

Abstract
Tandem mass spectral library search (MS/MS) is the fastest way to correctly annotate MS/MS spectra from screening small molecules in fields such as environmental analysis, drug screening, lipid analysis, and metabolomics. The confidence in MS/MS-based annotation of chemical structures is impacted by instrumental settings and requirements, data acquisition modes including data-dependent and data-independent methods, library scoring algorithms, as well as post-curation steps. We critically discuss parameters that influence search results, such as mass accuracy, precursor ion isolation width, intensity thresholds, centroiding algorithms, and acquisition speed. A range of publicly and commercially available MS/MS databases such as NIST, MassBank, MoNA, LipidBlast, Wiley MSforID, and METLIN are surveyed. In addition, software tools including NIST MS Search, MS-DIAL, Mass Frontier, SmileMS, Mass++, and XCMS2 to perform fast MS/MS search are discussed. MS/MS scoring algorithms and challenges during compound annotation are reviewed. Advanced methods such as the in silico generation of tandem mass spectra using quantum chemistry and machine learning methods are covered. Community efforts for curation and sharing of tandem mass spectra that will allow for faster distribution of scientific discoveries are discussed.
Keywords: compound identification, high-resolution mass spectrometry, library search, tandem mass spectrometry
1 |. INTRODUCTION
Spectral searching of tandem mass spectral data (MS/MS) against reference databases has been developed with the broader availability of tandem mass spectrometers since the early 1980s.1–5 In fact, searching MS/MS databases is currently the fastest approach for confident compound annotations in small molecule analysis including metabolomics,6,7 lipidomics,8 food, and environmental sciences.9 Despite advances in instrumentation from Fourier transform ion cyclotron resonance MS (FT-ICR)10 to orbital ion trap (Orbitrap), 3D ion trap, and time-of-flight/time-of-flight (TOF-TOF) mass spectrometers, the number of identified chemicals in profiling screens has remained limited because no large MS/MS database collections were historically available. However, during the last 10 years, small molecule MS/MS databases have been steadily growing in coverage and diversity. Recently, there has been an important shift from experimentally obtained reference spectral libraries to computationally generated (in silico) MS/MS databases. This review discusses MS/MS databases and software approaches for small molecules less than 2000 Da. Proteomics and glycomics MS/MS search strategies11–13 as well as multiple stage tandem mass spectrometry (MSn)14 are discussed elsewhere.
2 |. INSTRUMENTAL SETTINGS FOR TANDEM MASS SPECTROMETERS
Tandem mass spectrometers are becoming more accurate and are routinely operated within 1–5 ppm mass accuracy. Up to 100 MS/MS spectra per second can be acquired and instruments allow for operation with high mass resolving power ranging from 10 000 to 500 000 full width at half maximum (FWHM). Table 1 lists a selection of MS/MS capable instruments that can be utilized for generating MS/MS datasets. A review from 2012 lists additional instruments and their specifications.15 Many instrumental parameters influence the number of MS/MS spectra obtained as well as the quality of those tandem mass spectra. Such parameters include total acquisition speed, accumulation time per single MS/MS spectrum, precursor ion isolation width, intensity threshold, collision energy, and others.
TABLE 1.
A selection of instruments for accurate mass MS/MS analysis (2016) and their maximum mass resolving power and acquisition speed
| # | Instrument | MS1 mass resolving power | MS/MS acquisition speed (spectra/s) |
|---|---|---|---|
| 1 | Agilent 6550 iFunnel QTOF | 40 000 | 50 |
| 2 | Agilent 7200 GC/QTOF | 13 500 | 50 |
| 3 | Bruker GC-micrOTOF-Q II | 16 500 | 40 |
| 4 | Bruker impact II | 50 000 | 50 |
| 5 | JEOL SpiralTOF-TOF | 60 000 | 10 |
| 6 | Perkin Elmer AxION iQT GC/MS/MS | 12 000 | 50 |
| 7 | SCIEX TripleTOF 6600 | 35 000 | 100 |
| 8 | SCIEX TOF/TOF 5800 | 26 000 | 10 |
| 9 | SCIEX X500R | 35 000 | 100 |
| 10 | Shimadzu LCMS-IT-TOF | 10 000 | 10 |
| 11 | ThermoFisher Q Exactive HF | 240 000 | 18 |
| 12 | ThermoFisher Q Exactive GC Orbitrap GC-MS/MS | 140 000 | 12 |
| 13 | ThermoFisher Orbitrap Fusion Tribrid | 450 000 | 15 |
| 14 | Waters Synapt G2-Si HDMS | 60 000 | 30 |
| 15 | Waters Xevo G2-XS QToF | 40 000 | 30 |
Data were obtained from public sources and might vary slightly. Some instruments allow collecting data at different mass resolving power in MS1 and MS/MS in order to increase the sensitivity or duty cycle.
The “instrumental design” heavily influences the product ion masses and ion abundances in MS/MS spectra. Tandem mass spectrometry can be classified into tandem in-time (ion traps, FTICR) and tandem in-space (quadrupoles, TOFs) setups.16 Hybrid instrumentation can include various combinations of beam- or trap-type analyzers such as quadrupole/time-of-flight (QTOF), quadrupole/orbital ion trap, or quadrupole/linear ion trap (QLIT).17
The “ionization method” determines how sample material is transferred into the mass spectrometer.18 The most common ionization mode for small molecule LC-MS/MS is electrospray ionization (ESI).19,20 Other modes such as atmospheric pressure chemical ionization (APCI), atmospheric pressure photo ionization (APPI), and matrix-assisted laser desorption/ionization (MALDI)21 are covered to a lesser extend in tandem mass spectral databases.9 For GC-MS based experiments, it is possible to utilize 70 eV electron ionization (EI) as well as chemical ionization (CI) with different collision gases, APCI,22,23 or APPI.24
The “collision energy” plays an important role in MS/MS spectra generation. For collision-induced dissociation (CID), one can distinguish between low-energy collisions (0–100 eV range) observed in ion traps and high-energy collisions (keV range) utilized in sector instruments and TOF/TOFs.25,26 Most of the CID MS/MS libraries covered in this review were created using low-energy CID conditions. Very few examples of high-energy CID libraries exist, despite the advantage of creating fragment-rich and reproducible spectra.27,28 For low energy CID, one can measure energy resolved breakdown curves for specific ions, by ramping or increasing the collision energy.29,30 Energy ramps will allow for finding the optimum fragmentation energies for analysis.31 Low collision energies mostly preserve the precursor ion and only few product ions are observed. Increasing the collision energy will increase product ion abundances toward low m/z ranges and at the same time will lower the precursor ion abundance. Some instruments allow for ramped collision energies, which are then merged into a single MS/MS spectrum. For library searching, distinct individual voltages (0, 10, 20, 30, 60 eV) are preferential because they allow for more fine-grained library matching.
There are multiple “ion activation modes” that can be utilized in tandem mass spectrometry.26 The time scale of the different activation modes has an impact on the fragments and their abundances in measured tandem mass spectra.32,33 The most common ion activation and fragmentation modes for small biomolecule LC-MS/MS based experiments are collision-induced dissociation (CID)34,35 and higher energy collisional dissociation (HCD), the latter on orbital ion trap mass spectrometers.36 Both CID and HCD are commonly operated in low-energy collision modes (0–100 eV range) but can create fragmentation-rich MS/MS product ion spectra with sometimes overlapping fragments.37 HCD was originally introduced for proteomics experiments. Small molecule coverage for HCD MS/MS spectra has dramatically increased over the past years’ releases of the NIST14 and MassBank spectral libraries. Initial findings led to the conclusion that HCD MS/MS spectra can be searched in much larger CID spectral libraries.38 Currently, no comprehensive statistical analysis of fragment ions between HCD and CID modes has been performed for small molecule libraries. Both CID and HCD can be utilized complementary to increase compound identification rates.39,40 Electron-based dissociation techniques such as electron-induced dissociation (EID)41 have been successfully used in structure characterization of glycerophosphatidylcholines, specifically for determination of double-bond positions and localization of acyl chains.42 Other techniques would require various chemical derivatizations when combined with CID/HCD to fulfill the same task. Additional modes such as electron capture/transfer dissociation (ECD/ETD) are not commonly used for small molecule analysis but rather in proteomics.43
The influence of the “precursor ion isolation width” or precursor isolation window was discussed with a special focus on sensitivity and selectivity during MS/MS data acquisition.44 In general, selecting narrow precursor ion isolation windows (high resolution precursor isolation) lowers the sensitivity of the precursor ion and thus the intensity of fragment ions. This may lead to a lower number of acquired MS/MS spectra. However, this approach is less prone to co-isolation of potential interferences along with the targeted precursor ion. Widening the isolation window leads to the fragmentation of a larger number of different compounds and results in impure product ion spectra with interfering ions.45 Using data-dependent analysis (DDA), the current mass spectrometers permit relatively wide precursor ion isolation windows (0.7–9 Da), but the precursor ion isolation window is generally set between 1 and 3 Da, depending on instrumentation. On the other hand, using data-independent analysis (DIA), the precursor ion isolation window is much wider, depending on the settings for sequential window MS/MS acquisition. For example, the sequential window acquisition of all theoretical fragment-ion spectra, SWATH-MS/MS, (SCIEX) typically uses 20–50 Da windows. For other, all-fragment-ion approaches larger windows such as 600–2000 Da are used.46
The duty cycle and the “acquisition speed” determine how many tandem mass spectra can be acquired per scan event.47 With modern ultrahigh-performance liquid chromatography (UHPLC) setups, chromatographic peak widths may only last a few seconds. It is important to acquire a maximum number of product ion scans in untargeted metabolomics, as overall run-times for high-throughput experiments tend to get shorter while complexity in metabolomic experiments increase, for example, for analysis of fecal matter in microbiome studies. In such studies, many compounds are not completely resolved. Furthermore, acquiring maximum numbers of MS/MS spectra from the same precursor tremendously improves the signal/noise ratio and spectral quality. Modern quadrupole/time-of-flight (QTOF) instruments are able to acquire up to 50 MS/MS spectra per second. The latest SCIEX TripleTOF, a hybrid quadrupole time-of-flight mass spectrometer combining advantages of QTOF and QqQ systems, can acquire up to 100 MS/MS spectra per second which corresponds to 10 ms accumulation time per a single MS/MS spectrum (see Table 1). However, increasing the acquisition speed may lower the ion statistics and impact the quality of MS/MS spectra48 because fewer raw spectra are averaged. Additional parameters that influence MS/MS spectral quality and the total number of isolated precursor ions are charge state screening (maximum 2 for small molecules), dynamic exclusion parameters, and monoisotopic precursor selection.
For time-of-flight instruments, the “mass resolving power” is constant with increasing acquisition speed.49 On the other hand, for Fourier-transform based technology such as the orbital ion trap (Q Exactive HF instrument), the scan speed can be up to 18 Hz with a resolving power of 18 000 FWHM (m/z 200). However, if users want to increase the mass resolving power to 240 000 FWHM, the scan speed decreases to a mere 1.5 scans/s (1.5 Hz)50 which is too slow for fast UHPLC studies. For experiments that need a maximum number of annotated compounds with an existing LC-MS/MS protocol, it is recommended to limit the precursor ion mass range and split acquisitions into different runs. A simple alternative is of course to perform very long LC-MS/MS runs to allow for better chromatographic peak resolution, or to acquire dedicated MS/MS spectra acquisitions in multiple runs for a few select samples, but not for all the samples in a study.
Modern mass spectrometers and multimode ion sources allow for “voltage or polarity switching,” allowing the acquisition of data in positive and negative ionization mode in one run.51 This can increase sample throughput tremendously. Another option is to perform separate runs for profiling (with polarity switching) and identification (no polarity switching) to allow for enough time during the MS/MS acquisition.52 However, many compounds show radically different ionization efficiency in different ionization modes, depending on mobile-phase buffer systems.8,53 Therefore, using two different runs and optimized buffer systems in negative and positive electrospray mode may be preferred.
The hyphenation of tandem mass spectrometers with “ion mobility” provides an additional orthogonal dimension for better separation of isobaric compounds, stereoisomers, and challenging matrices.54 Many vendors provide plugin hardware solutions with short drift-tubes to improve peak separation; 1–2 m drift tubes within hybrid ion mobility QTOF instruments are also available. Using collision cross-section (CCS) information may help during compound deconvolution and compound identification, and better separation will likely yield cleaner product ion spectra.55
2.1 |. Data-dependent acquisition methods
During “data-dependent acquisition” (auto-MS/MS), a specific intensity threshold is used to trigger the acquisition of precursor ions48 (see Fig. 1A). Lowering the intensity threshold leads to more product ion peaks. However, the purity of spectra decreases due to the contribution of noise signals and data quality is hampered by lowered ion statistics for product ions.40 Tandem mass spectra of the same precursor and same ionization voltage can be merged to increase the signal-to-noise ratio and quality of a product ion scans. Furthermore, most instrument software also allow for a limit on reoccurring product ions during data acquisition (dynamic exclusion).
FIGURE 1.
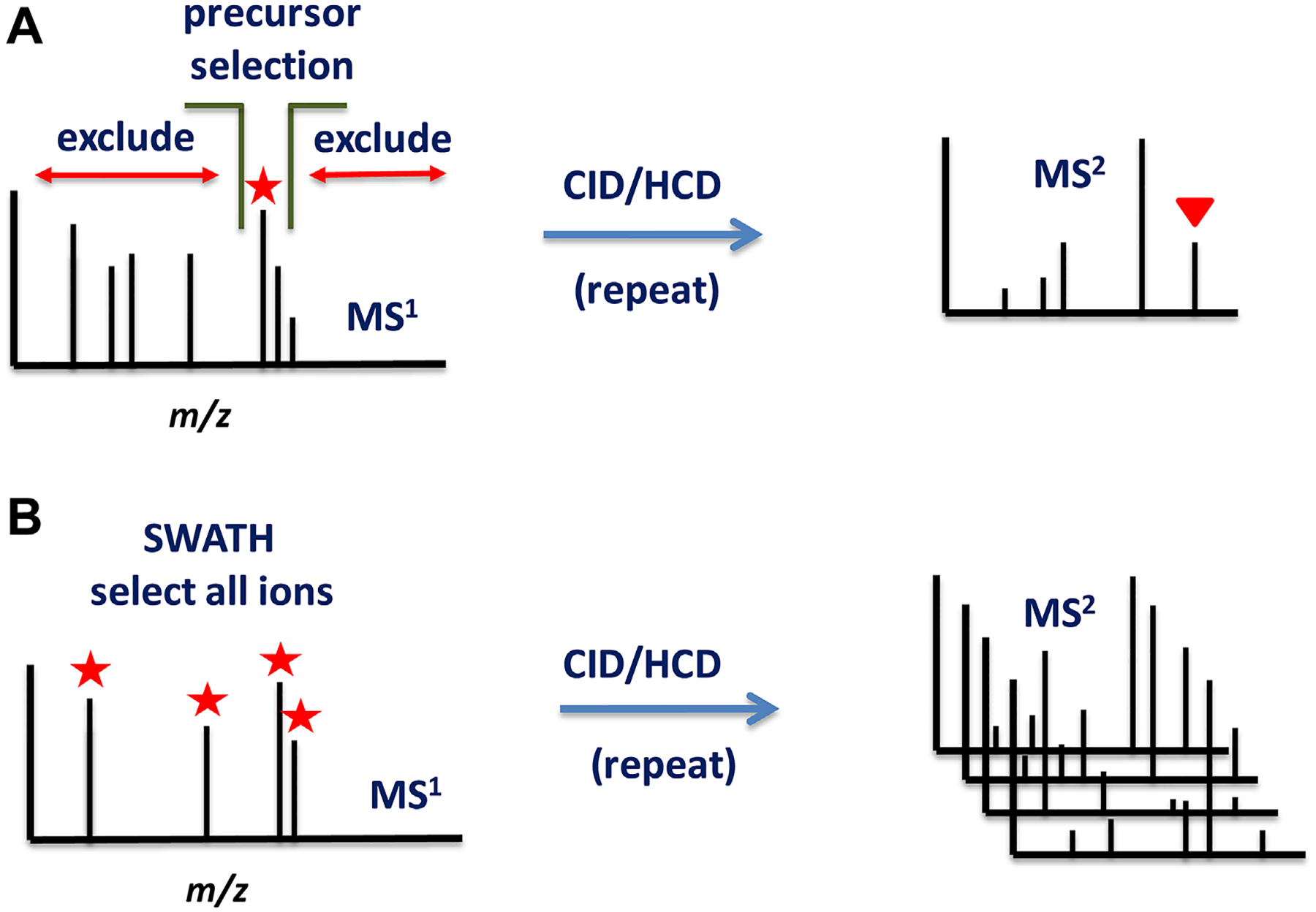
(A) During data-dependent MS/MS spectra acquisition, the instrument selects a highly abundant MS1 peak and discards all other peaks outside the selected precursor isolation window. The ions are fragmented during collision-induced dissociation (CID) or higher energy collisional dissociation (HCD) processes. The MS/MS contains information about the precursor ion. (B) During all-ion fragmentation or SWATH mode the instrument fragments all peaks indiscriminately of peak height. The spectra are information-rich collections but lack the precursor information. In order to perform MS/MS database search deconvolution software such as MS-DIAL has to be used to reconstruct the correct precursor ion
2.2 |. Data-independent acquisition methods
“Data-independent MS/MS acquisition”56 such as SWATH-MS/MS and all-fragment-ion techniques recently gained attention in the small molecule community46,57 (see Fig. 1B). Novel mass spectrometers with fast duty cycles and acquisition times with up to 100 MS/MS scans per second at 35 000 FWHM mass resolving power allowed for the development of these techniques.58 SWATH-MS/MS technique utilizes multiple cycles of large consecutive precursor ion isolation windows (20 Da or more).59 One advantage is that very low intensity precursor ions are fragmented, even if they would not trigger intensity thresholds (as in data-dependent MS/MS). Even if there are co-eluting molecules with higher intensities (that are usually triggered first in data-dependent MS/MS), low abundant ions are still fragmented. Hence, in principle, all molecules in data-independent MS/MS undergo fragmentations. The obvious disadvantage for SWATH-type analyses is that the direct link between a specific precursor ion and its corresponding product ions is broken. Therefore, mixed product ion spectra are generated, that originate from multiple precursor ions within each SWATH window. Moreover, precursor ions from isobaric overlapping components cannot be easily determined. Precursor determination in SWATH-MS/MS therefore requires mass-spectral deconvolution on the MS2 level and retention time information.
The recently introduced MS-DIAL software (http://prime.psc.riken.jp/) allows for automatic mass spectral deconvolution and MS/MS-based library search.60 Since compound identifications in metabolomics are based on overall similarity between experimental and reference spectra, the DIA-MS/MS spectra must be purified (ie, deconvoluted) from co-eluting compounds and from noise ions to increase library-matching scores of true positive compounds. Importantly, MS-DIAL requires at least two scan differences in the peak apex of co-eluting compounds to be purified. Therefore, it is important for the deconvolution to acquire a sufficiently large number of MS/MS data points across chromatographic peaks. Other software tools such as OpenSWATH,61 DIANA,62 pSMART,63 Biognosys Spectronaut, or DIA-Umpire64 are targeted toward the proteomics community and cannot be directly used for small molecule identifications for two reasons: first, in proteomics, SWATH-MS/MS-based identification relies solely on the MS/MS data. The fact that precursor ions are isolated with narrow Q1 isolation windows helps reducing the complexity of MS/MS spectra but MS1 information about the precursor ions is not used at all. Second, experimental libraries are used to do targeted data extraction followed by peak group scoring and false discovery rate (FDR) calculation to figure out which is the correct annotation of the peptide. For small molecule analysis we therefore recommend performing data-dependent and SWATH-MS/MS analysis in a combined way using both techniques.
3 |. CREATION OF MS/MS DATABASES
3.1 |. Creation of experimental MS/MS reference libraries
Traditionally, MS/MS databases were acquired by analysis of authentic reference standards (see Fig. 2A). For rapid creation of experimental MS/MS reference databases, a number of vendors now offer compound standards in 96 well plate formats. IROA Technologies LLC, Bolton, MA (www.iroatech.com) offers 619 unique small molecule metabolites on plates and MicroSource Discovery Systems Inc., Gaylordsville, CT (www.msdiscovery.com) offers plated natural products and drugs. These compound spectra should be acquired under different CID or HCD voltage settings (10, 20, 40, and 60 eV) in positive and negative ionization mode to acquire rich mass spectral fragmentations. Different molecular species beyond simply [M + H]+ and [M − H]− should be included for each compound to capture the whole width and breadth of possible adducts.65 That includes a variety of mobile phase modifiers and solvent related ions as well as sodium and ammonium adducts or commonly observed water loss.59,66 To allow for high spectral quality, a high enough number of spectra for each adducts type has to be obtained and later averaged. Additionally, improper data acquisition methods can hinder compound identification based on MS/MS spectral comparisons. For example, using a high fragmentor or cone voltage can result in loss of the parent ion due to in source fragmentation, and MS/MS spectra would be acquired on fragment ions instead of the parent ion. Careful consideration and planning should go into MS/MS library acquisition in order to increase identification quality of small molecules in a matrix of interest.
FIGURE 2.
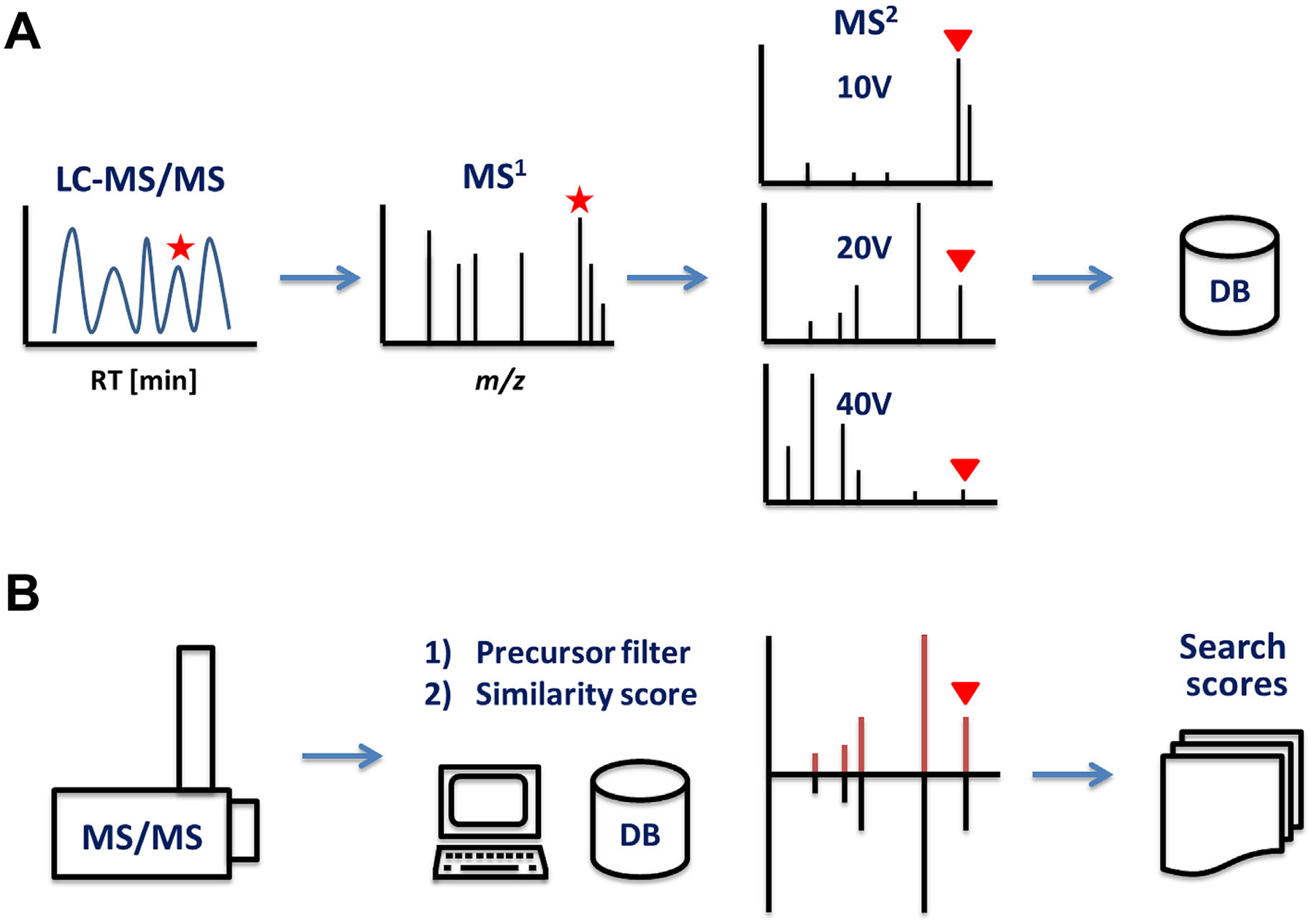
(A) MS/MS database creation: a MS1 ion (precursor) is picked from an LC-MS/MS run and undergoes fragmentation in the tandem mass spectrometer under different collision energies to cover a broad range of characteristic fragments (product ions). (B) MS/MS search: the precursor filter (from 0.1 to 0.001 Da) removes most of the candidates outside a mass accuracy search window. The similarity algorithm ranks the remaining spectra against all database spectra and creates a similarity score
3.2 |. Creation of in silico MS/MS libraries
A rising trend is the generation of purely computationally derived mass spectral libraries. Large compound libraries such as PubChem or Chemspider can then be utilized for input structures. The generated in silico MS/MS spectra can fill the large gap of missing experimental MS/MS spectra. Examples are databases utilizing the LipidBlast templates,67,68 the Greazy/LipidLama platform,69 or the CFM-ID computational software.70,71 The heuristic LipidBlast approach can only be used for compounds with reoccurring neutral losses and fragments with consistent fragmentation pattern such as lipids. CFM-ID is more flexible because it can create in silico spectra of any given spectrum type that was used during training. The training spectra for CFM-ID MS/MS spectra were based on QTOF tandem mass spectra from the METLIN database. Hence the output from CFM-ID for ESI MS/MS mimics the 10, 20, 40 eV MS/MS spectra from a QTOF instrument. A recent approach utilized CFM-ID to create a computational derived in silico MS/MS database of 170 000 natural products to be used for natural product dereplication.72
The development of quantum chemistry based methods for in silico generation of CID-MS/MS mass spectra will be one of the next grand challenges in computational mass spectrometry. So far only electron ionization mass spectra can be modeled with good accuracy.73–75 The jump to the creation of in silico ESI-MS/MS spectra will require a substantial innovative and intellectual input from the quantum chemical community, mostly due to the variability of low-energy CID spectra and the required fragmentation voltage spreads. Larger molecular weight compounds also will have higher degrees of freedom for conformational movements which will render computational approaches very expensive and time-consuming.
For the generation of in silico based MS/MS databases, it is extremely important to validate the computational method for accuracy and precision and to determine sensitivity and specificity based on experimental reference compounds. Most importantly, the structural domain of the training compounds has to be observed. LipidBlast would not be able to model fragmentations and rearrangements of small nucleotides. CFM-ID would not be able to accurately model large molecular weight lipids, because they were not adequately covered in the original training set of small metabolites. Once the algorithm is retrained by relevant input spectra, the structural scaffold has changed and spectra of a different structural domain can be created, as exemplified with the CFM-ID peptide set.76
One of the latest trends is the use of computational compound databases that were created using the most common enzymatic transformation reactions.77,78 These virtual compound collections can be converted into in silico MS/MS databases and unknown experimental MS/MS spectra can then be searched against them.79 However, with potentially millions of structurally very similar compounds, millions of very similar in silico tandem mass spectra will be created. Such an effect, called database poisoning, can only be overcome by novel search algorithms or orthogonal filtering strategies.
3.3 |. Curation and cleaning of mass spectra
The “manual curation of mass spectra” was historically performed by groups that built mass spectral collections such as NIST and Wiley and with that corrected spectra, added structures and CAS numbers and created value-enhanced products.80–82 Such curated and high-quality libraries are used by almost all mass spectrometry labs worldwide. Curation efforts include manual inspection of mass spectra by experienced mass spectrometrists, noise removal and artifact removal, building of consensus spectra and peak annotations,82 as well as inter-library comparisons.83 One caveat is that such approach affords high acquisition costs that can reach more than $10 000 USD per library. Especially the creation of consensus spectra in NIST and MassBank has gained attention because many MS/MS spectra of the same compound have been added over the years. Automation of specific curation aspects is now required for building high-quality spectral collections. For example, consensus building may involve combining multiple MS/MS spectra from the same instrument at the same collision energy into to a single high-quality spectrum. There are a number of R-language computational packages hosted on BioConductor (http://www.bioconductor.org/) that can be used to manipulate and process MS/MS spectra. That includes MSnID,84 MSnbase,85 msPurity,86 RMassBank,87 SwathXtend,88 and RAMClustR.89
The “automatic curation” of MS/MS spectra in order to create high-quality and high-accuracy data has been a focus especially for MassBank and recently the MoNA database (http://mona.fiehnlab.ucdavis.edu). Such automated cleaning processes include formula and substructure annotations for precursor and product ions, noise removal, the calculation of spectral quality codes, and annotations with metadata including InChIKey, SMILES, compound names, as well as experimental settings.87 The advantage of using recalibrated and cleaned spectra in MS/MS databases is that higher match scores can be obtained during database search. All modern MS/MS databases such as NIST, MassBank, mzCloud, or LipidBlast contain rich meta-data annotations such as compound structure, instrument type, collision energy, type of fragmentation technique, adduct ion type, and product ion annotations. There have been approaches in the past to create reproducible tandem mass spectra across instruments from multiple manufacturers.90 Furthermore, all databases subsequently undergo benchmark evaluations and quality checks.81
3.4 |. MS/MS data formats and software tools
There are a number of data exchange formats that can be used for MS/MS data transfer and import/export options. However, there is no standardized format for MS/MS data file storage. The three most frequently used formats in small molecule research and their associated file converter tools are described below.
The “Mascot Generic Format (*.MGF)” from Matrix Science (http://www.matrixscience.com) is the oldest and most commonly used format for storing tandem mass spectra. Originally developed for the proteomics community it is widely available as export format on almost all vendor platforms. External converter tools such as ProteoWizard91 allow the conversion from Agilent Technologies (Santa Clara, CA), Bruker (Billerica, MA), SCIEX, Thermo Fisher Scientific (Waltham, MA), Shimadzu (Pleasanton, CA), and Waters (Milford, MA) raw files to MGF format. MGF files in their simplest format provide a name, the precursor information as well as the product ion m/z and abundances. Multiple tandem mass spectra can be attached to each other resulting in files with thousands of precursors and their associated product ion spectra. One caveat is that the format widely differs in terms of additional defined meta-data options such as ionization information, MS level, retention time, and voltage settings. This can result in software tools easily crashing or refusing to import should such options be used or missing during the import/export of MGF files. One solution to that problem is to utilize the diverse public data files from the MetaboLights repository92 or the Metabolomics Data Repository and Coordination Center (DRCC) metabolomics workbench93 to validate an error-free MGF import. The associated *.dta and *.pkl which are single storage and container files did not obtain much traction and are used to a lesser extent.
The “NIST MSP” format is mostly used by the NIST MS Search software, the MS-DIAL application, and a number of databases such as LipidBlast and MoNA. The ASCII-based format is very simple and has been available for many years. The format includes meta-tags that describe important MS/MS spectral parameters, such as adduct type, collision energy, instrument type, and more than 30 other conditions. However, these MS/MS special tags were not completely documented until recently, which limited the broader use among the community. In order to convert MSP files into searchable NIST libraries, for use in NIST MS search GUI and the batch search software MSPepSearch, the freely available LIB2NIST library conversion tool (http://chemdata.nist.gov) has to be used. For import of MassBank records into the NIST format, the MassBank2NIST (https://github.com/MassBank/MassBank2NIST) or the MassBank to NIST MSP format converter (http://prime.psc.riken.jp/Metabolomics_Software/MassBankToMsp/index.html) can be utilized.
The “MassBank record format” is a well-documented format for storage of MassBank records and follows ontology rules and defined parameter sets. Around 40 parameters describe information about the chemical compound including mass, SMILES code, InChI code, and formula as well as information about analytical methods and settings such as instrument type and parameters, and additional parameters such as the type of biological sample or instrumental chromatography settings. Despite the excellent and detailed definition of the MassBank format, it is currently only used by MassBank itself and a number of external applications including RMassBank.87
4 |. MS/MS SEARCH ALGORITHMS AND SCORING
An “MS/MS database search” is a two-step procedure: (1) precursor ion matching and (2) similarity matching of the remaining candidates (see Fig. 2B). The first and most powerful filter, the precursor ion filter can remove up to 99.9% of the false candidates, depending on database size and distribution. Precursor search windows can be either set in absolute m/z values or in ppm. The second filter is the classical similarity-based filter that takes m/z values and product ion abundances into account. Such similarity filters have been mostly developed for electron ionization 70 eV mass spectra in the past.94
Typical precursor search windows range from 0.4 Da for low resolution instruments (unit mass) to 0.005 Da for high-resolution QTOF or orbital ion trap instruments. Also the product ion mass accuracy window can be adjusted which will exclude additional non-matching candidates from the search results. For product ion peaks, the mass accuracy settings during database search are usually relaxed. The lower mass accuracies observed on the product ion level are related to unresolved interferences such as competitive fragmentation pathways or simultaneous fragmentation of isobaric precursor ions.95
The obtained search score after a database search represents the likelihood of a search spectrum corresponding to a reference spectrum in a mass spectral reference database. Score-based equations typically include the m/z-intensity pairs of the search spectrum and library spectra as well as additional parameters such as weighing functions. Classical and established mass spectral scoring algorithms include, for example, the probability match algorithm (PBM)96 and the dot-product97 algorithm. The McLafferty PBM algorithm was introduced in 1974 and works especially well for very reproducible electron ionization (EI) spectra. The PBM scores range from 0% to 100% and a high value represents a high confidence that the spectrum was identified during database search. It is a linear combination of four probability measures: the uniqueness of m/z values of a specific peak, the peak abundance contributions, a window factor that integrates peak abundances, and a dilution factor for mixture spectra. The dot-product algorithm uses the cosine of the angle between the unknown and library spectral vectors97,98 and is now commonly used during accurate mass MS/MS database search. The composite equations include the dot-product function and terms that use intensity scaling based on mass as well as non-scaled intensity ratios of neighboring peaks.97 These library “match scores” range from 0 to 999. A low score indicates that compound is not found in the database, scores from 200 to 650 indicate few matching peaks, scores higher than 850 represent good matches and scores of 999 would present a perfect hit. However, these estimate rules are historically based on electron ionization spectra and may not hold true for the large diversity of CID or HCD based MS/MS spectra.99
Other “similarity measures” such as the Jaccard,70 Pearson,89,100 Jeffries-Matusita distance,101 and random projection102 can be used as well. A number of modified search algorithms with the aim to improve scoring results have been published.103–105 Many of the modified methods introduce correction factors to increase hit scores and sensitivity and specificity of search results. Thresholds for good hit scores and useful cutoff-values have to be based on statistical probability estimations. Such threshold must differ for EI and CID MS/MS mass spectra.98 Cutoff values are also depending on the software and algorithm used as well as library sizes, compound diversity, and MS instrument platform. Many ESI MS/MS spectra have sparse product ion peaks, sometimes less than five ions. In such a case, the match scores and the probability scores can be very low. Here, it would be better to utilize the reverse dot-product algorithm. However, there is currently no large scale statistical investigation published to shed light on the use of cut-off values or use of these scores.
A number of different “search options” are implemented in the freely available NIST MS Search program. Classical EI identity search, MS/MS search, high-resolution in-source search, and neutral loss search are available. The following search options are offered: forward, reverse, hybrid, neutral loss, similarity, and probability search. Related specific match factors are all shown after each search in the result hit list. For MS/MS identification purposes, the dot-product as well as the reverse dot-product search are commonly recommended. The “reverse search” ignores non-matching peaks in the search spectrum and the score is not penalized for peaks that are not found in the library spectrum. It has been observed that the reverse search option is particularly helpful when MS/MS spectra with sparse peaks are searched, such as in silico spectra generated for certain lipid classes.106 The “hybrid matching” search combines normal search and neutral loss search which is important for detecting compound classes with similar fragmentation patterns. The simple “similarity search mode,” without precursor ion search, should not be used for MS/MS database search alone. However, it is useful for compound identification purposes if the compound or precursor itself is not contained in the MS/MS database. The premise under such circumstances is that the core fragments still result in the same MS/MS fragmentations, even if additional modifications are missing. For example, a similarity mass spectral search of 4-acetamidoantipyrine would reveal hits with 4-formylaminoantipyrine and 4-aminoantipyrine because both molecules share the aminoantipyrine core structure. A neutral loss calculation would reveal a difference of 42.01 Da or an additional acetyl group (C2H2O).
Parameters that can influence the result search scores are the precursor ion search width, the acquisition mode of the product ion spectra (profile mode and centroided or stick spectra) and the number of peaks in the product ion spectra. A short example using 200 000 spectra from the NIST14 database and MassBank shows the powerful impact of the precursor filter. A histogram based analysis of NIST and MassBank precursor ions reveals that many of the molecules range between 250 and 450 Da. A search of the phenoxybenzamine MS/MS spectrum with a 100 ppm (0.03 Da) precursor window results in 81 diverse molecules, including many false positive candidates. Minimizing the window to 50 ppm (0.015 Da) lowers the result list to 68 candidates. A more realistic value for modern high-resolution QTOFs or orbital ion trap instruments is a 5 ppm (0.0015 Da) precursor window which returns the correct single phenoxybenzamine hit.
Reports from several Critical Assessment of Small Molecule Identification (CASMI) challenges provided a deeper insight into software, tools, and approaches used by multiple groups.107,108 By introducing a binning system, it might be possible to allow low resolution instruments obtain similar performances like searching spectra from high-resolution instrumentations.109 Recently, a new spectral identifier for mass spectra the SPLASH was developed.110 It allows for a one-way encoding of a mass spectrum into a fixed-length identifier and can be considered an analog of the InChIKey. It employs a binning technique to allow for efficient pre-filtering during tandem mass spectral search and is currently implemented in the MassBank of North America (MoNA) database.
Until recently MS/MS decoy databases were not available for small molecule research.5,112 It is now possible to estimate FDR values for small molecule MS/MS spectra.111 The authors compared naïve Bayes as well as three different target-decoy approaches. The proposed method is a spectrum-based approach, circumventing the use of decoy structures. Based on P-value and q-value calculations the authors concluded that for unfiltered spectral data the empirical Bayes approach resulted in good estimates. For noise-filtered data the tree-based decoy strategy using a re-rooted fragmentation tree can be recommended for FDR estimations. The Passatutto software (https://bio.informatik.uni-jena.de/software/) provides source code scripts for small molecule MS/MS decoy library generation, as well downloads for target-decoy MS/MS spectra.
4.1 |. Practical prerequisites for MS/MS search
Performing MS/MS database searches is relatively straightforward but has certain prerequisites. First, it has to be established if the database itself and the tandem mass spectra that are to be searched contain “unit masses or accurate masses.” For example, NIST14 and MassBank contain both unit mass and accurate mass spectra. In some cases, the relevant digits of precursors are truncated, leading from seemingly accurate to relatively inaccurate mass data. In cases where an accurate precursor mass is used, no spectra with unit mass precursors will be found, even if the substance is contained in the database.
The “instrument types” of MS/MS spectra have to be taken into consideration. Large diverse spectral databases such as NIST and MassBank contain ion trap, QqQ, QTOF, and other MS/MS spectra. If QTOF data are searched against an ion trap library, the hit scores will be different from an approach where QTOF spectra are searched in QTOF libraries.104,113,114 It is always best practice to search similar instrument types against their reference spectra.115
Second, the true “experimental mass accuracy” for each run or set of experiments has to be determined with internal or external reference compounds or quality check mixtures that contain known compounds. This approach leads to the responsibility of each user to accurately tune and calibrate the mass spectrometer independently in positive and negative mode before any batch experiment. A commonly observed practical error is to simply assume excellent mass accuracy without adequate tests. Experimentally obtained mass spectra actually may contain large m/z errors for precursors and product ions. Such errors will lead to non-existing or false annotations during MS/MS database search.
Third, in preparation of the actual search, the positive and negative “ionization mode” MS/MS data need to be separated. Positive ionization mode spectra are only searched against positive mode databases and negative ionization mode MS/MS spectra against negative mode databases. This lowers false positive rates and avoids post-curation steps that are otherwise needed. If libraries for specific adduct types such as formate, acetate (from LC mobile phase modifiers), chlorine adducts (from extraction solvents), or residues of salts (sodium, potassium) from sample preparation are available, the matching adduct library must be chosen as fragmentations and the resulting product ions are different for diverse adduct types.9 For example, false annotations can be expected if the MS/MS library covering acetate adducts is selected while ammonium formate is used as mobile-phase modifier leading to formation of formate adducts for some lipids such as phosphatidylcholines.
Tandem mass spectra can be collected in “profile mode or centroid” (stick) mode. MS/MS data collected in profile mode can result in very large data files (up to several gigabytes). Resulting search times can be extremely long, several minutes in profile mode versus a few seconds in centroid mode. The impact of different centroiding algorithms on search scores has been investigated on peptide databases. The authors state that different software tools create “surprisingly large intensity differences for even the most prominent peaks of a fragment ion spectrum.”116 Our own preliminary investigations have shown that profile mode MS/MS spectra create slightly higher match scores due to the fact that multiple very tightly binned m/z values (often with few mDa distance) have a higher probability of matching a library reference spectrum than a single peak. There are, however, practical advantages in the use of centroided spectra because of smaller file sizes and much faster library search speed.
The use of “multiple collision energies” (CID/HCD) for data acquisition is highly recommended (see Fig. 3) because it increases the confidence in compound annotations by multiple independent verifications. MS/MS databases such as METLIN, NIST, and MassBank cover multiple collision energies. Most new instruments allow for high speed data acquisition of MS/MS. Hence, either acquisition at discrete collision energies (eg, 10, 20, and 40 eV) or using collision energy spread or ramp (eg, 35 ± 15 eV) and providing a combined spectrum can be used. Distinct small step voltages (5 eV) are recommended to capture the whole width of mass spectral fragmentation and rearrangement reactions.34 Low CID voltage MS/MS spectra contain dominant precursor ions and few low m/z fragments, whereas high CID voltage spectra show extensive fragmentation in low m/z ranges. In the unfortunate event that a library was only created using a single collision energy, the same (or close) collision energy must be used to create high score values. The use of collision energy spreads or ramps (as well as reversed ramps) is advisable to create information-rich product ion spectra.117 Some ion trap instruments use normalized collision energies to compensate for mass dependencies during fragmentation. This allows for the creation of reproducible MS/MS spectra especially for library creation purposes.118 The use of wideband excitation on select ion traps is useful for the application of resonance energy on ions that are below 20 Da of the precursor ion. This allows for low energy fragmentation of molecules that undergo water loss but with the specificity of the precursor ion retained.119 Additional parameters such as in-source voltage and RF voltage can also influence peak abundances of product ion spectra.120
FIGURE 3.
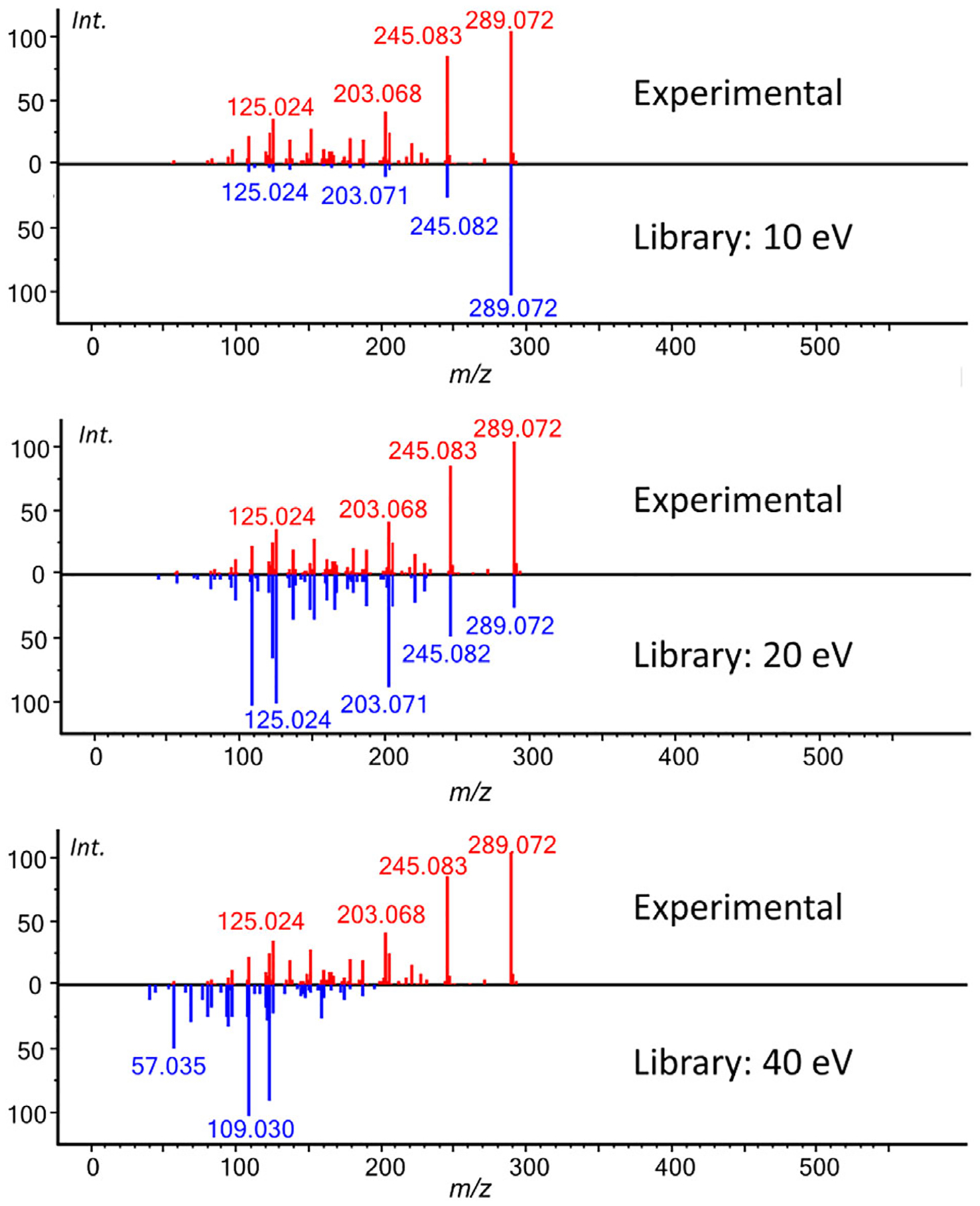
Modern mass spectrometers can record multiple CID voltages for each scan event. Therefore modern MS/MS libraries are now created with multiple distinct CID voltages, such as 10, 20, and 40 eV to increase compound identification probabilities. An example of matching an experimental MS/MS spectrum of catechin acquired in negative ionization mode ESI(−). It is matched against the 10, 20, and 40 eV reference spectra. If the library would only contain 40 eV spectra or single voltages a very low hit score would be obtained
There are around 300 “adduct ions” and in-source fragments that are covered in the NIST14 MS/MS database. The most prominent molecular ion species include [M + H]+, [M + H − H2O]+, [M − H]−, [M + Na]+, and [M + NH4]+. Many solvents and mobile-phase modifiers lead to dedicated adducts, for example formate [M + HCOO]− and acetate adducts [M + CH3COO]− commonly observed during lipidomics profiling in negative ESI mode.8 In-source fragments such as water gain or loss [M + H − H2O]+ or [M − H − H2O]− are commonly observed during small molecule ionizations.121 Certain substance classes such as flavonoids and nucleotides have a higher probability of in-source fragmentations.122,123 Product ion artifacts from the reaction of arylium ions with nitrogen124 and unexpected product ions from reactions with residual water can also influence spectral quality.125 In principle, all adduct ions, in-source fragments, breakdown products, dimers, and multimers need to be considered for the creation and collection of libraries as well for the library search process itself.126
From a practical point of view also “taxonomy restrictions” or restrictions on the compound space can be made during MS/MS search. Although taxonomy restrictions or molecular phylogenetics127 have been a long-standing feature in peptide search engines,128 small database sizes hindered acceptance in traditional analytical MS/MS searches for small molecule research. Moreover, metabolites cannot be captured on a single platform, as they include volatiles, polar, nonpolar and lipid compounds. For example, when investigating blood plasma samples, compounds only found in green algae should be excluded. Although such restrictions can lower false positive and false negative annotations, they may also hinder the discovery of unexpected compounds.
4.2 |. Post-processing of MS/MS search results
After an MS/MS search, a list of all spectral matches is returned. This list can contain multiple scores, the names of the retrieved compounds and additional meta-data such as accurate mass differences or links to traditional compound databases. Subsequently, the list must be inspected for false positive or false negative compounds.5
A recurring problem is “feature combination” of different adducts or different isomers at different retention times.129–131 The identification of multiple adducts for the same compound can be used as an additional confirmation of the substance, as formed ions usually show different fragmentation patterns for different adduct types. For metabolic profiling experiments, such adducts originating from the same compound sometimes need to be unified to allow for subsequent biological interpretations. This can lead to problems, because based on mobile phase composition and modifiers used, they are also represented by different chromatographic peak heights. The simplest solution would be to sum different adduct peak heights originating from the same compound and to report a single value for the peak abundance.132
Instruments that add an additional ion mobility separation dimension based on drift-time ion mobility spectrometry (DTIMS) or traveling-wave ion mobility spectrometry (TWIMS) may increase post-processing requirements, because MS/MS spectra for stereoisomers might be differentiated from each other. For clustering of millions of tandem mass spectra several algorithms from the proteomics community are available.133,134 The use of retention times or retention indices as orthogonal filters is highly recommended for high-confidence compound identifications.135,136 A few MS/MS databases such as ReSpect DB or the Agilent METLIN Personal Compound Database and Library (PCDL), also contain retention times to be used with a specific LC column, mobile phase composition, and separation gradient.
5 |. TANDEM MASS SPECTRAL DATABASES
Tandem mass spectral databases contain mostly CID- and HCD-based MS/MS spectra for LC-MS/MS settings. Because GC-MS/MS instruments are still niche products, no large GC-MS/MS databases are currently available. However, GC-MS/MS spectra are useful to gain additional insights into molecular fragmentations (see Fig. 4). Interestingly, the use of unit mass (inaccurate mass) instruments for small molecule MS/MS search never gained much attention, even though LC-MS/MS platforms have been available for more than 15 years. Only a number of smaller libraries were ever derived.118,137–139 An excellent review from 2004 covers most of the available libraries and search techniques at the time140 and a more recent review covers the structural overlap of some of the prominent MS/MS databases.9 The collection of multiple-stage MSn libraries (mass spectral tree libraries) has been covered in other publications141–143 and is not further discussed here. While large commercial libraries such as NIST and Wiley are available with curated spectra and enriched contents, new databases such as MassBank, MoNA, or GNPS have evolved that specifically focus on data sharing and community efforts (see Table 2). The following section gives an overview of existing public and commercial MS/MS databases.
FIGURE 4.
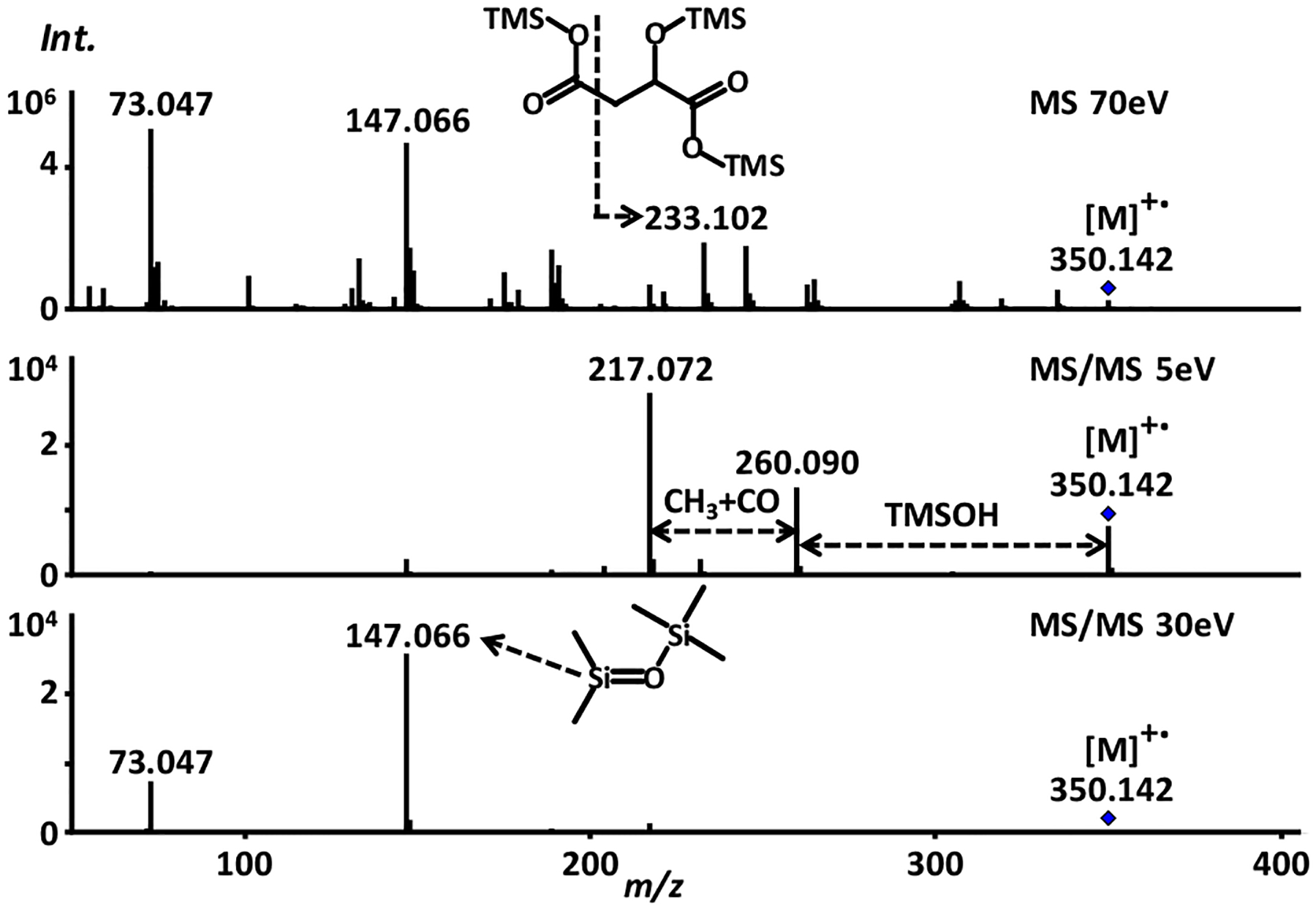
GC-EI-MS and GC-EI-MS/MS of malic acid (3-TMS, trimethylsilyl) with precursor ion m/z 350.142. Different ionization voltages (5 and 30 eV) for product ion spectra can create specific fragments. Such information is important for neutral loss and substructure analysis. Current GC-MS/MS databases currently contain only a small number of compounds in comparison to LC-MS/MS databases
TABLE 2.
Tandem mass spectral databases for small molecule identifications (2016)
| # | Name | Number of MS/MS spectra | Number of compounds | Online search | Freely available | Instrument diversity | In silico |
|---|---|---|---|---|---|---|---|
| 1 | NIST14 MS/MS | 193 120 | 9344 | − | $ | +++ | − |
| 2 | MassBank | 22 000 | 2800 | + | + | +++ | − |
| 3 | METLIN | 72 268 | 14 034 | + | +/$ | ++ | +/− |
| 4 | LipidBlast | 212 516 | 119 200 | − | + | ++ | + |
| 5 | MoNA | 194 000 | 68 700 | + | + | +++ | +/− |
| 6 | mzCloud | 182 000 | 2800 | + | + | + | − |
| 7 | MetaboBASE | 26 000 | 13 000 | − | $ | + | − |
| 8 | GNPS | 212 230 | 12 694 | + | + | ++ | − |
| 9 | Spektraris | 2626 | 487 | + | + | + | − |
| 10 | ReSpect | 9000 | 4000 | + | + | + | − |
| 11 | MSforID | 20 000 | 1200 | − | $ | + | − |
| 12 | HMDB | 5773 | 3729 | + | + | ++ | +/− |
| 13 | MetaMS | 150 | 150 | + | + | + | − |
| 14 | Sumner Library | 1734 | 289 | − | + | + | − |
| 15 | ChemicalSoft | 1619 | 6476 | − | +/$ | + | − |
| 16 | UNPD-ISDB | 170 602 | 170 602 | − | + | + | + |
| 17 | Designer drugs | 10 000 | 750 | − | $ | + | − |
| 18 | Drugs/poisons | 10 000 | 6816 | − | $ | + | − |
Databases that are not commercially or publicly available are not included. Additional vendor specific databases, not listed here, are described in the text. Data were obtained from public sources and might vary.
The “NIST14 MS/MS” (http://chemdata.nist.gov/) commercial database was one of the earliest available and highly curated databases. It covers 193 119 spectra of 43 912 precursor ions from 8351 small molecules.82 Many newly added spectra contain multiple collision energies for CID and HCD mode, covering 2–5 eV steps across the entire collision energy range available. Therefore, the number of MS/MS spectra is much larger than the number of unique compounds. Additionally, NIST14 also contains over 40 000 MS/MS spectra from peptides. All MS/MS spectra were derived from reference standards and over 300 different ionization species (adducts) are included. Data were derived from more than 100 different instrument types and several ionization techniques (ESI, APCI) are covered. All compounds are annotated with their structures, their InChiKeys, and metadata that gives detailed information about the conditions the spectra were acquired. A number of MS/MS spectra were collected from ion trap instruments and are unit mass based. The majority of entries are accurate mass MS/MS spectra from orbital ion traps (HCD MS/MS) and QTOF (CID MS/MS) instruments.
The “MassBank” online database (http://www.MassBank.jp/) is a large public repository of mass spectra from different instruments and multiple contributors.144 Originally developed in Japan starting in 2006, it has become one of the most popular community resources for mass spectral data. Around thirty laboratories contributed to MassBank and consortium members are from all over the world. The Norman MassBank (http://MassBank.normandata.eu/) is a mirror of spectra from the European network of reference laboratories (NORMAN). All MassBank spectra are annotated with structures, database links and additional metadata. All tandem mass spectra are searchable online and can be downloaded. Most spectra are available with a very distribution friendly open data license (Creative Commons by Attribution, CC-BY) and can be easily incorporated into independent software tools and databases.
The “METLIN” online database (http://metlin.scripps.edu/index.php) is one of the most long-standing and largest databases for QTOF tandem mass spectra.145,146 Experimental MS/MS spectra for 14 034 compounds were originally measured on an Agilent QTOF instrument in positive and negative ESI mode using different collision energies (0, 10, 20, and 40 eV CID). METLIN has a special focus on metabolites and the associated online structure database contains more than 240 000 metabolites and additional in silico spectra. The IsoMETLIN database (http://isometlin.scripps.edu) contains mass spectral data from isotopically labeled compounds147 as well as in silico generated fragments. METLIN is also available as METLIN PCDL library for Agilent QTOF instruments with a smaller selection of tandem mass spectra covering 2300 compounds. This allows for direct MS/MS and retention time search within the Agilent MassHunter vendor software. Recently vendor libraries for Bruker (MetaboBASE), WATERS (Progenesis QI), and SCIEX instruments have been released.
The “Wiley MSforID” or Wiley registry of tandem mass spectral data contains around 20 000 mass spectra from 1200 drugs, pharmaceuticals, pesticides, and other small compounds.107,148,149 Spectra were acquired in positive and negative ionization mode with ten different collision energies in the range of 5–50 eV. The library is available in three vendor formats and contains independent MS/MS search software.150
The “ChemicalSoft” libraries (http://www.chemicalsoft.de/) of drugs and toxic compounds were developed on QTRAP instruments151,152 by several researchers in the toxicological community. The provided databases contain MS/MS spectra at multiple collision energies and retention times for over 1200 compounds. The library was also utilized for a comprehensive overview of the fragmentation behavior of the selected compounds in positive and negative ionization mode.153,154
The “Maurer/Wissenbach/Weber” “LCMSn Library of Drugs, Poisons, and Their Metabolites” includes more than 10 000 spectra from 6816 compounds. The database contains MS2 and MS3 wideband spectra from an LXQ linear ion trap in ESI mode. It covers 1500 parent compounds and additional phase I and phase II metabolites as well as related artifacts and impurities.155,156
The “Mass Spectra of Designer Drugs” library (http://www.designer-drugs.de) traditionally contained only GC-MS spectra, but the latest editions added 10,000 MS/MS spectra from 750 compounds. The constantly updated database is available online with a large variety of meta-data and commercially covering different vendor formats.
The “MoNA” (MassBank of North America) database (http://mona.fiehnlab.ucdavis.edu) is a centralized repository that uses software based curation of mass spectra and depends on crowd sourcing (user based) input. Over 190 000 mass spectra are publicly available for download. Spectra were sourced from MassBank, LipidBlast, and in-house spectra. At the moment, mass spectra published in the peer-reviewed literature are only available on paper or PDF, preventing the broader use and applicability of such important information.157 MoNA provides programming interfaces such as REST (representational state transfer architecture) to easily allow automated upload of spectra from any software that can utilize such interfaces. Users can submit novel spectra for direct online access, to allow for broader sharing within the community. Furthermore, all curated and cleaned public spectra can be downloaded in MSP or JSON format to allow for independent in-house use.
The “mzCloud” online database (https://www.mzcloud.org/) focuses on searchable spectral trees (MSn) data. The idea is that multi-stage spectra allow for additional information even if the unknown spectrum is not found in the database.142,143 Multiple MS2 and MSn spectra with various collision energies are organized in spectral nodes that make investigating and searching spectra very intuitive and easy. Associated product ion structures for each peak were calculated using Mass Frontier and additional quantum chemical methods provide the most probable gas phase structure.
The “GNPS” library (http://gnps.ucsd.edu) is a platform focused on natural products.158,159 The Global Natural Product Social Molecular Networking (GNPS) website aims to let natural product researchers work together and share spectra. Users are able to contribute spectral collections and can search, view and download all shared spectra. Currently, around 9000 MS/MS spectra from the GNPS core and community collection are publicly available as well as MS/MS spectra from MassBank, ReSpect, and HMDB. The associated MassIVE website also hosts experimental LC-MS/MS runs and data analysis workflows from a large user base. With a growing database of novel MS/MS spectra, these datasets can be auto-searched and prior unknown compounds can be annotated.
The “ReSpect” database (http://spectra.psc.riken.jp/) is a plant-specific MS/MS database compiled by researchers at the RIKEN institute.160 The ReSpect database was one of the earliest freely available MS/MS databases. The database is derived from literature spectra as well as reference compound MS/MS spectra. The database also contains MS/MS fragmentation association rules for product ions and such information can be used to obtain compound class information. The associated MS/MS database also contains retention times and is especially helpful for plant-based metabolic profiling.161
The “Spektraris AMT Database” (http://langelabtools.wsu.edu/amt/) was developed for plant based metabolite screening and covers 2626 MS/MS spectra from 487 compounds.162 Compounds were measured under ESI and APCI conditions at three different collision energies (10, 20, and 40 eV). All annotated compounds and retention times as well as their associated adducts are freely available on the website and at MassBank.
The “Sumner plant natural product library” (http://metabolomics.missouri.edu/) is an MS/MS library of 289 flavonoids, isoflavonoids, and phenylpropanoids covering 1734 tandem mass spectra collected at different collision energies (10, 20, 30, 40, 50, and 60 eV) on an Agilent 6430 triple quadrupole mass spectrometer. The library can be used for plant metabolomics identifications.163
The “GC-MAXIS/MetaMS” (http://metams.lumc.nl/) GC-APCI-QqToF online spectral library is the only publicly available collection of GC-QTOF based MS/MS spectra. The compounds were acquired on a Bruker maXis 4G QTOF mass spectrometer equipped with an atmospheric pressure chemical ionization (APCI) interface.164
The “WeizMass” database is a collection of 3309 high-resolution MSE spectra measured on an UHPLC-QTOF system (HDMS Synapt, Waters). The database covers positive and negative ionization mode spectra from 3540 plant-based metabolites.165
The “DTU Mycotoxin-Fungal” Secondary Metabolite MS/HRMS library (http://www.bio.dtu.dk/english/Research/Platforms/Metabolom/MSMSLib) contains 836 MS/MS spectra of 277 compounds in Agilent PCDL format166 and was obtained on a Agilent 6550 UPLC-QTOF. The library covers and positive and negative ionization modes as well as multiple collision energies (10, 20, and 40 eV) per compound.
The “MyCompoundID MS/MS” (http://www.mycompoundid.org/) database is an in silico generated database of MS/MS spectra based on enzymatic reactions.79 The library covers 8021 metabolites and 375 809 predicted metabolites that were created by heteroatominitiated bond breakage rules. The library can be searched online in single and batch mode. All structures are visualized and rank scores are provided after search.
The “UNPD-ISDB MS/MS” (http://oolonek.github.io/ISDB/) is a freely available database consisting of 170 602 in silico MS/MS spectra covering 170 602 natural products from the UNPD (Universal Natural Products Database).72 All MS/MS spectra were generated with CFM-ID76 and were part of a natural product dereplication workflow.
The “LipidBlast” library (http://fiehnlab.ucdavis.edu/projects/LipidBlast) is a large in silico generated MS/MS database specifically for lipid identifications.106 LipidBlast employed a heuristic model for fragment and ion abundance calculations. Tandem mass spectra were modeled according to authentic reference spectra and then large numbers of phospholipids with changing side chain fatty acyl lengths and degree if unsaturation were modeled accordingly. Around 400–800 lipids can be annotated using LipidBlast and LC-MS/MS methods. Utilizing the freely available LipidBlast development templates, libraries for new lipid classes can be easily created.67 Originally developed for ion trap and FT-ICR-MS instrumentations LipidBlast has now been optimized for QTOF and Q/orbital ion trap based instruments by Tsugawa et al (http://prime.psc.riken.jp/Metabolomics_Software/). All MS/MS spectra are available under a Creative-Commons-By-Attribution (CC-BY) license that allows for commercial and non-commercial use.
The “Human Metabolome DataBase” (HMDB) (http://www.hmdb.ca/) provides coverage of 41 993 metabolites as well as 5774 experimental MS/MS and 27 999 predicted in silico MS/MS spectra. HMDB contains MS/MS spectra from a variety of instruments with ramped voltage settings and provides links to the MoNA DB. HMDB offers a convenient and fast online MS/MS search with precursor filter and product ion match and head-to-tail view of search versus reference spectrum.
6 |. SOFTWARE FOR MS/MS SEARCH
Mass spectral database software mainly provided search possibilities for single stage mass spectrometry data (MS1) and historically focused on GC-MS electron ionization (70 eV) spectra. Over the last 20 years, the focus has changed toward MS/MS data including CID and HCD spectra obtained from a variety of LC-MS/MS instruments. The following section only covers software applications that are focused on small molecule MS/MS search (see Table 3).
TABLE 3.
Software programs for small molecule MS/MS or MSn search (2016)
| # | Name | Operating system | Freely (+) or commercially ($) available |
|---|---|---|---|
| 1 | NIST MS Search | Windows | + |
| 2 | NIST MSPepSearch | Windows | + |
| 2 | MS-DIAL | Windows | + |
| 3 | MSforID | Windows | $ |
| 4 | SMILEMS | Windows | $ |
| 5 | ACD/Spectrus | Windows | $ |
| 6 | MSPepSearch | Windows | + |
| 7 | Mass Frontier | Windows | $ |
| 8 | Mass++ | Windows | + |
| 9 | Progenesis QI | Windows | $ |
| 10 | Refiner MS | All | $ |
| 11 | OpenMS | All | + |
| 12 | XCMS2 | All | + |
All vendor based software and additional details are described in the manuscript section for each of the software tools. Proteomics tools are not covered.
The “NIST MS Search GUI” (http://chemdata.nist.gov) can be considered the gold standard in mass spectral searching. It is a freely available search program with a graphical user interface (GUI). The program can visualize all structures and spectra in head-to-tail view for easy visual comparison. Furthermore, it provides an easy to navigate result list with match scores, reverse-match, and dot product and probability search. All additional meta-data from libraries can be searched and investigated with constraint search. Precursor and product ion tolerances can be set in mDa or ppm and an unlimited number of custom libraries can be imported using the LIB2NIST library converter.
The “NIST MSPepsearch” software (http://chemdata.nist.gov) is the batch software related to NIST MS for processing hundred thousands of MS/MS product ions scans. In order to create a NIST compatible library, MS/MS reference spectra have to be converted with Lib2NIST software from MSP to NIST format. These libraries can then be searched with traditional MGF or MSP files. The precursor or product ion search window can be defined in a GUI or on the command line (CLI). The database internally uses an indexed and memory cached non-relational database, which makes compound searches extremely fast with up to 5000 spectra per second. The investigation of an average LC-MS/MS run with 10 000 MS/MS spectra typically takes 5–10 sec to search, meaning thousands of LC-MS/MS chromatograms can be processed in a few hours. The results are presented in tab-separated values files (TSV) which include the search spectrum, precursor tolerance, library name found, formula, match scores, reverse match scores, and library hits. The result reports can be imported and filtered very conveniently with the Microsoft Office Excel application.
The “ACD/Spectrus” and “ACD/MS Workbook Suite” (http://www.acdlabs.com/) provides search support for MS/MS spectra and can read and extract a large number different vendor file formats.
The “XCMS2” software is a publicly available software that can be used within the R statistics language.167 It directly searches METLIN online data (acquired on an Agilent QTOF with multiple collision energies (0, 10, 20, and 40 eV). XCMS2 also intelligently matches the experimental collision energy if multiple energies available. It uses the traditional precursor ion selection window and additionally a distance matrix score to obtain good spectral matches.
The Wiley “MSforID” search algorithm is available with the MSforID library and uses a relative average match probability (ramp) score.105,168 The software calculates the similarity of fragment ions from the unknown MS/MS spectrum versus the library spectra. From the matching fragments a reference spectrum-specific match probability (mp) is then calculated. The match probability values from multiple reference compounds are averaged and the compound specific ramp score is subsequently obtained.
The “Mass++” software (http://masspp.jp) is an open source LC-MS/MS software with multiple vendor support and includes proteomics and metabolomics data processing tools.169 Mass++ supports MS/MS search of MassBank spectra and additional data analysis and visualization tools. Mass++ can search MassBank directly via the Simple Object Access Protocol Application Programming Interface (SOAP API).
The “Progenesis QI” software (http://www.nonlinear.com/progenesis/) allows for MS/MS search and import of external reference libraries such as MassBank, METLIN, or LipidBlast. Support for DDA and DIA workflows is provided. After the deconvolution, it allows for selection of different databases, selection of precursor and product ion accuracy and presents the output in a sortable identification sheet. The search score, mass error, and isotope similarity are also taken into account. Additional retention time matching is also supported. Progenesis QI also provides multi-vendor support for Agilent, Bruker, SCIEX, and Thermo as well as for mzML and mzXML data.
The “OpenMS” software (http://www.openms.de) is an open source software workflow for LC-MS/MS data processing.170,171 It supports open formats such as mzML and it is possible to perform accurate mass search. The precursor mass tolerance can be set by utilizing individual workflow nodes. The similarity scores are calculated by utilizing a number of provided matching algorithms.
The “Expressionist Refiner MS” software (http://www.genedata.com) is a workflow system that allows the processing of LC-MS/MS and direct-infusion MS/MS data. The software integrates different workflow modules that can be used to search local and online MS/MS databases and to perform additional retention time matching.
The “SMILEMS” software (http://www.genebio.com/) permits the utilization of multiple MS/MS and MSn databases. The software allows the import of data from multiple vendors and it can utilize different reference libraries. SmileMS utilizes the X-rank algorithm and retention time filters, which can aid during the removal of false positive identifications.103 The X-Rank does not take absolute or relative intensities into account, but sorts the peak intensities of a spectrum and subsequently calculates a correlation between the sorted spectra.
The “MS-DIAL” software (http://prime.psc.riken.jp/Metabolomics_Software/) can be used for alignment of multiple runs and subsequent MS/MS identification based on DDA and DIA data.60 The latter approach is more complicated because it requires a mass spectral deconvolution step to obtain clean mass spectra. A number of external libraries such as LipidBlast and MassBank can be imported and the MS-DIAL program and bundled MS/MS libraries are freely available.
7 |. VENDOR-BASED SOFTWARE AND DATABASES
Interestingly, many mass spectrometry vendors have backed away from tightly restricting their software and now allow the integration of third-party plugins and library import from multiple sources. This trend positively supports users, who are now able to access a wider product range and a larger number of libraries. Discussed below are only those vendors who distribute tandem mass spectrometers as well as software and databases for MS/MS search.
“Agilent” provides the MassHunter Personal Compound Database and Library software (PCDL) for searching MS/MS spectra. The PCDL Manager is software that allows for the creation and browsing of MS/MS databases. The import/export functions are limited to TXT, comma-separated values (CSV), and MOL files. No common mass spectral file formats are currently supported. The PCDL databases can be directly accessed from the MassHunter Workstation software or the ProFinder software. Both MS/MS search and retention time matching can be employed. A series of commercial databases such as Agilent/METLIN PCDL (2278 compounds) developed in collaboration with Gary Siuzdak at the Scripps Research Institute, a toxicology derived database including the Broecker/Herre/Pragst PCDL (2400 compounds) and a pesticide PCDL library (740 compounds) for LC-MS/MS and GC-MS/MS are available. The extractables and leachables (E/L) PCDL contains MS/MS spectra for 300 UV stabilizers, packaging contaminants, silicones, and nitrosamines measured with Electrospray and APCI on a QTOF instruments.
“Bruker” currently provides the MetaboBASE personal library with 30 000 MS/MS spectra measured at different collision energies from 13 000 compounds including di- and tri-peptides. The library was created in collaboration with Paul Benton and Gary Siuzdak at the Scripps Research Institute. The library covers 13 000 synthetic and isolated standards as well as di- and tri-peptides. The smaller Bruker Human Metabolome Database (HMDB) Metabolite Library with 800 compounds selected from the original HMDB (www.hmdb.ca) specifically covers human metabolites. Spectra were acquired at different collision energies (10, 20, 30, and 40 eV). The database also includes ramped spectra (20–50 eV) as well as different isolation windows (1 and 6 Da). The MetaboBASE Plant Libraries contains around 1000 MS/MS spectra of secondary plant metabolites and was created in collaboration with Lloyd Sumner at the University of Missouri. All compounds are annotated with compound structures and metadata as well as external database identifiers. Tandem mass spectral databases can be queried using the Bruker Compass DataAnalysis and edited with the LibraryEditor software. The Bruker ToxTyper Library of Drugs contains 830 compounds and their related MS1, MS2, and MS3 spectra measured on an ion trap instrument. Additional solutions are provided with the ToxScreener and ToxTyper software, which allow for the forensic analysis of compounds and drugs of abuse.
“SCIEX” provides diverse customized libraries for metabolites, forensic drugs (517 MS/MS spectra of 370 compounds), antibiotics (259 MS/MS spectra of 244 compounds), pesticides (1310 MS/MS spectra of 557 compounds), and food environmental analysis (2148 MS/MS spectra of 1189 compounds).172 The newer libraries are supported in high-resolution mode and contain positive and negative ionization mode spectra. The spectra can be used with the TripleTOF and QTRAP instruments. Support for mass spectral library search is provided within the Analyst package, the MasterView, LibraryView, and LightSight software.
“Shimadzu” provides the Profiling Solution software program to search MS/MS spectra. A number of MRM based libraries are provided, mostly for their QqQ line of instruments. The structural analysis of 256 glycosphingolipids can be performed with a new MS2 and MS3 Library. The integrated LCMSsolution software provides similarity search. Shimadzu also supported the open source software Mass++ which can be used for independent MS/MS search.
“ThermoFisher” now provides several software tools including TraceFinder, ToxFinder, and the Compound Discoverer Software to search high-resolution accurate mass MS/MS spectral libraries. The programs also allow for seamless integration into the mzCloud online repository. Historically, tandem mass spectral search was performed by NIST integration into ThermoFisher Xcalibur or the use of the Mass Frontier software. The “Library of Toxicological Compounds” includes 900 clinical and forensic toxicology related compounds with 4500 spectra with multiple collision energies. The “Library of Food Safety and Environmental Compounds” includes 1600 compounds and 8000 MS/MS spectra. The high-resolution spectra allow a baseline resolution of carbon, nitrogen, oxygen, and sulfur isotopes and the multiple collision energies allow for increased probabilities during compound identifications.
“Waters” allows for spectral library creation within the Waters ChromaLynx software. ChromaLynx also uses the NIST based search algorithm and can be used across all Waters instruments. The new Waters Progenesis QI software (Nonlinear Dynamics) allows for third party import of publicly available libraries such as MassBank or LipidBlast. The Waters METLIN MS/MS Library covers spectra for 13 900 measured compounds as well as 220 000 in silico spectra.
8 |. GLOBAL APPLICATIONS FOR SMALL MOLECULE MS/MS SEARCH
Fields of applications for MS/MS matching include nearly all scientific areas that make use of LC-MS/MS, GC-MS/MS, and tandem mass spectrometry data.173–175 This includes metabolic profiling,126 dereplication of complex natural extracts,176,177 fungal metabolites,178,179 marine products,166 plant based metabolic profiling160,162,180,181 and plant metabolomics,180 lipid analysis,182 toxicology analysis,183–186 environmental analysis,187,188 food contaminants,189 forensics,190,191 drugs,192,193 and pesticide screening,194–196 as well as statistical MS/MS fragmentation analysis197 and network maps for visualizations.198 For those cases where no reference spectrum is found in any database, purely computational approaches have to be taken. These in silico tools are extensively covered elsewhere.199–201
9 |. OUTLOOK AND CONCLUSIONS
Instrumental prerequisites to generate MS/MS databases have been available for a quarter of a century. Data-sharing and collaborative research projects such as MassBank gave a huge boost to promoting the wider use of MS/MS database search within the small molecule community. Still, there are many examples where research groups publish papers using in-house libraries that are neither publicly nor commercially available. Large European, Japanese, and US funding organizations such as US National Science Foundation (NSF) and the National Institutes of Health (NIH) clearly have an interest in data-sharing and novel databases such as MassBank, MoNA, or GNPS show that such projects can be very successful in supporting a large research community.
There is also a strong need for better and improved scoring algorithms. These algorithms must be validated by using purely statistical validations from large and highly diverse MS/MS databases. There are currently no decoy databases for small molecule MS/MS available, which would be necessary for calculations of false positive estimates. False discovery rates can now be calculated with recently developed MS/MS decoy databases for small molecules.111 MS/MS databases will grow in diversity and size and will include experimental spectra from reference compounds as well as in silico generated MS/MS spectra. Especially in silico generated libraries will gain in importance and accuracy, but have to be carefully validated to avoid the distribution of inaccurately modeled spectra.
Online services that provide easy-to-use search interfaces will allow researchers to annotate large LC-MS/MS runs in a convenient way. The post-processing of search results and library hit tables will require approaches such as adduct removal, peak merging from multiple spectra, and connections to compound databases for easy investigation. Here, we see enormous development potential to improve such database services on an academic and commercial level. MS/MS database search is a fast-lane for compound annotations and fortunately there is a lot of positive momentum to widen the use and distribution for the benefits of the broader community.
ACKNOWLEDGMENTS
Funding for T.K. and O.F. was supported by NSF MCB 1139644, NSF MCB 1153491, NSF MCB 1611846, NSF CBET 1438211, NSF IOS 1340058, and NIH U24 DK097154. Additional funding for T.K. was provided by American Heart Association 15SDG25760020 (Irvin) and NIH 7R01HL091357-06 (Arnett). The use of the Jane Journal Estimator (jane.biosemantics.org),202 Google Scholar, and the detailed online information about MS/MS databases from Scientific Instrument Services (http://www.sisweb.com) were greatly appreciated.
Funding information
NSF MCB, Grant numbers: 1139644, 1153491, 1611846; NSF CBET, Grant number: 1438211; NSF IOS, Grant number: 1340058; NIH, Grant number: U24 DK097154; American Heart Association, Grant number: 15SDG25760020; NIH, Grant number: 7R01HL091357-06
REFERENCES
- 1.Crawford RW, Brand HR, Wong CM, Gregg HR, Hoffman PA, Enke CG. Instrument database system and application to mass spectrometry/mass spectrometry. Anal Chem. 1984;56:1121–1127. [DOI] [PubMed] [Google Scholar]
- 2.Cross K, Enke C. A spectral matching system for MS/MS data. Comput Chem. 1986;10:175–181. [Google Scholar]
- 3.McLafferty FW. Tandem mass spectrometry. Science. 1981;214: 280–287. [DOI] [PubMed] [Google Scholar]
- 4.McLafferty FW, Hirota A, Barbalas MP. Library of collisional activation mass spectra. Organic Mass Spectrom. 1980;15:327–328. [Google Scholar]
- 5.Stein S Mass spectral reference libraries: an ever-expanding resource for chemical identification. Anal Chem. 2012;84: 7274–7282. [DOI] [PubMed] [Google Scholar]
- 6.Johnson SR, Lange BM. Open-access metabolomics databases for natural product research: present capabilities and future potential. Front Bioeng Biotechnol. 2015;3:22. 10.3389/fbioe.2015.00022. eCollection 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Milman BL, Zhurkovich IK. Mass spectral libraries: a statistical review of the visible use. Trends Anal Chem. 2016;80:636–640. [Google Scholar]
- 8.Cajka T, Fiehn O. Comprehensive analysis of lipids in biological systems by liquid chromatography-mass spectrometry. Trends Anal Chem. 2014;61:192–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vinaixa M, Schymanski EL, Neumann S, Navarro M, Salek RM, Yanes O. Mass spectral databases for LC/MS and GC/MS-based metabolomics: State of the field and future prospects. TrAC Trends Anal Chem. 2015;78:23–35. [Google Scholar]
- 10.Forcisi S, Moritz F, Kanawati B, Tziotis D, Lehmann R, Schmitt-Kopplin P. Liquid chromatography-mass spectrometry in metabolomics research: mass analyzers in ultra high pressure liquid chromatography coupling. J Chromatogr A. 2013;1292:51–65. [DOI] [PubMed] [Google Scholar]
- 11.Griss J Spectral library searching in proteomics. Proteomics. 2016;16. 10.1002/pmic.201500296 [DOI] [PubMed] [Google Scholar]
- 12.Kapp E, Schütz F. Overview of tandem mass spectrometry (MS/MS) database search algorithms. Curr Protoc Protein Sci. 2001. 10.1002/0471140864.ps2502s49 [DOI] [PubMed] [Google Scholar]
- 13.Yates JR III. Pivotal role of computers and software in mass spectrometry–SEQUEST and 20 years of tandem MS database searching. J Am Soc Mass Spectrom. 2015;26:1804–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vaniya A, Fiehn O. Using fragmentation trees and mass spectral trees for identifying unknown compounds in metabolomics. Trends Anal Chem. 2015;69:52–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holčapek M, Jirásko R, Lísa M. Recent developments in liquid chromatography-mass spectrometry and related techniques. J Chromatogr A. 2012;1259:3–15. [DOI] [PubMed] [Google Scholar]
- 16.Brunnée C The ideal mass analyzer: fact or fiction? Int J Mass Spectrom. 1987;76:125–237. [Google Scholar]
- 17.Glish GL, Burinsky DJ. Hybrid mass spectrometers for tandem mass spectrometry. J Am Soc Mass Spectrom. 2008;19:161–172. [DOI] [PubMed] [Google Scholar]
- 18.Kandiah M, Urban PL. Advances in ultrasensitive mass spectrometry of organic molecules. Chem Soc Rev. 2013;42:5299–5322. [DOI] [PubMed] [Google Scholar]
- 19.Weckwerth W Unpredictability of metabolism—the key role of metabolomics science in combination with next-generation genome sequencing. Anal Bioanal Chem. 2011;400:1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Werner E, Heilier J-F, Ducruix C, Ezan E, Junot C, Tabet J-C. Mass spectrometry for the identification of the discriminating signals from metabolomics: current status and future trends. J Chromatogr B. 2008;871:143–163. [DOI] [PubMed] [Google Scholar]
- 21.Prentice BM, Chumbley CW, Caprioli RM. High-speed MALDI MS/MS imaging mass spectrometry using continuous raster sampling. J Mass Spectrom. 2015;50:703–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li D-X, Gan L, Bronja A, Schmitz OJ. Gas chromatography coupled to atmospheric pressure ionization mass spectrometry (GC-API-MS): review. Anal Chim Acta. 2015;891:43–61. [DOI] [PubMed] [Google Scholar]
- 23.Van Bavel B, Geng D, Cherta L, et al. Atmospheric-pressure chemical ionization tandem mass spectrometry (APGC/MS/MS) an alternative to high-resolution mass spectrometry (HRGC/HRMS) for the determination of dioxins. Anal Chem. 2015;87:9047–9053. [DOI] [PubMed] [Google Scholar]
- 24.Lee YJ, Smith EA, Jun JH. Gas chromatography-high resolution tandem mass spectrometry using a GC-APPI-LIT orbitrap for complex volatile compounds analysis. Mass Spectrom Lett. 2012;3:29–38. [Google Scholar]
- 25.Cotter RJ. High energy collisions on tandem time-of-flight mass spectrometers. J Am Soc Mass Spectrom. 2013;24:657–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sleno L, Volmer DA. Ion activation methods for tandem mass spectrometry. J Mass Spectrom. 2004;39:1091–1112. [DOI] [PubMed] [Google Scholar]
- 27.Satoh T, Kubo A, Shimma S, Toyoda M. Mass spectrometry imaging and structural analysis of lipids directly on tissue specimens by using a spiral orbit type tandem time-of-flight mass spectrometer, spiral TOF-TOF. Mass Spectrom (Tokyo). 2012;1. A0013. 10.5702/massspectrometry.A0013. Epub 2012 Nov 16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Subramaniam R, Östin A, Nygren Y, Juhlin L, Nilsson C, Åstot C. An isomer-specific high-energy collision-induced dissociation MS/MS database for forensic applications: a proof-of-concept on chemical warfare agent markers. J Mass Spectrom. 2011;46:917–924. [DOI] [PubMed] [Google Scholar]
- 29.Weinmann W, Stoertzel M, Vogt S, Svoboda M, Schreiber A. Tuning compounds for electrospray ionization/in-source collision-induced dissociation and mass spectra library searching. J Mass Spectrom. 2001;36:1013–1023. [DOI] [PubMed] [Google Scholar]
- 30.Yost RA, Fetterolf DD. Tandem mass spectrometry (MS/MS) instrumentation. Mass Spectrom Rev. 1983;2:1–45. [Google Scholar]
- 31.McClellan JE, Murphy JP, Mulholland JJ, Yost RA. Effects of fragile ions on mass resolution and on isolation for tandem mass spectrometry in the quadrupole ion trap mass spectrometer. Anal Chem. 2002;74:402–412. [DOI] [PubMed] [Google Scholar]
- 32.Cunningham C Jr, Glish GL, Burinsky DJ. High amplitude short time excitation: a method to form and detect low mass product ions in a quadrupole ion trap mass spectrometer. J Am Soc Mass Spectrom. 2006;17:81–84. [DOI] [PubMed] [Google Scholar]
- 33.McLuckey SA, Goeringer DE. Slow heating methods in tandem mass spectrometry. J Mass Spectrom. 1997;32:461–474. [Google Scholar]
- 34.Demarque DP, Crotti AE, Vessecchi R, Lopes JL, Lopes NP. Fragmentation reactions using electrospray ionization mass spectrometry: an important tool for the structural elucidation and characterization of synthetic and natural products. Nat Prod Rep. 2016;33:432–455. [DOI] [PubMed] [Google Scholar]
- 35.Johnson AR, Carlson EE. Collision-induced dissociation mass spectrometry: a powerful tool for natural product structure elucidation. ACS Publications. 2015;87:10668–10678. [DOI] [PubMed] [Google Scholar]
- 36.Eliuk S, Makarov AA. Evolution of orbitrap mass spectrometry instrumentation. Ann Rev Anal Chem. 2015;8:61–80. [DOI] [PubMed] [Google Scholar]
- 37.Ichou F, Schwarzenberg A, Lesage D, et al. Comparison of the activation time effects and the internal energy distributions for the CID, PQD and HCD excitation modes. J Mass Spectrom. 2014;49:498–508. [DOI] [PubMed] [Google Scholar]
- 38.Tang H, Wang X, Xu L, et al. Establishment of local searching methods for orbitrap-based high throughput metabolomics analysis. Talanta. 2016;156:163–171. [DOI] [PubMed] [Google Scholar]
- 39.Bushee JL, Argikar UA. An experimental approach to enhance precursor ion fragmentation for metabolite identification studies: application of dual collision cells in an orbital trap. Rapid Commun Mass Spectrom. 2011;25:1356–1362. [DOI] [PubMed] [Google Scholar]
- 40.Mullard G, Allwood JW, Weber R, et al. A new strategy for MS/MS data acquisition applying multiple data dependent experiments on Orbitrap mass spectrometers in non-targeted metabolomic applications. Metabolomics. 2015;11:1068–1080. [Google Scholar]
- 41.Yoo HJ, Liu H, Håkansson K. Infrared multiphoton dissociation and electron-induced dissociation as alternative MS/MS strategies for metabolite identification. Anal Chem. 2007;79:7858–7866. [DOI] [PubMed] [Google Scholar]
- 42.Jones JW, Thompson CJ, Carter CL, Kane MA. Electron-induced dissociation (EID) for structure characterization of glycerophosphatidylcholine: determination of double-bond positions and localization of acyl chains. J Mass Spectrom. 2015;50:1327–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McLuckey SA, Mentinova M. Ion/neutral, ion/electron, ion/photon, and ion/ion interactions in tandem mass spectrometry: do we need them all? Are they enough? J Am Soc Mass Spectrom. 2011;22:3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nikolskiy I, Mahieu NG, Chen Y-J, Tautenhahn R, Patti GJ. An untargeted metabolomic workflow to improve structural characterization of metabolites. Anal Chem. 2013;85:7713–7719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gallien S, Duriez E, Demeure K, Domon B. Selectivity of LC-MS/MS analysis: implication for proteomics experiments. J Proteomics. 2013;81:148–158. [DOI] [PubMed] [Google Scholar]
- 46.Arnhard K, Gottschall A, Pitterl F, Oberacher H. Applying ‘Sequential Windowed Acquisition of All Theoretical Fragment Ion Mass Spectra’ (SWATH) for systematic toxicological analysis with liquid chromatography-high-resolution tandem mass spectrometry. Anal Bioanal Chem. 2015;407:405–414. [DOI] [PubMed] [Google Scholar]
- 47.Lacorte S, Agüera A, Cortina-Puig M, Gómez-Canela C. 2015. Recent developments in liquid chromatography–mass spectrometry. Mass spectrometry for the analysis of pesticide residues and their metabolites. Hoboken, NJ: John Wiley & Sons, Inc. 131–159. [Google Scholar]
- 48.Benton HP, Ivanisevic J, Mahieu NG, et al. Autonomous metabolomics for rapid metabolite identification in global profiling. Anal Chem. 2014;87:884–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Junot C, Fenaille F, Colsch B, Bécher F. High resolution mass spectrometry based techniques at the crossroads of metabolic pathways. Mass Spectrom Rev. 2014;33:471–500. [DOI] [PubMed] [Google Scholar]
- 50.Michalski A, Damoc E, Lange O, et al. Ultra high resolution linear ion trap Orbitrap mass spectrometer (Orbitrap Elite) facilitates top down LC MS/MS and versatile peptide fragmentation modes. Mol Cell Proteomics. 2012;11:O111.013698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yamada M, Kita Y, Kohira T, et al. A comprehensive quantification method for eicosanoids and related compounds by using liquid chromatography/mass spectrometry with high speed continuous ionization polarity switching. J Chromatogr B. 2015;995:74–84. [DOI] [PubMed] [Google Scholar]
- 52.Skibiński R, Komsta Ł. Optimization of data acquisition and sample preparation methods for LC-MS urine metabolomic analysis. Cancer Res. 2015;1:3. [Google Scholar]
- 53.Cajka T, Fiehn O. Increasing lipidomic coverage by selecting optimal mobile-phase modifiers in LC-MS of blood plasma. Metabolomics. 2016;12:34. [Google Scholar]
- 54.Tang K, Shvartsburg AA, Lee H-N, et al. High-sensitivity ion mobility spectrometry/mass spectrometry using electrodynamic ion funnel interfaces. Anal Chem. 2005;77:3330–3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhou Z, Shen X, Tu J, Zhu Z-J. Large-scale prediction of collision cross-section values for metabolites in ion mobility-mass spectrometry. Anal Chem. 2016;88:11084–11091. [DOI] [PubMed] [Google Scholar]
- 56.Broeckling CD, Heuberger AL, Prince JA, Ingelsson E, Prenni JE. Assigning precursor-product ion relationships in indiscriminant MS/MS data from non-targeted metabolite profiling studies. Metabolomics. 2013;9:33–43. [Google Scholar]
- 57.Zhu X, Chen Y, Subramanian R. Comparison of information-dependent acquisition, SWATH, and MSAll techniques in metabolite identification study employing ultrahigh-performance liquid chromatography–quadrupole time-of-flight mass spectrometry. Anal Chem. 2014;86:1202–1209. [DOI] [PubMed] [Google Scholar]
- 58.Gillet LC, Navarro P, Tate S, et al. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics. 2012;11:O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cajka T, Fiehn O. Toward merging untargeted and targeted methods in mass spectrometry-based metabolomics and lipidomics. Anal Chem. 2015;88:524–545. [DOI] [PubMed] [Google Scholar]
- 60.Tsugawa H, Cajka T, Kind T, et al. MS-DIAL: data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat Methods. 2015;12:523–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Röst HL, Rosenberger G, Navarro P, et al. OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data. Nat Biotechnol. 2014;32:219–223. [DOI] [PubMed] [Google Scholar]
- 62.Teleman J, Röst HL, Rosenberger G, et al. DIANA—algorithmic improvements for analysis of data-independent acquisition MS data. Bioinformatics. 2015;31:555–562. [DOI] [PubMed] [Google Scholar]
- 63.Prakash A, Peterman S, Ahmad S, et al. Hybrid data acquisition and processing strategies with increased throughput and selectivity: pSMART analysis for global qualitative and quantitative analysis. J Proteome Res. 2014;13:5415–5430. [DOI] [PubMed] [Google Scholar]
- 64.Tsou C-C, Avtonomov D, Larsen B, et al. DIA-Umpire: comprehensive computational framework for data-independent acquisition proteomics. Nat Methods. 2015;12:258–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Alechaga É, Moyano E, Galceran MT. Ion-molecule adduct formation in tandem mass spectrometry. Anal Bioanal Chem. 2016;408: 1269–1277. [DOI] [PubMed] [Google Scholar]
- 66.Zhang X, Clausen MR, Zhao X, Zheng H, Bertram HC. Enhancing the power of liquid chromatography-mass spectrometry-based urine metabolomics in negative ion mode by optimization of the additive. Anal Chem. 2012;84:7785–7792. [DOI] [PubMed] [Google Scholar]
- 67.Kind T, Okazaki Y, Saito K, Fiehn O. Lipid blast templates as flexible tools for creating new in-silico tandem mass spectral libraries. Anal Chem. 2014;86:11024–11027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ma Y, Kind T, Vaniya A, Gennity I, Fahrmann JF, Fiehn O. An in silico MS/MS library for automatic annotation of novel FAHFA lipids. J Cheminform. 2015;7:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kochen MA, Chambers MC, Holman JD, et al. Greazy: open-source software for automated phospholipid MS/MS identification. Anal Chem. 2016;88:5733–5741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Allen F, Greiner R, Wishart D. Competitive fragmentation modeling of ESI-MS/MS spectra for putative metabolite identification. Metabolomics. 2014;11:98–110. [Google Scholar]
- 71.Allen F, Pon A, Wilson M, Greiner R, Wishart D. CFM-ID: a web server for annotation, spectrum prediction and metabolite identification from tandem mass spectra. Nucleic Acids Res. 2014;42: W94–W99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Allard P-M, Péresse T, Bisson J, et al. Integration of molecular networking and in-silico MS/MS fragmentation for natural products dereplication. Anal Chem. 2016;88:3317–3323. [DOI] [PubMed] [Google Scholar]
- 73.Bauer CA, Grimme S. How to compute electron ionization mass spectra from first principles. J Phys Chem A. 2016;120:3755–3766. [DOI] [PubMed] [Google Scholar]
- 74.Cautereels J, Claeys M, Geldof D, Blockhuys F. Quantum chemical mass spectrometry: ab initio prediction of electron ionization mass spectra and identification of new fragmentation pathways. J Mass Spectrom. 2016;51:602–614. [DOI] [PubMed] [Google Scholar]
- 75.Grimme S Towards first principles calculation of electron impact mass spectra of molecules. Angew Chem Int Ed Engl. 2013;52: 6306–6312. [DOI] [PubMed] [Google Scholar]
- 76.Allen F, Greiner R, Wishart D. Competitive fragmentation modeling of ESI-MS/MS spectra for putative metabolite identification. Metabolomics. 2015;11:98–110. [Google Scholar]
- 77.Jeffryes JG, Colastani RL, Elbadawi-Sidhu M, et al. MINEs: open access databases of computationally predicted enzyme promiscuity products for untargeted metabolomics. J Cheminform. 2015;7:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Menikarachchi LC, Hill DW, Hamdalla MA, Mandoiu II, Grant DF. In silico enzymatic synthesis of a 400 000 compound biochemical database for nontargeted metabolomics. J Chem Inf Model. 2013;53:2483–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Huan T, Tang C, Li R, Shi Y, Lin G, Li L. MyCompoundID MS/MS Search: metabolite identification using a library of predicted fragment-ion-spectra of 383,830 possible human metabolites. Anal Chem. 2015;87:10619–10626. [DOI] [PubMed] [Google Scholar]
- 80.Ausloos P, Clifton C, Lias S, et al. The critical evaluation of a comprehensive mass spectral library. J Am Soc Mass Spectrom. 1999;10:287–299. [DOI] [PubMed] [Google Scholar]
- 81.Oberacher H, Weinmann W, Dresen S. Quality evaluation of tandem mass spectral libraries. Anal Bioanal Chem. 2011;400:2641–2648. [DOI] [PubMed] [Google Scholar]
- 82.Yang X, Neta P, Stein SE. Quality control for building libraries from electrospray ionization tandem mass spectra. Anal Chem. 2014;86:6393–6400. [DOI] [PubMed] [Google Scholar]
- 83.Wallace WE, Ji W, Tchekhovskoi DV, Phinney KW, Stein SE. Mass spectral library quality assurance by inter-library comparison. J Am Soc Mass Spectrom. 2017;28:733–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Breckels LM, Gibb S, Petyuk V, Gatto L. R for ProteomicsProteome informatics. Royal Soc Chem. 2016;321–364; Chapter 14. 10.1039/9781782626732 [DOI] [Google Scholar]
- 85.Gatto L, Lilley KS. MSnbase—an R/bioconductor package for isobaric tagged mass spectrometry data visualization, processing and quantitation. Bioinformatics. 2012;28:288–289. [DOI] [PubMed] [Google Scholar]
- 86.Lawson TN, Weber RJM, Jones MR, et al. MsPurity: automated evaluation of precursor ion purity for mass spectrometry based fragmentation in metabolomics. Anal Chem. 2017;89:2432–2439. [DOI] [PubMed] [Google Scholar]
- 87.Stravs MA, Schymanski EL, Singer HP, Hollender J. Automatic recalibration and processing of tandem mass spectra using formula annotation. J Mass Spectrom. 2013;48:89–99. [DOI] [PubMed] [Google Scholar]
- 88.Wu JX, Song X, Pascovici D, et al. SWATH mass spectrometry performance using extended peptide MS/MS assay libraries. Mol Cell Proteomics. 2016;15:2501–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Broeckling CD, Afsar F, Neumann S, Ben-Hur A, Prenni J. RAMClust: a novel feature clustering method enables spectral-matching-based annotation for metabolomics data. Anal Chem. 2014;86:6812–6817. [DOI] [PubMed] [Google Scholar]
- 90.Hopley C, Bristow T, Lubben A, et al. Towards a universal product ion mass spectral library-reproducibility of product ion spectra across eleven different mass spectrometers. Rapid Commun Mass Spectrom. 2008;22:1779–1786. [DOI] [PubMed] [Google Scholar]
- 91.Kessner D, Chambers M, Burke R, Agus D, Mallick P. ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics. 2008;24:2534–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Haug K, Salek RM, Conesa P, et al. MetaboLights—an open-access general-purpose repository for metabolomics studies and associated meta-data. Nucleic Acids Res. 20132;41: D781–D786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sud M, Fahy E, Cotter D, et al. Metabolomics workbench: an international repository for metabolomics data and metadata, metabolite standards, protocols, tutorials and training, and analysis tools. Nucleic Acids Res. 2016;44:D463–D470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Samokhin A, Sotnezova K, Lashin V, Revelsky I. Evaluation of mass spectral library search algorithms implemented in commercial software. J Mass Spectrom. 2015;50:820–825. [DOI] [PubMed] [Google Scholar]
- 95.Hopfgartner G, Chernushevich IV, Covey T, Plomley JB, Bonner R. Exact mass measurement of product ions for the structural elucidation of drug metabolites with a tandem quadrupole orthogonal-acceleration time-of-flight mass spectrometer. J Am Soc Mass Spectrom. 1999;10:1305–1314. [Google Scholar]
- 96.McLafferty F, Hertel R, Villwock R. Probability based matching of mass spectra. Rapid identification of specific compounds in mixtures. Organic Mass Spectrom. 1974;9:690–702. [Google Scholar]
- 97.Stein SE, Scott DR. Optimization and testing of mass spectral library search algorithms for compound identification. J Am Soc Mass Spectrom. 1994;5:859–866. [DOI] [PubMed] [Google Scholar]
- 98.Stein SE. Estimating probabilities of correct identification from results of mass spectral library searches. J Am Soc Mass Spectrom. 1994;5:316–323. [DOI] [PubMed] [Google Scholar]
- 99.Champarnaud E, Hopley C. Evaluation of the comparability of spectra generated using a tuning point protocol on twelve electrospray ionisation tandem-in-space mass spectrometers. Rapid Commun Mass Spectrom. 2011;25:1001–1007. [DOI] [PubMed] [Google Scholar]
- 100.Yilmaz Ş, Vandermarliere E, Martens L. Methods to calculate spectrum similarity. In: Keerthikumar S, Mathivanan S, editors. Proteome bioinformatics. New York, NY: Springer New York; 2017. pp. 75–100. [DOI] [PubMed] [Google Scholar]
- 101.Hansen ME, Smedsgaard J, Petras D, et al. Significance estimation for large scale untargeted metabolomics annotations. bioRxiv 109389; 10.1101/109389 [DOI] [Google Scholar]
- 102.Zhang J, Wei X-L, Zheng C-H, Wang B, Wang F, Chen P. Compound identification using random projection for gas chromatography-mass spectrometry data. Int J Mass Spectrom. 2016;407:16–21. [Google Scholar]
- 103.Mylonas R, Mauron Y, Masselot A, et al. X-Rank: a robust algorithm for small molecule identification using tandem mass spectrometry. Anal Chem. 2009;81:7604–7610. [DOI] [PubMed] [Google Scholar]
- 104.Oberacher H, Pavlic M, Libiseller K, et al. On the inter-instrument and the inter-laboratory transferability of a tandem mass spectral reference library: 2. Optimization and characterization of the search algorithm. J Mass Spectrom. 2009;44: 494–502. [DOI] [PubMed] [Google Scholar]
- 105.Oberacher H, Whitley G, Berger B, Weinmann W. Testing an alternative search algorithm for compound identification with the ‘Wiley Registry of Tandem Mass Spectral Data, MSforID’. J Mass Spectrom. 2013;48:497–504. [DOI] [PubMed] [Google Scholar]
- 106.Kind T, Liu K-H, Lee DY, DeFelice B, Meissen JK, Fiehn O. LipidBlast in silico tandem mass spectrometry database for lipid identification. Nat Methods. 2013;10:755–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Oberacher H Applying tandem mass spectral libraries for solving the critical assessment of small molecule identification (CASMI) LC/MS challenge 2012. Metabolites. 2013;3:312–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ridder L, van der Hooft JJ, Verhoeven S. Automatic compound annotation from mass spectrometry data using MAGMa. Mass Spectrom. 2014;3:S0033–S0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Spalding JL, Cho K, Mahieu NG, et al. Bar coding MS2 spectra for metabolite identification. Anal Chem. 2016;88:2538–2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wohlgemuth G, Mehta SS, Mejia RF, et al. SPLASH, a hashed identifier for mass spectra. Nat Biotechnol. 2016;34:1099–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Scheubert K, Hufsky F, Petras D, et al. Significance estimation for large scale untargeted metabolomics annotations. bioRxiv 109389. 10.1101/109389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Schrimpe-Rutledge AC, Codreanu SG, Sherrod SD, McLean JA. Untargeted metabolomics strategies—challenges and emerging directions. J Am Soc Mass Spectrom. 2016;27:1897–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Östman P, Ketola RA, Ojanperä I. Application of electrospray ionization product ion spectra for identification with atmospheric pressure matrix-assisted laser desorption/ionization mass spectrometry—a case study with seized drugs. Drug Test Anal. 2013;5:68–73. [DOI] [PubMed] [Google Scholar]
- 114.Wissenbach DK, Meyer MR, Weber AA, et al. Towards a universal LC-MS screening procedure—can an LIT LC-MSn screening approach and reference library be used on a quadrupole-LIT hybrid instrument? J Mass Spectrom. 2012;47:66–71. [DOI] [PubMed] [Google Scholar]
- 115.Bazsó FL, Ozohanics O, Schlosser G, Ludányi K, Vékey K, Drahos L. Quantitative comparison of tandem mass spectra obtained on various instruments. J Am Soc Mass Spectrom. 2016;27:1357–1365. [DOI] [PubMed] [Google Scholar]
- 116.Schubert OT, Gillet LC, Collins BC, et al. Building high-quality assay libraries for targeted analysis of SWATH MS data. Nat Protoc. 2015;10:426–441. [DOI] [PubMed] [Google Scholar]
- 117.Bott G, Ogden S, Leary JA. Collision-energy ramp. A modification to an RF-only quadrupole collision cell. Rapid Commun Mass Spectrom. 1990;4:341–344. [Google Scholar]
- 118.Baumann C, Cintora MA, Eichler M, et al. A library of atmospheric pressure ionization daughter ion mass spectra based on wideband excitation in an ion trap mass spectrometer. Rapid Commun Mass Spectrom. 2000;14:349–356. [DOI] [PubMed] [Google Scholar]
- 119.Bristow AW, Webb KS, Lubben AT, Halket J. Reproducible production tandem mass spectra on various liquid chromatography/mass spectrometry instruments for the development of spectral libraries. Rapid Commun Mass Spectrom. 2004;18:1447–1454. [DOI] [PubMed] [Google Scholar]
- 120.Dubey R, Hill DW, Lai S, Chen M-H, Grant DF. Correction of precursor and product ion relative abundances in order to standardize CID spectra and improve Ecom50 accuracy for non-targeted metabolomics. Metabolomics. 2015;11:753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Varghese RS, Zhou B, Ranjbar MRN, Zhao Y, Ressom HW. Ion annotation-assisted analysis of LC-MS based metabolomic experiment. Proteome Sci. 2012;10:S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Abrankó L, García-Reyes JF, Molina-Díaz A. In-source fragmentation and accurate mass analysis of multiclass flavonoid conjugates by electrospray ionization time-of-flight mass spectrometry. J Mass Spectrom. 2011;46:478–488. [DOI] [PubMed] [Google Scholar]
- 123.Xu Y-F, Lu W, Rabinowitz JD. Avoiding misannotation of in-source fragmentation products as cellular metabolites in liquid chromatography-mass spectrometry-based metabolomics. Anal Chem. 2015;87:2273–2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Liang Y, Neta P, Simón-Manso Y, Stein SE. Reaction of arylium ions with the collision gas N2 in electrospray ionization mass spectrometry. Rapid Commun Mass Spectrom. 2015;29:629–636. [DOI] [PubMed] [Google Scholar]
- 125.Neta P, Farahani M, Simón-Manso Y, Liang Y, Yang X, Stein SE. Unexpected peaks in tandem mass spectra due to reaction of product ions with residual water in mass spectrometer collision cells. Rapid Commun Mass Spectrom. 2014;28:2645–2660. [DOI] [PubMed] [Google Scholar]
- 126.Evans AM, DeHaven CD, Barrett T, Mitchell M, Milgram E. Integrated, nontargeted ultrahigh performance liquid chromatography/electrospray ionization tandem mass spectrometry platform for the identification and relative quantification of the small-molecule complement of biological systems. Anal Chem. 2009;81:6656–6667. [DOI] [PubMed] [Google Scholar]
- 127.Ohana D, Dalebout H, Marissen R, et al. Identification of meat products by shotgun spectral matching. Food Chem. 2016;203:28–34. [DOI] [PubMed] [Google Scholar]
- 128.Shao W, Lam H. Tandem mass spectral libraries of peptides and their roles in proteomics research. Mass Spectrom Rev. 2016. 10.1002/mas.21512 [DOI] [PubMed] [Google Scholar]
- 129.Calderón-Santiago M, Fernández-Peralbo MA, Priego-Capote F, de Castro MDL. MSCombine: a tool for merging untargeted metabolomic data from high-resolution mass spectrometry in the positive and negative ionization modes. Metabolomics. 2016;12:1–12. [Google Scholar]
- 130.Garg N, Kapono CA, Lim YW, et al. Mass spectral similarity for untargeted metabolomics data analysis of complex mixtures. Int J Mass Spectrom. 2015;377:719–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sandra K, dos Santos Pereira A, Vanhoenacker G, David F, Sandra P. Comprehensive blood plasma lipidomics by liquid chromatography/quadrupole time-of-flight mass spectrometry. J Chromatogr A. 2010;1217:4087–4099. [DOI] [PubMed] [Google Scholar]
- 132.Defelice BC, Mehta SS, Samra S, et al. Mass Spectral Feature List Optimizer (MS-FLO): a tool to minimize false positive peak reports in untargeted LC-MS data processing. Anal Chem. 2017;89: 3250–3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Frank AM, Monroe ME, Shah AR, et al. Spectral archives: extending spectral libraries to analyze both identified and unidentified spectra. Nat Methods. 2011;8:587–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Griss J, Perez-Riverol Y, Lewis S, et al. Recognizing millions of consistently unidentified spectra across hundreds of shotgun proteomics datasets. Nat Methods. 2016;13:651–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Herrera-Lopez S, Hernando M, García-Calvo E, Fernández-Alba A, Ulaszewska M. Simultaneous screening of targeted and non-targeted contaminants using an LC-QTOF-MS system and automated MS/MS library searching. J Mass Spectrom. 2014;49:878–893. [DOI] [PubMed] [Google Scholar]
- 136.Lynn K-S, Cheng M-L, Chen Y-R, et al. Metabolite identification for mass spectrometry-based metabolomics using multiple types of correlated ion information. Anal Chem. 2015;87:2143–2151. [DOI] [PubMed] [Google Scholar]
- 137.Fredenhagen A, Derrien C, Gassmann E. An MS/MS library on an ion-trap instrument for efficient dereplication of natural products. Different fragmentation patterns for [M+ H]+ and [M+ Na]+ ions. J Nat Prod. 2005;68:385–391. [DOI] [PubMed] [Google Scholar]
- 138.Josephs JL, Sanders M. Creation and comparison of MS/MS spectral libraries using quadrupole ion trap and triple-quadrupole mass spectrometers. Rapid Commun Mass Spectrom. 2004;18:743–759. [DOI] [PubMed] [Google Scholar]
- 139.Kienhuis P, Geerdink R. A mass spectral library based on chemical ionization and collision-induced dissociation. J Chromatogr A. 2002;974:161–168. [DOI] [PubMed] [Google Scholar]
- 140.Halket JM, Waterman D, Przyborowska AM, Patel RK, Fraser PD, Bramley PM. Chemical derivatization and mass spectral libraries in metabolic profiling by GC/MS and LC/MS/MS. J Exp Bot. 2005;56:219–243. [DOI] [PubMed] [Google Scholar]
- 141.Rojas-Cherto M, Peironcely JE, Kasper PT, et al. Metab olite identification using automated comparison of high-resolution multistage mass spectral trees. Anal Chem. 2012;84:5524–5534. [DOI] [PubMed] [Google Scholar]
- 142.Sheldon MT, Mistrik R, Croley TR. Determination of ion structures in structurally related compounds using precursor ion fingerprinting. J Am Soc Mass Spectrom. 2009;20:370–376. [DOI] [PubMed] [Google Scholar]
- 143.Vaniya A, Fiehn O. Using fragmentation trees and mass spectral trees for identifying unknown compounds in metabolomics. Trends Anal Chem. 2015;69:52–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Horai H, Arita M, Kanaya S, et al. MassBank: a public repository for sharing mass spectral data for life sciences. J Mass Spectrom. 2010;45:703–714. [DOI] [PubMed] [Google Scholar]
- 145.Smith CA, O’Maille G, Want EJ, et al. METLIN: a metabolite mass spectral database. Ther Drug Monit. 2005;27:747–751. [DOI] [PubMed] [Google Scholar]
- 146.Zhu Z-J, Schultz AW, Wang J, et al. Liquid chromatography quadrupole time-of-flight mass spectrometry characterization of metabolites guided by the METLIN database. Nat Protoc. 2013;8:451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Cho K, Mahieu N, Ivanisevic J, et al. IsoMETLIN: a database for isotope-Based metabolomics. Anal Chem. 2014;86:9358–9361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Oberacher H, Whitley G, Berger B. Evaluation of the sensitivity of the ‘Wiley registry of tandem mass spectral data, MSforID’with MS/MS data of the ‘NIST/NIH/EPA mass spectral library’. J Mass Spectrom. 2013;48:487–496. [DOI] [PubMed] [Google Scholar]
- 149.Würtinger P, Oberacher H. Evaluation of the performance of a tandem mass spectral library with mass spectral data extracted from literature. Drug Test Anal. 2012;4:235–241. [DOI] [PubMed] [Google Scholar]
- 150.Oberacher H, Pitterl F, Siapi E, et al. On the inter-instrument and the inter-laboratory transferability of a tandem mass spectral reference library. 3. Focus on ion trap and upfront CID. J Mass Spectrom. 2012;47:263–270. [DOI] [PubMed] [Google Scholar]
- 151.Dresen S, Gergov M, Politi L, Halter C, Weinmann W. ESI-MS/MS library of 1,253 compounds for application in forensic and clinical toxicology. Anal Bioanal Chem. 2009;395:2521–2526. [DOI] [PubMed] [Google Scholar]
- 152.Dresen S, Kempf J, Weinmann W. Electrospray-ionization MS/MS library of drugs as database for method development and drug identification. Forensic Sci Int. 2006;161:86–91. [DOI] [PubMed] [Google Scholar]
- 153.Niessen W Fragmentation of toxicologically relevant drugs in positive-ion liquid chromatography-tandem mass spectrometry. Mass Spectrom Rev. 2011;30:626–663. [DOI] [PubMed] [Google Scholar]
- 154.Niessen W Fragmentation of toxicologically relevant drugs in negative-ion liquid chromatography-tandem mass spectrometry. Mass Spectrom Rev. 2012;31:626–665. [DOI] [PubMed] [Google Scholar]
- 155.Wissenbach DK, Meyer MR, Remane D, Philipp AA, Weber AA, Maurer HH. Drugs of abuse screening in urine as part of a metabolite-based LC-MSn screening concept. Anal Bioanal Chem. 2011;400:3481–3489. [DOI] [PubMed] [Google Scholar]
- 156.Wissenbach DK, Meyer MR, Remane D, Weber AA, Maurer HH. Development of the first metabolite-based LC-MS n urine drug screening procedure-exemplified for antidepressants. Anal Bioanal Chem. 2011;400:79–88. [DOI] [PubMed] [Google Scholar]
- 157.Kind T, Scholz M, Fiehn O. How large is the metabolome? A critical analysis of data exchange practices in chemistry. PLoS ONE. 2009;4: e5440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Luzzatto-Knaan T, Melnik AV, Dorrestein PC. Mass spectrometry tools and workflows for revealing microbial chemistry. Analyst. 2015;140:4949–4966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Wang M, Carver JJ, Phelan VV, et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat Biotechnol. 2016;34:828–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Sawada Y, Nakabayashi R, Yamada Y, et al. RIKEN tandem mass spectral database (ReSpect) for phytochemicals: a plant-specific MS/MS-based data resource and database. Phytochemistry. 2012;82: 38–45. [DOI] [PubMed] [Google Scholar]
- 161.Matsuda F, Yonekura-Sakakibara K, Niida R, Kuromori T, Shinozaki K, Saito K. MS/MS spectral tag-based annotation of non-targeted profile of plant secondary metabolites. Plant J. 2009;57:555–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Cuthbertson DJ, Johnson SR, Piljac-Žegarac J, et al. Accurate mass-time tag library for LC/MS-based metabolite profiling of medicinal plants. Phytochemistry. 2013;91:187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Lei Z, Jing L, Qiu F, et al. Construction of an ultrahigh pressure liquid chromatography-tandem mass spectral library of plant natural products and comparative spectral analyses. Anal Chem. 2015;87:7373–7381. [DOI] [PubMed] [Google Scholar]
- 164.Pacchiarotta T, Derks RJ, Hurtado-Fernandez E, et al. Online spectral library for GC-atmospheric pressure chemical ionization-ToF MS. Bioanalysis. 2013;5:1515–1525. [DOI] [PubMed] [Google Scholar]
- 165.Shahaf N, Rogachev I, Heinig U, et al. The WEIZMASS spectral library for high-confidence metabolite identification. Nat Commun. 2016;7:12423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kildgaard S, Mansson M, Dosen I, et al. Accurate dereplication of bioactive secondary metabolites from marine-derived fungi by UHPLC-DAD-QTOFMS and a MS/HRMS library. Marine Drugs. 2014;12:3681–3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Benton HP, Wong DM, Trauger SA, Siuzdak G. XCMS2: processing tandem mass spectrometry data for metabolite identification and structural characterization. Anal Chem. 2008;80:6382–6389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Pavlic M, Libiseller K, Oberacher H. Combined use of ESI-QqTOF-MS and ESI-QqTOF-MS/MS with mass-spectral library search for qualitative analysis of drugs. Anal Bioanal Chem. 2006;386:69–82. [DOI] [PubMed] [Google Scholar]
- 169.Tanaka S, Fujita Y, Parry HE, et al. Mass++: a visualization and analysis tool for mass spectrometry. J Proteome Res. 2014;13:3846–3853. [DOI] [PubMed] [Google Scholar]
- 170.Röst HL, Sachsenberg T, Aiche S, et al. OpenMS: a flexible open-source software platform for mass spectrometry data analysis. Nat Methods. 2016;13:741–748. [DOI] [PubMed] [Google Scholar]
- 171.Sturm M, Bertsch A, Gröpl C, et al. OpenMS—an open-source software framework for mass spectrometry. BMC Bioinformatics. 2008;9:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Zhang K, Wong JW, Yang P, et al. Protocol for an electrospray ionization tandem mass spectral product ion library: development and application for identification of 240 pesticides in foods. Anal Chem. 2012;84:5677–5684. [DOI] [PubMed] [Google Scholar]
- 173.Dunn WB, Erban A, Weber RJ, et al. Mass appeal: metabolite identification in mass spectrometry-focused untargeted metabolomics. Metabolomics. 2013;9:44–66. [Google Scholar]
- 174.Kind T, Fiehn O. Advances in structure elucidation of small molecules using mass spectrometry. Bioanal Rev. 2010;2:23–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Milman BL. General principles of identification by mass spectrometry. Trends Anal Chem. 2015;69:24–33. [Google Scholar]
- 176.Pérez-Victoria I, Martín J, Reyes F. Combined LC/UV/MS and NMR strategies for the dereplication of marine natural products. Planta Med. 2016;82:857–871. [DOI] [PubMed] [Google Scholar]
- 177.Wolfender J-L, Marti G, Thomas A, Bertrand S. Current approaches and challenges for the metabolite profiling of complex natural extracts. J Chromatogr A. 2015;1382:136–164. [DOI] [PubMed] [Google Scholar]
- 178.El-Elimat T, Figueroa M, Ehrmann BM, Cech NB, Pearce CJ, Oberlies NH. High-resolution MS, MS/MS, and UV database of fungal secondary metabolites as a dereplication protocol for bioactive natural products. J Nat Prod. 2013;76:1709–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Nielsen KF, Larsen TO. The importance of mass spectrometric dereplication in fungal secondary metabolite analysis. Front Microbiol. 2015;6:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Matsuda F, Hirai MY, Sasaki E, et al. AtMetExpress development: a phytochemical atlas of Arabidopsis development. Plant Physiol. 2010;152:566–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Lee JS, Kim DH, Liu KH, Oh TK, Lee CH. Identification of flavonoids using liquid chromatography with electrospray ionization and ion trap tandem mass spectrometry with an MS/MS library. Rapid Commun Mass Spectrom. 2005;19: 3539–3548. [DOI] [PubMed] [Google Scholar]
- 182.Byrdwell W, Neff WE. Dual parallel electrospray ionization and atmospheric pressure chemical ionization mass spectrometry (MS), MS/MS and MS/MS/MS for the analysis of triacylglycerols and triacylglycerol oxidation products. Rapid Commun Mass Spectrom. 2002;16:300–319. [DOI] [PubMed] [Google Scholar]
- 183.Broecker S, Herre S, Wüst B, Zweigenbaum J, Pragst F. Development and practical application of a library of CID accurate mass spectra of more than 2,500 toxic compounds for systematic toxicological analysis by LC-QTOF-MS with data-dependent acquisition. Anal Bioanal Chem. 2011;400: 101–117. [DOI] [PubMed] [Google Scholar]
- 184.Liu HC, Liu RH, Lin DL, Ho HO. Rapid screening and confirmation of drugs and toxic compounds in biological specimens using liquid chromatography/ion trap tandem mass spectrometry and automated library search. Rapid Commun Mass Spectrom. 2010;24: 75–84. [DOI] [PubMed] [Google Scholar]
- 185.Mueller C, Weinmann W, Dresen S, Schreiber A, Gergov M. Development of a multi-target screening analysis for 301 drugs using a QTrap liquid chromatography/tandem mass spectrometry system and automated library searching. Rapid Commun Mass Spectrom. 2005;19:1332–1338. [DOI] [PubMed] [Google Scholar]
- 186.Renaud JB, Sumarah MW. Data independent acquisition-digital archiving mass spectrometry: application to single kernel mycotoxin analysis of Fusarium graminearum infected maize. Anal Bioanal Chem. 2016;408:3083–3091. [DOI] [PubMed] [Google Scholar]
- 187.del Mar Gómez-Ramos M, Pérez-Parada A, García-Reyes JF, Fernández-Alba AR, Agüera A. Use of an accurate-mass database for the systematic identification of transformation products of organic contaminants in wastewater effluents. J Chromatogr A. 2011;1218:8002–8012. [DOI] [PubMed] [Google Scholar]
- 188.Schymanski EL, Singer HP, Slobodnik J, et al. Non-target screening with high-resolution mass spectrometry: critical review using a collaborative trial on water analysis. Anal Bioanal Chem. 2015;407:6237–6255. [DOI] [PubMed] [Google Scholar]
- 189.Lehotay SJ, Sapozhnikova Y, Mol HG. Current issues involving screening and identification of chemical contaminants in foods by mass spectrometry. Trends Anal Chem. 2015;69:62–75. [Google Scholar]
- 190.DeTata D, Collins P, McKinley A. A fast liquid chromatography quadrupole time-of-flight mass spectrometry (LC-QToF-MS) method for the identification of organic explosives and propellants. Forensic Sci Int. 2013;233:63–74. [DOI] [PubMed] [Google Scholar]
- 191.Liu H-C, Liu RH, Ho H-O, Lin D-L. Development of an information-rich LC-MS/MS database for the analysis of drugs in postmortem specimens. Anal Chem. 2009;81:9002–9011. [DOI] [PubMed] [Google Scholar]
- 192.Lynch KL, Breaud AR, Vandenberghe H, Wu AH, Clarke W. Performance evaluation of three liquid chromatography mass spectrometry methods for broad spectrum drug screening. Clin Chim Acta. 2010;411:1474–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Thoren KL, Colby JM, Shugarts SB, Wu AH, Lynch KL. Comparison of information-dependent acquisition on a tandem quadrupole TOF vs a triple quadrupole linear ion trap mass spectrometer for broad-spectrum drug screening. Clin Chem. 2016;62:170–178. [DOI] [PubMed] [Google Scholar]
- 194.Milman BL, Zhurkovich IK. Towards a full reference library of MSn spectra. II: A perspective from the library of pesticide spectra extracted from the literature/Internet. Rapid Commun Mass Spectrom. 2011;25:3697–3705. [DOI] [PubMed] [Google Scholar]
- 195.Núñez O, Gallart-Ayala H, Ferrer I, Moyano E, Galceran MT. Strategies for the multi-residue analysis of 100 pesticides by liquid chromatography-triple quadrupole mass spectrometry. J Chromatogr A. 2012;1249:164–180. [DOI] [PubMed] [Google Scholar]
- 196.Wang Z, Cao Y, Ge N, et al. Wide-scope screening of pesticides in fruits and vegetables using information-dependent acquisition employing UHPLC-QTOF-MS and automated MS/MS library searching. Anal Bioanal Chem. 2016;408:7795–7810. [DOI] [PubMed] [Google Scholar]
- 197.Weissberg A, Dagan S. Interpretation of ESI (+)-MS-MS spectra—towards the identification of “unknowns”. Int J Mass Spectrom. 2011;299:158–168. [Google Scholar]
- 198.Nguyen DD, Wu C-H, Moree WJ, et al. MS/MS networking guided analysis of molecule and gene cluster families. Proc Natl Acad Sci USA. 2013;110:E2611–E2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Hufsky F, Scheubert K, Böcker S. New kids on the block: novel informatics methods for natural product discovery. Nat Prod Rep. 2014;31:807–817. [DOI] [PubMed] [Google Scholar]
- 200.Misra BB, der Hooft JJ. Updates in metabolomics tools and resources: 2014–2015. Electrophoresis. 2015. [DOI] [PubMed] [Google Scholar]
- 201.Neumann S, Böcker S. Computational mass spectrometry for metabolomics: identification of metabolites and small molecules. Anal Bioanal Chem. 2010;398:2779–2788. [DOI] [PubMed] [Google Scholar]
- 202.Schuemie MJ, Kors JA. Jane: suggesting journals, finding experts. Bioinformatics. 2008;24:727–728. [DOI] [PubMed] [Google Scholar]