∼300 host membrane proteins expressed preferentially in myeloid cells (i.e., monocyte, macrophage, and dendritic cell [DC] populations; Table S1). We applied a receptor overexpression and detection system modified from our previous report (Wang et al., 2019) and tested the binding of human immunoglobulin Fc-tagged SARS-CoV-2 S, S1, and RBD recombinant proteins to HEK293T cells transfected with individual cDNA (Figure 1 A). ACE2 and Fc receptors served as positive controls. We identified six proteins that interacted with the SARS-CoV-2 S protein or its subunits, including five C-type lectins (DC-SIGN, L-SIGN, LSECtin, ASGR1, and CLEC10A) and Tweety family member 2 (TTYH2) (Figure 1B). All six candidate genes were expressed at levels comparable with ACE2 (Figure S1A). In contrast to ACE2, which bound strongly to RBD-Fc, S-Fc, and S1-Fc, C-type lectins interacted primarily with S-Fc and S1-Fc, with weak or no binding to RBD-Fc (Figure 1C). These data suggest that C-type lectins likely interact with the SARS-CoV-2 S protein via non-RBD epitopes (e.g., the N-terminal domain [NTD] and C-terminal domain [CTD]) within the S1 region. TTYH2 interacted weakly with RBD-Fc but not with S-Fc or S1-Fc (Figure 1C).
Figure 1.

Discovery of C-type lectins and TTYH2 as interacting partners of the SARS-CoV-2 S protein
(A) Schematic of the myeloid cell receptor discovery approach. Individual plasmids of genes encoding myeloid cell receptors were transfected into HEK293T cells, and a human Fc-tagged SARS-CoV-2 S protein mixture (S-Fc, S1-Fc, and RBD-Fc) and anti-human immunoglobulin G (IgG) Fc detection antibody were added to the cell culture to assess binding (see STAR Methods for more details). S protein subunits and subdomains relative to ACE2 binding (RBD) are also shown.
(B) Other than ACE2 (purple), DC-SIGN, L-SIGN, LSECtin, ASGR1, CLEC10A, and TTYH2 (all in red) were identified as binding partners for the SARS-CoV-2 S protein (n = 2). Fc receptors (blue) served as positive controls.
(C) Representative images of the binding of the Fc-tagged S protein, its subunits, or Fc control (Fc Ctr) to the indicated receptors, captured by the cellular detection system (CDS) (n = 3).
(D) Quantification of the interaction between S protein subdomains/subunits and different receptors, indicated on the x axis. Normalized binding capacity is shown on the y axis (the sum of the total fluorescence intensity to the indicated receptor was set to 100).
(E) Binding between HEK293T cells expressing the indicated receptors and HIV-GFP virus pseudotyped with the SARS-CoV-2 S protein was detected by an anti-S polyclonal antibody and analyzed by flow cytometry (n = 4).
Data are presented as the mean ± SEM of five pooled independent experiments (D) or a representative experiment (E); ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 by one-way ANOVA (E). n refers to the number of independent experiments.
See also Figure S1.
To map the interaction domain, we generated recombinant NTD-Fc, RBD-Fc, CTD-Fc, and S2-Fc proteins. Consistent with the screening results, DC-SIGN, L-SIGN, and LSECtin associated predominantly with CTD-Fc and, to a lesser extent, NTD-Fc. ASGR1 and CLEC10A interacted mainly with NTD-Fc, whereas ACE2 and TTYH2 bound primarily to RBD-Fc (Figure 1D; Figure S1B). We next used an HIV-based lentivirus backbone pseudotyped with the SARS-CoV-2 S protein (Crawford et al., 2020) to validate these interactions in the context of a viral particle. SARS-CoV-2 pseudovirus bound strongly to HEK293T cells overexpressing ACE2, DC-SIGN, L-SIGN, or ASGR1 and exhibited weaker but significant binding to cells expressing LSECtin, CLEC10A, or TTYH2 (Figure 1E; Figure S1C). Finally, we assessed the direct protein-protein binding of the S1 subunit from SARS-CoV-2 to the recombinant receptor ectodomains by an enzyme-linked immunosorbent assay (ELISA), except for TTYH2, which has multiple extracellular domains. Analogous to the results with the pseudovirus, there was a clear, albeit weaker, interaction between the S1 recombinant protein and Fc-tagged ectodomain of these C-type lectins compared with ACE2-Fc (Figure S1D). These data indicate that DC-SIGN, L-SIGN, LSECtin, ASGR1, CLEC10A, and TTYH2 can associate directly with the SARS-CoV-2 S protein and mediate pseudovirus attachment to cells.
C-type lectins bind to the SARS-CoV-2 S protein via residues distinct from ACE2 and TTYH2
Next we characterized the interaction interfaces of the SARS-CoV-2 S protein with these myeloid receptors in comparison with ACE2. Pre-incubation of the His-tagged soluble ectodomain of ACE2 recombinant protein (ACE2-His) with S-Fc or RBD-Fc completely blocked their binding to ACE2-overexpressing cells (Figure 2 A; Figure S2A). In addition, soluble ACE2-His blocked binding of RBD-Fc to TTYH2 (Figure 2A; Figure S2A), likely because of potentially shared binding motifs between ACE2 and TTYH2 in the RBD. Unexpectedly, S protein binding to LSECtin was abolished in the presence of pre-bound ACE2-His (Figure 2A; Figure S2A), suggesting that LSECtin may associate with an RBD-proximal region of the S protein that is sterically hindered upon binding of ACE2. In contrast, binding of the S protein to other C-type lectins was not affected by soluble ACE2, except for a weak reduction of L-SIGN interaction (Figure 2A; Figure S2A). Based on the RBD-ACE2 structure (Shang et al., 2020), we generated an S1-Fc mutant bearing three point mutations in the RBD (T500A/N501A/G502E), resulting in loss of two hydrogen bonds between RBD and ACE2 interaction (Figure 2B). This S1 protein mutant retained the capacity to bind to all five C-type lectins but not to ACE2 (Figure 2C; Figure S2B), confirming that the C-type lectin interface is distinct from that of ACE2.
Figure 2.

Glycan-dependent SARS-CoV-2 S interaction with C-type lectins via residues distinct from ACE2
(A) Quantification of S-Fc or RBD-Fc binding to HEK293T cells expressing the indicated receptors in the presence or absence of a 100× excess (in the mass ratio) of His-tagged ACE2 ectodomain recombinant protein (ACE2-His) (n = 3).
(B) The hydrogen bond formation between the Lys352-Asp353, Tyr41 of human ACE2 (green) and the Thr500-Asn501-Gly502 loop segment of the wild-type (WT) SARS-CoV-2 receptor binding motif (RBM) (PDB: 6M0J; left, cyan) or the Ala500-Ala501-Glu502 SARS-CoV-2 RBM mutant (right, cyan).
(C) Quantitative comparison of binding to the indicated receptors of Fc-tagged WT S1 and the T500A/N501A/G502A mutant S1 recombinant protein (n = 3).
(D) Representative images (left) and quantification (right) of binding of S-Fc or RBD-Fc (for TTYH2) to the indicated receptors in the presence or absence of 20 μg/mL mannan (n = 3).
(E) Glycosylated SARS-CoV-2 S protein model highlighting Asn(N)165, N282, N343, and N603 glycans. The SARS-CoV-2 RBD is colored cyan, and the ACE2 N-terminal peptidase domain is shown in green.
(F) Quantification of representative S1 Asn mutants that enhanced interaction with ACE2 (N282Q, left) or some of the C-type lectins (N165Q, right) (n = 3).
(G) Quantification of representative S1 Asn mutants that reduced interaction with ACE2 (N343Q, left) or C-type lectins (N603Q, right) (n = 3).
(H) Schematic of the distribution of N- or O-glycosylation sites in the S1 subunit (top panel) and existence of natural mutations related to some of these sites among ∼5,000 SARS-CoV-2 viral genomes as well as quantitative analysis of the effect of individual mutations of these sites on S protein binding to the indicated receptors (bottom panel).
All fluorescence images were captured by CDS and analyzed by CellProfiler software. Data are presented as the mean ± SEM of a representative experiment (A, C, D, F, and G) or three pooled independent experiments (H). ns, not significant. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 by two-tailed unpaired Student’s t test (A, C, D, F, and G) or one-way ANOVA (H). n refers to the number of independent experiments.
See also Figure S2.
The SARS-CoV-2 S protein is heavily glycosylated, with 22 N-linked glycans located mainly outside of the RBD (within the NTD, CTD, and S2) and 2 O-linked glycans in the RBD (Shajahan et al., 2020; Watanabe et al., 2020). Previous studies suggest that glycosylation serves as a molecular shield for viruses to reduce immunogenicity and facilitate viral entry and immune escape (Gramberg et al., 2005; Han et al., 2007; Jeffers et al., 2004; Londrigan et al., 2011). HIV gp120, influenza hemagglutinin, Ebola GP protein, and the SARS-CoV S protein contain high-mannose glycans that can be recognized by DC-SIGN and L-SIGN (Alvarez et al., 2002; Han et al., 2007; Jeffers et al., 2004; Londrigan et al., 2011; Pöhlmann et al., 2001). Therefore, we tested whether S protein glycosylation affects interaction with the myeloid cell receptors identified in our screen. We found that addition of mannan (a mannose polymer) competitively blocked binding of the S protein to DC-SIGN and L-SIGN but not to ACE2, LSECtin, ASGR1, CLEC10A, or TTYH2 (Figure 2D). Similarly, endoglycosidase H (Endo H), which removes high-mannose oligosaccharides on N-linked glycans (Trimble and Maley, 1984), only reduced S protein binding to DC-SIGN, L-SIGN, and, to a weaker extent, LSECtin (Figure S2C). In contrast, treatment of the SARS-CoV-2 S protein with peptide:N-glycosidase F (PNGase F), which removes all types of N-linked glycans, significantly reduced S-Fc protein binding to all myeloid cell receptors (Figure S2C). PNGase F treatment also reduced S-Fc binding to ACE2 (Figure S2C), suggesting involvement of glycans in the interaction with ACE2 and the immune receptors.
To pinpoint the specific glycosylated residues important for receptor binding, we next performed a mutagenesis screen by disrupting the individual 13 N-glycosylation sites and 2 O-glycosylation sites within the S1 subunit. We observed functional mutation sites that could be categorized into an inhibitory group whose single mutation led to significant loss of S1-Fc binding to the host receptors (N343 to ACE2, N603 to almost all C-type lectins, and the majority of the mutants to ASGR1 and CLEC10A). We also identified an enhancing group; single mutation of these sites led to significant increases in S1-Fc interactions (N74, N149, N282, N603, N616, and N657 to ACE2; N165 to DC-SIGN/L-SIGN/LSECtin; N234, N343, and N657 to DC-SIGN; and N122 to LSECtin) (Figures 2E–2H; Figure S2D). Although the N282Q mutation showed the most enhanced (3-fold) binding to ACE2 among all glycosylation mutations, we observed that the N165Q mutation greatly enhanced binding to DC-SIGN, L-SIGN, and LSECtin but not ACE2 (Figure 2F; Figure S2D). The N343 mutation in the RBD (Figure 2E) completely abolished ACE2 binding (Figure 2G; Figure S2D), which is consistent with a recent report showing that the N343 mutation leads to reduced infectivity (Li et al., 2020). The N343 mutation also reduced binding of S1-Fc to ASGR1 and CLEC10A. However, it had no effect on L-SIGN and LSECtin binding but enhanced S1-Fc binding to DC-SIGN. Another N603 mutant in the CTD (Figure 2E) showed decreased binding to most of the C-type lectins, which contrasted with its effects on ACE2 binding (Figure 2G; Figure S2D). These data suggest that N603 may be a key glycosylation site supporting SARS-CoV-2 S binding to C-type lectins but interfering with its ACE2 interaction.
Although N-glycosylation greatly affects S1 interaction with ACE2 and myeloid receptors, O-glycosylation may not be required for binding of S1-Fc to most of the host receptors, except for CLEC10A (Figure S2D). In parallel, we analyzed over 5,000 genomes of SARS-CoV-2 natural variants documented as of May 2020 (Zhu et al., 2020), which revealed natural mutations at the majority of N-linked glycosylation sites (Figure 2H). Our findings support a critical role of SARS-CoV-2 S protein N-linked glycosylation in interacting with ACE2 and myeloid cell receptors.
C-type lectins and TTYH2 are expressed by myeloid cells from individuals with hyperinflammatory COVID-19
Macrophages and DCs are susceptible to SARS-CoV and MERS-CoV infections (Perlman and Dandekar, 2005; Zhou et al., 2014), and the SARS-CoV-2 viral genome has been detected in SPP1+ lung macrophages (Bost et al., 2020). We re-analyzed published scRNA-seq data of the BAL fluid from individuals with severe COVID-19 (Liao et al., 2020). Although SARS-CoV-2-positive cells accounted for 22% of general cell populations, cells expressing ACE2, the C-type lectins, and TTYH2 were associated with a higher frequency of viral RNA positivity (51% for ACE2, 40% for DC-SIGN, 67% for L-SIGN, 33% for LSECtin, 26% for ASGR1, 27% for CLEC10A, and 42% for TTYH2) (Figure S3A). We verified the presence of SARS-CoV-2 viral RNA in SPP1+, CD169+, or CD11C+ myeloid cell lineages that also expressed DC-SIGN, LSECtin, ASGR1, CLEC10A, and TTYH2 but not ACE2 (Figure 3 A; Figures S3B and S3C). Although the mRNA level of L-SIGN in scRNA-seq was relatively low, we confirmed its expression on the surface of peripheral blood mononuclear cell (PBMC)-derived myeloid cells, which had little ACE2 expression, as seen by flow cytometry analysis (Figure S3D). We also validated the expression of these myeloid receptors in BAL samples isolated from individuals with severe COVID-19. Within CD45+CD14loCD11c+CD206+ myeloid cells, DC-SIGN was expressed preferentially by the HLA-DRhi subsets, L-SIGN was expressed principally by the HLA-DRlo subsets, and LSECtin, ASGR1, and CLEC10A were expressed by both subsets (Figures S3E and S3F). The expression of TTYH2 was verified by western blot because of the lack of commercially available antibodies for flow cytometry (Figure S3G).
Figure 3.

Myeloid cell-associated expression of C-type lectins and TTYH2 in individuals with COVID-19
(A) Uniform manifold approximation and projection (UMAP) visualizations of single cells isolated from BAL fluid from six individuals with severe COVID-19, with color-coded clusters. Levels of SARS-CoV-2 viral RNA and the indicated host transcripts were plotted individually.
(B) Gene Ontology (GO) pathway enrichment connectivity diagram displaying pathways enriched in infected cells expressing the indicated receptors. The color shades of dots in each diagram denote the p value of the hypergeometric test (the darker, the smaller the p value), the size of the dots represents the number of genes associated in the indicated pathway (the larger, the more associated genes), and functionally related dots are connected by lines.
(C) Heatmaps of gene expression of myeloid receptors and pro-inflammatory cytokine/chemokines in BAL myeloid cells isolated from individuals without pneumonia and those with bacterial pneumonia and COVID-19.
See also Figure S3.
Beyond viral attachment factors, host furin protease and cysteine proteases (such as cathepsin L, CTSL) were expressed in myeloid cells (Figure 3A; Figures S3B and S3C). In contrast, TMPRSS2, similar to ACE2, was restricted to epithelial cells (Figure 3A; Figures S3B and S3C). Moreover, immune receptor-expressing myeloid cells in BAL fluid were associated with low levels of type I, II, and III interferons (IFNA2, IFNB1, IFNG, and IFNL1) but high levels of cytokines and chemokines, such as IL1B, IL8, CXCL10, CCL2, and tumor necrosis factor (TNF), as well as interferon (IFN)-stimulated genes with possible antiviral activity (IFI27 and IFI30) (Figure 3A; Figure S3C). Gene Ontology pathway analysis revealed that, unlike ACE2-positive epithelial cells, genes involved in proinflammatory immune responses were enriched in DC-SIGN-, LSECtin-, TTYH2-, and CLEC10A-positive myeloid cells with viral infection compared with those without (Figure 3B). Through reanalysis of another published scRNA-seq database (Grant et al., 2021), we also found specific upregulation of these immune receptors and pro-inflammatory cytokine/chemokines in BAL samples from individuals with COVID-19 compared with those from individuals without pneumonia or those with bacterial pneumonia (Figure 3C), highlighting their possible pathogenic role in COVID-19.
Myeloid cell receptors mediate SARS-CoV-2 S protein binding independent of ACE2
To test whether these C-type lectins and TTYH2 mediate SARS-CoV-2 interaction with myeloid cells, we used a GFP-encoding HIV-based lentivirus pseudotyped with SARS-CoV-2 S protein (SARS-CoV-2 pseudovirus). In this system, the GFP signals were enhanced by, but not strictly dependent on, productive infection. Therefore, the GFP signal was used as a surrogate for virus attachment and/or replication. In the presence of the SARS-CoV-2 pseudovirus, we detected robust GFP signals in HEK293T cells that ectopically expressed DC-SIGN or L-SIGN at levels comparable with those with ACE2 ectopic expression (Figure 4 A). The low level of SARS-CoV-2 pseudovirus engagement through LSECtin, ASGR1, CLEC10A, and TTYH2 was enhanced by co-expression of furin and/or TMPRSS2 (Figure S4A), which mediate S protein cleavage for viral entry (Hoffmann et al., 2020a, 2020b; Zang et al., 2020). Consistent with our S protein mannan blockade results (Figure 2D), pseudovirus-derived GFP signals through DC-SIGN and L-SIGN were inhibited by mannan treatment (Figure 4A). The SARS-CoV-2 pseudovirus with a mutated S protein (T500A/N501A/G502E) retained the ability to engage with DC-SIGN- or L-SIGN- but not ACE2-expressing cells (Figure 4A). We also assessed the interaction of the SARS-CoV-2 pseudovirus with the human myeloid cell line THP-1, which gained surface expression of DC-SIGN and L-SIGN upon myeloid differentiation (Figure S4B). Gene silencing of DC-SIGN or L-SIGN expression in differentiated THP-1 cells by short hairpin RNA (shRNA) led to reduced GFP signals (Figures S4B and S4C). In contrast to ACE2, the HIV-based SARS-CoV-2 pseudovirus GFP signals mediated by the myeloid cell receptors were not affected by nevirapine, a non-nucleoside viral reverse transcriptase inhibitor (Merluzzi et al., 1990) (Figure S4D). Similarly, nevirapine did not inhibit the GFP signals generated from PBMC-derived myeloid cells after culture with the SARS-CoV-2 pseudovirus (Figure S4E). These data suggest that these myeloid cell receptors can engage with the SARS-CoV-2 S protein on the pseudotyped virus, but this engagement may not lead to active virus infection or replication.
Figure 4.

Myeloid cell receptors mediate ACE2-independent SARS-CoV-2 virus-immune interactions
(A) HEK293T cells transfected with the indicated receptors or vector control were co-cultured with HIV-GFP virus pseudotyped with SARS-CoV-2 WT or mutant (T500A/N501A/G502E) S protein (SARS-CoV-2 pseudovirus) in the presence or absence of mannan (100 μg/mL) for 48 h, followed by flow cytometry analysis of GFP expression (n = 3).
(B) Human PBMC-derived myeloid cells were co-cultured with SARS-CoV-2 pseudovirus with WT S protein in the presence or absence of mannan (100 μg/mL) for 48 h, followed by flow cytometry analysis (n = 3).
(C) HEK293T cells with or without ACE2 overexpression (left) and human PBMC-derived myeloid cells (right) were co-cultured with SARS-CoV-2 pseudovirus in the presence of Fc Ctr or anti-ACE2 antibody (20 μg/mL). GFP-positive cells were quantified by flow cytometry after 48-h incubation (n = 3).
(D and E) Human PBMC-derived myeloid cells were co-cultured with SARS-CoV-2 pseudovirus with WT S protein (D) or a clinical isolate of SARS-CoV-2 (MOI = 1) (E), with or without Fc-tagged S-interacting decoy receptor(s) (25 μg/mL of each). Cells were analyzed by flow cytometry after 48-h incubation (D) or lysed for RNA extraction and RT-PCR analysis after 24-h incubation (E). The viral mRNA level was normalized to the host GAPDH level, and the average value of the Fc Ctr group was set to 100 (E) (n = 3).
Representative flow cytometry plots (left) and statistical analysis (right) are shown in (A), (B), and (D). Data are presented as mean ± SEM of a representative experiment. ∗p < 0.05, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 by two-way ANOVA (A), two-tailed unpaired Student’s t test (B and right panel in C), or one-way ANOVA (left panel in C, D, and E). n refers to the number of independent experiments.
See also Figure S4.
SARS-CoV-2 pseudovirus engagement of the more physiologically relevant primary human PBMC-derived myeloid cells was blocked partially by mannan treatment (Figure 4B). In contrast to the ACE2-expressing HEK293T cells, myeloid cell engagement by the SARS-CoV-2 pseudovirus was not blocked by ACE2 antibody treatment, again highlighting an alternative use of cell surface molecules in myeloid cells (Figure 4C). Our data suggest ACE2-independent SARS-CoV-2 S-myeloid cell interactions.
We next tested a clinical isolate of the authentic SARS-CoV-2 virus (2019-nCoV/USA-WA1/2020 strain) on HEK293T cells expressing ACE2 or individual myeloid cell receptors. Ectopic expression of the myeloid cell receptors in HEK293T cells supported SARS-CoV-2 engagement, as indicated by viral RNA levels after incubation with authentic SARS-CoV-2 (Figure S4F). However, viral RNA detected in HEK293T cells expressing individual myeloid receptors was significantly lower than that seen in cells expressing ACE2 (Figure S4F). To corroborate these findings, we tested the effect of soluble decoy receptors, alone or in combination, in blockade of SARS-CoV-2 binding to HEK293T cells expressing individual myeloid cell receptors. We found that ACE2-Fc alone could block the majority of the S-receptor interactions, except for DC-SIGN or L-SIGN. However, DC-SIGN-Fc or L-SIGN-Fc in combination with ACE2-Fc led to nearly complete blockade of ACE2, TTYH2, and DC-SIGN and around ∼70%–80% blockade of L-SIGN, LSECtin, and ASGR1 binding (Figure S4G). L-SIGN-Fc combined with ACE2-Fc also decreased pseudotyped SARS-CoV-2 (Figure 4D) and authentic SARS-CoV-2 engagement with human myeloid cells (Figure 4E). Our data suggest abortive viral infection after interaction with myeloid receptors and that this interaction could be blocked by soluble receptor decoy proteins.
SARS-CoV-2-myeloid cell interactions promote hyperinflammatory responses
To understand the effect of SARS-CoV-2 engagement with myeloid cell receptors, we also used a recombinant SARS-CoV-2 virus whose ORF7 was replaced with an mNeonGreen reporter as an indicator of active virus replication (Xie et al., 2020). We did not detect an mNeonGreen signal in primary human PBMC-derived myeloid cells or HEK293T cells overexpressing individual myeloid cell receptors co-cultured with mNeonGreen SARS-CoV-2, in contrast to positive control Vero E6 cells and HEK293T-ACE2 cells (Figure S5). The data further support that myeloid cell receptors do not facilitate active SARS-CoV-2 replication. To explore possible immune signaling effects after SARS-CoV-2 virus engagement with myeloid cells, we incubated human PBMC-derived myeloid cells with authentic SARS-CoV-2 and analyzed pro-inflammatory cytokines by RT-PCR (Figure 5 A). We observed induction of proinflammatory cytokines and chemokines, including IL-1A, IL-1B, IL-8, CXCL10, CCL2, and CCL3 (Figure 5A), after exposure to the authentic SARS-CoV-2 virus. Several of these molecules are elevated in the plasma of individuals critically ill with COVID-19 (Merad and Martin, 2020; Patterson et al., 2020). A delayed or impaired type I IFN response is thought to be a critical mechanism for COVID-19 pathogenesis (Blanco-Melo et al., 2020; Zhou et al., 2020b). In the presence of SARS-CoV-2, we found little induction of type I and III IFNs in myeloid cells, which is different from the general virus uptake and sensing mechanism that stimulates strong IFN responses (Park and Iwasaki, 2020; Zhou et al., 2020b).
Figure 5.

SARS-CoV-2 viral-immune interactions promote hyperinflammatory responses
(A) Human PBMC-derived myeloid cells from four healthy donors were incubated with a clinical isolate of SARS-CoV-2 (MOI = 0.5). Mock control was performed using conditioned medium. Cells were lysed, and RNA was harvested at 24 h after incubation. mRNA levels of the SARS-CoV-2 N protein and the indicated cytokines and chemokines were measured by RT-PCR and normalized to that of GAPDH. Data were presented as mean ± SEM of four pooled cohorts. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001 by two-tailed paired Student’s t test.
(B and C) Human PBMC-derived myeloid cells of one representative donor were incubated with a clinical isolate of SARS-CoV-2 (MOI = 0.5) or conditioned medium, followed by bulk RNA-seq analysis to dissect upregulated gene expression triggered by the virus. A volcano plot of gene expression (B) and a GO pathway enrichment connectivity diagram (C) are shown.
(D) Heatmaps of proinflammatory gene expression in BAL myeloid cell subsets from healthy control (HC) individuals and those with mild or severe COVID-19.
(E) Proinflammatory gene expression in BAL myeloid cell subsets was plotted for healthy individuals (control) or individuals with COVID-19 with different disease statuses. Data are presented as boxplots with the indicated mean line (black). ∗∗∗p < 0.001 by Wilcoxon rank-sum test.
(F) Correlation between the expression of myeloid receptors and the cytokines/chemokines in the myeloid cell subsets from COVID-19 BAL samples.
(E) and (F) were from the same scRNA-seq dataset.
To profile the global transcriptomic responses of myeloid cells to SARS-CoV-2, we performed bulk RNA-seq on primary human myeloid cells incubated with SARS-CoV-2 or conditioned medium. Incubation with SARS-CoV-2 led to robust amplification of a large number of pro-inflammatory cytokines (IL-1B, IL-8, CCL3, CCL2, CCL8, CCL4L2, CXCL2, CXCL14, and TNF) as well as inflammation-related genes (EGR1, THBD, C4A, and SOCS3) compared with conditioned medium alone (Figure 5B). Gene Ontology analysis revealed that the top enriched pathways were associated with cytokine activity, immune regulation, immune responses, and chemotaxis (Figure 5C). To determine correlations between myeloid cell inflammatory responses and COVID-19, we examined the gene expression of inflammatory cytokines using two published scRNA-seq datasets of BAL samples isolated from healthy control (HC) individuals and those with mild and severe COVID-19 (Liao et al., 2020; Ren et al., 2021). These proinflammatory genes were closely associated with disease severity (Figure 5D) as well as clinical progression or convalescence (Figure 5E). Furthermore, we found a positive correlation between the expression of proinflammatory genes and myeloid receptor genes (Figure 5F). These data support the hypothesis that SARS-CoV-2-myeloid cell interactions can induce pathogenic proinflammatory responses in individuals with COVID-19.
Screening of nanobodies that block SARS-CoV-2 viral-immune interactions
The majority of therapeutic antibodies currently under development prevent RBD-ACE2 interactions (Brouwer et al., 2020; Cao et al., 2020; Chen et al., 2020; Ju et al., 2020; Pinto et al., 2020; Wu et al., 2020b). Our data suggest that (1) the SARS-CoV-2 S protein interacts with C-type lectins and TTYH2 through interfaces distinct from ACE2, and (2) SARS-CoV-2 interaction with myeloid cells might promote pathological inflammation. Therefore, we hypothesized that interference of virus-myeloid receptor interactions via blocking antibodies could have therapeutic effects. To test this hypothesis, we first examined a panel of 15 previously reported human monoclonal antibodies (mAbs) against the NTD, RBD, CTD, and S2 domain that were derived from individuals recovered from COVID-19 (Figure S6A; Chi et al., 2020). However, none of these 15 antibodies elicited robust effects in blocking binding of S1-Fc or RBD-Fc to myeloid cell receptors (Figure S6B). In fact, many of these mAbs enhanced interaction of the S protein with myeloid cell receptors. To identify nanobodies that block all SARS-CoV-2 virus-host interactions (ACE2 and myeloid cell receptors), we screened two sets of naive or synthetic humanized llama VHH libraries (Dong et al., 2020; Figure 6 A). Eighty-eight S protein-specific single-domain nanobodies without an Fc tag were clustered on the basis of VHH-blocking patterns to ACE2 and individual immune receptors. We identified a close relationship between LSECtin, ACE2, and TTYH2 (Figure 6B), in accordance with soluble ACE2 blocking data (Figure 2A). Most of the VHH nanobodies bind to the RBD in the S1 protein in our S1 protein panning (Figure 6B). Some of the RBD-interacting VHH nanobodies showed broad and potent inhibition of S1-Fc binding to ACE2 and all myeloid cell receptors (RBD-Fc to TTYH2) (Figure 6B; Figure S6C). Despite lack of the Fc region, some nanobodies enhanced binding to host myeloid cell receptors (Figure 6B; Figure S6C). Two nanobody clones, A8 and G11, were selected for development of Fc-tagged nanobodies, based on their complementary capacity to broadly block all interactions between the S1 protein and host interaction partners (ACE2 and all six myeloid cell receptors) (Figure 6B). These two nanobodies recognized different S epitopes, and the Fc-tagged versions of A8 and G11 also showed broad and potent blocking activity (Figure S6D). We generated a bispecific nanobody, A8-G11-Fc, that demonstrated a higher affinity for the S protein (Figure 6C) as well as increased inhibition of SARS-CoV-2 pseudovirus interaction with human PBMC-derived myeloid cells compared with A8-Fc or G11-Fc alone (Figure 6D). These findings were corroborated in HEK293T cells overexpressing ACE2, DC-SIGN, or L-SIGN (Figure S6E). A8-G11-Fc not only blocked SARS-CoV-2 infection of HEK293T cells expressing ACE2 (Figure 6E) but also reduced pro-inflammatory responses, more so than the soluble Fc-tagged receptor cocktails (ACE2-Fc/L-SIGN-Fc) (Figures 6F and 6G). These data highlight an important role of SARS-CoV-2-myeloid receptor interactions in virus immunopathogenesis and suggest that nanobodies can be potent modalities to broadly neutralize these activities beyond ACE2-mediated virus infection.
Figure 6.

Identification of nanobodies capable of blocking SARS-CoV-2-induced hyperinflammatory responses
(A) Schematic of the nanobody screening program to develop bispecific nanobodies that block S protein interaction with ACE2 and myeloid cell receptors (see STAR Methods for details).
(B) Clustered heatmap of the relative blocking score for each VHH nanobody. Clones A8 and G11 are highlighted in the rectangular areas, and non-RBD interacting nanobodies were labeled with an asterisk (n = 3).
(C) Probelife kinetics analysis of binding of A8-Fc, G11-Fc, and A8-G11-Fc to the S protein. KD, Kon, and Koff values of individual nanobodies are shown in the table. A representative experiment is shown.
(D) Human PBMC-derived myeloid cells were incubated with HIV-GFP virus pseudotyped with SARS-CoV-2 S protein in the presence of Fc Ctr, A8-Fc, G11-Fc, or A8-G11-Fc (50 μg/mL) for 48 h, followed by flow cytometry (n = 3).
(E) Neutralization of mNeonGreen SARS-CoV-2 reporter virus (MOI = 1) infection of HEK293T cells expressing ACE2 by an S-interacting decoy receptor cocktail (ACE2-Fc/L-SIGN-Fc, 25 μg/mL of each) or a bi-specific nanobody (A8-G11-Fc, 50 μg/mL).
(F and G) Human PBMC-derived myeloid cells from two healthy cohorts were incubated with a clinical isolate of SARS-CoV-2 (MOI = 0.5) in the presence of Fc Ctr protein (50 μg/mL), an ACE2-Fc/L-SIGN-Fc cocktail (25 μg/mL of each), or an A8-G11-Fc bi-specific nanobody (50 μg/mL). Mock control was performed using conditioned medium. Cells were lysed for RNA extraction, and the supernatant was harvested 24 h after incubation. mRNA levels of the indicated cytokines and chemokines were measured by RT-PCR and normalized to that of GAPDH (F), and cytokines in the supernatant were quantified by a multiplex magnetic bead assay (G).
Data are presented as mean ± SEM of a representative experiment (D and E) or four independent pooled experiments (F and G). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 by one-way ANOVA (D, F, and G). n refers to the number of independent experiments.
See also Figure S6.
Discussion
In this study, we aimed to define ACE2-independent SARS-CoV-2 binding partners as a potential molecular basis of immune dysregulation in COVID-19. We identified and characterized five C-type lectins and TTYH2 as new SARS-CoV-2 S interaction partners on myeloid cells. DC-SIGN was first identified as a binding receptor for HIV-1 (Curtis et al., 1992), which can also use L-SIGN for entry (da Silva et al., 2011). Both receptors are engaged by SARS-CoV for entry into DCs (Jeffers et al., 2004; Marzi et al., 2004). Other than its role as a virus attachment factor, the intracellular signaling triggered by DC-SIGN has been involved in multiple aspects of immune function, such as DC maturation, myeloid cell cytokine response, and T cell priming (Konieczna et al., 2015; Svajger et al., 2010). LSECtin is enriched in myeloid cells as a ligand that suppresses T cells (Tang et al., 2009). In the case of Ebola virus, recombinant LSECtin protein triggers downstream inflammatory responses (Zhao et al., 2016). ASGR1 has been shown to bind directly to HBV and HEV, which facilitate viral entry (Yang et al., 2010; Zhang et al., 2011, 2016). CLEC10A is capable of promoting Ebola virus and filovirus infection (Brudner et al., 2013; Takada et al., 2004). ASGR1 and CLEC10A are associated with inflammatory diseases (Kanemaru et al., 2019). In particular, CLEC10A ligation enhances cytokine production induced by Toll-like receptor 7/8 (TLR7/8) (Heger et al., 2018). TTYH2, a relatively undercharacterized protein, currently has no known role in virus infection or immune signaling. However, it can be involved in regulation of immune responses, given its dominant expression on myeloid cells and potential function as an ion channel (Suzuki and Mizuno, 2004). These studies suggest that C-type lectins and TTYH2 may act as possible signaling receptors of SARS-CoV-2 in myeloid cells, which may initiate subsequent cytokine production and promote pathological inflammation.
Our data reveal a role of non-RBD epitopes in the S protein (NTD and CTD) in binding to myeloid receptors. The NTD and CTD appear to be more glycosylated than the RBD, with unclear implications. The SARS-CoV-2 S trimer has been suggested to adopt a “closed” structure or an “open” conformation, and it is possible that the NTD or CTD allosterically affect the conformation of the RBD required for proper interaction with ACE2 (Walls et al., 2020). We found that S2 also bound weakly to the C-type lectins. Given that co-expression of furin enhances SARS-CoV-2 pseudovirus engagement with LSECtin, ASGR1, and CLEC10A, this S2 binding may play a functional role at the fusion step in addition to initial viral attachment.
Our results also highlight an important role of glycosylation in repressing and enhancing SARS-CoV-2 receptor interactions. The N343 residue, whose mutation to Q completely blocked binding of the S protein to ACE2, is conserved between SARS-CoV-2 and SARS-CoV (equivalent to N330), implying selective pressure to maintain ACE2 interaction (Li et al., 2020; Watanabe et al., 2020). A recent structural study of the S protein showed that the N343 glycan is proximal to and surrounds the ACE2-interacting motif in the RBD (Watanabe et al., 2020). An antibody targeting the core fucose moiety of N343 neutralizes SARS-CoV-2 infection without affecting ACE2 binding, although the mechanism of neutralization is not clearly understood (Pinto et al., 2020). The three ACE2-binding motifs are buried at the interface in the closed structure, and the open conformation is expected to be necessary for ACE2 interaction (Walls et al., 2020). We hypothesize that the N343 glycan may be responsible for the conformational switch or maintenance of the open structure, but why removal of this glycan only enhances binding to DC-SIGN is unclear. A detailed study of other glycan mutants, particularly those that occur naturally (N282, N165, etc.), would yield additional insights into the mechanism of viral engagement at a structural level and help determine the interfaces to be targeted for disruption of viral entry and myeloid cell dysfunction. Additionally, although we did not define a functional role of the two purported O-glycan sites except for CLEC10A, the role of O-glycans, particularly in combination with other mutations, requires further investigation.
Our data support that SARS-CoV-2 can engage ectopically expressed myeloid cell receptors and PBMC-derived myeloid cells, but neither leads to active viral infection or replication. Why these receptors, unlike ACE2, do not support active infection warrants further investigation. We also have not yet assessed whether SARS-CoV-2 is endocytosed in these cells and whether this is required for activation of innate immune signaling. Our results suggest that SARS-CoV-2 directly stimulates pro-inflammatory cytokine production from myeloid cells. This, however, does not exclude the possibility that these myeloid cell receptors support SARS-CoV-2 entry and infection in other cell types and/or in the presence of some unknown cofactors. It will be important to test whether myeloid cells from individuals with severe COVID-19 are susceptible to SARS-CoV-2 infection and prone to induction of immune responses. Other than direct virus engagement through receptors, it is also possible that myeloid cells may acquire SARS-CoV-2 via uptake of apoptotic or necrotic infected epithelial cells. In response to virus uptake or infection, some immune cells may prevent or stall virus replication through cell-intrinsic innate sensing mechanisms that can be beneficial to the host.
We observed a strong induction of cytokine/chemokine expression in primary myeloid cells co-cultured with SARS-CoV-2. Unlike the general uptake of many other viruses that strongly stimulates type I/III IFN responses, we did not see changes in these IFN genes’ transcription, which may reflect no productive virus internalization or little viral RNA recognized by RIG-I/MDA5 and other endosome-associated RNA sensing molecules to trigger downstream IFN production. This observation is consistent with our data showing no bona fide SARS-CoV-2 infection in myeloid cells. However, we observed specific upregulation of IL-1A, IL-1B, IL-8, CXCL10, CCL2, and CCL3, a proinflammatory gene program associated closely with COVID-19 disease severity. Therefore, it is likely that these myeloid-cell expressing SARS-CoV-2 S-interacting proteins can serve as signaling receptors to trigger specific hyperinflammatory responses and play important roles in the immune dysregulation and immunopathogenesis of COVID-19.
The current vaccine or neutralization antibody programs mainly target SARS-CoV-2 RBD or RBD-ACE2 interaction. Most, if not all, of those antibody strategies may not block viral-immune cell interactions or alleviate the hyperimmune responses of COVID-19. To begin to explore the therapeutic utility of our findings, we employed a nanobody discovery approach to identify candidates that interfere with SARS-CoV-2 interactions beyond ACE2. Although several vaccine programs have shown some encouraging initial data in generating combinations of neutralizing antibodies in humans (Pinto et al., 2020; Weisblum et al., 2020), the quality and quantity of this repertoire can be difficult to control. With a lower cost of production and enhanced stability, nanobodies have major advantages over traditional antibodies, including a small size, which allows enhanced penetration of the lungs via aerosolization (Gai et al., 2021; Nambulli et al., 2021). Our data show that nanobodies can broadly block SARS-CoV-2 S/ACE2 and myeloid cell receptor interactions. Therefore, although ACE2 and myeloid cell receptors engage the S protein through different epitopes, an individual nanobody can still broadly block those interactions, possibly through the specific features of nanobodies in binding of discontinuous epitopes (McMahon et al., 2018) and/or possible structural changes on the S protein upon engagement with nanobodies. Because of the large and complex structure of S trimers, we also performed bispecific nanobody engineering, which showed potent activity in neutralization of virus-induced inflammatory responses. We envision that simultaneous blockade of SARS-CoV2 targeting ACE2-positive epithelial cells and reducing myeloid cell hyperactivation is a promising therapeutic strategy for COVID-19 treatment.
Limitations of study
Our myeloid receptor discovery screen was solely based on the interaction between soluble Fc-tagged S/S1/RBD protein and the receptors expressed by HEK293T, followed by transfection. Thus, it only captured plasma membrane proteins with good surface expression that do not require additional factors for attachment of the S protein or its subunits. In this study, we identified myeloid cell-associated expression of these C-type lectins and TTYH2 in BAL samples from individuals with severe COVID-19. Whether expression of these receptors can be associated with COVID-19 severity (for example, mild versus severe diseases) remains unclear. Moreover, the detailed mechanism of action and in vivo therapeutic potential of our nanobodies warrant further investigation, especially in animal models of COVID-19 infection and pathology and, more importantly, in individuals with severe COVID-19.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-SARS-CoV-2 spike antibody | Sino Biological | Cat#: 40589-T62 |
| Alexa Fluor 647 goat anti-human IgG (H+L) | Invitrogen | Cat#: A-21445; RRID: AB_2535862 |
| Alexa Fluor 647 mouse anti-human IgG Fc, clone JDC-10 | SouthernBiotech | Cat#: 9040-31; RRID: AB_2796603 |
| Alexa Fluor 647 goat anti-mouse Ig | SouthernBiotech | Cat#: 1010-31; RRID: AB_2794131 |
| Alexa Fluor 647 donkey anti-rabbit IgG | BioLegend | Cat#: 406414; RRID: AB_2563202 |
| Human TruStain FcX | BioLegend | Cat#: 422302; RRID: AB_2818986 |
| Mouse anti-GAPDH monoclonal antibody, clone 1E6D9 | Proteintech | Cat#: 60004-1-lg |
| Mouse anti-FLAG monoclonal antibody, clone M2 | Sigma Aldrich | Cat#: F3165; RRID: AB_259529 |
| HRP-conjugated anti-mouse IgG | Cell Signaling Technology | Cat#: 7076; RRID: AB_330924 |
| Rabbit anti-human DC-SIGNR monoclonal antibody, clone EPR11211 | Abcam | Cat#: ab169783 |
| Rabbit anti-human DC-SIGN antibody | Proteintech | Cat#: 25404-1-AP; RRID: AB_2880062 |
| HRP-conjugated anti-rabbit IgG | Cell Signaling Technology | Cat#: 7074; RRID: AB_2099233 |
| HPR-conjugated anti-human IgG | Sino Biological | Cat#: SSA001 |
| BUV737 Mouse anti-human CD11c antibody, Clone B-ly6 | BD Biosciences | Cat#: 741827; RRID: AB_2871162 |
| Brilliant Violet 785 anti-human CD14 antibody, Clone M5E2 | BioLegend | Cat#: 301840; RRID: AB_2563425 |
| Brilliant Violet 711 anti-human CD16 antibody, Clone 3G8 | BioLegend | Cat#: 302044; RRID: AB_2563802 |
| PerCP/Cyanine5.5 anti-human CD24 antibody, Clone ML5 | BioLegend | Cat#: 311116; RRID: AB_10960741 |
| BUV496 Mouse anti-human CD45 antibody, Clone HI30 | BD Biosciences | Cat#: 750179; RRID: AB_2868405 |
| Brilliant Violet 650 anti-human CD123 antibody, Clone 6H6 | BioLegend | Cat#: 306020; RRID: AB_2563827 |
| Brilliant Violet 605 anti-human CD169 (Sialoadhesin, Siglec-1) antibody, Clone 7-239 | BioLegend | Cat#: 346010; RRID: AB_2721539 |
| PE/Dazzle 594 anti-human CD206 (MMR) antibody, Clone 15-2 | BioLegend | Cat#: 321130; RRID: AB_2616867 |
| PE-Cyanine7 anti-human HLA-DR monoclonal antibody, Clone LN3 | eBioscience | Cat#: 25-9956-42; RRID: AB_1582284 |
| Brilliant Violet 510 anti-human CD3 antibody, Clone OKT3 | Biolegend | Cat#: 317332; RRID: AB_2561943 |
| Human ACE2 Alexa Fluor 750-conjugated antibody, Clone 535919 | R&D Systems | Cat#: FAB9332S |
| Brilliant Violet 421 anti-human CD209 (DC-SIGN) antibody, Clone 9E9A8 | BioLegend | Cat#: 330118; RRID: AB_2734324 |
| Human DC-SIGNR/CD299 Alexa Fluor 700-conjugated antibody, Clone 120604 | R&D Systems | Cat#: FAB162N |
| Human LSECtin/CLEC4G Alexa Fluor 488-conjugated antibody, Clone 845404 | R&D Systems | Cat#: FAB2947G |
| PE Mouse Anti-human ASGPR1 antibody, Clone 8D7 | BD Biosciences | Cat#: 563655; RRID: AB_2687910 |
| APC anti-human CD301 (CLEC10A) antibody, Clone H037G3 | BioLegend | Cat#: 354705; RRID: AB_11218803 |
| Mouse IgG2A Alexa Fluor 750-conjugated isotype control, Clone 20102 | R&D Systems | Cat#: IC003S |
| Brilliant Violet 421 Mouse IgG2A isotype control, Clone MOPC-173 | BioLegend | Cat#: 400259; RRID: AB_10895919 |
| Mouse IgG2B Alexa Fluor 700-conjugated isotype control, Clone 133303 | R&D Systems | Cat#: IC0041N; RRID: AB_10973174 |
| Mouse IgG2A Alexa Fluor 488-conjugated isotype control, Clone 20102 | R&D Systems | Cat#: IC003G; RRID: AB_10718683 |
| PE Mouse IgG1 isotype control, Clone MOPC-21 | BD Biosciences | Cat#: 551436; RRID: AB_394195 |
| LIVE/DEAD Fixable Blue Dead Cell Stain Kit | Invitrogen | Cat#: L23105 |
| Rabbit anti-TTYH2 polyclonal antibody | Invitrogen | Cat#: PA5-34395; RRID: AB_2551747 |
| Anti-SARS-CoV-2 spike VHH-Fc, Clone A8 | This paper | N/A |
| Anti-SARS-CoV-2 spike VHH-Fc, Clone G11 | This paper | N/A |
| Anti-SARS-CoV-2 spike VHH-Fc, Clone A8-G11 | This paper | N/A |
| Bacterial and virus strains | ||
| SARS-CoV-2 (2019-nCoV/USA-WA1/2020 strain) | Centers for Disease Control and Prevention | BEI Resources, Cat#: NR-52281 |
| SARS-CoV-2-mNeonGreen reporter virus | Xie et al., 2020 | N/A |
| Biological samples | ||
| Healthy human buffy coat blood | NYBC | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| SARS-CoV-2 spike S, hFc-tagged | KACTUS Biosystems | Cat#: COV-VM5SS |
| SARS-CoV-2 spike S1, hFc-tagged | KACTUS Biosystems | Cat#: COV-VM5S1 |
| SARS-CoV-2 spike S1, mFc-tagged | Sino Biological | Cat#: 40591-V05H1 |
| SARS-CoV-2 spike S1, mFc-tagged | Acrobiosystems | Cat#: S1N-C5257-100ug |
| SARS-CoV-2 spike RBD, hFc-tagged | KACTUS Biosystems | Cat#: COV-VM5BD |
| SARS-CoV-2 spike RBD, mFc-tagged | Sino Biological | Cat#: 40592-V05H |
| SARS-CoV-2 spike S1, His-tagged | KACTUS Biosystems | Cat#: COV-VM4S1 |
| SARS-CoV-2 spike S1, hFc-tagged | This paper | N/A |
| SARS-CoV-2 spike NTD, hFc-tagged | This paper | N/A |
| SARS-CoV-2 spike RBD, hFc-tagged | This paper | N/A |
| SARS-CoV-2 spike CTD, hFc-tagged | This paper | N/A |
| SARS-CoV-2 spike S2, hFc-tagged | This paper | N/A |
| Human IgG1-Fc protein | Sino Biological | Cat#: 10702-HNAH |
| Human ACE2, His-tagged | KACTUS Biosystems | Cat#: ACE-HM401 |
| Human ACE2, hFc-tagged | Sino Biological | Cat#: 10108-H02H |
| Human DC-SIGN, hFc-tagged | Sino Biological | Cat#: 10200-H01H |
| Human CD299, hFc-tagged | Sino Biological | Cat#: 10559-H01H |
| Human LSECtin, hFc-tagged | Acro Biosystems | Cat#: CLG-H5250 |
| Human ASGR1, hFc-tagged | R&D Systems | Cat#: 10255-AS |
| Human CLEC10A, His-tagged | R&D Systems | Cat#: 4888-CL |
| Human CLEC10A, hFc-tagged | Sino Biological | Cat#: 10821-H01H |
| DNase I | Roche | Cat#: 11284932001 |
| Recombinant human GM-CSF | R&D Systems | Cat#: 215-GM |
| Recombinant human IL-4 | R&D Systems | Cat#: 204-IL |
| Recombinant human TNF-α | PeproTech | Cat#: 300-01A |
| Lipofectamine 2000 | Invitrogen | Cat#:11668019 |
| Polyethylenimine (PEI), Linear, MW 25000 | Polysciences | Cat#: 23966 |
| TMB ELISA Substrate Solution | Sino Biological | Cat#: SEKCR01 |
| Pierce ECL Western Blotting Substrate | Thermo Fisher Scientific | Cat#: 32209 |
| Bovine Serum Albumin | Sigma | Cat#: B4287-25G |
| Ionomycin (free acid), Ca2+ ionophore | Abcam | Cat#: ab120370 |
| Critical commercial assays | ||
| PureLink RNA Mini Kit | Invitrogen | Cat#: 12183025 |
| RNeasy Mini Kit | QIAGEN | Cat#: 74106 |
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat#: 4368813 |
| SYBR Green PCR Master Mix | Applied Biosystems | Cat#: 4309155 |
| TaqMan Universal PCR Master Mix | Applied Biosystems | Cat#: 4304437 |
| MILLIPLEX MAP Human Cytokine/Chemokine Magnetic Bead Panel - Premixed 30 Plex - Immunology Multiplex Assay | Millipore | Cat#: HCYTMAG-60K-PX30 |
| HIV-1 p24 ELISA Assay | XpressBio | Cat#: XB-1000 |
| Q Buffer | Probelife | Cat#: 120010 |
| HFC (Anti-HIgG Fc) Probe | Probelife | Cat#: 160003 |
| Deposited data | ||
| RNA-sequencing data of human dendritic cells or macrophages infected with SARS-CoV-2 | GEO | Accession#: GSE155106 |
| Experimental models: Cell lines | ||
| HEK293T | ATCC | Cat#: CRL-3216; RRID: CVCL_0063 |
| Expi293F | Thermo Fisher Scientific | Cat#: A14527; RRID: CVCL_D615 |
| 293FT | Thermo Fisher Scientific | Cat#: R70007; RRID: CVCL_6911 |
| Vero CCL81 | ATCC | Cat#: CCL-81; RRID: CVCL_0059 |
| Vero E6 | ATCC | Cat#: CRL-1586; RRID: CVCL_0574 |
| THP-1 | ATCC | Cat#: TIB-202; RRID: CVCL_0006 |
| Oligonucleotides | ||
| See Table S2 for primer sequences for RT-PCR | This paper | N/A |
| Non-targeting control shRNA: Forward:CCGGC AACAAGATGAAGAGCACCAAC TCGAGTTGGTGCTCTTCATCTT GTTGTTTTTG; Reverse:AAT TCAAAAACAACAA GATGAAGAGCACCAACTCGAG TTGGTGCTCTTCATCTTGTTG |
This paper | N/A |
| DC-SIGN shRNA#1: Forward:CCGG ACTGGTTGCAAGAGCTCATTTCT CGAGAAATGAGCTCTTGCAACCA GTTTTTTG; Reverse:AATTCAAAAAACTG GTTGCAAGAGCTCATTTCT CGAGAAATGAGCTCTTGCAACCAGT |
This paper | N/A |
| DC-SIGN shRNA#3: Forward:CCGGGC TTCCAGAGAAATCTAAGATCTCGAGAT CTTAGATTTCTCTGGAAGCTTTTTG; Reverse:AATTCAAAAAGCTTCCAGAG AAATCTAAGATCTCGAGATCTTAGAT TTCTCTGGAAGC |
This paper | N/A |
| L-SIGN shRNA#2: Forward:CCGGATC GATGTGACGTTGACAATTCTCGAGA ATTGTCAACGTCACATCGATTTTTTG; Reverse:AATTCAAAAAATCGATGTGA CGTTGACAATTCTCGAGAATTGTCA ACGTCACATCGAT |
This paper | N/A |
| L-SIGN shRNA#4: Forward:CCGGATC CGAGCAAGACGCAATCTACTCGAGT AGATTGCGTCTTGCTCGGATTTTTTG; Reverse:AATTCAAAAAATCCG AGCAAGACGCAATCTACTCGAG TAGATTGCGTCTTGCTCGGAT |
This paper | N/A |
| Recombinant DNA | ||
| cDNA library | GeneCopoeia | Cat#: OC-13865-M02-300-5 |
| cDNA library | DNASU | N/A |
| pcDNA3.1-SARS-CoV-2-S | Laboratory of Dr. Lu Lu, Fudan University | N/A |
| pcDNA6B-FLAG-ACE2 | Laboratory of Dr. Peihui Wang, Shandong University | N/A |
| pCMV-dR8.2 dvpr | Stewart et al., 2003 | Addgene, Cat#: 8455 |
| pCDH-GFP | System Bioscience | Cat#: CD511B-1 |
| pENTER-CD209 | vigenebio | Cat#: CH827318 |
| pENTER-CLEC4G | vigenebio | Cat#: CH899337 |
| pENTER-CLEC4M | vigenebio | Cat#: CH891751 |
| pENTER-TTYH2 | vigenebio | Cat#: CH886758 |
| pENTER-ASGR1 | vigenebio | Cat#: CH830386 |
| pENTER-CLEC10A | vigenebio | Cat#: CH822531 |
| pENTER | vigenebio | Cat#: PD88001 |
| pcDNA3.1/nV5-DEST-ACE2 | Zang et al., 2020 | N/A |
| pLX304-TMPRSS2 | Zang et al., 2020 | N/A |
| pLenti6.3/V5-DEST-FURIN | This paper | N/A |
| psPAX2 | Laboratory of Dr. Didier Trono, EPFL | Addgene, Cat#: 12260 |
| pMD2.G | Laboratory of Dr. Didier Trono, EPFL | Addgene, Cat#: 12259 |
| pLKO.1-puro | Stewart et al., 2003 | Addgene, Cat#: 8453 |
| Software and algorithms | ||
| CellProfiler version 4.0.5 | Broad Institute | https://cellprofiler.org/ |
| R version 4.0.2 | The R project | https://www.r-project.org |
| Python version 3.6.9 | Python software foundation | https://www.python.org |
| FlowJo version 10.6.1 | FlowJo | https://www.flowjo.com/ |
| GraphPad Prism 8 version 8.4.3 | GraphPad Software | https://www.graphpad.com/ |
| Viral-Track | Bost et al., 2020 | https://github.com/PierreBSC/Viral-Track |
| Cytoscape version 3.7.2 | Shannon et al., 2003 | https://cytoscape.org/ |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| xPONENT version 4.3.229.0 | Luminex | https://www.luminexcorp.com/xponent/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jun Wang (jun.wang@nyulangone.org).
Materials availability
Unique material requests should be directed to the Lead Contact and will be released with a Material Transfer Agreement for non-commercial usage. There are restrictions on the availability of VHH nanobodies due to limited stock and continued consumption. We are glad to share the remaining antibodies with reasonable compensation for processing and shipping upon completion of a Material Transfer Agreement for non-commercial usage. Further discussion with NYU Grossman of Medicine and Ab Studio may be required if there is potential for commercial application of these nanobodies.
Data and code availability
The accession number for the RNA-seq raw and processed datasets related to Figure 5B is Gene Expression Omnibus (GEO) database: GSE155106.
Experimental model and subject details
Cell lines
Human embryonic kidney cell line HEK293T cells (ATCC, CRL-3216, RRID: CVCL_0063, Female) were cultured in complete Dulbecco’s Modified Eagle Medium (DMEM), supplemented with 10% heat-inactivated fetal bovine serum (FBS), 2 mM L-Glutamine, 1X Penicillin/Streptomycin, 1mM Sodium Pyruvate, and 20 mM HEPES. THP-1 cells (ATCC, TIB-202, RRID: CVCL_0006, Male) were cultured in RPMI 1640 medium supplemented with 10% FBS, 2 mM L-Glutamine, 20 mM HEPES, 1 mM Sodium Pyruvate, and 1X Penicillin/Streptomycin. To perform THP-1 differentiation, THP-1 monocytes were resuspended in complete media supplemented with 200 ng/mL rhIL-4 (R&D Systems), 100 ng/mL rhGM-CSF (R&D Systems), 20 ng/mL rhTNF-a (PeproTech), and 200 ng/mL ionomycin (Abcam) at a concentration of 2 × 105 cells/mL, followed by incubation at 37°C for 72 hr. Vero E6 (ATCC, CRL-1586, RRID: CVCL_0574, Female) and Vero (ATCC, CCL-81, RRID: CVCL_0059, Female) cells were cultured in DMEM supplemented with 10% FBS, 2 mM L-Glutamine, 10 mM HEPES, 1 mM Sodium Pyruvate, 0.1 mM non-essential amino acids (NEAA), and 1X Penicillin/Streptomycin. 293FT cells (Thermo Fisher, R70007, RRID: CVCL_6911, Female) were cultured in DMEM supplemented with 4.5 g/L Glucose, 10% FBS, 0.1 mM NEAA, 6 mM L-glutamine, 1 mM Sodium Pyruvate, and 1X Penicillin/Streptomycin. Expi293F cells (Thermo Fisher Scientific, A14527, RRID: CVCL_D615, Female) were cultured in Expi293 Expression Medium (Thermo Fisher Scientific, A1435101). All cells were maintained at 37°C and in a 5% CO2 atmosphere, except for Expi293F cells that were cultured at 37°C and in an 8% CO2 atmosphere with 125 rpm orbital shaking.
Viruses
A clinical isolate of SARS-CoV-2 (2019-nCoV/USA-WA1/2020 strain) was obtained from the Centers for Disease Control and Prevention (Harcourt et al., 2020). An mNeonGreen SARS-CoV-2 reporter virus was used as previously described (Xie et al., 2020). SARS-CoV-2 viruses were passaged in Vero CCL81 cells and titrated using a plaque assay on Vero E6 cells (Zang et al., 2020).
Method details
Primary cell cultures
Human primary myeloid cells were generated as described before (Lu et al., 2018). Briefly, human PBMCs were isolated from buffy coat blood obtained by the New York Blood Center (NYBC) from healthy donors. PBMCs were allowed to attach to the bottom of a T75 flask in RPMI 1640 media, supplemented with 2mM L-Glutamine, 0.5% FBS, and 30 μg/mL DNase I (Roche), at 37°C for 1 hr. Cell culture was aspirated and attached cells were gently washed with 30 mL PBS for 3 times. Cells were then cultured in RPMI 1640 media, supplemented with 2mM L-Glutamine, 10% FBS, 1X Penicillin/Streptomycin, 30 ng/mL human GM-CSF (R&D Systems), and 10 ng/mL human IL-4 (R&D Systems) for 5-7 days to promote myeloid cell differentiation. Cells were maintained at 37°C in a 5% CO2 atmosphere.
Plasmids and fusion proteins
The human cDNA library constructs encoding ∼300 full-length myeloid cell-associated membrane proteins were purchased from Genecopoeia (Rockville, MD), DNASU (Tempe, AZ), or were individually cloned by the Wang laboratory at the New York University Grossman School of Medicine. All the genes used for the myeloid cell receptor discovery were cloned into a mammalian expression vector. Human fusion proteins were provided by KACTUS Biosystems (Woburn, MA), purchased from Sino Biological (Wayne, PA) and ACRO Biosystems (Newark, DE), or were individually generated by tagging the extracellular domain with human IgG1 Fc and expressed in Expi293F cells. Human ACE2 (in a pcDNA3.1/nV5-DEST vector) and TMPRSS2 (in a pLX304 lentiviral vector) were used as previously described (Zang et al., 2020). FURIN was cloned into a pLenti6.3/V5-DEST vector with a C-terminal V5 tag and a blasticidin selection marker.
The myeloid cell receptor interaction assay
The myeloid cell receptor discovery approach was modified from a previous study (Wang et al., 2019). ∼300 genes encoding transmembrane proteins were selected based on the myeloid cell-related expression profiles (bioinformatics analysis using BioGPS databases). To express the transmembrane proteins, cDNA library plasmids were individually transfected into HEK293T cells in a 384-well plate (Applied Biosystems, 4307723). Briefly, an equal volume of plasmid (pre-diluted to 4 μg/mL in OptiMEM) and Lipofectamine 2000 (pre-diluted to 7 μL/mL in OptiMEM) were mixed and incubated at room temperature (RT) for 15 minutes (min). 20 μL of the mixture were added to each well of the 384-well plate followed by 30 min incubation at RT. 10,000 HEK293T cells in 40 μL complete medium were then added into each well and incubated at 37°C for 18 hr. For the initial screening experiment, 10 ng of human IgG1 Fc (hFc)-tagged S, S1, and RBD (in total 30 ng) (KACTUS Biosystems) or 30 ng control human IgG1 Fc (Sino Biological), and 45 ng of Alexa Fluor 647 Mouse Anti-Human IgG secondary antibody (SouthernBiotech) were added into each well. The plates were read 24 hr later in the Applied Biosystems (Foster City, CA) 8200 cellular detection system (CDS) and analyzed by the CellProfiler software. Human Fc receptors served as internal positive controls for this assay.
To validate and characterize the S protein binding domains of the identified receptors, HEK293T cells were transfected with individual receptors as described above, followed by the addition of 10 ng of S-hFc (KACTUS Biosystems), S1-hFc (KACTUS Biosystems or this paper), NTD-hFc (this paper), RBD-hFc (KACTUS Biosystems or this paper), CTD-hFc (this paper), or S2-hFc (this paper), as well as 15 ng of Alexa Fluor 647 goat anti-human IgG secondary antibody (Invitrogen) into each well. Data were collected and analyzed as described above.
For the glycan mutagenesis screening, HEK293T cells were transfected with individual receptors or Fc receptor (FCGR2A) as described above. 50 μL WT S1-hFc (this paper) or mutant S1-hFc (this paper) in Expi293F media was added to each well together with 15 ng of Alexa Fluor 647 mouse anti-human IgG secondary antibody (SouthernBiotech). The fluorescence intensity was calculated by CellProfiler software. Binding of S1-hFC (WT or mutant) to receptors was normalized to the protein amount determined by its binding to FCGR2A expressing cells.
ACE2-His competition assay
To test whether ACE2-His could compete for the S protein binding to the host receptors, HEK293T cells were transfected with individual receptors as described above. 10 ng of S-hFc (KACTUS Biosystems) or RBD-hFc (KACTUS Biosystems) (to check the binding to TTYH2) were incubated with 1 μg of ACE2-His (KACTUS Biosystems) overnight at 4°C and then added to each well in a 384-well plate. 15 ng of AF647-conjugated goat anti-human IgG secondary antibody (Invitrogen) was added to each well and incubated for 24 hr. Data was then collected by CDS and analyzed by CellProfiler software.
Soluble receptor blocking assay
To examine the blocking of S protein binding in the presence or absence of soluble receptors, HEK293T cells were transfected with individual receptors as described above. 15 ng of mouse Fc (mFc)-tagged S1-mFc (Sino Biological) or RBD-mFc (Sino Biological) (to check the binding to TTYH2) were incubated with 750 ng of soluble receptors overnight at 4°C and then added to each well in a 384-well plate. 20 ng of AF647-conjugated goat anti-mouse IgG secondary antibody (SouthernBiotech) was added to each well and incubated for 24 hr. Data was then collected by CDS and analyzed by CellProfiler software.
Pseudovirus experiments
HIV-GFP virus pseudotyped with SARS-CoV-2 S protein were generated by co-transfecting pcDNA3.1-SARS-CoV-2-S (a gift from Dr. Lu Lu at Fudan University), pCMV-dR8.2 dvpr (Addgene, 8455) (encoding HIV backbone) and pCDH-GFP (System Bioscience, CD-511B-1) (encoding the GFP reporter) at 1:2:3 mass ratio into Expi293F cells using PEI (Polysciences) at 3:1 (PEI:DNA) mass ratio. Four days post-transfection, the supernatant was harvested and filtered through a 0.45 μm sterile syringe filter, followed by concentration using Amicon Ultra-15 Centrifugal Filter Unit (Millipore, UFC910024). Pseudoviruses were titrated by HIV-1 p24 ELISA Assay (XpressBio) according to the manufacturer’s protocol and stored at −80°C. Cells were co-cultured with the pseudovirus (p24 = ∼60 ng/mL) for 12 hr and resuspended in fresh media, followed by an additional 36 hr incubation before analysis by flow cytometry to determine the GFP expression.
SARS-CoV-2 virus experiments
SARS-CoV-2 virus co-culture
Human PBMC-derived myeloid cells, Vero E6 cells, or HEK293T cells transfected with the indicated receptors were co-cultured with the authentic SARS-CoV-2 (Harcourt et al., 2020) (MOI = 0.5 - 1) or the mNeoGreen SARS-CoV-2 reporter virus (Xie et al., 2020) (MOI = 0.1 - 10) with or without centrifugation at 1200 x g for 2 hr at RT. Media were then changed and cells were incubated at 37°C until analysis.
RNA extraction and quantitative PCR
Total RNA was extracted from cells using Trizol or lysis buffer from commercially available RNA extraction Kit (Thermo Fisher Scientific or QIAGEN) at 24 hr after incubation at 37°C. Reverse transcription was performed with High-Capacity RT kit (Applied Biosystems). RT-PCR was performed using the CFX96 Touch Real-Time PCR Detection System (Bio-Rad) or StepOne Real-Time PCR System (Applied Biosystems) with a 20 μL reaction, composed of 50 ng of cDNA, 10 μL Power SYBR Green master mix (Applied Biosystems) for IL1A, IL1B, IL6, IL8, IL10, IL18, TNF, IFNA2, IFNB, IFNG, TGFB, CXCL3, CXCL10, CXCL11, CCL2, CCL3, CCL5, GAPDH or Taqman master mix (Applied Biosystems) for SARS-CoV-2 N protein, and 200 nM of each forward and reverse primer. All SYBR Green primers and Taqman probes used in this study are listed in Table S2.
RNA-sequencing
Total RNA was extracted from human PBMC-derived myeloid cells in culture with conditioned media (mock) or a clinical isolate of SARS-CoV-2 using Trizol. RNA sample quality was examined by the NanoDrop spectrophotometer (Thermo Fisher) and Bioanalyzer 2100 (Agilent). Libraries were sequenced on the BGISEQ-500 platform. The SE reads were aligned to the hg19 build using Bowtie2 (Langmead and Salzberg, 2012) to map clean reads to reference gene and using HISAT2 to reference genome with the following parameters:–phred64–sensitive -I 1 -X 1000. Reads were counted using Subread and differential gene expression analysis was performed using DESeq2BGI. Data were analyzed using DESeq2.
shRNA knockdown assay
For DC-SIGN and L-SIGN knockdown experiments, shRNAs were cloned into the lentiviral pLKO.1-puro vector (Addgene, 8453). Lentivirus carrying shRNAs were generated by co-transfecting plasmids encoding non-targeting control shRNA or targeting shRNAs, psPAX2 (Addgene, 12260), and pMD2.G (Addgene, 12259) at 3:2:1 mass ratio into 293FT cells using PEI (Polysciences). Four days post-transfection, the supernatant was harvested and filtered through a 0.45 μm sterile syringe filter, followed by concentration using Amicon Ultra-15 Centrifugal Filter Unit (Millipore, UFC910024). Viruses were stored at −80°C. THP-1 cells were infected with the viruses and the medium was changed at 12 hr post-infection. Cells were selected in the presence of 0.5-1 μg/mL puromycin for an additional 48 hr. Cells were collected and knockdown efficiency were analyzed by western blot.
Flow cytometry
To validate the binding of S protein to the myeloid cell receptors, HIV-GFP virus pseudotyped with SARS-CoV-2 S protein were first incubated with HEK293T cells expressing different receptors at 4°C for 45 min. Cells were then incubated with rabbit anti-SARS-CoV-2 S protein antibody (Sino Biological, 40589-T62, dilution 1:200) at 4°C for 45 min, and stained with AF647 donkey anti-rabbit IgG antibody (Biolegend, 406414, dilution 1:500) at 4°C for 45 min. Data were acquired on a ZE5 Cell Analyzer (Bio-Rad) and analyzed by the FlowJo software.
For checking the infection by the mNeonGreen SARS-CoV-2 reporter virus, the cells were washed in PBS followed by fixation using 10% formalin and then resuspended in FACS buffer containing 1X PBS with 0.5% bovine serum albumin and 0.05% sodium azide. Cells were analyzed by an LSRII flow cytometer (BD) or an Accuri C6 (BD, CA) and data was analyzed by FlowJo software.
To check the myeloid receptor expression by PBMC-derived myeloid cells, and cells in BAL fluid isolated from COVID-19 patients, an immuno-profiling antibody set was used as described before to distinguish different cell populations (Yu et al., 2016). Human TruStain FcX (Biolegend, 422302, dilution 1:100) was used for Fc receptor blocking. Cells were stained using the following fluorescently labeled anti-human antibodies: from Biolegend (San Diego, CA): Brilliant Violet 785 anti-human CD14 Antibody (301840, dilution 1:100), Brilliant Violet 711 anti-human CD16 Antibody (302044, dilution 1:100), PerCP/Cyanine5.5 anti-human CD24 Antibody (311116, dilution 1:100), Brilliant Violet 650 anti-human CD123 Antibody (306020, dilution 1:100), Brilliant Violet 605 anti-human CD169 (Sialoadhesin, Siglec-1) Antibody (346010, dilution 1:100), PE/Dazzle 594 anti-human CD206 (MMR) Antibody (321130, dilution 1:100), Brilliant Violet 510 anti-human CD3 Antibody (317332, dilution 1:100), Brilliant Violet 421 anti-human CD209 (DC-SIGN) Antibody (330118, dilution 1:100), and APC anti-human CD301 (CLEC10A) Antibody (354705, dilution 1:100); from eBioscience (San Diego, CA): PE-Cyanine7 anti-human HLA-DR Monoclonal Antibody (25-9956-42, dilution 1:100); from BD Biosciences (San Jose, CA): BUV737 anti-human CD11c (741827, dilution 1:100), BUV496 anti-human CD45 (750179, dilution 1:100), and PE Anti-Human ASGPR 1 (563655, dilution 1:100); and from R&D Systems (Minneapolis, MN); Alexa Fluor® 750 anti-human ACE2 antibody (FAB9332S, dilution 1:100), Alexa Fluor® 700 anti-human DC-SIGNR/CD299 antibody (FAB162N, dilution 1:100), and Alexa Fluor® 488 anti-human LSECtin/CLEC4G antibody (FAB2947G, dilution 1:100). Cells were fixed in 1% paraformaldehyde overnight. Data were acquired on a Cytek® Aurora (Cytek Biosciences) and analyzed by the FlowJo software.
Western blot
HEK293T cells were transfected with FLAG-tagged receptors (Shandong University, P.Wang: pcDNA6B-FLAG-ACE2; vigenebio: CH827318, CH891751, CH899337, CH830386, CH822531, CH886758 for DC-SIGN, L-SIGN, LSECtin, ASGR1, CLEC10A, and TTYH2, respectively) or vector control (vigenebio, PD88001). 24 hr post-transfection, cells were lysed, and the lysates were harvested for western blot using the following antibodies: from Proteintech (Rosemont, IL): mouse anti-GAPDH antibody (60004-1-lg, dilution 1:5,000) and from Sigma-Aldrich (St. Louis, MO): mouse anti-FLAG antibody (F3165, dilution 1:10,000). To check the expression of DC-SIGN and L-SIGN in the undifferentiated or differentiated THP1 cells, cell lysates were harvested for western blot using the following antibodies: from Proteintech (Rosemont, IL): mouse anti-GAPDH antibody (60004-1-lg, dilution 1:5,000) and rabbit anti-DC-SIGN antibody (25404-1-AP, dilution 1:1,000) and from Abcam (Cambridge, MA): rabbit anti-L-SIGN antibody (ab169783, dilution 1:1,000). HRP-conjugated secondary antibodies were purchased from Cell Signaling Technology (Danvers, MA): HRP-conjugated anti-mouse IgG (7076, dilution 1:5,000) and HRP-conjugated anti-rabbit IgG (7074, dilution 1:5,000).
To confirm the expression of TTYH2 in HEK293T, HEK293T transfected with TTYH2 cDNA, and PBMC-derived myeloid cells with or without differentiation, these cells were directly lysed in Laemmli Sample Buffer followed by centrifugation. Supernatant was used for gel electroporation. To assess the expression of TTYH2 in BAL samples isolated from COVID-19 patients, the BAL samples were first lysed in Trizol followed by protein extraction. Briefly, chloroform was added to the Trizol-lysed samples, followed by thorough mix and centrifugation. The top aqueous phase containing RNA was carefully removed by pipetting. DNA was precipitated by adding 100% ethanol to the remaining samples followed by centrifugation. The supernatant containing proteins was collected and proteins were precipitated by adding isopropanol followed by centrifugation. Precipitated proteins were washed twice by 0.3 M guanidine hydrochloride and once by 100% ethanol. Protein pallet was dissolved in 1% SDS solution. Anti-TTYH2 antibody (Invitrogen, PA5-34395) was used at 1:1,000 dilution, and anti-GAPDH and HRP-conjugated secondary antibody were used as described above.
VHH, VHH-Fc, and bispecific nanobody
88 VHH nanobodies were generated from a naive llama VHH library and a humanized VHH nanobody library and selected for high-affinity binding to SARS-CoV-2 S protein through three rounds of S1 panning as described before (Dong et al., 2020). ELISA was performed to examine the binding to S1 and RBD. Selected VHH nanobodies were further tested for their capability to block S protein binding to ACE2, DC-SIGN, L-SIGN, LSECtin, ASGR1, CLEC10A, and TTYH2, by cellular detection system (for ACE2, LSECtin, ASGR1, CLEC10A, and TTYH2) or ELISA (for DC-SIGN and L-SIGN). VHH-Fc was constructed by fusing VHH domain with human IgG1 Fc domain. Based on the results from competitive binding assay and blocking assay, two clones, A8 and G11, were picked to construct A8-G11-Fc bispecific nanobody. A8 and G11 were connected by a Gly-Ser linker. All the Fc-fusion VHH nanobodies were expressed in Expi293F cell and purified on a Protein A column.
To examine the blocking of S protein binding in the presence or absence of VHH nanobody, HEK293T cells were transfected with individual receptors as described above. 10 ng of S1-hFc (KACTUS Biosystems) (to check the binding to ACE2, ASGR1, CLEC10A and LSECtin) or RBD-hFc (KACTUS Biosystems) (to check the binding to TTYH2) were incubated with 50 μL VHH (various amount from 10 μg to 400 μg) overnight at 4°C to allow the binding of VHH to S protein and then added to each well in a 384-well plate. 15 ng of AF647-conjugated anti-human IgG secondary antibody (Invitrogen) were added to each well and incubated for 24 hr. Data was then collected by CDS and analyzed by CellProfiler software.
To examine the blocking of S protein binding in the presence or absence of human Fc-tagged VHH nanobodies, HEK293T cells were transfected with individual receptors as described above. 15 ng of S1-mFc (Sino Biological) or RBD-mFc (Sino Biological) (to check the binding to TTYH2) were incubated with 750 ng of VHH-hFc overnight at 4°C to allow the binding of VHH-hFc to S1-mFc or RBD-mFc and then added to each well in a 384-well plate. 20 ng of AF647-conjugated anti-mouse IgG secondary antibody (SouthernBiotech) was added to each well and incubated for 24 hr. Data was then collected by CDS and analyzed by CellProfiler software.
ELISA
96-well plates (Nunc-468667) were coated with 2 μg/mL recombinant SARS-CoV-2-S1 protein with His tag (Kactus Biosystems, China) overnight. The plates were washed with TBST (TBS + 0.1% Tween-20) three times and blocked using 2% BSA (Sigma) in TBST for 1 hr at 37°C. The wells were incubated with recombinant ACE2 (Sino Biological), DC-SIGN (Sino Biological), L-SIGN (Sino Biological), LSECtin (Acro Biosystems), ASGR1 (R&D Systems), and CLEC10A (Sino Biological) with human Fc tag at 37°C for 6 hr. Following three times washes with TBST, HRP conjugated anti-human Fc tag antibody (Sino Biological, dilution 1:5,000) was added and incubated at 37°C for 1 hr. Plates were washed three times with TBST and 200 μL of the mixture of TMB Substrate Solution (Sino Biological) was added to each well. While protected from light, plates were incubated at room temperature for 20 min. Stop solution (2M H2SO4) was added and the microplate was read at OD450.
VHH was generated from the bacterial culture containing mannan that can block S protein interaction with DC-SIGN and L-SIGN. To overcome this problem, we switched to ELISA and were able to check the capability of VHH to block S protein binding to DC-SIGN and L-SIGN. S1-His (KACTUS Biosystems) were added at 50 ng per well for plate coating at 4°C overnight. The S1-His-coated plate was first blocked using 10% FBS in PBS at RT for 1 hr and then incubated with 100 μL VHH at 37°C for 4 hr. Following washes using PBS supplemented with 0.1% Tween-20 (American Bio, AB02038-00500) to remove free mannan and unbound VHH, 2 μg/mL hFc-tagged DC-SIGN (Sino Biological), 5 μg/mL L-SIGN (Sino Biological) or equal concentration of recombinant hIgG1 (Sino Biological), served as the negative control, were added and incubated at 37°C for 2 hr. Results were assessed by spectrophotometric measurement of absorbance at 450 nm using a FlexStation 3 Multi-Mode Microplate Reader (Molecular Devices, San Jose, CA).
Probelife Kinetic Assay
Fc-tagged antibodies were each diluted in Q Buffer (Probelife) to 10 μg/mL to serve as the loading protein (enough for 8 wells each) for human Fc probes (Probelife). For the association/dissociation steps, the spike S1-mFc Recombinant Protein (Acrobiosystems) was serially diluted (1:2) in Q Buffer starting at 49.02 nM and ending at 0.766 nM (6 serial dilutions), with 0 mM being the negative control. The kinetic assay program was set as: 120 s Baseline, 100 s Loading, 180 s Baseline, 180 s Association, and 900 s dissociation. The shake speed was set to 1,000 rpm and the temperature was set to 30°C.
Structural analysis of SARS-CoV-2 S protein
The site-specific Swiss-Model homology model based on PDB: 6VSB (Wrapp et al., 2020) was used in Figure 2E. Structures of the glycans attached at each glycosylation site are based on the mass spectrometry data (Watanabe et al., 2020). N343 FA2, N603 M5, N165 FA2, and N282 FA2B glycans (glycan naming system from (Watanabe et al., 2020)) are shown as atom-type colored space-filling models, with carbons colored blue for N-acetyl-glucosamine, green for mannose and red for fucose moieties. The position of the bound human ACE2 N-terminal peptidase domain was modeled using the structure of the complex (PDB: 6MOJ) (Lan et al., 2020).
Multiplex cytokine and chemokine assay
Human PBMC-derived myeloid cells were co-cultured with authentic SARS-CoV-2 in the presence of Fc Control protein, ACE2-Fc/L-SIGN-Fc (25 μg/mL for each), or A8-G11-Fc (50 μg/mL) for 24hr. Supernatant was then collected, and the secreted cytokine and chemokine protein levels were measured using a 30-plex array assay (Millipore, HCYTMAG-60K-PX30) according to the manufacturer’s protocol. Briefly, 25 μL of cell culture supernatant were incubated with the magnetic capture beads, washed, and incubated with the biotinylated detection antibodies and PE-conjugated streptavidin. Cytokines and chemokines were recorded on a MAGPIX machine (Luminex) and quantitated via comparison to a standard curve. xPONENT software was used for the data collection and analysis.
Quantification and statistical analysis
Analysis of the COVID-19 BAL fluid dataset
Detection of SARS-CoV-2 expression
The single-cell RNA-seq raw data was collected from the GEO database through the accession number GSE145926. The SARS-CoV-2 and human reference genome were downloaded from the viruSITE website (http://www.virusite.org/archive/2020.2/genomes.fasta.zip) and Ensembl database (http://www.ensembl.org//useast.ensembl.org/info/data/ftp/index.html?redirectsrc=//www.ensembl.org%2Finfo%2Fdata%2Fftp%2Findex.html). The computational framework of Viral-track (Bost et al., 2020) was applied to detect SARS-CoV-2 RNA reads in each cell with default parameters.
Data quality control
The following criteria were applied to filter all the cells: gene number between 200 and 6,000, UMI count > 1,000, and mitochondrial gene percentage < 10%. After filtering, a total of 37,308 cells were left for the downstream analysis.
Dimensionality reduction
The filtered gene barcode matrix was first normalized and logarithmic converted using ‘normalized_total’ and ‘log1p’ methods in the preprocess function of scanpy (https://github.com/theislab/scanpy; Wolf et al., 2018). The top 2,000 highly variable genes were then identified using the ‘highly_variable_genes’ function. PCA was performed on the gene expression of these 2,000 genes. BBKNN (Batch balanced k-nearest neighbors) was performed on the low dimension data to compute the weighted adjacency matrix that removed the batch effect to describe the distance between cells. Then UMAP was performed on the matrix for visualization.
Statistical analysis
To determine SARS-CoV-2 infection, we calculated the number of infected cells, the intersection of infected cells, and the positive cells of the indicated gene. The hypergeometric test was adopted to calculate the p value of the enrichment of SARS-CoV-2 to selected cells on the last step. ‘rank_genes_groups’ in scanpy tools function was invoked to perform differential gene expression analysis. For each set of selected cells, DEGs (differential expression genes) of infected cells and uninfected cells were calculated using the ‘rank_genes_groups’ method, parameter ‘n_genes’ is set to the number of all genes, with other parameters using the default value. A gene was considered significant with a p value < 0.05. Pathway enrichment bubble plots were generated using the BiNGO Application (Maere et al., 2005) and Cytoscape (Shannon et al., 2003).
Statistical analysis of non-scRNAseq data
Two tailed Student’s t test, one-way ANOVA, or two-way ANOVA were applied for statistical analysis in GraphPad Prism 8. P values were calculated and reported as follows: ns, not significant; ∗, p < 0.05; ∗∗, p < 0.01; ∗∗∗, p < 0.001, ∗∗∗∗, p < 0.0001. The error bars in each figure represented standard error of the mean (SEM). n referred to the number of independent experiment unless specified.
Acknowledgments
We thank Dr. Pratip Chattopadhyay and Dr. Nathaniel Landau from New York University Grossman School of Medicine for providing human PBMCs from healthy donors and help with the anti-ACE2 blocking antibody, respectively; Dr. Peihui Wang from Shandong University for providing the pcDNA6B-FLAG-ACE2 plasmid; Dr. Lu Lu from Fudan University for providing the SARS-CoV-2 pseudovirus plasmids; and members of the laboratories of New York University Grossman School of Medicine and Ab Studio for helpful discussions and technical assistance. This work is supported by internal funds provided by the Office of Science & Research (OSR) and the Department of Pathology of New York University Grossman School of Medicine (to J.W.); the Westlake Education Foundation, the Tencent Foundation (XHTX202001008), and the Hangzhou Science and Technology Development Foundation (20202013A05) (to Q.X.); National Institutes of Health (NIH) DDRCC grant P30 DK052574, NIH grants R00 AI135031 and R01 AI150796, and COVID-19 Fast Grants funding (to S.D.); NIH grants R01 AI157155 (to M.S.D.); NIH grants R01 AI143861 and AI143861S1 (to K.M.K.); NIH grants R01-AI059167 (to P.C.); and a Cancer Research Institute Irvington postdoctoral fellowship (to J.H.).
Author contributions
Conceptualization, J.W.; interaction discovery and mutation studies, J.W., Q.L., and J.L.; interaction analysis, J.W., Q.L., Q.X., J.L., and S.Z.; pseudovirus studies, J.L., S.Z., Q.L., J.W., and Q.X.; authentic virus studies, M.F.G.C., M.L.-R., J.S., M.E.K., K.A.S., P.D.-Y., J.L., Q.L., J.C.C.H., S.D., P.C., B.T.M., R.E.C., M.S.D., and K.M.K.; BAL samples, L.N.S.; structural analysis, M.T.; bioinformatics, H.T., X. Ran, X. Ren, F.T., Q.L., J.M., Q.X., A.H., S.B.K., J.W., J.Z., T.W., and Zemin Zhang; nanobodies, Q.L., J.D., J.W., J.L., B.H., and Y.L.; data analysis, Q.L., J.L., Ze Zhang, T.K., X.C., H.D., Z.L., T.M., S.J., S.T.Y., K.M.K., P.L., J.D., and R.Z.; resources, X.C., J.C., I.A., R.Y., and Q.Z.; writing – review & editing, J.W., Q.L., S.D., T.T.S., and Q.X.; visualization, J.W., Q.L., J.L., Q.X., and S.D.; supervision, J.W., Q.X., and S.D.; project administration, J.W., Q.L., Q.X., and S.D.; funding acquisition, J.W., Q.X., and S.D.
Declaration of interests
J.W., Q.L., J.L., B.H., J.D., and Y.L. are named inventors on a patent application that describes the anti-SARS-CoV-2 blocking nanobodies. J.W. is a consultant for Lilly Asia Ventures and is on the Scientific Advisory Board of Rootpath Genomics, which is not relevant to this work. M.S.D. is a consultant for Inbios, Vir Biotechnology, and NGM Biopharmaceuticals and is on the Scientific Advisory Board of Moderna and Immunome. The Diamond laboratory has received unrelated funding support in sponsored research agreements from Moderna, Vir Biotechnology, and Emergent BioSolutions.
Published: May 9, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.immuni.2021.05.006.
Supplemental information
References
- Alvarez C.P., Lasala F., Carrillo J., Muñiz O., Corbí A.L., Delgado R. C-type lectins DC-SIGN and L-SIGN mediate cellular entry by Ebola virus in cis and in trans. J. Virol. 2002;76:6841–6844. doi: 10.1128/JVI.76.13.6841-6844.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Melo D., Nilsson-Payant B.E., Liu W.-C., Uhl S., Hoagland D., Møller R., Jordan T.X., Oishi K., Panis M., Sachs D., et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell. 2020;181:1036–1045.e9. doi: 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bost P., Giladi A., Liu Y., Bendjelal Y., Xu G., David E., Blecher-Gonen R., Cohen M., Medaglia C., Li H., et al. Host-Viral Infection Maps Reveal Signatures of Severe COVID-19 Patients. Cell. 2020;181:1475–1488.e12. doi: 10.1016/j.cell.2020.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwer P.J.M., Caniels T.G., van der Straten K., Snitselaar J.L., Aldon Y., Bangaru S., Torres J.L., Okba N.M.A., Claireaux M., Kerster G., et al. Potent neutralizing antibodies from COVID-19 patients define multiple targets of vulnerability. Science. 2020;369:643–650. doi: 10.1126/science.abc5902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brudner M., Karpel M., Lear C., Chen L., Yantosca L.M., Scully C., Sarraju A., Sokolovska A., Zariffard M.R., Eisen D.P., et al. Lectin-dependent enhancement of Ebola virus infection via soluble and transmembrane C-type lectin receptors. PLoS ONE. 2013;8:e60838. doi: 10.1371/journal.pone.0060838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantuti-Castelvetri L., Ojha R., Pedro L.D., Djannatian M., Franz J., Kuivanen S., van der Meer F., Kallio K., Kaya T., Anastasina M., et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science. 2020;370:856–860. doi: 10.1126/science.abd2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y., Su B., Guo X., Sun W., Deng Y., Bao L., Zhu Q., Zhang X., Zheng Y., Geng C., et al. Potent Neutralizing Antibodies against SARS-CoV-2 Identified by High-Throughput Single-Cell Sequencing of Convalescent Patients’ B Cells. Cell. 2020;182:73–84.e16. doi: 10.1016/j.cell.2020.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.T., Lin Y.L., Huang M.T., Wu M.F., Cheng S.C., Lei H.Y., Lee C.K., Chiou T.W., Wong C.H., Hsieh S.L. CLEC5A is critical for dengue-virus-induced lethal disease. Nature. 2008;453:672–676. doi: 10.1038/nature07013. [DOI] [PubMed] [Google Scholar]
- Chen X., Li R., Pan Z., Qian C., Yang Y., You R., Zhao J., Liu P., Gao L., Li Z., et al. Human monoclonal antibodies block the binding of SARS-CoV-2 spike protein to angiotensin converting enzyme 2 receptor. Cell. Mol. Immunol. 2020;17:647–649. doi: 10.1038/s41423-020-0426-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi X., Yan R., Zhang J., Zhang G., Zhang Y., Hao M., Zhang Z., Fan P., Dong Y., Yang Y., et al. A neutralizing human antibody binds to the N-terminal domain of the Spike protein of SARS-CoV-2. Science. 2020;369:650–655. doi: 10.1126/science.abc6952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen T.M., Sandoval D.R., Spliid C.B., Pihl J., Perrett H.R., Painter C.D., Narayanan A., Majowicz S.A., Kwong E.M., McVicar R.N., et al. SARS-CoV-2 Infection Depends on Cellular Heparan Sulfate and ACE2. Cell. 2020;183:1043–1057.e15. doi: 10.1016/j.cell.2020.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford K.H.D., Eguia R., Dingens A.S., Loes A.N., Malone K.D., Wolf C.R., Chu H.Y., Tortorici M.A., Veesler D., Murphy M., et al. Protocol and Reagents for Pseudotyping Lentiviral Particles with SARS-CoV-2 Spike Protein for Neutralization Assays. Viruses. 2020;12:513. doi: 10.3390/v12050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis B.M., Scharnowske S., Watson A.J. Sequence and expression of a membrane-associated C-type lectin that exhibits CD4-independent binding of human immunodeficiency virus envelope glycoprotein gp120. Proc. Natl. Acad. Sci. USA. 1992;89:8356–8360. doi: 10.1073/pnas.89.17.8356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva R.C., Segat L., Crovella S. Role of DC-SIGN and L-SIGN receptors in HIV-1 vertical transmission. Hum. Immunol. 2011;72:305–311. doi: 10.1016/j.humimm.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly J.L., Simonetti B., Klein K., Chen K.-E., Williamson M.K., Antón-Plágaro C., Shoemark D.K., Simón-Gracia L., Bauer M., Hollandi R., et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science. 2020;370:861–865. doi: 10.1126/science.abd3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Witte L., Abt M., Schneider-Schaulies S., van Kooyk Y., Geijtenbeek T.B. Measles virus targets DC-SIGN to enhance dendritic cell infection. J. Virol. 2006;80:3477–3486. doi: 10.1128/JVI.80.7.3477-3486.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao B., Wang C., Tan Y., Chen X., Liu Y., Ning L., Chen L., Li M., Liu Y., Wang G., et al. Reduction and Functional Exhaustion of T Cells in Patients With Coronavirus Disease 2019 (COVID-19) Front. Immunol. 2020;11:827. doi: 10.3389/fimmu.2020.00827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J., Huang B., Jia Z., Wang B., Gallolu Kankanamalage S., Titong A., Liu Y. Development of multi-specific humanized llama antibodies blocking SARS-CoV-2/ACE2 interaction with high affinity and avidity. Emerg. Microbes Infect. 2020;9:1034–1036. doi: 10.1080/22221751.2020.1768806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z., Diao B., Wang R., Wang G., Wang C., Tan Y., Liu L., Wang C., Liu Y., Liu Y., et al. The Novel Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Directly Decimates Human Spleens and Lymph Nodes. medRxiv. 2020 doi: 10.1101/2020.2003.2027.20045427. [DOI] [Google Scholar]
- Gai J., Ma L., Li G., Zhu M., Qiao P., Li X., Zhang H., Zhang Y., Chen Y., Ji W., et al. A potent neutralizing nanobody against SARS-CoV-2 with inhaled delivery potential. MedComm (Beijing) 2021;2:101–113. doi: 10.1002/mco2.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giamarellos-Bourboulis E.J., Netea M.G., Rovina N., Akinosoglou K., Antoniadou A., Antonakos N., Damoraki G., Gkavogianni T., Adami M.E., Katsaounou P., et al. Complex Immune Dysregulation in COVID-19 Patients with Severe Respiratory Failure. Cell Host Microbe. 2020;27:992–1000.e3. doi: 10.1016/j.chom.2020.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong J., Dong H., Xia Q.-S., Huang Z.Y., Wang D.K., Zhao Y., Liu W.H., Tu S.H., Zhang M.M., Wang Q., Lu F.E. Correlation analysis between disease severity and inflammation-related parameters in patients with COVID-19: a retrospective study. BMC Infect. Dis. 2020;20:963. doi: 10.1186/s12879-020-05681-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gramberg T., Hofmann H., Möller P., Lalor P.F., Marzi A., Geier M., Krumbiegel M., Winkler T., Kirchhoff F., Adams D.H., et al. LSECtin interacts with filovirus glycoproteins and the spike protein of SARS coronavirus. Virology. 2005;340:224–236. doi: 10.1016/j.virol.2005.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant R.A., Morales-Nebreda L., Markov N.S., Swaminathan S., Querrey M., Guzman E.R., Abbott D.A., Donnelly H.K., Donayre A., Goldberg I.A., et al. NU SCRIPT Study Investigators Circuits between infected macrophages and T cells in SARS-CoV-2 pneumonia. Nature. 2021;590:635–641. doi: 10.1038/s41586-020-03148-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grein J., Ohmagari N., Shin D., Diaz G., Asperges E., Castagna A., Feldt T., Green G., Green M.L., Lescure F.X., et al. Compassionate Use of Remdesivir for Patients with Severe Covid-19. N. Engl. J. Med. 2020;382:2327–2336. doi: 10.1056/NEJMoa2007016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han D.P., Lohani M., Cho M.W. Specific asparagine-linked glycosylation sites are critical for DC-SIGN- and L-SIGN-mediated severe acute respiratory syndrome coronavirus entry. J. Virol. 2007;81:12029–12039. doi: 10.1128/JVI.00315-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harcourt J., Tamin A., Lu X., Kamili S., Sakthivel S.K., Murray J., Queen K., Tao Y., Paden C.R., Zhang J., et al. Severe Acute Respiratory Syndrome Coronavirus 2 from Patient with Coronavirus Disease, United States. Emerg. Infect. Dis. 2020;26:1266–1273. doi: 10.3201/eid2606.200516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heger L., Balk S., Lühr J.J., Heidkamp G.F., Lehmann C.H.K., Hatscher L., Purbojo A., Hartmann A., Garcia-Martin F., Nishimura S.I., et al. CLEC10A Is a Specific Marker for Human CD1c+ Dendritic Cells and Enhances Their Toll-Like Receptor 7/8-Induced Cytokine Secretion. Front. Immunol. 2018;9:744. doi: 10.3389/fimmu.2018.00744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillaire M.L., Nieuwkoop N.J., Boon A.C., de Mutsert G., Vogelzang-van Trierum S.E., Fouchier R.A., Osterhaus A.D., Rimmelzwaan G.F. Binding of DC-SIGN to the hemagglutinin of influenza A viruses supports virus replication in DC-SIGN expressing cells. PLoS ONE. 2013;8:e56164. doi: 10.1371/journal.pone.0056164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M., Kleine-Weber H., Pöhlmann S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell. 2020;78:779–784.e5. doi: 10.1016/j.molcel.2020.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.H., Nitsche A., et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020;181:271–280.e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffers S.A., Tusell S.M., Gillim-Ross L., Hemmila E.M., Achenbach J.E., Babcock G.J., Thomas W.D., Jr., Thackray L.B., Young M.D., Mason R.J., et al. CD209L (L-SIGN) is a receptor for severe acute respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. USA. 2004;101:15748–15753. doi: 10.1073/pnas.0403812101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju B., Zhang Q., Ge J., Wang R., Sun J., Ge X., Yu J., Shan S., Zhou B., Song S., et al. Human neutralizing antibodies elicited by SARS-CoV-2 infection. Nature. 2020;584:115–119. doi: 10.1038/s41586-020-2380-z. [DOI] [PubMed] [Google Scholar]
- Kanemaru K., Noguchi E., Tahara-Hanaoka S., Mizuno S., Tateno H., Denda-Nagai K., Irimura T., Matsuda H., Sugiyama F., Takahashi S., et al. Clec10a regulates mite-induced dermatitis. Sci. Immunol. 2019;4:eaax6908. doi: 10.1126/sciimmunol.aax6908. [DOI] [PubMed] [Google Scholar]
- Konieczna P., Schiavi E., Ziegler M., Groeger D., Healy S., Grant R., O’Mahony L. Human dendritic cell DC-SIGN and TLR-2 mediate complementary immune regulatory activities in response to Lactobacillus rhamnosus JB-1. PLoS ONE. 2015;10:e0120261. doi: 10.1371/journal.pone.0120261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan J., Ge J., Yu J., Shan S., Zhou H., Fan S., Zhang Q., Shi X., Wang Q., Zhang L., Wang X. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581:215–220. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q., Wu J., Nie J., Zhang L., Hao H., Liu S., Zhao C., Zhang Q., Liu H., Nie L., et al. The Impact of Mutations in SARS-CoV-2 Spike on Viral Infectivity and Antigenicity. Cell. 2020;182:1284–1294.e9. doi: 10.1016/j.cell.2020.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao M., Liu Y., Yuan J., Wen Y., Xu G., Zhao J., Cheng L., Li J., Wang X., Wang F., et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020;26:842–844. doi: 10.1038/s41591-020-0901-9. [DOI] [PubMed] [Google Scholar]
- Liu L., Chopra P., Li X., Wolfert M.A., Tompkins S.M., Boons G.J. SARS-CoV-2 spike protein binds heparan sulfate in a length- and sequence-dependent manner. bioRxiv. 2020 doi: 10.1101/2020.05.10.087288. [DOI] [Google Scholar]
- Londrigan S.L., Turville S.G., Tate M.D., Deng Y.M., Brooks A.G., Reading P.C. N-linked glycosylation facilitates sialic acid-independent attachment and entry of influenza A viruses into cells expressing DC-SIGN or L-SIGN. J. Virol. 2011;85:2990–3000. doi: 10.1128/JVI.01705-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Q., Grotzke J.E., Cresswell P. A novel probe to assess cytosolic entry of exogenous proteins. Nat. Commun. 2018;9:3104. doi: 10.1038/s41467-018-05556-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R., Zhao X., Li J., Niu P., Yang B., Wu H., Wang W., Song H., Huang B., Zhu N., et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maere S., Heymans K., Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21:3448–3449. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- Marzi A., Gramberg T., Simmons G., Möller P., Rennekamp A.J., Krumbiegel M., Geier M., Eisemann J., Turza N., Saunier B., et al. DC-SIGN and DC-SIGNR interact with the glycoprotein of Marburg virus and the S protein of severe acute respiratory syndrome coronavirus. J. Virol. 2004;78:12090–12095. doi: 10.1128/JVI.78.21.12090-12095.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon C., Baier A.S., Pascolutti R., Wegrecki M., Zheng S., Ong J.X., Erlandson S.C., Hilger D., Rasmussen S.G.F., Ring A.M., et al. Yeast surface display platform for rapid discovery of conformationally selective nanobodies. Nat. Struct. Mol. Biol. 2018;25:289–296. doi: 10.1038/s41594-018-0028-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta P., Porter J.C., Manson J.J., Isaacs J.D., Openshaw P.J.M., McInnes I.B., Summers C., Chambers R.C. Therapeutic blockade of granulocyte macrophage colony-stimulating factor in COVID-19-associated hyperinflammation: challenges and opportunities. Lancet Respir. Med. 2020;8:822–830. doi: 10.1016/S2213-2600(20)30267-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merad M., Martin J.C. Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat. Rev. Immunol. 2020;20:355–362. doi: 10.1038/s41577-020-0331-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merluzzi V.J., Hargrave K.D., Labadia M., Grozinger K., Skoog M., Wu J.C., Shih C.K., Eckner K., Hattox S., Adams J., et al. Inhibition of HIV-1 replication by a nonnucleoside reverse transcriptase inhibitor. Science. 1990;250:1411–1413. doi: 10.1126/science.1701568. [DOI] [PubMed] [Google Scholar]
- Moore J.B., June C.H. Cytokine release syndrome in severe COVID-19. Science. 2020;368:473–474. doi: 10.1126/science.abb8925. [DOI] [PubMed] [Google Scholar]
- Morniroli D., Giannì M.L., Consales A., Pietrasanta C., Mosca F. Human Sialome and Coronavirus Disease-2019 (COVID-19) Pandemic: An Understated Correlation? Front. Immunol. 2020;11:1480. doi: 10.3389/fimmu.2020.01480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nambulli S., Xiang Y., Tilston-Lunel N.L., Rennick L.J., Sang Z., Klimstra W.B., Reed D.S., Crossland N.A., Shi Y., Duprex W.P. Inhalable Nanobody (PiN-21) prevents and treats SARS-CoV-2 infections in Syrian hamsters at ultra-low doses. bioRxiv. 2021 doi: 10.1101/2021.2002.2023.432569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park A., Iwasaki A. Type I and Type III Interferons - Induction, Signaling, Evasion, and Application to Combat COVID-19. Cell Host Microbe. 2020;27:870–878. doi: 10.1016/j.chom.2020.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson B.K., Seethamraju H., Dhody K., Corley M.J., Kazempour K., Lalezari J.P., Pang A.P., Sugai C., Francisco E.B., Pise A., et al. Disruption of the CCL5/RANTES-CCR5 Pathway Restores Immune Homeostasis and Reduces Plasma Viral Load in Critical COVID-19. medRxiv. 2020 doi: 10.1101/2020.05.02.20084673. [DOI] [Google Scholar]
- Perlman S., Dandekar A.A. Immunopathogenesis of coronavirus infections: implications for SARS. Nat. Rev. Immunol. 2005;5:917–927. doi: 10.1038/nri1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto D., Park Y.J., Beltramello M., Walls A.C., Tortorici M.A., Bianchi S., Jaconi S., Culap K., Zatta F., De Marco A., et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature. 2020;583:290–295. doi: 10.1038/s41586-020-2349-y. [DOI] [PubMed] [Google Scholar]
- Pöhlmann S., Baribaud F., Lee B., Leslie G.J., Sanchez M.D., Hiebenthal-Millow K., Münch J., Kirchhoff F., Doms R.W. DC-SIGN interactions with human immunodeficiency virus type 1 and 2 and simian immunodeficiency virus. J. Virol. 2001;75:4664–4672. doi: 10.1128/JVI.75.10.4664-4672.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren X., Wen W., Fan X., Hou W., Su B., Cai P., Li J., Liu Y., Tang F., Zhang F., et al. COVID-19 immune features revealed by a large-scale single-cell transcriptome atlas. Cell. 2021;184:1895–1913.e19. doi: 10.1016/j.cell.2021.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shajahan A., Supekar N.T., Gleinich A.S., Azadi P. Deducing the N- and O-glycosylation profile of the spike protein of novel coronavirus SARS-CoV-2. Glycobiology. 2020;30:981–988. doi: 10.1093/glycob/cwaa042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang J., Ye G., Shi K., Wan Y., Luo C., Aihara H., Geng Q., Auerbach A., Li F. Structural basis of receptor recognition by SARS-CoV-2. Nature. 2020;581:221–224. doi: 10.1038/s41586-020-2179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart S.A., Dykxhoorn D.M., Palliser D., Mizuno H., Yu E.Y., An D.S., Sabatini D.M., Chen I.S., Hahn W.C., Sharp P.A., et al. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA. 2003;9:493–501. doi: 10.1261/rna.2192803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M., Mizuno A. A novel human Cl(-) channel family related to Drosophila flightless locus. J. Biol. Chem. 2004;279:22461–22468. doi: 10.1074/jbc.M313813200. [DOI] [PubMed] [Google Scholar]
- Svajger U., Anderluh M., Jeras M., Obermajer N. C-type lectin DC-SIGN: an adhesion, signalling and antigen-uptake molecule that guides dendritic cells in immunity. Cell. Signal. 2010;22:1397–1405. doi: 10.1016/j.cellsig.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada A., Fujioka K., Tsuiji M., Morikawa A., Higashi N., Ebihara H., Kobasa D., Feldmann H., Irimura T., Kawaoka Y. Human macrophage C-type lectin specific for galactose and N-acetylgalactosamine promotes filovirus entry. J. Virol. 2004;78:2943–2947. doi: 10.1128/JVI.78.6.2943-2947.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang L., Yang J., Liu W., Tang X., Chen J., Zhao D., Wang M., Xu F., Lu Y., Liu B., et al. Liver sinusoidal endothelial cell lectin, LSECtin, negatively regulates hepatic T-cell immune response. Gastroenterology. 2009;137:1498–1508.e1–5. doi: 10.1053/j.gastro.2009.07.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassaneetrithep B., Burgess T.H., Granelli-Piperno A., Trumpfheller C., Finke J., Sun W., Eller M.A., Pattanapanyasat K., Sarasombath S., Birx D.L., et al. DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells. J. Exp. Med. 2003;197:823–829. doi: 10.1084/jem.20021840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimble R.B., Maley F. Optimizing hydrolysis of N-linked high-mannose oligosaccharides by endo-beta-N-acetylglucosaminidase H. Anal. Biochem. 1984;141:515–522. doi: 10.1016/0003-2697(84)90080-0. [DOI] [PubMed] [Google Scholar]
- Vabret N., Britton G.J., Gruber C., Hegde S., Kim J., Kuksin M., Levantovsky R., Malle L., Moreira A., Park M.D., et al. Sinai Immunology Review Project Immunology of COVID-19: Current State of the Science. Immunity. 2020;52:910–941. doi: 10.1016/j.immuni.2020.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls A.C., Park Y.J., Tortorici M.A., Wall A., McGuire A.T., Veesler D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell. 2020;181:281–292.e6. doi: 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Sanmamed M.F., Datar I., Su T.T., Ji L., Sun J., Chen L., Chen Y., Zhu G., Yin W., et al. Fibrinogen-like Protein 1 Is a Major Immune Inhibitory Ligand of LAG-3. Cell. 2019;176:334–347.e12. doi: 10.1016/j.cell.2018.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K., Chen W., Zhang Z., Deng Y., Lian J.-Q., Du P., Wei D., Zhang Y., Sun X.-X., Gong L., et al. CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Signal Transduct. Target. Ther. 2020;5:283. doi: 10.1038/s41392-020-00426-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y., Allen J.D., Wrapp D., McLellan J.S., Crispin M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science. 2020;369:330–333. doi: 10.1126/science.abb9983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisblum Y., Schmidt F., Zhang F., DaSilva J., Poston D., Lorenzi J.C.C., Muecksch F., Rutkowska M., Hoffmann H.-H., Michailidis E., et al. Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. eLife. 2020;9:e61312. doi: 10.7554/eLife.61312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen W., Su W., Tang H., Le W., Zhang X., Zheng Y., Liu X., Xie L., Li J., Ye J., et al. Immune cell profiling of COVID-19 patients in the recovery stage by single-cell sequencing. Cell Discov. 2020;6:31. doi: 10.1038/s41421-020-0168-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf F.A., Angerer P., Theis F.J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19:15. doi: 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrapp D., Wang N., Corbett K.S., Goldsmith J.A., Hsieh C.-L., Abiona O., Graham B.S., McLellan J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367:1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M.F., Chen S.T., Yang A.H., Lin W.W., Lin Y.L., Chen N.J., Tsai I.S., Li L., Hsieh S.L. CLEC5A is critical for dengue virus-induced inflammasome activation in human macrophages. Blood. 2013;121:95–106. doi: 10.1182/blood-2012-05-430090. [DOI] [PubMed] [Google Scholar]
- Wu F., Zhao S., Yu B., Chen Y.M., Wang W., Song Z.G., Hu Y., Tao Z.W., Tian J.H., Pei Y.Y., et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y., Wang F., Shen C., Peng W., Li D., Zhao C., Li Z., Li S., Bi Y., Yang Y., et al. A noncompeting pair of human neutralizing antibodies block COVID-19 virus binding to its receptor ACE2. Science. 2020;368:1274–1278. doi: 10.1126/science.abc2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia S., Lan Q., Su S., Wang X., Xu W., Liu Z., Zhu Y., Wang Q., Lu L., Jiang S. The role of furin cleavage site in SARS-CoV-2 spike protein-mediated membrane fusion in the presence or absence of trypsin. Signal Transduct. Target. Ther. 2020;5:92. doi: 10.1038/s41392-020-0184-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X., Muruato A., Lokugamage K.G., Narayanan K., Zhang X., Zou J., Liu J., Schindewolf C., Bopp N.E., Aguilar P.V., et al. An Infectious cDNA Clone of SARS-CoV-2. Cell Host Microbe. 2020;27:841–848.e3. doi: 10.1016/j.chom.2020.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Wang F., Tian L., Su J., Zhu X., Lin L., Ding X., Wang X., Wang S. Fibronectin and asialoglyprotein receptor mediate hepatitis B surface antigen binding to the cell surface. Arch. Virol. 2010;155:881–888. doi: 10.1007/s00705-010-0657-5. [DOI] [PubMed] [Google Scholar]
- Yu Y.R., Hotten D.F., Malakhau Y., Volker E., Ghio A.J., Noble P.W., Kraft M., Hollingsworth J.W., Gunn M.D., Tighe R.M. Flow Cytometric Analysis of Myeloid Cells in Human Blood, Bronchoalveolar Lavage, and Lung Tissues. Am. J. Respir. Cell Mol. Biol. 2016;54:13–24. doi: 10.1165/rcmb.2015-0146OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang R., Gomez Castro M.F., McCune B.T., Zeng Q., Rothlauf P.W., Sonnek N.M., Liu Z., Brulois K.F., Wang X., Greenberg H.B., et al. TMPRSS2 and TMPRSS4 promote SARS-CoV-2 infection of human small intestinal enterocytes. Sci. Immunol. 2020;5:eabc3582. doi: 10.1126/sciimmunol.abc3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Lin S.M., Chen T.Y., Liu M., Ye F., Chen Y.R., Shi L., He Y.L., Wu L.X., Zheng S.Q., et al. Asialoglycoprotein receptor interacts with the preS1 domain of hepatitis B virus in vivo and in vitro. Arch. Virol. 2011;156:637–645. doi: 10.1007/s00705-010-0903-x. [DOI] [PubMed] [Google Scholar]
- Zhang L., Tian Y., Wen Z., Zhang F., Qi Y., Huang W., Zhang H., Wang Y. Asialoglycoprotein receptor facilitates infection of PLC/PRF/5 cells by HEV through interaction with ORF2. J. Med. Virol. 2016;88:2186–2195. doi: 10.1002/jmv.24570. [DOI] [PubMed] [Google Scholar]
- Zhao D., Han X., Zheng X., Wang H., Yang Z., Liu D., Han K., Liu J., Wang X., Yang W., et al. The Myeloid LSECtin Is a DAP12-Coupled Receptor That Is Crucial for Inflammatory Response Induced by Ebola Virus Glycoprotein. PLoS Pathog. 2016;12:e1005487. doi: 10.1371/journal.ppat.1005487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J., Chu H., Li C., Wong B.H., Cheng Z.S., Poon V.K., Sun T., Lau C.C., Wong K.K., Chan J.Y., et al. Active replication of Middle East respiratory syndrome coronavirus and aberrant induction of inflammatory cytokines and chemokines in human macrophages: implications for pathogenesis. J. Infect. Dis. 2014;209:1331–1342. doi: 10.1093/infdis/jit504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P., Yang X.L., Wang X.G., Hu B., Zhang L., Zhang W., Si H.R., Zhu Y., Li B., Huang C.L., et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T., Su T.T., Mudianto T., Wang J. Immune asynchrony in COVID-19 pathogenesis and potential immunotherapies. J. Exp. Med. 2020;217:e20200674. doi: 10.1084/jem.20200674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Fu B., Zheng X., Wang D., Zhao C., Qi Y., Sun R., Tian Z., Xu X., Wei H. Pathogenic T-cells and inflammatory monocytes incite inflammatory storms in severe COVID-19 patients. Natl. Sci. Rev. 2020;7:998–1002. doi: 10.1093/nsr/nwaa041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J., Kim J., Xiao X., Wang Y., Luo D., Chen R., Xu L., Zhang H., Xiao G., Schoggins J.W., et al. The immune vulnerability landscape of the 2019 Novel Coronavirus, SARS-CoV-2. bioRxiv. 2020 doi: 10.1101/2020.2002.2008.939553. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA-seq raw and processed datasets related to Figure 5B is Gene Expression Omnibus (GEO) database: GSE155106.