

Abstract
Objectives
The increasing success of Chimeric Antigen Receptor (CAR) T cell therapy in haematological malignancies is reinvigorating its application in many other cancer types and with renewed focus on its application to solid tumors. We present a novel CAR against glioblastoma, an aggressive, malignant glioma, with a dismal survival rate for which treatment options have remained unchanged for over a decade.
Methods
We use the human Retained Display (ReD) antibody platform (Myrio Therapeutics) to identify a novel single‐chain variable fragment (scFv) that recognises epidermal growth factor receptor mutant variant III (EGFRvIII), a common and tumor‐specific mutation found in glioblastoma. We use both in vitro functional assays and an in vivo orthotopic xenograft model of glioblastoma to examine the function of our novel CAR, called GCT02, targeted using murine CAR T cells.
Results
Our EGFRvIII‐specific scFv was found to be of much higher affinity than reported comparators reverse‐engineered from monoclonal antibodies. Despite the higher affinity, GCT02 CAR T cells kill equivalently but secrete lower amounts of cytokine. In addition, GCT02‐CAR T cells also mediate rapid and complete tumor elimination in vivo.
Conclusion
We present a novel EGFRvIII‐specific CAR, with effective antitumor functions both in in vitro and in a xenograft model of human glioblastoma.
Keywords: Chimeric Antigen Receptor (CAR), T cells, CAR T cells, EGFRvIII, glioblastoma, immunotherapy, brain cancer
We have made novel chimeric antigen receptor T (CART) cells specific for glioblastoma, a devastating brain tumor with poor survival. This receptor is of extremely high affinity, and the CAR T cells can completely clear brain tumors in mice.

Introduction
Glioblastoma (GBM) is the most aggressive and lethal form of glioma. 1 Glioblastoma diagnoses account for approximately 60% of all adult primary brain tumors, 2 and patients have a 5% 5‐year survival rate. 3 Concomitant treatment with temozolomide and radiotherapy provides a small but significant extension of survival by an average of 2.5 months. 4 Clearly, this has not sufficiently decreased mortality in the majority of patients, and new treatments are critically needed.
Immunotherapeutics are a class of treatments, which provide a targeted tumor‐specific approach, as compared to chemo‐ and radiotherapy. Clinical trials targeting a non‐tumor‐specific protein such as CD19 have been successful; however, such an approach is not appropriate for many tumor types. Current immunotherapy approaches have been focussed on the identification of exquisitely tumor‐specific target antigens 5 ; however, these are rare. The epidermal growth factor receptor (EGFR) is an endogenous, growth‐promoting cell surface protein and has been shown to be highly expressed in glioblastoma. 6 EGFRvIII is the most common EGFR mutation in primary glioblastoma present in approximately 30% of newly diagnosed patients. 7 , 8 , 9 The mutation is caused by the deletion of exons 2–7 (267 amino acids) from the extracellular domain of EGFR, and the resulting conformational change exposes a therapeutically targetable binding region. 10 Importantly, although the expression of EGFRvIII is highly heterogeneous and dynamic even within a tumor, 11 its expression is restricted to malignant tissue. 12
Early studies indicated that the expression of EGFRvIII may associate with chemotherapy 13 and radiotherapy resistance 14 ; however, the recent literature surrounding the use of EGFRvIII expression as a prognostic factor is somewhat controversial 15 , 16 with a more recent meta‐analysis of 14 678 patients finding EGFRvIII expression not to be prognostic. 17 In any case, EGFRvIII has been shown to exert pro‐tumorigenic functions via a combination of enhanced tumor growth via oncogenic signalling pathways such as RAS 18 and PI3K, 19 and a reduction in apoptosis, mediated by increased expression of Bcl family member Bcl‐XL. 20
To date, there have been several different therapeutic approaches to targeting EGFRvIII including vaccine and immunotherapy interventions for glioblastoma. The peptide vaccine rindopepimut (CDX‐110) takes advantage of the novel sequence created between EGFR exons 1 and 8, specifically targeting the peptide sequence at the mutant junction site. The vaccine showed initial therapeutic promise and advanced through phase II trials 21 before failing to significantly enhance patient survival in phase III trials. 22 Bispecific T cell engagers (BiTEs), specific for EGFRvIII, have also been developed, designed to cross‐link EGFRvIII‐expressing tumor cells and T cells via dual single‐chain variable fragment (scFv) binders. Preclinically, these EGFRvIII‐specific BiTEs, in conjunction with human peripheral blood mononuclear cells, significantly controlled the in vivo growth of a patient‐derived xenograft tumor, 23 and a clinical trial is ongoing (NCT03296696).
The final modality investigated for EGFRvIII targeting in glioblastoma is Chimeric Antigen Receptor (CAR) T cells. CAR T cell immunotherapy has enabled the elimination of malignant cells, previously ‘invisible’ to the immune system, and provided excellent therapeutic results in patients with certain relapsed or refractory haematological tumors. 24 , 25 , 26 , 27 CAR T immunotherapy utilises binders such as scFvs to target surface‐expressed tumor antigens and facilitate malignant cell death. The fusion of these scFv antibody binding domains to the endogenous T cell signalling protein CD3ζ redirects the T cell specificity to tumor‐expressed antigens and induces functional T cell responses. Second‐ and third‐generation CARs have evolved to include various components of costimulatory molecules such as CD28 and CD137, or other inducible elements, with receptor design being the focus of intense research in recent times. 28
Multiple CAR T cells specific for the EGFRvIII mutation have been described, in which the scFv binders have been derived from pre‐existing monoclonal antibodies. One therapeutic antibody clone developed by the University of Pennsylvania and Novartis utilises the scFv Clone 2173 (C2173), a humanised construct derived from the murine antibody 3C10. This developed CAR is a second‐generation construct containing a CD137 costimulatory domain with a CD8α hinge and transmembrane domain. 29 Maus and colleagues have reported C2173 CAR T cells demonstrated successful trafficking to the brain and persistence in glioblastoma patients following peripheral infusion. Reassuringly, the vast majority of reported adverse responses were considered grade 2 toxicities or below. Some patients (16%) experienced grade 3 or above neurological toxicities. 30 It is possible these toxicities may have been associated with the disease itself, or in the case of seizures, localised cytokine release was not ruled out.
Another EGFRvIII CAR that has made it to the clinic was developed at the National Cancer Institute (NCI) using Clone 139 (C139) derived from the human 139 antibody. This CAR was designed with a third‐generation signalling tail containing both CD28 and CD137 domains. 31 Both constructs have completed phase I clinical trial testing. In the majority of patients, there were no dose‐limiting toxicities associated with CAR T cell infusion in either trial, with the exception of the C139 clone trial by Goff, Rosenberg and colleagues, who recently reported one patient fatality after administration of a very high dose of greater than 1010 CD3+ cells. 32 Neither trial reported objective responses in secondary measures; however, it should be noted that these measures are difficult to quantify in brain cancer patients.
We now demonstrate the identification and function of the first EGFRvIII‐targeting scFv discovered via screening of a human scFv retained‐display library. These novel EGFRvIII‐specific murine CAR T cells have very high affinity, perform antigen‐dependent killing and cytokine release in vitro, and mediate rapid and complete regression of intracranial EGFRvIII‐expressing glioblastoma human tumors.
Results
Validation of novel scFv specificity to EGFRvIII Protein
A fully human germline scFv library (Ruby) was screened in the Retained Display (ReD) platform for binding to the extracellular domain (ECD) of recombinant EGFRvIII protein. Those that bound were counter‐screened against the wild‐type (WT) EGFR ECD recombinant protein to remove scFvs with cross‐reactivity to EGFR, which is expressed widely on healthy tissues (Human Protein Atlas). A high‐affinity scFv designated GCT02 from the remaining pool was selected for further validation.
We determined the specificity and affinity of the scFv to both recombinant EGFR and EGFRvIII using surface plasmon resonance (SPR) (Figure 1a). Recombinant biotinylated GFP (negative control) (Figure 1a, left) or the biotinylated recombinant GCT02 scFv (Figure 1a, right) was immobilised to a streptavidin‐coupled GLC sensor chip, and the titrated, recombinant EGFR or EGFRvIII protein was injected over the chip at 10–0.12 nm concentrations. There was no detectable ligand binding of either EGFR or EGFRvIII to GFP. The GCT02‐EGFRvIII scFv showed clear specific binding to the EGFRvIII recombinant protein, even at the lowest concentration of 0.12 nm, with no binding of GCT02 to EGFR detected. The affinity of the GCT02 scFv to EGFRvIII was calculated to be 3.27e−10M.
Figure 1.
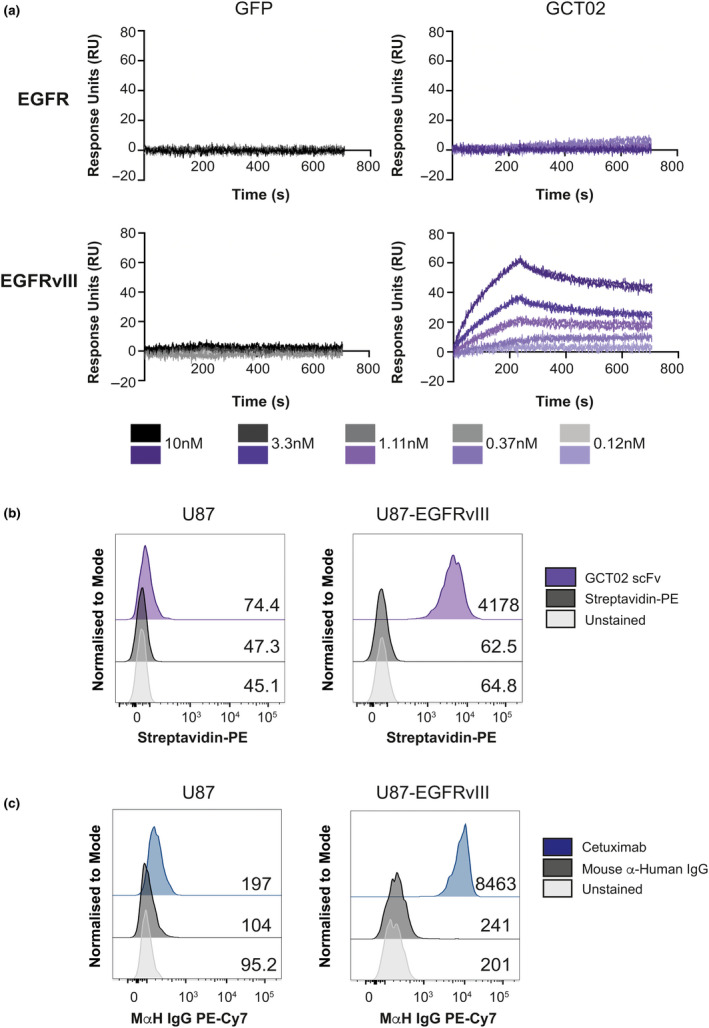
Characterisation of an EGFRvIII‐specific scFv. (a) Surface plasmon resonance (SPR) shows titratable binding of the GCT02 scFv to the recombinant EGFRvIII protein compared with the negative GFP control and no binding to the EGFR wild‐type (WT) protein at the concentrations tested. Shown on each graph is duplicate injection of protein per concentration. (b) Representative flow cytometry histograms of the human glioblastoma cells U87 and U87‐EGFRvIII labelled with recombinant biotinylated GCT02 scFv‐PE binding or streptavidin‐PE control. The mean fluorescence intensity for each plot is shown. (c) Binding of cetuximab to U87 and U87‐EGFRvIII cells. The mean fluorescence intensity for each plot is shown. The flow cytometry backgating is shown in Supplementary figure 8.
To determine whether this highly specific binding of the GCT02 scFv to EGFRvIII was maintained on cells, we utilised the human glioblastoma U87 and generated U87‐EGFRvIII GFP‐Luc cell lines. The cells were labelled with the biotinylated GCT02 scFv and analysed by flow cytometry (Figure 1b). The GCT02 scFv bound to U87‐EGFRvIII, but not the U87 cell line, confirming the specificity of the novel single‐chain antibody. As verification, the U87 and U87‐EGFRvIII cells were also labelled with cetuximab, which binds EGFR proximal to the cell membrane and is thus cross‐reactive with wild‐type EGFR and EGFRvIII. 33 , 34 The U87 parental cells show a slight increase in fluorescence; however, a clear shift was observed with the U87‐EGFRvIII cell line (Figure 1c). The combined results from these experiments verify the specificity of the GCT02 scFv for EGFRvIII protein with no reactivity to EGFR protein.
Generation and in vitro validation of EGFRvIII‐targeted CAR
To determine whether GCT02 scFv would function in a CAR format, second‐generation CAR constructs were generated containing either the GCT02 scFv (Figure 2a, top) or the positive control C2173 scFv 29 (Figure 2a, bottom). These constructs contain a human CD8α hinge, human CD28 transmembrane and costimulatory domains, human CD3ζ T cell signalling domains and an IRES/mCherry for transduction detection. A protein tag (MYC‐tag for GCT02 and FLAG‐tag for C2173) was incorporated into the extracellular stalk region, allowing the direct detection of cell surface CAR expression using antibody labelling 35 . We transduced primary, purified CD4+ or CD8+ murine T cells and evaluated transduction efficiency using flow cytometry by detection of the intracellular fluorophore mCherry (Figure 2b). Both populations of CD4+ and CD8+ T cells were labelled with antibodies to the cell surface CAR, either GCT02‐anti‐MYC (Figure 2c) or C2173‐anti‐FLAG (Figure 2d), and displayed excellent concordance with intracellular mCherry expression.
Figure 2.
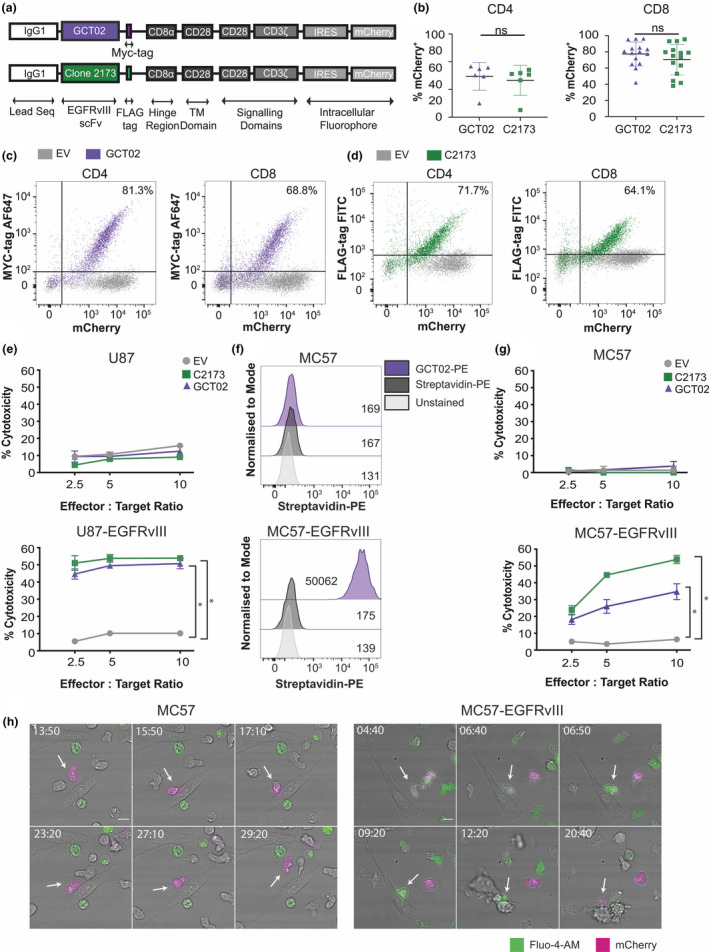
Novel GCT02‐generated chimeric antigen receptor T cells are functional and EGFRvIII‐specific. (a) Schematic of the chimeric antigen receptor (CAR) construct design. CAR constructs are designed with an IgG1 leader sequence, either the GCT02 or C2173 scFv, a cell surface detectable tag (GCT02‐MYC and C2173‐FLAG), CD8α hinge region, CD28 transmembrane domains and CD28‐CD3ζ signalling domains, linked by IRES to the intracellular fluorophore mCherry. (b) Individual transduction efficiencies of the primary CD4+ (left) and CD8+ (right) GCT02 and C2173 murine CAR T cells, as determined by the expression of the intracellular fluorophore mCherry, as a marker of transduction. Transduction efficiency was measured by flow cytometry at days 4–7 post‐T cell activation. Each symbol is representative of one experiment. The mean ± SD transduction efficiency is shown. Statistical analysis was determined by the unpaired two‐tailed t‐test. Representative flow cytometry dotplots of CD4+ and CD8+ T cells at 6 days post‐activation, labelled with anti‐MYC for GCT02 (purple) or anti‐FLAG antibodies for C2173 (green), expressing either empty vector (EV, grey), or (c) GCT02‐MYC EGFRvIII CAR or (d) C2173‐FLAG EGFRvIII CAR. Plots show the expression of the cell surface detectable tag (MYC or FLAG, Y‐axis) and correlation with the intracellular fluorophore mCherry (X‐axis). Percentages in the top right quadrant indicate cells double positive for mCherry and the extracellular protein tag in cells transduced with the EGFRvIII CAR. (e) Cytotoxicity induced by CD8+ GCT02 or C2173 CAR T cells coincubated with chromium‐labelled human glioblastoma cell line U87 or U87‐EGFRvIII for 24 h. Shown is the mean ± SD, n = triplicate samples, representative of three independent experiments. The percentage cytotoxicity was normalised for mCherry expression as determined by flow cytometry. Statistical analysis was determined by the unpaired t‐test. *P‐value < 0.05. (f) Flow cytometry histograms of murine fibrosarcoma cell line MC57 and MC57‐EGFRvIII labelled with recombinant biotinylated GCT02‐PE or streptavidin‐PE control. The mean fluorescence intensity for each peak is shown. (g) Chromium release assay showing percentage cytotoxicity induced by CD8+ GCT02 or C2173 CAR T cells coincubated with chromium‐labelled murine fibrosarcoma cell line MC57‐EGFRvIII and MC57 for 4 h, at varying effector‐to‐target ratios. The mean ± SD is shown, n = triplicate samples, representative of three independent experiments. The percentage cytotoxicity was normalised for mCherry expression as determined by flow cytometry. Statistical analysis was determined by the unpaired t‐test. * P‐value < 0.05. (h) Representative time‐lapse microscopy montages of CD8+ GCT02 CAR T cell coincubated with MC57 (left) or MC57‐EGFRvIII (right) targets. T cells (mCherry+, magenta) are labelled with calcium flux indicator Fluo‐4 (green). Timestamps show min: sec. The scale bar is 10 μm. Note (left) T cells do not initiate cell death in MC57 culture, but (right) T cells induce target cell apoptosis against MC57‐EGFRvIII cells. The flow cytometry backgating is shown in Supplementary figures 9 and 10.
Next, to determine whether the GCT02 CAR maintained specificity in T cells, cytotoxicity was evaluated by chromium lysis killing assay, using parental and EGFRvIII‐expressing human U87 glioblastoma cells (Figure 2e). The murine GCT02 and C2173 CAR T cells effectively mediated the death of the human U87‐EGFRvIII targets potently and equivalently (Figure 2e, bottom) without killing the parental U87 cells (Figure 2e, top), demonstrating that the specificity to EGFRvIII of the GCT02 scFv was maintained in CAR format. Importantly, the human tumor target cells were not affected by this short culture in murine T cell media as shown in the empty vector control conditions and in Supplementary figure 1. To further validate the specificity of the GCT02 CAR T cells for EGFRvIII, we generated an EGFRvIII‐expressing variant of the MC57 mouse fibrosarcoma cell line and labelled them with the recombinant, biotinylated GCT02 scFv, which demonstrated strong, specific binding (Figure 2f). We further demonstrated potent killing by murine GCT02 CAR T cells against the MC57‐EGFRvIII cells (Figure 2g). To further our functional characterisation of GCT02 CAR, we show titratable levels of cytotoxicity with low levels of EGFRvIII expression (Supplementary figure 2).
To investigate that this cytotoxicity was indeed specific and apoptotic, we used live‐cell microscopy to visualise the GCT02 CAR T cell behaviour when recognising and killing EGFRvIII‐expressing targets (Figure 2h Supplementary video 1, 2). CAR T cells, identified by expression of mCherry (magenta), were loaded with the calcium indicator Fluo‐4 AM (green), which increases fluorescence intensity upon antigen recognition. 36 No calcium flux or apoptosis was observed in the murine GCT02 CAR T and parental murine MC57 cultures, indicating no CAR‐independent mechanisms of killing. However, GCT02 CD8+ CAR T cells were observed with increased intracellular calcium upon target recognition, followed by rapid death of the EGFRvIII‐expressing target cells, indicating CAR antigen‐specific recognition. Target cells displayed a morphology consistent with apoptosis, including cell membrane blebbing (Figure 2h right timestamped 20:40).
While most studies examining cytokine release syndrome after CAR T immunotherapy have centred on haematological cancers, and there are nuances when comparing side effects between blood and solid tumors, there can be no debate that the brain is a dangerous location for inflammation. Studies have shown that the biomarkers most associated with severe and serious side effects of CAR T cells include T cell production of IFN‐γ and production of IL‐6, which is thought to be mostly produced by bystander and myeloid cells. 37 A previous study has examined the effect of decreasing the scFv binding affinity in order to prevent on‐target off‐tumor toxicities. 38 Here, we report an approximate 300‐fold increase in the affinity of the novel GCT02, compared with what has been previously reported for C2173 29 and that this increase of affinity does not detrimentally effect the capacity of the scFv to function (by cytotoxicity) as a CAR. Therefore, we examined the secreted cytokine profile of the GCT02 CAR T cells and compared it to the profile of the C2173 CAR T cells (or empty vector (EV) T cells). Coculture supernatant of the GCT02 or C2173 CAR T cells incubated with either U87 or U87‐EGFRvIII cells was analysed for the presence of cytokines and chemokines using a cytokine bead array (Figure 3). Generally, the concentrations of cytokine and chemokines secreted by T cells cocultured with an anti‐CD3 agonist were broadly equivalent between CAR constructs (Figure 3, Supplementary figure 3) and were similar to the levels of cytokine and chemokine secretion when GCT02 CAR T cells were stimulated with plate‐bound anti‐MYC (Supplementary figure 3). This indicated that the GCT02‐CAR T cells had the capacity to secrete cytokine and chemokines at levels equivalent to those induced by CD3 activation.
Figure 3.
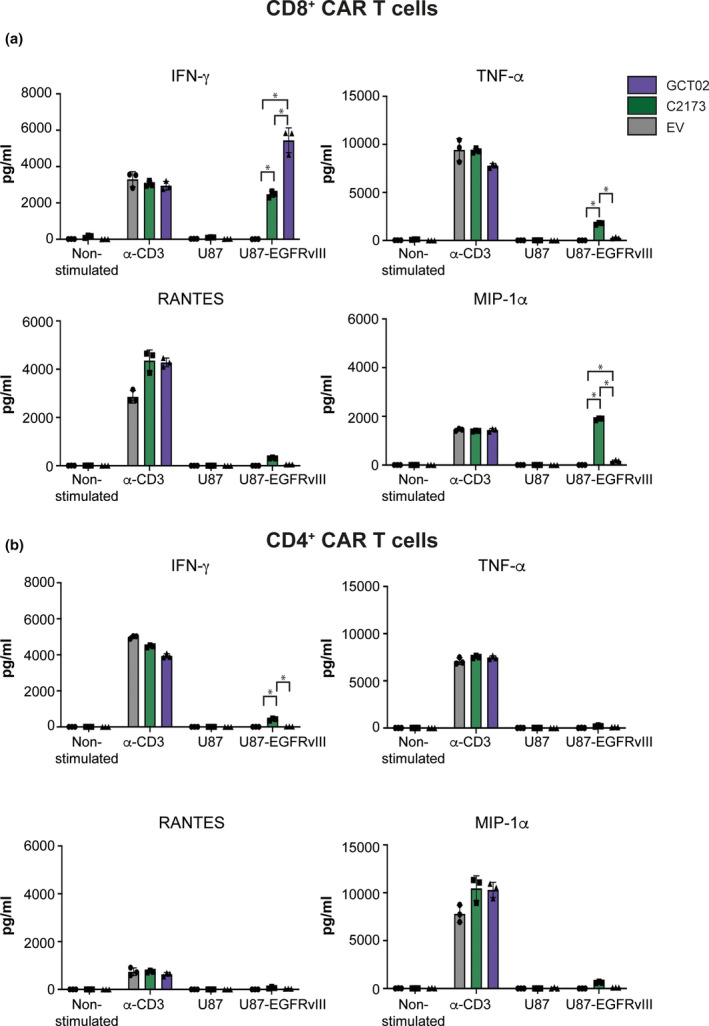
Cytokine and chemokine secretion by CAR T cells. Cytokine bead array quantification after 24 h as assayed from cultures of purified (a) CD8+ or (b) CD4+ CAR T cells that were either activated by agonistic plate‐bound anti‐CD3 antibody as a maximum control, or cocultured with U87 or U87‐EGFRvIII target cells. Shown is the secreted amount of IFN‐γ, TNF‐α, RANTES and MIP‐1α in pg mL−1 of culture. Data are shown as mean ± SD, n = 3 technical replicates, and are representative of 4 experiments. Statistical analysis was determined by 2‐way ANOVA, multiple comparisons. * P‐value < 0.05.
Interestingly, when activated via the CAR, the murine GCT02 CD8+ CAR T cells displayed enhanced secretion of IFN‐γ (Figure 3a) compared with the C2173 CAR T cells when cocultured with the U87‐EGFRvIII cells. The CD8+ GCT02 CAR T cells also secreted significantly less TNF‐α (Figure 3a) and Mip‐1α (Figure 3a) than the C2173 CAR T cells. Also of note, the CD4+ GCT02 CAR T cells showed a reciprocal response with a reduction in IFN‐γ secretion, in response to EGFRvIII (Figure 3b). Critically, there was negligible cytokine secretion when either C2173 or GCT02 CAR T cells were cocultured with U87 cells (Figure 3), further affirming a lack of cross‐reactivity to the parental antigen‐negative tumors. Murine CD4+ and CD8+ GCT02 CAR T cells showed negligible levels of IL‐2 when cocultured with human U87‐EGFRvIII target cells (Supplementary figure 4). However, activation with plate‐bound anti‐MYC or anti‐CD3 induced IL‐2 secretion by GCT02 CAR T cells, indicating that these CAR T cells have the capacity to secrete IL‐2 (Supplementary figure 4). Cytokines such as IL‐4, IL‐10, IL‐6 and MCP‐1 were assayed for, they were not detected (data not shown). Generally, both GCT02 and C2173 CAR T cells had the capacity to produce cytokine as shown by culturing in the presence of anti‐CD3. However, with the exception of IFN‐γ from CD8+ cells, CAR T cells generally produced less cytokine in response to EGFRvIII than when activated via CD3 (Figure 3, Supplementary figure 4). The data from these in vitro assays demonstrate the capacity of CD8+ EGFRvIII‐specific GCT02 CAR T cells to effectively kill EGFRvIII‐expressing target cells (Figure 2e, g, h), and both CD4+ and CD8+ GCT02 CAR T cells generally display reduced cytokine secretion in response to EGFRvIII compared with C2173 CAR T cells (Figure 3, Supplementary figure 4), with the exception of IFN‐γ in CD8+ T cells.
We next evaluated the survival of GCT02 and C2173 CAR T cells after coculture either with anti‐CD3 stimulation or with U87 or U87‐EGFRvIII cells. We found that in response to stimulation by the U87‐EGFRvIII cells, a significant proportion of C2173 CAR but not the GCT02 CAR T cells were lost, indicating superior survival by the GCT02 CAR T cells after ligation through the CAR (Figure 4a–c). Using mCherry as a marker of transduction efficiency and taking into account any subtle differences in transduction efficiency, we quantitated the proportion of surviving CAR T cells after activation. CAR T cell proportions did not change in response to anti‐CD3 stimulation; however, when cocultured with U87‐EGFRvIII targets, there was a substantial specific loss of C2173 CAR T cells, and only minimal loss of the GCT02 CAR T cells, as shown both by the frequency (Figure 4b) and by the absolute number (Figure 4c) of mCherrypos CAR T cells relative to total CD8+ T cells. No differential loss of mCherryneg cell numbers was observed after U87 or U87‐EGFRvIII coculture when comparing C2173 or GCT02 wells, confirming that this cell death was CAR activation‐specific (Supplementary figure 5a). Superior survival of GCT02 CAR T cells was also demonstrated as measured by the absence of DAPI uptake, in contrast to C2173 CAR T cells (Supplementary figure 5b). The mechanism of enhanced death of C2173 CAR T cells is unlikely to be apoptotic, as it was not blocked by the pan‐caspase inhibitor Q‐VD‐OPh (QVD) (Supplementary figure 5c).
Figure 4.
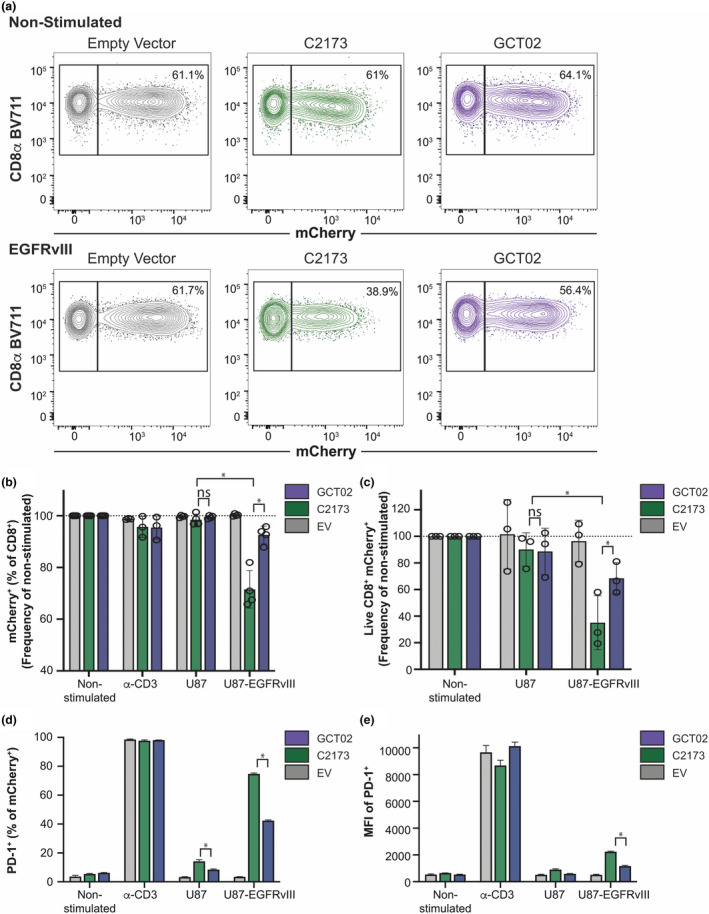
CD8+ GCT02 CAR T cells show increased survival and decreased PD‐1 expression upon exposure to U87‐EGFRvIII cells. Murine CD8+ CAR T cells were cocultured with plate‐bound agonistic CD3 antibody, U87 or U87‐EGFRvIII cells, or left unstimulated (NS) for 16 h before analysis by flow cytometry. Samples were loaded with equivalent counting beads prior to collection, staining and acquisition, to allow direct enumeration where required. (a) Representative flow cytometry plots comparing the percentage of live CD8+/mCherry+ cells in empty vector, C2173‐ and GCT02‐non‐stimulated cultures (top) or U87‐EGFRvIII cocultures (bottom). (b) The proportion of surviving mCherry+ T cells within all CD8+, normalised to non‐stimulated CAR T cells, in each stimulation condition. Each data point represents the mean + SD of triplicate normalised samples within one experiment, with n = 4 independent experiments displayed. Statistical analysis was determined by the unpaired t‐test, *P‐value < 0.05. (c) Direct enumeration of mCherry+ subset survival. CD8+mCherry+ events were counted and normalised to counts per 1 × 104 counting beads. Each normalised count was expressed as a percentage of mean non‐stimulated counts. Each data point represents the mean + SD of triplicate normalised samples within one experiment, with n = 3 independent experiments displayed. Statistical analysis was determined by the unpaired t‐test, *P‐value < 0.05. (d) The percentage and (e) mean fluorescence intensity (MFI) of mCherry+ CD8+ T cells expressing PD‐1 per stimulation condition. Data are shown as mean ± SD, n = 3 replicates, and are representative of 2 experiments. Statistical analysis was determined by 2‐way ANOVA, multiple comparisons. *P‐value < 0.05. The flow cytometry backgating is shown in Supplementary figure 11.
We evaluated the GCT02 and C2173 CAR T cells for the expression of the immune checkpoint marker programmed cell death protein 1 (PD‐1), to look for evidence of potential T cell exhaustion by increased expression of PD‐1. The coincubation of U87‐EGFRvIII cells and either the C2173 or the GCT02 CAR T cells induced PD‐1 expression by both GCT02 and C2173 T cell populations, compared with CARneg T cells (EV) with U87‐EGFRvIII stimulation. However, this level of PD‐1 expression was always to a lesser extent than that induced by anti‐CD3 stimulation (Figure 4d, e). However, the C2173 CAR T cells upregulated PD‐1 to a greater extent than GCT02 (percentage Figure 4d and mean fluorescence intensity (MFI) Figure 4e).
EGFRvIII CAR T cell treatment induces rapid and complete regression of intracranial human tumors
Next, we examined the ability of the GCT02 CAR T cells to eliminate EGFRvIII‐expressing glioblastoma tumors in vivo. We stably transduced the U87‐EGFRvIII cell line with GFP‐Firefly luciferase to enable visualisation using bioluminescence imaging (BLI). We established intracranial U87‐EGFRvIII GFP‐Luc tumors in NOD. Cg‐PrkdcscidIL2rgtmWjl/SzJ (NSG) mice. Tumors were measured at day 7 post‐implantation using IVIS BLI imaging, and the mice were distributed to each treatment group by ranking tumor size and then distributing mice evenly into groups. Mice received a single intravenous dose of 10 million T cells harbouring either empty vector (EV), GCT02 CAR or C2173 CAR T cells at a CD4‐to‐CD8 ratio of 1:1 (Figure 5a). Mice were imaged weekly by bioluminescence (Figure 5b, Supplementary figure 6a), and in vivo tumor cell growth was quantified in response to treatment (Figure 5c, Supplementary figure 6b).
Figure 5.
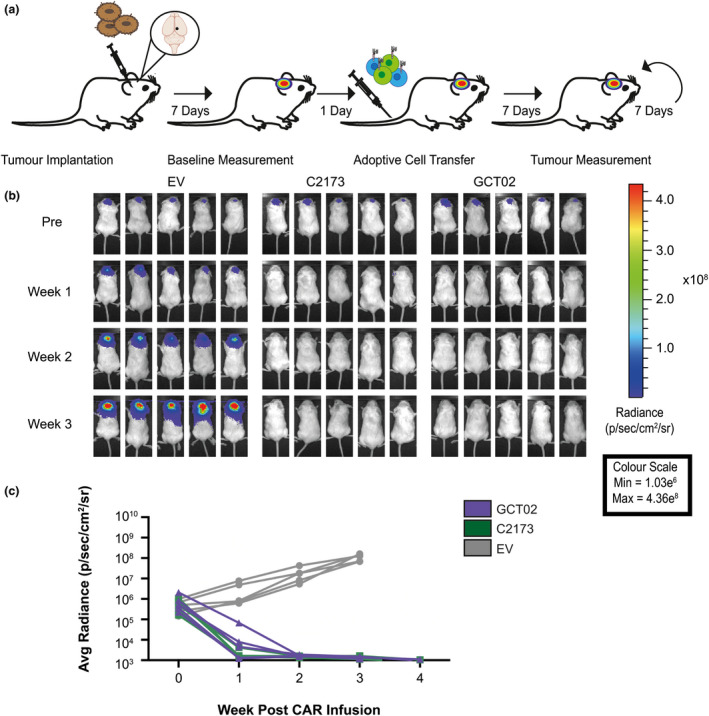
GCT02 CAR T cells effectively induce regression of intracranial tumors. (a) Schematic of the experimental protocol to evaluate the in vivo function of CAR T cells against EGFRvIII‐expressing intracranial tumors. Mice were intracranially injected with U87‐EGFRvIII GFP‐Luc tumor cells and 7 days later were imaged using bioluminescence. The mice were allocated to treatment groups before delivery of a single intravenous dose of 5 × 106 CD4+: 5 × 106 CD8+ T cells day 8 post‐activation GCT02 or C2173 CAR T cells. Empty vector T cells (EV) were injected as a negative control. Bioluminescence was examined weekly to monitor tumor size over time. The tumor injection site is indicated by black circle. (b) Bioluminescence imaging of U87‐EGFRvIII GFP‐Luc tumor‐bearing NSG mice, treated with either empty vector (EV), C2173 or GCT02 CAR T cells. Individual mice from each treatment group are shown for up to 3 weeks after CAR T cell infusion. Representative of two independent experiments. (c) Quantification of tumor growth in the mice in panel b. Tumor size was quantitated in radiance (photons/sec/area/sr). Each line represents a single mouse. n = 5 mice per group. Data are one representative of two independent biological replicates.
As expected, and in accordance with previous studies 39 mice treated with EV T cells failed to control tumor growth (Figure 5b, c). In contrast, strikingly, one week after the single peripheral infusion of CAR T cells, tumors in mice treated with either GCT02 CAR T cells or C2173 CAR T cells were significantly reduced in size, and two weeks post‐CAR T cell infusion, tumors were below the limit of detection. This remained stable for an additional 2 weeks (Figure 5c). These data clearly show that our novel GCT02 CAR T cells can mediate rapid and complete clearance of intracranial EGFRvIII‐expressing tumors. Given that the CAR T cells were delivered intravenously and were able to traffic to the brain as evidenced by tumor regression, we next evaluated the brains of the treated mice using histology to confirm CAR T cell trafficking into the site and validate the complete tumor regression (Figure 6), as indicated by the BLI in live mice.
Figure 6.
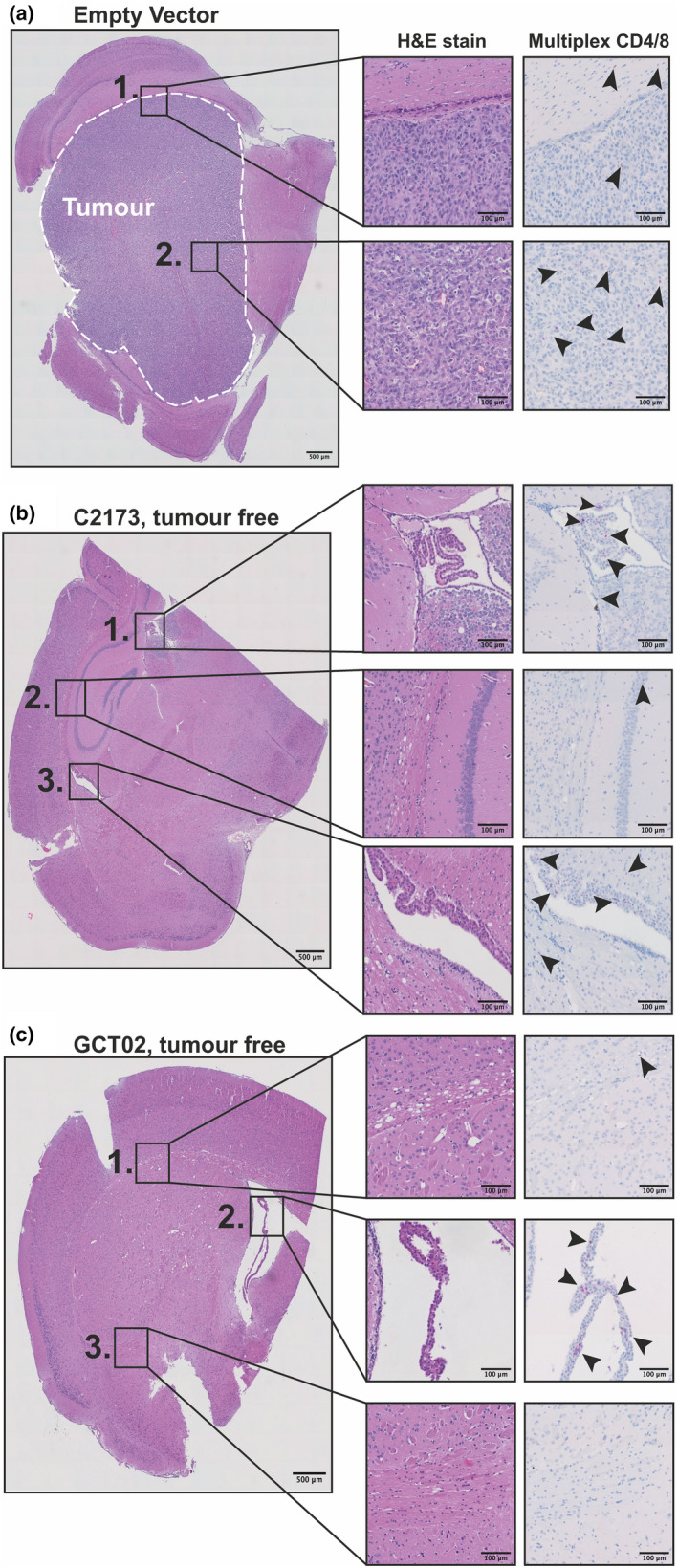
Histological analysis of murine brains treated with CAR T cells. Immunohistochemistry sections of murine brains from same experiment presented in Figure 5. H&E staining of hemisphere of U87‐EGFRvIII‐bearing mouse brain treated with either (a) Empty vector (EV) T cells, (b) C2173 CAR T cells or (c) GCT02 CAR T cells. Inset boxes show H&E staining (left) and multiplex labelling for CD4 and CD8 T cells (right) of (a) EV; Box 1, tumor margin; Box 2, tumor centre. (b) C2173; Box 1, third ventricle; Box 2, hippocampus margin; Box 3, lateral ventricle with choroid plexus. (c) GCT02; Box 1, cerebral white matter showing vacuolation; Box 2, lateral ventricle with choroid plexus; Box 3, central brain tissue. Arrows indicate the location of T cells, CD4+ (magenta) and CD8+ DAB (Brown). One representative image selected per group. The scale bar is 500 μm in images on left and 100 μm smaller boxes on right.
Histologic examination of haematoxylin‐and‐eosin (H&E)‐stained sections of the brains of the EV CAR T cell‐treated group showed that there was large visible tumor present, with morphology consistent with high‐grade glioma as determined by pathologist examination (Figure 6a). Comparatively, the sections from the C2173 (Figure 6b) and GCT02 (Figure 6c) CAR T cell‐treated mice revealed a complete absence of tumor cells. We further labelled brain sections with antibodies to murine CD4 and CD8 to detect the T cells in the brains of mice treated with EV, C2173 and GCT02 CAR T cells 19 days after CAR T cell injection (Figure 6, right inset boxes). Both populations of CD4+ and CD8+ T cells were visible, mainly being detected within periventricular white matter and in the choroid plexus, indicating effective CAR T cell trafficking into the brain.
Despite administering a 1:1 CD4:CD8 CAR T cell ratio intravenously, flow cytometric analysis of organs from CAR T cell‐treated mice 19 days after T cell transfer revealed a significantly enhanced ratio of CD4+:CD8 + CAR T cells across the organs compared with the pre‐infusion ratio (Supplementary figure 7). These data suggest that the infused CD4+ T cells displayed increased survival or proliferation compared with the CD8+ T cells or that a greater fraction of the infused CD8+ T cells had trafficked to an unexamined tissue location.
The field is moving to incorporating various strategies to improve the T cell fitness and diminished propensity for exhaustion of CAR T cells. Therefore, we further investigated CAR T cell trafficking and phenotype in the brain tissue. Pre‐infusion, the majority of both CD4+ and CD8+ CAR T cells retained a CD44+CD62L+ phenotype (Figure 7a, b). Nineteen days after T cell transfer, we identified CD4+ and CD8+ T cells in harvested spleens, lymph nodes, blood and brains of surviving mice and determined changes to phenotype and markers of exhaustion. A majority of both C2173 and GCT02 CAR T cells exhibited a CD44+CD62L+ central memory phenotype prior to infusion, but predominantly exhibited differentiation into a CD44+CD62L− effector/effector memory phenotype in vivo, with the CD44+CD62L− frequency being the highest in the brain and with a greater retention of a CD44+CD62L+ CAR T subset in the spleen (Figure 7a, b).
Figure 7.
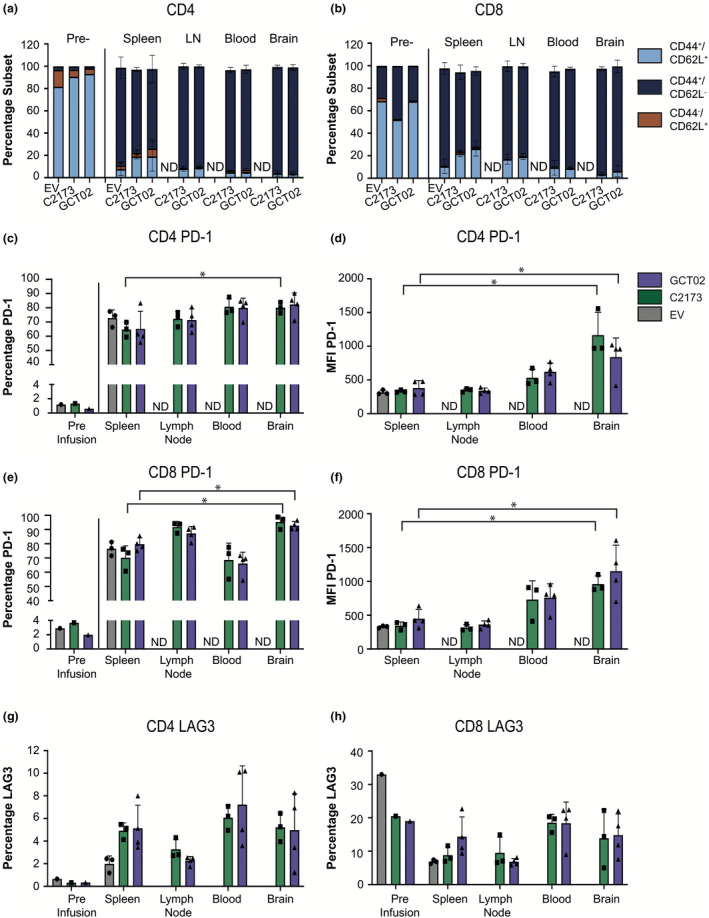
Phenotypic analysis of CAR T cells post‐in vivo infusion shows skewing towards an effector memory phenotype. Quantification of flow cytometric analysis of CAR T cells pre‐infusion (7 days post‐activation) and mice at ethical endpoint post‐CAR infusion. Flow cytometry showing (a) CD3+ CD4+ mCherry+ CAR T cells and (b) CD3+ CD8+ mCherry+ CAR T cells showing the frequency of each phenotypic subset: CD44+/CD62L+ (light blue), CD44+/CD62L− (dark blue) and CD44−/CD62L+ (orange), in each tissue. The presence of EV cells in the lymph node, blood and brain was not determined. Data are shown as mean ± SD for n = 3 mice per group for empty vector and C2173, and n = 4 mice per group for GCT02. (c) The percentage of PD‐1‐positive and (d) PD‐1 surface expression as indicated by MFI of CD4+ mCherry+ CAR T cells. (e) The percentage of PD‐1‐positive and (f) PD‐1 surface expression levels as indicated by MFI of CD8+ mCherry+ CAR T cells. (g) The percentage of CD4+ and (h) CD8+ mCherry+ CAR T cells expressing LAG3. Data are shown as mean ± SD for n = 3 or 4 mice per CAR group. Statistical analysis was determined by using the unpaired t‐test, *P‐value < 0.05. The flow cytometry backgating is shown in Supplementary figures 12 and 13.
T cell failure is commonly associated with a reduced fitness and expression of the coinhibitory markers PD‐1 40 and LAG3, 41 indicators of T cell exhaustion. 42 We examined expression of PD‐1 and LAG3 on GCT02 and C2173 CAR T cells 19 days after their transfer into tumor‐bearing mice, when tumors had been cleared. The CD4+ and CD8+ CAR T cell populations showed a large increase in the proportion of cells expressing PD‐1 compared with the pre‐infusion expression (Figure 7c,e), with equivalent percentages and levels between the GCT02 and C2173 CAR T cell groups in both CD4+ CAR T cells (Figure 7c, d) and CD8+ CAR T cells (Figure 7e, f). Interestingly, there was an equivalent expression of PD‐1 on the EV CAR T cells in the spleen, indicating that the upregulation of PD‐1 was not antigen‐specific. However, the levels of PD‐1 (as determined by MFI) expressed on CD4+ and CD8+ CAR T cells were significantly enhanced in the brain compared with the spleen, indicating that the presence of antigen in this site influenced PD‐1 expression in both CD4+ and CD8+ GCT02 and C2173 CAR T cells (Figure 7d, f). There was a greater variation in the expression of LAG3 by both CD4+ and CD8+ CAR T cells in vivo; however, the CD4+ CAR T cells displayed an enhanced expression of LAG3 upon in vivo infusion (Figure 7g), in contrast to CD8+ T cells (Figure 7h) that remained similar. In summary, strikingly, we have found these novel EGFRvIII‐specific GCT02 CAR T cells to effectively traffic into the brain and mediate complete tumor clearance.
Discussion
Recent advancements in immunotherapy have allowed some of the more difficult tumors to be targeted using antibody redirection strategies. High‐precision CAR T cell targeting of tumors is providing further avenues to patients with glioblastoma, a group with few clinical options. Despite EGFRvIII expression in glioblastoma being heterogeneous, EGFRvIII is a very attractive target for cancer immunotherapy because of its restricted expression on tumor cells, thereby limiting any off‐tumor effects. In fact, in recent CAR T trials targeting EGFRvIII, low levels of neurotoxicities have been observed, with the exception of one patient who received a very high dose of CAR T cells. 30 , 32 In this study, we have identified the first de novo human EGFRvIII‐specific scFv using a fully germline human scFv library. This scFv, GCT02, was found to be EGFRvIII‐specific and importantly displayed no binding to the wild‐type EGFR protein. We generated second‐generation CAR T cells, containing a CD28 costimulation and CD3ζ signalling tail and obtained high cell surface expression of the CAR with up to 85% of CD4+ and CD8+ T cells expressing the receptor. Importantly, in this study we compared the function of our novel CAR with another EGFRvIII‐specific clone, C2173, 29 which has been shown to be also highly specific for EGFRvIII, yet has an approximately 300‐fold lower reported affinity than the GCT02 EGFRvIII‐specific CAR (GCT02 scFv, KD = 0.327 nm (Figure 1a) compared with the reported affinity of the C2173 scFv, KD = 101 nm 29 ).
It has been hypothesised that a high‐affinity scFv limits the capacity of the CAR T cells to discriminate between high antigen expression on tumors and low antigen expression on healthy tissue. 43 Rational design and affinity tuning of single‐chain antibodies have reported affinities of 1–16 nm to be optimal when targeting ErbB2 38 , 44 . Affinity tuning the single chain of the CAR may be an important consideration when targeting antigens with a wide tissue distribution and a low level of expression on healthy tissue, such as ErbB2 (HER‐2) and EGFR. 38 It is possible that a higher affinity antibody may offer functional gains in potency, given the tumor‐specific nature of EGFRvIII. 45
The large differences in affinity did not influence capacity for cytotoxicity, with the GCT02 CAR T cells mediating efficient lysis of tumor targets, both in vitro and in vivo. Previous studies have examined the role of scFv affinity in tumor lysis and found affinity tuning the binding domains may influence T cell signalling strength, as determined by ERK phosphorylation. 46 High‐affinity scFvs have been associated with stronger T cell signalling and enhanced cytokine production, but off‐tumor effects are not a major concern when targeting a highly tumor‐specific protein such as EGFRvIII. 47 High signal strength associated with affinity and the integration of additional stimulatory signals received by the T cells, including costimulation and cytokine inputs, have all been shown to influence T cell fate and functional responses. 48 Given that cytokine release syndrome is a major adverse response associated with CAR T cell therapy, we examined the capacity of the GCT02 CAR T cells to secrete inflammatory cytokines and found no correlation of increased cytokine or chemokine release with enhanced affinity. In fact, with the exception of CD8+ T cell secretion of IFN‐γ, the murine GCT02 CAR T cells displayed a reproducible reduction in the secretion of TNF‐α and RANTES compared with C2173. There was also an 11‐fold reduction in secretion of the MIP‐1α, which chemoattracts macrophages, monocytes and neutrophils. To our knowledge, the role of MIP‐1α in the context of glioblastoma is not well understood, although it may have an important role in modulating central nervous system inflammatory states and the regulation of inflammatory responses across the blood–brain barrier, as reviewed by Schaller and colleagues. 49 Macrophages and brain microglia are responsive to chemotactic signals provided by MIP‐1α. 50 As these cells can have a significant immunosuppressive role in the brain and have been shown to have a suppressive effect on T cells, it is tempting to speculate that a decrease in circulating MIP‐1α may decrease the trafficking of immunosuppressive cells into the tumor environment and influence T cell interaction with microglia. In the current model, it is difficult to dissect the influence of decreased MIP‐1α secretion by murine GCT02 CAR T cells on the anti‐tumor response to U87‐EGFRvIII glioblastoma and future studies will focus on the examination of murine GCT02 and C2173 CAR T cells in an immune‐competent model.
The modest cytokine production profiles combined with robust anti‐tumor killing are key features of a desirable CAR T cell response. Another key feature of optimal CAR T cell therapy is T cell persistence. After coculture with U87‐EGFRvIII tumor targets cells, murine GCT02 CAR T cells demonstrated enhanced survival, as compared to the C2173 CAR T cells. Activation‐induced cell death (AICD) has been previously observed in T cells undergoing repeated antigen stimulation. 51 In this study, antigen density was equivalent on target cells, and therefore, a direct comparison between GCT02 and C2173 CAR T cells could be made, with the scFv being the only structural difference. Interestingly, we did see superior survival of our novel GCT02 CAR T cells in response to EGFRvIII, in relation to the comparator C2173 clone. Previous studies have shown that perhaps de‐tuning the affinity of CARs can increase therapeutic index against tumors in mice. 38 Work by Maus, Sadelain and colleagues has shown that a MHC‐restricted antibody‐based CAR requires a lower (TCR‐like) affinity for function. 52 In a recent Nature paper from Sadelain and colleagues, CAR T cell trogocytosis (a process by which target antigen is transferred to T cells upon detachment) was shown to regulate reversible antigen loss and drive escape of low antigen tumors, and also promote fratricide of T cells and T cell exhaustion. 53 Whether or not trogocytosis, and subsequent fratricide, is influenced by the affinity of the CAR–target interaction has not, to our knowledge, ever been examined. We also do not know how the affinity of these two CARs influences their on–off rate on the target cells, and we anticipate that the influence of target antigen density and functional avidity on CAR function and future CAR design will be the subject of intense future research for the field.
In this study, we utilised a model of U87‐EGFRvIII glioblastoma to evaluate murine GCT02 CAR T cell function. With only one intravenous injection of the GCT02‐CAR T cells, complete tumor regression was observed, indicating efficient T cell infiltration into the brain. Immunohistochemical analysis revealed no evidence of residual tumor after GCT02 CAR T cell treatment, as opposed to the empty vector‐treated groups, which displayed very large tumors. In the murine GCT02 CAR T cell‐treated brains, there was some white matter vacuolation, thought to be a reactive change related to CAR T cell treatment. Even after tumor clearance, CD4+ and CD8+ T cells could be detected in the brain indicating infiltration and evidence of T cell persistence. The complete tumor regression was compelling. Greater insight would be gained in future studies upon evaluation of GCT02 CAR T cell function in an immunocompetent host, with a full complement of immune cells, including glial cancer stem cells and microglial cells, which strongly contribute to the immunosuppressive tumor microenvironment by secretion of cytokines and chemokines. 54
The choice of costimulation domain expressed in the CAR has also been shown to influence persistence, with CD137 costimulatory domain containing CAR T cells found to persist longer than CARs containing CD28 costimulatory domains in haematological cancers. 25 , 55 However, the choice of CAR design is likely to be influenced by the nature of the tumor type. Future studies of GCT02 CAR T cells will examine various costimulation domains and designs in the evaluation of long‐term tumor regression.
Expression of inhibitory receptor checkpoint markers has been associated with T cell exhaustion and a decrease in CAR T cell efficacy. There was a significantly lower proportion of murine GCT02 CAR T cells expressing the immune checkpoint marker PD‐1 compared with C2173 T cells in vitro; however, PD‐1 expression was equivalent in vivo. Interestingly, even non‐transduced T cells (mCherry−) upregulated PD‐1 post‐infusion (data not shown), indicating that PD‐1 upregulation can occur independent of antigen exposure in vivo and that some upregulation of PD‐1 is imprinted upon initial T cell activation. This is not surprising given that PD‐1 is upregulated after primary activation and the T cells are activated with CD3 and CD28 prior to transduction with the CAR, and even non‐tumor‐specific bystander T cells have been shown to express PD‐1. 56 Consequently, adoptive cell transfer therapies may benefit from combination with an anti‐PD‐1 or other immune checkpoint blocking monoclonal antibodies to enhance T cell responses. The first checkpoint inhibitor trials for glioblastoma (CheckMate 143) found that although anti‐PD‐1 blocking antibody nivolumab was well tolerated by patients, the results published from phase III trials demonstrated no increase in overall survival with nivolumab, compared with the control cohort receiving an anti‐angiogenic antibody bevacizumab. 57 The first clinical trial investigating combination therapy of CAR T cells and checkpoint inhibition for glioblastoma is in progress. The University of Pennsylvania and Novartis is evaluating EGFRvIII‐specific CAR T cell therapy in combination with PD‐1 inhibition for glioblastoma (NCT03726515). Whether or not an enhanced T cell response with high‐affinity GCT02 CAR T cells in combination with checkpoint inhibition would offer a survival advantage will be the focus of future studies.
In this study, we also report an apparent survival advantage of CD4+ CAR T cells over the CD8+ CAR T cells in vivo. We identified a higher proportion of CD4+ CAR T cells in the organs of mice receiving CAR T cells (GCT02, C2173 and EV) compared with CD8+ CAR T cells, despite an infusion ratio of 1:1. In 2005, Moeller and colleagues published the first evidence that both antigen‐specific CD4+ and CD8+ T cells were required to mediate effective tumor clearance in vivo. 58 The skewing towards a remaining pool of CD4+ T cells could be attributed to either the accumulation of CD8+ T cells in a location not investigated, or preferential survival or proliferation of the CD4+ T cell population compared with CD8+ T cells. Refining CAR T cell therapies, particularly when targeting the brain, does require a deeper analysis of the effective T cell subsets. In a recent study, Brown and colleagues have reported superior CD4+ T cell‐mediated clearance of glioblastoma, with the mechanism proposed to be superior potency and survival of the CD4+ subset and rapid exhaustion of CD8+ T cells. 59
In this study, we have identified and developed a novel and highly specific EGFRvIII‐targeted CAR. The tumor‐specific expression of the EGFRvIII mutation makes this protein an excellent target for immunotherapeutic intervention, and we have shown that our GCT02‐EGFRvIII‐targeted CAR T cells mediate very effective tumor clearance. In glioblastoma, antigen specificity, limiting the secretion of pro‐inflammatory cytokines and chemokines, avoiding antigen escape and overcoming immunosuppression will be the key focus of CAR T cell design and combination therapies. The future of glioblastoma treatment regimens is likely to include a combination of targeted approaches such as CAR T therapy, other genetically engineered products and small molecules, with the standard treatments of chemotherapy and radiotherapy. This multifactorial approach may prove to more effectively eliminate malignant cells, reducing the likelihood of antigen escape and refractory disease, and, more importantly, improve the survival rates for glioblastoma patients for whom survival rates are currently so poor.
Methods
Cell lines and culture
The human glioblastoma cell line U87 was kindly provided by Rodney Luwor (Royal Melbourne Hospital, the University of Melbourne). The murine MC57 cell line was kindly provided by researchers at the Peter MacCallum Cancer Centre. The U87 and MC57 cell lines were lentivirally transduced to stably express a non‐signalling variant of the EGFRvIII mutation (EGFRvIIIns, amino acids 1‐26, G mutation, 298‐671 cloned into pFUGW addgene # 14883, kind gift from Marco Herold, Walter and Eliza Hall Institute of Medical Research (WEHI), Melbourne). We refer to these cells as U87‐EGFRvIII or MC57‐EGFRvIII throughout the manuscript. Five days post‐double transduction, the transduced cells were surface‐labelled with the GCT02‐biotinylated scFv (Myrio Therapeutics, Melbourne), and streptavidin‐PE secondary (Becton Dickinson (BD) Pharmingen, New Jersey) and EGFRvIII positive cells were sort‐purified using flow cytometry.
The human U87‐EGFRvIII cells were transduced with a lentivirus encoding GFP‐Firefly luciferase (pFUGW‐Luc‐T2A‐GFP) to enable in vivo tracking of tumor cells.
The U87‐EGFRvIIIHigh and U87‐EGFRvIIILow cell lines were generated by lentiviral transduction of pFUGW plasmid containing EGFRvIII non‐signalling virus (as mentioned above). The EGFRvIIIHigh and EGFRvIIILow cells were FACS‐sort‐purified using GCT02 IgG monoclonal antibody (Myrio Therapeutics, Melbourne), followed by mouse anti‐human IgG (BV711, Clone G18‐145, BD Biosciences, New Jersey).
The human U87 panel of cell lines was maintained in Roswell Park Memorial Institute Media (RPMI) (WEHI, Melbourne) supplemented with 10% heat‐inactivated foetal calf serum (FCS) (Sigma‐Aldrich, St Louis), 100 U mL−1 penicillin and 100 μg mL−1 streptomycin.
The human embryonic kidney cells (HEK293T), the murine MC57 parental cells and MC57‐EGFRvIII cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen, Carlsbad) supplemented with 10% heat‐inactivated foetal calf serum (FCS) (Sigma‐Aldrich, St Louis).
The selected murine T cells were maintained in RPMI media (Gibco, Life Technologies, Waltham) supplemented with 10% heat‐inactivated foetal calf serum (FCS) (Sigma‐Aldrich, St Louis), 2 mm glutamax, 100 U mL−1 penicillin and 100 μg mL−1 streptomycin, 0.1 mm MEM non‐essential amino acids, 10 mm HEPES buffer solution, 1 mm sodium pyruvate and 50 μm 2‐mercaptoethanol. This media also contained 100 IU mL−1 recombinant human interleukin‐2 (rhIL‐2) (PeproTech, Rocky Hill) (referred to as T cell media).
Cell lines were tested, and STR profile was authenticated by CellBank Australia.
Vectors and constructs
The CAR vectors for GCT02‐MYC‐CD28‐CD3ζ‐IRES‐mCherry and C2173‐FLAG‐CD28‐CD3ζ‐IRES‐mCherry CAR constructs were generated using Gibson Assembly cloning of gene blocks containing GCT02‐MYC or C2173‐FLAG inserted into the retroviral vector pMSCV‐IRES‐mCherry (a kind gift from the Call Lab, WEHI, Melbourne) containing human CD8αhinge, human CD28 transmembrane domain and human CD28‐CD3ζ signalling domains.
The EGFRvIII non‐signalling gene construct was generated using Gibson Assembly cloning of a gBlock designed to contain amino acids 1–26, G mutation and amino acids 298–671 of EGFR into a lentiviral pFUGW vector. This non‐signalling EGFRvIII was truncated intracellularly, proximally to the transmembrane domain by removing the signalling tail (539 amino acids).
Human retained display screen
Recombinant soluble EGFRvIII protein (Cat# EGI‐H52H4) and the wild‐type EGFR (Cat# EGR‐H5222) consisting of the extracellular domains, and without the transmembrane or internal domains, were purchased from ACROBiosystems (Newark). Recombinant EGFRvIII was labelled with the chemical fluorophores DyLight 405 NHS Ester (Thermo Fisher, Waltham Cat# 46400) and ATTO 488 NHS ester (ATTO‐TEC, Cat# AD 488‐31) according to the manufacturer’s instructions. Recombinant EGFR was labelled with DyLight 405 NHS ester. The Retained Display (ReD) protein display platform 60 and Ruby libraries 61 were used to screen for scFv antibodies with human germline scaffolds that would selectively target the EGFRvIII form over the wild‐type EGFR protein. ReD entails expression of the Ruby scFv libraries in the Escherichia coli cytoplasm as either lambdoid bacteriophage‐linked or capsid‐linked fusions. ReD enables logic‐gated clone selection, which was employed here to select for binders that bound to EGFRvIII and not EGFR. Specifically, following two rounds of panning of the Ruby libraries using EGFRvIII as the target protein, the panning output was switched to the ReD modality and clones specific to EGFRvIII‐ATTO488 were isolated by FACS. In later rounds of FACS, an EGFR labelled with Dy405 was added along with EGFRvIII‐ATTO488 to select for EGFRvIII‐specific scFv clones. Individual clones were expressed in the E. coli cytoplasm with a N‐terminal His6 tag and a C‐terminal fusion to the AviTag 62 peptide to respectively purify and biotinylate the soluble protein.
Surface plasmon resonance
The experiment was performed using ProteOn XPR36 instrument (Bio‐Rad, Hercules) at 25°C in phosphate‐buffered saline (PBS) containing 0.05% Tween (PBS‐T). Streptavidin was diluted into 10 mm sodium acetate (pH 4.0), and 300 response units (RU) were immobilised to three flow cells of the GLC sensor chip by amine coupling. 200–400 RU of biotinylated GCT02 and GFP scFvs were captured onto separate flow cells of the streptavidin‐coupled sensor chip. A separate flow cell containing immobilised streptavidin alone served as a control channel.
Recombinant EGFR and EGFRvIII (ACROBiosystems, Newark HEK293T source, HIS tag) protein was reconstituted at 1 μg μL−1 in PBS before dilution into PBS with 0.05% Tween and injection over the sensor chip at the concentrations indicated (Figure 1a) (flow rate 30 μL per min). GFP protein controlled for non‐specific scFv binding. 10 mm glycine HCl buffer (pH 3) was used to strip injected protein from the chip between injections. Results from at least two independent injections were analysed. After subtraction of data from control cells, interactions were analysed with ProteOn Manager software (version 2.1). The equilibrium dissociation constant (KD) values were derived from kinetic fit analysis using 1:1 Langmuir binding model.
Flow cytometry
For determination of recombinant scFv or cetuximab (Erbitux, Clifford Hallam Healthcare, Keysborough) binding to the target tumor cells, cell pellets of U87 and U87‐EGFRvIII GFP‐Luc cells were labelled with 10 μg mL–1 of biotinylated GCT02 scFv, or 10 μg mL–1 of cetuximab for 25 min at 4°C, in the dark. For determination of EGFRvIII expression on U87‐EGFRvIIIHigh and U87‐EGFRvIIILow cell lines, 5 μg mL–1 of GCT02 monoclonal IgG antibody (generated by Myrio Therapeutics, Melbourne) was used to label the pellets of target cells for 40 min at 4°C, in the dark. Cells were washed in phosphate‐buffered saline (PBS) containing 2% bovine serum albumin and 0.4% EDTA (FACS Buffer), and centrifuged at 548 RCF for 5 min, before labelling with secondary antibodies: streptavidin‐PE for scFv labelling (Becton Dickinson (BD) Pharmingen, New Jersey), mouse anti‐human IgG‐PE‐Cy7 (Clone G18‐145, BD Biosciences, New Jersey) for cetuximab‐labelled cells or mouse anti‐human IgG‐BV711 (Clone G18‐145, BD Biosciences, New Jersey) for GCT02 IgG‐labelled cells for 25 min at 4°C in the dark. Cells were washed in FACS buffer, pelleted at 548 RCF for 5 min and resuspended in FACS buffer for analysis using a BD Fortessa X20 flow cytometer. Data were analysed using FlowJo™ software (version 10).
Transduction efficiency and CAR cell surface expression were assessed by flow cytometry. 1x105 transduced murine T cells were labelled with antibodies to MYC (GCT02) (Clone 9B11, Cell Signaling Technology, Danvers) or FLAG‐tag (C2173) (Clone M2, Sigma‐Aldrich, St Louis) for 1 hour at 4°C. Cells were washed in FACS buffer and resuspended in FACS buffer containing anti‐CD3ζ‐AF700 (Clone 500A2, BD Pharmingen, New Jersey), anti‐CD8‐BV711 (Clone 53‐6.7, BioLegend, San Diego) and anti‐CD4‐FITC (Clone GK1.5, WEHI‐Bundoora, Melbourne), for 25 min at 4°C, in the dark. Cells were washed in FACS buffer and resuspended in FACS buffer for analysis using a BD Fortessa X20 flow cytometer. Cell surface CAR expression was determined by gating on CD3+/CD4+ or CD3+/CD8+ cells, then evaluating concordance between mCherry and either MYC‐tag (GCT02) or FLAG‐tag (C2173) expression.
In vitro T cell transduction and culture
Human embryonic kidney cells (HEK293T) were retrovirally transfected using Fugene transfection reagent following the manufacturer’s protocols (Promega, Madison). Lymph nodes from 6‐ to 8‐week‐old C57BL/6 mice were processed to a single‐cell suspension, and CD4+‐positive or CD8+‐positive EasySep mouse isolation kits (Stemcell Technologies, Vancouver) were used, according to the manufacturer’s protocol. The purified T cells were stimulated with murine α‐CD3/α‐CD28 Dynabeads (Life Technologies, Carlsbad) at a bead‐to‐cell ratio of 1:1 for 24 h, in T cell media containing 100 IU recombinant human interleukin (IL)‐2 (rhIL‐2) (PeproTech, Rocky Hill). Spinoculation of T cells was performed at 24 and 48 h post‐activation using retrovirus from 293T cells and retronectin‐coated plates (Takara Bio, Kusatsu). Transduced T cells were maintained in T cell media containing 100 IU rhIL‐2.
Chromium release assay
CAR T cell cytotoxicity was assessed using a chromium release assay, eight days post‐activation. Target cells were labelled with 100μCi 51Cr for 1 h at 37°C. After labelling, target cells were coincubated with effector T cells to a total volume of 200 μL at effector‐to‐target ratios (E:T) i.e. 20:1 – 2.5:1, reducing by twofold. Target and effector cells were coincubated for 4 or 24 h in murine T cell media at 37°C after which cells were centrifuged and supernatant (~25 μL) was harvested for analysis using a ‐emission counter (Perkin‐Elmer, Waltham). The percentage lysis was determined as (sample counts – spontaneous counts)/ (maximum counts – spontaneous counts) × 100. Percentage lysis was normalised to mCherry expression as pre‐determined by flow cytometry.
Cytometric bead array
In cytometric bead array experiments, seven to eight days post‐activation, antigen‐specific CAR T cells were quantitated based on Tag+ and mCherry expression and corrected for viable antigen‐specific cells before activation, either by culturing on antibody‐coated plates or with antigen‐expressing cells, and cytokine production was assessed. The agonistic antibody against CD3 (Clone 145‐2C11, BioXcell, Lebanon, USA, and MYC‐tag (Clone 9B11, Cell Signaling Technology, Danvers, USA) was pre‐coated into 96‐well flat bottom plates (10 μg mL–1 2C11, 1/1000 MYC‐tag in PBS), at 4°C overnight, before washing with PBS. 1 × 105 antigen‐specific CAR T cells were added to each well, in triplicate, and incubated for 4 h at 37°C, 5% CO2. The CAR T cells were seeded into 96‐well plates at 1:1 E:T with U87WT or U87‐EGFRvIII GFP‐Luc cells and coincubated in murine T cell media at 37°C, 5% CO2. Culture supernatants were collected at 4 and 24 h and stored at –20°C until analysis.
Analysis of secretion of cytokine and chemokine was measured from 10 μL of culture supernatant using a Cytokine Bead Array (CBA) Flex sets (Becton Dickinson), for mouse IFN‐γ, TNF‐α, IL‐2, IL‐4, IL‐6, IL‐10, MIP‐1α (CCL3), MIP‐1β (CCL4), MCP‐1 (CCL2) and RANTES (CCL5) according to the manufacturer’s instructions. Samples were analysed on a FACS VERSE using FCAP Array software version 3.0 (Becton Dickinson (BD) Biosciences, New Jersey).
CAR T cell and target coculture assay
Murine CAR T cells were cocultured for 16 h in sextuple with one of four stimulation conditions: non‐stimulated (media‐alone), agonistic CD3 plate‐bound antibody (Clone 145‐2C11, BioXcell, Lebanon, USA), 1 × 104 human U87 cells or 1 × 104 human U87WT.EGFRvIIIns GFP‐Luc cells. After 16 h, the cultures were loaded with equivalent numbers of non‐antibody‐binding counting beads (BD, New Jersey Negative Control CompBead Plus), dissociated, collapsed into triplicate wells and transferred to a new plate for antibody labelling. Cells were incubated with either MYC‐tag (GCT02) or FLAG‐tag (C2173) for 1 h on ice. Cells were washed with FACS buffer and labelled with a cell surface murine antibody cocktail for 30 min containing anti‐CD3‐AF700 (BD Pharmingen New Jersey), anti‐CD8‐BV711 (BioLegend, San Diego), anti‐PD‐1 PE‐Cy7 (BioLegend, San Diego), anti‐LAG‐3 (CD223) APC (BD Pharmingen, New Jersey), anti‐CD69 BV786 (BD Horizon, New Jersey) and DAPI (live/dead). The expression of these surface markers on each population was determined by flow cytometry using a BD Fortessa X20 flow cytometer, and data analysis was performed in FlowJo™ software v10 (BD, New Jersey). Where relevant, CAR T cells were directly enumerated by determining cell subset and counting bead numbers, and normalising CAR T cell number to 1 × 104 counting beads to allow comparison across samples.
Live‐cell imaging
Interactions between CD8+ GCT02 EGFRvIII‐specific murine CAR T cells and human tumor cells were assessed by time‐lapse live‐cell microscopy in a 37°C and 5% CO2‐controlled chamber, using a previously published protocol. 63 T cells were labelled with 1 µm Fluo‐4‐AM + 0.02% (wt/vol) Pluronic F‐127 carrier (Thermo Fisher, Waltham) at 37°C, 5% CO2 for 20 min. T cells were then added to Ibidi µ‐slide chambers (Ibidi®, Martinsried) containing target cells at 1:1 E:T ratio. Chamber slides were mounted on a heated stage within a temperature‐controlled chamber maintained at 37°C and constant CO2 concentration of 5% using a gas incubation system (Zeiss, Stuttgart). Optical sections were acquired through the centre of the cells by sequential scans of Fluo‐4 (excitation 488 nm), mCherry (excitation 561 nm) and transmitted light on a Leica SP8 using the HC PL APO 40×/1.30 Oil CS2 objective using the LASX 5.1 software. For the 488‐nm and 561‐nm channels, the pinhole was set to 4.2 AU, resulting in the XY pixel size of 227.27 nm. Images were acquired between frames every 10–20 s for up to 90 min. Post‐acquisition analysis was performed on FIJI software. 64
Mice
The 6‐ to 8‐week‐old C57BL/6 mice or NOD. Cg‐PrkdcscidIL2rgtmWjl/SzJ (NSG) mice were bred under specific pathogen‐free conditions at WEHI Kew facility, and maintained in the animal facility at the Walter and Eliza Hall Institute of Medical Research (WEHI) (Parkville, Victoria, Australia). All mouse experiments were conducted with ethical approval from the WEHI animal ethics committee (approval 2019.020).
Xenograft models
For orthotopic xenograft model of human glioblastoma, 5 × 104 U87‐EGFRvIII GFP‐Luc cells were implanted intracranially using a stereotactic frame into 5‐ or 6‐week‐old NSG mice, with 5 mice per group. The injection site was 2 mm right of bregma at the coronal suture to a depth of 3 mm into the brain. Prior to surgery, mice were intraperitoneally injected with analgesics and antibiotics, and the antibiotics was continued for three days post‐surgery. Mice were monitored and weighed once daily for 2 weeks post‐surgery, then once per week for the remainder of the experiment, in accordance with ethical guidelines. Mice were assigned to treatment groups based on tumor measurements biased against the experimental group and injected intravenously via the tail vein with day 8 post‐activation 5 × 106 CD4+: 5 × 106 CD8+ murine CAR T cells 7–8 days post‐surgery.
In vivo imaging
Bioluminescent imaging was used as a surrogate measure for intracranial tumor size. Approximately 7 days post‐intracranial tumor cell delivery, mice were imaged using the IVIS imaging system (IVIS Lumina III Series Hardware, Perkin‐Elmer, Waltham). Each mouse received an intraperitoneal injection of D‐luciferin (Promega, Madison), and 6 min later, mice were anaesthetised with isoflurane inhalant (initial 4%, maintained at 2%). Mice were placed onto the platform in the IVIS system, and metal dividers were placed between the mice to reduce signal being detected in neighbouring mice. Anaesthetised mice were imaged over a maximum of 2 min, before removing mice and monitoring recovery. Tumor growth was monitored over time, and the experiment was repeated twice. Image analysis was performed using the Living Image Software (Perkin‐Elmer, Waltham, software version 4.7.2), and average radiance values (Radiance/cm/sec/sr) were plotted in GraphPad Prism.
Histology
At experimental endpoint, mice were euthanised and the brains fixed for 24 h in 10% formalin and transferred to 80% ethanol. Brains were embedded in paraffin, and tissue was sliced into 5‐μm sections. These paraffin sections were stained with haematoxylin and eosin (H&E) or murine anti‐CD4 (Clone 4SM95, Invitrogen, Carlsbad) and anti‐CD8 antibodies (Synaptic Systems, Goettingen). The slides were scanned at ×20 magnification using the Vectra Polaris (Akoya Biosciences®, Marlborough). Murine brain tissue sectioning and staining was performed by the Histology Facility at the Walter and Eliza Hall Institute. Blinded pathological analysis of H&E‐, CD4‐ and CD8‐stained sections was performed by a certified pathologist (CD, Royal Children’s Hospital).
Flow cytometric analysis at experimental endpoint
At experimental endpoint of the intracranial tumor experiments in which mice were treated with CAR T cells, some organs were taken for analysis by flow cytometry. The brain, spleen, lymph nodes and blood (in the form of a cardiac bleed) were isolated from mice from each treatment group (n = 3 or 4 mice per group). Organs were processed through a 70‐µm filter to a single‐cell suspension and prepared for antibody staining. Blood was collected in a heparin/EDTA tube. Red blood cells were lysed by incubation in red cell removal buffer (156 mm ammonium chloride, 11.9 mm sodium bicarbonate, 0.097 mm EDTA, WEHI, Melbourne) and centrifugation at 548RCF for 5 min at 4°C. Cells were labelled with a cell surface murine antibody cocktail containing anti‐CD3‐AF700 (BD Pharmingen, New Jersey), anti‐CD4‐BV421 (BD Horizon, New Jersey), anti‐CD8‐BV711 (BioLegend, San Diego), anti‐PD‐1 (BD Pharmingen, New Jersey), LAG‐3 (CD223)‐APC (BD Pharmingen, New Jersey), anti‐CD27‐BV605 (BioLegend, San Diego), anti‐CD28‐PE‐Cy7 (eBioscience, San Diego), CD44‐BV786 (BD Horizon, New Jersey) and anti‐CD62L‐BV510 (BD Horizon, New Jersey). Samples were analysed using a BD Fortessa X20 flow cytometer and FlowJo™ software v10 (BD, New Jersey).
Incucyte assay
To monitor the morphology of the U87‐EGFRvIII cells over time in mouse T cell media, the live‐cell‐based assay system Incucyte was used. The Incucyte platform images the cell culture repeatedly and measures target cell update of propidium iodide (PI, Calbiochem), as a surrogate marker of cell death. In these assays, 7.5 × 103 target cells were seeded at an identical density in triplicate in the presence of 50 μm PI. Assay wells were imaged every hour for 24 hr, and PI fluorescence was monitored.
Study design and statistical analysis
Each experiment was performed multiple times in biological replicates with the numbers of replicate experiments listed in the corresponding figure caption. Data are presented as mean ± SD, or as stated in the figure captions. All statistical analysis was performed using GraphPad Prism software version 8.4.3 (GraphPad Software). Asterisks refer to P < 0.05. Statistical significance was determined by various statistical tests, as indicated in the figure captions.
Author Contribution
Rebecca C Abbott: Formal analysis; Investigation; Methodology; Visualization; Writing‐original draft; Writing‐review & editing. Daniel J Verdon: Formal analysis; Investigation; Methodology; Project administration; Supervision; Visualization; Writing‐original draft; Writing‐review & editing. Fiona M Gracey: Formal analysis; Investigation; Methodology. Hannah E Hughes‐Parry: Investigation; Methodology; Writing‐review & editing. Melinda Iliopoulos: Formal analysis; Funding acquisition; Investigation; Writing‐review & editing. Katherine Watson: Investigation; Methodology; Writing‐review & editing. Matthias Mulazzani: Investigation; Methodology; Writing‐review & editing. Kylie Luong: Formal analysis; Funding acquisition; Investigation; Writing‐review & editing. Colleen D'Arcy: Data curation; Formal analysis; Investigation; Methodology; Writing‐review & editing. Lucy Sullivan: Data curation; Formal analysis; Investigation; Methodology; Writing‐review & editing. Ben R Kiefel: Data curation; Formal analysis; Investigation; Methodology; Resources; Supervision; Writing‐review & editing. Ryan S Cross: Conceptualization; Data curation; Formal analysis; Investigation; Methodology; Resources; Supervision; Validation; Visualization; Writing‐review & editing. Misty Rayna Jenkins: Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Resources; Software; Supervision; Validation; Visualization; Writing‐original draft; Writing‐review & editing.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
Acknowledgments
We thank the following for financial support: NHMRC, Robert Connor Dawes Foundation, Carrie’s Beanies for Brain Cancer and Isabella and Marcus Foundation. We thank Rodney Luwor (Royal Melbourne Hospital, the University of Melbourne) for providing cell lines. MRJ is funded by NHMRC Investigator Grant (APP1172858). RSC is funded by Cure Brain Cancer Foundation Fellowship. We thank Dr Ruth Mitchell, Professor Kate Drummond, Professor Mark Rosenthal, Pete Smith and Matt Beasley for helpful discussions.
References
- 1. Lim M, Xia Y, Bettegowda C, Weller M. Current state of immunotherapy for glioblastoma. Nat Rev Clin Oncol 2018; 15: 422–442. [DOI] [PubMed] [Google Scholar]
- 2. Chistiakov DA, Chekhonin IV, Chekhonin VP. The EGFR variant III mutant as a target for immunotherapy of glioblastoma multiforme. Eur J Pharmacol 2017; 810: 70–82. [DOI] [PubMed] [Google Scholar]
- 3. Brain and Other Central Nervous System Cancers. Canberra: Australian Institute of Health and Welfare; 2017; p. 80.
- 4. Stupp R, Mason WP, van den Bent MJ et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. New Eng J Med 2005; 352: 987–996. [DOI] [PubMed] [Google Scholar]
- 5. Abbott RC, Cross RS, Jenkins MR. Finding the keys to the CAR: identifying novel target antigens for T cell redirection immunotherapies. Int J Mol Sci 2020; 21: 515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schwechheimer K, Huang S, Cavenee WK. EGFR gene amplification ‐ rearrangement in human glioblastomas. Int J Cancer 1995; 62: 145–148. [DOI] [PubMed] [Google Scholar]
- 7. Faulkner C, Palmer A, Williams H et al. EGFR and EGFRvIII analysis in glioblastoma as therapeutic biomarkers. Br J Neurosurg 2015; 29: 23–29. [DOI] [PubMed] [Google Scholar]
- 8. Heimberger AB, Suki D, Yang D, Shi W, Aldape K. The natural history of EGFR and EGFRvIII in glioblastoma patients. J Transl Med 2005; 3: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Feldkamp MM, Feldkamp MM, Lala P et al. Expression of activated epidermal growth factor receptors, ras‐guanosine triphosphate, and mitogen‐activated protein kinase in human glioblastoma multiforme specimens. Neurosurgery 1999; 45: 1442–1453. [DOI] [PubMed] [Google Scholar]
- 10. Frederick L, Wang XY, Eley G, James CD. Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res 2000; 60: 1383–1387. [PubMed] [Google Scholar]
- 11. Del Vecchio CA, Giacomini CP, Vogel H et al. EGFRvIII gene rearrangement is an early event in glioblastoma tumorigenesis and expression defines a hierarchy modulated by epigenetic mechanisms. Oncogene 2013; 32: 2670–2681. [DOI] [PubMed] [Google Scholar]
- 12. Moscatello DK, Holgado‐Madruga M, Godwin AK et al. Frequent expression of a mutant epidermal growth factor receptor in multiple human tumors. Cancer Res 1995; 55: 5536–5539. [PubMed] [Google Scholar]
- 13. Nagane M, Levitzki A, Gazit A, Cavenee WK, Huang HJS. Drug resistance of human glioblastoma cells conferred by a tumor‐specific mutant epidermal growth factor receptor through modulation of Bcl‐XL and caspase‐3‐like proteases. Proc Nat Acad Sci USA 1998; 95: 5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mukherjee B, McEllin B, Camacho CV et al. EGFRvIII and DNA double‐strand break repair: a molecular mechanism for radioresistance in glioblastoma. Cancer Res 2009; 69: 4252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Montano N, Cenci T, Martini M et al. Expression of EGFRvIII in glioblastoma: prognostic significance revisited. Neoplasia 2011; 13: 1113–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Felsberg J, Hentschel B, Kaulich K et al. Epidermal growth factor receptor variant III (EGFRvIII) positivity in EGFR‐amplified glioblastomas: prognostic role and comparison between primary and recurrent tumors. Clin Cancer Res 2017; 23: 6846–6855. [DOI] [PubMed] [Google Scholar]
- 17. Thuy MN, Kam JK, Lee GC et al. A novel literature‐based approach to identify genetic and molecular predictors of survival in glioblastoma multiforme: Analysis of 14,678 patients using systematic review and meta‐analytical tools. J Clin Neurosci 2015; 22: 785–799. [DOI] [PubMed] [Google Scholar]
- 18. Prigent SA, Nagane M, Lin H et al. Enhanced tumorigenic behavior of glioblastoma cells expressing a truncated epidermal growth factor receptor is mediated through the Ras‐Shc‐Grb2 pathway. J Biol Chem 1996; 271: 25639–25645. [DOI] [PubMed] [Google Scholar]
- 19. Moscatello DK, Holgado‐Madruga M, Emlet DR, Montgomery RB, Wong AJ. Constitutive activation of phosphatidylinositol 3‐kinase by a naturally occurring mutant epidermal growth factor receptor. J Biol Chem 1998; 273: 200–206. [DOI] [PubMed] [Google Scholar]
- 20. Nagane M, Coufal F, Lin H, Bögler O, Cavenee WK, Huang HJS. A Common Mutant Epidermal Growth Factor Receptor Confers Enhanced Tumorigenicity on Human Glioblastoma Cells by Increasing Proliferation and Reducing Apoptosis. Cancer Res 1996; 56: 5079. [PubMed] [Google Scholar]
- 21. Schuster J, Lai RK, Recht LD et al. A phase II, multicenter trial of rindopepimut (CDX‐110) in newly diagnosed glioblastoma: the ACT III study. Neuro Oncol 2015; 17: 854–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Weller M, Butowski N, Tran DD et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII‐expressing glioblastoma (ACT IV): a randomised, double‐blind, international phase 3 trial. Lancet Oncol 2017; 18: 1373–1385. [DOI] [PubMed] [Google Scholar]
- 23. Gedeon PC, Schaller TH, Chitneni SK et al. A rationally designed fully human EGFRvIII:CD3‐targeted bispecific antibody redirects human T cells to treat patient‐derived intracerebral malignant glioma. Clin Cancer Res 2018; 24: 3611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Maude SL, Barrett DM, Rheingold SR et al. Efficacy of humanized CD19‐targeted chimeric antigen receptor (CAR)‐modified T cells in children and young adults with relapsed/refractory acute lymphoblastic leukemia. Blood 2016; 128: 217.27207794 [Google Scholar]
- 25. Maude SL, Frey N, Shaw PA et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. New Engl J Med 2014; 371: 1507–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Brentjens RJ, Davila ML, Riviere I et al. CD19‐targeted T cells rapidly induce molecular remissions in adults with chemotherapy‐refractory acute lymphoblastic leukemia. Sci Trans Med 2013; 5: 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Park JH, Rivière I, Gonen M et al. Long‐term follow‐up of CD19 CAR therapy in acute lymphoblastic leukemia. New Engl J Med 2018; 378: 449–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hughes‐Parry HE, Cross RS, Jenkins MR. The evolving protein engineering in the design of chimeric antigen receptor T cells. Int J Mol Sci 2019; 21: 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Johnson LA, Scholler J, Ohkuri T et al. Rational development and characterization of humanized anti‐EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci Trans Med 2015; 7: 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. O'Rourke DM, Nasrallah MP, Desai A et al. A single dose of peripherally infused EGFRvIII‐directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Trans Med 2017; 9: 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Morgan RA, Johnson LA, Davis JL et al. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Human Gene Ther 2012; 23: 1043–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Goff SL, Morgan RA, Yang JC et al. Pilot trial of adoptive transfer of chimeric antigen receptor‐transduced T cells targeting EGFRvIII in patients with glioblastoma. J Immunother 2019; 42: 126–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Aboud‐Pirak E, Hurwitz E, Pirak ME, Bellot F, Schlessinger J, Sela M. Efficacy of antibodies to epidermal growth factor receptor against KB carcinoma in vitro and in nude mice. J Natl Cancer Inst 1988; 80: 1605–1611. [DOI] [PubMed] [Google Scholar]
- 34. Jutten B, Dubois L, Li Y et al. Binding of cetuximab to the EGFRvIII deletion mutant and its biological consequences in malignant glioma cells. Radiother Oncol 2009; 92: 393–398. [DOI] [PubMed] [Google Scholar]
- 35. Yong CS, Westwood JA, Schroder J et al. Expression of a chimeric antigen receptor in multiple leukocyte lineages in transgenic mice. PLoS One 2015; 10: e0140543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jenkins MR, Rudd‐Schmidt JA, Lopez JA et al. Failed CTL/NK cell killing and cytokine hypersecretion are directly linked through prolonged synapse time. J Exp Med 2015; 212: 307–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Norelli M, Camisa B, Barbiera G et al. Monocyte‐derived IL‐1 and IL‐6 are differentially required for cytokine‐release syndrome and neurotoxicity due to CAR T cells. Nat Med 2018; 24: 739–748. [DOI] [PubMed] [Google Scholar]
- 38. Liu X, Jiang S, Fang C et al. Affinity‐tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res 2015; 75: 3596–3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ohno M, Ohkuri T, Kosaka A et al. Expression of miR‐17‐92 enhances anti‐tumor activity of T‐cells transduced with the anti‐EGFRvIII chimeric antigen receptor in mice bearing human GBM xenografts. J Immunother Cancer 2013; 1: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Freeman GJ, Long AJ, Iwai Y et al. Engagement of the Pd‐1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 2000; 192: 1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Grosso JF, Kelleher CC, Harris TJ et al. LAG‐3 regulates CD8+ T cell accumulation and effector function in murine self‐ and tumor‐tolerance systems. J Clin Invest 2007; 117: 3383–3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Verdon DJ, Mulazzani M, Jenkins MR. Cellular and molecular mechanisms of CD8+ T cell differentiation, dysfunction and exhaustion. Int J Mol Sci 2020; 21: 7357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Park S, Shevlin E, Vedvyas Y et al. Micromolar affinity CAR T cells to ICAM‐1 achieves rapid tumor elimination while avoiding systemic toxicity. Sci Rep 2017; 7: 14366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chmielewski M, Hombach A, Heuser C, Adams GP, Abken H. T Cell Activation by antibody‐like immunoreceptors: increase in affinity of the single‐chain fragment domain above threshold does not increase T cell activation against antigen‐positive target cells but decreases selectivity. J Immunol 2004; 173: 7647. [DOI] [PubMed] [Google Scholar]
- 45. Hudecek M, Lupo‐Stanghellini MT, Kosasih PL et al. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1‐specific chimeric antigen receptor T cells. Clin Cancer Res 2013; 19: 3153–3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Caruso HG, Hurton LV, Najjar A et al. Tuning sensitivity of CAR to EGFR density limits recognition of normal tissue while maintaining potent antitumor activity. Cancer Res 2015; 75: 3505–3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Drent E, Themeli M, Poels R et al. A rational strategy for reducing on‐target off‐tumor effects of CD38‐chimeric antigen receptors by affinity optimization. Mol Ther 2017; 25: 1946–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Marchingo JM, Kan A, Sutherland RM et al. Antigen affinity, costimulation, and cytokine inputs sum linearly to amplify T cell expansion. Science 2014; 346: 1123. [DOI] [PubMed] [Google Scholar]
- 49. Schaller TH, Batich KA, Suryadevara CM, Desai R, Sampson JH. Chemokines as adjuvants for immunotherapy: implications for immune activation with CCL3. Expert Rev Clin Immunol 2017; 13: 1049–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wu Y, Li Y‐Y, Matsushima K, Baba T, Mukaida N. CCL3‐CCR5 axis regulates intratumoral accumulation of leukocytes and fibroblasts and promotes angiogenesis in murine lung metastasis process. J Immunol 2008; 181: 6384. [DOI] [PubMed] [Google Scholar]
- 51. Gargett T, Yu W, Dotti G et al. GD2‐specific CAR T cells undergo potent activation and deletion following antigen encounter but can be protected from activation‐induced cell death by PD‐1 blockade. Mol Ther 2016; 24: 1135–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Maus MV, Plotkin J, Jakka G et al. An MHC‐restricted antibody‐based chimeric antigen receptor requires TCR‐like affinity to maintain antigen specificity. Mol Ther Oncolytics 2016; 3: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hamieh M, Dobrin A, Cabriolu A et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature 2019; 568: 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wu A, Wei J, Kong LY et al. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol 2010; 12: 1113–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lee DW, Kochenderfer JN, Stetler‐Stevenson M et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose‐escalation trial. Lancet 2015; 385: 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Simoni Y, Becht E, Fehlings M et al. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 2018; 557: 575–579. [DOI] [PubMed] [Google Scholar]
- 57. Reardon DA, Brandes AA, Omuro A et al. Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: the CheckMate 143 phase 3 randomized clinical trial. JAMA Oncol 2020; 6: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Moeller M, Haynes NM, Kershaw MH et al. Adoptive transfer of gene‐engineered CD4+ helper T cells induces potent primary and secondary tumor rejection. Blood 2005; 106: 2995–3003. [DOI] [PubMed] [Google Scholar]
- 59. Wang D, Aguilar B, Starr R et al. Glioblastoma‐targeted CD4+ CAR T cells mediate superior antitumor activity. JCI Insight 2018; 3: e99048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Beasley MD, Niven KP, Winnall WR, Kiefel BR. Bacterial cytoplasmic display platform Retained Display (ReD) identifies stable human germline antibody frameworks. Biotechnol J 2015; 10: 783–789. [DOI] [PubMed] [Google Scholar]
- 61. Affinity Biosciences Pty Ltd. Soluble Polypeptides. In: Ltd ABP, editor. WO2013023251A12013.
- 62. Tsao K‐L, Debarbieri B, Michel H, Waugh DS. A versatile plasmid expression vector for the production of biotinylated proteins by site‐specific, enzymatic modification in Escherichia coli . Gene 1996; 169: 59–64. [DOI] [PubMed] [Google Scholar]
- 63. Lopez JA, Susanto O, Jenkins MR et al. Perforin forms transient pores on the target cell plasma membrane to facilitate rapid access of granzymes during killer cell attack. Blood 2013; 121: 2659–2668. [DOI] [PubMed] [Google Scholar]
- 64. Schindelin J, Arganda‐Carreras I, Frise E et al. Fiji: an open‐source platform for biological‐image analysis. Nat Methods 2012; 9: 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials