

Abstract
Terpenoids, also called isoprenoids, are the largest and most structurally diverse family of natural products. Found in all domains of life, there are over 80,000 known compounds. The majority of characterized terpenoids, which include some of the most well known, pharmaceutically relevant, and commercially valuable natural products, are produced by plants and fungi. Comparatively, terpenoids of bacterial origin are rare. This is counter-intuitive to the fact that recent microbial genomics revealed that almost all bacteria have the biosynthetic potential to create the C5 building blocks necessary for terpenoid biosynthesis. In this review, we catalogue terpenoids produced by bacteria. We collected 1062 natural products, consisting of both primary and secondary metabolites, and classified them into two major families and 55 distinct subfamilies. To highlight the structural and chemical space of bacterial terpenoids, we discuss their structures, biosynthesis, and biological activities. Although the bacterial terpenome is relatively small, it presents a fascinating dichotomy for future research. Similarities between bacterial and non-bacterial terpenoids and their biosynthetic pathways provides alternative model systems for detailed characterization while the abundance of novel skeletons, biosynthetic pathways, and bioactivies presents new opportunities for drug discovery, genome mining, and enzymology.
Graphical Abstract
We highlight the current state of the bacterial terpenome, emphasizing the discoveries, structures, biosynthetic pathways, and biological activities of these terpenoid natural products.
1. Introduction
Terpenoids, also called isoprenoids, are the largest family of natural products (NPs) with over 80,000 known compounds.1 Found in all domains of life, but particularly prevalent in plants, fungi, and marine invertebrates,1 terpenoids are essential constituents of both primary and secondary metabolism. They are some of the most studied and well-known NPs with steroids (e.g., cholesterol), vitamins (classes A, D, E, and K), flavors and fragrances (e.g., menthol, limonene, pinene), plant hormones and photosynthetic pigments (e.g., gibberellins and chlorophylls), and highly successful drugs (e.g., Taxol, artemisinin) all highlighting this superfamily.
This chemical library of compounds, coined the terpenome,2 possesses an extraordinary amount of structural and stereochemical diversity. These diversities arise through an array of complex biosynthetic mechanisms including prenyltransfers, regio- and stereoselective cyclizations, skeleton rearrangements, attachments to a multitude of other scaffolds, and additional tailoring reactions. All terpenoids are built from two simple C5 building blocks, the allylic dimethylallyl diphosphate (DMAPP) and the homoallylic isopentenyl diphosphate (IPP) (Scheme 1).3 These activated isoprene units are either condensed to generate linear C5n allylic diphosphates or used as prenyl donors to alkylate other chemical scaffolds (i.e., prenylation); in rare cases, two C5 isoprene units are condensed to form branched or cyclic C10 diphosphates.2 The nomenclature for terpenoid subfamilies is based on the number of isoprene units in the parent terpene: hemi- (C5), mono- (C10), sesqui- (C15), di- (C20), sester- (C25), tri- (C30), sesquar- (C35), and tetraterpenoids (C40). Hybrid natural products partially derived from terpenoid precursors are termed meroterpenoids; the prefix mero- means “part, partial, or fragment”.4 Meroterpenoids may be the result of direct prenylation or the attachment of a terpenoid skeleton to another moiety via an alternative mechanism. Cyclization and rearrangement reactions occur on linear or cyclized prenyl diphosphates, linear prenyl chains lacking diphosphates, and meroterpenoids. As would be expected, the immensity of the terpenome and its vast structural and stereochemical diversity coincides with a wide range of biological activities.
Scheme 1.
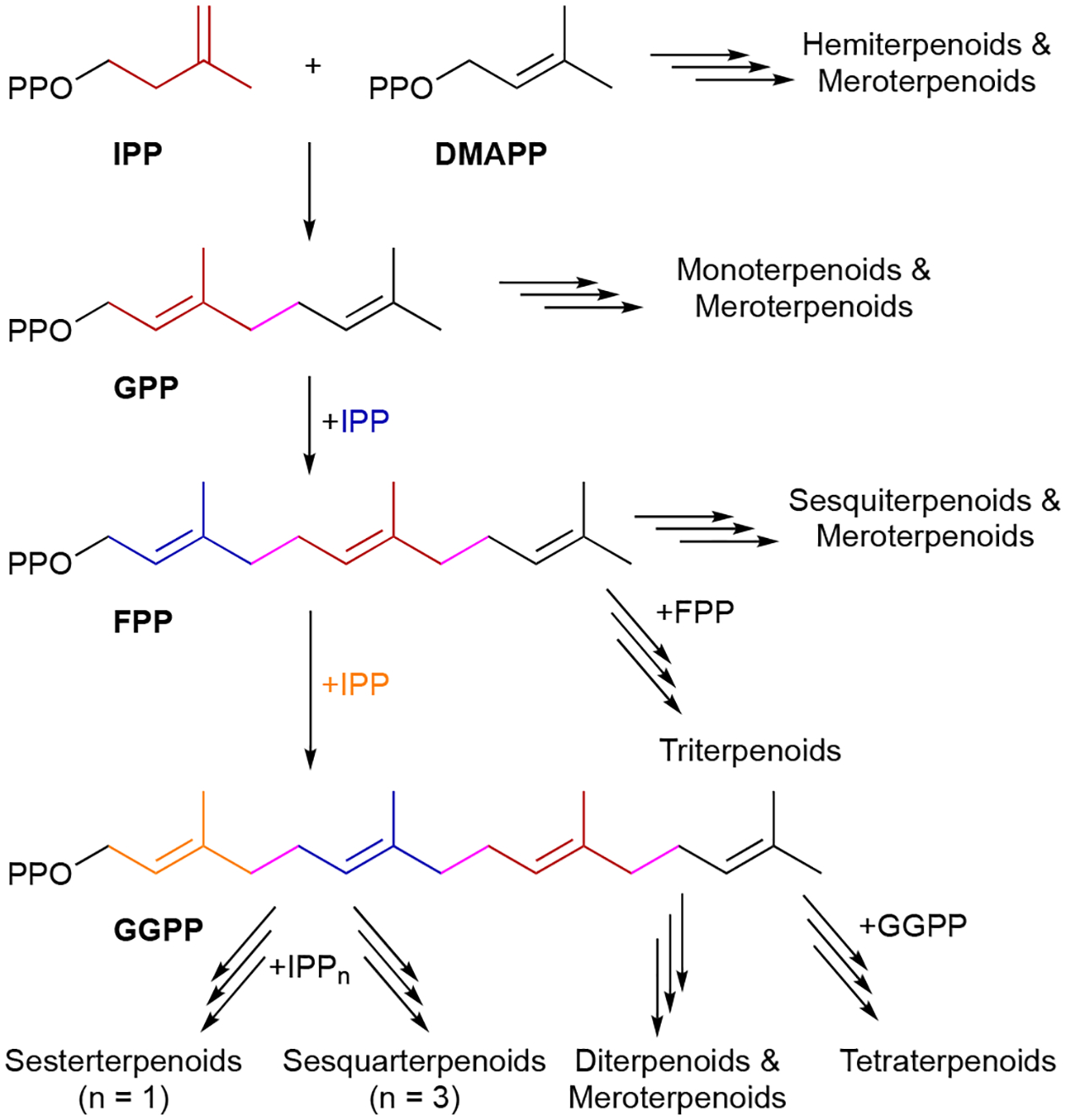
The biosynthesis of terpenoids.
Bacterial terpenoids remain a relatively small family of compounds. This reality is in spite of the fact that the origins of microbial terpenoids date back to the late 19th century with the study of the characteristic odor of soil.5 This odor was later determined to be a combination of the degraded sesquiterpenoid geosmin (121) and the methylated monoterpenoid 2-methylisoborneol (2-MIB, 23).6–8 Traditional NP programs utilizing structure- or activity-guided screening of bacterial extracts have been notably deficient in terpenoids.9 The discrepancy in the number of total terpenoids versus those found in bacteria may suggest that bacteria (i) have not evolved terpenoid secondary metabolism on the same scale to that of other organisms and therefore do not have expansive biosynthetic machinery for terpenoid biosynthesis, (ii) have strict regulatory control of terpenoid biosynthetic pathways that does not translate well to traditional laboratory fermentation conditions, or (iii) that the NP community at large has not focused on or developed efficient means of bacterial terpenoid discovery.
Biochemical and genomics studies revealed that almost all bacteria biosynthesize terpenoids. Most bacteria solely employ the methylerythritol phosphate (MEP) pathway for terpenoid precursor biosynthesis, while some bacteria use the mevalonate (MVA) pathway, and some exploit both pathways.10–13 For example, while all Streptomyces, well-known producers of NPs, use the MEP pathway for essential terpenoids, some strains also use the MVA pathway to supplement NP biosynthesis.14,15 There are a few cases of bacteria, such as the parasitic Mycoplasma, that do not biosynthesize terpenoids de novo, instead relying on their host to supply any necessary terpenoids.16
Microbial genomics, in correlation with enzymatic studies, also indicate the enormous potential for terpenoid biosynthesis in bacteria. Well before the first genome sequences of actinomycetes were sequenced, it was clear that many actinomycetes, with Streptomyces in particular, produced volatile sesquiterpenoids.17,18 Once the complete genomes of Streptomyces coelicolor and Streptomyces avermitilis were reported in the early 2000s,19–21 a wealth of biosynthetic potential was revealed. Both known and novel terpene synthases (TSs), the enzymes responsible for the multitude of cyclization reactions,22,23 as well as carotenoid biosynthetic enzymes were found in both species.19–21,24 A decade later, bioinformatics analysis of 20 actinomycete genomes revealed over 120 candidate bacterial TSs; approximately six TSs per strain.9 Three years later, in a seminal study confirming the prevalence of TSs in bacteria, 262 candidate TSs were identified from public genomic data.25 It should be noted that while a significant portion of these TSs were from Gram-positive actinomycetes, likely reflecting their importance and dominance in the NP community, putative TSs were also identified in a variety of Gram-negative bacteria. Solidifying that these TSs were not all functionally redundant, heterologous expression of 29 selected TSs in S. avermitilis resulted in 13 novel cyclic sesqui- and diterpenes.25 At the time of writing this introduction, there were ~2000 Streptomyces genome assemblies in the NCBI database. Assuming an average of six TSs per genome, there are over 12,000 TSs in Streptomyces alone (a search for “terpene synthase Streptomyces” in UniProt gave 3890)! To further underscore the biosynthetic potential for terpenoids in bacteria, this estimate does not include prenyltransferases (PTs)26 or any of the 10 non-canonical TSs found in bacteria.27
In this review, we aim to describe the current state of the bacterial terpenome. We focus on the discoveries, structures, biosynthesis, and known biological activities of these unique NPs. This review does not address total synthetic efforts and does not emphasize the structural and mechanistic characterization of bacterial TSs. TSs will be described for certain classes of terpenoids for clarity when describing their discoveries or biosynthesis. For an in-depth examination of bacterial TSs and terpenoid biosynthesis in general, we direct readers to the excellent reviews cited here.9,15,22,23,28–31
To identify and collect terpenoids of bacterial origin, we initially utilized four main NP databases: (i) the Dictionary of Natural Products,1 (ii) the Natural Products Atlas,32 (iii) StreptomeDB,33 and (iv) TeroKit.34 This consolidated database was then checked for redundancy, structures were validated by examination of the primary literature, and terpenoids reported in the primary literature but not included in any of these NP databases were added. We included naturally occurring terpenoids, excluding biosynthetic intermediates or shunt pathway products that were only isolated through genetic mutation of biosynthetic genes; however, NPs isolated through genetic manipulation of regulatory genes were included. Products of heterologous expression of full biosynthetic gene clusters (BGCs) were also included. We did not include terpenoids isolated only from in vitro enzyme reactions or biotransformations as these are either biosynthetic intermediates or not known to be produced in vivo. Our analysis resulted in a total of 1062 bacterial terpenoids (Fig. 1).1
Fig. 1.
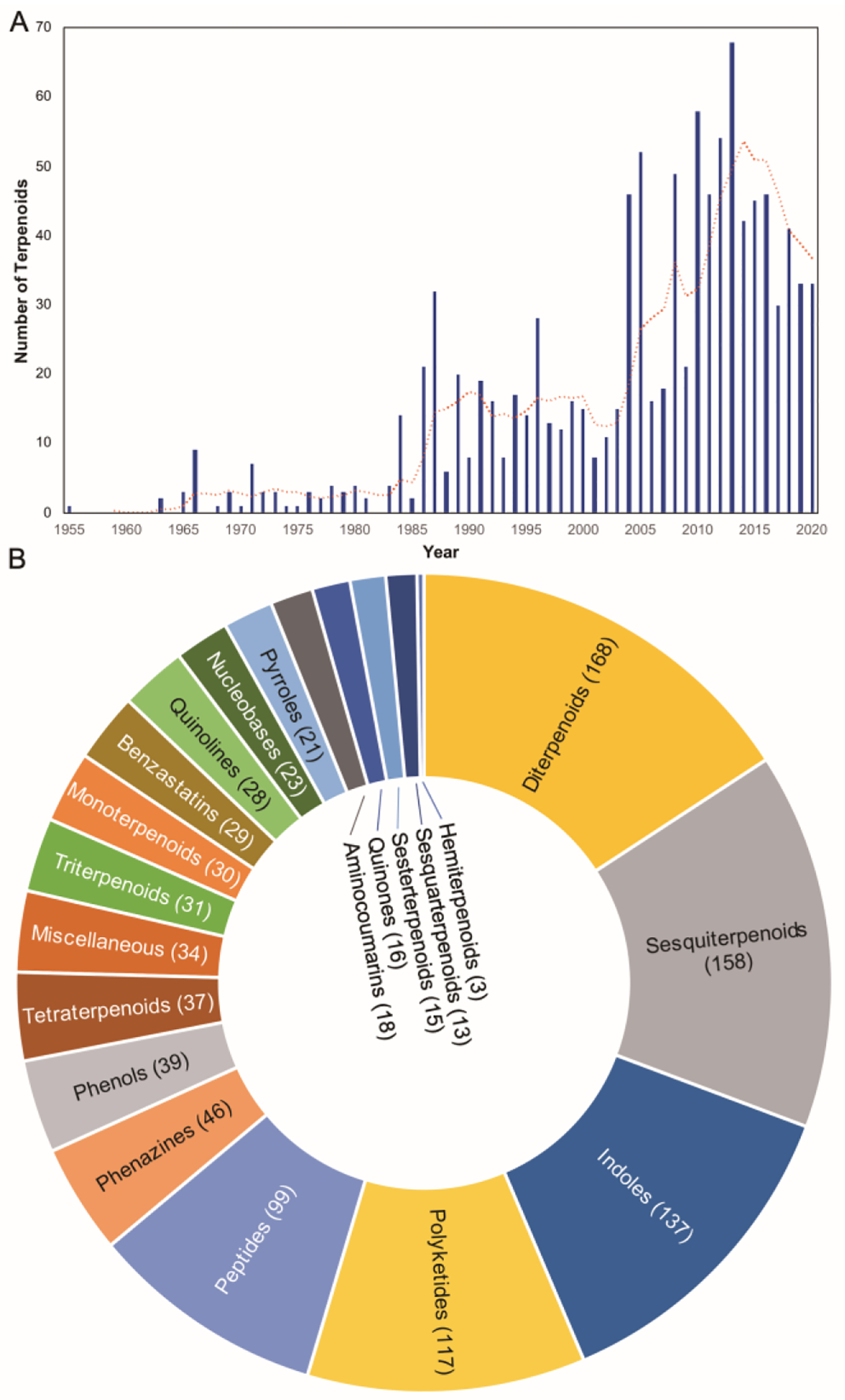
Summary of bacterial terpenoids in the literature. (A) Number of terpenoids discovered in bacteria per year. The orange line is a moving average of five years. Count for 2020 only includes up to mid-2020. (B) Distribution of terpenoids. The parenthetical numbers represent the number of bacterial terpenoids compiled in this review.
We organized these 1062 NPs into two major categories, Terpenoids and Meroterpenoids, further subdivided them into eight and 12 subfamilies, respectively. Those subfamilies were even further divided into 26 and 29 distinct categories, respectively. It should be noted that lines between these divisions are not always clear. The NPs found in the Terpenoids category (chapter 2) are mainly based on the condensation of multiple C5 prenyl units to each other and in most cases, a single or multiple subsequent cyclization reactions. These foundational hydrocarbon skeletons are then extensively modified to produce the variety of structural, chemical, and functional diversities described below. The NPs found in the Meroterpenoids category (chapter 3) are hybrid molecules consisting of a terpenoid portion appended onto another structural motif. This addition provides an entirely different suite of chemical entities that can be structurally and functionally diversified through the addition of an electron-rich appendage. Prenyl groups can add hydrophobicity, provide an electron-rich alkyl chain for further modifications, or supply the framework for additional cyclization reactions. Prenylation is most commonly seen on aromatic rings and can naturally occur (on small molecules) on C, N, and O atoms.
In this review, only a selection of highlighted terpenoids are discussed in detail with their structures shown (compounds with in-text structures are italicized). Selected biosynthetic pathways of bacterial terpenoids are also depicted in schemes (in order to differentiate between isolated NPs discussed in this review and biosynthetic intermediates, known intermediates discussed in the text and shown in schemes are labelled with names and are not numbered). The full bacterial terpenoid database and all structures are available in ESI documents associated with this review. This database was also deposited in the Open Access Natural Products Atlas.32
2. Terpenoids
2.1. Hemiterpenoids
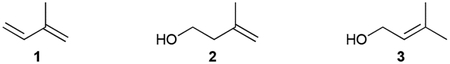
Hemiterpenoids are the smallest terpenoid NPs as they are generated directly from the C5 building blocks IPP and DMAPP without the addition of any other chemical moieties. There are only three known hemiterpenoids that have been isolated from bacteria: isoprene (1), isoprenol (2), and prenol (3). Both Gram-positive and Gram-negative bacteria emit 1, or 2-methyl-1,3-butadiene, with Bacilli and actinobacteria particularly prevalent producers.35–37 1, the majority of which is produced by plants, is the most abundant natural volatile organic compound (VOC) on Earth and influences atmospheric chemistry.38,39 Although the biosynthesis of 1 in plants is known to be the result of diphosphate elimination of DMAPP by isoprene synthase,40 the biogenesis in bacteria remains unclear.41,42 2 (3-methyl-3-buten-1-ol or isopentenol) and 3 (3-methyl-2-buten-1-ol), which were initially detected in Streptomyces,43 are the hydrolysis products of IPP and DMAPP, respectively.
2.2. Monoterpenoids
Bacteria also produce a significant number of terpenoid-based VOCs that can be categorized as monoterpenoids, sesquiterpenoids, diterpenoids, or various degradation products. Taxonomic and environmental differences in bacteria correspond to vast diversities in VOCs. Although VOCs are produced by most bacteria (it was estimated that 50–80% of bacteria produce VOCs in laboratory conditions44), each combination of emitted volatiles is different with some Streptomyces emitting up to 80 different volatile components.37 The literature on VOCs is expansive and we direct readers to impressive reviews that focused solely on bacterial volatiles.44,45
Monoterpenoids are all derived from geranyl diphosphate (GPP) and can be in linear or cyclic form. Geraniol (4) and linalool (5) are linear hydrolysis products of GPP and its rearranged isomer linalyl diphosphate (LPP); β-myrcene (6) is the diphosphate elimination product of LPP. They have been found in Streptomyces.37,43 Methyl geranate (7), was the only volatile terpenoid identified from Salinispora tropica.46 Other bacterial monoterpenoid VOCs, most of which are also found in plants, include the monocyclic compounds limonene (8),43 menthol (9), p-meth-1-en-4-ol (10), and α-terpineol (11),47 the [2.2.1]bicyclic α- and β-pinenes (12, 13),48 borneol (14) and endo-bornyl acetate (15),49,50 camphor (16)49,51 the [3.1.0]bicyclic thujene (17), thujanol (18), and isothujone (19)37,52 and the tetrahydrofuran-containing cis- and trans-linalool oxides (20, 21).53 The ether containing 1,8-cineole (22, eucalyptol) was initially found as a product of a type I TS in Streptomyces clavuligerus ATCC 27094 and later found as an emitted VOC.54,55
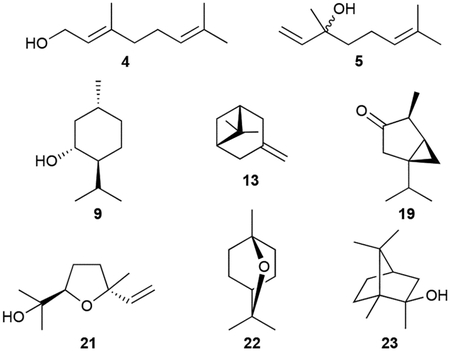
Due to its history, characteristic smell, and abundance in actinomycetes, 2-methylisoborneol (2-MIB, 23) is the most renowned (homo)monoterpenoid. Its structure was characterized after isolation from three different Streptomyces spp. but has also been found in myxobacteria and cyanobacteria.7,56,57 Its [2.2.1]bicyclic structure only deviates from that of 14 with a C2 methyl group. Biosynthetic studies revealed that numerous linear and cyclic C2-methylated monoterpenoids (24–32) were also produced by various actinomycetes, suggesting they were biosynthetically related to 23.37,56,58 A highly oxidized derivative of 23, 2-methyl-2,5,6-bornantriol (33), was also found.59
Biosynthesis.
The biosynthesis of most of the bacterial monoterpenoids can be envisaged by a single type I TS acting on GPP. For the cyclic monoterpenoids, GPP must first be isomerized to LPP during catalysis (Scheme 2).23,60 Cyclization of (E)-configured GPP after diphosphate abstraction would result in a highly strained (E)-cyclohexene intermediate. Instead, GPP is first converted into LPP, a prenyl diphosphate with a freely rotatable C2/C3 single bond.23,60 The diphosphate of LPP is then abstracted and cyclization ensues. Until recently, only two bacterial mono-TSs were characterized: 1,8-cineole synthase (Scheme 2) and linalool synthase from S. clavuligerus;54,61 another linalool synthase was also identified from Chryseobacterium polytrichastri.62 A recent enzymological study screened 22 type I TSs from bacteria and revealed many of the NPs listed above.63 The ability of several TSs to accept prenyl diphosphates of different lengths and therefore producing distinct products, some of which have not been seen in bacteria before, suggests that bacteria are likely a richer source of monoterpenoids than previously assumed.
Scheme 2.
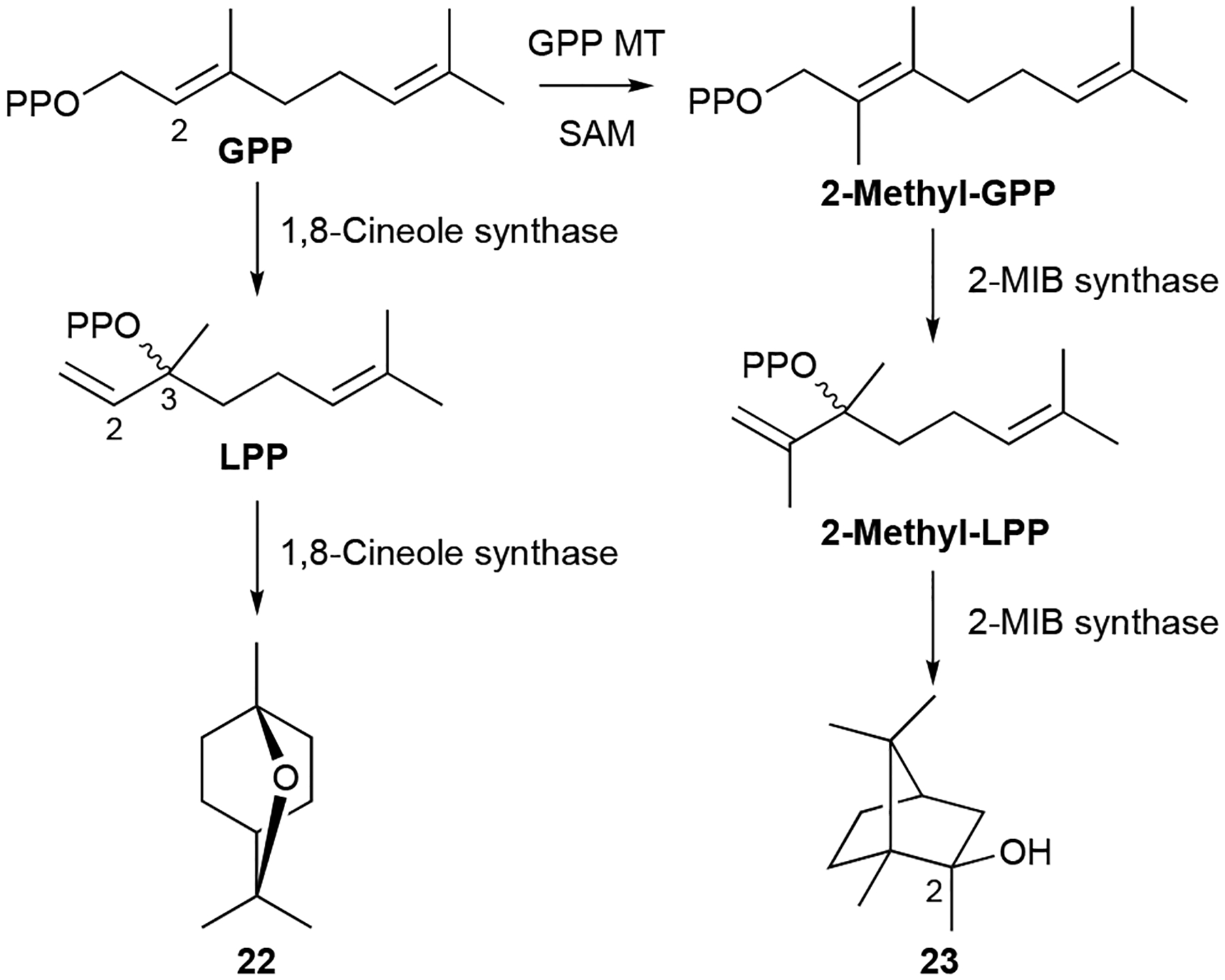
Biosynthesis of 1,8-cineole (22) and 2-MIB (23).
The biosynthesis of 23, which possesses 11 carbons, has an added biosynthetic wrinkle. Early isotopically labeled precursor feeding experiments revealed that the extra carbon on 23 originated from S-adenosylmethionine (SAM) and that methyl incorporation likely happens prior to cyclization.8,56 The 2-MIB BGC from S. coelicolor revealed a C-methyltransferase (MT) encoded adjacent to a type I TS.64 In vitro studies confirmed that GPP is C2-methylated prior to cyclization by 2-MIB synthase; isomerization of 2-methyl-GPP into 2-methyl-LPP is also required for cyclization (Scheme 2). The mechanism of GPP methylation, i.e., alkene methylation resulting in a carbocation that is quenched by proton elimination, hinted at the future discovery of a family of SAM-dependent noncanonical TSs.27
Scheme 11.
Biosynthesis of PTM (265), PTN (266), and their thioacid analogues (278 and 279).
It should also be mentioned that the cyclization of GPP or 2-methyl-GPP via LPP or 2-methyl-LPP, respectively, can occur with both enantiomers of LPP or 2-methyl-LPP.60,65 This would result in enantiomeric intermediates and thus enantiomeric terpenoids. Some terpenoid-producing organisms biosynthesize just one set of enantiomers, but there are examples where both enantiomers are produced by the same organism.65 The stereoselective binding of GPP, in either its right-handed or left-handed helical conformation, suggests that the TSs that control these reactions are structurally different.
Biological activity.
The biological activities of monoterpenoids detected or isolated from bacteria are not commonly reported per se; however, as most of these NPs were originally isolated from plant source material and are constituents of essential oils, their bioactivities have been extensively studied. Many of these volatile monoterpenoids have anti-inflammatory properties, typically mediated by the reduction in levels of tumor necrosis factor (TNF)-α, interleukins, and nitric oxide.66 Other bioactivities, too numerous to exhaustively list here, include antimicrobial and insecticidal, anticancer, analgesic and the antitussive and cooling properties synonymous with camphor and menthol.67–72 The caveat here is that it is not known if many of the monoterpenoids produced by bacteria are the same enantiomeric forms as those produced by plants.
2.3. Sesquiterpenoids
Sesquiterpenoids are derived from farnesyl diphosphate (FPP) and are much more numerous in bacteria than their monoterpenoid counterparts. Given its C15 alkyl length is 50% longer than the monoterpene precursor GPP, as well as the fact that its additional C5 unit possesses a third double bond, FPP can fold and cyclize into a myriad of skeletons creating significantly more structural diversity than monoterpenoids. As with monoterpenoids, some of the cyclization reactions for sesquiterpenoids require the isomerization of FPP into nerolidyl diphosphate (NPP), the C15 equivalent of LPP; both enantiomers of NPP are also possible. For simplicity, we consolidated several subclasses of sesquiterpenoids into larger families based on structural similarity. Most bacterial sesquiterpenoids are VOCs and their structures are in linear or cyclic form with the latter form more prevalent. We again point readers to reviews detailing bacterial VOCs.44,45 As most of the biosynthesis of bacterial sesquiterpenoids is dependent solely on TSs, we will not discuss these in detail and direct readers to the cited reviews.9,22,23
2.3.1. Simple sesquiterpenoids
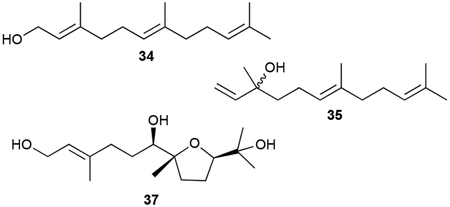
Although likely prevalent in many bacteria, the linear sesquiterpenoid volatiles farnesol (34), nerolidol (35), β-farnesene (36), hydrolysis and dehydration products of FPP and NPP, have been identified from myxobacteria.47,73 Two monocyclic sesquiterpenoids, 7,10-epoxy-2-farnesene-1,6,11-triol (37) and its 6,10-epoxy analogue (38), were isolated from Streptomyces scopuliridis.74 The tetrahydrofuran triol 37 is conspicuously similar to the terpenoid fragment of heronapyrrole (vide infra chapter 3.9).
2.3.2. Cadinanes
The cadinene, muurolane, and amorphane skeletons all have 6/6 bicyclic frameworks with a 3,9-dimethyl-6-isopropyl substitution pattern; their skeletons only differ in their stereochemical configurations. About half of the bacterial cadinanes are purely hydrocarbons with 14 members being diene isomers. These VOCs, which are produced by various types bacteria, are cadinenes (39, 40–45), muurolenes (46–49), zonarene (50), and bicyclosesquiphellandrene (51).37,43,50,73 There are three aromatic ‘trienes,’ namely cadinatriene (52) and the cis- and trans-calamenenes (53, 54).37,47,73 There is the usual assortment of mono-, di-, and trihydroxylated diastereomeric terpenoids (55, 56–61, 62) with hydroxyl groups commonly being positioned at C9 (i.e., muurolols, amorphenols, and cadinols) or C10 (i.e., cubenols).17,37,50,73,75–77
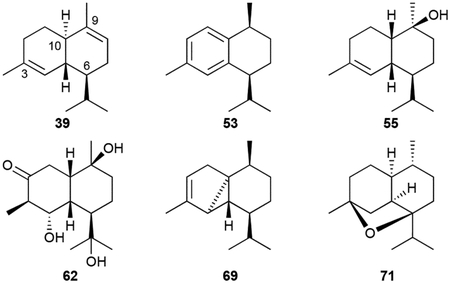
There are also three types of volatile tricyclic cadinene-like sesquiterpenoids. The cubebols (63–67) and cubebenes (68, 69) from Sorangium cellulosum So ce56 have a cyclopropane moiety constructed between C4–C5–C10;73 α-copaene (70) has a cyclobutene ring between C4–C5–C10–C9;48 and corvol ether A (71) is a tetrahydrofuran-containing 4,7-epoxy from Kitasatospora setae KM-6054.78 Finally, although not detected as VOCs in S. cellulosum, T-cadinol (72), α-cadinol (73), 1,10-di-epi-cubenol (74), epi-zonarene (75) and cubebol (76) were all detected and/or isolated after the heterologous expression of a TS encoded by sce6369.73
Biological activity.
Most of the bacterial cadinane sesquiterpenoids do not have reported bioactivities. Only 57 was cytotoxic with a mean IC50 value of ~28 μM.76
2.3.3. Eudesmanes
Eudesmanes have a 6/6 bicyclic core motif with a 4,10-dimethyl-7-isopropyl substitution pattern. The basic hydrocarbon structures of eudesma-5,11-diene (77), selina-3,7(11)-diene (78), β-ylangene (79), and β-copaene (80) as well as the single oxygen-bearing α-eudesmol (81), β-eudesmol (82), rosifoliol (83), (+)-intermedeol (84), dihydroagarofuran (85), and selina-4(14),7(11)-diene-9-ol (86) are all VOCs.18,37,43,47,50,79,80 Numerous diols and triols (87–96) have also been isolated from a variety of actinobacteria.77,81–88
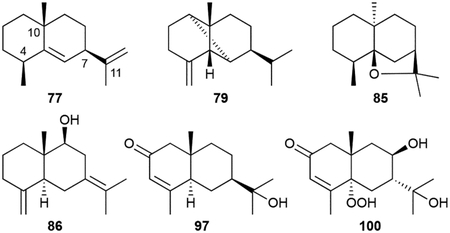
Although there are no congeners with hydroxy groups at C2 or C3, there are 2-oxo and 3-oxo eudesmane derivatives. The 2-oxo eudesmanes include isopterocarpolone (97), previously known as a plant metabolite,89 and kandenols A–E (98–102).90 Kandenols 100 and 101 possess hydroperoxides at C5. 6,12-Dihydroxy-1,4-eudesmadien-3-one (103), produced by the Gram-positive Lentzea violacea AS 08, is the only 3-oxo derivative, although its absolute configuration was not determined.91 Hydroxyl or alkene functional groups can also be modified with phenylacetate (104) and aminobenzoate (105) derivatives seen.85,92
Biological activity.
As with the cadinanes described above, most of the eudesmane bacterial sesquiterpenoids do not have reported bioactivities. Those that showed some activity, such as 98–104, only have weak to moderate antibacterial or anticancer properties.85,90,91
2.3.4. Germacranes
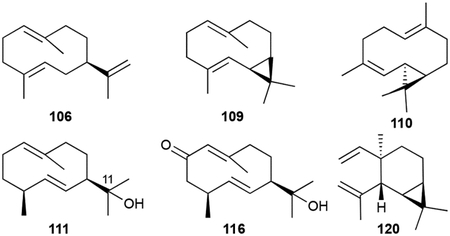
The germacrene sesquiterpenoid skeleton is a cyclodecadiene monocycle. This skeleton is particularly relevant in sesquiterpenoid biosynthesis as the germacradienyl cation is an important intermediate in the formation of various hydrocarbon scaffolds including the geosmins and eudesmanes.23 The bacterial germacrene VOCs, produced by a variety of Streptomyces and myxobacteria, include germacrene A (106), germacrene D (107), iso-germacrene D (108), and the bicyclo[8.1.0]undecane bicyclogermacrene (109) and lepidozene (110).37,43,50 Oxygenated germacranes (111, 112–115, 116), all possessing at least one hydroxyl group at C11, are common in actinobacteria.43,93–95 The monocyclic elemene VOCs (117–120) are commonly reported but may not be legitimate NPs as they are seen during routine GC analysis due to the thermal degradation of germacrenes.43,50
2.3.5. Geosmins
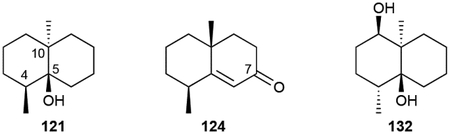
Geosmin (121), the volatile, earthy odor (literally!) present in actinomycetes and in some cyanobacteria, is a C12 6/6 bicyclic norsesquiterpenoid.6 Its trans-decalin ring is decorated as a 4S,10R-dimethyl-5S-alcohol. We categorized structurally similar C12 4,10-dimethylbicycles as geosmin terpenoids. Volatile geosmins from the myxobacterium Chondromyces crocatus and Streptomyces sp. JMRC:ST027706 include the 3- (122) or 7-ketones (123–126, 124) and 7-alcohols (127, 128).50,88 A panel of diols, triols, and tetrols (128–136, 132) have also been isolated from a variety of Streptomyces spp.50,86,88,96,97 Octalins 137 and 138, likely intermediates or shunt products in the cyclization cascade of 121, were detected in the headspace extracts of several myxobacteria and Streptomyces strains.98
Biosynthesis.
The biosynthesis of geosmins has been extensively studied in Streptomyces, cyanobacteria, and plants.9,23,99,100 Given the irregularity of the C12 scaffold, an isotopically-labeled precursor feeding study was required to determine that 121 is a degraded sesquiterpenoid.8 Cloning and deletion of a single gene, sco6073, from S. coelicolor and subsequent biochemical studies revealed that a single protein is responsible for the totality of geosmin biosynthesis.101–104 The didomain and multifunctional germacradienol-geosmin synthase synthesizes 121 from FPP via the intermediate germacradienol (111). Several mechanisms have been postulated including pathways consisting of only cationic and neutral intermediates99 and pathways with additional oxidative101 or both oxidative and reductive steps.100 The currently valid model makes use of the cationic and neutral pathway with this unique TS catalyzing three distinct reactions: a type I cyclization of FPP into the germacradienyl cation via the neutral intermediate hedycaryol, a type II cyclization and subsequent retro-Prins fragmentation releasing acetone, and a final sequence of octalin 137 protonation, 1,2-hydride shift, and water quench to yield 121 (Scheme 3).98,103,105 An alternative pathway through isolepidozene, a bicyclogermacrene isomeric intermediate, is also possible.55,103
Scheme 3.
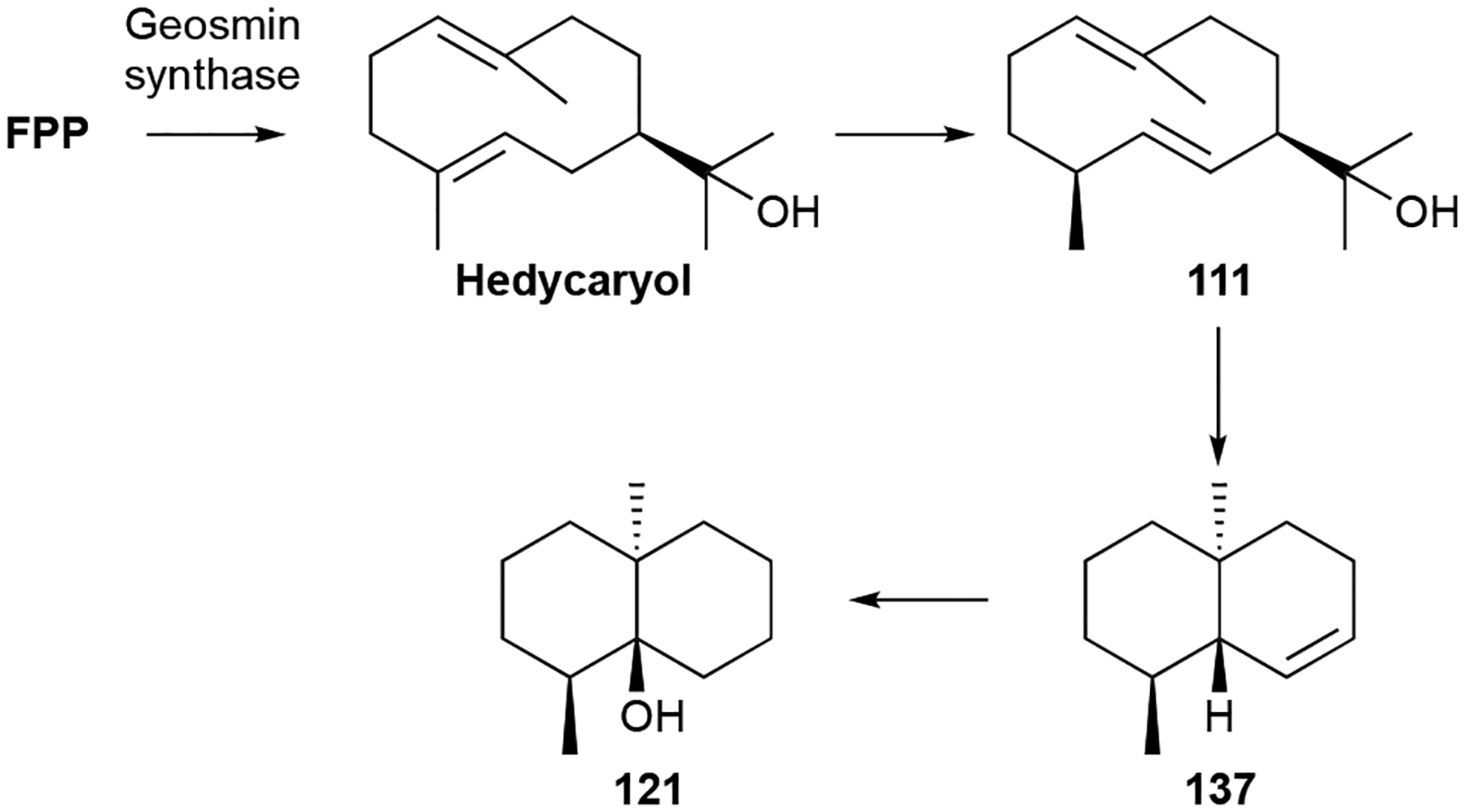
Biosynthesis of geosmin (121).
Biological activity.
The multi-hydroxy geosmins show weak to moderate antimicrobial and cytotoxic activities with 11,12,13-trinor-1,5-eudesmanediol (132) having the most potent activity with a minimum inhibitory concentration (MIC) against Candida of 3.13 μg mL−1.77,86,88,96
2.3.6. Pentalenolactones
The pentalenolactone (PNT) family of tricyclic sesquiterpenoids is a structurally unique group of NPs produced by numerous Streptomyces spp. Known as an antibiotic since the mid 1950s,106 the structure of PNT (139), also named arenaemycin after its reisolation,107,108 was not fully elucidated until 1970.109,110 The tricyclic scaffold of 139 is constructed from two fused cyclopentenes and a 6-membered lactone and functionalized with a C9–C10 epoxide and C13 carboxylic acid. Fifteen additional naturally produced PNT family members have been identified and can now be split into three categories: biosynthetic intermediates, shunt products (in regard to PNT biosynthesis), and proposed isolation artifacts.
The biosynthetic intermediates include pentalenene and PNTs D–F. Pentalenene (140), first isolated from Streptomyces griseochromogenes, is a 5/5/5 tricyclic hydrocarbon core with four methyl groups, two of which are found in the gem-dimethyl group on C2 that is absent in PNT.111 PNTs D (141) and E (142) are carboxyl- and lactone-carrying congeners of 140 with PNT F (143) also possessing the epoxide.112,113
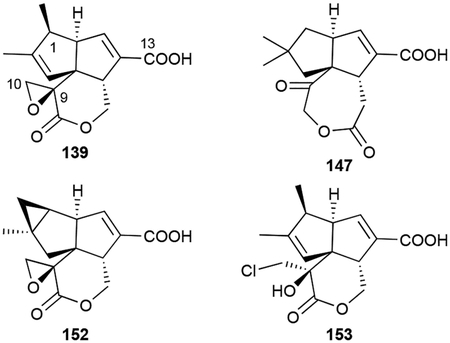
Shunt products include pentalenic acid (144),114 the 9-epimer epi-PNT F (145), which was initially reported as PNT F prior to structural revision,115–117 a glucuronidate of 1-deoxypentalenic acid (146),118 and neopentalenoketolactone (147), an unusual 7-membered ketolactone.119 147 was produced through the heterologous expression of the entire ptl BGC in the engineered host S. avermitilis SUKA5.120 The 1-hydroxylated and 1-keto congeners 144, PNT G (148), and PNT H (149) were initially proposed as intermediates, but later found to be the result of adventitious oxidation.114,121 Sesquiterpenoids with rearranged carbon skeletons, namely PNTs A, B, and P (150–152),113,122 were also found to be by-products of a unique rearrangement step.
Finally, the chlorohydrin PNT C/I (153, also named AA-57) and diol PNT O (154) are likely isolation artifacts due to the epoxide opening under acidic conditions.107,122–124 We included these compounds in this review as they have distinct biological properties worth mentioning.
Biosynthesis.
Understanding PNT biosynthesis has been a focus of several research groups for 30 years and may be considered as one of the quintessential examples of the rationale for studying terpenoid biosynthetic pathways in bacteria.9 Prior to the release of the full genome of S. avermitilis, which provided the ptl BGC,120 PNT was known to be of mevalonate origin125 and the sesqui-TS pentalenene synthase was found to form pentalenene.126 The ptl BGC,120 as well as the subsequently identified pen and pnt BGCs from Streptomyces exfoliatus and Streptomyces arenae, respectively,127 provided the opportunity to use biochemical and genetic techniques to elucidate the full biosynthetic pathway (Scheme 4). After cyclization of FPP into 140, the cytochrome P450 PenI/PntI/PtlI first transforms the C13 methyl into a carboxylic acid by triple hydroxylation.128 Then C11 hydroxylation and oxidation to ketone by the α-ketoglutarate (KG)-dependent dioxygenase PenH/PntH/PtlH and dehydrogenase PenF/PntF/PtlF prepare the scaffold for lactone formation.129,130 141 formation occurs via a Baeyer-Villiger reaction, catalyzed by the FAD-dependent PenE/PntE;119 the homologous PtlE also is a Baeyer-Villiger monooxygenase but inserts its oxygen on the other side of the ketone, diverging its pathway towards 147.131 Another α-KG-dependent dioxygenase, PenD/PntD/PtlD, performs sequential oxidation and epoxidation of the C9–C10 bond yielding the penultimate products 143 or the proposed neo-PNT F;127,131 neo-PNT F is not stable and rearranges to 147.131 The final step in 139 biosynthesis is a unique TS-like rearrangement catalyzed by the P450 PenM/PntM (Scheme 4).132 This P450 performs an carbocation-based oxidative rearrangement of the sesquiterpenoid scaffold due to its innate steric hindrance precluding typical oxygen radical rebound.133 The preclusion of oxygen rebound allows a typically kinetically silent electron transfer to occur, thus forming the C1 carbocation.133 A 1,2-methyl shift and subsequent deprotonation yield 139. The constitutional isomers 150–152 are proposed to be competing by-products of the generated carbocation intermediate.132 Shunt pathway products with oxygens at C1 are the result of CYP105D7, a P450 that is encoded elsewhere in the genome.134
Scheme 4.
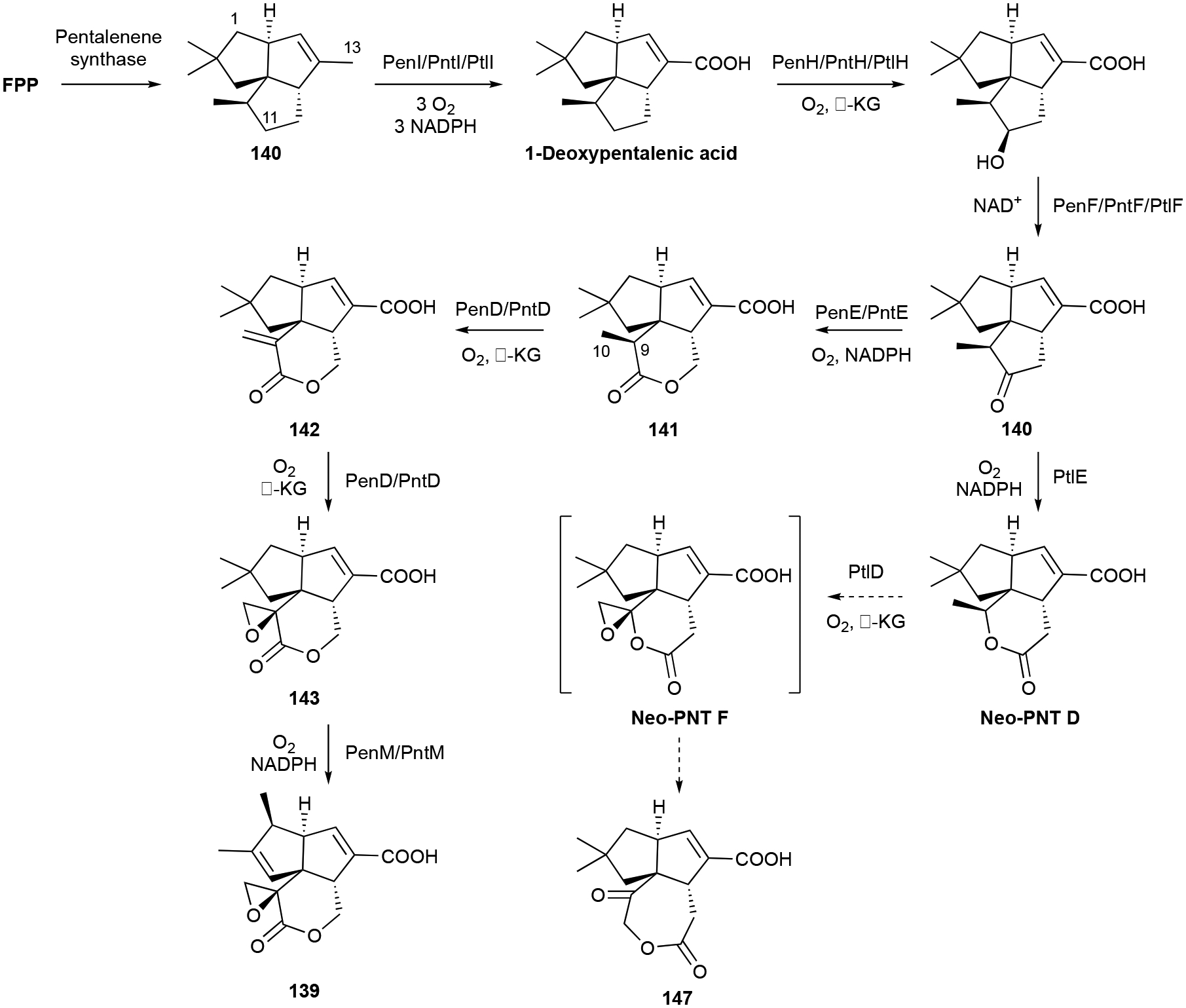
Biosynthesis of PNT (139) and neopentalenoketolactone (147).
Biological activity.
The PNT antibiotics are active against both Gram-positive and Gram-negative bacteria as well as fungi.135 139 is quite potent with MICs approaching 100 ng mL−1.136 The mechanism of action was determined to be inhibition of the glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH).107,108 PNTs are competitive, covalent inhibitors of GAPDH, specifically alkylating Cys149 via attack on the C9–C10 epoxide.137,138 The inactivation of glucose metabolism is also evident in mammalian cells as 139 inhibits glycolysis of various cell types at 18–90 μM.139
139 and some variants also have antitumor properties.118,140 139 was seen to inhibit vascular smooth muscle cell proliferation mediated in part through its effect on the mitogen-activated protein kinase (MAPK) signalling pathway.140 As 139 inhibited glycolysis at a concentration 10-fold higher (IC50 = 7.4 μM) than that of cell proliferation, the inhibition of GAPDH is likely not the mechanism for the inhibition of cell proliferation.
139 and the diol artifact 154, which did not have antibiotic activity, were also found to be effective against DNA viruses including herpes simplex viruses-1 and −2, having EC50 values in the sub- to low μM range.136 The chlorohydrin artifact 153, which retained antibacterial activity (MICs as low as 2 μg mL−1), also acted as an immunosuppressant by inhibiting interleukin (IL)-2 production at an IC50 of ~1.5 μM.123,124
2.3.7. Zizaanes
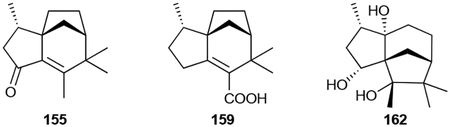
Zizaanes are a small family of [6.2.1.01,5] tricyclic undecanes. The first bacterial zizaane sesquiterpenoid found was albaflavenone (155), a 5/6/5 tricyclic antibiotic with an α,β-unsaturated ketone isolated from Streptomyces albidoflavus DSM 5415.141,142 After the identification of the BGC for albaflavenone,143 several additional zizaane sesquiterpenoids have been reported from both native strains and heterologous hosts harboring the albaflavenone BGC. These include the albaflavenols (156–158), albaflavenoid (159), 4β,5β-epoxy-2-epi-zizaan-6β-ol (160), and antartin (161), an anthrilinic acid derivative of albaflavenone.144–147 Strepsesquitriol (162), identified in the deep-sea Streptomyces sp. SCSIO 10355, has a rearranged 5/6/5 tricycle.148
Biosynthesis.
Genome mining led to the discovery of epi-isozizaene synthase, a widespread type I TS in bacteria that is responsible for constructing the zizaane scaffold (Scheme 5).143,149 The albaflavenone BGC from S. coelicolor is a two gene operon consisting of an epi-isozizaene synthase and a co-transcribed P450, CYP170A1.150 CYP170A1 catalyzes two sequential allylic oxidations at C4 of epi-isozizaene, going through both stereoisomeric alcohols 156 and 157, to yield 155 (Scheme 5). Upon further investigation, CYP170A1 was seen to have moonlighting TS activity in vitro, converting FPP into several farnesene isomers including 36.151 This function was traced back to a secondary active site within the P450 structure that had an unusual TS-like α-helical barrel and signature TS sequence motifs.151 It is unclear if this non-canonical TS activity is biologically relevant in vivo.27
Scheme 5.
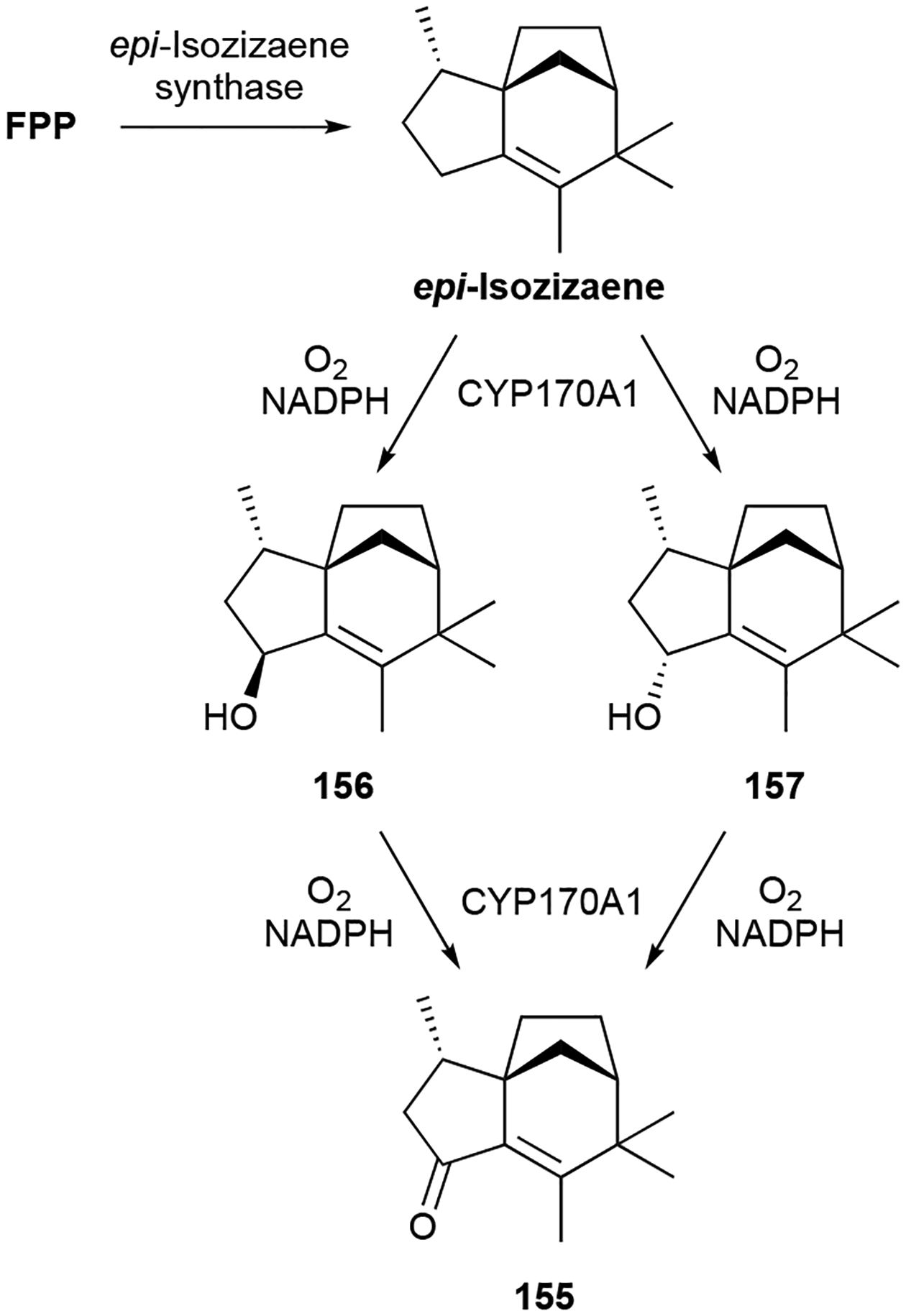
Biosynthesis of albaflavenone (155).
Biological activity.
155 was identified as an antibiotic against Bacillus subtilis with a modest MIC value of ~10 μg mL−1.141 Other zizaane sesquiterpenoids are not antimicrobials but affect eukaryotic cells. 162 inhibited lipopolysaccharide-induced tumor necrosis factor (TNF)-α production in macrophages.148 Albaflavenol B (158), 159, and 161 were weakly cytotoxic with IC50 values of >20 μM, with the latter causing cell cycle arrest at the G1 phase.146,147
2.3.8. Miscellaneous Polycyclic Sesquiterpenoids
There are many other polycyclic sesquiterpenoids produced by bacteria, many of which are VOCs and were originally discovered from plants. The volatile (+)-eremophilene (163), valerianol (164), and β-gurjunene (165) are similar to the eudesmanes but have a 4,5-dimethyl-7-isopropyl substitution pattern on their 6/6 cores.37,47,50,152 β-Caryophyllene (166), clovene (167), and isolongifolene (168) were identified as volatiles in Flavobacteria.49 Sesquiterpenoids with 5/7 hydrocarbon cores include α-gurjunene (169), γ-gurjunene (170), guaioxide (171), 8-daucen-11-ol (172), neomeranol B (173), isoafricanol (174), and africantriol (175).37,80,153,154 172 was initially named isodauc-8-en-11-ol,80 but the position of its methyl group on the cycloheptene ring represents the daucane sesquiterpene skeleton and therefore 172 should be renamed 8-daucen-11-ol. Sesquiterpenoids with 5/6 hydrocarbon cores include the aromatic anmindenols (176, 177),155 sodorifen (178), an unusual symmetrical C16 volatile from Serratia spp.,156 and the tetrahydrofuran-containing corvol ether B (179).78
The caryolanes, represented by (+)-caryolan-1-ol (180), caryolanediols (181–183), and bacaryolanes (184–186) are 6/7/4 tricyclic alcohols produced by Streptomyces spp.84,157–159 Additional tricyclic sesquiterpenoids include the 5/4/5 bourbonenes (187, 188), the 5/5/4 kelsoene (189), and the 5/5/5 triquinane isohirsutenes (190, 191).37,4325,160
Biosynthesis.
The biosynthesis of 178 revealed another example of a MT acting as a TS (see teleocidins in chapter 3.5.3 for first discovered example).27 A genome mining and systematic genetic knockout approach was required to identify the sod BGC.161,162 The simplicity of the BGC, only encoding two biosynthetic enzymes, a TS (SodD) and MT (SodC), contrasted with the complexity of the structure of 178, where every carbon has a methyl or methylidene substituent. However, FPP is methylated and cyclized by SodC into pre-sodorifen, a hexasubstituted cyclopentene diphosphate whose absolute configuration has not yet been solved, prior to an extraordinarily complex type I cyclization reaction catalyzed by SodD (Scheme 6).163,164 The proposed cyclization cascade for 178 formation involves two highly unlikely primary cations and is thus an intriguing system for future mechanistic studies.164
Scheme 6.
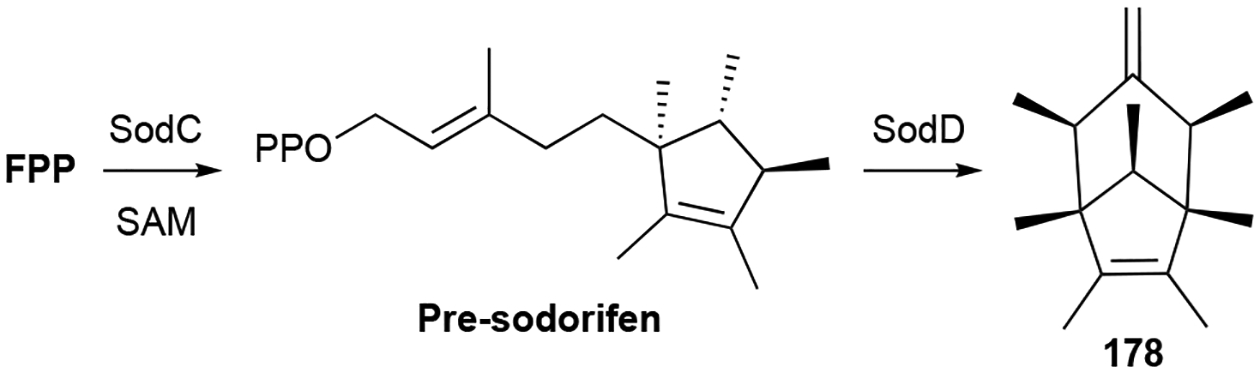
Biosynthesis of sodorifen (178).
The TS responsible for the formation of 180 was found in Streptomyces griseus.157 The gene encoding this (+)-caryolan-1-ol synthase would serve as an excellent probe for the identification of the BGCs responsible for 181–186; however, the genomes of these producing strains have not been reported. Similarly, related TSs in Streptomyces violaceusniger and Streptomyces malaysiensis were determined to be produce 174,153,165 the likely parent compound of 175, although the genome of the producing strain of 175 is not yet known.
Biological activity.
As with most of the other sesquiterpenoids, this group had either no or unreported biological properties, with the exception of several of the plant sesquiterpenoids that have been extensively studied.66 Exceptions include the weak antibacterial activities of the bacaryolanes,159 the moderate antifungal activity of 180 (IC50 = 26 μM for Botrytis cinerea,166 and the ability of anmindenols 176 and 177 to inhibit nitric oxide (NO) production in macrophages (IC50 values of ~20 μM).155
2.4. Diterpenoids
Diterpenoids are derived from geranylgeranyl diphosphate (GGPP) and while they have been extensively studied in plants and fungi,22,167–169 they are comparably rare in bacteria.30 In terms of total numbers, bacterial diterpenoids rival that of bacterial sesquiterpenoids, although the number of structural families encompassed by the diterpenoid category is smaller. Given a total of four prenyl units in the C20 alkyl chain of GGPP, one biosynthetic advantage that diterpenoids possess is the ability to be cyclized solely by type I TSs or by a type II TS in combination with a type I TS or PT. This provides the potential for an extraordinary amount of structural diversity that has been seen in other organisms but not yet fully realized in bacteria.30,169 Unlike the mono- and sesquiterpenoids, most isolated bacterial diterpenoids are functionalized and therefore are not VOCs, although there are a few volatile hydrocarbon skeletons that were identified from bacterial cultures. The TSs responsible for diterpenoid skeletal formation were recently reviewed and we direct readers to these excellent review articles.23,28,30
2.4.1. Brasilicardins, phenalinolactones, and tiancilactones
The brasilicardin, phenalinolactone, and tiancilactone family of diterpenoids contain perhydrophenanthrene scaffolds with various peripheral decorations. Brasilicardin A (192), the first member of this family to be discovered, was isolated from Nocardia brasiliensis IFM 0406 (later named N. terpenica).170,171 Its anti/syn/anti-perhydrophenanthrene diterpenoid skeleton has an amino acid moiety appended to C15 and a tripartite structure on the C2 hydroxyl group. A 3-hydroxybenzoate and an N-acetylglucosamine are attached to the diterpenoid via a rhamnose linker. Other brasilicardins (193–197) have been isolated from both the native strain and by heterologous expression of the entire bra BGC in Amycolatopsis japonicum.172,173
Phenalinolactones (198–201), isolated from Streptomyces sp. Tü 6071, are also terpenoglycosides.174 Their perhydrophenanthrene backbone is anti/anti/syn-configured (only its relative configuration is known) and in place of the amino acid moiety on C15 in the brasilicardins, the phenalinolactones have an uncommonly oxidized and unsaturated γ-butyrolactone. Its other peripheral decorations include l-amicetose and 5-methylpyrrole-2-carboxylic acid groups located on the gem dimethyls of the A-ring and a C3 acetyl unit.
The tiancilactones (202–209), structurally very similar to the phenalinolactones, were recently discovered by genome mining for atypical type II di-TSs in Streptomyces.175 Tiancilactone A (202) had the same γ-butyrolactone, but a different oxygenation pattern on its anti/anti/syn-perhydrophenanthrene tricyclic core and a chloroanthranilate on C20. Other natural tiancilactones included a dechloro analogue (203), an isomerized chloroanthranilate ester (204), congeners with various γ-butyrolactone degradations (205–208), and a unique C11–C15 ether containing diterpenoid with a carboxylic acid tail (209). The concurrently discovered trinulactones A–D (210–213) from Streptomyces sp. S006 are also tiancilactones; trinulactones C and D are the methylated derivatives of tiancilactones A and C.176
Biosynthesis.
Early precursor feeding experiments confirmed that the terpenoid skeletons of the brasilicardins and phenalinolactones were derived from the MEP pathway;177,178 the exact building block for the additional three carbon atoms that end up as the amino acid or γ-butyrolactone moieties is unknown, although glucose is a definite precursor and pyruvate or phosphoenol pyruvate have both been suggested.177,179
The pla BGC, responsible for the production of 198, was identified by screening for genes encoding an NDP-glucose-4,6-dehydratase as it is involved in the formation of l-amicetose.178 Bioinformatics, in vivo inactivation of select genes, extensive tandem MS detection, and a few in vitro experimental confirmations provided a biosynthetic proposal for the phenalinolactones.178–180 GGPP is epoxidized at its terminal olefin leading to cyclization by the type II TS PlaT2 and subsequent prenylation onto an unknown C3 unit is catalysed by the UbiA-like PT PlaT3. The next steps are isomerization of the C3 hydroxyl and oxidation of the C15-C16 bond into an olefin; however, the responsible enzymes are undetermined. Lactone formation, which is completed by the α-KG-dependent dioxygenase PlaO1, occurs prior to oxygenation of the terpene scaffold by various P450s at C1, C20, and C21 and the attachment of decorations at C3, C20, and C21 decorations (Scheme 7).178,179 The biosynthetic proposal for the tiancilactones closely follows the above proposal and is supported by bioinformatics, in vivo knockouts, and isolated congeners.175,176
Scheme 7.
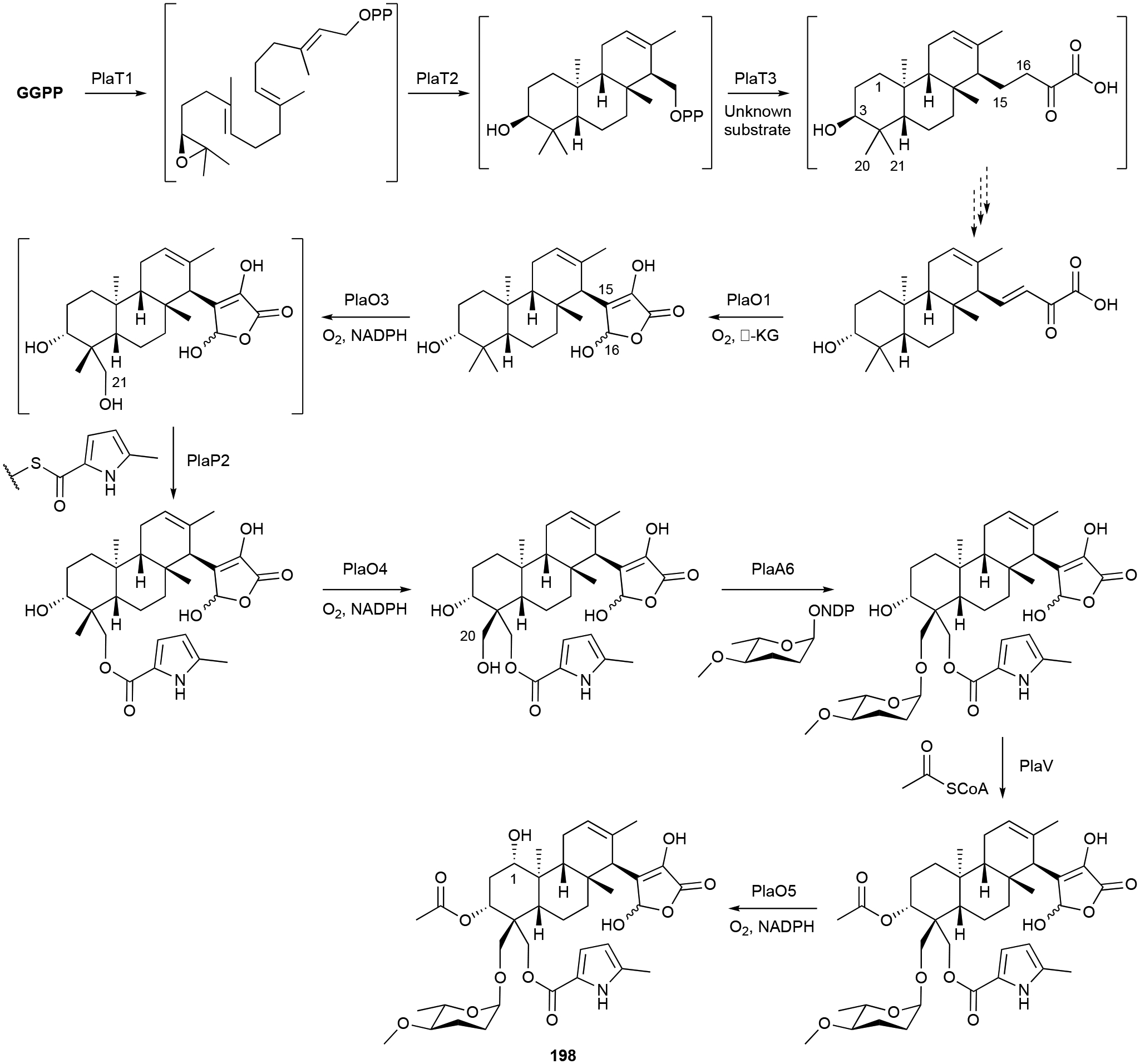
Biosynthesis of phenalinolactone A (198).
The brasilicardin BGC was later found after searching for GGPP synthases in the genome of N. terpenica.170 After an initial biosynthetic proposal, the heterologous expression of the bra BGC produced four biosynthetic intermediates and prompted a revised pathway.173 Hydroxylation at C2 by P450 Bra6 precedes C17 amination by Bra1. Although Bra0, a homologue of PlaO1, catalyzes C16 hydroxylation, the lack of the C15-C16 olefin precludes the rearrangement to the γ-butyrolactone of the phenalinolactones and tiancilactones. Ensuing methylation and glycosyltransfer reactions finalize the structure.173
The proposed terpene epoxidation, cyclization, and prenylation reactions have not been experimentally confirmed. Their structural and BGC similarity to KS-505a (vide infra chapter 3.7) suggests that prenylation onto the C3 unit may occur prior to epoxidation and cyclization. In addition, the differences in the configurations of the tricyclic diterpenoid scaffolds of the brasilicardins and tiancilactones implies that these type II di-TSs do not all generate the same stereoisomers.175
Biological activity.
The phenalinolactones and tiancilactones are antibacterials with moderate levels of activity. Phenalinolactones 198 and 199 inhibited Gram-positive bacteria (MICs ≤10 μg mL−1), but was inactive against Gram-negative bacteria, fungi, and human cells.174 The tiancilactones had MICs of 8–64 μg mL−1 for Gram-positive and some Gram-negative strains.175
Conversely, brasilicardins are unique and potent immunosuppressors with no antibacterial activity. 192 had an IC50 value of 67 nM in a mouse mixed lymphocyte assay and was cytotoxic against a variety of cancer cells; its most potent activity was an IC50 value of 87 nM against Adriamycin-resistant leukemia P388 cells.181,182 By targeting the amino acid transport system L, inhibiting the uptake of amino acids, and arresting cells in the G1 phase, its mechanism of action is different from that of the well-known immunosuppressants cyclosporin A and FK-506.183 Brasilicardins 193 and 194 were 50 times less potent than 192 asserting that the C2 decorations and C16 methoxy group are important for activity; brasilicardin 195 was inactive.172
2.4.2. Cyclooctatins
The cyclooctatins are named for their central 8-membered ring in their 5/8/5 fused tricyclic ring system. These bacterial diterpenoids are structurally very similar to the fusicoccin-type fungal toxins.184 The namesake NP, cyclooctatin (214), was first identified as a lysophospholipase inhibitor from Streptomyces melanosporofaciens and is a 5,7,18-triol of the cyclooctat-9(10)-ene carbon skeleton.185,186 Seven analogues (215–221), all with varying combinations of hydroxyl groups, have since been discovered from various Streptomyces spp.187–190 The hydroxyl groups at C17 and C18 are prone to esterification as evidenced by the isolation of 18-acetylcyclooctatin (222) and the fusicomycins (223–225).187,191
Biosynthesis.
214 is produced through the action of four enzymes, a GGPP synthase, TS, and two P450s, encoded within the cot BGC (Scheme 8).192 CotB2, a type I di-TS, cyclizes GGPP into cyclooctat-9-en-7-ol, providing one of the three hydroxyl groups in cyclooctatin through a water quench of the carbocation at C7.193 Successive P450 hydroxylations by CotB3 and CotB4 at C5 and C18, respectively, complete the biosynthesis.192 Is it currently unknown what enzymes further modify cyclooctatin.
Scheme 8.
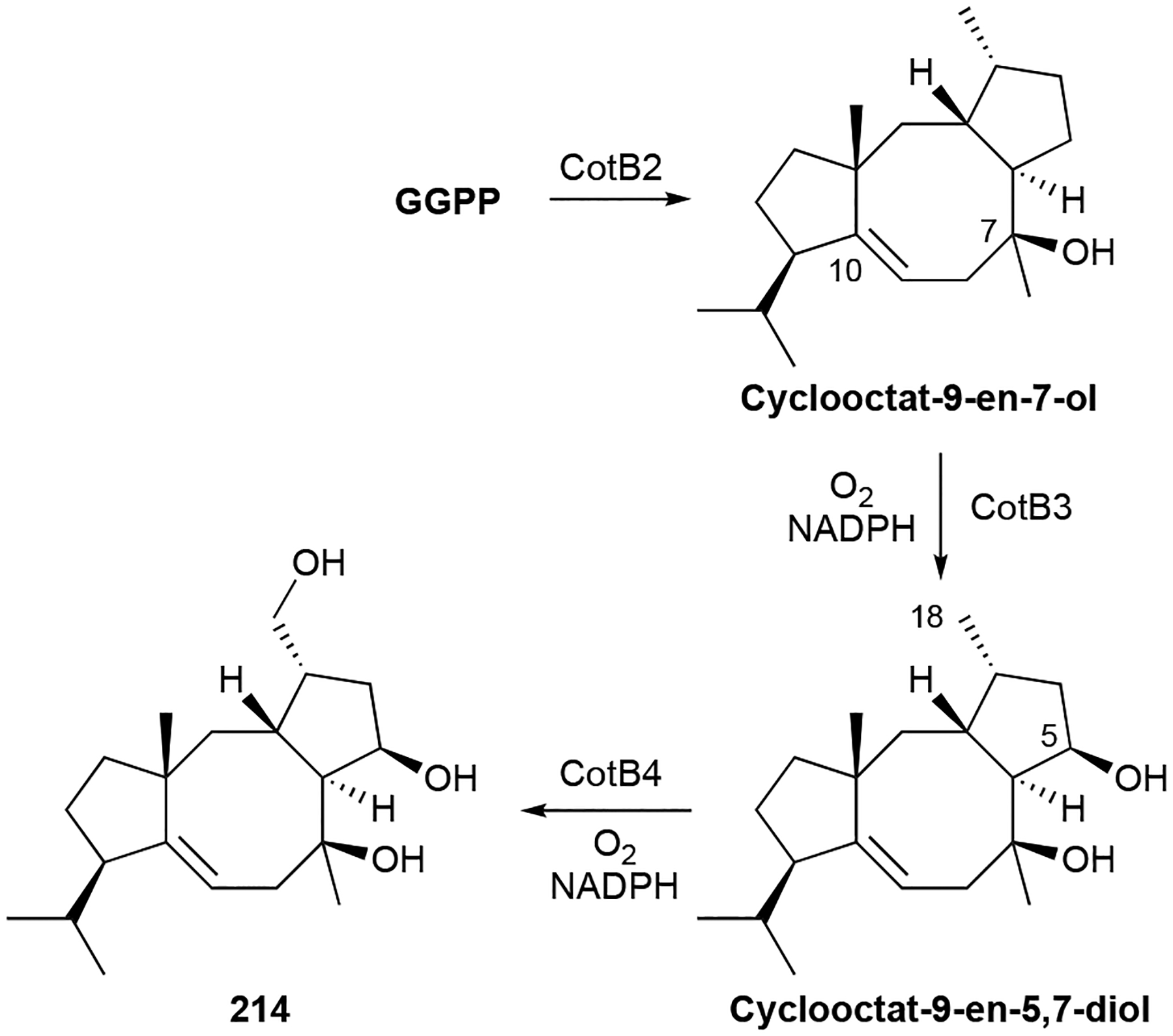
Biosynthesis of cyclooctatin (214).
Biological activity.
214 was first reported as a single digit μM inhibitor of lysophospholipase with no antimicrobial or cytotoxic activities;185 it was later shown to be antiplasmodic with an IC50 value of 20 μM.194 While some of the variants do have weak antibiotic activities against both Gram-positive and Gram-negative bacteria,189,191 the fusicomycins showed cytotoxicities with low μM IC50 values.187 The mode of action for fusicomycin cytotoxicity appears to be the inhibition of matrix metalloproteinases, resulting in the suppression of cell proliferation, cancer migration, and invasion.187
2.4.3. Cyslabdans
Cyslabdans, aptly named for their labdane diterpene skeleton and appended N-acetylcysteine moiety, are unique bacterial NPs. Cyslabdan A (226), the first representative isolated from Streptomyces sp. K04–0144, is a 7,8-dihydroxy-trans-decalin ring with the Cys unit attached via a thioether linkage at C17.195,196 Cyslabdans B and C (227, 228) and the 2-hydroxy (229) and 17-hydroxy (230) congeners of 226 were later isolated from the same strain and after the heterologous expression of the entire cld BGC in S. avermitilis SUKA22.197,198 An 8,17-epoxy-labdadiene intermediate (231) was also isolated from this cld-expressing host.198 Parallel heterologous expression of a highly homologous rmn BGC from Streptomyces anulatus GM95 only produced raimonol (232), a known plant NP.198
Biosynthesis.
Genome mining for the cyslabdan BGC was achieved by searching for genes encoding prenyl diphosphate synthases. The target gene, named cldA, was transcriptionally coupled with three other biosynthetic genes.198 Heterologous expression of the cld BGC confirmed its role in cyslabdan biosynthesis and supported CldB and CldD as (+)-copalyl diphosphate (CPP) and labda-8(17),12E,14-triene synthases. CldC, a P450, is proposed to incorporate two oxygens, a hydroxyl and epoxide at C7 and C8/C17, respectively, forming 231 (Scheme 9). The N-acetylcysteine moiety is added by mycothiol-S-conjugation and subsequent hydrolysis. Mycothiol conjugation was proposed to occur non-enzymatically in vivo,198 although it is feasible to consider an enzyme controls this crucial biosynthetic step. The divergence in 232 biosynthesis is due to the sole C7 hydroxylation of labda-8(17),12E,14-triene.198
Scheme 9.
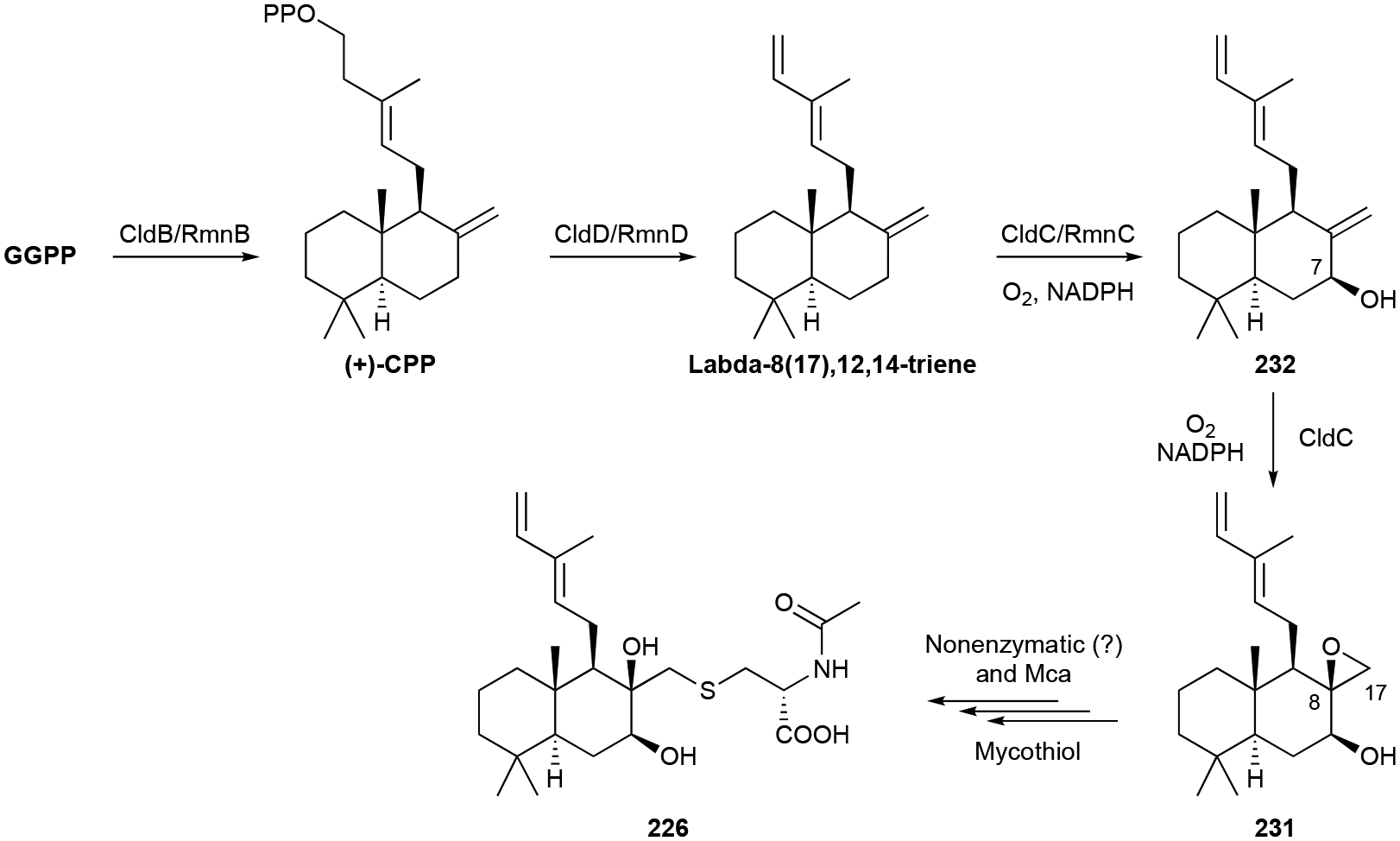
Biosynthesis of cyslabdan (226).
Biological activity.
The cyslabdans do not have antibacterial activity themselves but are strong potentiators of imipenem activity against methicillin-resistant Staphylococcus aureus (MRSA). At 10 μg mL−1 of 226, a non-lethal concentration, the MIC of imipenem is 0.015 μg mL−1; this corresponds to a >1000-fold decrease from its monotherapeutic MIC of 16 μg mL−1.199 227 and 228 also enhanced imipenem activity, albeit at lower levels (123-fold and 533-fold, respectively) than 226.197 The cellular target of cyslabdans is the binding and inhibition of FemA, an enzyme involved in the formation of the pentaglycine interpeptide bridge in peptidoglycan biosynthesis.200 While the inhibition of FemA by the cyslabdans is apparently not detrimental enough to prevent cell wall synthesis and therefore cause cell death, it significantly improves the potency of β-lactam antibacterial activity.
2.4.4. Gibberellins
Gibberellins (GAs) are 6/5/6/5 tetracyclic diterpenoids that are well known plant and fungal NPs.201,202 These phytohormones, at least the few that are bioactive (i.e., GA1, GA3, GA4, and GA7), are important in developmental and physiological processes of plants including seed germination and stem, leaf, flower, and fruit growth. The GA family, first identified in the fungal rice pathogen Gibberella fujikuroi, now includes over 100 different structural members.1,202 These tetracyclic diterpenoids are derived from ent-kaurene, have rearranged B-rings, are highly oxidized, and are found as either C20 diterpenoids or C19 norditerpenoids.
Over 30 years ago, bacteria were also found to produce gibberellins after GA1 (233), GA4 (234), GA9 (235), and GA20 (236) were identified in the nitrogen-fixing Gram-negative bacterium Rhizobium phaseoli.203 Now there are a total of 16 known GAs in bacteria (237–248) produced by a variety of Gram-negative and Gram-positive genera including Acinetobacter, Azospirillum, Bacillus, Bradyrhizobium, Burkholderia, Enterococcus, Pseudomonas, Rhizobium, and Sphingomonas.203–206 GAs in bacteria have been extensively reviewed.168,201,202,206
Biosynthesis.
The biosynthetic pathways of GAs in plants, fungi, and now bacteria are well understood.206 Bacteria evolved their own pathway for GA biosynthesis, although it partially follows the pathway found in plants. The ent-kaurene skeleton is initially formed from GGPP by two distinct ent-CPP and ent-kaurene synthases (Scheme 10).207 The GA operon was later identified in Bradyrhizobium diazoefficiens and functional characterization of five unknown genes led to a proposal for GA9 biosynthesis.208 Along with GGPP synthase and the two di-TSs, three P450s, a short chain dehydrogenase/reductase (SDRGA), and a ferredoxin were encoded nearby. Using heterologous expression and in vivo knockouts, CYP117, CYP114, CYP112 and SDRGA were all characterized as oxidases. CYP117 oxidizes ent-kaurene into ent-kauren-19-oic acid; CYP114 catalyzes C7 β-hydroxylation and B-ring contraction to form the aldehyde form of GA12; SDRGA completes C7 oxidation to the carboxylic acid 239; and finally, CYP112 catalyzes C20 oxidation and lactone ring closure with the C19 acid group via loss of C20 to yield 235.208 A fourth P450 catalyzes C3 β-hydroxylation to yield the phytoactive 234 (Scheme 10).209
Scheme 10.
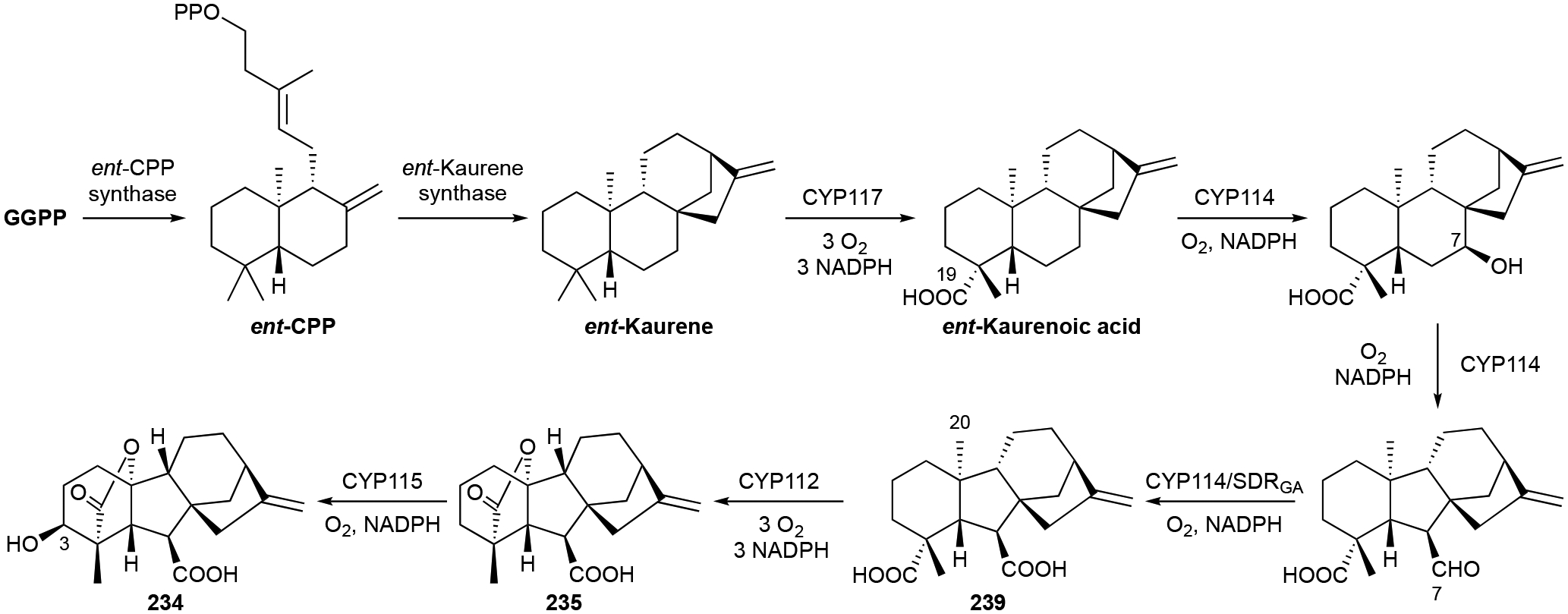
Biosynthesis of GA9 (235) and GA4 (234).
2.4.5. Oxaloterpins
Viguiepinol, or 3-hydroxypimara-9(11),15-diene (249), was first identified in bacteria after it was heterologously produced, along with (–)-pimara-9(11),15-diene (250), from the expression of a four gene operon from Streptomyces sp. KO-3988 in Streptomyces lividans TK23.210 249 was originally isolated from plants.211 Re-fermentation of Streptomyces sp. KO-3988 not only yielded 249, but an additional six related diterpenoids, viguiepinone (251) and oxaloterpins A–E (252–256).212 Oxaloterpins are 3-acylated derivatives of viguiepinol with moieties including N-hydroxyoxalyl amides. The related chloroxaloterpins A and B (257, 258), also from a Streptomyces sp., had unique 2-chloroaniline-containing side chains.213
Other isopimaradienols include 259, 260, and 261.83,214,215 We also include here three C19 norditerpenoids, gifhorneolone A (262), actinomadurol (263), and JBIR-65 (264), that are proposed to originate from C20 decarboxylation of pimaradiene NPs.83,216,217
Biosynthesis.
Prior to the discovery of any oxaloterpin members from bacteria, a four gene operon containing a P450, two DTSs, and a GGPP synthase was found near MVA-encoding genes.218 In vitro characterization of the type II di-TS, ORF2, revealed it to be a CPP synthase. Heterologous expression of the entire operon in S. lividans TK23 yielded both 249 and 250.210 Subsequent in vitro characterization of the type I di-TS ORF3, confirmed the pimaradiene scaffold is constructed prior to hydroxylation by the P450 ORF1.219 With the exception of CYP1051A1-catalyzed C19 hydroxylation in isopimara-8,15-dien-19-ol (259) biosynthesis,214 the other functionalities seen in the oxaloterpins, including the norditerpenoids, are biosynthetically uncharacterized.
Biological activity.
252 and 263 are antibacterials with the latter effective against S. aureus and Proteus hauseri with MICs 0.39–0.78 μg mL−1;212,216 264 was not active and the other oxaloterpins were not tested.212,217 Chloroxaloterpins 257 and 258 were found to inhibit spore germination in Botrytis cinerea with EC50 values of ~10 μM.213
2.4.6. Platensimycin and platencin
Platensimycin (PTM, 265) and platencin (PTN, 266) are unique hybrid natural products derived from labdane intermediates.220 Discovered from Streptomyces platensis MA7327 and S. platensis MA7339 by Merck in 2006 and 2007, respectively, using an innovative antisense differential sensitivity whole-cell assay, 265 and 266 received worldwide attention due to their novel chemical structures and antibacterial mode of action.221,222 Both 265 and 266 possess 3-amino-2,4-dihydroxybenzoic acid (ADHBA) moieties linked to diterpene-derived aliphatic cages via a flexible propionamide linker.223,224 These polycyclic enone acids were initially called ketolides and while technically incorrect,225 the term is still used in the literature. The ‘ketolides’ of 265 and 266 are 17-carbon polycyclic enones with differences in the polycyclic moieties. The polycyclic enone of 265 is a tetracyclic scaffold with a fused cyclohexyl-cyclopentyl-furan; the polycyclic enone of 266 is a tricyclic scaffold with an exocyclic methylene. Their structural similarities to ent-kaurene and ent-atiserene,224 labeling studies,225,226 the isolation of homoplatensimide A (267),227 a 20-carbon congener of 265, and subsequent biosynthetic studies confirmed their diterpenoid origins.228–230
An additional 54 naturally occurring PTM and PTN congeners have been isolated so far, mostly through the use of large-scale fermentations of the wild-type (WT) strains and the utilization of high-producing genetic mutants.229,231 Given its hybrid nature, analogues have modified ADHBA moieties, polycyclic enones, or both. Variations in the ADHBA moiety include a carboxy amide (268), a cyclic carbamate (269), a decarboxyaniline (270), 5´-O-glucosides (271–277) and thiocarboxylic acids (278, 279) .232–236 Biosynthetic studies support that the thiocarboxylic acids are the genuine, genetically encoded natural products.236 Most congeners, like 280, have also been isolated as their methyl benzoate derivatives. Although there is evidence that PTM and PTN congeners with methyl benzoates are artifacts of methanol-based isolations, a few congeners (281, 282) were only isolated in their methyl benzoate forms and one glucosylated congener (277) was isolated as a methyl thiobenzoate.234,237,238 Variations in the polycyclic enones mainly consisted of overoxidation products proposed to be generated from adventitious oxygenases (275–277, 283–301).234,235,237–240 One unique variant, and the initial clue for the discovery of the thiocarboxylic acids 278 and 279, is the pseudo-dimer 302, which connects 265 and 278 via a thioester linkage at C7 of 265; this product was hypothesized to be the result of a non-enzymatic hetero-Michael addition.241
Shunt products of biosynthetic intermediates, both putative and confirmed, have also been isolated from native and overproducing strains. These include the C17 platensic or platencinic acids congeners (281, 282, 303–307) as well as the C20 enones, most of which are overoxidized and/or derivatized by amino acids (267, 308–314) or glycerol moieties (315).227,234,237–239,242
Heterologous expression of the entire ptn gene cluster in S. lividans K4–114 produced 266 and six other PTN congeners including both isomers of the C20 enones (316, 317), ent-agathic acid (313), 12R-hydroxy-ent-atiseren-19-oic acid (318), an N-acetylcysteamine thioester of 7R-hydroxy-ent-atiseren-19-oic acid (319), and 14,15-dinor-13-oxo-8(17)-ent-labden-19-oic acid (320).243 A glutaminylated ent-agathic acid derivative (314) was also isolated from a PTN overproducing strain.234 Finally, 7-(3-butanonyl)-6,6-dimethyl-2-cyclohex-4-enone-2-carboxylic acid (321) appears to be either a diterpenoid-derived acid that was oxidatively cleaved in a manner similar to that of 320 or an apocarotenoid (vide infra chapter 2.8.3).240
Biosynthesis.
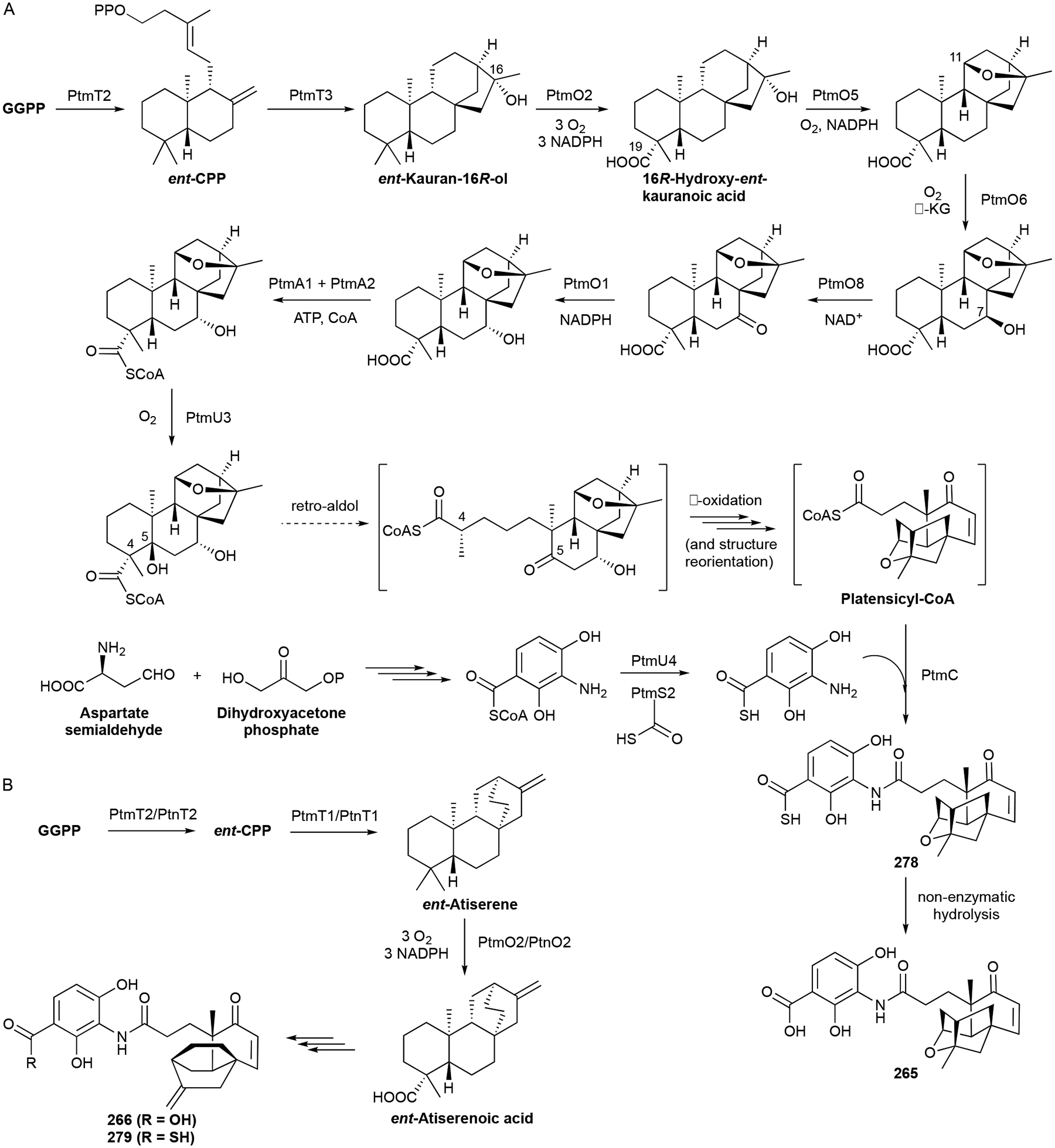
265 and 266 are heavily modified diterpenoids and their biosynthesis has been expansively studied and recently reviewed.220 The overall biosynthetic logic is a divergent, but unified, model that requires construction of the diterpenoid skeleton, maturation of the C20 unit into a highly oxidized C17 coenzyme A (CoA) thioester, and condensation with the separately biosynthesized ADHBA moiety (Scheme 11). After GGPP is converted into ent-CPP by the type II TS PtmT2,230 the two polycyclic diterpene scaffolds, ent-kaurenol and ent-atiserene, are formed from ent-CPP by two different type I TSs, PtmT3 and PtmT1, respectively.228,244 A group of enzymes then processes both ent-kaurenol and ent-atiserene to the penultimate precursors platensicyl-CoA and platencinyl-CoA (Scheme 11). These enzymes, which perform reactions including oxygenations, epimerizations, CoA ligations, oxidations, and C–C bond cleavages,229,245–247 must be flexible enough to accommodate both scaffolds; only one enzyme, a P450 that initiates ether formation in the 265 polycyclic enone, is specific to one pathway.248 It was recently discovered that ADHBA is a precursor to the genetically encoded ADHBSH, a thiocarboxylic acid analogue of ADHBA, and that ADHBSH is likely the bona fide substrate for the final coupling reaction with platensicyl-CoA and platencinyl-CoA (Scheme 11).236 There is one major question left to be answered in PTM and PTN biosynthesis: which enzyme controls the C4–C5 retro-aldol cleavage of the A ring?
PTM and PTN are truly a showcase for the importance of studying and understanding terpenoid biosynthesis in bacteria.
It was the discovery of PtmT1 that brought about the realization that noncanonical UbiA-like di-TSs from bacteria and fungi were an alternative to the canonical TSs and provided new opportunities for terpene enzymology and genome mining.27,249 The identification and characterization of the thioacid cassette in the ptm and ptn BGCs led to the detection of 160 additional thioacid cassettes in bacteria, suggesting that thiocarboxlic acid-containing NPs are underrepresented in current NP databases.236 PtmA1 and PtmA2, two acyl-CoA ligases, are the first example of a natural separation of the acyl-CoA ligation reaction247 while PtmU3 is the first member of a triosephosphate isomerase (TIM)-barrel fold diiron monooxygenase.246
Biological activity.
265 and 266 were originally discovered as potent bacteriostatics that targeted bacterial type II fatty acid synthesis.221,222 265 selectively inhibited S. aureus FabF (IC50 = 48 nM), an elongation condensing enzyme in the FASII cycle.221 Conversely, 265 was a weak inhibitor of S. aureus FabH (IC50 = 67 μM), the initiation condensing enzyme. 266 was found to be a dual inhibitor of FabF (IC50 = 4.6 μM) and FabH (IC50 = 9.2 μM).222 The MIC values for 265 and 266 were 0.1–1 μg mL−1 and <0.06–4 μg mL−1 against common drug-resistant Gram-positive pathogens including macrolide-, linezolid-, vancomycin- and MRSA, macrolide- and vancomycin-resistant enterococci (VRE), and Streptococcus pneumoniae.221,222 265 also showed moderate antibacterial activity against Mycobacterium tuberculosis (MIC = 12 μg mL−1) due to its inhibition of mycolic acid biosynthesis.250 265 and 266 do not exhibit antibacterial activity against Gram-negative bacteria, except against the efflux-negative Escherichia coli (ΔtolC).221,222 Although both 265 and 266 show efficacy in vivo when administering via continuous intravenous infusion,221,222 they are limited by poor pharmacokinetics due to rapid renal clearance.251 Recent studies aimed to address these limitations.252–254
Most PTM and PTN congeners have diminished or completely abolished antibacterial activity. While modest variations on the polycyclic enones can be tolerated with some loss of activity, minor modifications on the ADHBA moiety cause drastic negative effects on activity.220 The exceptions are 278 (1–4 μg mL−1), 279 (0.5–1 μg mL−1), and, 302 (0.25–0.5 μg mL−1), which retain the strong potencies of PTM and PTN.236,241
265 is also a promising drug lead for the treatment of diabetes in animal models. Studies in both mice and non-human primates support that 265 inhibits mammalian fatty acid synthase, selectively inhibits de novo lipogenesis, decreases glucose levels while reducing, or at least not significantly increasing, liver triglyceride levels, and leads to improved insulin sensitivity.255,256 These studies substantiate that mammalian FAS is a viable target for a host of metabolic disorders.
2.4.7. Terpentecins
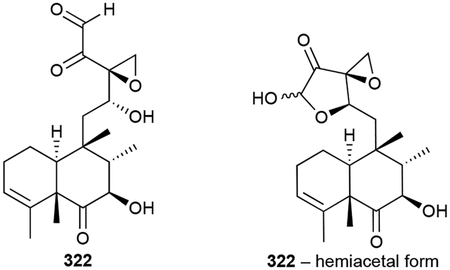
Terpentecin (322) is a highly functionalized clerodane antitumor antibiotic with a trans-decalin core.257,258 Its heavily oxidized side chain consists of hydroxyl, epoxide, ketone, and aldehyde functional groups. Interestingly, a structurally very similar fungal antibiotic, clerocidin, was isolated the previous year from Oidiodendron truncatum.259 Terpentecin are clerocidin are tautomeric in nature and can be found in their monomeric, hemiacetal, or dimeric forms depending on their solvent.259 Other bacterial forms of terpentecin, which are all produced by actinomycetes, include the 18-hydroxy congener UCT4B (323) and the spirocardins (324, 325), C14 and C15 reduced derivatives.260–262
Biosynthesis.
Early labelling studies supported that 322 is formed from MVA-derived isoprene units,263 a finding that was reinforced after several MVA genes were found in its BGC.264,265 The remaining biosynthetic enzymes included two TSs, two P450s and a ferredoxin.265 In vitro studies confirmed that the type II di-TS Cyc1 converts GGPP into terpentedienyl diphosphate, a trans-trans clerodane, and the type I di-TS Cyc2 abstracts the diphosphate to yield terpentetriene.266 The numerous oxygenation steps have yet to be revealed.
Biological activity.
The bacterial terpentecins, along with clerocidin, are potent antitumor antibiotics with the same mode of action. 322 was initially found to be a broad-spectrum antibiotic with MIC values as low as 0.05 μg mL−1;257 the other analogues have similar potencies.260,262 Investigation into its mode of action in bacteria showed that 322 inhibits DNA synthesis and causes cell elongation at sub-MIC levels.267 In mammals, 322 targets topoisomerase II, leading to DNA damage and cellular death. 322, along with clerocidin, induce the formation of a heat-stable complex and irreversibly inhibit the resealing of DNA.268,269 The IC50 for 322 cytotoxicity is 82 nM, about 10 times more potent than that of clerocidin, suggesting the C6 and C7 functional groups contribute to its potency.268
2.4.8. Miscellaneous Diterpenoids
In this section, we combined 16 distinct diterpenoid scaffolds (23 total NPs) that do not fit into the categories above. Volatiles phomopsene (326), allokutznerene (327), spiroviolene (328), and cattleyene (329) are tetracyclic hydrocarbon ring systems and the volatile micromonocyclol (330) has a rare C15 monocyclic structure.270–272 The neoverrucosanes (331–335) and (–)-verruconsan-2β-ol (336), well known constituents of plants, are 3/6/6/5 tetracycles; the neoverruconsanes were identified from marine gliding bacterium Saprospira grandis ATCC 23116 while 336 was found in the phototrophic Chloroflexus aurantiacus.273,274 331–336 were later found to be synthesized by UbiA-like TSs.249
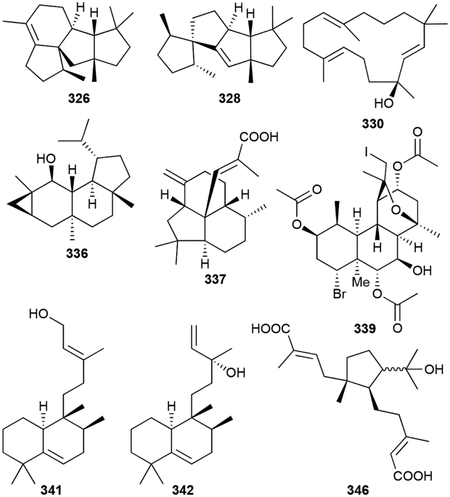
Other tricyclic diterpenoids include enhygromic acid (337), a decahydroacenaphthylene fused 6/6/5 tricyclic skeleton with an acrylic acid on the central C atom;275 isoagathenediol (338), which resembles the 6/6/6 tricyclic perhydrophenanthrene skeleton of the phenalinolactones, from the purple bacterium Rhodospirillum rubrum,276 and the extensively functionalized cyanobacterial tasihalides A and B (338, 340).277 The tasihalides are extremely rare examples of natural iodinated diterpenoids and represent a new structural class of terpenoids with a cis-decalin core fused to an oxabicyclic system.
There are four known halimane diterpenoids in bacteria. Tuberculosinol (341) and isotuberculosinol (342, originally named edaxadiene), first identified from the in vitro characterization of the type II and type I DTSs, Rv3377c and Rv3378c, respectively, from M. tuberculosis,278–280 were later produced and detected in the membranes of M. tuberculosis.281 These alcohols, although seen in vivo, appear to be shunt products in prenylated adenosine biosynthesis (vide infra chapter 3.4) via tuberculosinyl diphosphate hydrolysis.282,283 Micromonohalimanes A and B (343, 344) retain the halimane scaffold but have a significantly oxygenated prenyl chain at C9 of the decalin core, somewhat reminiscent of terpentecin.284 (+)-O-Methylkolavelool (345), a diterpenoid with a clerodane scaffold yet similar to 342 and also proposed to be attached to adenosine, was first identified through in vitro experiments and later detected in vivo from Herpetosiphon aurantiacus ATCC 23779.285
Finally, cystodienoic acid (346) is a unique monocyclic diacid from myxobacterium Cystobacter sp. Cbfe23 and two C19 abietadiene norditerpenoids with distinctive naphthalene cores (347, 348) were reported from the cyanobacterium Microcoleous lacustris.286,287
Biological activity.
The abietadiene norditerpenoids are active against various Staphylococci with MICs of 14–20 μg mL−1 while micromonohalimanes 343 and 344 are weak (>40 μg mL−1) bacteriostatic agents against MRSA.284,287 337, which enhances nerve growth factor-induced neurite outgrowth of rat adrenal cells, is an antitumor antibiotic with cytotoxicity against B16 melanoma cells (IC50 = 46 μM) and antibacterial against B. subtilis (MIC = 8 μg mL−1).275 346 is cytostatic with a GI50 value of 1.33 μM against HCT-116.286
2.4.9. Heterologously expressed diterpenoids
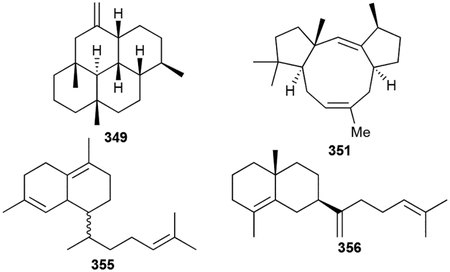
In this review, we elected not to include most terpenoids that were the result of heterologous expression of only the terpene synthase-encoding genes as they may not be genuine NPs. However, as we discussed the importance of the following study in the introduction, we feel it would be remiss to not include the novel skeletons identified from the heterologous expression of 29 TSs in S. avermitilis.25
Hydropyrene (349) and hydropyrenol (350) have novel 6/6/6/6 tetracyclic skeleton; the tricyclic tsukubadiene (351), odyverdienes A and B (352, 353), and cyclooctat-7(8),10(14)-diene (354) have 5/9/5, 6/8/4, 6/7/5, and 5/8/5 ring systems, respectively; isoelisabethatriene B (355), and the clavulatrienes (356, 357) are eudesmane-like 6/6 bicycles; and prenyl-β-elemene (358) and prenylgermacrene B (359) are the diterpenoid versions of 118 and 106, respectively.160 It is highly likely that these TS products are precursors for further transformations given that many are encoded in genetic proximity to other putative NP biosynthetic enzymes.
2.5. Sesterterpenoids
Compared with the widespread mono-, sesqui-, di- and triterpenoids, the C25 sesterterpenoids are much more rare. Of the approximately 1000 known sesterterpenoids,1 most of which have been discovered from plants and marine sponges,288,289 there are only 15 of bacterial origin. Scytoscalarol (360), the first bacterial sesterterpenoid, was found in the cyanobacterium Scytonema sp. (UTEX 1163).290 360 has a tetracyclic scalarane skeleton, which is commonly found in marine sponges, with a guanidinium functional group attached to the methyl group on C18. Two additional guanidinium-containing sesterterpenoids were recently isolated from Nostoc sp. (BEA-0956).291 The cybastacines A (361) and B (362) are pentacycles with a cyclic guanidino group composing the fifth six-membered ring.
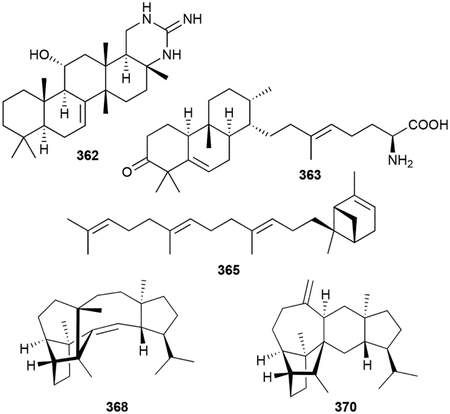
Two additional sesterterpenoids, atolypenes A (363) and B (364), fall into the same family as the brasilicardin diterpenoids. Using a clever strategy of BGC disassembly and reassembly with inserted synthetic promoters, the ato BGC from Amycolatopsis tolypomycina NRRL B-24205 was heterologously expressed in S. albus.292 The atolypene sesterterpenoids have the 6/6/6 tricyclic scaffold of their diterpenoid counterparts but are less oxidized, possess different methyl substitution patterns, and have an extended tail. The tails of atolypenes 363 and 364 resemble the γ-butyrolactone and amino acid moieties of the phenalinolactones and brasilicardins, respectively. The ato BGC was initially identified as terpenoid given its inclusion of a type II TS.292
The somaliensenes A (365) and B (366) are bicyclo[3.1.1]heptene and cyclohexene terpenoids, respectively, with oligoprenyl linear chains.293 These were discovered by heterologous expression of a TS gene from Streptomyces somaliensis in E. coli. Although we have not included most terpenoids that are the result of heterologous expression of only the TS gene, we included these two terpenoids given how few known members there are.
While genome mining for the TS responsible for the formation of the sesquarterpenoid tetraprenyl-β-curcumene (vide infra chapter 2.7), β-geranylfarnesene (367), a new linear sesterterpenoid, was identified.294
Very recently, novel sesterterpenoids were discovered from Streptomyces mobaraensis.295 Sestermobaraenes A–C (368–370) are complex pentacycles that are released as VOCs from bacterial cultures on solid medium. A type I sester-TS (SmTS1) was identified in S. mobaraensis and shown to be responsible for the production of 368–370, as well as several other sestermobaraenes (371–373) and sestermobaraol (374) that were not detected in the headspace extracts of the bacterial culture.295 A detailed mechanistic study was performed using extensive isotope labeling experiments revealing the plausible cationic intermediates leading to each of these sesterterpenoids.295 SmTS1 is encoded next to a geranylfarnesyl diphosphate (GFPP) synthase, polyketide synthase (PKS), and glycosyltransferase. While the C25 polyprenyl synthase is the first characterized bacterial GFPP synthase, it is unclear if the other two enzymes are involved in the biosynthesis of a more complex NP.295
Biosynthesis.
The biosynthetic pathways of 360 and cybastacines 361 and 362 are unknown. The formation of somaliensenes 365 and 366 arises from the cyclization of GFPP by the UbiA-like TS StsC.293 Given that a type II SHC-like TS is encoded only two genes away from stsC, it is likely that 365 and 366 are not the genetically encoded NP of the sts BGC. 367 is the result of diphosphate elimination of GFPP by the ‘large’ TS Bcl-TS.27,294
Biological activity.
The guanidinium-sesterterpenoids are antimicrobials. 360 was active against Gram-positive (MICs as low as 0.8 μg mL−1) and Gram-negative bacteria (MIC against E. coli = ~12 μg mL−1), as well as the fungus Candida albicans (MIC = 1.6 μg mL−1).290 362 was most potent against Gram-positive bacteria with an MIC range of 2–4 μg mL−1.291 The atolypenes were moderately toxic to several human cell lines with 363 exhibiting IC50 values at ~15 μM.292
2.6. Triterpenoids
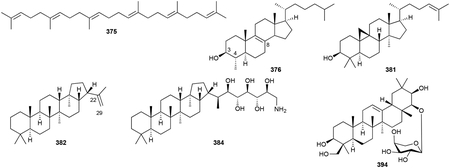
Triterpenoids are C30 NPs and include perhaps the most famous of all terpenoids, cholesterol. Unlike all of the terpenoids described above, which utilize precursors that are biosynthesized by adding successive C5 units onto linear prenyl chains in a head-to-tail fashion, the common precursor for all triterpenoids is squalene (375), a linear isoprenoid created by a head-to-head fusion of two molecules of FPP. The lack of a diphosphate moiety on squalene necessitates that a type II TS catalyzes any ensuing cyclization reaction on squalene or its epoxidized derivative oxidosqualene. While the skeletal diversity of triterpenoids is expansive, including linear carotenoids (vide infra chapter 2.8.1), mono-, bi-, tri-, tetra-, and pentacyclic systems,296 triterpenoids in bacteria generally fall into three main categories: pentacyclic 6/6/6/6/5 and 6/6/6/6/6 hopanoids, and tetracyclic 6/6/6/5 sterols.297–300
Triterpenoids are widely distributed in nature and have important roles in hormone signaling and membrane rigidity, stabilization, and organization.301 Hopanoids, the most common triterpenoids in bacteria, structurally resemble the relatively planar tetracyclic sterols that are incorporated into eukaryotic membranes. Accordingly, hopanoids have long been proposed to be the functional equivalents of sterols in bacteria, although their differences in structure and chemical properties ensure that their functional abilities are not identical.300,302–304 Other putative roles of hopanoids, and bacterial triterpenoids in general, such as lipid raft formation, stress tolerance, nitrogen fixation, and plant-bacteria communication should not be discounted305
In this section, we will not catalogue the multitude of tetracyclic and pentacyclic triterpenoids (our initial count of bacterial triterpenoids was ~200) or describe their well characterized biosynthesis or biological activities. This is for two main reasons: (i) triterpenoids and their biosynthesis are frequently and extensively reviewed296,297,300,302,305–310 and (ii) there is still some debate whether many of the sterols isolated or detected from bacteria are biosynthesized de novo. While it is conclusive that certain types of bacteria do biosynthesize sterols,311 very low quantities of reported sterols from some bacteria suggest that they were actually contaminants from media components, laboratory conditions, or other organisms.312,313 Instead, we here introduce the triterpenoid skeletons most commonly found in bacteria by featuring some of the early discoveries as well as including some of the more exotic skeletons.
2.6.1. Hopanoids and sterols
The first hint of triterpenoid presence in bacteria came when squalene (375) and an unknown polycyclic derivative were identified in a few cyanobacteria.314 Unequivocal data supporting the ability of bacteria to produce cyclic triterpenoids came a year later when Methylococcus capsulatus produced 375, 4-methyl-, and 4,4-dimethylcholesterol variants (376–379) on media only containing methane as its carbon source.315,316 Myxobacteria were among the first type of bacteria found to produce true sterols; cholest-8(9)-en-3β-ol (380) was the main sterol of Nannocystis exedens and a later survey of other myxobacteria revealed several other sterols including cycloartenol (381).317,318 A recent bioinformatics and lipid analysis study found that various bacteria including myxobacteria, methanotrophs, bacteriodetes, and α-proteobacteria all produce at least one sterol NP.311
Hopanoids, which were first known as ubiquitous constituents in sedimentary rocks, are produced by many different types of bacteria including proteobacteria, actinobacteria, cyanobacteria, and acidobacteria.298,305,319 The first bacterial hopanoid detected was diploptene [hop-22(29)-ene, 382].320,321 The most common modification to hopanoids is the addition of a polyol alkyl side chain attached to C29. This C5 addition, resulting in C35 hopanoids or homohopanoids, arises from the attachment and degradation of adenosine to the terminal olefin of 382.305,309 The first bacterial homohopanoid, bacteriohopanetetrol (383), was isolated from Acetobacter xylinum.322,323 This family of triterpenoids, which can be diversified by core methylations or alkyl chain oxidations, aminations, esterifications, or glycosylations, is exemplified by members such as 384–389.324–327 The subclass of 6/6/6/6/6 pentacyclic hopanoids, including the tetrahymanols (390–393), MK800–62F1 (394), β-amyrin (395), and the soyasapogenols (396, 397) have core scaffolds that are much more highly oxidized than the 6/6/6/6/5 hopanoids.328–331
2.6.2. Miscellaneous triterpenoids
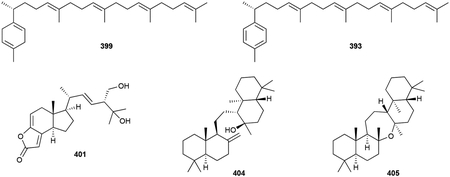
Other classes of triterpenoids are quite rare in bacteria. The linear β-hexaprene (398) and monocyclic triprenylcurcumenes (399, 400) are structurally related to the sesterterpenoids 367 and 366, respectively.294,332 The salimyxin antibiotics (401, 402) are the first NPs found in the marine myxobacteria genus Enhygromyxa.333 Their base structure, with a 6/5 hexhydroindane core fused to a 5-membered lactone, appears to be an unusually degraded sterol.
Onoceroids are a set of three unique triterpenoids first identified through heterologous expression of a TS, BmeTC, in E. coli and later identified from the hexane extracts of Bacillus megaterium.334 All derived from squalene (375), the bicyclic 8α-hydroxypolypoda-13,17,21-triene (403) and tetracyclic 14β-hydroxyonocera-8(26)-ene (404) are on-pathway intermediates to the symmetrical and oxepane-containing pentacycle onoceranoxide (405). BmeTC is an unusual type II TS that appears to independently cyclize the two termini of squalene.334
2.7. Sesquarterpenoids
Sesquarterpenoids, NPs derived from linear C35 precursors, are a recent addition to the superfamily of natural terpenoids with their name being coined about 10 years ago.335 There are two types of bacterial sesquarterpenoids: the monocyclic and polycyclic curcumenes and sporulenes from Bacillus spp. and the monocyclic heptaprenylcyclines from Mycobacterium spp.336
Tetraprenyl-β-curcumene (406) and tetraprenyl-α-curcumene (407, originally named tetraprenyl-ar-curcumene for its aromaticity), were also isolated from various Gram-positive and Gram-negative bacteria, are the sesquarterpenoid versions of triprenyl-β-curcumene (399) and triprenyl-ar-curcumene (400).332 Subsequent studies exposed the seemingly related pentacyclic isoprenoids in the spores of B. subtilis;337 these NPs, named sporulenes A–C (408–410), were structurally characterized after overproducing the putative TS responsible for cyclization.338 After two C35 pentacyclic terpenols were discovered, it was proposed that the non-aromatic alcohol (411) was the legitimate NP and the aromatic analogue and dehydrated sporulenes were artifacts of autooxidation and thermal dehydration, respectively;339 these alcohols were later named baciterpenols A (411) and B (412).336
Lipid analysis of various nonpathogenic Mycobacterium spp. revealed the presence of C35 terpenoids heptaprenylcycline (413) and octahydroheptaprenol (414).340 414 is proposed to be the hydrolysis product of heptaprenyl diphosphate analogue or the mycolic acids. Various heptaprenylcyclines have also been identified including congeners with additional E or Z olefins (415–417) or a C18 ketone (418).341,342
Biosynthesis.
The biosynthesis of the Bacilli sesquarterpenoids revealed a new class of ‘large’ TSs. Using a genome mining and systematic knockout approach to identify the TS responsible for initial cyclization reaction, YtpB was found to cyclize heptaprenyl diphosphate into the monocyclic 406.335 The type II TS SqhC then acts to yield the pentacyclic 411. Heptaprenyl diphosphate was later found to be formed by HepS/HepT, two (all-E)-prenyl diphosphate synthases.343 At the time, YtpB was an unprecedented type I TS and had a primary sequence unlike any other characterized proteins.335 Now, a new family of noncanonical ‘large’ TSs is beginning to be revealed and mechanistically characterized.27
The mycobacterial sesquarterpenoids rely on substrate flexible prenyltransferases and TSs. Synthetic preparation of octahydroheptaprenyl diphosphate and biotransformation in cell-free extracts of Myobacterium chlorophenolicum demonstrated it was the substrate of heptaprenylcycline formation.340 Although the enzyme responsible for this transformation was unknown, this was the first example of a cyclization of a linear C35 terpenoid and suggested the presence of a natural Z-terpene cyclase. The discovery of numerous E and Z isomers of heptaprenylcyclines implied that linear E or Z precursors are distinctly formed by elongation of E,E-FPP or E,E,E,-GGPP and that the polyprenyl reductases can likely act on both substrate configurations.336 Functional characterization of three Z-prenyl transferases supported this former proposal.341 The heptaprenylcycline synthase has not been identified yet, but the ketone moiety on 418 is a plausible P450 functionalization.344
2.8. Tetraterpenoids
Tetraterpenoids are C40 NPs composed of two C20 units fused together in the same head-to-head manner as that of the triterpenoids. Although they are abundant in number, currently characterized tetraterpenoids are not very structurally diverse in comparison to the other terpenoids; tetraterpenoids are almost exclusively represented by C40 carotenoids. Carotenoids contain extended conjugated polyene systems that are found in linear or 6-membered mono- or bicyclic form. The vast number of carotenoids arises from countless combinations of oxidation, oxygenation, cyclization, and glycosylation. Formation of polycyclic (not including the bicyclic carotenoids) tetraterpenoids is not yet known to occur, although given the capability of TSs to cyclize sesquarterpenoids, it would be reasonable to consider the possibility.
The polyenic nature of carotenoids imparts the ability to absorb light in the blue-green (450–570 nm) range of the visible spectrum, thus acting as accessory pigments that can absorb light in the absorption gap of chlorophyll.345,346 The chemical properties of carotenoids also provide protection to the cell as scavengers of triplet chlorophyll and singlet oxygen.345,347,348 In addition, these lipids may be incorporated into membranes to increase membrane rigidity.349 Therefore, all photosynthetic organisms, including cyanobacteria and purple photosynthetic bacteria, as well as some non-photosynthetic bacteria, archaea, algae, and fungi, also produce C40 carotenoids. Animals require and utilize carotenoids, mainly as oxidatively degraded products (e.g., vitamin A), but they are not biosynthesized de novo and must be obtained from their diet.350
In this section, we will not describe the excessive numbers of carotenoids (our initial count of bacterial carotenoids was ~300) or describe their well characterized biosynthesis or biological activities. In fact, there is a database, ProCarDB, of 304 unique bacterial carotenoids available online.351 Instead, we focus on introducing the most common scaffolds in bacteria and highlighting some of the tetraterpenoids and apocarotenoids that are not produced by other organisms. For comprehensive reviews on carotenoids, their biosynthesis, and biological activities, we highly recommend the reviews cited herein.345,347,350,352
2.8.1. Carotenoids
Common representatives of linear, monocyclic, and bicyclic C40 carotenoids include lycopene (419), γ-carotene (420), and β-carotene (421), respectively. The 6-membered rings are located terminally with the two rings in bicyclic carotenoids analogous to each other. These rings are most prevalent as cyclohexenes, but aromatic rings such as the benzoates seen in synechoxanthin (422) from Syncechococcus sp. PCC7002 have been identified.353 Oxygens are typically incorporated at the termini with hydroxyl, ketone, and carboxylic acids commonly seen on the rings, as in canthaxanthin (423).354 Carotenoids are also commonly modified as glucosides, as evidenced by the cyanobacterial myxol 2´-fucoside (424).355
2.8.2. Other carotenoids
Although not technically tetraterpenoids, we thought it was logical to include here examples of C30 carotenoids and C45 and C50 homocarotenoids given their structural, biosynthetic (i.e., the head-to-head fusion of two isoprenoid precursors), and functional similarities. Some bacteria, such as S. aureus, do not biosynthesize C40 carotenoids and instead utilize C30 carotenoids. Certainly, the most famous C30 carotenoid is the virulence factor staphyloxanthin (425), a linear and terminally oxidized C30 unit linked to an acylated glucose.356 Other bacterial C45 and C50 homocarotenoids are elongated tetraterpenoids that have one or two dimethylallyl units appended onto the cyclohexene ring. Like the C40 carotenoids, these can be found as oxidized linear, monocyclic, or bicyclic variants exemplified by flavuxanthin (426), nonaprenoxanthin (427), and decaprenoxanthin (428), the first known natural C50 carotenoid.357–360
2.8.3. Norcarotenoids
Carotenoids are degraded into a variety of NPs and are commonly referred to as apocarotenoids;345 vitamin A is the archetype apocarotenoid.361 Carotenoid degradation, which may be the result of nonenzymatic or enzymatic oxidation, can occur at any of the numerous double bonds on linear, monocyclic, or bicyclic carotenoids yielding both linear and monocyclic apocarotenoids of various lengths.345,362 Some apocarotenoids are pigments as they retain their polyene nature, but most of the detected apocarotenoids in bacteria are VOCs due to their small size and hydrophobic characteristics.44
Most bacterial apocarotenoids have been detected as VOCs from cyanobacteria,44,363,364 but others have also been seen in Streptomyces, myxobacteria, and bacteroidetes.37,44,47,49 Linear apocarotenoids, derived from oxidative cleavage from the uncyclized end of carotenoids or menaquinones, include C8 (429, 430), C13 (431–433), C18 (434), and C33 (435) alcohols and acetones;37,47,49,365,366 the C11 homoterpenoid 436 was also identified in Streptomyces sp. GWS-BW-H5.37
Monocyclic apocarotenoids are much more common, or at least are more commonly identified, in cyanobacteria. Being derived from the terminal ring of carotenoids, these all have the trimethyl substituted 6-membered ring in common. Most structures are either simply oxidized 6-membered rings (439–441, 439) or derivatives of the β-ionone (442–449) scaffold.49,57,363,365,367,368 Variations lie in the position and type of added oxygen atoms on the ring and the level of olefin saturation. Recently, two abscisic acid sesquiterpenoids were reported from Amycolatopsis alba DSM 44262, but are likely just isophorones with extended acrylic acid tails (450, 451).369
There are a couple of examples where the bacteria appear to utilize apocarotenoids as building blocks for additional modifications. A megastigmane diglycoside (452) from Streptomyces sp. YIM 6334 exhibits a trihydroxylated β-ionol core that has been adapted with a β-d-apiofuranosyl(1→2)-β-d-glucopyranoside.370 There are two 2-hydroxymethylindole-3-carboxylic esters with linear apocarotenoid tails. Sphestrin (453) and the phytohormone rhodestrin (454) are metabolites from Rhodobacter sphaeroides produced when grown on media containing anthranilate.371,372 The most exotic apocarotenoid is nostocionone (455).373 This antioxidant from Nostoc commune is a formal aldol condensation product of 442 and syringic aldehyde.
3. Meroterpenoids
3.1. Aminocoumarins
Aminocoumarins are a family of antibiotics discovered during the Golden Age of Antibiotic Discovery.374 They are categorized by the presence of a 3-amino-4,7-dihydroxycoumarin bicyclic nucleus and are all produced by Streptomyces. The three “classical” aminocoumarins, novobiocin, clorobiocin, and coumermycin A1 have been thoroughly studied since their discoveries, revealing a wealth of knowledge on their biosynthesis and biology.375 Not all aminocoumarins have isoprenoid moieties, but 18 family members have 3-prenyl-4-hydroxybenzoyl moieties linked to the aminocoumarin via an amide bond. Being the first antibiotic discovered with a side chain of terpenoid origin,13 novobiocin, and the later discovered clorobiocin, have not only been used as probes to discover new modes of action for antibiotics and to understand the use of MVA and MEP pathways in NP biosynthesis, but biosynthetic studies revealed a novel family of PTs that opened up a new avenue for bacterial and fungal terpenoid enzymology and genome mining.
Novobiocin (456), also known early on as streptonivicin, cathomycin, and trademarked by Upjohn Company as Albamycin,376–379 was discovered from Streptomyces niveus in 1955. Its structure is a composite of the aminocoumarin core, a 3-dimethylallyl-4-hydroxylbenzoyl moiety, and a 3-carbamoyl-4-O-methyl-5-C-methyl-l-rhamnose (l-noviose).380,381 Clorobiocin (457), found in several Streptomyces species, differs from novobiocin only in its 8´-chloroaminocoumarin moiety and replacement of the acylated rhamnose with a 5-methyl-pyrrole-2-carboxyl moiety.382 Most of the novobiocin analogues are either desmethyl (458–460) or descarbamoyl variations (461), or both (462); the C5 position of the benzoyl moiety is occasionally hydroxylated (463–465).383–385 Clorobiocin (457) is the only aminocoumarin-terpenoid hybrid with a chlorine atom and novclobiocin 101 (466), the deschloro variant initially isolated from a halogenase mutant, was later found as a genuine NP.386,387 Isonovobiocin (467), a 2´´-carbamoyl isomer of 456, was also isolated,385 although it was previously shown to be a non-enzymatic isomerization product from novobiocin in the presence of dilute alkali.388 Coumabiocin A–F (468–473) from Streptomyces sp. L-4–4 are the only analogues with modified prenylbenzoyl groups resulting in coumaran (A and B) or chromane (C and D) rings and/or hydroxylated units.389
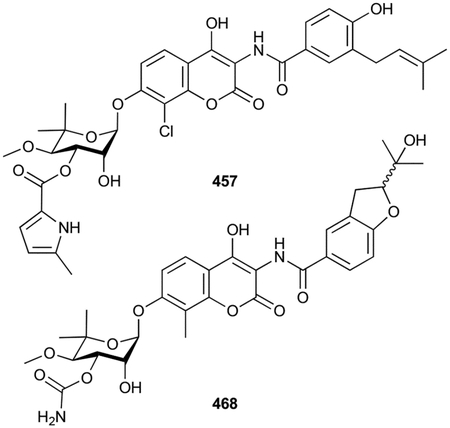
Biosynthesis.
The biosynthesis of the aminocoumarins is comprehensive. A myriad of studies has detailed the construction of the aminocoumarin moiety, the noviose sugar, the 3-prenyl-4-hydroxybenzoyl group, the linkages of these three entities, as well as the regulatory and resistance mechanisms.375 Studies of 456 and 457 prenylation led to the revelation of a new family of microbial soluble aromatic PTs. Prior to the report of any aminocoumarin BGCs, feeding studies using isotopically labeled precursors exposed that the dimethylallyl moiety of novobiocin originated via the MEP pathway, confirming its isoprenoid origin.390,391 Almost simultaneously, a soluble protein fraction contained an unknown protein that catalyzed the prenylation of DMAPP onto 4-hydroxyphenylpyruvate, suggesting that prenylation occurred prior to the maturation of 4-hydroxybenzoic acid and its attachment to the nitrogen of the aminocoumarin scaffold (Scheme 12).392 Bioinformatics analysis of the clorobiocin BGC revealed there were no gene candidates that had homology to known PTs.393 CloQ, a soluble protein with no similarity to known PTs, no DDxxD motif, and no dependence on divalent cations, was later exposed as an aromatic PT.394 The crystal structure of NphB, a CloQ homologue, revealed this unique family also had a novel structural fold, a β/α barrel with ten antiparallel β strands.395 The five α-β-β-α secondary structure repeat elements led to the term ABBA PT.396 The identification of CloQ led to a series of discoveries of homologous soluble aromatic PTs, including NovQ from the novobiocin BGC and a variety of fungal indole PTs.394,396–399 These enzymes tend to have relaxed substrate specificities for both the prenyl donor and aromatic acceptor, making determination of its natural substrates via in vitro assays challenging while providing impressive chemoenzymatic tools.26
Scheme 12.
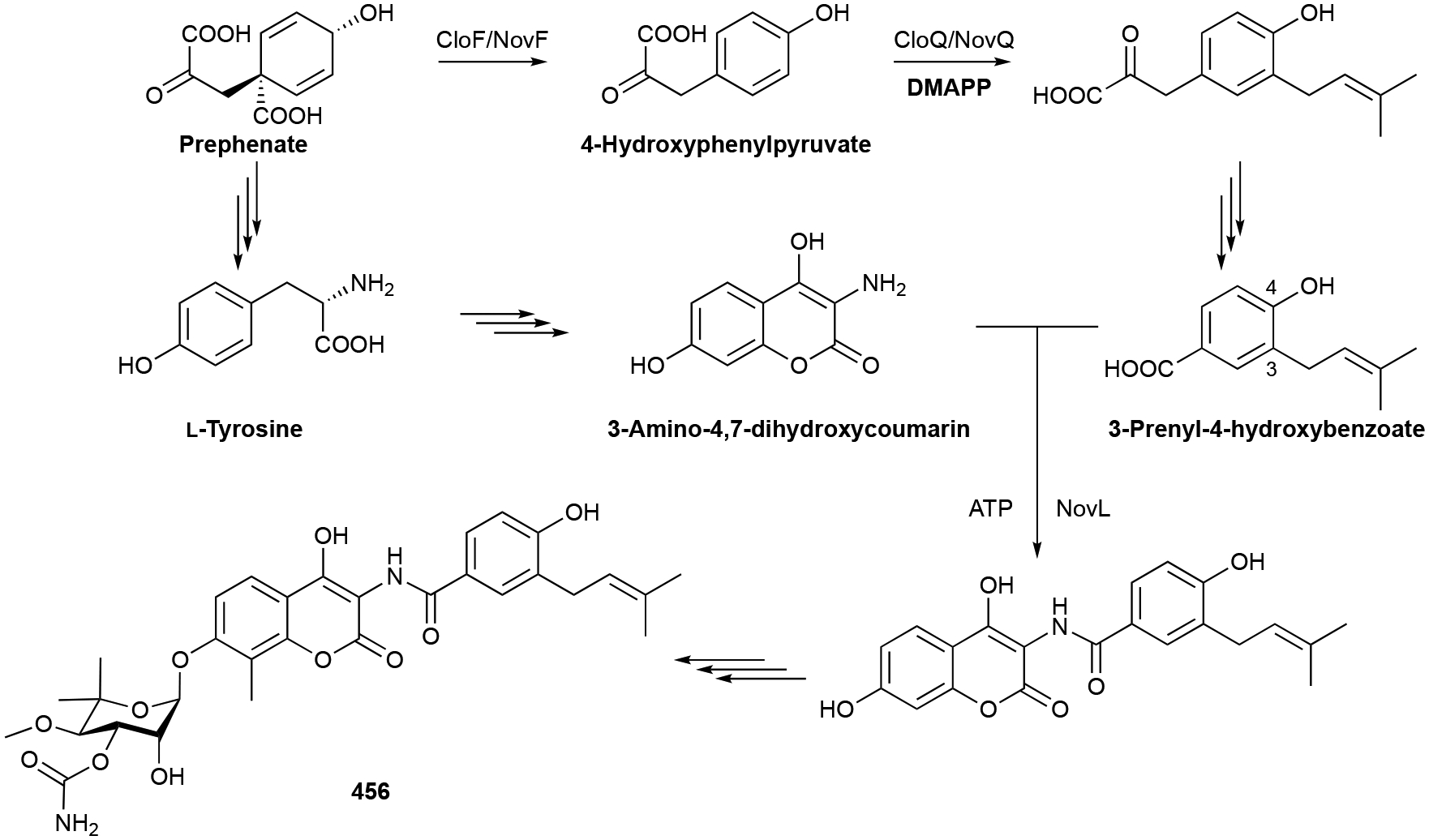
Biosynthesis of novobiocin (456).
Biological activity.
The aminocoumarin family of antibiotics inhibit bacterial topoisomerase.400 The fluoroquinolones are the quintessential antibiotic for topoisomerase inhibition, but novobiocin was discovered well before the quinolones and was licensed for a brief period as a commercial antibiotic for MRSA before being pulled due to its poor pharmacokinetic properties and toxicity.375,401 Aminocoumarins, which bind to the ATP pocket of GyrB and ParE,402 are potent inhibitors of DNA gyrase with IC50 values in the range of 10 nM.400 Extensive SAR studies have been performed on the aminocoumarins and most modifications, including those on the prenyl group, have deleterious effects on antibacterial activity.389 It was proposed that the prenylbenzoyl moiety may contribute to bacterial uptake and provide enhanced affinity to DNA gyrase.403,404 In comparison with novobiocin, clorobiocin is 10-fold more active against DNA gyrase and 70-fold more active in the inhibition of topoisomerase IV;405 even loss of just the chlorine atom results in 8-fold less activity.386 Interestingly, the clorobiocin BGC possesses a resistant form of topoisomerase IV while the novobiocin BGC does not.406 It is therefore expected, due to the targeting of two distinct essential enzymes, that clinical use of clorobiocin may be resilient to the development of antibiotic resistance.375
In accordance with the toxicity of aminocoumarins, 456 and 465 also have antiproliferative activities. These NPs, along with coumermycin A1, bind to the C-terminal domain of 90 kDa heat shock protein (Hsp90) causing Hsp90 complex destabilization and client protein degradation.407 Hsp90 plays crucial roles in stabilizing and folding proteins involved in oncogenic processes and is therefore an emerging target for anticancer treatments.408 Some aminocoumarin analogues, such as 466, also showed moderate antifungal activities.387
3.2. Benzastatins
Benzastatins are unique NPs derived from geranylated p-aminobenzoic acid (PABA) and produced by Streptomyces. Like the prenylated phenazines and pyrroles (vide infra chapters 3.6 and 3.9), they are radical scavengers with an assortment of other biological activities. The origin of the 29 benzastatins, in their four distinct forms, was expertly determined in a single biosynthetic study and led to the discovery of a novel nitrene transfer P450.409
Virantmycin (474), a unique chlorine-containing tetrahydroquinoline skeleton with a tetrasubstituted terminal olefin, was the first member of the benzastatin family to be discovered.410,411 This bacterial alkaloid was initially discovered after screening for antiviral NPs from Streptomyces nitrosporeus.412 Subsequent microbial screening programs led to the isolation of benzastatins A and B (475, 476), p-aminobenzamides with linear geranyl chains, and the tetrahydroquinoline benzastatins C (477) and D (478).413 Benzastatin derivatives (479–481) with indoline, rather than tetrahydroquinoline scaffolds, were discovered soon after;414 total synthesis of benzastatin E revealed the absolute configuration of the indolines is 9S,10R.415 Other natural monocyclic, tetrahydroquinoline, and indoline benzastatins are modified with hydroxyl groups at various positions (482–486), chlorines at C9 or C10 (477, 487), an ergothioneine moiety at C9 (488), or a hydroxycyclopentenone moiety on the C7 amide group (489, 490).416–419 The unpublished ramthacins include didehydro (491) and hydroxyl variations (492, 493) of 465.420
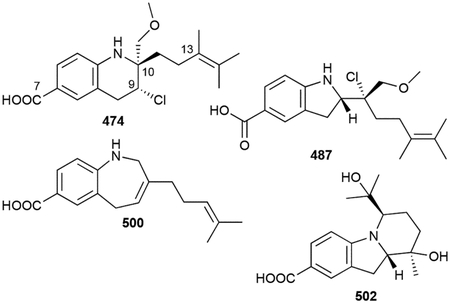
Heterologous expression of the entire bez gene cluster in S. lividans produced 474, 484, 486, and eight new benzastatin congeners.409 Six of the eight new compounds were variants of monocyclic, tetrahydroquinoline, and indoline benzastatins (494–499); the other two had novel benz[b]azepine scaffolds (500, 501). The production of new benzastatins was proposed to be the result of using inducible promoters rather than the native promoters found in the bez BGC.409 One additional benzastatin-like NP, isolated from Streptomyces sp. SP301, is a 6/5/6 cyclized version of JBIR-67 (484) and was named aminobenzoate D (502).421
Biosynthesis.
The biosynthetic pathway of the benzastatins was recently revealed in an admirable study using heterologous expression, genetics, and in vitro biochemical experiments (Scheme 13).409 The structural components of the benzastatin scaffold are created from modified PABA and geranyl units. GPP is C6 methylated and C10 hydroxylated; PABA is N-acetoxylated. Prenylation of the modified GPP onto the meta position of N-acetoxy-PABA is likely performed by a UbiA PT. The cyclization of this reactive intermediate into the tetrahydroquinoline or indoline bicycles is mediated by an unusual P450. BezE controls cyclization by successively forming iron nitrenoid and aziridine intermediates via acetic acid elimination and nitrene transfer to the nearby double bond of the geranyl moiety, respectively. The generation of tetrahydroquinoline and indoline is dependent on how the aziridine ring is opened, either at C9 by Cl– or C10 by OH– to give 494 or 499, respectively. BezE is expected to control the use of the chloride nucleophile in its active site. The biosynthesis of 474 is completed by O-methylation (Scheme 13).409 The monocyclic benzastatins result from premature prenylation of PABA while the many hydroxyl and methyl variations stem from nonspecific use of prenyl donors. Given the innate reactivity of N-acetoxy-PABA, cyclization can also occur nonenzymatically resulting in the 5-, 6-, or 7-membered heterocycles. The ergothioneine moiety of 488 is also nonenzymatically formed as the C9 chlorine of 474 is a good leaving group.409 The discovery of BezE as the first natural P450 nitrene transferase—mutants of the biotechnologically useful BM3 have previously been engineered for nitrene transfer422—supports the rationale for continued discovery of and biosynthetic studies on bacterial terpenoids.
Scheme 13.
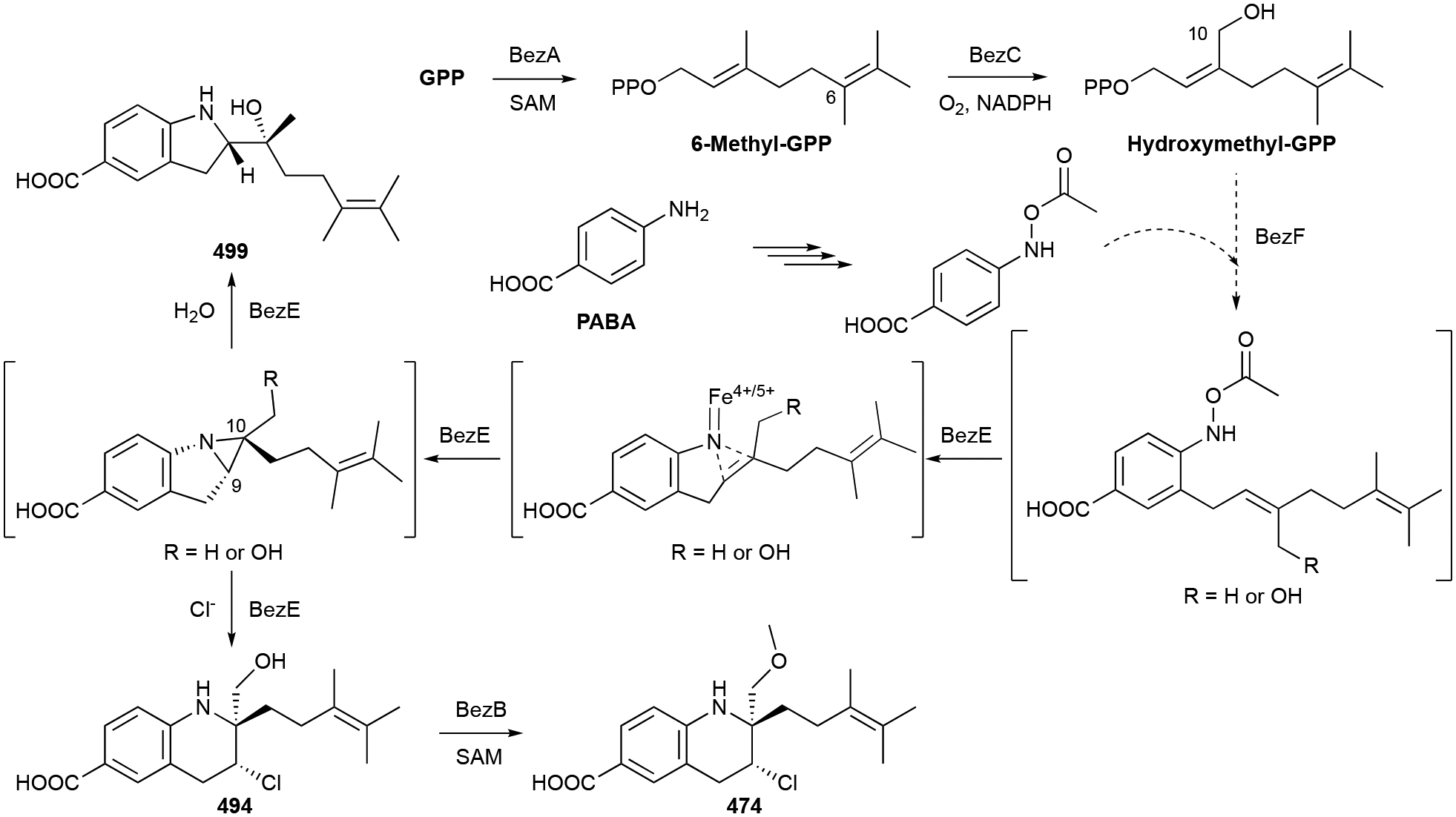
Biosynthesis of virantmycin (474) and 7-hydroxylbenzastatin F (499).
Biological activity.
Benzastatins are known to have antibacterial, antifungal, antiviral, radical scavenging, and neuronal protection properties. The biological selectivities appear to be highly dependent on the nature of the bicyclic moiety and the presence of the C9 chlorine atom. 474 is an antiviral antibiotic with potent inhibitory activities against both RNA and DNA viruses. The inhibition of viral plaque formation was seen at low concentrations (>0.01 μg mL−1).412 The tetrahydroquinoline structure, terpene chain, stereochemistry, and chorine atom of 474 and other benzastatins are all important for antiviral activity.411,419,423
Benzastatins have shown radical scavenging abilities by inhibiting lipid peroxidation in rat liver microsomes (IC50 values as low as 3.3 μM) and glutamate toxicity in neuronal cells (EC50 values as low as 1.7 μM).413,414,416,424 Both 474 and 487 also potently activated hypoxia-induced factor (HIF) with EC50 values of 8 and 17 ng mL−1 (23 and 48 nM), respectively.418 The dechlorinated analogues 485 and 486 lost this activity but retained their radical scavenging abilities suggesting that HIF induction requires the formation of a covalent bond. Later studies showed that 487 does not function as an iron chelator as most natural HIF activators do, but rather through interaction with an unidentified and likely metal-dependent target.424
3.3. Indoles
Indole is an electron-rich heteroatom-containing bicycle perhaps best known as part of the side chain of the proteinogenic amino acid Trp. It is a versatile nucleophile and functionalized indoles are prevalent throughout NPs. The nucleophilicity of indole is a perfect partner for the electrophilic prenyl diphosphates; in fact, prenylation of the indole core has been seen at each of the seven non-bridgehead atoms, N1 and C2–C7.399,425 In this chapter, we separated prenylated indoles from peptides containing prenylated Trp residues (vide infra chapter 3.5).
3.3.1. Simple indoles
There are 28 prenylated indoles that we categorized as ‘simple’ indoles, as opposed to the more complex polycyclic NPs below. The majority of these possess dimethylallyl moieties, or variants, appended to C4–C7 in a normal prenylation orientation. As would be expected given their origin from Trp, many of these simple indoles also have various functional groups at C3. The simplest prenylated indoles known in bacteria are 6-prenylindole (503, i.e., dimethylallylindole) and 6-dimethylallyltryptophan (504), first discovered from Streptomyces sp. TP-A0595 and Actinoplanes missouriensis NBRC 102363, respectively.426,427
Chloroindole, or 3-chloro-4-dimethylallylindole (505) from Streptomyces sp. SN0280 is the simplest C4 prenylated indole.428 Other C4 prenylated indoles include amycolactam (506) from a sponge-associated Amycolatopsis sp. and indiacens A and B (507, 508), C3-aldehydes with modified isoprenyl moieties from the myxobacterium Sandaracinus amylolyticus NOSO-4T.429,430
A family of related disubstituted indoles is differentiated by their prenylation pattern, being alkylated at either C5, C6, or C7, and their C3 functional groups. This family includes tryptophols (509–513), indole acetaldoximes (514, 515), indole acetonitriles (516–519), and indole carboxylic acids (520–522) or esters (523).191,431–437 The dimethylallyl groups are mostly unmodified, but a few oxidized variations exist, including the uniquely modified but-1E-en-3-one side chain (511).433 Many of these NPs are from Streptomyces or myxobacteria.
Finally, there are also several 6- and 7-prenylated 2-oxindoles and isatins. These include 6-dimethylallylisatin (524) and its 3-hydroxyl congener (525), the 3-isopropylidene-2-oxindoles (526–528), 7-prenylisatin (529), and 3-acetonylidene-7-prenylindolin-2-one (530).427,436,438–440
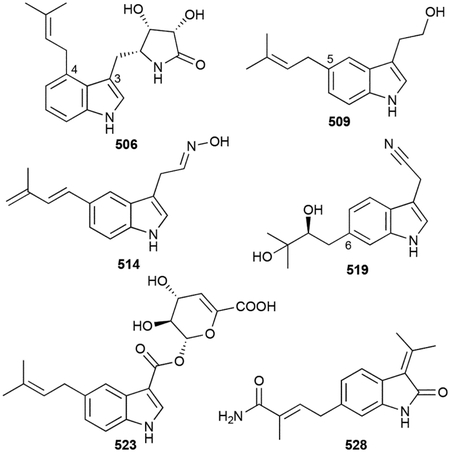
Biosynthesis.
The full biosynthetic pathways of most of the simple indoles are unknown, although the prenylation reactions for many of them can be reasoned to be the result of prenylation of Trp by ABBA PTs. IptA, a 6-dimethylallyl-Trp synthase, was the first indole PT identified in bacteria.441 Using iptA as a probe, multiple BGCs were identified containing nearby iptA-like genes next to tryptophanase-encoding genes.427,440,442 PriB, from the BGC responsible for the production of 523, was shown to be a 6-dimethylallyl-Trp synthase and occurs prior to the action of tryptophanase. Although the pri BGC has two P450s, the conversion of 6-prenylindole into 523 is still not understood, as are most of the indole transformations in this family of NPs.440,442
Biological activity.
Most of the simple prenylated indoles do not possess significant biological activities. Several (503, 507, 508, 514, 524, 526, 529) had weak to moderate antimicrobial activities (MICs >20 μg mL−1).426,430,433,439,440 Only a few (506, 512, 515, 526, 530) were moderately cytotoxic with IC50 values in the low to mid μM range, with 5-prenyltryptophol (509) also exhibiting bone morphogenetic protein-induced alkaline phosphatase inhibition (IC50 = 81 μM).429,431,432,436,439
3.3.2. Hapalindole-like indole alkaloids
Over 80 natural members of the hapalindole family of cyanobacterial indole alkaloids are known. The structures, chemistry, biosynthesis, and biological activities of these prenylated indole alkaloids from the order Stigonematales have been extensively reviewed.443,444
There are four major classes of stigonematalean indole alkaloids: hapalindoles, fischerindoles, ambiguines, and welwitindolinones. These four classes can be further subdivided into nine structural groups.444 Structural elements that are conserved amongst all nine groups include a polycyclic scaffold with an indole or oxindole core, a highly functionalized cyclohexane unit fused to the (ox)indole at C3, and a terminal vinyl group. Most members also possess isonitrile or isothiocyanate functional groups and a site-specific chlorine atom.
Hapalindoles are the largest class of these indole alkaloids. First isolated in 1984 from Hapalosiphon fontinalis while searching for the NP(s) responsible for its antialgal properties, hapalindoles A (531) and B (532) were identified as novel 10R,11R,12R,13R,15S-tetracyclic indole-containing scaffolds with 12-chloro and 11-isonitrile or -isothiocyanate functionalities, respectively.445 Since then, 20 additional ‘Group 1’ tetracyclic hapalindoles, all possessing 3,4-fused indole cores, have been isolated from Hapalosiphon (533–552, 538, 539, 541). Most of these hapalindoles have variations in their stereochemical configurations at C10, C12, and C15, and are dechlorinated, hydroxylated, or epoxidized.446–449 Hapalindoles T (549) and X (550) are unique with a cyclic thiocarbamate and a shifted allyl moiety, respectively.446,449 Hapalindole formamides A (536) and J (537) were recently reported as NPs from Hapalosiphon sp. CBT1235, although a similar welwitindolinone formamide was earlier reported as an isolation artifact.450,451 Hapaloxindoles (553–555), hapalonamides (556–559), and fontonamides (560–562) were also isolated as minor constituents and are likely oxidation products formed by singlet oxygen reactivity.449,452,453
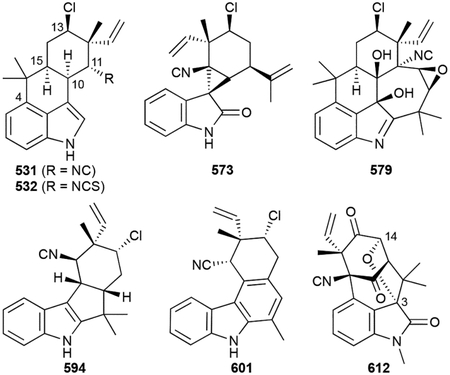
The ‘Group 2’ tricyclic hapalindoles (563–572, 567) are very similar to the tetracyclic versions with the exception that the C4–C16 bond is not formed.446,447,454 The ‘Group 3’ hapalindolinones, of which only two have been isolated from Fischerella ATCC 53558, are tetracyclic alkaloids with an unusual C3-spiro-fused cyclopropane connecting the 2-oxindole and cyclohexane rings (573, 574).455
The ambiguine class of indole alkaloids, first isolated from Fischerella ambigua UTEX 1903, are categorized based on an additional dimethylallyl group that resides at C2 of the indole ring.456 ‘Group 4’ tetracyclic ambiguines (575–577, 578) are 2-reverse prenylated tetracyclic hapalindoles.456,457 The 2-prenyl moieties of the ‘Group 5’ pentacyclic ambiguines are also attached to C11 to form a 7-membered ring (579–590, 591). Along with the expected isonitrile groups, these new rings have one, two, or three double bonds and are commonly substituted with oxygens.456–460 Ambiguine G nitrile (584) was the first nitrile-containing analogue discovered.458 Two ‘Group 6’ fischambiguines were later isolated from Fischerella ambigua UTEX 1903.460 Fischambiguines A and B (592, 593) are also 2-prenylated pentacyclic alkaloids, but instead of a 7-membered ring, cyclization results in a new 6-membered ring.
Fischerindoles have similarity to the tetracyclic hapalindoles, but final cyclization onto the indole ring occurs at C2. These ‘Group 7’ tetracycles have been identified in both Fischerella and Hapalosiphon and have 6/5/5/6 (594–600) or 6/5/6/6 scaffolds (601, 602).454,461,462 The C rings in 601 and 602 are aromatic.462
The welwitindolinones combine to form the ‘Group 8’ and ‘Group 9’ tetracyclic indole alkaloids. ‘Group 8’ is a single unique molecule, welwitindolinone A (603), produced by Hapalosiphon welwitschii that displays a C3-spirocyclobutane ring on its 2-oxindole core.454 The welwitindolinones in ‘Group 9’ have a cycloheptaenone connected to C3 and C4 of the 2-oxindole core and have differences in stereochemical configuration at C3, levels of oxidation, presence or absence of a methyl group on the indole N, and are either isonitriles or isothiocyanates (604–612).451,454 Adding to the structural complexity of the welwitindolinones, is the C3–C14 cyclic ether-containing 612.
Biosynthesis.
Early biosynthetic proposals of the stigonematalean indole alkaloids, based on their structural similarities and the fact that most were isolated from the same strain, suggested a unified pathway that shared common intermediates.454 A series of concurrent and successive genomic and biochemical studies revealed the amb/fam and wel BGCs from F. ambigua UTEX1903 and H. welwitschii UTEX B1830, respectively, and provided a new proposal with (3R)-3-geranyl-3-isocyanovinyl indolenine as a conserved and cryptic intermediate (Scheme 14);463–465 additional BGCs were later identified.466 This intermediate is generated by the Mg2+-dependent C3-geranylation of cis-indolylvinyl isonitrile by the ABBA PT FamD2/AmbP1 or its homologues.465,467 A family of Stig cyclases, a type of non-canonical TSs,27 then catalyze regio- and stereoselective cyclization cascades to yield the tri- and tetracyclic scaffolds of the hapalindoles or fischerindoles. These fascinating terpene cyclization reactions follow a Cope rearrangement, stereoselective 6-exo-trig cyclization, and regioselective carbocation quench by electrophilic aromatic substitution (at C2 or C4 of indole) or deprotonation (Scheme 14).465,466,468,469 Other characterized tailoring enzymes include a non-heme iron aliphatic halogenase, AmbO5, for C13 chlorination,464,470 another ABBA PT, AmbP3, for ambiguine C2 prenylation,463 and an N-MT for the welwitindolinones.464 Other cyclization reactions, such as the C-3–C-11 fusion in ‘Group 3’ hapalindolinones, the unusual ring expansions in 601 and 602, and the remaining tailoring steps including isothiocyanate formation and various oxidations are currently undetermined.
Scheme 14.
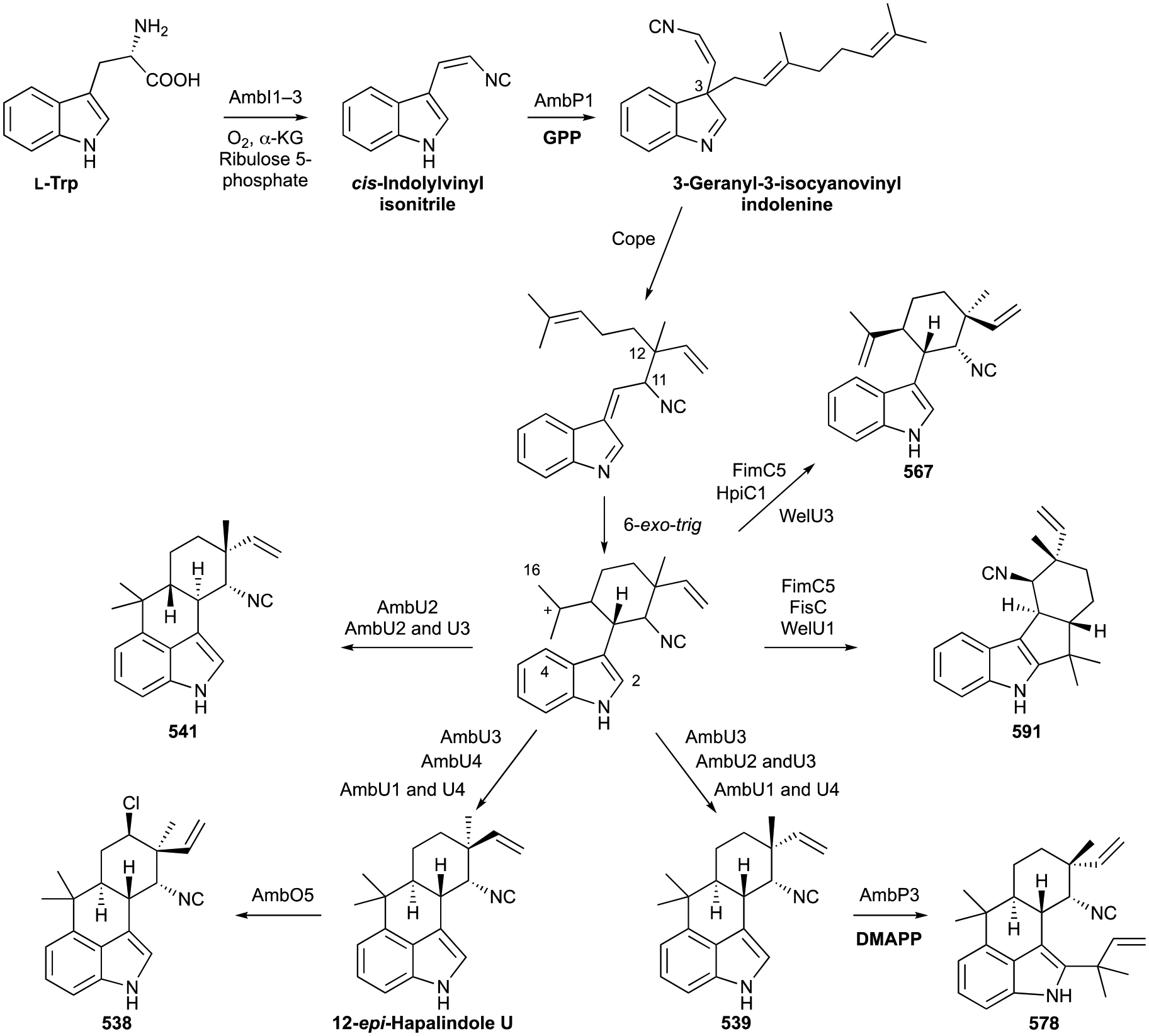
Biosynthesis of hapalindoles 538, 539, 541, and 567 and ambiguines 578 and 591.
Biological activity.
The stigonematalean indole alkaloids have an amazing abundance of biological activities. They are known to have antimicrobial (antibacterial, antifungal, and antialgal) and anticancer properties, and have toxicity to plants, insects, and animals. The biological activities of these hapalindole-like alkaloids have been extensively reviewed.443,444
First isolated for their antialgal properties,445 most of the hapalindoles and ambiguines have antimicrobial properties.445,449,454,456,457,459,460,462 Active against mainly Gram-positive bacteria including M. tuberculosis, Bacillus anthracis, and S. aureus, as well as fungi C. albicans and Saccharomyces cerevisiae, most MIC values range from low to sub mM. It was later found that 569 competitively inhibits bacterial RNA polymerase, although given its lack of potency (Ki = ~1 mM) additional targets are expected.471,472
All four classes of indole alkaloids have members with weak to moderate levels (IC50 values of ~10–100 μM) of human cell cytotoxicity.449,459,460 Several molecular targets and modes of action have been described for these activities. 582 inhibited NF-κB at an IC50 value of 30 nM, arrested cells in the G1 phase, and led to apoptosis in breast cancer cells.473 Welwitindolinones inhibit P-glycoprotein-induced multi-drug resistance in human ovarian adenocarcinoma at concentrations as low as 100 nM and have antiproliferative effects associated with microtubule depolymerization.474,475 Some hapalindoles are sodium channel-modulating neurotoxins with a recent report showing that hapalindoles also inhibit T cell proliferation, with 531 being the most potent (IC50 = 1.56 μM).450,476
Some alkaloids were also reported to be insecticides, phytotoxins, and teratogens. Hapalindoles killed fly and mosquito larvae at μM concentrations and 548 and 576 were toxic to developing zebrafish embryos.444,448,477 579 caused oxidative stress in lettuce via reactive oxygen species (ROS) generation, which led to lipid peroxidation and inhibited plant mitosis.478
3.3.3. Xiamycins
A structurally and biosynthetically unique family of indolosesquiterpenoids are the xiamycins. Carbazoles, tricyclic alkaloids with two benzenes flanking a pyrrole, are mainly found in plants, although they have also been isolated from algae, fungi, ascidians, and bacteria.479 Most xiamycins have 6/5/6/6/6 pentacyclic skeletons and are found in either monomeric or dimeric form. Xiamycin A (613) and its methyl ester (614) from Streptomyces sp. GT2002/1503 and oridamycins A and B (615, 616) from Streptomyces sp. KS84, essentially discovered concurrently, were the first examples of this class of bacterial NPs.480,481 Fused to the carbazole of 613 is a dimethyl decalin ring functionalized with hydroxyl and carboxylic acid groups. 615 is a C16 epimer of 613. Other monomeric xiamycins, all from Streptomyces spp., have added hydroxyl (617–619), keto (620), or chlorine (621) functionalities at positions C17 or C19.482–484 Indosespene (622) sespenine (623), and oxiamycin (624) are all xiamycin variants, but do not possess the 6/5/6/6/6 pentacyclic scaffold.482,483 622 is a seco xiamycin intermediate with only a single connection between the indole and decalin ring systems; 624 has a seven-membered 2,3,4,5-tetrahydrooxepine ring fused to the carbazole; 623 has a rearranged skeleton with a tetrahydroquinoline-containing 6/6/6/6/6 pentacycle similar to the fungal NPs nominine and aspernomine.485,486
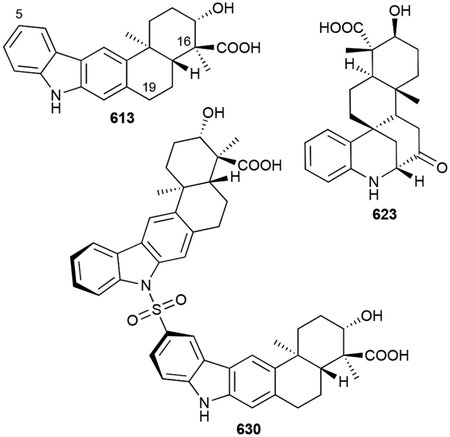
Numerous xiamycin dimers have also been isolated from both native strains and in heterologous hosts harboring the entire xia BGC. Dixiamycins A and B (625, 626), N-N coupled dimers of 613, were originally discovered in the deep-sea Streptomyces sp. SCSIO 02999; this was the first example of a naturally occurring atropdiastereomeric N-N axis between carbazole nitrogens.483,487 These two dimers were reisolated, along with N-C19 atropodiastereomers (627, 628) and a non-atropisomeric dimer bearing an N-C6 (C5 of indole ring) linkage (629), from xia-expressing Streptomyces albus.488,489 Further MS analysis revealed the presence of three sulfonyl-containing dimers and ensuing full spectroscopic characterization confirmed the sulfonyl bridges of sulfadixiamycins A–C (630–632) were located between N-C6, C6-C6, and C6-C19, respectively.490
Biosynthesis.
The biosynthetic pathway of the xiamycins was systematically unveiled through a series of genetic and biochemical experiments, revealing several unique features. Two xia BGCs, simultaneously identified and independently named, were reported.488,491 Initial bioinformatic analysis revealed the xia BGC did not encode canonical PT or TS genes. XiaM/P, which by protein sequence appears to be a polyprenyl synthase, is proposed to farnesylate indole or a related precursor. Terminal epoxidation sets up a type II TS cyclization reaction catalyzed by the integral membrane cyclase XiaE/H (Scheme 15).488,491 Triple hydroxylation of the methyl on preindosespene by the P450 XiaJ/M yields 622492 and suggests that a homologous reaction in oridamycin biosynthesis may occur on the methyl group on the opposite face of the decalin. A second cyclization reaction, catalyzed by the flavin-dependent oxidocyclase XiaF/I, another noncanonical TS,27 occurs via cryptic hydroxylation at the C3 position of indole and nucleophilic attack by the exocyclic methylene.493 Divergent phenyl migration or dehydration and spontaneous aromatization yield 623 or 613, respectively (Scheme 15).488,491,493 Dimerization, for both the xiamycins and the sulfadixiamycins, occurs due to a suspected radical mechanism facilitated by the flavoenzyme XiaH/K; the role of XiaH/K was confirmed by gene knockout, genetic complementation, and biotransformation experiments.489,490 Endogenous sulfur dioxide is suspected to react with the initial carbazole radical, forming a sulfonyl radical that can pair with another carbazole radical.490 624 is also a product of XiaH/K acting on 613, perhaps occurring via a hydroperoxide radical rearrangement (Scheme 15).489
Scheme 15.
Biosynthesis of xiamycin A (613), oxiamycin (624), dixiamycin A (625), and sespenine (623).
Biological activity.
Xiamycins are antibacterial antivirals. 613 and 614 were discovered as selective anti-HIV NPs (IC50 values >10 μM);480 619 and 620 were later seen to inhibit the replication of coronaviruses with EC50 values between 1 and 3 μM.484 As this activity was reported prior to the severe acute respiratory syndrome–coronavirus 2 (SARS-CoV-2) outbreak in 2019, it is unclear if xiamycins would be effective against SARS-CoV-2. The antibacterial properties of the monomeric xiamycins are fairly weak, although the dixiamycins and sulfadixiamycins were a little more effective (MIC range of 4–25 μg mL−1) against S. aureus, B. subtilis, E. coli, and Mycobacterium vaccae.482,483,490 615 was also initially reported as an antifungal against the freshwater mold Saprolegnia parasitica with an MIC value of 3 μg mL−1, although most xiamycins are not cytotoxic to other eukaryotes.481
3.3.4. Miscellaneous polycyclic indoles
Another family of terpenoid-carbazole NPs are the tricyclic neocarazostatins, carquinostatins, and lavanduquinocin. These carbazoles, however, are biosynthetically distinct from the xiamycins. Their carbazole skeleton does not include an isoprenoid component; instead, their prenyl units are located at C5 of the indole core.
Neocarazostatins A–C (633–635), from Streptomyces sp. GP 38, were the first of this family to be discovered.494 They are 5-dimethylallylcarbazoles with an alkylated ortho-hydroquinone unit. The carquinostatins A and B (636, 637) were structurally identical to the neocarazostatins except for their ortho quinone functionality.495,496 Lavanduquinocin (638), isolated from S. viridochromogenes 2942-SVS3, was 636 with cyclolavandulyl replacing the dimethylallyl group.497
An unusual bacterial indole alkaloid, amycocyclopiazonic acid (639) from the sponge-associated Amycolatopsis sp., is a 6/5/6/5/5 pentacycle with a C4-dimethylallyl moiety contributing to the polycyclic skeleton.429 Prior to this discovery, cyclopiazonic acids were a class of indole alkaloids known in fungi, but not in bacteria.
Biosynthesis.
The neocarazostatin class of carbazoles are built from four building blocks: Trp, pyruvate, polyketide, and isoprenoid. Based on the C5-dimethylallyl group, the BGC was first identified after mining for IptA homologues.498 However, after no homologues were found in the producing strain Streptomyces sp. MA37, a thiamine pyrophosphate-dependent enzyme responsible for C–C bond formation using pyruvate was used to identify the nzs BGC. Directly adjacent to this target gene was a gene bioinformatically predicted to encode a phytoene synthase, NzsG. NzsG is the C5-carbazole PT and suggested an emerging type of phytoene synthase-like PTs.498 Later biosynthetic studies on the carquinostatin BGC completed the pathway by the realization of an unprecedented oxidative cyclization reaction catalyzed by a novel enzyme following the condensation of an indole-3-(α-hydroxyl-β-keto)-butanoic acid and 3-hydroxylbutyryl-ACP.499 Prenylation and P450 hydroxylation both occur as late-stage tailoring reactions.498
Biological activity.
Like the benzastatins, the neocarazostatin, carquinostatin, and lavanduquinocin family of carbazoles are potent radical scavengers. They showed strong sub-μM inhibitory activities against lipid peroxidation and prevented glutamate toxicity in neuronal cells (EC50 values as low as 3.1 nM).494–497
3.4. Nucleobases and nucleosides
Prenylated nucleobases and nucleosides are a relatively small family of NPs and can be classified into cytokinins, cyclic terpenoid adenosines, and uridine ethers. Cytokinins are important phytohormones that regulate plant growth, development, and reproductive competence.500 Isoprenoid cytokinins, which all have a dimethylallyl moiety attached at N6 of adenine or adenosine, are one of the most abundant classes of natural cytokinins and are ubiquitous in plants, but also produced in certain bacteria.500 Isoprenoid cytokinins have had many names and abbreviations throughout the last 65 years; for naming simplicity, we chose to use the zeatin nomenclature in this review.
Bacterial zeatins were first found and structurally characterized in the Gram-positive phytopathogen Corynebacterium fascians (now named Rhodococcus fascians). Deoxyzeatin (640), the simplest zeatin with an unmodified dimethylallyl moiety attached to adenine, was the first zeatin identified in bacteria.501 Numerous other zeatins, many of which were originally identified in plants, were later identified in species including R. fascians, Pseudomonas amygdali, and Pseudomonas syringae. The dimethylallyl moiety can be present in its hydroxylated, reduced, and/or methylated forms giving cis- or trans-zeatins (641, 642), dihydrozeatin (643), or 1´-methylzeatin (644).502–504 The C2 position on adenine can also possess a 2-methylthio functional group (645).505 These terpenoid-adenine hybrids are also commonly present in bacteria as terpenoid-adenosines, named here zeatin ribosides (646–654).505–511
There are three additional N6-prenylated adenosines. Sorangiadenosine (655) and its 2-hydroxyl analogue (656), produced by the myxobacterium S. cellulosum KM 1003, have bicyclic eudesmane sesquiterpenoid moieties.512,513 N6-Tuberculosinyladenosine (657) has an appended tuberculosinyl skeleton (vide supra chapter 2.4.8) and is one of two diterpenoid-adenosine hybrids found in M. tuberculosis.283 The other, 1-tuberculosinyladenosine (658), is found in significantly higher quantities and is likely the major biosynthetic product.514
Farnesides A and B, JBIR-68, and simamycin (659–662) compose the final class of 5´-O-prenylated nucleosides.515–517 All found in Streptomyces, they have linear geranyl or oxidized farnesyl units on uridine or dihydrouridine nucleosides.
Biosynthesis.
Isoprenoid cytokinins can be biosynthesized de novo or from the degradation of prenylated tRNA molecules.518 De novo biosynthesis in bacteria is only known to occur via the N-prenylation of AMP, catalyzed by isopentenyltransferases such as the isozymes Tmr and Tzs from Agrobacterium tumefaciens.519–522 Interestingly, these enzymes can utilize DMAPP or its 5-hydroxyl variant to directly produce deoxy-or trans-zeatin glycosides.521 The cytokinin BGC in R. fascians also encodes a P450 and phosphoribohydrolase that facilitate hydroxylation of deoxyzeatin and ribosyl hydrolysis, respectively, although the former has not been biochemically characterized.523,524 Plant cytokinin biosynthesis is quite similar, although the PTs prefer ATP and ADP as prenyl acceptors.525 This suggests that prenylation of ATP or ADP in bacteria is also a possibility.
Posttranscriptionally modified tRNA molecules can also be degraded into their respective monophosphate nucleotides.518 The prenylation of preformed tRNA molecules, specifically the adenine adjacent to the 3´-end of the anticodon, is catalyzed by a tRNA dimethylallyltransferase.526–528 This tRNA modification stabilizes the anticodon loop, enhances codon:anticodon recognition, and has been linked to the fidelity and speed of translation.529 Other modifications on prenylated tRNA substrates, including isoprenyl hydroxylation and 2-methylthiolation, provide the precursors for the structural diversity seen in the zeatin family of NPs.530,531
The biosynthesis of the four cyclic terpenoid-adenosines follows a two-step process of initial terpene cyclization followed by prenylation. To account for the discovery of 658, after the identification of the Rv3377c and Rv3378c TSs for the production of 341, the biosynthetic pathway was revised.514 Rv3378c, which is required for the biosynthesis of 658, does not simply perform an elimination reaction after diphosphate abstraction as first reported;280 Rv3378c is in fact a unique cis-PT with two substrate binding pockets that catalyzes the prenylation of adenosine at N1.514 Sorangiadenosines 655 and 656 are proposed, based on bioinformatics, to be formed from the condensation of adenosine with a preformed 3,11-eudesmadiene, although in light of the activity of Rv3378c, it would be reasonable to consider that one enzyme catalyzes both terpene cyclization and prenylation of adenosine via attack on a cationic intermediate.513 Supplementation of labeled acetate in S. cellulosum, which mainly utilizes the MVA pathway for terpene precursor biosynthesis, did not result in isotopically-labeled sorangiadenosines.513 However, labeled dimethyl acrylic acid was incorporated into 655 and 656 supporting that their terpenoid moieties originate from the myxobacteria leucine degradation pathway, a pathway that branches off the MVA pathway; the genome of S. cellulosum was found to encode all proteins required for this alternative route.513,532
Biological activity.
The cytokinin phytohormones work in concert with the auxins to promote plant cell proliferation and development.533,534 Cytokinins target a signal transduction pathway and are thus active at very low concentrations. In phytopathogenic bacteria, they act as virulence factors directing plant growth into a preferred infectious niche.535
658, a virulence factor from M. tuberculosis, has the unique ability to disrupt phagolysosomes.283 Acting as an antacid because of its basic properties, 658 selectively accumulates in the acidic compartment, neutralizing the pH and causing swelling and structural remodeling of the lysosome. N6-Tuberculosinyladenosine (657) is a weak base and thus not effective.283
The remaining terpenoid-nucleosides have weak to moderate activities against bacteria, influenzae plaque formation, and Plasmodium falciparum.512,513,517,536
3.5. Peptides
The posttranslational prenylation of proteins is a well understood and biologically important transformation537 and therefore it is not surprising that there is a family of prenylated peptidyl NPs. There are at least three types of peptide NPs in bacteria: ribosomally synthesized and posttranslationally modified peptides (RiPPs), nonribosomal peptides, and cyclodipeptides constructed by tRNA-dependent cyclodipeptide synthases (CDPSs). Prenylation has been seen on all three types of peptides and can be the result of C, N, or O-prenylation in either normal or reverse fashion. The prenylation of peptides increases the lipophilicity of the compounds, provides additional structural aspects for further modifications and cyclizations, and impacts both biological and pharmacological properties.538
3.5.1. Ribosomal peptides
The ComX pheromones are a group of quorum-sensing signals produced by Bacillus subtilis (663–669).539–542 These ribosomally synthesized peptide NPs stimulate natural competence in crowded microenvironments.543,544 The structures of the ComX pheromones are divergent in their polypeptide sequences, but typically range from six to twelve amino acids.545 Conserved amongst all ComX pheromones is an internal Trp residue that is C3 prenylated and cyclized into a 6/5/5 tricyclic structure. The ComXRO-E-2 pheromone (GIFW*EQ, 663) was the first to be structurally defined and had a normal geranyl moiety;539 the Trp residue in ComXRO-C-2 (TREW*DG, 666) was farnesylated.540
Cyanobactins are ribosomally-derived peptides, either in macrocyclic or linear form, and are estimated to be produced by up to 30% of all cyanobacteria.546 There are over 100 known cyanobactins, but only a handful of prenylated cyanobactins. Linear cyanobactins consist of the muscorides, aeruginosamides, and viridisamide. Discovered from Nostoc muscorum, muscoride A (670), a linear tetrapeptide containing two contiguous methyloxazoles, was the first reported linear cyanobactin.547 Both its N- and C-termini were protected with reverse and normal dimethylallyl moieties, respectively. Later genome mining and heterologous expression of the mus BGC revealed muscoride B (671), an oxazole-containing pentapeptide with normal dimethylallyl moieties on both its termini.548 The aeruginosamides (672–674) from Microcystis aeruginosa and viridisamide A (675) from Oscillatoria nigro-viridis PCC 7112 are all prenylated peptides with a methyl thiazole-4-carboxylate moiety.549,550 674 has a unique diisoprenyl N-terminal amine.
Macrocyclic cyanobactins have prenyl groups appended onto aromatic side chains. Kawaguchipeptin A (676) is a diprenylated cyclic undecapeptide from M. aeruginosa (NIES-88).551 Like the ComQ pheromone, its two Trp residues have prenylated 6/5/5 tricyclic skeletons, although both Trps were modified with dimethylallyl units. Prenylagaramides A and B (677, 678), isolated from cyanobacteria Oscillatoria agardhii (NIES-205 and NIES-596), are cyclic nona- and heptapeptides that have the rarely seen O-dimethylallyltyrosine side chain;552 aestuaramide A (679) is a heptamer with a reverse prenyl unit on its Tyr.553 Later genome mining afforded the related prenylagaramide C (680) and sphaerocyclamide (681).554,555 Croissamide (682) and trikoramide A (683) from the Symploca genus are Pro-rich cyclic peptide with N1-reverse and C3-normal prenylated Trp units.556,557 Unsurprisingly, the C3 prenylated Trp in 683 formed a 6/5/5 tricycle.
Biosynthesis.
The ComX pheromones require two posttranslational maturation steps. The com BGC consists of two key biosynthetic genes.543,558 The precursor peptide, encoded by comX, is modified by ComQ.558 ComQ is not homologous to ABBA PTs; instead it shows homology to polyprenyl diphosphate synthases. The internal Trp residue of the immature ComX peptide is prenylated at C3 attendant with a cyclization between the amide N and C2 forming a hexahydropyrrolloindole moiety (Scheme 16). The N–C2 cyclization is the result of an intramolecular capture of an imine tautomer created by the prenylation of C3.425 A proteolysis reaction must occur to generate the mature ComX hexapeptide containing the geranylated hexahydropyrrolloindole, although the timing and responsible enzyme(s) for this posttranslational modification is unclear.
Scheme 16.
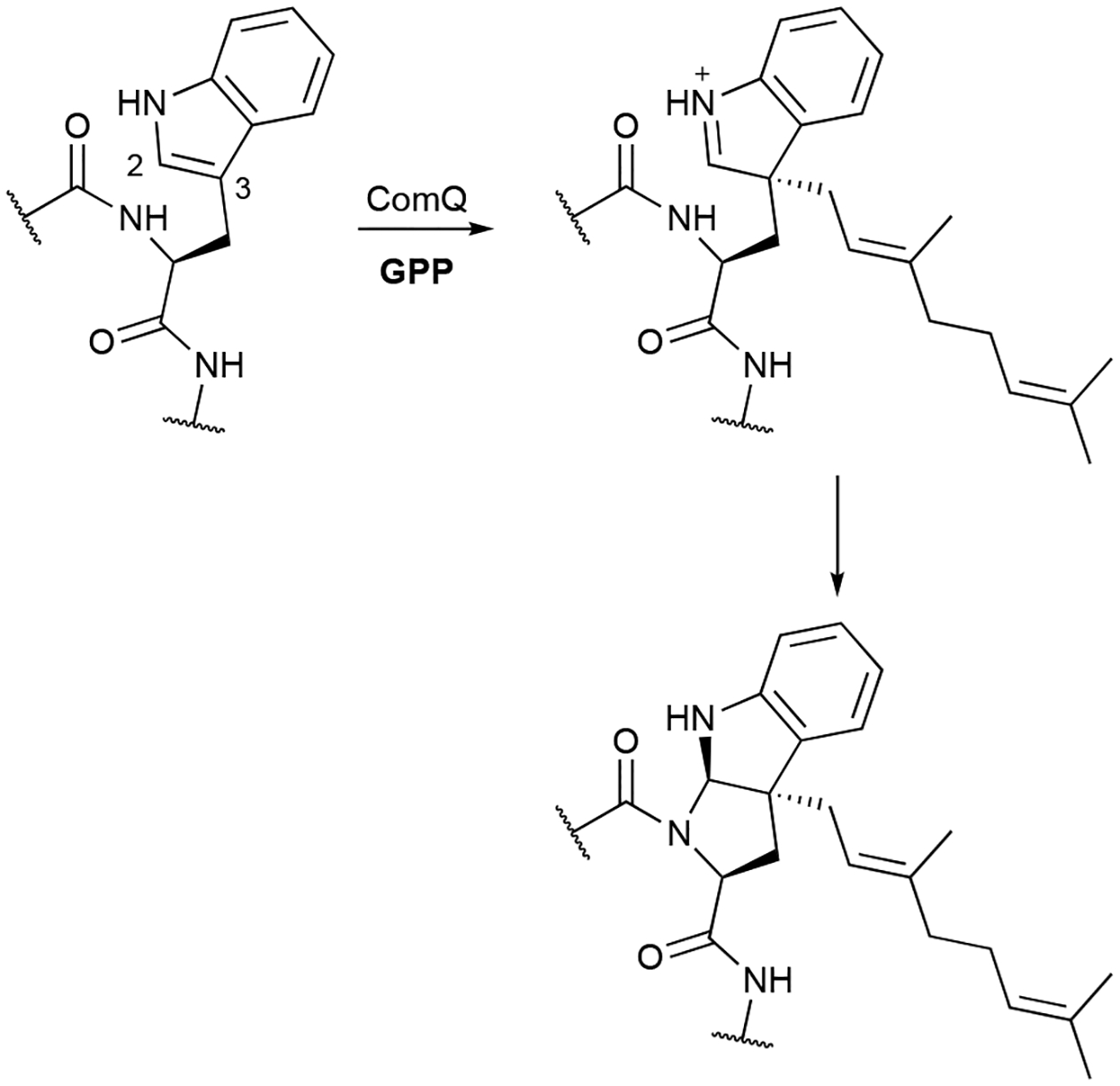
Prenylation and cyclization of ComX pheromones.
The related cyanobactin PT, KgpF, was found to catalyze a similar C3 normal prenylation reaction on two Trp residues of kawaguchipeptin B, yielding 676.559 KgpF can prenylate both linear and cyclic peptides, but only diprenylates kawaguichipeptin B. Although the reaction and subsequent cyclizations of the Trp residues are the same for KgpF and ComQ, the stereochemistry of the tricyclic structures differs.560
The discovery of the prenylagaramide BGC (pag) led to the realization that cyanobactin BGCs can encode many different precursor peptides.554 A later systematic survey of cyanobacteria expanded on the impressive biosynthetic potential of cyanobactin production.550 Regarding peptide prenylation, a majority of cyanobactin BGCs have at least one PT, which are phylogenetically related to the ABBA PTs, encoded within. LynF, a cyanobactin PT homologue from Lyngbya aestuarii, was the first ribosomal peptide prenyltransferase to be biochemically characterized.561 At the time, there were no known cyanobactin NPs from the lyn pathway, yet LynF performed a reverse O-prenylation on a Tyr-containing substrate mimic. A spontaneous Claisen rearrangement then provided a C3-dimethylallyl-Tyr moiety. The later discovered 679, along with several other aestuaramides that were only tentatively identified by MS/MS, appear to support this initial reverse O-prenylation followed by Claisen rearrangement.553 On a side note and perhaps biosynthetically related, 3-prenyl-l-tyrosine (684) and its acetylated derivative (685) were isolated as NPs from Streptomyces sp. IFM 10937.562 A structural study of PagF, a LynF homologue from the pag BGC, revealed that the cyanobactin PTs are modified ABBA PTs with a truncated barrel fold, paving the way for the large peptide substrates to bind and help create a catalytically competent active site.563
This subfamily of ABBA PTs also N- and O-prenylates the termini of linear cyanobactins. Two PTs from the muscoride BGC (mus), MusF1 and MusF2, have substrate and reaction specificity.548 MusF1 catalyzes normal prenylation on the C-terminus, while MusF2 catalyzes either normal or reverse prenylation on the N-terminus, depending on its peptide substrate. A unique bifunctional MT-PT protein, AgeMTPT, is responsible for the protection of both termini in aeruginosamide biosynthesis by catalyzing both N-terminal prenylation and C-terminal methylation reactions.564
Biological activity.
Cyanobactins have potential as drug leads based on their antimalarial and antitumor activities, as well as their ability to reverse multidrug resistance, although not all cyanobactins have known bioactive properties.546,552 Aeruginosamides 672–674 and trikoramide 683 both have low μM IC50 values against various cancer cell lines.549,557 Muscorides 670 and 671 are weak antibacterial agents against Gram-positive strains.547 685 had mild synergistic activity in sensitizing TNF-related apoptosis-inducing ligand (TRAIL)-resistant human gastric adenocarcinoma cells.562
3.5.2. Nonribosomal peptides
Both macrocyclic and linear peptides are also formed by non-ribosomal peptide synthetases (NRPSs). These NPs commonly have non-proteinogenic amino acid components, including prenylated tryptophans. Cyclomarins A–D (686–689) from Streptomyces sp. CNB-982 and Salinispora arenicola CNS-205 are cyclic heptapeptides with four unusual amino acids.565,566 One of these residues is a N-reverse prenyl β-hydroxytryptophan, which is either epoxidized in A and B or left unmodified in C and D.
The ilamycins are cycloheptapeptides containing non-proteinogenic amino acids including 2-amino-4-hexenoic acid, 3-nitro-Tyr, and N-dimethyallyl-Trp. Several members of this family, including B1, B2, C1, C2, and D (690–694), were originally discovered as ilamycins from Streptomyces islandicus and Streptomyces atratus ATCC 14046 and later isolated as rufomycins from Streptomyces macrosporeus DSM-12818.567–573 As with the cyclomarins, the reverse N-prenyl group was found in both unmodified (B1) or epoxidized forms (B2, C1, C2, D). The absolute configurations of the epoxidized ilamycins were not confirmed until they were rediscovered, along with ilamycin E1 (695) from S. atratus SCSIO ZH16 in a genomic and biosynthetic study.574 Eight new rufomycins, NBZ1–NBZ8 (696–703), and a chlorohydrin analogue (704) from S. atratus MJM3502 were found.575
The cyanobacterial aeruginoguanidines and microguanidines are exceptional NPs. They are linear peptides containing prenylated N-methyl-Arg residues and a trisubstituted phenyl trisulfate. Aeruginoguanidines 98-A–98-C (705–707) from M. aeruginosa (NIES-98) have two Arg residues that have dimethylallyl, neryl, or modified neryl units attached to either one or both guanidium side chains.576 The microguanidines, in either ester (708, 709) or amide (710, 711) form, only have one prenylated Arg with dimethylallyl, geranyl, or modified geranyl units.577,578
Biosynthesis.
The biosynthesis of prenylated nonribosomal peptides follows canonical megasynthetase assembly line biosynthesis. The reverse N-dimethylallyl-Trp residues are biosynthesized prior to incorporation into the growing polypeptide, or in the case of cyclomarins and ilamycins/rufomycins, prior to being used as the starter unit.566,574 CymD, the ABBA PT from cyclomarin and cyclomarazine biosynthesis, confirmed its N1 prenylation of free l-Trp.579 The epoxidation of the dimethylallyl moiety occurs late, if not last, in biosynthesis by associated P450s.566,574
Currently, the biosynthesis of aeruginoguanidines and microguanidines is only proposed based on bioinformatics analysis of the several homologous BGCs.578 An NRPS condenses either one or two l-Arg residues with the unusual phenyl building block, which is proposed to be biosynthesized from isochorimate through a substituted phenylpyruvate intermediate.578 Both prenylation, likely catalyzed by the undecaprenyl diphosphate synthase-like PT AgdJ, and sulfation are proposed to occur after the (depsi)peptide is formed.578
Biological activity.
Prenylated nonribosomal peptides of bacterial origin are antitubercular and cytotoxic NPs. Cyclomarins 686–689, whichh are cytotoxic to human cells with a mean IC50 of ~2 μM,565,566 do not show antibacterial activity against any tested bacteria except M. tuberculosis. 686 kills Mtb persistor cells by targeting and activating the ClpC1 subunit of caseinolytic protease.580,581 This activation leads to uncontrolled protein degradation and thus cell death;582 it is bacteriocidal at 0.3 μM.580 The ilamycin family of peptides are also potent antitubercular compounds, with MICs as low as 10 nM.574,575,583 Ironically, their activity is due to inhibition of ClpC1.584 The cytotoxicity of ilamycins, which is also in the low μM range, is at least partially controlled by the induction of apoptosis by the down-regulation of the anti-apoptotic protein Bcl-2; ilamycin C additionally suppresses the IL-6/STAT3 pathway leading to the inhibition of migration and invasion in triple-negative breast cancer cells.585,586 Aeruginoguanidines 705–707 and microguanidines 708–711 have, at best, moderate cytotoxicities (IC50 = 25–50 μM).576,577
686 was also shown to have anti-inflammatory and quite potent antiplasmodial activity;565,587 686 prevents growth of P. falciparum (IC50 = 40 nM) by inhibiting the formation of the enzyme-substrate complex of diadenosine triphosphate hydrolase PfAp3Aase.587
3.5.3. Teleocidins
The teleocidins are indolactam-terpenoid hybrid NPs isolated from both cyanobacteria and actinobacteria. The overall structure of teleocidins consists of three components: an indole ring, an l-Trp-l-Val derived nine-membered lactam, and a terpenoid moiety appended to the indole. The teleocidins can be further divided into three major categories depending on its terpenoid moiety. Teleocidins A and lyngbyatoxins have linear geranyl (C10) substituents at C7 of the indole ring, teleocidins B and olivoretins have cyclic C11 terpenoid units fused to C6 and C7, and pendolmycins have linear dimethylallyl (C5) moieties at C7. Although the teleocidins are nonribosomal peptide meroterpenoids, we distinguish them in their own section to highlight their structures, biosynthesis, and activity.
‘Teleocidin’ was discovered from Streptomyces mediocidicus over 60 years ago as a toxic substance to fish.588,589 The structural and stereochemical complexity of ‘teleocidin’ resulted in a series of reports culminating in an X-ray analysis of a hydrogenated bromoacetate derivative that provided full structural elucidation of ‘teleocidin B’.590,591 Later chromatographic studies revealed that ‘teleocidin’ is a complex mixture of six distinct compounds.592 Teleocidins A-1 (712), which was also isolated from the blue-green alga Lyngbya majuscula, named lyngbyatoxin A, and previously determined to have a reverse geranyl moiety at C7,593 and teleocidin A-2 (713) are C19 epimers.594 Teleocidins B-1–B-4 (714–717), which all contain 1,4-tetrasubstituted cyclohexanes fused to C6 and C7 of indole at its C2 and C3 positions, are diastereomers with the four possible stereochemical combinations at C19 and C25.592 Olivoretins A–C (718–720) were concurrently reported as methyl ether variants from Streptoverticillium olivoreticuli; olivoretin D (717) is identical to teleocidin B-4.595,596 720, its desmethyl analogue (721), and olivoretin E (722) were structurally distinct as their terpenoid connection patterns were reversed.596–598 Blastmycetin E (723) from Streptoverticillium blastmyceticum NA34–17 is also structurally divergent.599 Although it has a C11 group at C7, the other end is fused to N1 creating another nine-membered ring; it also possesses a Z olefin and its C11 unit has an isobutyl functionality, similar to that of 722.
Pendolmycin (724) and (–)-7-geranylindolactam-V (725) are teleocidin A analogues with reverse dimethylallyl and normal geranyl moieties, respectively.600,601 There are only a few derivatives with variations in the peptidic core: methylpendolmycin (726) has an l-Ile in place of l-Val and 12-epi-lyngbyatoxin A (727) has a d-Val residue.602,603 As would be expected, there are many other oxidized (728–735), N-desmethyl (736–738), O-methyl (739), O-acetyl (A2-740, B3-741), and O-glycosylated (742, 743) teleocidins.515,598,599,604–613
Biosynthesis.
The biosynthesis of the teleocidins has been extensively studied and was recently reviewed, although some questions still remain.614,615 Given the variety of prenyl groups (i.e., C5 and C10) attached at C7 of the indole core, it was evident that these moieties, even those with an additional carbon (C11), were of terpenoid origin; these groups were later confirmed to be of MEP origin.616 The lyngbyatoxin A BGC, ltx, from L. majuscula was found by probing a fosmid library for putative NRPS adenylation domains.617 Three enzymes, an NRPS (LtxA), an MbtH/P450 (LtxB), and an ABBA PT (LtxC), were proposed to form the N-methyl-l-valine-l-tryptophanol dipeptide, catalyze macrocyclization to form indolactam V, and reverse geranylation at C7 yields 712, respectively (Scheme 17).617 Follow-up biochemical experiments later confirmed that LtxA uses a four-electron reduction to release the dipeptide,618 LtxB is a C–N bond forming P450,619,620 and LtxC selectively forms 712.617 When ltx was reported, LtxD, an oxidase/reductase was proposed to hydroxylate the geranyl moiety giving lyngbyatxins 729 and 730, although this has not been confirmed.
Scheme 17.
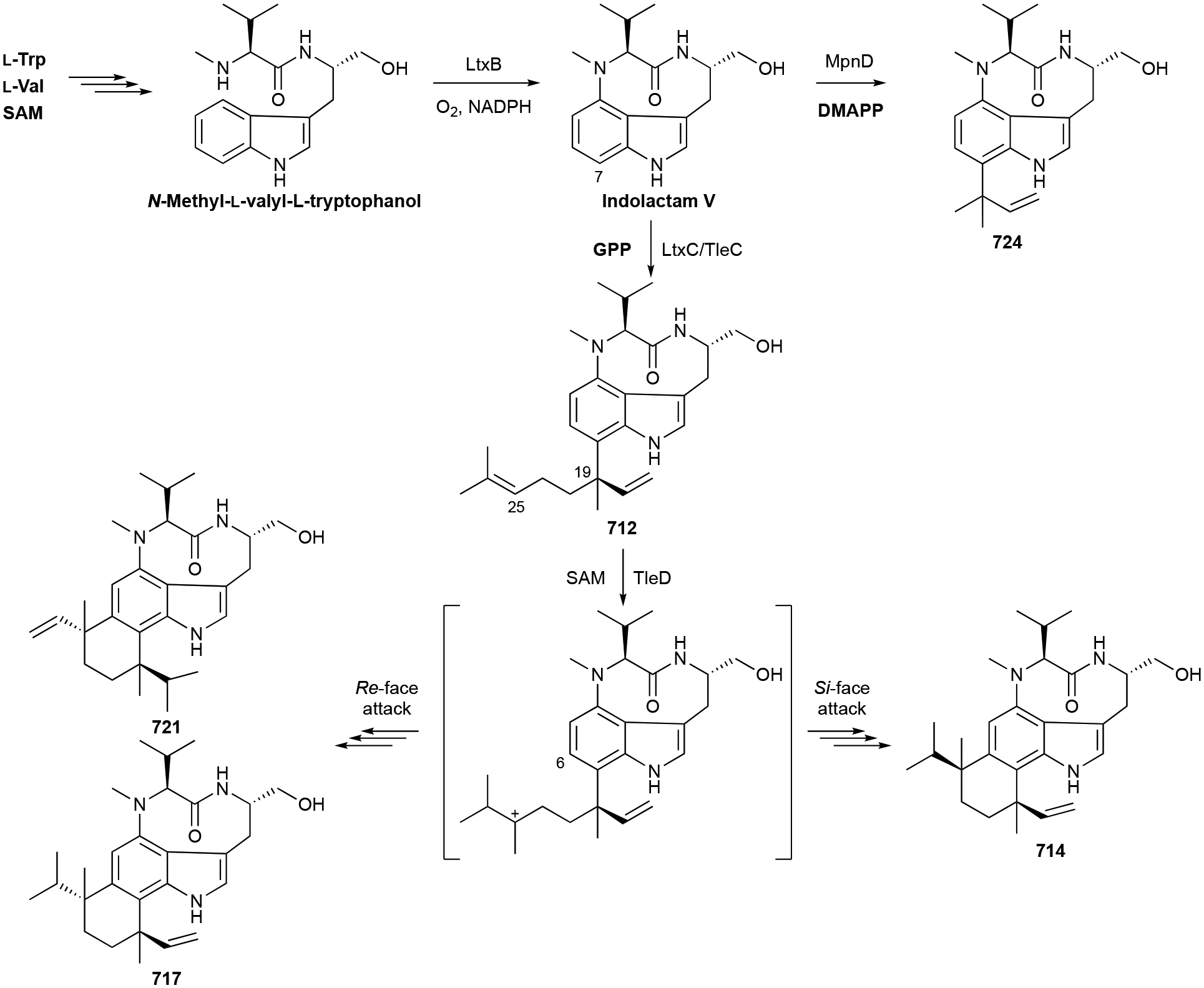
Biosynthesis of lyngbyatoxin A (712), teleocidin B-1 (714), teleocidin B-4 (717), des-O-methylolivoretin C (721), and pendolmycin (724).
With 712 proposed as the precursor to teleocidins B (i.e., 714–717),617 the search for the MT and TS responsible for the modification of the C7 geranyl unit began. The tle BGC from Streptomyces blastmyceticus NBRC 12747 was identified, had the same three biosynthetic proteins from the ltx BGC, and heterologous expression of tleABC produced 712.621 With no MTs or TSs encoded nearby, a genome-wide search for MTs resulted in the discovery of TleD. Although located outside of the tle BGC, TleD catalyzed both C25 methylation and subsequent ring closure via fusion to C6 of indole (Scheme 17). This unusual mechanism of methylation-induced terpene cyclization is proposed to include a 1,2-hydride shift, C7 nucleophilic attack, and a final 1,2-alkyl shift to C6.614,615,621 The various fusion patterns and diastereomers (e.g., 714, 717, 721) are formed depending on which carbon shifts and which face of the carbocation is attacked, although a post-methylation nonenzymatic cyclization has not been ruled out.614
During the search for TleD, another MT encoded outside of the tle BGC was found to be responsible for the methyl ether of olivoretins.621 The (methyl)pendolmycin BGC, mpn, was also found to be highly homologous to the ltx and tle BGCs, with the major differences being that the NRPS can incorporate both l-Val and l-Ile and the ABBA PT MpnD is selective for C5 prenylation; 724 is formed directly from indolactam V by MpnD (Scheme 15).622 Enzymes responsible for other variations, whether in terpenoid attachment or teleocidin modification, remain enigmatic.
Biological activity.
Teleocidins are potent activators of protein kinase C (PKC). PKC isozymes play important roles in both normal and disease physiologies by controlling the phosphorylation states of protein within signal transduction cascades.623 Overactivation of PKC leads to tumor cell proliferation as well as numerous other diseases including psoriasis, diabetes, heart disease, and autoimmune and neurological diseases.624
The original ‘teleocidin’ was initially found as a toxic substance to aquatic organisms, nematodes, and mice; it was also a mammalian skin irritant.588,589 Teleocidins are not toxic to microorganisms,588,589 although the methylpendolmycins are active against P. falciparum with IC50 values ranging from 5 to 20 μM.613 The mammalian toxicities of 712 and ‘teleocidin B’ were later determined to be approximately equal with LD100 values in mice of ~0.3 mg kg−1.593 Soon after, the teleocidins were found to be potent tumor promoters with concentrations required for induction only 2–10 nM.625,626 Tumor stimulation occurs via PKC activation at nM to μM concentrations.627–629 While an intact indolactam ring is required for the activation of PKC,603,610,611,628 longer terpenoid moieties improve potency, as evidenced by the various analogues.601,606,628,630 713 was also found to be a proteinase-activated receptor 2 antagonist in the low nM range, interfering with various cellular events including cancer cell migration.629
3.5.4. Diketopiperazines
Another structural family of bioactive peptides produced by microorganisms are the 2,5-diketopiperazines (DKPs). These cyclic dipeptides are known to be synthesized by either NRPS or CDPS systems and commonly have associated tailoring enzymes such as PTs and P450s.631 As of now, all bacterial prenylated DKPs have tryptophan residues. Cyclomarazines A and B (744, 745), produced by S. arenicola, are likely premature NRPS truncation products of the cyclomarins.566 Rufomyazine (746), found in Streptomyces sp. MJU3502, is also a likely truncation product of the ilamycin/rufomycin family.632 The DKPs lansai A and B (747, 748) from Streptomyces sp. SUC1 are heptacyclic Trp dimers with a reverse dimethylallyl moiety on C5 of one indole ring.633 Structurally similar, nocardioazines A and B (749, 750) from Nocardiopsis sp. (CMB-M0232) also are heptacyclic Trp dimers, but their C5 units are placed on C3 of one indole.634 The absolute configurations of 749 and 750 were revised after their total syntheses were completed,635,636 revealing that both structures possessed two d-Trp units. In 749, this normal prenyl group, which is also epoxidized, forms a novel bridge with N1 of the other Trp forming an 11-membered ring.
Drimentines are a novel class of terpenylated DKPs as they have sesquiterpenoid-derived drimane units (751–759). These NPs of actinomycete origin are unique as all other bacterial prenylated peptides have C5 prenyl units, with the exception of the ComX pheromones (vide supra chapter 3.5.1). Natural drimentines are either Trp-Leu (751, 754–757), Trp-Val (758), or Trp-Pro (751, 753, 759) DKPs and have been identified with drimane moieties singly attached to the C3 of indole (751–756) or doubly attached to the C3 and N1 of indole (757–759).637–640 The double fusion of drimane to the indole-containing DKP creates a unique heptacyclic skeleton. Two related compounds, named indotertines A and B (760, 761) are drimane-possessing Trp-Val DKPs but a second fusion event between the C2 of indole and the exocyclic methylene of drimane creates a 6/5/6/6/6 indole-drimane pentacycle.638,639
Biosynthesis.
CDPSs, which employ amino-acyl-tRNAs as substrates to build dipeptide DKPs, are commonly associated with a variety of tailoring enzymes that act after the DKP core is built.631 These enzymes create chemical diversity by the addition of structural units or functional groups or by modifying the amino acid components. The BGC responsible for the nocardioazines is located in two genetic loci where the MT NozB and PT NozC are not in genetic proximity to the associated CDPS, P450s, and regulatory genes.641 Since 749 and 750 feature d-amino acids and CDPS are known to use charged aminoacyl-tRNA from primary metabolism (i.e., l-amino acids), an isomerase is expected to isomerize cyclo-l-Trp-l-Trp into cyclo-d-Trp-d-Trp.641 Two methylation steps, at C3 and N1´, and prenylation at C3´ completes 750 biosynthesis (Scheme 18); alkylations at C3 and C3´ allow the formation of two pyrroloindoline moieties (as in ComX, vide supra Chapter 3.5.1). The timing of the C3´ prenylation and N1´ methylation steps is still unknown. In addition, it is unclear how the epoxidized dimethylallyl bridge is formed in 749, but two P450s are likely candidates.
Scheme 18.
Biosynthesis of nocardioazine A (749).
The terpenoid biosynthetic machinery responsible for the drimane unit of the drimentines consists of two unique enzymes. The dmt BGC from Streptomyces sp. CHQ-64, now named Streptomyces youssoufiensis OUC6819, consists of the CDPS DmtB1, a phytoene synthase-like PT DmtC1, and an integral membrane cyclase DmtA1.642 The Trp-containing DKP is, as above, first prenylated at the C3 position of the indole ring, which leads to pyrroloindoline formation. The resultant farnesylated 6/5/5/6/5 pentacycle is the substrate for terpene cyclization by the noncanonical type II TS DmtA1.27,642 The second fusion event between the drimane unit and the indole ring seen in drimentines 757–759 or indotertines 760 and 761 is less clear. Drimentines 757–759 may be isolation artifacts resulting from acidic conditions; in fact, 757 and 759 were converted into 751 and 752 in the presence of acid.637 It is conceivable that 760 and 761 can be formed by the attachment of the drimenyl unit and subsequent attack by the drimenyl alkene at C3 and C2 of the indole ring, respectively. This would explain the lack of the pyrroloindoline moiety in 760 and 761 but require a different biosynthetic pathway where drimenyl diphosphate is formed prior to prenylation.
Biological activity.
Cyclomarazines 744 and 745 were moderately effective against MRSA and VRE with MIC values of <20 μg mL−1 while some of the drimane-containing drimentines and indotertines 760 and 761 showed anticancer activities ranging from 1–17 μM.566,638–640 Conversely, the nocardioazines were not cytotoxic antibiotics, but 749 was shown to inhibit the membrane efflux pump P-glycoprotein responsible for some of the multidrug resistance in cancer cells.634
3.6. Phenazines
Phenazines are a well-known class of colorful and redox-active NPs. Their study dates back to the late 1850s when the blue pigment pyocyanin was observed in the discharged pus of patients infected by Pseudomonas aeruginosa.643,644 Phenazines, or 9,10-diazaanthracenes (it should be noted that atom numbering for phenazines differ from that of anthracenes), are produced by Gram-positive and Gram-negative bacteria as well as some archaea.644,645 The phenazine pigments are able to both donate and accept electrons and thereby act as free radical scavengers, generate reactive oxygen species, decrease intracellular glutathione, act as virulence factors by interfering with host cell functions, and regulate genes.644,646 As discussed below, prenylated phenazines are typically produced by the Gram-positive actinomycetes.
The first phenazine-terpenoid hybrid natural product was discovered from Streptomyces cinnamonensis ATCC 15413 and reported as 6-prenylphenazine-1-carboxylic acid.647,648 The preponderance of C9-prenylated 1-phenazinecarboxylic acid (PCA) analogues subsequently discovered, however, including endophenazine A (762), which had NMR, MS, UV, and IR data identical to that of 6-prenylphenazine-1-carboxylic acid, suggests that the first prenylated phenazine was indeed 9-prenyl-1-PCA.649 Other close relatives of 762 produced by various Streptomyces and Kitasatospora spp. include the 5,10-dihydrophenazine-N5-methylated endophenazine C (763),649,650 the N5-alkylated phenazine-7-ones endophenazines B and F (764, 765), the hydroxylated endophenazines A1, F1, and G (766–768), and the heterologously produced endophenazine E (769), an l-glutaminylated congener of 762.649–653
The benthocyanins, isolated from Streptomyces prunicolor 1884-SVT2, were early examples of prenylated phenazines with extended core structures and the first examples of geranylated phenazines. Benthocyanin A (770) is an N5-geranylated PCA with a fused phenyl-substituted γ-lactone.654 Benthocyanin B (771) is a regioisomer of 770 with the carboxylic acid connected to C9 instead of C1 and benthocyanin C (772) displays a conjugated C8-keto-C7-phenylacetonitrile group.655 Benthophoenin (773), also isolated from S. prunicolor, has benzoyl moieties at C3 and C7.656 The chromophenazines (774–779), isolated 20 years later from Streptomyces sp. Ank315, revealed additional modifications to the phenazine core. While chromophenazine C (776) is an N5-prenyl-7-keto-phenazine-1-carboxamide and chromophenazines D–F (777–779) are dimethylallyl variations of benthophoenin, chromophenazines A and B (774, 775) have unique methylbenzene D rings.657 Although the biosynthesis of this unique D ring is unknown, the planar structure suggests cyclization and aromatization of a C9-prenyl group.
Although glycosylated phenazines are rare, there are seven known structures encompassing both the endo- and chromophenazine scaffolds. Phenazoviridin (780) is a 6-deoxy-α-l-talopyranose ester of 762 although it was initially reported as a glycosylated analogue of 6-prenyl-PCA.649,658 The endophenasides B and C (781, 782) are l-rhamnosyl esters of 766; endophenaside D (783) is rhamnosylated 768.652 As the names suggest, aestivophoenins A and B (784, 785) have the same benthophoenin-like benzoyl additions to the phenazine core with additional decorations including l-rhamnosyl esters and in the case of B, a C9 prenyl group.497 Aestivophoenin C (786) is debenzoyl 785.659
Prenylated phenazines of bacterial origin have also been identified in their hydroxyphenazine and pyocyanin forms. JBIR-46, −47, and −48 (787–789) are C1,C6-dihydroxyphenainzes with either C9-prenyl or C4,C9-diprenyl groups, were isolated from Streptomyces spp. after using a PCR screen to identify potential terpenoid producers that possess 3-hydroxy-3-methylglutaryl-CoA reductase genes.660,661 787 and 789 are modified to the N5 oxide.661 C4-Monoprenylated hydroxy- and methoxyphenazines (790–793) were also recently reported.662 Other variants of hydroxyphenazines include the C9-alkylated geranylphenazinediol (794) and the O-geranylated phenaziterpenes (795, 796) and phenazine SC (797).662–664 An N-oxide form of 795, named marinophenazine A (798), was isolated from Streptomyces sp. CNQ-509.665,666
The pyocyanin-terpenoid hybrids were initially discovered after screening bacterial extracts for cytotoxicity. Lavanducyanin (799), named due to the presence of its rarely found in nature cyclolavandulyl moiety, and phenazinomycin (800), which exhibits an unusual (S)-trans-γ-monocyclofarnesol group, were both found in 1989 from Streptomyces sp. CL190 and S. sp. WK-2057, respectively.667–669 Various halogenated (801, 802), hydroxylated (803–805), and esterified analogues (806) of 799 also exist.670–672 One halogenated pyocyanin (807) has a simple dimethylallyl group on N5.671 Overall, phenazines can be modified by C-, N-, and O-prenylation reactions at a variety of positions with hemi-, mono-, and sesquiterpene precursors that are in either their linear or cyclic forms.
Biosynthesis.
Phenazine biosynthesis is well documented.644 Studies on the prenylation reactions catalyzed on phenazine NPs have revealed new enzymes in terpenoid biosynthesis. After the discovery of the ABBA PTs, PpzP and EpzP, ABBA PTs found within phenazine BGCs of S. anulatus and S. cinnamonensis, respectively, were determined to prenylate 5,10-dihydro-PCA.673–675 Identification of EpzP was initially confounding given part of the epz BGC was found next to the fnq BGC (vide infra chapter 3.8.2), which contained the flaviolin ABBA PT Fnq26 and a seemingly nonfunctional ABBA PT Fnq28;398,674 EpzP resided in a partial phenazine BGC found >40 kb away from fnq and epz.675 Understandably, ABBA PTs were then used to genome mine for other phenazine PTs with mixed results.665,676–678 Phm7, a putative ABBA N-PT found in the phenazinomycin BGC was identified next to the putative type II TS Phm1.676 After no ABBA PTs were identified for the biosynthesis of 787–789 or 795 and 798, the membrane-bound PTs Mpz10 and CnqPT1 were found to be responsible for the C-and O-prenylation reactions, respectively.665,677 These proteins are more similar to UbiA-like PTs than the ABBA PTs. Finally, the N-prenylated 799 has a novel cis-PT-type cyclase that condenses two molecules of DMAPP in a “head-to-middle” fashion into cyclolavandulyl diphosphate (CLPP) prior to prenylation (Scheme 19).27,679 The PT responsible for the attachment of CLPP to the phenazine core is still unknown.679
Scheme 19.
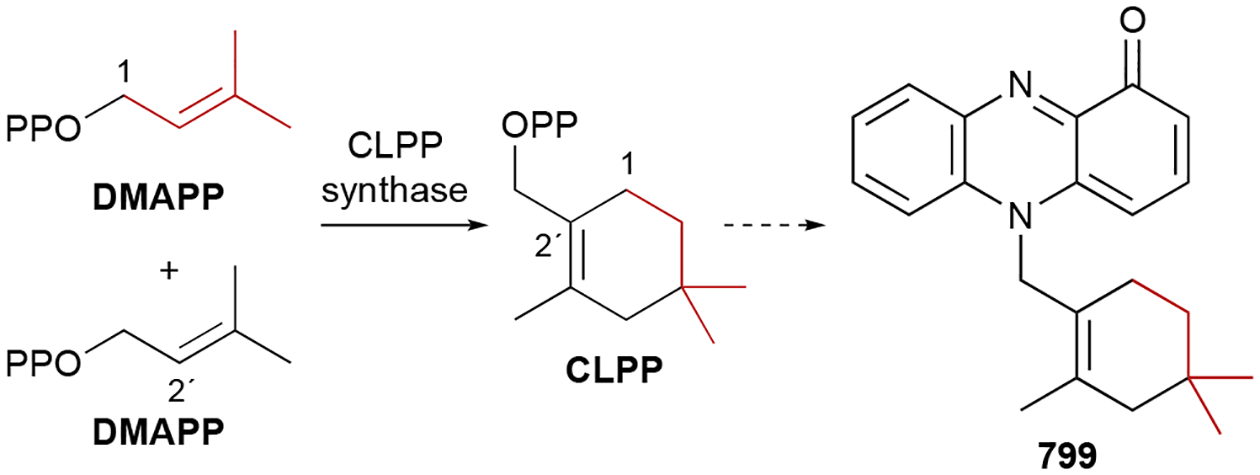
Biosynthesis of lavanducyanin (799).
Biological activity.
In general, most biological activities of phenazines, which include antibacterial, antifungal, antitumor, insecticidal, and antiparasitic activities, stems from their ability to negotiate redox systems, although there is no clear correlation between types of prenylated phenazines and their biological activities.644,645
Phenazines are well known as radical scavengers and some of their bioactive properties can be attributed to this ability.644,646 770 was initially reported to be an order of magnitude more potent than vitamin E.654 771 and 772, along with 773, inhibited lipid peroxidation in rat microsomes 30–70x stronger than vitamin E with IC50 values of 0.16, 0.29, and 0.15 μg mL−1, respectively.655,656 780 inhibited lipid peroxidation in rat brain homogenate at IC50 = 6.6 μg mL−1. This radical scavenging ability also translated to protective attributes with 780 and aestivophoenins 784–786 showing protective activities against KCN-induced hypoxia in mice and glutamate toxicity in the brain, respectively.658,659,680
Many of the prenylated phenazines possess weak to moderate antibacterial and antifungal activities. Endophenazines 762, 763, and 767 all exhibited weak activity against Gram-positive bacteria with endophenazines 766 and 768 also showing activity against Mycobacteria.648,650–652,681 Interestingly, the glycosylated endophenazines were active while the glutaminylated 769 was not.652,653 Mode of action studies on 768 revealed that it is bacteriostatic against MRSA and that its interference on redox processes do not exclusively lead to its antibacterial activity.681 775 and 794 were also reported as weak antibacterials.657,663 799, its 2-chloro analogue 801, and 800 are currently the most potent antibacterials with MIC values against Gram-positive strains as low as 1–2 μg mL−1.668,670 Several prenylated phenazines killed various fungal strains with 802 particularly potent with an MIC of 0.39 μg mL−1 against C. albicans.650,657,672
800 also exhibited a potent anti-Trypanosoma brucei IC50 value of 230 ng/mL with 23 times selectivity over cytotoxicity.682 In an infection mouse model, 800 extended the mean survival days 2.7-fold, suggesting that it may be a lead candidate for new antitrypanosomal drugs.
Phenazines have been associated as potential anticancer agents since 1959 and prenylated phenazine-terpenoid hybrids continue that tradition. 799 was initially discovered during a cytotoxicity screen and was active against P388 and L1210 leukemia cells with IC50 values of 0.27 and 0.30 μM, respectively.667,683 Likewise, marinocyanins 802–807, brominated lavanducyanin analogues, were also potently cytotoxic with IC50 values ranging from 0.029–17.14 μM; compounds with modifications on the terpenoid ring had diminished potencies.672 799 and marinocyanins 802 and 807 induce apoptosis, possibly by their inhibition of TNF-α-induced NFκB (IC50 = 4.1–24.2 μM), COX-1 (IC50 = 5.6–30.0 μM), and COX-2 (IC50 = 4.0–34.0 μM) activities, and their reduction of PGE2 (IC50 = 0.63–7.5 μM) and LPS-induced nitric oxide (IC50 = 8.0–>48.6 μM) production.671 799, at subinhibitory concentrations (0.01–10 ng mL−1), also has been shown to stimulate cell proliferation.684,685 The prenylated phenazine diols 787–789 also possessed cytotoxic activities, although they were >300-fold less active than 799.660
The inhibition of specific targets in humans have also been associated with prenylated phenazines. 794 and phenazines SA–SC possessed moderate inhibition (IC50 = ~2–3 μM) of human acetylcholinesterase.662,663 The 1,6-phenazinediol core, but not the geranyl moiety, appears to be essential for this bioactivity. 799 inhibits testosterone 5α-reductase activity. Both 799 and 801 were reported to have IC50 values of 0.5 and 10 μM, respectively.670
3.7. Phenols
Phenols are common organic molecules found throughout NPs. Many of the prenylated phenolic NPs found in this review are described in other sections (e.g., aminocoumarins); however, a selection of phenolic meroterpenoids required a discrete section. During the biosynthesis of the universal benzoquinones (vide infra chapter 3.11.1), 4-hydroxybenzoate is prenylated and subsequently decarboxylated. This two-step process provides bacteria with 3-prenyl-4-hydroxybenzoates and 2-prenylphenols, which can be processed to benzoquinones or diverted to form other secondary metabolites.
The ortho prenylphenol substitution pattern is common in this group of terpenoids; hydroxyl groups are strong ortho/para directors in electrophilic aromatic substitution reactions (e.g., prenylations). Various 2-polyprenylphenols, direct precursors of benzoquinones, have been isolated from WT bacteria (808–811).686,687 Xiamenmycins A, C, and D (812–814) from Streptomyces xiamenensis are benzopyrans generated by cyclization of a geranylated moiety onto the phenolic oxygen.688–690 A related hemiterpenoid and aminocoumarin precursor, 3-dimethylallyl-4-hydroxybenzamide (815), as well as p-dimethylallylbenzamide (816), were also isolated from Streptomyces.387,691
Erythrolic acids A–E (817–821) are unique prenylated NPs due to their unusual terpenoid lengths.692 Produced by the Erythrobacter, a Gram-negative bacterium not known for producing NPs, the erythrolic acids are 4-hydroxybenzoate analogues with terminally oxidized prenyl moieties. Erythrolic acids 817 and 818, 819, and 820 have prenyl chain lengths of C22, C17, and C12, respectively; erythrolic acids 817, 818, and 820 also have terminal Z olefins.692 Erythrazoles A and B (822, 823), discovered prior to 817–821, are structurally related but contain rare tetrasubstituted benzothiazole core scaffolds.693 Xanthomonic acid (824), produced by the plant pathogen Xanthomonas citri pv. Mangiferaeindicae and discovered using a differential isotope labeling experiment, has the extended and terminally oxidized prenyl moiety, which is appended to PABA at C3.694 Pseudomonol (825), which was activated in Pseudomonas sp. SZ57 using a clever chimeric LuxR transcriptional activation system, is a C45 polyhydrated polyprenyl NP also at C3 of PABA.695
Another family of 3-prenylated 4-hydroxybenozates include the cyanobacterial noscomins, comnostins, and tolypodiols. These diterpenoid NPs are structurally, and likely biosynthetically, similar to the phenalinolactone-like diterpenoids. They are included in this section given that they are clearly the result of initial prenylation of 4-hydroxybenzoate. Noscomin (826) from N. commune Vaucher (EAWAG 122b) has a geranylgeranyl-derived dodecaphenanthrene 6/6/6 tricyclic scaffold.696 The unnamed noscomin variant (827) has an additional tetrahydrobenzooxepane formed between the phenol oxygen and C7 of dodecaphenanthrene core.697 The tolypodiols (828–830), of which 828 was the first diterpenoid NP found in cyanobacteria, has a benzopyran fused to a perhydrophenanthrene core at C13.698,699 Comnostins A–E (831–835) are similar but have a 5/6/6 dodecahydro-1H-benz[e]indene scaffold. The comnostins have different states of oxidation on C23; interestingly, 835 has an acetyl group on C3 and the acetal in 834 may be an artifact due to the use of methanol during extraction.700
KS-505a (836), also called longestin and produced by Streptomyces argenteolus A-2, is a unique decacyclic tetraterpenoid with unusually placed branched methyl groups, a 2-O-methylglucuronic acid, and a γ-hydroxy-γ-lactone that is in equilibrium with a ring-opened γ -ketocarboxylic acid.701 The terpenoid octacycle is fused to succinylbenzoate in a similar manner to that of the tolypodiols.
The merosterols and habiterpenols are steroid-like phenol-diterpenoid hybrids. Habiterpenol (837), produced by Phytohabitans suffuscus 3787_5, has the familiar 6/6/6/5-tetracyclic scaffold fused to C3 and C4 of phenol.702,703 Merosterols A and B (838 and 839), which were found in cyanobacteria, share the same carbon skeleton and are highly functionalized.704
Nocarasin A (840) is simply 6-geranyl-3-hydroxymethylphenol; nocarasins B and C (841, 842), also produced by N. brasiliensis IFM 0667, are 3-hydroxybenzoate analogues.705 Finally, farnesylbenzenediol (843), the reduced form of farnesylquinone (vide infra chapter 3.11.1), and the bicyclic nitrosporeunols E and F (844, 845) were isolated from S. nitrosporeus YBH10–5; geranylbenzenediol (846) was also identified.706
Biosynthesis.
The biosynthesis for most of these compounds is unknown, although UbiA and ABBA PTs are presumed culprits for the prenyltransfers. A five-gene BGC for xiamenmycins 812–814 was reported confirming the role of a UbiA-like 4-hydroxybenzoate PT (XimB).707 Epoxidation of the 2´,3´-olefin leads to benzopyran formation by the SnoaL-like cyclase XimE with threonine derivatization by XimA concluding its biosynthesis.708
Currently, erythrolic acids 817–821 and erythrazoles 822 and 823 only have biosynthetic proposals.692,693 A C2 extension of prenyl chains may occur as the result of a Claisen condensation with acetyl-CoA and a terminally oxidized methyl group followed by dehydration and olefin isomerization. The isolation of 821 may support this proposal since its C15 chain is terminally oxidized.692 Alternatively, oxidative cleavage of a longer prenyl chain at the terminal double bond could result in the loss of three carbons; olefin isomerization would be required in this case as well. If acetyl-CoA is used, the erythrazoles would be a rare case of a NP derived from four biosynthetic pathways: terpenoid, shikimate, nonribosomal peptide, and polyketide.693
The BGC for 836 was identified by PCR-guided gene discovery of its octaprenyl diphosphate synthase Lon12 and confirmed by gene inactivation of the putative TS Lon15.709 Further studies revealed the unique methylation pattern of the cyclic tetraterpenoid skeleton.710 Lon23 methylates the terminal vinyl carbon of IPP to form Z-homoIPP, which is elongated by two polyprenyl synthases, Lon22 and the aforementioned Lon12, with DMAPP and IPP to form dimethyloctaprenyl diphosphate (Scheme 20).710 Although no other biosynthetic work has been reported, the structural similarity of 836 to the phenalinolactone diterpenoids suggests that the UbiA-like PT Lon13 first prenylates 4-hydroxy-2-succinylbenzoate at C3. Then, epoxidation at the terminal olefin and cyclization by the type II TS Lon15 is speculated to form the terpenoid octacyclic scaffold in one step. Oxygenations and glucuronidation at C3 completes the biosynthesis (Scheme 20). The biosyntheses of noscomins 826 and 827, comnostins 831–835, and tolypodiols 828–830 are also unknown, however their pathways can be inferred based on their likely similarities to 836 and the phenalinolactone-like diterpenoids.
Scheme 20.
Biosynthesis of KS-505a (836).
In merosterol (838 and 839) biosynthesis, only prenylation and terpene cyclization is understood. First, the UbiA PT MstC catalyzes the geranylgeranylation of 3,4-dihydroxybenzoate at C5.704 A subsequent type II TS, MstE, constructs the pentacyclic merosterol scaffold.
Biological activity.
The noscomins and nocarasins are antibacterial agents. Noscomins 826 and 827 are moderate to weak inhibitors of Bacillus cereus, Staphylococcus epidermidis, and E. coli.696,697 Nocarasins 840 and 842 have MICs of 0.39–6.25 μg mL−1 against Gram-positive strains;705 the 3-phenol is important for activity.
Comnostin B (832), which also has 47 μM molluscidial activity, 820, 823, and 838 and 839 all had low μM IC50 values against various cancer cell lines.692,693,700,704 The lack of the terminal Z olefin or oxidation on the middle of the terpene chains of the Erythrobacter NPs appears to diminish their activities. The related xanthomonic acid (824) is also modestly cytotoxic and induces cell death via activation of autophagy.694 While not cytotoxic, 837 selectively abrogates bleomycin-induced G2 arrest in Jurkat cells with an IC50 of 3.55 μM.702
Xiamenmycins 812–814) showed anti-fibrotic activity with multiple effects including the inhibition of proliferation, cell adhesion, and contractile capacity of human lung fibroblasts.688,689 The benzopyran, but not the affixed Thr is important for activity.
The tolypodiols are potential anti-inflammatory compounds with 830 inhibiting thromboxane B2 at an apparent IC50 of 100 nM.698,699
836, which was not antibacterial or antifungal, was a novel and selective in vitro and in vivo inhibitor of bovine brain calmodulin-dependent cyclic-nucleotide phosphodiesterase with an IC50 range of 65–170 nM.701,711 836 was also shown to have ~1 μM anti-trypansomal activity without cytotoxicity.712
3.8. Polyketides
Bacteria biosynthesize an immense diversity of polyketide NPs. Polyketides are built by megasynthethases, which are termed polyketide synthases (PKSs) and classified into three main types according to their protein makeup and how they utilize acyl-CoA precursors as building blocks.713 Due to the modular biosynthetic logic of PKSs, polyketides have astonishing structural complexity and are commonly found in linear, macrocyclic, polycyclic, and aromatic forms.713–715 Polyketide-derived meroterpenoids in bacteria are all the result of prenylation of an aromatic core. The most common aromatic polyketide that is used as the framework for initial prenylation is 1,3,6,8-tetrahydroxynaphthalene (THN). Chemical and biosynthetic studies of these unique bacterial meroterpenoids have revealed some of the most mechanistically intriguing enzymes involved in bacterial terpenoid biosynthesis. This family of meroterpenoids was recently reviewed.716
3.8.1. Flavonoids
The only prenylated flavonoids, which are very common type III polyketide NPs in plants,717,718 of bacterial origin are the C6-prenyl (847), C8-prenyl (848), and 7,3´-O-diprenyl flavonoids (849) and the lavandulylflavanones (850, 851, 852) and lavandulylchalcones (853, 854).436,719–721 These are all produced by Streptomyces spp. and have not been biosynthetically studied as of yet.
Biological activity.
The flavanones and chalcones with lavandulyl moieties are broad-spectrum antibiotics with activity against both Gram-positive and Gram-negative bacteria as well as fungi.719–721 The lavadulyltetrahydroxyl analogue 850 is the most potent with IC90 values as low as 1 μg mL−1. 847 and 851 and were effective against M. tuberculosis activity with IC90 values of 6 and 11 μg mL−1.720 These two flavonoids, along with isoxanthohumol (848), are also mildly cytotoxic.720–722
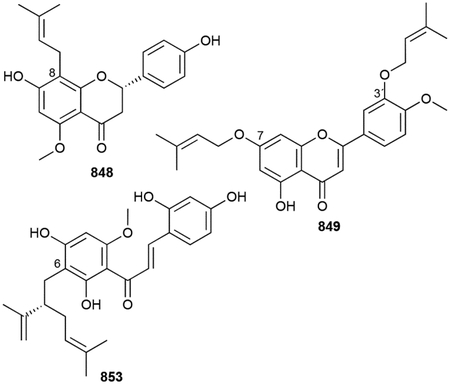
3.8.2. Furaquinocins
A screen for cytotoxic antibiotics resulted in the identification of furaquinocins A and B (855, 856) from Streptomyces sp. KO-3988.723 These NPs had a 6/6/5 tricyclic naphto[1,2-b]-furan-6,9-dione core that appeared to be a highly modified 1,4-naphthoquinone chromophore with a prenyl-like side chain consisting of ten carbons. At the time of discovery, these polyketide-derived meroterpenoids were quite unique, but have since become part of a larger family of meroterpenoids. In this subfamily, we included NPs with prenyl chains attached to C6 of the dihydroxybenzene ring.
Furaquinocins C, D, F, and H (857–860) are analogues of 855 with different hydroxylation patterns, E (861) has a conjugated E,E-diene on its prenyl chain, and the prenyl moiety on G (862) is a cyclic hemiacetal.724 The absolute stereochemical configurations were later determined.725 Furaquinocins I and J (863, 864) have C14 carboxyl and carboxamide functional groups at the end of the prenyl chain.726 JBIR-136 (865) is a C3-demethyl and reduced form of furaquinocin D (858) and PI-220 (866) contains an 11E olefin and C13 hydroxyl group, although only its relative stereochemistry was determined.727,728 Furanonaphthoquinone I (867), produced by S. cinnamonensis ATCC 15413, is very similar to 857, although its 2,3-dihydrofuran ring is attached to the oxygen on C7.729
All of the meroterpenoids above are monoterpene-derived, but there are also known hemi- and sesquiterpene furaquinocin analogues. Fumaquinone (868) has a C6 noncyclized dimethylallyl moiety while neomarinone (869) and the marfuraquinocins A–D (870–873) have cyclized sesquiterpene chains.664,730,731 869, a product of the marine Streptomyces sp. MAR4 CNH-099, was initially reported to possess trisubstituted cyclopentane connected to the cis dihydrofuran ring by an 11Z-olefin.731 Following biosynthetic studies and total synthesis, the prenyl moiety of 869 was later revised to a tetrasubstituted cyclohexene.732,733 The tetrasubstituted cyclohexane rings in 870–873 likely originate from a different cyclization mechanism as that in 869.664
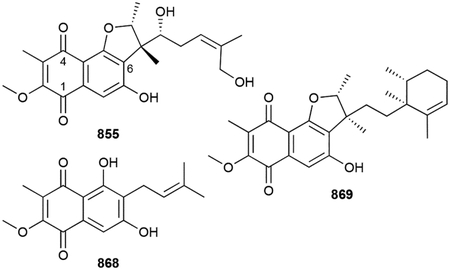
Biosynthesis.
The BGCs for furaquinocin A (855, fur) and furanonaphthoquinone (867, fnq) have been known for 15 years. Prior to any genetic studies, early precursor labeling studies confirmed that 855 and 856 were of mixed polyketide and MVA-derived terpenoid origin.734 fur, the first reported hybrid polyketide-terpenoid BGC, was identified by genome mining for MVA pathway genes.735 The structure of the furaquinocins and bioinformatics analysis supported a biosynthetic proposal of THN formation by a type III PKS, reverse prenylation of GPP by an ABBA PT (Fur7), an unknown cyclization reaction, and two methylation and multiple hydroxylation tailoring steps (Scheme 21). The fnq BGC, published in rapid succession, supported a similar biosynthetic proposal, with 2-O and C3 methylation being catalyzed by Fnq9 and Fnq27, respectively.674
Scheme 21.
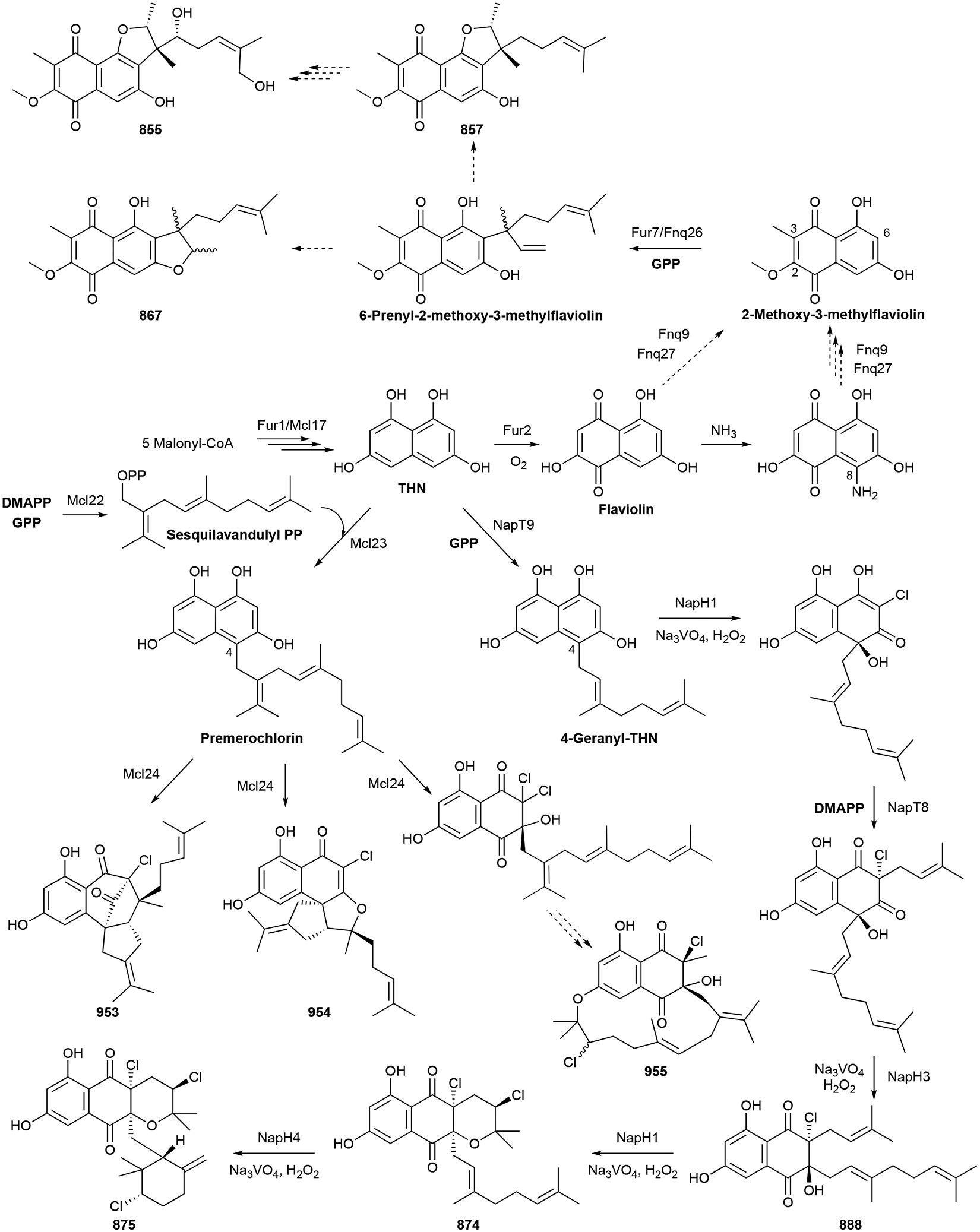
Biosynthesis of furaquinocin A (855), furanonaphthoquinone I (867), napyradiomycin B1 (875), and merochlorins A–C (953–955).
Both in vivo knockouts and in vitro enzyme reactions support Fur7 as a C6-reverse PT that acts on 2-methoxy-3-methylflaviolin, a likely intermediate in both furaquinocin and 867 biosynthesis (Scheme 21).736 It should be noted here that both Fur7 and the homologous Fnq26 catalyze C3-reverse prenylation of flaviolin.674,736 Further biochemical studies revealed a cryptic 8-amino-flaviolin intermediate, synthesized by the aminotransferase Fur3 prior to demethylation, although its role and the fate of the amino group is unclear (Scheme 21).737
Two major terpene-related questions remain: (i) how is ether formation controlled in 855 and 867 biosynthesis and (ii) how and when does terminal cyclization of the sesquiterpene moiety occur in 869 and the marfuraquinocins (870–873) biosynthesis?
Biological activity.
Although furaquinocin meroterpenoids are considered as cytotoxic antibiotics, most of these NPs are cytotoxic without antibacterial or antifungal activities. Only marfuraquinocins 870–873 had antibacterial activities (MIC values ≥8.0 μg mL−1) against Gram-positive S. aureus and methicillin-resistant S. epidermidis (MRSE).664
Furaquinocins 855–858 and 860–862 were active against B16 melanoma and HeLa S3 cells with 860 being the most active (IC50 values of 0.19 and 0.52 μM, respectively).724,738 Furaquinocins 859 and 863–865 were not cytotoxic,724,726,727 but 869, 870, 872 were active with IC50 values of ~19, 3.7, and 4.4 μM, respectively, against various cancer cell lines.664,731 866, which was not cytotoxic, inhibited rabbit platelet aggregation with an IC50 value of 1.0–2.1 mM.728 Furaquinocins 855–862 were also mentioned, but not published, to have antihypertensive, anticoagulative, and antiplatelet activities.725
3.8.3. Napyradiomycins
Napyradiomycins are unique halogenated polyketide-derived meroterpenoids with a naphthoquinone chromophore. The napyradiomycins are categorized into three types depending on how the prenyl groups are appended to the naphthoquinone core. Type A compounds have linear terpenoid side chains; type B have 6/6/6 tricyclic scaffolds where the C2-dimethylallyl group cyclizes onto 3-O to form a tetrahydropyran; type C members have monoterpenoid-derived 14-membered macrocycles between C3 and C7 (numbering based on naphthoquinone core).739 There are 58 characterized napyradiomycin analogues, mostly produced by marine Streptomyces spp., with various regio- and stereoselective patterns of halogenation, oxygenations, and terpene cyclizations adding complexity.
The first napyradiomycins, A1 (originally named A), B1, B2, B3, C1, and C2 (874–876, 877–879) were reported in 1986 from the actinomycete Chainia rubra MG802-AF1 (now classified in the Streptomyces genus).740 874 was a dichloro-3-geranyl type B napyradiomycin; 875–877 were also type B, but their geranyl moieties were cyclized into chlorinated cyclohexanes; 878 and 879 also had the 6/6/6 core but the geranyl groups were found to also be connected to the naphthoquinone core at C7 thus erecting additional 14-membered rings. The first A-type napyradiomycins found were SF2415A1, A2, B1, and B2 (880–883) in Streptomyces aculeolatus, along with napyradiomycin A1 derivatives SF2415 A3 and B3 (884, 885).741 880, 881, and 884 had additional α-diazoketones on C5/C6 of their naphthoquinone cores.
There are seven additional type A napyradiomycins (886–892) including naphthomevalin (888) and the C2-deprenyl analogues phosphatoquinones A and B (889, 890).435,742–746 Other type B napyradiomycins with noncyclized geranyl moieties (893–905) and cyclohexyl geranyl moieties (906–922) have been identified from native Streptomyces spp.739,742,747–754 Heterologous expression of the Streptomyces sp. CNQ-525-based nap BGC produced at least seven B-type napyradiomycins including the new 2-deschloro-2-hydroxy-A80915C (923).755
The C-type napyradiomycins have garnered significant interest due to their unusual structures. Variations of C-type compounds include the “strained-ring” napyradiomcyin SR (924) that has a C3-, 6-O-, and C7 trifused geranyl moiety, the C16/17 E variant of 924, napyradiomycin D (925), and the 6-O-linked geranyl macrocycle napyradiomycin D1 (926).739,748,753 Other type C napyradiomycins include 16-dechloro-16-hydroxynapyradiomycin C2 (927), napyradiomycins A–C (928–930).748,753
Azamerone (931) from Streptomyces sp. CNQ-766 has a chloropyranophthalazinone core with a 3-chloro-6-hydroxy-2,2,6-trimethylcyclohexylmethyl side chain.756 Its overall structure resembles that of the B-type napyradiomycins but with a conspicuous pyridazine ring.
Biosynthesis.
With early stable isotope feeding studies757 and a large variety of natural A-, B-, and C-type napyradiomycins isolated prior to the discovery of the nap BGCs from S. aculeolatus NRRL 18422 and Streptomyces sp. CNQ-525,755 it was clear that these hybrid polyketide-terpenoids originated from prenylated THN (Scheme 21).758 Cloning of the nap BGCs revealed a type III PKS synthase, multiple PTs, and three vanadium-dependent haloperoxidases (VHPOs). It was clear that these VHPOs, typically known to introduce halides into NP scaffolds through either nonselective or enzyme-direct regio- and stereoselective halogenations,759 were involved in both chlorination and terpene cyclization,755 but their exact roles were not delineated for another decade.
An illuminating study, conceived by questioning how the electron poor C3 carbon of THN is geranylated in the napyradiomycins, as well as some of the merochlorins and naphterpins (vide infra chapter 3.8),758 revealed a key non-chlorinating role for the VHPO NapH3. Naphthomevalin (888), the C2,C3-diprenylnaphthoquinone, is the result of an unprecedented enzyme-catalyzed α-hydroxyketone rearrangement and suggests that other related meroterpenoids arise through a similar VHPO-catalyzed mechanism.760
After the key finding of the activity of NapH3, the total enzyme syntheses of 888, 874, and 875 were conducted.761 THN is first C4-geranylated by the ABBA PT NapT9, NapH1 then oxidatively dearomatizes and chlorinates the eventual dihydroquinone ring (Scheme 21). Another ABBA PT, NapT8, prenylates the chlorinated C2 position before the α-hydroxyketone rearrangement to form 888.760 NapH1 then acts again to complete the cyclization of the tetrahydropyran ring forming 874.761,762 NapH4 then mediates a chloronium-induced cyclization of the geranyl moiety to yield 875 (Scheme 21).761 Given this biosynthetic pathway, one can appreciate that many of the napyradiomycin congeners are biosynthetic intermediates and by-products, or the result of non-enzymatic transformations; although differences in the associated BGCs may purposefully biosynthesize some variants. It is currently unclear how the diazo functional groups or the 14-membered macrocycles of the C-type napyradiomycins are installed.
Biosynthesis of 931 is proposed to occur through a diazonapyradiomycin intermediate that undergoes oxidative rearrangement, decarboxylation, and dehydration to form the aromatic pyridazine ring.756 Labeling studies biosynthetically link 931 to the napyradiomycins as 884 was converted into 931 in a precursor feeding experiment.763
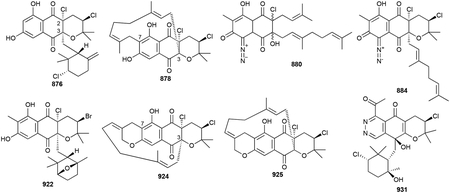
Biological activity.
The napyradiomycins were initially found as antibacterials but are now known for their broad spectrum antibacterial, anticancer, and antiogenic activities. Most A- and B-type napyradiomycins have respectable activities against both aerobic and anaerobic Gram-positive bacteria with potencies as low as 0.015 μg mL−1.435,739,742,743,748,749,752,754,764,765 905 (0.78 μg mL−1), 877 (0.25 μg mL−1), and 908 (15 ng mL−1) were particularly potent analogues.742,749,765 906 and 908 also had strong activities (MICs <8 ng mL−1) against the Gram-negative Haemophilus influenzae.742 C-type napyradiomycins are less potent with MICs >12.5 μg mL−1.740,753 The mode of action of antibiotic activity is uncertain, yet DNA, RNA, protein, and cell wall biosynthesis are all inhibited.742
All three types of napyradiomycins also show moderate cytotoxicities against a variety of mammalian cells. Most antitumor IC50 values were in the low micromolar range (2–20 μM).739,749,750,752,753,764 Type B napyradiomycins were shown to cause cell death via activation of apoptosis with 921 inducing apoptosis at 4 μM.750 921 and 907 localized in the ER of HCT-116 colon adenocarcinoma cells and targeted the human Hsp90 paralogue Grp97.766
Napyradiomycins hit a variety of targets in human cells. 906 was also found to be a noncovalent ~2 μM IC50 inhibitor of the gastric (H+-K+)-ATPase.767 874 and 875 act as estrogen receptor antagonists with IC50 values of 4.2 and 35 μM, respectively.768 A1 also has various antiangiogenic effects including the suppression of blood vessel formation, endothelial cell proliferation and migration, and improved permeability of cell membranes.769 The A-type phosphatoquinones 889 and 890 inhibited protein tyrosine phosphatase with IC50 values of 28 μM and 2.9 μM, respectively while 891 is an inhibitor of bovine aldolase reductase with an IC50 value of 0.2 μM.744,745
3.8.4. Naphterpins
We classified the naphterpin family of polyketide-derived meroterpenoids as those with 6/6/6/6 tetracyclic ring systems where geranyl or farnesyl prenylation at C3 of THN sets up formation of the additional two rings. Naphterpin A (932), isolated from Streptomyces sp. CL190 four years after the napyradiomycins, had a core similar to the 6/6/6 chromophore of B-type napyradiomycins but with an additional methylcyclohexene ring fused to the dihydropyran.770 As with the napyradiomycins, various naphterpin analogues, naphterpins 933–936 and naphthgeranines A–F (937–942) were subsequently isolated with assorted functional group modifications, mostly on the terpenoid moiety.771–775 Naphthgeranines 941 and 942 have aromatized D rings. The naphthoquinone core is sometimes reverse prenylated at C7, yielding naphthablins 943–945 and JBIR-79 and −80 (946, 947).776–778 Naphthablins 944 and 945 have extended 5/5/6/6/6/6 hexacyclic scaffolds after cyclization of the dimethylallyl moiety on C7.777
Prenylation with FPP and subsequent cyclization results in the marinones (948–952), sesquiterpenoid versions of naphterpins.731,779 There is some ambiguity of the absolute configuration of these compounds in the literature as the relative stereochemistry was initially assigned, but a study on the total synthesis later depicted the enantiomeric forms.780
Biosynthesis.
Unsurprisingly, given their structural similarities to the napyradiomycins, the biosynthesis of naphterpins and marinones mirrors that of the napyradiomycins. An elegant biomimetic total synthesis led to the identification, through use of testing synthetic intermediates as substrates for biosynthetic enzymes, of two VHPOs that are responsible for formation of putative biosynthetic intermediates.780 Initial geranylation (or farnesylation) of THN at C4 is likely and supported by early labelling studies.781 MarH1 and MarH3, found in Streptomyces sp. CNQ-509,782 successively catalyze the same reactions as NapH1 and NapH3, oxidative dearomatization and C2 chlorination and C4-to-C3 α-hydroxyketone rearrangement, respectively (vide supra Scheme 21).780 The major difference being that MarH3 also adds an additional chlorine on C2 supporting that geminal disubstitution is required for alkyl migration.760 The remaining biosynthetic steps have yet to be revealed; the current proposal includes 2,3-epoxidation, reductive dehalogenation, oxidation and alkene isomerization, and intramolecular hetero-Diels-Alder cyclization to form 7-demethylnaphterpin (or debromomarinone, 950).780 Bromination of 950 completes the biosynthesis of 948 or 949.
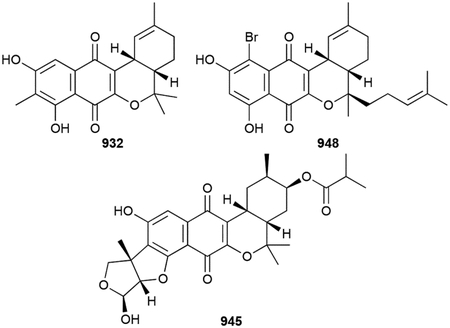
Biological activity.
Contrary to the napyradiomycins, most naphterpins did not show significant antibiotic activities;770,773,775 only marinones 948 and 950 had MIC values as low as 1–2 μg mL−1 against Gram-positive pathogens.779 A few analogues, including naphthgeranines 937 and 938, marinones 950 and 951, and naphthablins 944 and 945,731,773,777 were cytotoxic at a level similar to that of the napyradiomycins. Naphthablin 943 was initially found in a screen for inhibitors of the tyrosine-protein kinase oncogene abl. 943 prevented Abl-induced morphological transformation in v-Abl-expressing NIH3T3 cells at 30 μg mL−1 and was shown to specifically inhibit RNA synthesis.776
The naphterpins also showed antioxidative properties with reported radical scavenging abilities.770–772,774,778 946 and 947 prevented glutamate toxicity in neuronal cells, but at a significantly decreased efficacy compared to the benzastatins.778
3.8.5. Merochlorins
Another family of structurally intriguing chlorinated naphthoquinone terpenoids are the merochlorins. Although the four members all have different molecular architectures, they share common features and a single BGC. Merochlorin A (953) has a unique benzo-bicyclo[3.2.1]-octadione ring system with a propan-2-ylidenecyclopentane moiety.783 Merochlorin B (954) had the same molecular formula as that of A, but with a completely different carbon connectivity: a propan-2-ylidenecyclopentane-containing 6/5/5 fused tricycle with an α-chloroenone. Both 953 and 954 have their prenyl units attached at C4 of the naphthoquinone core.784 Merochlorins C and D (955, 956) had sesquilavandulyl moieties attached on the naphthyl unit at C3 with merochlorin C forming a 15-membered macrocycle by additional attachment at 6-O. The sesquilavandulyl moiety is a rearranged C15 unit that was unprecedented in bacteria at the time of discovery.
Merochlorins E and F (957, 958) were recently identified as variants of merochlorin D with cyclized sesquilavandulyl groups.785 Meroindenon (959) has an indanedione core, perhaps as the result of a pinacol-type ring contraction.785
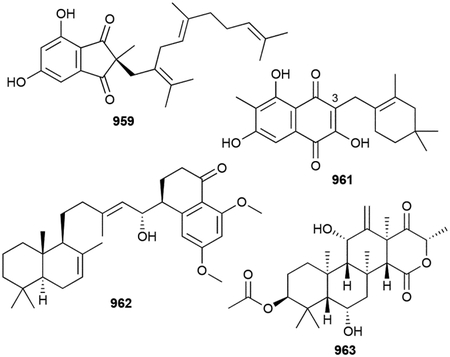
Biosynthesis.
A single PKS-terpenoid hybrid BGC, similar to the BGCs described above, was found responsible for the production of merochlorins in Streptomyces sp. CNH-189. The differing prenylation patterns of 953/954 and 955/956 initially hinted at two sites of direct prenylation.784 Follow up studies confirmed the ABBA PT Mcl23 exclusively prenylates C4 of THN with sesquilavandulyl diphosphate, which is synthesized by Mcl22,786 to give the oxygen-sensitive pre-merochlorin (Scheme 21).787 Mcl24, a VHPO, performs tandem chloronium-induced dearomatization and terpene cyclization to yield both merochlorins 953 and 954.788 Mcl24 can also catalyze α-hydroxyketone rearrangement on pre-merochlorin to an intermediate with a scaffold identical to 956.760 Mcl40 is proposed to then catalyze the chloronium-induced macrocyclization converting merochlorin 956 into 955 (Scheme 21).758 It is fascinating that a single enzyme, Mcl24, is responsible for generating the vast structural diversities of the merochlorins.
Biological activity.
Merochlorins 953, 954, 957, and 958 are effective Gram-positive antibacterials with an MIC range of 1–4 μg mL−1 against MRSA, VRE, and Streptococci.783–785 Clostridium difficile is also inhibited by 953 (0.15–0.3 4 μg mL−1).783
3.8.6. Miscellaneous Polyketides
A few other naphthoquinone meroterpenoids have been isolated. Flaviogeranin (960) is simply a 7-O-geranyl substituted naphthoquinone.789 Arromycin (961) has a C3 cyclolavandulyl moiety and was identified after BioMAP antibiotic activity profile screening.790 The menaquinone-type naphthoquinones are discussed in section 3.11.2 (vide infra).
Actinoranone (962), produced by the marine bacterium Streptomyces sp. CNQ-027, is an unusual polyketide meroterpenoid.791,792 Although not a true prenylated naphthoquinone, 962 has a bicyclic labdane diterpenoid moiety attached to its substituted dihydronaphthalenone core.
Terretonins are polyketide meroterpenoid mycotoxins known in fungi for their structurally complex and highly functionalized ring systems.793 Interestingly, the first bacterial member, terretonin N (963), was recently identified from a culture of Nocardiopsis sp. LGO5.794 Its biosynthesis was not investigated but presumably follows biosynthesis in fungi with an initial farnesylation of the polyketide-based 3,5-dimethylorsellinic acid followed by a noncanonical TS cyclization by an integral membrane cyclase.27,795,796
Biosynthesis.
Nothing is known about the biosyntheses of these meroterpenoids, although reasonable proposals can be formed based on related bacterial, or fungal in the case of 963, meroterpenoids.
Biological activity.
961 and 963 have antibacterial activities with only mild to no cytotoxic activity.790,794 960 shows nanomolar inhibition of glutamate toxicity in rat brain cancer cells with no cytotoxicity.789 962 was cytotoxic against HCT-116 with an LD50 value of 4 μM.791
3.9. Pyrroles
Pyrroles are five-membered nitrogen-containing heteroaromatic rings that are planar and electron rich. From a biological perspective, they are most renowned for their presence in the tetrapyrrole scaffolds of heme, chlorophyll, and vitamin B12.797 The pyrrole ring, not including those contributing to the indole skeleton, is also found in many families of NPs, some of which are described in this review. The mixed pyrroloterpene class of NPs is small with only 21 known variants all produced by Streptomyces.
The first pyrrole-terpenoid hybrid NP discovered was also the simplest in structure. Pyrrolostatin (964) is a C4 geranylated pyrrole-2-carboxylic acid produced by Streptomyces chrestomyceticus EC50 and was identified in the same screen for free radical scavengers that resulted in the discovery of carazostatin and neocarazostatin.798 Geranylpyrrol A (965), the only other pyrrolomonoterpenoid, is pyrrolostatin-3-acetamide methyl ester.799
The 19 other prenylated pyrroles are all farnesylated at either the ortho (i.e., C2 of pyrrole) or meta (i.e., C4 of 2-nitropyrrole) positions. The glaciapyrroles (966–968), named after the Alaskan origin of their producer Streptomyces sp. NPS008187, and six unnamed analogues 969–974 are C2-farnesylated pyrroles with a 2´Z,4´E-dien-1-one modified sesquiterpene chain, each with a different state of oxidation. 966 contains a tetrahydrofuran (THF) ring and a 6´,11´-diol, 967 is a 6´,7´-diol, and 968 is a 6´,7´-epoxide.800,801 The unnamed glaciapyrrole analogues 969–974 were all isolated from Streptomyces sp. Hd7–21. These terpenoid tails had different levels of oxidation at C9´–C12´.802–805
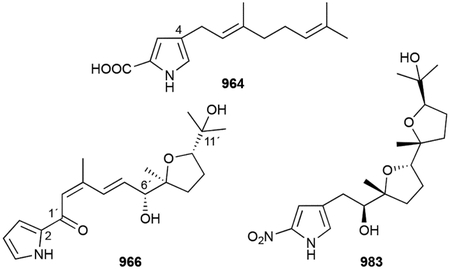
The nitropyrrolins and heronapyrroles both have the 2-nitro-4-prenyl pyrrole core. The nitropyrrolins A–E (975–979) all have linear farnesyl chains with various epoxy, hydroxyl, or chlorine modifications.806 978 was distinct with an additional E double bond between C3´–C4´. Chlorination at C3´ of 977 and 979 was unsurprising given the marine origin of their producer, Streptomyces sp. CNQ-509. 10´,11´-Epoxynitropyrrolin A (980) was initially reported in a perspective prior to the publication of 975–979 and later re-identified by UV, but its structural characterization, including stereochemistry, was never published.807,808 The heronapyrroles (981–984), named for their isolation from a Streptomyces sp. collected off of Heron Island, Australia, are variations of the nitropyrrolins.809 While 981 and 982 have oxidized linear terpenoid chains 983 is a cyclized version of A with two THF rings. In an interesting example of synthesis-guided NP discovery, the synthesis of a proposed biosynthetic alternative to heronapyrroles led to the identification of 984 as a genuine NP.810 Although this monoether was produced in quantities too low for structural elucidation, comparison with synthetic 984 clearly confirmed its presence in Streptomyces sp. CMB-M0423.
Biosynthesis.
The biosynthesis of the pyrroloterpenoids is unknown. Their structures suggest that a relatively simple biosynthesis including the prenylation of pyrrole, oxidations and/or epoxidations on the terpene chain, and cyclization or epoxide hydrolysis via epoxide hydrolases. Acylation of pyrrol-2-carboxyl thioester with farnesol or farnesic acid followed by decarboxylation is an alternative proposal;801 however, the pyrroloterpenoids without a ketone at C1´ suggests direct m-prenylation on pyrroles is likely. ABBA PTs are probable candidates for the prenylation of pyrroles, but no activity with pyrroles have been established.678
Biological activity.
Similar to the phenazines, 964 was initially discovered by searching for small molecule radical scavengers.798 964 inhibited lipid peroxidation in rat brain homogenate, showing an IC50 of 49 μM, and protected mice from KCN-induced acute hypoxia. It was speculated at the time that the mode of action of 964 is intrinsic Fe2+ chelation resulting in a reduction of hydroxyl radicals.798
The nitropyrrole-terpenoid heronapyrroles 981–984 were active against Gram-positive bacteria with MICs ranging from 0.7–6 μg mL−1; they had no activity against Gram-negative bacteria and were not cytotoxic.809,810 Conversely, the structurally related nitropyrrolins (975–979), which are moderately cytotoxic (vide infra), were not active against MRSA, although the synthesized 3-farnesylpyrrole had an MIC of 2.8 μg μL−1.806 It is unclear what leads to the difference in antibacterial activity for these two families of prenylated nitropyrroles.
The glaciapyrrole congeners and nitropyrrolins showed moderate to weak antitumor activity. 965 inhibited the cell growth of both colorectal adenocarcinoma HT-29 and melanoma B16-F10 with an IC50 value of 180 μM.800 The oxidized analogues 970–973 were 5–22-fold more cytotoxic than 966.803–805 The nonhydrolyzed 874 was inactive, suggesting that the presence of the oxirane precluded cytotoxicity.805 Only the non-chlorinated nitropyrrolins 975, 976, and 978 showed activity against HCT-116 cells with 978 being the most potent (IC50 = 5.7 μM).806
3.10. Quinolines
Quinolines, also known as 1-aza-naphthalenes, are heterocyclic and aromatic 6/6 scaffolds that have been established as pharmaceutically relevant drugs.811,812 Most prenylated quinolines of bacterial origin fall into one family of compounds known as aurachins. They are unique isoprenoid alkaloids with farnesyl residues at C4 (A-type) or C3 of the quinoline nucleus (C-type); the position that does not have farnesyl attached is oxygenated. Aurachins have been isolated as quinolines, 4-quinolones, or quinoline N-oxides.
Aurachins were first identified from myxobacteria over 30 years ago.813 Eleven aurachins, A–I, K, and L (985–995, 994), were identified from Stigmatella aurantiaca SG a15 with aurachins 985–988 being the major constituents.813–815 A-type aurachins include 987, 986, and 990 and C-type aurachins include 987–989 and 991–995. Aurachins have non-modified farnesyl groups or farnesyl moieties that are cyclized onto the ortho oxygen substituent resulting 5- or 6-membered ether rings. 989 is a structural outlier with a cyclic urea functional group between N1 and C8.
Several other C-type aurachins (996–998) have been isolated from various Rhodococcus.816,817 Aurachin SS (999), produced by Streptomyces sp. NA04227, is a 3-geranyl-4-methoxyquinoline N-oxide.818 Aurachin P (1000), produced by Stigmatella erecta Pd e32, is the only other A-type reported.819
Another subfamily of aurachins are structurally related to aurachin C but have linear geranyl moieties at C2 of their 4-quinolone cores. Eight CJ-13 analogues (1001–1008) are produced by Pseudonocardia sp. CL38489.820 Intervenolin (1009) from Nocardia sp. ML96–86F2 is another 2-geranylquinolone with a dimethyl carbonimidodithioate moiety attached to the N-methyl of the quinolone.821
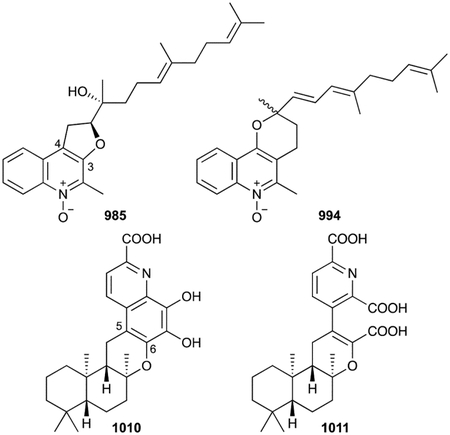
Saccharoquinoline (1010) and thallusin (1011), isolated from marine-derived Saccharomonospora sp. CNQ-490 and Cytophaga sp. YM2–23, respectively, appear to be related alkaloidal meroterpenoids.822–824 Saccharoquinoline, which has a 6/6/6/6/6 pentacycle, is clearly a 2-carboxyquinoline with a drimane sesquiterpenoid moiety fused to C5 and 6-O. Thallusin (1011) has a sclareol-like structure linked to a 2,6-pyridinedicarboxylic acid,823,824 although oxidative cleavage of the benzene ring of 1010 would yield 1011.822
An unnamed quinoline (1012) from Streptomyces sp. neau50 appears to be a prenylated quinoline as it has an 8-(3-methoxyisoprenyl) moiety on its 2-methylquinoline-4-carboxylic acid methyl ester scaffold, although no biosynthetic studies have confirmed its origin.825
Biosynthesis.
Based on early feeding studies and the aurachin BGC from S. aurantiaca, it was clear that the quinoline core is built from anthranilate and two polyketide units (Scheme 22).826 In fact, anthranilate priming for PKS processing requires two adenylating enzymes, the CoA ligase AuaEII and acyltransferase AuaE.827 A full biosynthetic pathway, including prenylation at C3 by a UbiA-like PT and a unique transposition of the farnesyl group from C3 to C4 (i.e., A-type aurachins are derived from C-type), was proposed based on feeding experiments828 and was updated based on a combination of bioinformatics, in vitro, and in vivo studies. Farnesylation at C3 of 2-methyl-4-hydroxyquinoline by AuaA and N-hydroxylation by the Rieske[2Fe–2S] oxygenase AuaF yields 988 and 987, respectively; AuaG then catalyzes an FAD-dependent epoxidation of the 2,3-double bond (Scheme 22).829,830 Reduction of the C4 keto group by AuaH forces a pinacol-type rearrangement after 4-O protonation to give 986. AuaJ epoxidation and cyclization catalysed by the epoxide hydrolase AuaI form the pyran of 985.830 The genetic work in S. aurantiaca was complicated by the fact that the aurachin biosynthetic genes were located across three different genetic loci. N-hydroxylation in aurachin RE biosynthesis is catalysed by a cytochrome P450, as opposed to a Rieske[2Fe–2S] oxygenase.831 Divergent pathways to the other aurachins have been postulated, but no definitive steps have been characterized.818,828
Scheme 22.
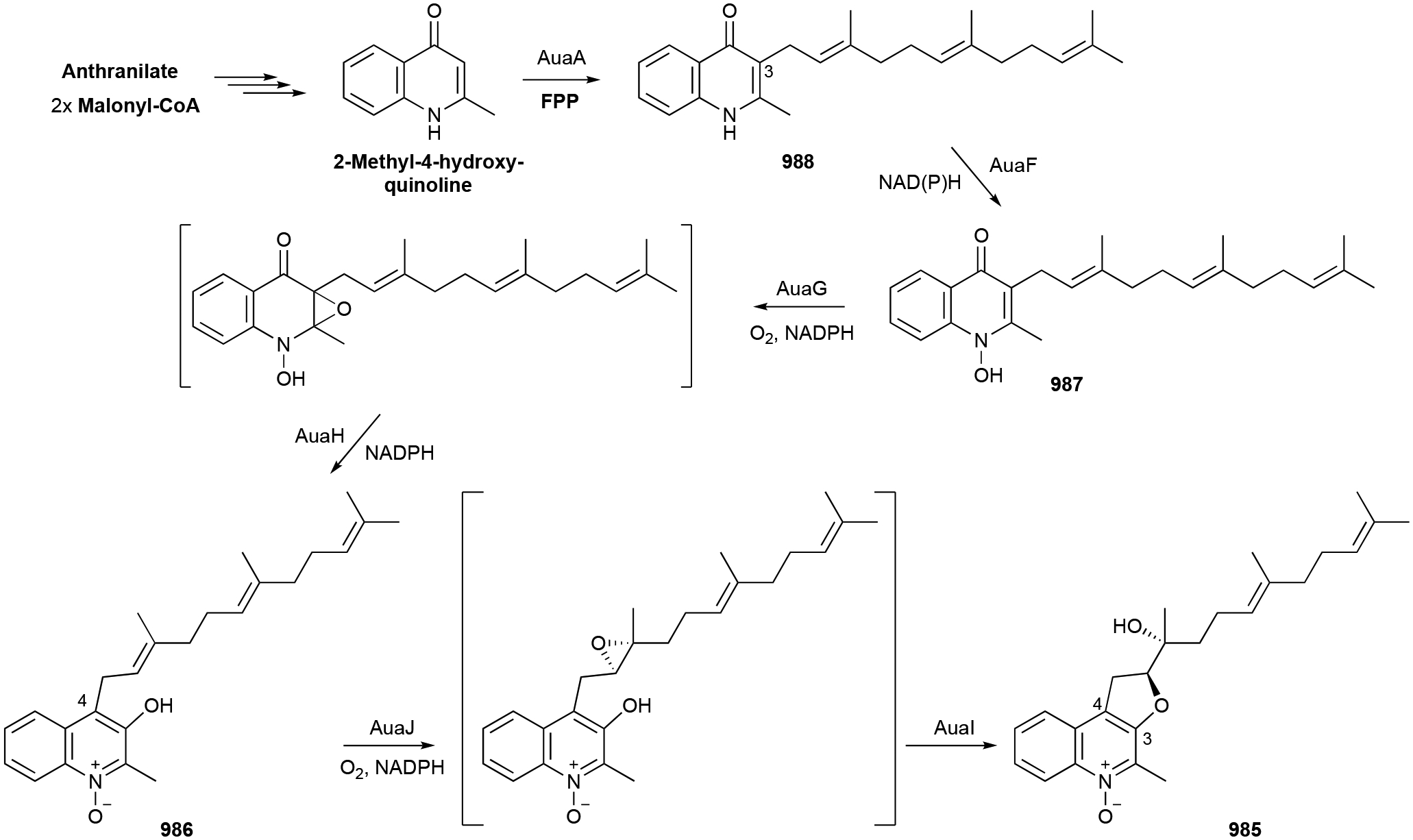
Biosynthesis of aurachins A (985), B (986), C (987), and D (988).
Biological activity.
Aurachins 985–988 were potent Gram-positive antibacterials and antifungals with MICs as low as 0.05 μg mL−1.814 Aurachin RE (996) was also active against some Gram-negative bacteria, although not E. coli suggesting terpene hydroxylation may positively affect solubility or its ability to transverse the cell membrane.816 The poor antibacterial activity of 999 hints that the shorter geranyl chain or 4-methoxy group negatively affects activity.818 The CJ-13 analogues (1001–1008) and 1009 were selective and potent antibiotics against Helicobacter pylori.820,821 For example, CJ-13,136 (1001) was bactericidal against H. pylori at 10 ng mL−1 and bacteriostatic at 0.1 ng mL−1.
Being structurally similar to menaquinones, the aurachins are potent inhibitors of the electron transport chain. Aurachins 985–988 were initially found to block NADH oxidation in beef heart submitochondrial particles and later found to be excellent inhibitors of photosystem II and cytochrome b6/f-complex (IC50 values of 63 and 100 nM, respectively, for 987).814,832 987 also inhibited the quinol oxidation sites of the terminal oxidases cytochrome bo and bd while 988 was selective for cytochrome bd.833
1009 and some of the CJ-13 analogues preferentially inhibited (~10-fold preference and IC50 = 0.4 μM) gastric and colorectal cancer cells when grown in the presence of their respective stromal cells compared to the cancer cells alone.821
Aurachin E (989), which had no antibacterial activity nor inhibited mitochondrial respiration, was a non-cytotoxic anti-plasmodial agent with IC50 values as low as 0.4 ng mL−1.815 Aurachins 986–988 also showed anti-plasmodial activity, but their antibacterial activities suggest a different mode of action. Aurachins 987 and 998 were also weak inhibitors of the glycogen synthase kinase 3β.817
1011 is an extremely potent algal morphogenesis inducer; in a cell differentiation assay, its minimum effective concentration was between 1 fg mL−1and 1 ag mL−1.823 Its presumed precursor, 1010, was found to induce G1 arrest in HCT-116 cells resulting in mild cytotoxicity.822
3.11. Quinones
Prenylated quinones are ubiquitous membrane-bound compounds that have essential physiological roles in electron transfer, energy generating processes, signaling, oxidative stress, antibiotic resistance, and virulence.834 These hybrid natural products have polar aromatic head groups with hydrophobic polyprenyl side chains appended at C3 of the aromatic core. The quinone ring can undergo reversible two-step redox reactions yielding the oxidized 1,4-benzoquinone and the reduced hydroquinone (quinol, 1,4-dihydroxybenzene). This redox potential allows prenylated quinones to function as membrane-bound electron and proton shuttles in the electron transport chain. There are two major families of isoprenoid quinones: benzoquinones and the bicyclic naphthoquinones. Members of these families, separated into subfamilies based on their quinone ring substitution patterns, commonly have prenyl chains of various lengths and degrees of saturation. The degree of prenyl saturation is organism and environment dependent.835 Each organism produces its own quinone pool, consisting of quinones of both major and minor abundances; these have been used as chemotaxonomic markers.835,836 In this section, we will not describe the numerous variants of isoprenoid quinones, but instead focus on introducing the most common scaffolds in bacteria and highlighting non-canonical linear and cyclized prenylated quinones. For a more in-depth look at isoprenoid quinones in bacteria, we direct readers to the excellent reviews cited herein.834,835,837,838
3.11.1. Benzoquinones
The most ubiquitous, and hence the name,839 benzoquinones are the ubiquinones (UQs). UQ-8, possessing eight prenyl units, is the most well-known UQ, but UQs (1013) have been found with 1–12 prenyl units, various hydrogenation or epoxidation modifications, and are mainly found in aerobic Gram-negative bacteria.835,837 Other commonly found benzoquinones are rhodoquinones (RQs), plastoquinones (PQs), and α-tocopherolquinone (α-TQ).835,838 RQs (1014) are amino analogues of UQs and present in the purple bacterium R. rubrum. PQ (1015) and α-TQ, (1016) are key components of cyanobacteria (and plant) photosynthesis. PQs are nonaprenyl dimethylbenzoquinones and 1016 is a trimethylbenzoquinone with a saturated and hydroxylated geranyl side chain.
Pseudoalteromone A (1017), produced in Pseudoalteromonas sp. CGH2XX, is a UQ-2-like benzoquinone, but has a unique oxidized C9 nor-monoterpenoid chain.840
Bioassay-guided NP discovery efforts using Streptomyces nitrosporeus YBH10–5 resulted in the isolation of unique prenylated benzoquinones.706 Farnesylquinone (1018) is the oxidized form of 2-farnesyl-5-methyl-1,4-benzoquinone and appears to be the precursor to the 7-deacetoxyyanuthone (1019) and nitrosporeunol G (1020). 1020 is a drimane-type epoxyquinol and structurally very similar to the fungal macrophorin A.841 Simple pyran formation of 1020 yielding a 6/6/6/6 tetracycle would give BE-40644 (1021), a compound isolated from Actinoplanes sp. A40644 almost 20 years earlier.842,843
Biosynthesis.
The bacterial biosynthesis of UQs is well understood while the pathways of RQs, PQs, and α-TQ are still incomplete.834,838,844 p-Hydroxybenzoate, a product from chorismate via the shikimate pathway, is prenylated by the PT UbiA. The UbiA superfamily of PTs are membrane-bound aromatic PTs distinct from the ABBA PTs.845 They typically have prenyl acceptor (i.e., quinone head group) specificity and prenyl donor flexibility. After prenylation, 3-polyprenol-4-hydroxybenzoic acid is decarboxylated by UbiD, a non-oxidative decarboxylase that requires a novel prenylated-FMN (1030, vide infra chapter 3.12.2) cofactor made by UbiX.846,847 The decarboxylation is facilitated via a 1,3-dipolar cycloaddition addition reaction with 1030.848 After decarboxylation, a series of hydroxylations and methylations are required to form UQ.834,838
No biosynthetic studies have ensued for 1017 or 1020, although their similarity to macrophorin A suggests cyclization occurs via a noncanonical integral membrane TS.27
Biological activity.
1021 was originally found to be a selective thioredoxin (TRX) system inhibitor with IC50 values of 0.12 and 0.80 μg mL−1 for E. coli and human TRX, respectively.842 The related, but non-cyclized, analogues 1018 and 1019 decreased lipid accumulation with 1018 comparable to lovastatin.706 1018 increased the production of PPAR-α controlled proteins by transcriptional upregulation.
1017 was cytotoxic against MOLT-4 human acute lymphoblastic leukemia cells (IC50 = 11.7 μM) and a possible anti-inflammatory agent as it inhibited the release of elastase by neutrophils.840
3.11.2. Naphthoquinones
Menaquinones (MKs, vitamin K2) are the most ancient type of prenylated quinones and are solely found in most Gram-positive and anaerobic Gram-negative bacteria.835,837 They have low redox potentials compared to ubiquinones and are highly reactive with molecular oxygen implying that their ancient use as respiratory quinones was due to the reducing character of the atmosphere at that time.838 MKs (1022) are 2-methyl-3-polyprenyl-1,4-naphthoquinones with 1–14 prenyl units, although 6–10 units are most common.835 Demethyl-MKs (DMKs, 1023) are also common in proteo- and Gram-positive bacteria.835 Most identified MKs and DMKs have fully unsaturated polyprenyl chains, but partially (e.g., phylloquinone, 1024) or fully saturated are possible. Naphthoquinone ring C- and O- methylations can also be present. Chlorobiumquinone (1025), is a uniquely 1´-oxidized MK found in the photosynthetic green sulfur bacterium Chlorobium thiosulphatophilum.849,850 Other unique bacterial MKs include methionaquinone (1026) from Hydrogenobacter thermophilus TK-6,851,852 the terminally sulfated sulfo-MK (1027) from Mycobacterium tuberculosis,853 and a carotenoid-like cyclic MK (1028) from Nocardia spp.854
Biosynthesis.
There are two biosynthetic pathways for the biosynthesis of MKs and both are well characterized (Scheme 23).838,855 Like the UQs, the precursor for MKs in both pathways is chorismate. In the classical pathway, deduced from studies in E. coli, chorismate is transformed into 1,4-dihydroxy-2-naphthoate via o-succinylbenzoyl-CoA by six enzymes from the men pathway. The UbiA-type PT, MenA, then catalyzes C2-decarboxylative prenylation before the biosynthesis is completed by methylation at C3.838,845,856 The alternative pathway, found in H. pylori, Campylobacter jejuni, and S. coelicolor, generates 1,4-dihydroxy-6-naphthoate through futalosine.855,857,858 The last two steps of the futalosine (mqn) pathway may parallel those in the men pathway,838,855 but there are proposals of a distinct PT.859 As opposed to the early prenylation in UQ biosynthesis, both MK pathways require a late prenylation reaction (Scheme 23). MK tailoring reactions, such as methylations or sulfations are catalysed by radical SAM MTs or tandem P450–sulfate transferase reactions, respectively.860,861
Scheme 23.
Biosynthesis of menaquinones (1022).
Biological activity.
In vivo studies of 1027 revealed that it is a negative regulator of virulence in mouse infections indicating a possible role in human-pathogen interactions.862
3.12. Miscellaneous
There are several other families of bacterial meroterpenoids that do not fit well into any of the categories mentioned above. These include the aromatic dibenzodiazepinones, flavins, and aminobenzoates and the nonaromatic phosphoglycolipids and saccharides. We also included the porpyrin-rich chlorophylls and hemes in this section. Although fairly scarce in number at the time of writing this review, it would be unwise to assume these NPs are not the beginning of expanding families of bacterial terpenoids.
3.12.1. Dibenzodiazepinones
Diazepinomicin (1029) is a unique microbial metabolite produced by Micromonospora spp.863 Its core is an exceptionally rare tricyclic dibenzodiazepinone alkaloid with the amide N carrying a farnesyl moiety.
Biosynthesis.
BGC analysis and labeled feeding experiments support that the dibenzodiazepinone core is built from the condensation of a 3-hydroxyanthranilic acid and 2-amino-6-hydroxy[1,4]benzoquinone (Scheme 24).864 Feeding experiments also confirmed that the 3-hydroxyanthranilic acid precursor is obtained via the degradation of indole. The MVA-derived farnesyl unit864 is fastened to the amide N by DzmP, an ABBA PT.865
Scheme 24.
Biosynthesis of diazepinomicin (1029).
Biological activity.
1029 is an antitumor antibiotic. It has modest (8–32 μg mL−1) antibacterial activity against Gram-positive bacteria and has low μM cytotoxic activity.866,867 Its cytotoxicity is the result of its effect on the Ras-MAPK signaling pathway, specifically inhibiting the phosphorylation of downstream effector proteins.867 Although it was an ineffective monotherapy in a glioblastoma clinical trial,868 it is currently under evaluation as a therapeutic for the treatment of Phelan-McDermid Syndrome and co-morbid epilepsy.869
3.12.2. Flavins
Prenylated flavin mononucleotide (1030, prenyl-FMN) is a new cofactor that is utilized by the family of UbiD decarboxylases to catalyze α,β-unsaturated decarboxylation via 1,3-dipolar cycloaddition.846,847 The UbiD family is most well-known for its role in ubiquinone biosynthesis, but recent studies show that this cofactor is also involved in microbial NP biosynthesis.870,871 The isopentenyl adduct of prenyl-FMN, which is connected at N5 and C6 of FMN, forms a 6-membered ring.846 In hindsight, the discovery of hunanamycin A (1031) from Bacillus hunanensis, a NP with a pyrido[1,2,3-de]quinoxaline-2,3-dione core, was a hint at the presence of 1030.872 It is very likely an oxidative degradation product of 1030.
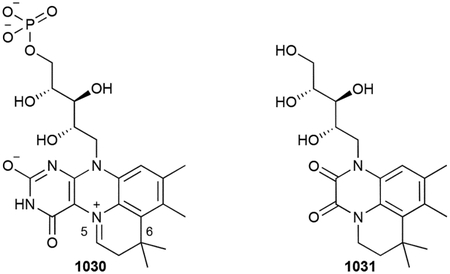
Biosynthesis.
One PT, UbiX or its homologues, acts as both a PT and TS to produce 1030.847,873 N-prenylation sets up an ensuing TS-like mechanism, where protonation by the discarded inorganic pyrophosphate creates a prenyl carbocation that is quenched by the electron-rich C5 of FMN. Interestingly, some of these PTs are selective for dimethylallyl monophosphate or DMAPP as the prenyl donor, while some can utilize both.
Biological activity.
1031 was a selective antibiotic active against Salmonella enterica with an MIC of 12.4 μM.872
3.12.3. Phosphoglycolipids
Phosphoglycolipds are important components of bacterial cell walls; the lipid portions are derived from fatty acids. However, a C55 isoprenoid lipid is used to shuttle the glycopeptide fragment of lipid II, the peptidoglycan building block, across the membrane; the C55 isoprenoid also serves as a membrane anchor for the growing peptidoglycan chain.874 The C55 alcohol, bactoprenol (1032, undecaprenol), is found in high concentrations in Gram-positive bacteria and was structurally reported to be composed of eight cis and two trans double bonds.875,876 As bactoprenol is utilized in its phosphorylated form, its concentration is regulated by an undecaprenol kinase/phosphatase.877 Although bactoprenol is not typically seen in Gram-negative bacteria, an undecaprenyl phosphogalactosamine (1033) was isolated from the Gram-negative Francisella novicida.878
By far, the most structurally interesting and biologically relevant phosphoglycolipid NPs are the moenomycins. Members of the moenomycin class of antibiotics have three structural elements in common: a 3-phosphoglycerate (3-PG) backbone, an unusual C25 isoprenoid chain connected to 3-PG as an ether, and a tetrasaccharide tethered to 3-PG via a phosphodiester linkage. Most structural differences seen in the moenomycins reside in the tetrasaccharide component.
Moenomycin A (1034) was originally discovered in 1965 as an antibiotic and has been since produced from a variety of Streptomyces spp.879–881 The complexity of its structure as well as its physicochemical properties delayed its complete structural determination for 25 years.882,883 1034 is a pentasaccharide with a 2-aminocyclopentane-1,3-dione unit (ring A) attached by an amide to the terminal sugar (ring B). Its isoprenoid chain was determined to be moenocinol, an irregularly linked C25 linear chain with a quaternary carbon at C8.882,884
There are several variants of 1034, some of which have still not been fully structurally characterized.884–887 Those that have been characterized include the moenomycins A12, C1, C3, and C4 (1035–1038), nosokomycins A–D (1039–1042), and pholipomycin (1043).888–893 These phosphoglycolipids all have the moenocinol terpenoid unit and differ in the presence or absence of the 2-aminocyclopentane-1,3-dione (ring A) and the branching glucose (ring D) units, as well as the structure and functionalization of the sugar units C–F. AC326-α (1044), isolated from Actinomyces sp. AC326, is identical to that of moenomycin A with the exception that its terpenoid chain is the monocyclic diumycinol.894 Nosokophic acid (1045), a predicted intermediate in nosokomycin biosynthesis bearing a Z,E-farnesyl moiety, was also isolated from Streptomyces sp. K04–0144.895
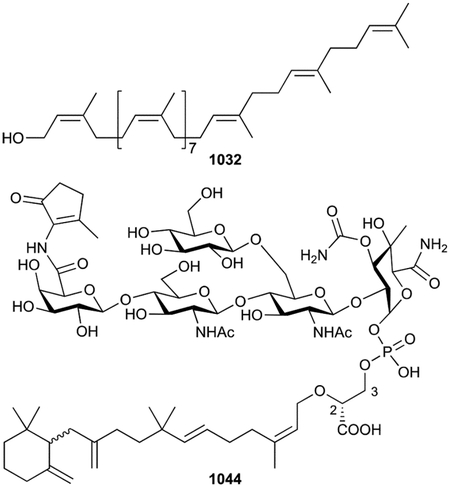
Biosynthesis.
The biogenic origins of moenomycins from Streptomyces ghanaensis has been extensively studied resulting in a proposed 17 step biosynthetic pathway.896 Complicating the discovery of the moe BGC was the fact that all required precursors, except for the ring A chromophore, could be directly obtained from primary metabolism. In addition, not only was the BGC located across two genetic locations in the chromosome, the low sequence similarity of most of the moe genes to known NP biosynthetic genes at the time made for a challenging problem.896
The biosynthesis and attachment of the moenocinol moiety is of most relevance to this review (Scheme 25). Early tracer experiments supported that the unique C25 moenocinol moiety was of MEP origin and led to a mechanistic proposal of the joining and rearrangement of distinct farnesyl and geranyl units.897 In vitro characterization of MoeO5, a TIM barrel PT of the geranylgeranylglycerol phosphate synthase family, revealed that the prenylation of 3-PG with E,E-FPP yielded 2-(Z,E)-farnesyl-3-PG as the first committed step in moenomycin biosynthesis.896,898 The transformation of the C15 tail into the C25 moenocinol unit occurs at the trisaccharide stage and is catalyzed by MoeN5.896,899 Detailed mechanistic experiments have not been performed but an unusual head-to-middle geranylation reaction must occur between the two terpenoid components followed by two rearrangements through cyclopentyl and cyclohexyl carbocations to produce the linear moenocinol sidechain-containing trisaccharide (Scheme 25).881,897
Scheme 25.
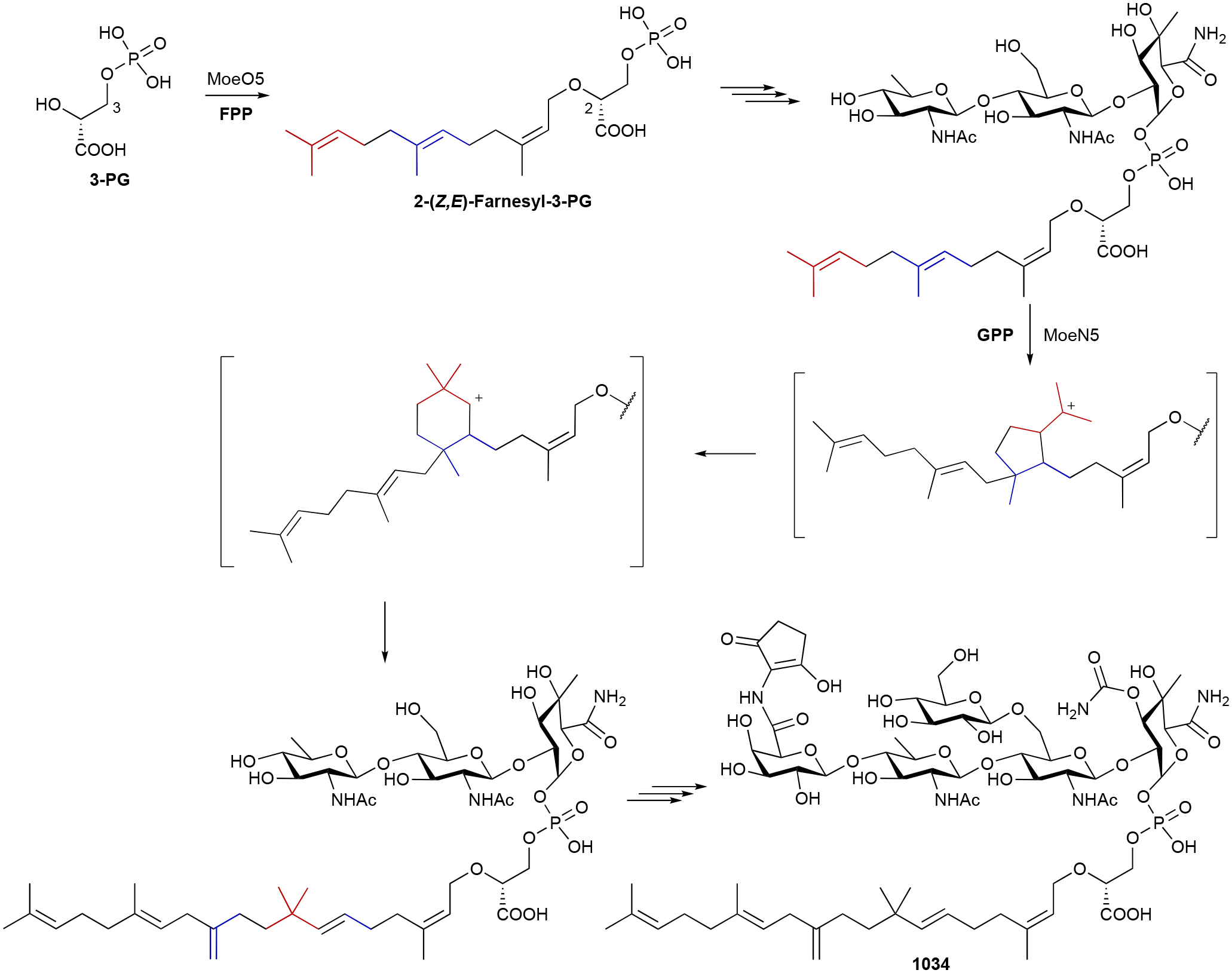
Biosynthesis of moenomycin A (1034).
Biological activity.
The moenomycins are potent antibiotics. Their MICs range from 1–100 ng mL−1 for Gram-positive bacteria and 0.3–150 μg mL−1 against Gram-negative strains.879,894,900,901 They are bacteriostatic at low concentrations (1 μg mL−1) and bactericidal at high concentrations (10 μg mL−1).874 Being the only natural antibiotics that directly inhibit the transglycosylases responsible for the polymerization of the bacterial peptidoglycan layer, the moenomycins have been extensively studied and reviewed.881,901 Unfortunately, these antibiotics suffer from poor pharmacokinetics and therefore are not clinically relevant in humans; they have, however, been successfully introduced as animal growth promoters. Interestingly, nosokophic acid, which does not have antibacterial activity, potentiates imipenem activity against MRSA 512-fold.895
3.12.4. Saccharides
While there are many NPs that contain both terpenoid and sugar moieties, there are only three bacterial NPs with prenyl groups attached directly onto sugars. Lentztrehaloses A–C (1046, 1047, 1048), isolated from the actinomycete Lentzea sp. ML457 mF8, are prenylated versions of the disaccharide trehalose.902–904 1047 retains an unmodified dimethylallyl group, while 1046 and 1048 appear to be the result of epoxide ring opening. The dimethylallyl moiety of 1048 was cyclized onto the trehalose forming an additional 1,4-dioxane ring. The biosynthesis of these terpenoid sugars has not been studied.
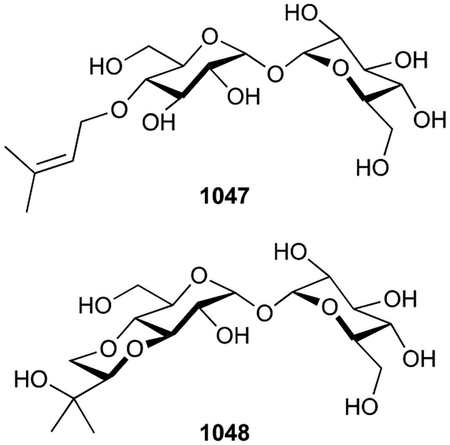
Biological activity.
Trehalose is used as a sweetener and is metabolized into glucose in vivo.905 The lentztrehaloses are potential artificial sweetener substitutes for trehalose as they are nonhydrolyzable variants.902,904 They do not appear to be toxic to microbes or mammalian cells, although they were shown to have antitumor activity in mice bearing S-180 sarcoma and induce autophagy in human cancer cells on a level comparable to that of trehalose.902,904
3.12.5. Chlorophylls
Chlorophylls (Chls) are pigments that are essential for photosynthesis and are widely distributed throughout nature, being present in plants, algae, photosynthetic bacteria, and even some animals.906,907 While all Chls have a conserved central porphyrin core which coordinate a Mg2+ ion, there are over 100 characterized Chls with differences occurring in their ring structures, substituent patterns, and esterifying alcohols.907,908 All photosynthetic Chls have a fifth isocyclic pentanone ring as exemplified by Chl a (1049), the most widely distributed Chl in nature;907 bacteriochlorophylls (BChls), which is a misnomer considering they are in fact chlorins with two reduced pyrrole rings are particularly prevalent in bacteria.909 The major functional difference between Chls and BChls is whether they partake in oxygenic photosynthesis (i.e., plants and cyanobacteria) or anoxygenic photosynthesis (e.g., green sulfur bacteria, purple bacteria), respectively.908,909 We will not dwell on all the variations of Chls in this review as they have been exhaustively reviewed.907–910 However, it should be noted here that many of the major Chls, including those found in bacteria, are esterified with terpenoid moieties making them de facto meroterpenoids.
Most chlorophylls, such as 1049, have a C20 phytanyl moiety attached to their C173 carboxylic acid.907,910 BChls, on the other hand, have one of six prenyl chain variations; this prenyl preference appears to be species specific. The predominant esterifying alcohol of the archetypal BChls a and b (1050, 1051) is phytol while that of BChls c, d, e, and g (1052–1055) is farnesol.911–917 Some BChls have been isolated with geranylgeranyl, dihydrogeranylgeranyl, tetrahydrogeranylgeranyl, or Δ2,6-phytadienyl moieties (1056–1060).911,914,918–921
Biosynthesis.
We will leave discussion regarding the biosynthesis of the porphyrin core to the numerous reviews currently in the literature.907,908,910,922–924 We will note, however, that the final steps in (B)Chl biosynthesis is the esterification of the C173 carboxylic acid and any reduction of the poly-olefin side chain. Attachment of the C15 or C20 isoprenoid moiety occurs via a subfamily of UbiA PTs, named chlorophyll or bacteriochlorophyll synthases. ChlG and BchG from Synechocystis and Rhodobacter spp., respectively, are Chl a and BChl a synthases; based on total conversion in vitro, they both appear to minorly (~2-fold) prefer phytyl diphosphate over GGPP.925 Other (B)Chl PTs responsible for geranylgeranylation and farnesylation have been identified and characterized.922,926,927 Reduction of GGPP to phytyl diphosphate, or geranylgeranylated (B)Chls to the various reduced counterparts is catalyzed by the NADPH-dependent reductase BchP and its homologues.922,928 It is unclear whether reduction occurs prior to or after prenylation as in vitro experiments with various PTs can accept diverse prenyl diphosphate substrates, although the range of isolated reduced C20 esters on Chls suggests reduction occurs after prenylation.907,924
3.12.6. Hemes
Hemes are another biological scaffold that contains a metallated porphyrin core. These redox active prosthetic groups play essential roles as cofactors for a variety of essential cellular functions in most living organisms.929–931 In bacteria, heme plays an important role in respiration as the prosthetic group in cytochrome oxidases, membrane-bound heme-copper oxidative proton pumps in the electron transport chain that catalyze electron transfer from quinols or cytochrome c to molecular oxygen.932 While there are several types of hemes, there are two prenylated hemes, heme A (1061) and heme O (1062).933 Structurally, hemes A and O only differ in the presence of a formyl group on one of the pyrrole rings; both possess a hydroxyethylfarnesyl chain on C2 of pyrrole ring A.933,934 Heme A has long been known, in both eukaryotic mitochondria and bacteria (e.g., Paracoccus and Rhodobacter), to be present in aa3-type cytochrome c oxidase.935,936 Heme O was extracted from cytochrome o, one of two terminal ubiquinol oxidases in E. coli.934
Biosynthesis.
The biosynthesis of the porphyrin ring is similar to that in the Chls and is reviewed elsewhere.923,937 The farnesyl group on hemes A and O originate from the prenylation of farnesylation of the exocyclic vinyl group of pyrrole ring A in heme B (protoporphyrin IX). This PT reaction is catalyzed by heme O synthase,938 a UbiA PT that this phylogenetically divergent from those seen in Chl biosynthesis.845 This was confirmed by both in vivo knockout and complementation experiments as well as in vitro biochemical characterization.938,939 1062 is a direct precursor of 1061, only requiring two successive oxidations on a methyl group to provide the aldehyde functionality.940 Interestingly, heme O and heme A synthases directly interact, likely providing a direct route of formation from heme B to 1061.941
4. Conclusion
In this review, we described the current state of the bacterial terpenome. Our goal was to showcase the diversity of structures, biosynthetic enzymes, and biological activities of these natural products while providing access to this collection of compounds, structures, and references (all entries included in the ESI documents and were deposited into the open-access Natural Products Atlas).32
It is clear that bacteria are a major resource for novel terpenoids and terpenoid biosynthetic pathways and enzymes. Terpene cyclization and prenylation, fundamental steps in terpenoid biosynthesis, are known to be catalyzed by TSs and PTs. However, bacteria, as a whole, have more than one family of enzymes to perform these essential chemical reactions. Both canonical and non-canonical TSs cyclize linear prenyl diphosphates, multiple types of PTs can append prenyl groups onto a diverse array of scaffolds, and some families of enzymes (e.g., UbiA PTs) can perform both PT and TS reactions. With every new discovery and enzyme characterization, new opportunities for novel NP discovery will be presented.
Currently, genome mining for bacterial terpenoids is particularly appealing. Over the last two decades, genome mining-based NP discovery programs have mainly utilized the most recognizable TSs and PTs to target and quickly identify new terpenoids. These efforts clearly led to an overall improvement in bacterial terpenoid discovery and characterization (Fig. 1) and provide optimism for the future of bacterial terpenoid discovery. Imagine all of the exciting new avenues to pursue and NPs that will be discovered by using these new TSs, PT, and other functionalization enzymes as probes in future genome mining efforts!
NP research in bacteria has dramatically changed since advances in DNA sequencing technologies have provided the ability to easily obtain microbial genomes. Genome mining allows the simultaneous discovery of NPs and their BGCs and enzymes. Even if research groups continue to use traditional NP discovery methods, it is quick and fairly straight-forward to sequence the producing organisms genome and identify an associated BGC. These types of multidisciplinary studies not only facilitate future NP discovery efforts and provide opportunities for NP biosynthetic studies, they also inspire and accelerate orthogonal research directions. These may include the discovery of enzymes with novel functions,846,847 understanding new biological modes of action,221,222 and the exploitation of bacterial terpenoid oxygenases for the chemoenzymatic synthesis of non-bacterial NPs.942
Finally, while it is clear that bacteria have evolved their own systems to create terpenoid diversity, bacteria also biosynthesize some of the same classes of terpenoids, even some of the same compounds, found in plants, fungi, and other organisms. These relationships provide accessible and renewable prokaryotic systems for eukaryotic natural products biosynthesis and enzymology. Consider the phytohormone gibberellins or the sponge-associated sesterterpenoids. The discovery and characterization of gibberellins in bacteria provided an evolutionary perspective on gibberellin biosynthesis. Identification of a NP from a non-renewable resource precludes the ability to further its development, isolated congeners, or study its biosynthesis. Isolating similar NPs from a culturable bacterium provides opportunities that would otherwise be incredibly difficult.
In conclusion, we hope it is evident from this review that (i) novel terpenoids can and should still be targeted for discovery efforts, (ii) the biosynthetic pathways of bacterial terpenoids are far from complete, (iii) the pathways that have been extensively studied have revealed a fascinating array of cyclization, prenylation, and functionalization enzymes, (iv) and most of these NPs are biologically active with a few key scaffolds paving a path towards potential drug development.
Supplementary Material
6. Acknowledgements
Studies on natural products discovery, biosynthesis, and enzymology in the Rudolf laboratory are currently supported in part by a National Institutes of Health grant R00 GM124461 and the University of Florida.
Footnotes
Electronic Supplementary Information (ESI) available: See DOI: 10.1039/d0np00066c
Conflicts of interest
There are no conflicts of interest to declare.
7 References
- 1.Dictionary of Natural Products, http://dnp.chemnetbase.com, (accessed May–July 2020).
- 2.Christianson DW, Science, 2007, 316, 60–61. [DOI] [PubMed] [Google Scholar]
- 3.Sacchettini JC and Poulter CD, Science, 1997, 277, 1788–1789. [DOI] [PubMed] [Google Scholar]
- 4.IUPAC Gold Book, https://goldbook.iupac.org, (accessed May 15, 2020).
- 5.Berthelot M and Andre G, Comptes Rendus l’Académie des Sci, 1891, 112, 589–599. [Google Scholar]
- 6.Gerber NN, Tetrahedron Lett, 1968, 9, 2971–2974. [DOI] [PubMed] [Google Scholar]
- 7.Medsker LL, Jenkins D, Thomas JF and Koch C, Environ. Sci. Technol, 1969, 3, 476–477. [DOI] [PubMed] [Google Scholar]
- 8.Bentley R and Meganathan R, FEBS Lett, 1981, 125, 220–222. [DOI] [PubMed] [Google Scholar]
- 9.Cane DE and Ikeda H, Acc. Chem. Res, 2012, 45, 463–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoshino Y and Gaucher EA, Mol. Biol. Evol, 2018, 35, 2185–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frank A and Groll M, Chem. Rev, 2017, 117, 5675–5703. [DOI] [PubMed] [Google Scholar]
- 12.Miziorko HM, Arch. Biochem. Biophys, 2011, 505, 131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuzuyama T and Seto H, Nat. Prod. Rep, 2003, 20, 171–183. [DOI] [PubMed] [Google Scholar]
- 14.Takagi M, Kuzuyama T, Takahashi S and Seto H, J. Bacteriol, 2000, 182, 4153–4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuzuyama T, J. Antibiot, 2017, 70, 811–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Razin S, Yogev D and Naot Y, Microbiol. Mol. Biol. Rev, 1998, 62, 1094–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gerber NN, Phytochemistry, 1971, 10, 185–189. [Google Scholar]
- 18.Gerber NN, Phytochemistry, 1972, 11, 385–388. [Google Scholar]
- 19.Omura S, Ikeda H, Ishikawa J, Hanamoto A, Takahashi C, Shinose M, Takahashi Y, Horikawa H, Nakazawa H, Osonoe T, Kikuchi H, Shiba T, Sakaki Y and Hattori M, Proc. Natl. Acad. Sci. U. S. A, 2001, 98, 12215–12220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bentley SD, Chater KF, Cerdeño-Tárraga AM, Challis GL, Thomson NR, James KD, Harris DE, Quail MA, Kieser H, Harper D, Bateman A, Brown S, Chandra G, Chen CW, Collins M, Cronin A, Fraser A, Goble A, Hidalgo J, Hornsby T, Howarth S, Huang CH, Kieser T, Larke L, Murphy L, Oliver K, O’Neil S, Rabbinowitsch E, Rajandream MA, Rutherford K, Rutter S, Seeger K, Saunders D, Sharp S, Squares R, Squares S, Taylor K, Warren T, Wietzorrek A, Woodward J, Barrell BG, Parkhill J and Hopwood DA, Nature, 2002, 417, 141–147. [DOI] [PubMed] [Google Scholar]
- 21.Ikeda H, Ishikawa J, Hanamoto A, Shinose M, Kikuchi H, Shiba T, Sakaki Y, Hattori M and Omura S, Nat. Biotechnol, 2003, 21, 526–531. [DOI] [PubMed] [Google Scholar]
- 22.Christianson DW, Chem. Rev, 2017, 117, 11570–11648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dickschat JS, Nat. Prod. Rep, 2016, 33, 87–110. [DOI] [PubMed] [Google Scholar]
- 24.Ikeda H, Shin-ya K and Omura S, J. Ind. Microbiol. Biotechnol, 2014, 41, 233–250. [DOI] [PubMed] [Google Scholar]
- 25.Yamada Y, Kuzuyama T, Komatsu M, Shin-ya K, Omura S, Cane DE and Ikeda H, Proc. Natl. Acad. Sci. U. S. A, 2015, 112, 857–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mori T, J. Nat. Med, 2020, 74, 501–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rudolf JD and Chang CY, Nat. Prod. Rep, 2020, 37, 425–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dickschat JS, Angew. Chemie, Int. Ed, 2019, 58, 15964–15976. [DOI] [PubMed] [Google Scholar]
- 29.Dairi T, J. Antibiot, 2005, 58, 227–243. [DOI] [PubMed] [Google Scholar]
- 30.Smanski MJ, Peterson RM, Huang SX and Shen B, Curr. Opin. Chem. Biol, 2012, 16, 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Helfrich EJN, Lin GM, Voigt CA and Clardy J, Beilstein J. Org. Chem, 2019, 15, 2889–2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Van Santen JA, Jacob G, Singh AL, Aniebok V, Balunas MJ, Bunsko D, Neto FC, Castaño-Espriu L, Chang C, Clark TN, Cleary Little JL, Delgadillo DA, Dorrestein PC, Duncan KR, Egan JM, Galey MM, Haeckl FPJ, Hua A, Hughes AH, Iskakova D, Khadilkar A, Lee JH, Lee S, Legrow N, Liu DY, Macho JM, McCaughey CS, Medema MH, Neupane RP, O’Donnell TJ, Paula JS, Sanchez LM, Shaikh AF, Soldatou S, Terlouw BR, Tran TA, Valentine M, Van Der Hooft JJJ, Vo DA, Wang M, Wilson D, Zink KE and Linington RG, ACS Cent. Sci, 2019, 5, 1824–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klementz D, Döring K, Lucas X, Telukunta KK, Erxleben A, Deubel D, Erber A, Santillana I, Thomas OS, Bechthold A and Günther S, Nucleic Acids Res, 2016, 44, D509–D514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zeng T, Liu Z, Zhuang J, Jiang Y, He W, Diao H, Lv N, Jian Y, Liang D, Qiu Y, Zhang R, Zhang F, Tang X and Wu R, J. Chem. Inf. Model, 2020, 60, 2082–2090. [DOI] [PubMed] [Google Scholar]
- 35.Kuzma J, Nemecek-Marshall M, Pollock WH and Fall R, Curr. Microbiol, 1995, 30, 97–103. [DOI] [PubMed] [Google Scholar]
- 36.Schöller CEG, Gürtler H, Pedersen R, Molin S and Wilkins K, J. Agric. Food Chem, 2002, 50, 2615–2621. [DOI] [PubMed] [Google Scholar]
- 37.Dickschat JS, Martens T, Brinkhoff T, Simon M and Schulz S, Chem. Biodivers, 2005, 2, 837–865. [DOI] [PubMed] [Google Scholar]
- 38.Pacifico F, Harrison SP, Jones CD and Sitch S, Atmos. Environ, 2009, 43, 6121–6135. [Google Scholar]
- 39.McGenity TJ, Crombie AT and Murrell JC, ISME J, 2018, 12, 931–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Köksal M, Zimmer I, Schnitzler JP and Christianson DW, J. Mol. Biol, 2010, 402, 363–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sivy TL, Shirk MC and Fall R, Biochem. Biophys. Res. Commun, 2002, 294, 71–75. [DOI] [PubMed] [Google Scholar]
- 42.Hess BM, Xue J, Markillie LM, Taylor RC, Wiley HS, Ahring BK and Linggi B, PLoS One, 2013, 8, e66104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pollak FC and Berger RG, Appl. Environ. Microbiol, 1996, 62, 1295–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schulz S and Dickschat JS, Nat. Prod. Rep, 2007, 24, 814–842. [DOI] [PubMed] [Google Scholar]
- 45.Schulz S, Biwer P, Harig T, Koteska D and Schlawis C, in Comprehensive Natural Products III, eds. Liu H and Begley T, Elsevier, 3rd Ed., 2020, pp. 161–178. [Google Scholar]
- 46.Groenhagen U, Leandrini De Oliveira AL, Fielding E, Moore BS and Schulz S, ChemBioChem, 2016, 17, 1978–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dickschat JS, Bode HB, Wenzel SC, Müller R and Schulz S, ChemBioChem, 2005, 6, 2023–2033. [DOI] [PubMed] [Google Scholar]
- 48.Wilkins K, Chemosphere, 1996, 32, 1427–1434. [Google Scholar]
- 49.Dickschat JS, Helmke E and Schulz S, Chem. Biodivers, 2005, 2, 318–353. [DOI] [PubMed] [Google Scholar]
- 50.Schulz S, Fuhlendorff J and Reichenbach H, Tetrahedron, 2004, 60, 3863–3872. [Google Scholar]
- 51.Kiviranta H, Tuomainen A, Reiman M, Laitinen S, Liesivuori J and Nevalainen A, Cent. Eur. J. Public Health, 1998, 6, 296–299. [PubMed] [Google Scholar]
- 52.Rabe P, Citron CA and Dickschat JS, ChemBioChem, 2013, 14, 2345–2354. [DOI] [PubMed] [Google Scholar]
- 53.Citron CA, Barra L, Wink J and Dickschat JS, Org. Biomol. Chem, 2015, 13, 2673–2683. [DOI] [PubMed] [Google Scholar]
- 54.Nakano C, Kim HK and Ohnishi Y, ChemBioChem, 2011, 12, 1988–1991. [DOI] [PubMed] [Google Scholar]
- 55.Citron CA, Gleitzmann J, Laurenzano G, Pukall R and Dickschat JS, ChemBioChem, 2012, 13, 202–214. [DOI] [PubMed] [Google Scholar]
- 56.Dickschat JS, Nawrath T, Thiel V, Kunze B, Müller R and Schulz S, Angew. Chemie, Int. Ed, 2007, 46, 8287–8290. [DOI] [PubMed] [Google Scholar]
- 57.Tellez MR, Schrader KK and Kobaisy M, J. Agric. Food Chem, 2001, 49, 5989–5992. [DOI] [PubMed] [Google Scholar]
- 58.Brock NL, Ravella SR, Schulz S and Dickschat JS, Angew. Chemie, Int. Ed, 2013, 52, 2100–2104. [DOI] [PubMed] [Google Scholar]
- 59.Yu L, Dai HF, Zhao YX, Zeng YB, Jiang WT, Mei WL and Zeng HC, J. Asian Nat. Prod. Res, 2011, 13, 901–906. [DOI] [PubMed] [Google Scholar]
- 60.Dickschat JS, Nat. Prod. Rep, 2011, 28, 1917–1936. [DOI] [PubMed] [Google Scholar]
- 61.Nakano C, Kim HK and Ohnishi Y, ChemBioChem, 2011, 12, 2403–2407. [DOI] [PubMed] [Google Scholar]
- 62.Hou A, Lauterbach L and Dickschat JS, Chem. - A Eur. J, 2020, 26, 2178–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Reddy GK, Leferink NGH, Umemura M, Ahmed ST, Breitling R, Scrutton NS and Takano E, PLoS One, 2020, 15, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang CM and Cane DE, J. Am. Chem. Soc, 2008, 130, 8908–8909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Finefield JM, Sherman DH, Kreitman M and Williams RM, Angew. Chemie, Int. Ed, 2012, 51, 4802–4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kim T, Song B, Cho KS and Lee I-S, Int. J. Mol. Sci, 2020, 21, 2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Da Silva ACR, Lopes PM, De Azevedo MMB, Costa DCM, Alviano CS and Alviano DS, Molecules, 2012, 17, 6305–6316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen W, Vermaak I and Viljoen A, Molecules, 2013, 18, 5434–5454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tang X, Shao Y-L, Tang Y-J and Zhou W-W, Molecules, 2018, 23, 2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pereira I, Severino P, Santos AC, Silva AM and Souto EB, Colloids Surfaces B Biointerfaces, 2018, 171, 566–578. [DOI] [PubMed] [Google Scholar]
- 71.Sobral MV, Xavier AL, Lima TC and de Sousa DP, Sci. World J, 2014, 2014, 1–35. [Google Scholar]
- 72.Kamatou GPP, Vermaak I, Viljoen AM and Lawrence BM, Phytochemistry, 2013, 96, 15–25. [DOI] [PubMed] [Google Scholar]
- 73.Schifrin A, Khatri Y, Kirsch P, Thiel V, Schulz S and Bernhardt R, Org. Biomol. Chem, 2016, 14, 3385–3393. [DOI] [PubMed] [Google Scholar]
- 74.Li L, Liu R, Han L, Jiang Y, Liu J, Li Y, Yuan C and Huang X, Magn. Reson. Chem, 2016, 54, 606–609. [DOI] [PubMed] [Google Scholar]
- 75.Shao JW, Fotso S, Li F, Qin S and Laatsch H, J. Nat. Prod, 2007, 70, 304–306. [DOI] [PubMed] [Google Scholar]
- 76.Ding L, Pfoh R, Rühl S, Qin S and Laatsch H, J. Nat. Prod, 2009, 72, 99–101. [DOI] [PubMed] [Google Scholar]
- 77.Ding N, Jiang Y, Liu J, Li Q, Wang X, Mu Y, Han L and Huang X, Magn. Reson. Chem, 2016, 54, 930–932. [DOI] [PubMed] [Google Scholar]
- 78.Rabe P, Pahirulzaman KAK and Dickschat JS, Angew. Chemie, Int. Ed, 2015, 54, 6041–6045. [DOI] [PubMed] [Google Scholar]
- 79.Rabe P and Dickschat JS, Angew. Chemie, Int. Ed, 2013, 52, 1810–1812. [DOI] [PubMed] [Google Scholar]
- 80.Rabe P, Rinkel J, Klapschinski TA, Barra L and Dickschat JS, Org. Biomol. Chem, 2016, 14, 158–164. [DOI] [PubMed] [Google Scholar]
- 81.Zhao PJ, Li GH and Shen YM, Chem. Biodivers, 2006, 3, 337–342. [DOI] [PubMed] [Google Scholar]
- 82.Wu SJ, Fotso S, Li F, Qin S, Kelter G, Fiebig HH and Laatsch H, J. Antibiot, 2006, 59, 331–337. [DOI] [PubMed] [Google Scholar]
- 83.Shirai M, Okuda M, Motohashi K, Imoto M, Furihata K, Matsuo Y, Katsuta A, Shizuri Y and Seto H, J. Antibiot, 2010, 63, 245–250. [DOI] [PubMed] [Google Scholar]
- 84.Yang Z, Yang Y, Yang X, Zhang Y, Zhao L, Xu L and Ding Z, Chem. Pharm. Bull, 2011, 59, 1430–1433. [DOI] [PubMed] [Google Scholar]
- 85.Nie Y, Wang C, Zheng Y, Lin R, Xu L, Peng F, Fang D, Lian Y and Jiang H, Tianran Chanwu Yanjiu Yu Kaifa, 2011, 23, 797–800. [Google Scholar]
- 86.Yu L, Dai HF, Zhao YX, Zuo WJ, Dong WH, Mei WL and Zeng HC, Phytochem. Lett, 2013, 6, 110–112. [Google Scholar]
- 87.Shi N, Lu C, Ho CC and Shen Y, Rec. Nat. Prod, 2013, 7, 1–5. [Google Scholar]
- 88.Ding L and Hertweck C, J. Nat. Prod, 2020, 83, 2207–2211. [DOI] [PubMed] [Google Scholar]
- 89.Shaaban KA, Singh S, Elshahawi SI, Wang X, Ponomareva LV, Sunkara M, Copley GC, Hower JC, Morris AJ, Kharel MK and Thorson JS, Nat. Prod. Res, 2014, 28, 337–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ding L, Maier A, Fiebig HH, Lin WH, Peschel G and Hertweck C, J. Nat. Prod, 2012, 75, 2223–2227. [DOI] [PubMed] [Google Scholar]
- 91.Hussain A, Rather MA, Dar MS, Aga MA, Ahmad N, Manzoor A, Qayum A, Shah A, Mushtaq S, Ahmad Z and Hassan QP, Bioorganic Med. Chem. Lett, 2017, 27, 2579–2582. [DOI] [PubMed] [Google Scholar]
- 92.Le Qin L, Zhou B, Ding W and Ma Z, Phytochem. Lett, 2018, 23, 46–51. [Google Scholar]
- 93.Guan S, Grabley S, Groth I, Lin W, Christner A, Guo D and Sattler I, Magn. Reson. Chem, 2005, 43, 1028–1031. [DOI] [PubMed] [Google Scholar]
- 94.GanβEr D, Pollak FC and Berger RG, J. Nat. Prod, 1995, 58, 1790–1793. [Google Scholar]
- 95.Sun MW, Zhang XM, Bi HL, Li WJ and Lu CH, Nat. Prod. Res, 2017, 31, 77–83. [DOI] [PubMed] [Google Scholar]
- 96.Xie XC, Mei WL, Zhao YX, Hong K and Dai HF, Chinese Chem. Lett, 2006, 17, 1463–1465. [Google Scholar]
- 97.Ding N, Jiang Y, Han L, Chen X, Ma J, Qu X, Mu Y, Liu J, Li L, Jiang C and Huang X, J. Nat. Prod, 2016, 79, 799–805. [DOI] [PubMed] [Google Scholar]
- 98.Nawrath T, Dickschat JS, Müller R, Jiang J, Cane DE and Schulz S, J. Am. Chem. Soc, 2008, 130, 430–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dickschat JS, Bode HB, Mahmud T, Müller R and Schulz S, J. Org. Chem, 2005, 70, 5174–5182. [DOI] [PubMed] [Google Scholar]
- 100.Spiteller D, Jux A, Piel J and Boland W, Phytochemistry, 2002, 61, 827–834. [DOI] [PubMed] [Google Scholar]
- 101.Cane DE and Watt RM, Proc. Natl. Acad. Sci. U. S. A, 2003, 100, 1547–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.He X and Cane DE, J. Am. Chem. Soc, 2004, 126, 2678–2679. [DOI] [PubMed] [Google Scholar]
- 103.Jiang J, He X and Cane DE, Nat. Chem. Biol, 2007, 3, 711–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gust B, Challis GL, Fowler K, Kieser T and Chater KF, Proc. Natl. Acad. Sci, 2003, 100, 1541–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cane DE, He X, Kobayashi S, Omura S and Ikeda H, J. Antibiot, 2006, 59, 471–479. [DOI] [PubMed] [Google Scholar]
- 106.Koe BK, Sobin BA and Celmer WD, Antibiot. Annu, 1956, 672–675. [PubMed] [Google Scholar]
- 107.Keller-Schierlein W, Lemke J, Nyfeler R and Zähner H, Arch. Mikrobiol, 1972, 84, 301–316. [PubMed] [Google Scholar]
- 108.Hartmann S, Neeff J, Heer U and Mecke D, FEBS Lett, 1978, 93, 339–342. [DOI] [PubMed] [Google Scholar]
- 109.Takeuchi S, Ogawa Y and Yonehara H, Tetrahedron Lett, 1969, 10, 2737–2740. [DOI] [PubMed] [Google Scholar]
- 110.Martin DG, Slomp G, Mizsak S, Duchamp DJ and Chidester CG, Tetrahedron Lett, 1970, 11, 4901–4904. [DOI] [PubMed] [Google Scholar]
- 111.Seto H and Yonehara H, J. Antibiot, 1980, 33, 92–93. [DOI] [PubMed] [Google Scholar]
- 112.Cane DE and Rossi T, Tetrahedron Lett, 1979, 20, 2973–2974. [Google Scholar]
- 113.Cane DE, Sohng JK and Williard PG, J. Org. Chem, 1992, 57, 844–851. [Google Scholar]
- 114.Seto H, Sasaki T, Uzawa J, Takeuchi S and Yonehara H, Tetrahedron Lett, 1978, 19, 4411–4412. [Google Scholar]
- 115.Tillman AM and Cane DE, J. Antibiot, 1983, 36, 170–172. [DOI] [PubMed] [Google Scholar]
- 116.Seto H, Noguchi H, Sankawa U and Iitaka Y, J. Antibiot, 1984, 37, 816–817. [DOI] [PubMed] [Google Scholar]
- 117.Williard PG, Sohng JK and Cane DE, J. Antibiot, 1988, 41, 130–133. [DOI] [PubMed] [Google Scholar]
- 118.Takahashi S, Takeuchi M, Arai M, Seto H and Ŏtake N, J. Antibiot, 1983, 36, 226–228. [DOI] [PubMed] [Google Scholar]
- 119.Jiang J, Tetzlaff CN, Takamatsu S, Iwatsuki M, Komatsu M, Ikeda H and Cane DE, Biochemistry, 2009, 48, 6431–6440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tetzlaff CN, You Z, Cane DE, Takamatsu S, Omura S and Ikeda H, Biochemistry, 2006, 45, 6179–6186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Seto H, Sasaki T, Yonehara H and Uzawa J, Tetrahedron Lett, 1978, 19, 923–926. [Google Scholar]
- 122.Seto H, Sasaki T, Yonehara H, Takahashi S, Takeuchi M, Kuwano H and Arai M, J. Antibiot, 1984, 37, 1076–1078. [DOI] [PubMed] [Google Scholar]
- 123.Izawa S, Akutsu H, Atomi T, Sh. Kawabata and K. Sasaki, J. Antibiot, 1978, 31, 729–731. [DOI] [PubMed] [Google Scholar]
- 124.Uyeda M, Mizukami M, Yokomizo K and Suzuki K, Biosci. Biotechnol. Biochem, 2001, 65, 1252–1254. [DOI] [PubMed] [Google Scholar]
- 125.Cane DE, Rossi T, Tillman AM and Pachlatko JP, J. Am. Chem. Soc, 1981, 103, 1838–1843. [Google Scholar]
- 126.Cane DE, Sohng JK, Lamberson CR, Rudnicki SM, Wu Z, Lloyd MD, Oliver JS and Hubbard BR, Biochemistry, 1994, 33, 5846–5857. [DOI] [PubMed] [Google Scholar]
- 127.Seo MJ, Zhu D, Endo S, Ikeda H and Cane DE, Biochemistry, 2011, 50, 1739–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Quaderer R, Omura S, Ikeda H and Cane DE, J. Am. Chem. Soc, 2006, 128, 13036–13037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.You Z, Omura S, Ikeda H and Cane DE, J. Am. Chem. Soc, 2006, 128, 6566–6567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.You Z, Omura S, Ikeda H, Cane DE and Jogl G, J. Biol. Chem, 2007, 282, 36552–36560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Deng Q, Liu Y, Chen L, Xu M, Naowarojna N, Lee N, Chen L, Zhu D, Hong X, Deng Z, Liu P and Zhao C, Org. Lett, 2019, 21, 7592–7596. [DOI] [PubMed] [Google Scholar]
- 132.Zhu D, Seo MJ, Ikeda H and Cane DE, J. Am. Chem. Soc, 2011, 133, 2128–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Duan L, Jogl G and Cane DE, J. Am. Chem. Soc, 2016, 138, 12678–12689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Takamatsu S, Xu L-H, Fushinobu S, Shoun H, Komatsu M, Cane DE and Ikeda H, J. Antibiot, 2011, 64, 65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.English AR, McBride TJ and Lynch JE, Antibiot. Annu, 1956, 676–681. [PubMed] [Google Scholar]
- 136.Nakagawa A, Tomoda H, Van Hao M, Okano K, Iwai Y and Omura S, J. Antibiot, 1985, 38, 1114–1115. [DOI] [PubMed] [Google Scholar]
- 137.Cane DE and Sohng JK, Arch. Biochem. Biophys, 1989, 270, 50–61. [DOI] [PubMed] [Google Scholar]
- 138.Cane DE and Sohng JK, Biochemistry, 1994, 33, 6524–6530. [DOI] [PubMed] [Google Scholar]
- 139.Duszenko M, Balla H and Mecke D, Biochim. Biophys. Acta - Gen. Subj, 1982, 714, 344–350. [DOI] [PubMed] [Google Scholar]
- 140.Ikeda M, Fukuda A, Takagi M, Morita M and Shimada Y, Eur. J. Pharmacol, 2001, 411, 45–53. [DOI] [PubMed] [Google Scholar]
- 141.Gūrtler H, Pedersen R, Anthoni U, Christophersen C, Nielsen PH, Wellington EMH, Pedersen C and Bock K, J. Antibiot, 1994, 47, 434–439. [DOI] [PubMed] [Google Scholar]
- 142.Kobayashi T, Kon Y, Abe H and Ito H, Org. Lett, 2014, 16, 6397–6399. [DOI] [PubMed] [Google Scholar]
- 143.Lin X, Hopson R and Cane DE, J. Am. Chem. Soc, 2006, 128, 6022–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Takamatsu S, Lin X, Nara A, Komatsu M, Cane DE and Ikeda H, Microb. Biotechnol, 2011, 4, 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Raju R, Gromyko O, Fedorenko V, Luzketskyy A and Müller R, J. Antibiot, 2014, 68, 286–288. [DOI] [PubMed] [Google Scholar]
- 146.Zheng D, Ding N, Jiang Y, Zhang J, Ma J, Chen X, Liu J, Han L and Huang X, J. Antibiot, 2016, 69, 773–775. [DOI] [PubMed] [Google Scholar]
- 147.Kim D, Lee EJ, Lee J, Leutou AS, Shin YH, Choi B, Hwang JS, Hahn D, Choi H, Chin J, Cho SJ, Hong YD, Ko J, Seong CN, Maloney KN, Oh DC, Yang I, Hwang H and Nam SJ, Mar. Drugs, 2018, 16, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Yang XW, Peng K, Liu Z, Zhang GY, Li J, Wang N, Steinmetz A and Liu Y, J. Nat. Prod, 2013, 76, 2360–2363. [DOI] [PubMed] [Google Scholar]
- 149.Lin X and Cane DE, J. Am. Chem. Soc, 2009, 131, 6332–6333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zhao B, Lin X, Lei L, Lamb DC, Kelly SL, Waterman MR and Cane DE, J. Biol. Chem, 2008, 283, 8183–8189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Zhao B, Lei L, Vassylyev DG, Lin X, Cane DE, Kelly SL, Yuan H, Lamb DC and Waterman MR, J. Biol. Chem, 2009, 284, 36711–36719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Schifrin A, Ly TTB, Günnewich N, Zapp J, Thiel V, Schulz S, Hannemann F, Khatri Y and Bernhardt R, ChemBioChem, 2015, 16, 337–344. [DOI] [PubMed] [Google Scholar]
- 153.Riclea R, Citron CA, Rinkel J and Dickschat JS, Chem. Commun, 2014, 50, 4228–4230. [DOI] [PubMed] [Google Scholar]
- 154.Hu JF, Wunderlich D, Thiericke R, Dahse HM, Grabley S, Feng XZ and Sattler I, J. Antibiot, 2003, 56, 747–754. [DOI] [PubMed] [Google Scholar]
- 155.Lee J, Kim H, Lee TG, Yang I, Won DH, Choi H, Nam SJ and Kang H, J. Nat. Prod, 2014, 77, 1528–1531. [DOI] [PubMed] [Google Scholar]
- 156.Von Reuß SH, Kai M, Piechulla B and Francke W, Angew. Chemie, Int. Ed, 2010, 49, 2009–2010. [DOI] [PubMed] [Google Scholar]
- 157.Nakano C, Horinouchi S and Ohnishi Y, J. Biol. Chem, 2011, 286, 27980–27987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Bin Yang Y, Yang Z, Yang XQ, Zhang Y, Zhao LX, Xu LH and Ding ZT, Yaoxue Xuebao, 2012, 47, 364–366. [PubMed] [Google Scholar]
- 159.Ding L, Goerls H, Dornblut K, Lin W, Maier A, Fiebig HH and Hertweck C, J. Nat. Prod, 2015, 78, 2963–2967. [DOI] [PubMed] [Google Scholar]
- 160.Yamada Y, Arima S, Nagamitsu T, Johmoto K, Uekusa H, Eguchi T, Shin-Ya K, Cane DE and Ikeda H, J. Antibiot, 2015, 68, 385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Domik D, Magnus N and Piechulla B, FEMS Microbiol. Lett, 2016, 363, fnw139. [DOI] [PubMed] [Google Scholar]
- 162.Duell ER, D’Agostino PM, Shapiro N, Woyke T, Fuchs TM and Gulder TAM, Microb. Cell Fact, 2019, 18, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Domik D, Thürmer A, Weise T, Brandt W, Daniel R and Piechulla B, Front. Microbiol, 2016, 7, 737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Von Reuss S, Domik D, Lemfack MC, Magnus N, Kai M, Weise T and Piechulla B, J. Am. Chem. Soc, 2018, 140, 11855–11862. [DOI] [PubMed] [Google Scholar]
- 165.Rabe P, Samborskyy M, Leadlay PF and Dickschat JS, Org. Biomol. Chem, 2017, 15, 2353–2358. [DOI] [PubMed] [Google Scholar]
- 166.Cho G, Kim J, Park CG, Nislow C, Weller DM and Kwak YS, Open Biol, 2017, 7, 170075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Croteau R, Ketchum REB, Long RM, Kaspera R and Wildung MR, Phytochem. Rev, 2006, 5, 75–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Bömke C and Tudzynski B, Phytochemistry, 2009, 70, 1876–1893. [DOI] [PubMed] [Google Scholar]
- 169.Peters RJ, Nat. Prod. Rep, 2010, 27, 1521–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Hayashi Y, Matsuura N, Toshima H, Itoh N, Ishikawa J, Mikami Y and Dairi T, J. Antibiot, 2008, 61, 164–174. [DOI] [PubMed] [Google Scholar]
- 171.Shigemori H, Komaki H, Yazawa K, Mikami Y, Nemoto A, Tanaka Y, Sasaki T, In Y, Ishida T and Kobayashi JNI, J. Org. Chem, 1998, 63, 6900–6904. [DOI] [PubMed] [Google Scholar]
- 172.Komatsu K, Tsuda M, Shiro M, Tanaka Y, Mikami Y and Kobayashi J, Bioorganic Med. Chem, 2004, 12, 5545–5551. [DOI] [PubMed] [Google Scholar]
- 173.Schwarz PN, Roller L, Kulik A, Wohlleben W and Stegmann E, Synth. Syst. Biotechnol, 2018, 3, 56–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Gebhardt K, Meyer SW, Schinko J, Bringmann G, Zeeck A and Fiedler HP, J. Antibiot, 2011, 64, 229–232. [DOI] [PubMed] [Google Scholar]
- 175.Dong L-B, Rudolf JD, Deng MR, Yan X and Shen B, ChemBioChem, 2018, 19, 1727–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Wei F, Li W, Song R and Shen Y, Nat. Prod. Commun, 2018, 13, 1433–1436. [Google Scholar]
- 177.Shigemori H, Komaki H, Yazawa K, Mikami Y, Nemoto A, Tanaka Y and Kobayashi J, Tetrahedron Lett, 1999, 40, 4353–4354. [Google Scholar]
- 178.Dürr C, Schnell HJ, Luzhetskyy A, Murillo R, Weber M, Welzel K, Vente A and Bechthold A, Chem. Biol, 2006, 13, 365–377. [DOI] [PubMed] [Google Scholar]
- 179.Daum M, Schnell HJ, Herrmann S, Günther A, Murillo R, Müller R, Bisel P, Müller M and Bechthold A, ChemBioChem, 2010, 11, 1383–1391. [DOI] [PubMed] [Google Scholar]
- 180.Kiske C, Erxleben A, Lucas X, Willmann L, Klementz D, Günther S, Römer W and Kammerer B, Rapid Commun. Mass Spectrom, 2014, 28, 1459–1467. [DOI] [PubMed] [Google Scholar]
- 181.Komaki H, Nemoto A, Tanaka Y, Takagi H, Yazawa K, Mikami Y, Shigemori H, Kobayashi J, Ando A and Nagata Y, J. Antibiot, 1999, 52, 13–19. [DOI] [PubMed] [Google Scholar]
- 182.Komaki H, Tanaka Y, Yazawa K, Takagi H, Ando A, Nagata Y and Mikami Y, J. Antibiot, 2000, 53, 75–77. [DOI] [PubMed] [Google Scholar]
- 183.Usui T, Nagumo Y, Watanabe A, Kubota T, Komatsu K, Kobayashi J and Osada H, Chem. Biol, 2006, 13, 1153–1160. [DOI] [PubMed] [Google Scholar]
- 184.Camoni L, Visconti S, Aducci P and Marra M, Planta, 2019, 249, 49–57. [DOI] [PubMed] [Google Scholar]
- 185.Aoyagi T, Aoyama T, Kojima F, Hattori S, Honma Y, Hamada M and Takeuchi T, J. Antibiot, 1992, 45, 1587–1591. [DOI] [PubMed] [Google Scholar]
- 186.Aoyama T, Naganawa H, Muraoka Y, Takeuchi T and Aoyagi T, J. Antibiot, 1992, 45, 1703–1704. [DOI] [PubMed] [Google Scholar]
- 187.Zheng D, Han L, Qu X, Chen X, Zhong J, Bi X, Liu J, Jiang Y, Jiang C and Huang X, J. Nat. Prod, 2017, 80, 837–844. [DOI] [PubMed] [Google Scholar]
- 188.Zhao G, Drug Discov. Ther, 2013, 7, 185–188. [PubMed] [Google Scholar]
- 189.Kawamura A, Iacovidou M, Hirokawa E, Soll CE and Trujillo M, J. Nat. Prod, 2011, 74, 492–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Lee SR, Lee D, Park M, Lee JC, Park HJ, Kang KS, Kim CE, Beemelmanns C and Kim KH, J. Nat. Prod, 2020, 83, 354–361. [DOI] [PubMed] [Google Scholar]
- 191.Yi W, Li Q, Song T, Chen L, Li XC, Zhang Z and Lian XY, Tetrahedron, 2019, 75, 1186–1193. [Google Scholar]
- 192.Kim SY, Zhao P, Igarashi M, Sawa R, Tomita T, Nishiyama M and Kuzuyama T, Chem. Biol, 2009, 16, 736–743. [DOI] [PubMed] [Google Scholar]
- 193.Meguro A, Motoyoshi Y, Teramoto K, Ueda S, Totsuka Y, Ando Y, Tomita T, Kim SY, Kimura T, Igarashi M, Sawa R, Shinada T, Nishiyama M and Kuzuyama T, Angew. Chemie, Int. Ed, 2015, 54, 4353–4356. [DOI] [PubMed] [Google Scholar]
- 194.Supong K, Sripreechasak P, Tanasupawat S, Danwisetkanjana K, Rachtawee P and Pittayakhajonwut P, Appl. Microbiol. Biotechnol, 2017, 101, 533–543. [DOI] [PubMed] [Google Scholar]
- 195.Fukumoto A, Kim YP, Matsumoto A, Takahashi Y, Shiomi K, Tomoda H and Omura S, J. Antibiot, 2008, 61, 1–6. [DOI] [PubMed] [Google Scholar]
- 196.Ohtawa M, Hishinuma Y, Takagi E, Yamada T, Ito F, Arima S, Uchida R, Kim YP, Omura S, Tomoda H and Nagamitsu T, Chem. Pharm. Bull, 2016, 64, 1370–1377. [DOI] [PubMed] [Google Scholar]
- 197.Koyama N, Tokura Y, Takahashi Y and Tomoda H, Acta Pharm. Sin. B, 2011, 1, 236–239. [Google Scholar]
- 198.Ikeda H, Shin-ya K, Nagamitsu T and Tomoda H, J. Ind. Microbiol. Biotechnol, 2016, 43, 325–342. [DOI] [PubMed] [Google Scholar]
- 199.Fukumoto A, Kim YP, Hanaki H, Shiomi K, Tomoda H and Omura S, J. Antibiot, 2008, 61, 7–10. [DOI] [PubMed] [Google Scholar]
- 200.Koyama N, Tokura Y, Münch D, Sahl HG, Schneider T, Shibagaki Y, Ikeda H and Tomoda H, PLoS One, 2012, 7, e48981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.MacMillan J, J. Plant Growth Regul, 2001, 20, 387–442. [DOI] [PubMed] [Google Scholar]
- 202.Bottini R, Cassán F and Piccoli P, Appl. Microbiol. Biotechnol, 2004, 65, 497–503. [DOI] [PubMed] [Google Scholar]
- 203.Atzorn R, Crozier A, Wheeler CT and Sandberg G, Planta, 1988, 175, 532–538. [DOI] [PubMed] [Google Scholar]
- 204.Bottini R, Fulchieri M, Pearce D and Pharis RP, Plant Physiol, 1989, 90, 45–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Joo GJ, Kim YM, Lee IJ, Song KS and Rhee IK, Biotechnol. Lett, 2004, 26, 487–491. [DOI] [PubMed] [Google Scholar]
- 206.Salazar-Cerezo S, Martínez-Montiel N, García-Sánchez J, Pérez-y-Terrón R and Martínez-Contreras RD, Microbiol. Res, 2018, 208, 85–98. [DOI] [PubMed] [Google Scholar]
- 207.Morrone D, Chambers J, Lowry L, Kim G, Anterola A, Bender K and Peters RJ, FEBS Lett, 2009, 583, 475–480. [DOI] [PubMed] [Google Scholar]
- 208.Nett RS, Montanares M, Marcassa A, Lu X, Nagel R, Charles TC, Hedden P, Rojas MC and Peters RJ, Nat. Chem. Biol, 2017, 13, 69–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Nett RS, Contreras T and Peters RJ, ACS Chem. Biol, 2017, 12, 912–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Kawasaki T, Hayashi Y, Kuzuyama T, Furihata K, Itoh N, Seto H and Dairi T, J. Bacteriol, 2006, 188, 1236–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Soriano-García M, Guerrero C and Toscano RA, Acta Crystallogr. Sect. C Cryst. Struct. Commun, 1986, 42, 729–731. [Google Scholar]
- 212.Motohashi K, Ueno R, Sue M, Furihata K, Matsumoto T, Dairi T, Omura S and Seto H, J. Nat. Prod, 2007, 70, 1712–1717. [DOI] [PubMed] [Google Scholar]
- 213.Bi Y and Yu Z, J. Agric. Food Chem, 2016, 64, 8525–8529. [DOI] [PubMed] [Google Scholar]
- 214.Xu M, Hillwig ML, Lane AL, Tiernan MS, Moore BS and Peters RJ, J. Nat. Prod, 2014, 77, 2144–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Mullowney MW, hAinmhire EÓ, Tanouye U, Burdette JE, Van Pham C and Murphy BT, Mar. Drugs, 2015, 13, 5815–5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Shin B, Kim BY, Cho E, Oh KB, Shin J, Goodfellow M and Oh DC, J. Nat. Prod, 2016, 79, 1886–1890. [DOI] [PubMed] [Google Scholar]
- 217.Takagi M, Motohashi K, Khan ST, Hashimoto J and Shin-Ya K, J. Antibiot, 2010, 63, 401–403. [DOI] [PubMed] [Google Scholar]
- 218.Kawasaki T, Kuzuyama T, Kuwamori Y, Matsuura N, Itoh N, Furihata K, Seto H and Dairi T, J. Antibiot, 2004, 57, 739–747. [DOI] [PubMed] [Google Scholar]
- 219.Ikeda C, Hayashi Y, Itoh N, Seto H and Dairi T, J. Biochem, 2007, 141, 37–45. [DOI] [PubMed] [Google Scholar]
- 220.Rudolf JD, Dong L-B and Shen B, Biochem. Pharmacol, 2017, 133, 139–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Wang J, Soisson SM, Young K, Shoop W, Kodali S, Galgoci A, Painter R, Parthasarathy G, Tang YS, Cummings R, Ha S, Dorso K, Motyl M, Jayasuriya H, Ondeyka J, Herath K, Zhang C, Hernandez L, Allocco J, Basilio Á, Tormo JR, Genilloud O, Vicente F, Pelaez F, Colwell L, Lee SH, Michael B, Felcetto T, Gill C, Silver LL, Hermes JD, Bartizal K, Barrett J, Schmatz D, Becker JW, Cully D and Singh SB, Nature, 2006, 441, 358–361. [DOI] [PubMed] [Google Scholar]
- 222.Wang J, Kodali S, Sang HL, Galgoci A, Painter R, Dorso K, Racine F, Motyl M, Hernandez L, Tinney E, Colletti SL, Herath K, Cummings R, Salazar O, González I, Basilio A, Vicente F, Genilloud O, Pelaez F, Jayasuriya H, Young K, Cully DF and Singh SB, Proc. Natl. Acad. Sci. U. S. A, 2007, 104, 7612–7616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Singh SB, Jayasuriya H, Ondeyka JG, Herath KB, Zhang C, Zink DL, Tsou NN, Ball RG, Basilio A, Genilloud O, Diez MT, Vicente F, Pelaez F, Young K and Wang J, J. Am. Chem. Soc, 2006, 128, 11916–11920. [DOI] [PubMed] [Google Scholar]
- 224.Jayasuriya H, Herath KB, Zhang C, Zink DL, Basilio A, Genilloud O, Diez MT, Vicente F, Gonzalez I, Salazar O, Pelaez F, Cummings R, Ha S, Wang J and Singh SB, Angew. Chemie, Int. Ed, 2007, 46, 4684–4688. [DOI] [PubMed] [Google Scholar]
- 225.Herath KB, Attygalle AB and Singh SB, J. Am. Chem. Soc, 2007, 129, 15422–15423. [DOI] [PubMed] [Google Scholar]
- 226.Herath K, Attygalle AB and Singh SB, Tetrahedron Lett, 2008, 49, 5755–5758. [Google Scholar]
- 227.Jayasuriya H, Herath KB, Ondeyka JG, Zink DL, Burgess B, Wang J and Singh SB, Tetrahedron Lett, 2008, 49, 3648–3651. [Google Scholar]
- 228.Smanski MJ, Yu Z, Casper J, Lin S, Peterson RM, Chen Y, Wendt-Pienkowski E, Rajski SR and Shen B, Proc. Natl. Acad. Sci. U. S. A, 2011, 108, 13498–13503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Rudolf JD, Dong L-B, Huang T and Shen B, Mol. Biosyst, 2015, 11, 2717–2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Rudolf JD, Dong L-B, Cao H, Hatzos-Skintges C, Osipiuk J, Endres M, Chang CY, Ma M, Babnigg G, Joachimiak A, Phillips GN and Shen B, J. Am. Chem. Soc, 2016, 138, 10905–10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Smanski MJ, Peterson RM, Rajski SR and Shen B, Antimicrob. Agents Chemother, 2009, 53, 1299–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Zhang C, Ondeyka J, Zink DL, Burgess B, Wang J and Singh SB, Chem. Commun, 2008, 5034–5036. [DOI] [PubMed] [Google Scholar]
- 233.Zhang C, Ondeyka J, Guan Z, Dietrich L, Burgess B, Wang J and Singh SB, J. Antibiot, 2009, 62, 699–702. [DOI] [PubMed] [Google Scholar]
- 234.Yu Z, Smanski MJ, Peterson RM, Marchillo K, Andes D, Rajski SR and Shen B, Org. Lett, 2010, 12, 1744–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Yu Z, Rateb ME, Smanski MJ, Peterson RM and Shen B, J. Antibiot, 2013, 66, 291–294. [DOI] [PubMed] [Google Scholar]
- 236.Dong L-B, Rudolf JD, Kang D, Wang N, He CQ, Deng Y, Huang Y, Houk KN, Duan Y and Shen B, Nat. Commun, 2018, 9, 2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Singh SB, Ondeyka JG, Herath KB, Zhang C, Jayasuriya H, Zink DL, Parthasarathy G, Becker JW, Wang J and Soisson SM, Bioorganic Med. Chem. Lett, 2009, 19, 4756–4759. [DOI] [PubMed] [Google Scholar]
- 238.Zhang C, Ondeyka J, Herath K, Jayasuriya H, Guan Z, Zink DL, Dietrich L, Burgess B, Ha SN, Wang J and Singh SB, J. Nat. Prod, 2011, 74, 329–340. [DOI] [PubMed] [Google Scholar]
- 239.Singh SB, Jayasuriya H, Herath KB, Zhang C, Ondeyka JG, Zink DL, Ha S, Parthasarathy G, Becker JW, Wang J and Soisson SM, Tetrahedron Lett, 2009, 50, 5182–5185. [Google Scholar]
- 240.Zhang C, Ondeyka J, Dietrich L, Gailliot FP, Hesse M, Lester M, Dorso K, Motyl M, Ha SN, Wang J and Singh SB, Bioorganic Med. Chem, 2010, 18, 2602–2610. [DOI] [PubMed] [Google Scholar]
- 241.Dong L-B, Rudolf JD and Shen B, Bioorganic Med. Chem, 2016, 24, 6348–6353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Herath KB, Zhang C, Jayasuriya H, Ondeyka JG, Zink DL, Burgess B, Wang J and Singh SB, Org. Lett, 2008, 10, 1699–1702. [DOI] [PubMed] [Google Scholar]
- 243.Smanski MJ, Casper J, Peterson RM, Yu Z, Rajski SR and Shen B, J. Nat. Prod, 2012, 75, 2158–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Rudolf JD, Dong L-B, Manoogian K and Shen B, J. Am. Chem. Soc, 2016, 138, 16711–16721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Dong L-B, Zhang X, Rudolf JD, Deng MR, Kalkreuter E, Cepeda AJ, Renata H and Shen B, J. Am. Chem. Soc, 2019, 141, 4043–4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Dong L-B, Liu YC, Cepeda AJ, Kalkreuter E, Deng MR, Rudolf JD, Chang C, Joachimiak A, Phillips GN and Shen B, J. Am. Chem. Soc, 2019, 141, 12406–12412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Wang N, Rudolf JD, Dong L-B, Osipiuk J, Hatzos-Skintges C, Endres M, Chang CY, Babnigg G, Joachimiak A, Phillips GN and Shen B, Nat. Chem. Biol, 2018, 14, 730–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Rudolf JD, Dong L-B, Zhang X, Renata H and Shen B, J. Am. Chem. Soc, 2018, 140, 12349–12353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Yang YL, Zhang S, Ma K, Xu Y, Tao Q, Chen Y, Chen J, Guo S, Ren J, Wang W, Tao Y, Yin WB and Liu H, Angew. Chemie, Int. Ed, 2017, 56, 4749–4752. [DOI] [PubMed] [Google Scholar]
- 250.Brown AK, Taylor RC, Bhatt A, Fütterer K and Besra GS, PLoS One, 2009, 4, e6306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Dong L-B, Rudolf JD, Lin L, Ruiz C, Cameron MD and Shen B, Bioorganic Med. Chem, 2017, 25, 1990–1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Su M, Qiu L, Deng Y, Ruiz CH, Rudolf JD, Dong L-B, Feng X, Cameron MD, Shen B, Duan Y and Huang Y, Mol. Pharm, 2019, 16, 3065–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Liu X, Wang Z, Feng X, Bai E, Xiong Y, Zhu X, Zhu X, Shen B, Shen B, Duan Y, Duan Y, Duan Y, Huang Y and Huang Y, Bioconjug. Chem, 2020, 31, 1425–1437. [DOI] [PubMed] [Google Scholar]
- 254.Wang Z, Liu X, Peng Y, Su M, Zhu S, Pan J, Shen B, Duan Y and Huang Y, Mol. Pharm, 2020, 17, 2451–2462. [DOI] [PubMed] [Google Scholar]
- 255.Wu M, Singh SB, Wang J, Chung CC, Salituro G, Karanam BV, Lee SH, Powles M, Ellsworth KP, Lassman ME, Miller C, Myers RW, Tota MR, Zhang BB and Li C, Proc. Natl. Acad. Sci. U. S. A, 2011, 108, 5378–5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Singh SB, Kang L, Nawrocki AR, Zhou D, Wu M, Previs S, Miller C, Liu H, Hines CDG, Madeira M, Cao J, Herath K, Wang L, Kelley DE, Li C and Guan H-P, PLoS One, 2016, 11, e0164133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Tamamura T, Sawa T, Isshiki K, Masuda T, Homma Y, Iinuma H, Naganawa H, Hamada M, Takeuchi T and Umezawa H, J. Antibiot, 1985, 38, 1664–1669. [DOI] [PubMed] [Google Scholar]
- 258.Isshiki K, Tamamura T, Takahashi Y, Sawa T, Naganawa H, Takeuchi T and Umezawa H, J. Antibiot, 1985, 38, 1819–1821. [DOI] [PubMed] [Google Scholar]
- 259.Andersen NR and Rasmussen PR, Tetrahedron Lett, 1984, 25, 465–468. [Google Scholar]
- 260.Kawada SZ, Yamashita Y, Uosaki Y, Nakano H, Gomi K, Iwasaki T and Takiguchi T, J. Antibiot, 1992, 45, 1182–1184. [DOI] [PubMed] [Google Scholar]
- 261.Uosaki Y, Kawada SZ, Nakano H, Saitoh Y and Sano H, J. Antibiot, 1993, 46, 235–240. [DOI] [PubMed] [Google Scholar]
- 262.Nakajima M, Okazaki T, Iwado S, Kinoshita T and Haneishi T, J. Antibiot, 1989, 42, 1741–1748. [DOI] [PubMed] [Google Scholar]
- 263.Isshiki K, Tamamura T, Sawa T, Naganawa H, Takeuchi T and Umezawa H, J. Antibiot, 1986, 39, 1634–1635. [DOI] [PubMed] [Google Scholar]
- 264.Dairi T, Hamano Y, Kuzuyama T, Itoh N, Furihata K and Seto H, J. Bacteriol, 2001, 183, 6085–6094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Hamano Y, Dairi T, Yamamoto M, Kawasaki T, Kaneda K, Kuzuyama T, Itoh N and Seto H, Biosci. Biotechnol. Biochem, 2001, 65, 1627–1635. [DOI] [PubMed] [Google Scholar]
- 266.Hamano Y, Kuzuyama T, Itoh N, Furihata K, Seto H and Dairi T, J. Biol. Chem, 2002, 277, 37098–37104. [DOI] [PubMed] [Google Scholar]
- 267.Tamamura T, Tsuchiya M, Isshiki K, Sawa T, Takeuchi T, Hori M and Sakata N, J. Antibiot, 1988, 41, 648–654. [DOI] [PubMed] [Google Scholar]
- 268.Kawada SZ, Yamashita Y, Fujii N and Nakano H, Cancer Res, 1991, 51, 2922–2925. [PubMed] [Google Scholar]
- 269.Sehested M and Jensen PB, Biochem. Pharmacol, 1996, 51, 879–886. [DOI] [PubMed] [Google Scholar]
- 270.Lauterbach L, Rinkel J and Dickschat JS, Angew. Chemie, Int. Ed, 2018, 57, 8280–8283. [DOI] [PubMed] [Google Scholar]
- 271.Rinkel J, Steiner ST and Dickschat JS, Angew. Chemie, Int. Ed, 2019, 58, 9230–9233. [DOI] [PubMed] [Google Scholar]
- 272.Rinkel J and Dickschat JS, Org. Lett, 2019, 21, 9442–9445. [DOI] [PubMed] [Google Scholar]
- 273.Spyere A, Rowley DC, Jensen PR and Fenical W, J. Nat. Prod, 2003, 66, 818–822. [DOI] [PubMed] [Google Scholar]
- 274.Hefter J, Richnow HH, Fischer U, Trendel JM and Michaelis W, J. Gen. Microbiol, 1993, 139, 2757–2761. [Google Scholar]
- 275.Tomura T, Nagashima S, Yamazaki S, Iizuka T, Fudou R and Ojika M, Mar. Drugs, 2017, 15, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Chuck JA and Barrow KD, Microbiology, 1995, 141, 2659–2663. [Google Scholar]
- 277.Williams PG, Yoshida WY, Moore RE and Paul VJ, Org. Lett, 2003, 5, 4167–4170. [DOI] [PubMed] [Google Scholar]
- 278.Nakano C, Okamura T, Sato T, Dairi T and Hoshino T, Chem. Commun, 2005, 1016–1018. [DOI] [PubMed] [Google Scholar]
- 279.Mann FM, Xu M, Chen X, Fulton DB, Russell DG and Peters RJ, J. Am. Chem. Soc, 2009, 131, 17526–17527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Mann FM, Xu M, Chen X, Fulton DB, Russell DG and Peters RJ, J. Am. Chem. Soc, 2010, 132, 10953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Prach L, Kirby J, Keasling JD and Alber T, FEBS J, 2010, 277, 3588–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Young DC, Layre E, Pan SJ, Tapley A, Adamson J, Seshadri C, Wu Z, Buter J, Minnaard AJ, Coscolla M, Gagneux S, Copin R, Ernst JD, Bishai WR, Snider BB and Moody DB, Chem. Biol, 2015, 22, 516–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Buter J, Cheng TY, Ghanem M, Grootemaat AE, Raman S, Feng X, Plantijn AR, Ennis T, Wang J, Cotton RN, Layre E, Ramnarine AK, Mayfield JA, Young DC, Jezek Martinot A, Siddiqi N, Wakabayashi S, Botella H, Calderon R, Murray M, Ehrt S, Snider BB, Reed MB, Oldfield E, Tan S, Rubin EJ, Behr MA, van der Wel NN, Minnaard AJ and Moody DB, Nat. Chem. Biol, 2019, 15, 889–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Zhang Y, Adnani N, Braun DR, Ellis GA, Barns KJ, Parker-Nance S, Guzei IA and Bugni TS, J. Nat. Prod, 2016, 79, 2968–2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Nakano C, Oshima M, Kurashima N and Hoshino T, ChemBioChem, 2015, 16, 772–781. [DOI] [PubMed] [Google Scholar]
- 286.Raju R, Mohr KI, Bernecker S, Herrmann J and Müller R, J. Antibiot, 2015, 68, 473–475. [DOI] [PubMed] [Google Scholar]
- 287.Pérez Gutiérrez RM, Martínez Flores A, Vargas Solís R and Carmona Jimenez J, J. Nat. Med, 2008, 62, 328–331. [DOI] [PubMed] [Google Scholar]
- 288.Liu Y, Wang L, Jung JH and Zhang S, Nat. Prod. Rep, 2007, 24, 1401–1429. [DOI] [PubMed] [Google Scholar]
- 289.Wang L, Yang B, Lin XP, Zhou XF and Liu Y, Nat. Prod. Rep, 2013, 30, 455–473. [DOI] [PubMed] [Google Scholar]
- 290.Mo S, Krunic A, Pegan SD, Franzblau SG and Orjala J, J. Nat. Prod, 2009, 72, 2043–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Cabanillas AH, Tena Pérez V, Maderuelo Corral S, Rosero Valencia DF, Martel Quintana A, Ortega Doménech M and Rumbero Sánchez Á, J. Nat. Prod, 2018, 81, 410–413. [DOI] [PubMed] [Google Scholar]
- 292.Kim SH, Lu W, Ahmadi MK, Montiel D, Ternei MA and Brady SF, ACS Synth. Biol, 2019, 8, 109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Yang Y, Zhang Y, Zhang S, Chen Q, Ma K, Bao L, Tao Y, Yin W, Wang G and Liu H, J. Nat. Prod, 2018, 81, 1089–1092. [DOI] [PubMed] [Google Scholar]
- 294.Sato T, Yamaga H, Kashima S, Murata Y, Shinada T, Nakano C and Hoshino T, ChemBioChem, 2013, 14, 822–825. [DOI] [PubMed] [Google Scholar]
- 295.Hou A and Dickschat JS, Angew. Chemie, Int. Ed, 2020, DOI: 10.1002/anie.202010084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Xu R, Fazio GC and Matsuda SPT, Phytochemistry, 2004, 65, 261–291. [DOI] [PubMed] [Google Scholar]
- 297.Taylor RF, Microbiol. Rev, 1984, 48, 181–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Rohmer M, Bouvier-Nave P and Ourisson G, J. Gen. Microbiol, 1984, 130, 1137–1150. [Google Scholar]
- 299.Ourisson G, Rohmer M and Poralla K, Annu. Rev. Microbiol, 1987, 41, 301–333. [DOI] [PubMed] [Google Scholar]
- 300.Rezanka T, Siristova L, Melzoch K and Sigler K, Mini. Rev. Org. Chem, 2010, 7, 300–313. [Google Scholar]
- 301.Hillier SG and Lathe R, J. Endocrinol, 2019, 242, R9–R22. [DOI] [PubMed] [Google Scholar]
- 302.Kannenberg EL and Poralla K, Naturwissenschaften, 1999, 86, 168–176. [Google Scholar]
- 303.Sáenz JP, Grosser D, Bradley AS, Lagny TJ, Lavrynenko O, Broda M and Simons K, Proc. Natl. Acad. Sci. U. S. A, 2015, 112, 11971–11976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Mangiarotti A, Genovese DM, Naumann CA, Monti MR and Wilke N, Biochim. Biophys. Acta - Biomembr, 2019, 1861, 183060. [DOI] [PubMed] [Google Scholar]
- 305.Belin BJ, Busset N, Giraud E, Molinaro A, Silipo A and Newman Di. K., Nat. Rev. Microbiol, 2018, 16, 304–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Connolly JD and Hill RA, Nat. Prod. Rep, 2003, 20, 640–659. [DOI] [PubMed] [Google Scholar]
- 307.Connolly JD and Hill RA, Nat. Prod. Rep, 2010, 27, 79–132. [DOI] [PubMed] [Google Scholar]
- 308.Hill RA and Connolly JD, Nat. Prod. Rep, 2020, 37, 962–998. [DOI] [PubMed] [Google Scholar]
- 309.Sohlenkamp C and Geiger O, FEMS Microbiol. Rev, 2015, 40, 133–159. [DOI] [PubMed] [Google Scholar]
- 310.Hu D, Gao H and Yao X, in Comprehensive Natural Products III, eds. Liu HW and Begley TP, Elsevier, 3rd edn., 2020, pp. 577–612. [Google Scholar]
- 311.Wei JH, Yin X and Welander PV, Front. Microbiol, 2016, 7, 990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Volkman JK, Appl. Microbiol. Biotechnol, 2003, 60, 495–506. [DOI] [PubMed] [Google Scholar]
- 313.Volkman JK, Org. Geochem, 2005, 36, 139–159. [Google Scholar]
- 314.Gelpi E, Schneider H, Mann J and Oró J, Phytochemistry, 1970, 9, 603–612. [Google Scholar]
- 315.Bird CW, Lynch JM, Pirt FJ, Reid WW, Brooks CJW and Middleditch BS, Nature, 1971, 230, 473–474. [DOI] [PubMed] [Google Scholar]
- 316.Bouvier P, Rohmer M, Benveniste P and Ourisson G, Biochem. J, 1976, 159, 267–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Kohl W, Gloe A and Reichenbach H, J. Gen. Microbiol, 1983, 129, 1629–1635. [Google Scholar]
- 318.Bode HB, Zeggel B, Silakowski B, Wenzel SC, Reichenbach H and Müller R, Mol. Microbiol, 2003, 47, 471–481. [DOI] [PubMed] [Google Scholar]
- 319.Ourisson G, Albrecht P and Rohmer M, Pure Appl. Chem, 1979, 51, 709–729. [Google Scholar]
- 320.De Rosa M, Gambacorta A, Minale L and Bu’Lock JD, J. Chem. Soc. D Chem. Commun, 1971, 619–620. [Google Scholar]
- 321.Bird CW, Lynch JM, Pirt SJ and Reid WW, Tetrahedron Lett, 1971, 12, 3189–3190. [Google Scholar]
- 322.Forster HJ, Biemann K and Haigh WG, Biochem. J, 1973, 135, 133–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Rohmer M and Ourisson G, Tetrahedron Lett, 1976, 17, 3633–3636. [Google Scholar]
- 324.Simonin P, Jürgens UJ and Rohmer M, Eur. J. Biochem, 1996, 241, 865–871. [DOI] [PubMed] [Google Scholar]
- 325.Herrmann D, Bisseret P, Connan J and Rohmer M, Tetrahedron Lett, 1996, 37, 1791–1794. [Google Scholar]
- 326.Cvejic JH, Bodrossy L, Kovács KL and Rohmer M, FEMS Microbiol. Lett, 2000, 182, 361–365. [DOI] [PubMed] [Google Scholar]
- 327.Rosa-Putra S, Nalin R, Domenach AM and Rohmer M, Eur. J. Biochem, 2001, 268, 4300–4306. [DOI] [PubMed] [Google Scholar]
- 328.Bravo JM, Perzl M, Härtner T, Kannenberg EL and Rohmer M, Eur. J. Biochem, 2001, 268, 1323–1331. [DOI] [PubMed] [Google Scholar]
- 329.Hai T, Schneider B, Schmidt J and Adam G, Phytochemistry, 1996, 41, 1083–1084. [Google Scholar]
- 330.Kaneda M, Ishimaru K and Nakamura S, Chem. Pharm. Bull, 1984, 32, 1287–1293. [Google Scholar]
- 331.Yoshimoto Y, Sawa T, Naganawa H, Sugai T, Takeuchi T and Imoto M, J. Antibiot, 2000, 53, 575–578. [DOI] [PubMed] [Google Scholar]
- 332.Böröczky K, Laatsch H, Wagner-Döbler I, Stritzke K and Schulz S, Chem. Biodivers, 2006, 3, 622–634. [DOI] [PubMed] [Google Scholar]
- 333.Felder S, Kehraus S, Neu E, Bierbaum G, Schäberle TF and König GM, ChemBioChem, 2013, 14, 1363–1371. [DOI] [PubMed] [Google Scholar]
- 334.Ueda D, Hoshino T and Sato T, J. Am. Chem. Soc, 2013, 135, 18335–18338. [DOI] [PubMed] [Google Scholar]
- 335.Sato T, Yoshida S, Hoshino H, Tanno M, Nakajima M and Hoshino T, J. Am. Chem. Soc, 2011, 133, 9734–9737. [DOI] [PubMed] [Google Scholar]
- 336.Sato T, Biosci. Biotechnol. Biochem, 2013, 77, 1155–1159. [DOI] [PubMed] [Google Scholar]
- 337.Bosak T, Losick RM and Pearson A, Proc. Natl. Acad. Sci. U. S. A, 2008, 105, 6725–6729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Kontnik R, Bosak T, Butcher RA, Brocks JJ, Losick R, Clardy J and Pearson A, Org. Lett, 2008, 10, 3551–3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Takigawa H, Sugiyama M and Shibuya Y, J. Nat. Prod, 2010, 73, 204–207. [DOI] [PubMed] [Google Scholar]
- 340.Sato T, Kigawa A, Takagi R, Adachi T and Hoshino T, Org. Biomol. Chem, 2008, 6, 3788–3794. [DOI] [PubMed] [Google Scholar]
- 341.Sato T, Takizawa K, Orito Y, Kudo H and Hoshino T, ChemBioChem, 2010, 11, 1874–1881. [DOI] [PubMed] [Google Scholar]
- 342.Sato T, Ono E, Nakajima M, Nakano C and Hoshino T, Tetrahedron Lett, 2012, 53, 2522–2524. [Google Scholar]
- 343.Ueda D, Yamaga H, Murakami M, Totsuka Y, Shinada T and Sato T, ChemBioChem, 2015, 16, 1371–1377. [DOI] [PubMed] [Google Scholar]
- 344.Sato T, Takagi R, Orito Y, Ono E and Hoshino T, Biosci. Biotechnol. Biochem, 2010, 74, 147–151. [DOI] [PubMed] [Google Scholar]
- 345.Walter MH and Strack D, Nat. Prod. Rep, 2011, 28, 663. [DOI] [PubMed] [Google Scholar]
- 346.Mirkovic T, Ostroumov EE, Anna JM, Van Grondelle R, Govindjee and Scholes GD, Chem. Rev, 2017, 117, 249–293. [DOI] [PubMed] [Google Scholar]
- 347.Johnson EA and Schroeder WA, in Advances in Biochemical Engineering/Biotechnology, 1995, vol. 53, pp. 119–178. [DOI] [PubMed] [Google Scholar]
- 348.Sandmann G, Antioxidants, 2019, 8, 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Seel W, Baust D, Sons D, Albers M, Etzbach L, Fuss J and Lipski A, Sci. Rep, 2020, 10, 330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Torregrosa-Crespo J, Montero Z, Fuentes JL, García-Galbis MR, Garbayo I, Vílchez C and Martínez-Espinosa RM, Mar. Drugs, 2018, 16, 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Nupur LNU, Vats A, Dhanda SK, Raghava GPS, Pinnaka AK and Kumar A, BMC Microbiol, 2016, 16, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Moise AR, Al-Babili S and Wurtzel ET, Chem. Rev, 2014, 114, 164–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Graham JE, Lecomte JTJ and Bryant DA, J. Nat. Prod, 2008, 71, 1647–1650. [DOI] [PubMed] [Google Scholar]
- 354.Cooney JJ, Marks HW and Smith AM, J. Bacteriol, 1966, 92, 342–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Takaichi S, Mochimaru M, Maoka T and Katoh H, Plant Cell Physiol, 2005, 46, 497–504. [DOI] [PubMed] [Google Scholar]
- 356.Marshall JH and Wilmoth GJ, J. Bacteriol, 1981, 147, 900–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Krubasik P, Takaichi S, Maoka T, Kobayashi M, Masamoto K and Sandmann G, Arch. Microbiol, 2001, 176, 217–223. [DOI] [PubMed] [Google Scholar]
- 358.Weeks OB, Andrewes AG, Brown BO and Weedon BCL, Nature, 1969, 224, 879–882. [DOI] [PubMed] [Google Scholar]
- 359.Arpin N, Liaaen-Jensen S and Trouilloud M, Acta Chem. Scand, 1972, 26, 2524–2526. [DOI] [PubMed] [Google Scholar]
- 360.Andrewes AG, Liaaen Jensen S and Weeks OB, Acta Chem. Scand, 1975, 29, 884–886. [DOI] [PubMed] [Google Scholar]
- 361.Meléndez-Martínez AJ, Mol. Nutr. Food Res, 2019, 63. [DOI] [PubMed] [Google Scholar]
- 362.Jüttner F and Höflacher B, Arch. Microbiol, 1985, 141, 337–343. [Google Scholar]
- 363.Jüttner F, Zeitschrift fur Naturforsch. - Sect. C J. Biosci, 1976, 31, 491–495. [Google Scholar]
- 364.Höckelmann C, Moens T and Jüttner F, Limnol. Oceanogr, 2004, 49, 1809–1819. [Google Scholar]
- 365.Höckelmann C and Jüttner F, Water Sci. Technol, 2004, 49, 47–54. [PubMed] [Google Scholar]
- 366.Ueda D, Matsugane S, Okamoto W, Hashimoto M and Sato T, Angew. Chemie, Int. Ed, 2018, 57, 10347–10351. [DOI] [PubMed] [Google Scholar]
- 367.Höckelmann C and Jüttner F, Flavour Fragr. J, 2005, 20, 387–394. [Google Scholar]
- 368.Aguero J, Lora J, Estrada K, Concepcion F, Nunez A, Rodriguez A and Pino JA, J. Essent. Oil Res, 2003, 15, 114–117. [Google Scholar]
- 369.Li XM, Li XM and Lu CH, J. Asian Nat. Prod. Res, 2017, 19, 946–953. [DOI] [PubMed] [Google Scholar]
- 370.Yang XQ, Bin Yang Y, Zhou H, He GW, Zhao LX, Xu LH and Ding ZT, Nat. Prod. Res, 2013, 27, 1191–1196. [DOI] [PubMed] [Google Scholar]
- 371.Sunayana MR, Sasikala C and Ramana CV, J. Ind. Microbiol. Biotechnol, 2005, 32, 41–45. [DOI] [PubMed] [Google Scholar]
- 372.Sunayana MR, Sasikala C and Ramana CV, Biotechnol. Lett, 2005, 27, 1897–1900. [DOI] [PubMed] [Google Scholar]
- 373.Ninomiya M, Satoh H, Yamaguchi Y, Takenaka H and Koketsu M, Biosci. Biotechnol. Biochem, 2011, 75, 2175–2177. [DOI] [PubMed] [Google Scholar]
- 374.Walsh CT and Wencewicz TA, J. Antibiot, 2014, 67, 7–22. [DOI] [PubMed] [Google Scholar]
- 375.Heide L, Nat. Prod. Rep, 2009, 26, 1241–1250. [DOI] [PubMed] [Google Scholar]
- 376.Hoeksema H, Caron EL and Hinman JW, J. Am. Chem. Soc, 1956, 78, 2019–2020. [Google Scholar]
- 377.Harris DA, Ruger M, Reagan MA, Wolf FJ, Peck RL, Wallick H and Woodruff HB, Antibiot. Chemother, 1955, 5, 183–190. [PubMed] [Google Scholar]
- 378.Welch H and Wright WW, Antibiot. Chemother, 1955, 5, 670–673. [PubMed] [Google Scholar]
- 379.Hinman JW, Hoeksema H, Caron EL and Jackson WG, J. Am. Chem. Soc, 1956, 78, 1072–1074. [Google Scholar]
- 380.Shunk CH, Stammer CH, Kaczka EA, Walton E, Spencer CF, Wilson AN, Richter JW, Holly FW and Folkers K, J. Am. Chem. Soc, 1956, 78, 1770–1771. [Google Scholar]
- 381.Crow FW, Duholke WK, Farley KA, Hadden CE, Hahn DA, Kaluzny BD, Mallory CS, Martin GE, Smith RF and Thamann TJ, J. Heterocycl. Chem, 1999, 36, 365–370. [Google Scholar]
- 382.Ninet L, Benazet F, Charpentie Y, Dubost M, Florent J, Mancy D, Preud’Homme J, Threlfall TL and Vuillemin B, C.R.Acad.Sci., Ser.C, 1972, 275, 455–458. [Google Scholar]
- 383.Sasaki T, Igarashi Y, Saito N and Furumai T, J. Antibiot, 2001, 54, 441–447. [DOI] [PubMed] [Google Scholar]
- 384.Dalisay DS, Williams DE, Wang XL, Centko R, Chen J and Andersen RJ, PLoS One, 2013, 8, e77078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Gracheva IV and Severina VA, Antibiotiki, 1966, 11, 45–51. [PubMed] [Google Scholar]
- 386.Eustáquio AS, Gust B, Luft T, Li SM, Chater KF and Heide L, Chem. Biol, 2003, 10, 279–288. [DOI] [PubMed] [Google Scholar]
- 387.Dramae A, Nithithanasilp S, Choowong W, Rachtawee P, Prabpai S, Kongsaeree P and Pittayakhajonwut P, Tetrahedron, 2013, 69, 8205–8208. [Google Scholar]
- 388.Hinman JW, Caron EL and Hoeksema H, J. Am. Chem. Soc, 1957, 79, 5321–5322. [Google Scholar]
- 389.Cheenpracha S, Vidor NB, Yoshida WY, Davies J and Chang LC, J. Nat. Prod, 2010, 73, 880–884. [DOI] [PubMed] [Google Scholar]
- 390.Li SM, Hennig S and Heide L, Tetrahedron Lett, 1998, 39, 2717–2720. [Google Scholar]
- 391.Orihara N, Kuzuyama T, Takahashi S, Furihata K and Seto H, J. Antibiot, 1998, 51, 676–678. [DOI] [PubMed] [Google Scholar]
- 392.Steffensky M, Li SM, Vogler B and Heide L, FEMS Microbiol. Lett, 1998, 161, 69–74. [Google Scholar]
- 393.Pojer F, Li SM and Heide L, Microbiology, 2002, 148, 3901–3911. [DOI] [PubMed] [Google Scholar]
- 394.Pojer F, Wemakor E, Kammerer B, Chen H, Walsh CT, Li SM and Heide L, Proc. Natl. Acad. Sci. U. S. A, 2003, 100, 2316–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Kuzuyama T, Noel JP and Richard SB, Nature, 2005, 435, 983–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Tello M, Kuzuyama T, Heide L, Noel JP and Richard SB, Cell. Mol. Life Sci, 2008, 65, 1459–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Ozaki T, Mishima S, Nishiyama M and Kuzuyama T, J. Antibiot, 2009, 62, 385–392. [DOI] [PubMed] [Google Scholar]
- 398.Saleh O, Haagen Y, Seeger K and Heide L, Phytochemistry, 2009, 70, 1728–1738. [DOI] [PubMed] [Google Scholar]
- 399.Tanner ME, Nat. Prod. Rep, 2015, 32, 88–101. [DOI] [PubMed] [Google Scholar]
- 400.Heide L, Int. J. Med. Microbiol, 2014, 304, 31–36. [DOI] [PubMed] [Google Scholar]
- 401.Food Fed. Regist, 1996, 61, 3143–3144. [Google Scholar]
- 402.Maxwell A and Lawson D, Curr. Top. Med. Chem, 2005, 3, 283–303. [DOI] [PubMed] [Google Scholar]
- 403.Galm U, Heller S, Shapiro S, Page M, Li SM and Heide L, Antimicrob. Agents Chemother, 2004, 48, 1307–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Lafitte D, Lamour V, Tsvetkov PO, Makarov AA, Klich M, Deprez P, Moras D, Briand C and Gilli R, Biochemistry, 2002, 41, 7217–7223. [DOI] [PubMed] [Google Scholar]
- 405.Flatman RH, Eustaquio A, Li SM, Heide L and Maxwell A, Antimicrob. Agents Chemother, 2006, 50, 1136–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Schmutz E, Mühlenweg A, Li SM and Heide L, Antimicrob. Agents Chemother, 2003, 47, 869–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Marcu MG, Schulte TW and Neckers L, J. Natl. Cancer Inst, 2000, 92, 242–248. [DOI] [PubMed] [Google Scholar]
- 408.Hong DS, Banerji U, Tavana B, George GC, Aaron J and Kurzrock R, Cancer Treat. Rev, 2013, 39, 375–387. [DOI] [PubMed] [Google Scholar]
- 409.Tsutsumi H, Katsuyama Y, Izumikawa M, Takagi M, Fujie M, Satoh N, Shin-Ya K and Ohnishi Y, J. Am. Chem. Soc, 2018, 140, 6631–6639. [DOI] [PubMed] [Google Scholar]
- 410.Omura S and Nakagawa A, Tetrahedron Lett, 1981, 22, 2199–2202. [Google Scholar]
- 411.Morimoto Y and Shirahama H, Tetrahedron, 1996, 52, 10631–10652. [Google Scholar]
- 412.Nakagawa A, Iwai Y, Hashimoto H, Miyazaki N, ŌIwa R, Takahashi Y, Hirano A, Shibukawa N, Kojima Y and ŌMura S, J. Antibiot, 1981, 34, 1408–1415. [DOI] [PubMed] [Google Scholar]
- 413.Kim WG, Kim JP, Kim CJ, Lee KEHO and Yoo ID, J. Antibiot, 1996, 49, 20–25. [DOI] [PubMed] [Google Scholar]
- 414.Kim WG, Kim JP, Koshino H, Shin-ya K, Seto H and Yoo ID, Tetrahedron, 1997, 53, 4309–4316. [Google Scholar]
- 415.Toda N, Ori M, Takami K, Tago K and Kogen H, Org. Lett, 2003, 5, 269–271. [DOI] [PubMed] [Google Scholar]
- 416.Kim WG, Ryoo IJ, Parl JS and Yoo ID, J. Antibiot, 2001, 54, 513–516. [DOI] [PubMed] [Google Scholar]
- 417.Motohashi K, Nagai A, Takagi M and Shin-ya K, J. Antibiot, 2011, 64, 281–283. [DOI] [PubMed] [Google Scholar]
- 418.Hirota-Takahata Y, Kobayashi H, Kizuka M, Ohyama T, Kitamura-Miyazaki M, Suzuki Y, Fujiwara M, Nakajima M and Ando O, J. Antibiot, 2016, 69, 747–753. [DOI] [PubMed] [Google Scholar]
- 419.Kimura T, Suga T, Kameoka M, Ueno M, Inahashi Y, Matsuo H, Iwatsuki M, Shigemura K, Shiomi K, Takahashi Y, Ōmura S and Nakashima T, J. Antibiot, 2019, 72, 169–173. [DOI] [PubMed] [Google Scholar]
- 420.Al-Refa’i Mahmoud Hussien Ibrahim, PhD thesis, University of Göttingen, 2008.
- 421.Liu M, Shi P, Lu C and Zhong L, Nat. Prod. Commun, 2019, 14, 1934578X1986179. [Google Scholar]
- 422.Farwell CC, Zhang RK, McIntosh JA, Hyster TK and Arnold FH, ACS Cent. Sci, 2015, 1, 89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.Lee JG, Yoo ID and Kim WG, Biol. Pharm. Bull, 2007, 30, 795–797. [DOI] [PubMed] [Google Scholar]
- 424.Kobayashi H, Ohyama T, Kitamura-Miyazaki M, Hirota-Takahata Y and Ando O, J. Antibiot, 2016, 69, 754–758. [DOI] [PubMed] [Google Scholar]
- 425.Walsh CT, ACS Chem. Biol, 2014, 9, 2718–2728. [DOI] [PubMed] [Google Scholar]
- 426.Sasaki T, Igarashi Y, Ogawa M and Furumai T, J. Antibiot, 2002, 55, 1009–1012. [DOI] [PubMed] [Google Scholar]
- 427.Satou R, Izumikawa M, Katsuyama Y, Matsui M, Takagi M, Shin-ya K and Ohnishi Y, J. Antibiot, 2014, 67, 231–236. [DOI] [PubMed] [Google Scholar]
- 428.Tian H, Shafi J, Ji M, Bi Y and Yu Z, J. Nat. Prod, 2017, 80, 1015–1019. [DOI] [PubMed] [Google Scholar]
- 429.Kwon Y, Kim SH, Shin Y, Bae M, Kim BY, Lee SK, Oh KB, Shin J and Oh DC, Mar. Drugs, 2014, 12, 2326–2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430.Steinmetz H, Mohr KI, Zander W, Jansen R, Gerth K and Müller R, J. Nat. Prod, 2012, 75, 1803–1805. [DOI] [PubMed] [Google Scholar]
- 431.Uchida R, Nakai M, Ohte S, Onaka H, Katagiri T and Tomoda H, J. Antibiot, 2014, 67, 589–591. [DOI] [PubMed] [Google Scholar]
- 432.Sánchez López JM, Martinez Insua M, Pérez Baz J, Fernández Puentes JL and Cañedo Hernández LM, J. Nat. Prod, 2003, 66, 863–864. [DOI] [PubMed] [Google Scholar]
- 433.Jansen R, Mohr KI, Bernecker S, Stadler M and Müller R, J. Nat. Prod, 2014, 77, 1054–1060. [DOI] [PubMed] [Google Scholar]
- 434.Ma J, Lei H, Chen X, Bi X, Jiang Y, Han L and Huang X, J. Antibiot, 2017, 70, 991–994. [DOI] [PubMed] [Google Scholar]
- 435.Motohashi K, Irie K, Toda T, Matsuo Y, Kasai H, Sue M, Furihata K and Seto H, J. Antibiot, 2008, 61, 75–80. [DOI] [PubMed] [Google Scholar]
- 436.Zhang J, Wang JD, Liu CX, Yuan JH, Wang XJ and Xiang WS, Nat. Prod. Res, 2014, 28, 431–437. [DOI] [PubMed] [Google Scholar]
- 437.Wang X, Reynolds AR, Elshahawi SI, Shaaban KA, Ponomareva LV, Saunders MA, Elgumati IS, Zhang Y, Copley GC, Hower JC, Sunkara M, Morris AJ, Kharel MK, Van Lanen SG, Prendergast MA and Thorson JS, Org. Lett, 2015, 17, 2796–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Gräfe U and Radics L, J. Antibiot, 1986, 39, 162–163. [DOI] [PubMed] [Google Scholar]
- 439.Jang JP, Nogawa T, Uramoto M, Okano A, Futamura Y, Shimizu T, Takahashi S, Jang JH, Ahn JS and Osada H, J. Antibiot, 2014, 68, 293–295. [DOI] [PubMed] [Google Scholar]
- 440.Wu C, Du C, Gubbens J, Choi YH and Van Wezel GP, J. Nat. Prod, 2015, 78, 2355–2363. [DOI] [PubMed] [Google Scholar]
- 441.Takahashi S, Takagi H, Toyoda A, Uramoto M, Nogawa T, Ueki M, Sakaki Y and Osada H, J. Bacteriol, 2010, 192, 2839–2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Elshahawi SI, Cao H, Shaaban KA, Ponomareva LV, Subramanian T, Farman ML, Spielmann HP, Phillips GN, Thorson JS and Singh S, Nat. Chem. Biol, 2017, 13, 366–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443.Bhat V, Dave A, MacKay JA and Rawal VH, Alkaloids Chem. Biol, 2014, 73, 65–160. [DOI] [PubMed] [Google Scholar]
- 444.Walton K and Berry JP, Mar. Drugs, 2016, 14, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.Moore RE, Cheuk C and Patterson GML, J. Am. Chem. Soc, 1984, 106, 6456–6457. [Google Scholar]
- 446.Moore RE, Cheuk C, qiang X Yang G, Patterson GML, Bonjouklian R, Smitka TA, Mvnderse JS, Foster RS, Jones ND, Swartzendruber JK and Deeter JB, J. Org. Chem, 1987, 52, 1036–1043. [Google Scholar]
- 447.Klein D, Daloze D, Braekman JC, Hoffmann L and Demoulin V, J. Nat. Prod, 1995, 58, 1781–1785. [DOI] [PubMed] [Google Scholar]
- 448.Becher PG, Keller S, Jung G, Süssmuth RD and Jüttner F, Phytochemistry, 2007, 68, 2493–2497. [DOI] [PubMed] [Google Scholar]
- 449.Kim H, Lantvit D, Hwang CH, Kroll DJ, Swanson SM, Franzblau SG and Orjala J, Bioorganic Med. Chem, 2012, 20, 5290–5295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450.Chilczuk T, Steinborn C, Breinlinger S, Zimmermann-Klemd AM, Huber R, Enke H, Enke D, Niedermeyer THJ and Gründemann C, Planta Med, 2020, 86, 96–103. [DOI] [PubMed] [Google Scholar]
- 451.Jimenez JI, Huber U, Moore RE and Patterson GML, J. Nat. Prod, 1999, 62, 569–572. [DOI] [PubMed] [Google Scholar]
- 452.Moore RE, Yang XQG and Patterson GML, J. Org. Chem, 1987, 52, 3773–3777. [Google Scholar]
- 453.Moore RE, Yang XG, Patterson GML, Bonjouklian R and Smitka TA, Phytochemistry, 1989, 28, 1565–1567. [Google Scholar]
- 454.Stratmann K, Moore RE, Patterson GML, Bonjouklian R, Deeter JB, Shaffer S, Smitka TA and Smith CD, J. Am. Chem. Soc, 1994, 116, 9935–9942. [Google Scholar]
- 455.Schwartz RE, Hirsch CF, Pettibone DJ, Zink DL and Springer JP, J. Org. Chem, 1987, 52, 3704–3706. [Google Scholar]
- 456.Smitka TA, Bonjouklian R, Doolin L, Jones ND, Deeter JB, Yoshida WY, Prinsep MR, Moore RE and Patterson GML, J. Org. Chem, 1992, 57, 857–861. [Google Scholar]
- 457.Raveh A and Carmeli S, J. Nat. Prod, 2007, 70, 196–201. [DOI] [PubMed] [Google Scholar]
- 458.Huber U, Moore RE and Patterson GML, J. Nat. Prod, 1998, 61, 1304–1306. [DOI] [PubMed] [Google Scholar]
- 459.Mo S, Krunic A, Chlipala G and Orjala J, J. Nat. Prod, 2009, 72, 894–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Mo S, Krunic A, Santarsiero BD, Franzblau SG and Orjala J, Phytochemistry, 2010, 71, 2116–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461.Park A, Moore RE and Patterson GML, Tetrahedron Lett, 1992, 33, 3257–3260. [Google Scholar]
- 462.Kim H, Krunic A, Lantvit D, Shen Q, Kroll DJ, Swanson SM and Orjala J, Tetrahedron, 2012, 68, 3205–3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463.Hillwig ML, Zhu Q and Liu X, ACS Chem. Biol, 2014, 9, 372–377. [DOI] [PubMed] [Google Scholar]
- 464.Hillwig ML, Fuhrman HA, Ittiamornkul K, Sevco TJ, Kwak DH and Liu X, ChemBioChem, 2014, 15, 665–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465.Li S, Lowell AN, Yu F, Raveh A, Newmister SA, Bair N, Schaub JM, Williams RM and Sherman DH, J. Am. Chem. Soc, 2015, 137, 15366–15369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466.Li S, Lowell AN, Newmister SA, Yu F, Williams RM and Sherman DH, Nat. Chem. Biol, 2017, 13, 467–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467.Liu X, Hillwig ML, Koharudin LMI and Gronenborn AM, Chem. Commun, 2016, 52, 1737–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Zhu Q and Liu X, Chem. Commun, 2017, 53, 2826–2829. [DOI] [PubMed] [Google Scholar]
- 469.Zhu Q and Liu X, Angew. Chemie, Int. Ed, 2017, 56, 9062–9066. [DOI] [PubMed] [Google Scholar]
- 470.Hillwig ML, Zhu Q, Ittiamornkul K and Liu X, Angew. Chemie, Int. Ed, 2016, 55, 5780–5784. [DOI] [PubMed] [Google Scholar]
- 471.Doan NT, Rickards RW, Rothschild JM and Smith GD, in Journal of Applied Phycology, 2000, vol. 12, pp. 409–416. [Google Scholar]
- 472.Doan NT, Stewart PR and Smith GD, FEMS Microbiol. Lett, 2001, 196, 135–139. [DOI] [PubMed] [Google Scholar]
- 473.Acuña UM, Zi J, Orjala J and Carcache de Blanco EJ, Int. J. Cancer Res, 2015, 49, 1655–1662. [PMC free article] [PubMed] [Google Scholar]
- 474.Smith CD, Zilfou JT, Stratmann K, Patterson GML and Moore RE, Mol. Pharmacol, 1995, 47, 241–247. [PubMed] [Google Scholar]
- 475.Zhang X and Smith CD, Mol. Pharmacol, 1996, 49, 288–294. [PubMed] [Google Scholar]
- 476.Cagide E, Becher PG, Louzao MC, Espiña B, Vieytes MR, Jüttner F and Botana LM, Chem. Res. Toxicol, 2014, 27, 1696–1706. [DOI] [PubMed] [Google Scholar]
- 477.Walton K, Gantar M, Gibbs PDL, Schmale MC and Berry JP, Toxins, 2014, 6, 3568–3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478.Koodkaew I, Sunohara Y, Matsuyama S and Matsumoto H, Plant Growth Regul, 2012, 68, 141–150. [Google Scholar]
- 479.Schmidt AW, Reddy KR and Knölker HJ, Chem. Rev, 2012, 112, 3193–3328. [DOI] [PubMed] [Google Scholar]
- 480.Ding L, Münch J, Goerls H, Maier A, Fiebig HH, Lin WH and Hertweck C, Bioorganic Med. Chem. Lett, 2010, 20, 6685–6687. [DOI] [PubMed] [Google Scholar]
- 481.Takada K, Kajiwara H and Imamura N, J. Nat. Prod, 2010, 73, 698–701. [DOI] [PubMed] [Google Scholar]
- 482.Ding L, Maier A, Fiebig HH, Lin WH and Hertweck C, Org. Biomol. Chem, 2011, 9, 4029–4031. [DOI] [PubMed] [Google Scholar]
- 483.Zhang Q, Mándi A, Li S, Chen Y, Zhang W, Tian X, Zhang H, Li H, Zhang W, Zhang S, Ju J, Kurtán T and Zhang C, European J. Org. Chem, 2012, 2012, 5256–5262. [Google Scholar]
- 484.Kim SH, Ha TKQ, Oh WK, Shin J and Oh DC, J. Nat. Prod, 2016, 79, 51–58. [DOI] [PubMed] [Google Scholar]
- 485.Gloer JB, Rinderknecht BL, Wicklow DT and Dowd PF, J. Org. Chem, 1989, 54, 2530–2532. [Google Scholar]
- 486.Staub GM, Gloer JB, Wicklow DT and Dowd PF, J. Am. Chem. Soc, 1992, 114, 1015–1017. [Google Scholar]
- 487.Smyth JE, Butler NM and Keller PA, Nat. Prod. Rep, 2015, 32, 1562–1583. [DOI] [PubMed] [Google Scholar]
- 488.Xu Z, Baunach M, Ding L and Hertweck C, Angew. Chemie, Int. Ed, 2012, 51, 10293–10297. [DOI] [PubMed] [Google Scholar]
- 489.Baunach M, Ding L, Bruhn T, Bringmann G and Hertweck C, Angew. Chemie, Int. Ed, 2013, 52, 9040–9043. [DOI] [PubMed] [Google Scholar]
- 490.Baunach M, Ding L, Willing K and Hertweck C, Angew. Chemie, Int. Ed, 2015, 54, 13279–13283. [DOI] [PubMed] [Google Scholar]
- 491.Li H, Zhang Q, Li S, Zhu Y, Zhang G, Zhang H, Tian X, Zhang S, Ju J and Zhang C, J. Am. Chem. Soc, 2012, 134, 8996–9005. [DOI] [PubMed] [Google Scholar]
- 492.Zhang Q, Li H, Li S, Zhu Y, Zhang G, Zhang H, Zhang W, Shi R and Zhang C, Org. Lett, 2012, 14, 6142–6145. [DOI] [PubMed] [Google Scholar]
- 493.Kugel S, Baunach M, Baer P, Ishida-Ito M, Sundaram S, Xu Z, Groll M and Hertweck C, Nat. Commun, 2017, 8, 15804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 494.Kato S, Shindo K, Kataoka Y, Yamagishi Y and Mochizuki J, J. Antibiot, 1991, 44, 903–907. [DOI] [PubMed] [Google Scholar]
- 495.Shin-ya K, Tanaka M, Furihata K, Hayakawa Y and Seto H, Tetrahedron Lett, 1993, 34, 4943–4944. [Google Scholar]
- 496.Shin-ya K, Kunigami T, Kim JS, Furihata K, Hayakawa Y and Setot H, Biosci. Biotechnol. Biochem, 1997, 61, 1768–1769. [DOI] [PubMed] [Google Scholar]
- 497.Shin-ya K, Shimizu S, Kunigami T, Hayakawa Y, Seto H and Furihata K, J. Antibiot, 1995, 48, 1378–1381. [DOI] [PubMed] [Google Scholar]
- 498.Huang S, Elsayed SS, Lv M, Tabudravu J, Rateb ME, Gyampoh R, Kyeremeh K, Ebel R, Jaspars M, Deng Z, Yu Y and Deng H, Chem. Biol, 2015, 22, 1633–1642. [DOI] [PubMed] [Google Scholar]
- 499.Kobayashi M, Tomita T, Shin-ya K, Nishiyama M and Kuzuyama T, Angew. Chemie, Int. Ed, 2019, 58, 13349–13353. [DOI] [PubMed] [Google Scholar]
- 500.Mok DWS and Mok MC, Annu. Rev. Plant Biol, 2001, 52, 89–118. [DOI] [PubMed] [Google Scholar]
- 501.Helgeson JP and Leonard NJ, Proc. Natl. Acad. Sci. U. S. A, 1966, 56, 60–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502.Scarbrough E, Armstrong DJ and Skoog F, Proc. Natl. Acad. Sci. U. S. A, 1973, 70, 3825–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 503.Iacobellis NS, Evidente A and Surico G, Experientia, 1988, 44, 70–72. [Google Scholar]
- 504.Evidente A, Suricot G, Iacobellis NS and Randazzo G, Phytochemistry, 1986, 25, 525–526. [Google Scholar]
- 505.Armstrong DJ, Scarbrough E, Skoog F, Cole DL and Leonard NJ, Plant Physiol, 1976, 58, 749–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506.Einset JW and Skoog FK, Biochem. Biophys. Res. Commun, 1977, 79, 1117–1121. [DOI] [PubMed] [Google Scholar]
- 507.Murai N, Skoog F, Doyle ME and Hanson RS, Proc. Natl. Acad. Sci. U. S. A, 1980, 77, 619–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 508.Surico G, Evidente A, Iacobellis NS and Randazzo G, Phytochemistry, 1985, 24, 1499–1502. [Google Scholar]
- 509.Evidente A, Iacobellis NS, Vellone R, Sisto A and Surico G, Phytochemistry, 1989, 28, 2603–2607. [Google Scholar]
- 510.Evidente A, Di Maio E, Caponero A and Iacobellis NS, Experientia, 1995, 51, 990–993. [Google Scholar]
- 511.Omer ZS, Björkman PO, Nicander B, Tillberg E and Gerhardson B, Physiol. Plant, 2004, 121, 439–447. [Google Scholar]
- 512.Ahn JW, Jang KH, Chung SC, Oh KB and Shin J, Org. Lett, 2008, 10, 1167–1169. [DOI] [PubMed] [Google Scholar]
- 513.Okoth DA, Hug JJ, Garcia R, Spröer C, Overmann J and Müller R, Molecules, 2020, 25, 2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 514.Layre E, Lee HJ, Young DC, Martinot AJ, Buter J, Minnaard AJ, Annand JW, Fortune SM, Snider BB, Matsunaga I, Rubin EJ, Alber T and Moody DB, Proc. Natl. Acad. Sci. U. S. A, 2014, 111, 2978–2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 515.Izumikawa M, Khan ST, Komaki H, Takagi M and Shin-ya K, J. Antibiot, 2010, 63, 33–36. [DOI] [PubMed] [Google Scholar]
- 516.Igarashi Y, Kyoso T, Kim Y and Oikawa T, J. Antibiot, 2017, 70, 607–610. [DOI] [PubMed] [Google Scholar]
- 517.Ilan EZ, Torres MR, Prudhomme J, Le Roch K, Jensen PR and Fenical W, J. Nat. Prod, 2013, 76, 1815–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 518.Kamada-Nobusada T and Sakakibara H, Phytochemistry, 2009, 70, 444–449. [DOI] [PubMed] [Google Scholar]
- 519.Akiyoshi DE, Klee H, Amasino RM, Nester EW and Gordon MP, Proc. Natl. Acad. Sci. U. S. A, 1984, 81, 5994–5998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 520.Barry GF, Rogers SG, Fraley RT and Brand L, Proc. Natl. Acad. Sci, 1984, 81, 4776–4780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 521.Krall L, Raschke M, Zenk MH and Baron C, FEBS Lett, 2002, 527, 315–318. [DOI] [PubMed] [Google Scholar]
- 522.Sugawara H, Ueda N, Kojima M, Makita N, Yamaya T and Sakakibara H, Proc. Natl. Acad. Sci. U. S. A, 2008, 105, 2734–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 523.Crespi M, Vereecke D, Temmerman W, Van Montagu M and Desomer J, J. Bacteriol, 1994, 176, 2492–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 524.Pertry I, Václavíková K, Gemrotová M, Spíchal L, Galuszka P, Depuydt S, Temmerman W, Stes E, De Keyser A, Riefler M, Biondi S, Novák O, Schmülling T, Strnad M, Tarkowski P, Holsters M and Vereecke D, Mol. Plant-Microbe Interact, 2010, 23, 1164–1174. [DOI] [PubMed] [Google Scholar]
- 525.Kakimoto T, Plant Cell Physiol, 2001, 42, 677–685. [DOI] [PubMed] [Google Scholar]
- 526.Caillet J and Droogmans L, J. Bacteriol, 1988, 170, 4147–4152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 527.Moore JA and Poulter CD, Biochemistry, 1997, 36, 604–614. [DOI] [PubMed] [Google Scholar]
- 528.Xie W, Zhou C and Huang RH, J. Mol. Biol, 2007, 367, 872–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 529.Schweizer U, Bohleber S and Fradejas-Villar N, RNA Biol, 2017, 14, 1197–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 530.Mathevon C, Pierrel F, Oddou JL, Garcia-Serres R, Blondin G, Latour JM, Ménage S, Gambarelli S, Fontecave M and Atta M, Proc. Natl. Acad. Sci. U. S. A, 2007, 104, 13295–13300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531.Esberg B, Leung HCE, Tsui HCT, Bjork GR and Winkler ME, J. Bacteriol, 1999, 181, 7256–7265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 532.Bode HB, Ring MW, Schwär G, Altmeyer MO, Kegler C, Jose IR, Singer M and Müller R, ChemBioChem, 2009, 10, 128–140. [DOI] [PubMed] [Google Scholar]
- 533.Hwang I and Sakakibara H, Physiol. Plant, 2006, 126, 528–538. [Google Scholar]
- 534.Frébort I, Kowalska M, Hluska T, Frébortová J and Galuszka P, J. Exp. Bot, 2011, 62, 2431–2452. [DOI] [PubMed] [Google Scholar]
- 535.Pertry I, Václavíková K, Depuydt S, Galuszka P, Spíchal L, Temmerman W, Stes E, Schmülling T, Kakimoto T, Van Montagu MCE, Strnad M, Holsters M, Tarkowski P and Vereecke D, Proc. Natl. Acad. Sci. U. S. A, 2009, 106, 929–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 536.Takagi M, Motohashi K, Nagai A, Izumikawa M, Tanaka M, Fuse S, Doi T, Iwase K, Kawaguchi A, Nagata K, Takahashi T and Shin-ya K, Org. Lett, 2010, 12, 4664–4666. [DOI] [PubMed] [Google Scholar]
- 537.Palsuledesai CC and Distefano MD, ACS Chem. Biol, 2015, 10, 51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 538.Winkelblech J, Fan A and Li SM, Appl. Microbiol. Biotechnol, 2015, 99, 7379–7397. [DOI] [PubMed] [Google Scholar]
- 539.Okada M, Sato I, Cho SJ, Iwata H, Nishio T, Dubnau D and Sakagami Y, Nat. Chem. Biol, 2005, 1, 23–24. [DOI] [PubMed] [Google Scholar]
- 540.Okada M, Yamaguchi H, Sato I, Tsuji F, Dubnau D and Sakagami Y, Biosci. Biotechnol. Biochem, 2008, 72, 914–918. [DOI] [PubMed] [Google Scholar]
- 541.Hayashi S, Usami S, Nakamura Y, Ozaki K and Okada M, Biosci. Biotechnol. Biochem, 2015, 79, 1567–1569. [DOI] [PubMed] [Google Scholar]
- 542.Okada M, Yamaguchi H, Sato I, Tsuji F, Qi J, Dubnau D and Sakagami Y, Biosci. Biotechnol. Biochem, 2007, 71, 1807–1810. [DOI] [PubMed] [Google Scholar]
- 543.Magnuson R, Solomon J and Grossman AD, Cell, 1994, 77, 207–216. [DOI] [PubMed] [Google Scholar]
- 544.Luo P, Li H and Morrison DA, Mol. Microbiol, 2003, 50, 623–633. [DOI] [PubMed] [Google Scholar]
- 545.Tortosa P, Logsdon L, Kraigher B, Itoh Y, Mandic-Mulec I and Dubnau D, J. Bacteriol, 2001, 183, 451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 546.Sivonen K, Leikoski N, Fewer DP and Jokela J, Appl. Microbiol. Biotechnol, 2010, 86, 1213–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 547.Nagatsu A, Kajitani H and Sakakibara J, Tetrahedron Lett, 1995, 36, 4097–4100. [Google Scholar]
- 548.Mattila A, Andsten RM, Jumppanen M, Assante M, Jokela J, Wahlsten M, Mikula KM, Sigindere C, Kwak DH, Gugger M, Koskela H, Sivonen K, Liu X, Yli-Kauhaluoma J, Iwaï H and Fewer DP, ACS Chem. Biol, 2019, 14, 2683–2690. [DOI] [PubMed] [Google Scholar]
- 549.Lawton LA, Morris LA and Jaspars M, J. Org. Chem, 1999, 64, 5329–5332. [DOI] [PubMed] [Google Scholar]
- 550.Leikoski N, Liu L, Jokela J, Wahlsten M, Gugger M, Calteau A, Permi P, Kerfeld CA, Sivonen K and Fewer DP, Chem. Biol, 2013, 20, 1033–1043. [DOI] [PubMed] [Google Scholar]
- 551.Ishida K, Matsuda H, Murakami M and Yamaguchi K, Tetrahedron, 1996, 52, 9025–9030. [Google Scholar]
- 552.Murakami M, Itou Y, Ishida K and Shin HJ, J. Nat. Prod, 1999, 62, 752–755. [DOI] [PubMed] [Google Scholar]
- 553.McIntosh JA, Lin Z, Tianero MDB and Schmidt EW, ACS Chem. Biol, 2013, 8, 877–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 554.Donia MS and Schmidt EW, Chem. Biol, 2011, 18, 508–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 555.Martins J, Leikoski N, Wahlsten M, Azevedo J, Antunes J, Jokela J, Sivonen K, Vasconcelos V, Fewer DP and Leão PN, Sci. Rep, 2018, 8, 14537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 556.Iwasaki K, Iwasaki A, Sumimoto S, Sano T, Hitomi Y, Ohno O and Suenaga K, Tetrahedron Lett, 2018, 59, 3806–3809. [Google Scholar]
- 557.Phyo MY, Ding CYG, Goh HC, Goh JX, Ong JFM, Chan SH, Yung PYM, Candra H and Tan LT, J. Nat. Prod, 2019, 82, 3482–3488. [DOI] [PubMed] [Google Scholar]
- 558.Schneider KB, Palmer TM and Grossman AD, J. Bacteriol, 2002, 184, 410–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 559.Parajuli A, Kwak DH, Dalponte L, Leikoski N, Galica T, Umeobika U, Trembleau L, Bent A, Sivonen K, Wahlsten M, Wang H, Rizzi E, De Bellis G, Naismith J, Jaspars M, Liu X, Houssen W and Fewer DP, Angew. Chemie, Int. Ed, 2016, 55, 3596–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 560.Okada M, Sugita T, Akita K, Nakashima Y, Tian T, Li C, Mori T and Abe I, Org. Biomol. Chem, 2016, 14, 9639–9644. [DOI] [PubMed] [Google Scholar]
- 561.McIntosh JA, Donia MS, Nair SK and Schmidt EW, J. Am. Chem. Soc, 2011, 133, 13698–13705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 562.Ahmed F, Ohtsuki T, Aida W and Ishibashi M, J. Nat. Prod, 2008, 71, 1963–1966. [DOI] [PubMed] [Google Scholar]
- 563.Hao Y, Pierce E, Roe D, Morita M, McIntosh JA, Agarwal V, Cheatham TE, Schmidt EW and Nair SK, Proc. Natl. Acad. Sci. U. S. A, 2016, 113, 14037–14042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 564.Sardar D, Hao Y, Lin Z, Morita M, Nair SK and Schmidt EW, J. Am. Chem. Soc, 2017, 139, 2884–2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 565.Renner MK, Shen YC, Cheng XC, Jensen PR, Frankmoelle W, Kauffman CA, Fenical W, Lobkovsky E and Clardy J, J. Am. Chem. Soc, 1999, 121, 11273–11276. [Google Scholar]
- 566.Schultz AW, Oh DC, Carney JR, Williamson RT, Udwary DW, Jensen PR, Gould SJ, Fenical W and Moore BS, J. Am. Chem. Soc, 2008, 130, 4507–4516. [DOI] [PubMed] [Google Scholar]
- 567.Takita T, Ohi K, Okami Y, Maeda K and Umezawa H, J. Antibiot, 1962, 15, 46–48. [PubMed] [Google Scholar]
- 568.Higashide E, Shibata M and Yamamoto H, Agric. Biol. Chem, 1962, 26, 234–237. [Google Scholar]
- 569.Takita T, Naganawa H, Maeda K and Umezawa H, J. Antibiot, 1964, 17, 129–131. [PubMed] [Google Scholar]
- 570.Takita T, Naganawa H, Maeda K and Umezawa H, J. Antibiot, 1965, 18, 135–136. [PubMed] [Google Scholar]
- 571.Cary LW, Takita T and Ohnishi M, FEBS Lett, 1971, 17, 145–148. [DOI] [PubMed] [Google Scholar]
- 572.Iitaka Y, Nakamura H, Takada K and Takita T, Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem, 1974, 30, 2817–2825. [Google Scholar]
- 573.PCT WO 00/78798, 2000, 1–36.
- 574.Ma J, Huang H, Xie Y, Liu Z, Zhao J, Zhang C, Jia Y, Zhang Y, Zhang H, Zhang T and Ju J, Nat. Commun, 2017, 8, 391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 575.Zhou B, Shetye G, Yu Y, Santarsiero BD, Klein LL, Abad-Zapatero C, Wolf NM, Cheng J, Jin Y, Lee H, Suh J-W, Lee H, Bisson J, McAlpine JB, Chen S-N, Cho S-H, Franzblau SG and Pauli GF, J. Nat. Prod, 2020, 83, 657–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 576.Ishida K, Matsuda H, Okita Y and Murakami M, Tetrahedron, 2002, 58, 7645–7652. [Google Scholar]
- 577.Gesner-Apter S and Carmeli S, Tetrahedron, 2008, 64, 6628–6634. [Google Scholar]
- 578.Pancrace C, Ishida K, Briand E, Pichi DG, Weiz AR, Guljamow A, Scalvenzi T, Sassoon N, Hertweck C, Dittmann E and Gugger M, ACS Chem. Biol, 2019, 14, 67–75. [DOI] [PubMed] [Google Scholar]
- 579.Schultz AW, Lewis CA, Luzung MR, Baran PS and Moore BS, J. Nat. Prod, 2010, 73, 373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 580.Schmitt EK, Riwanto M, Sambandamurthy V, Roggo S, Miault C, Zwingelstein C, Krastel P, Noble C, Beer D, Rao SPS, Au M, Niyomrattanakit P, Lim V, Zheng J, Jeffery D, Pethe K and Camacho LR, Angew. Chemie, Int. Ed, 2011, 50, 5889–5891. [DOI] [PubMed] [Google Scholar]
- 581.Vasudevan D, Rao SPS and Noble CG, J. Biol. Chem, 2013, 288, 30883–30891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 582.Maurer M, Linder D, Franke KB, Jäger J, Taylor G, Gloge F, Gremer S, Le Breton L, Mayer MP, Weber-Ban E, Carroni M, Bukau B and Mogk A, Cell Chem. Biol, 2019, 26, 1169–1179.e4. [DOI] [PubMed] [Google Scholar]
- 583.PCT WO 00/78797, 2000, 1–92.
- 584.Choules MP, Wolf NM, Lee H, Anderson JR, Grzelak EM, Wang Y, Ma R, Gao W, McAlpine JB, Jin YY, Cheng J, Lee H, Suh JW, Duc NM, Paik S, Choe JH, Jo EK, Chang CL, Lee JS, Jaki BU, Pauli GF, Franzblau SG and Cho S, Antimicrob. Agents Chemother, 2019, 63, e02204–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 585.Xie Q, Yang Z, Huang X, Zhang Z, Li J, Ju J, Zhang H and Ma J, J. Hematol. Oncol, 2019, 12, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 586.Zhou W, Fang H, Wu Q, Wang X, Liu R, Li F, Xiao J, Yuan L, Zhou Z, Ma J, Wang L, Zhao W, You H, Ju J, Feng J and Chen C, Int. J. Biol. Sci, 2019, 15, 1723–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 587.Bürstner N, Roggo S, Ostermann N, Blank J, Delmas C, Freuler F, Gerhartz B, Hinniger A, Hoepfner D, Liechty B, Mihalic M, Murphy J, Pistorius D, Rottmann M, Thomas JR, Schirle M and Schmitt EK, ChemBioChem, 2015, 16, 2433–2436. [DOI] [PubMed] [Google Scholar]
- 588.Takashima M and Sakai H, Bull. Agric. Chem. Soc. Japan, 1960, 24, 647–651. [Google Scholar]
- 589.Takashima M and Sakai H, J. Agric. Chem. Soc. Japan, 1960, 24, 652–655. [Google Scholar]
- 590.Takashima M, Sakai H and Arima K, Agric. Biol. Chem, 1962, 26, 660–668. [Google Scholar]
- 591.Sakabe N, Harada H, Hirata Y, Tomiie Y and Nitta I, Tetrahedron Lett, 1966, 7, 2523–2525. [Google Scholar]
- 592.Hitotsuyanagi Y, Aimi N, Sakai SI, Fujiki H, Suganuma M, Sugimura T, Endo Y and Shudo K, Chem. Pharm. Bull, 1984, 32, 4233–4236. [DOI] [PubMed] [Google Scholar]
- 593.Cardellina JH, Marner FJ and Moore RE, Science, 1979, 204, 193–195. [DOI] [PubMed] [Google Scholar]
- 594.Sakai SI, Hitotsuyanagi Y, Aimi N, Fujiki H, Suganuma M, Sugimura T, Endo Y and Shudo K, Tetrahedron Lett, 1986, 27, 5219–5220. [Google Scholar]
- 595.Sakai SI, Aimi N, Yamaguchi K, Hitotsuyanagi Y, Watanabe C, Yokose K, Koyama Y, Shudo K and Itai A, Chem. Pharm. Bull, 1984, 32, 354–357. [DOI] [PubMed] [Google Scholar]
- 596.Hitotsuyanagi Y, Yamaguchi K, Ogata K, Aimi N, Sakai SI, Koyama Y, Endo Y, Shudo K, Itai A and Iitaka Y, Chem. Pharm. Bull, 1984, 32, 3774–3778. [DOI] [PubMed] [Google Scholar]
- 597.Horiuchi T, Fujiki H, Suganuma M, Hakii H, Nakayasu M, Hitotsuyanagi Y, Aimi N, Sakai S, Endo Y and Shudo K, Gann, Japanese J. Cancer Res, 1984, 75, 837–840. [PubMed] [Google Scholar]
- 598.Sakai SI, Oqata K, Fujiki H, Endo Y, Koyama Y, Hitotsuyanagi Y, Kuramochi T, Suganuma M, Shudo K, Yamaguchi K, Seki H, Sugimura T, Aimi N and Hara R, Chem. Pharm. Bull, 1986, 34, 4883–4886. [DOI] [PubMed] [Google Scholar]
- 599.Irie K, Funaki A, Koshimizu K, Hayashi H and Arai M, Tetrahedron Lett, 1989, 30, 2113–2116. [Google Scholar]
- 600.Yamashita T, Imoto M, Isshiki K, Sawa T, Naganawa H, Kurasawa S, Umezawa K and Zhu BQ, J. Nat. Prod, 1988, 51, 1184–1187. [Google Scholar]
- 601.Irie K, ichiro Kajiyama S, Okuno S, Kondo M, Koshimizu K, Hayashi H, Arai M, Nishino H and Iwashima A, J. Nat. Prod, 1994, 57, 363–368. [DOI] [PubMed] [Google Scholar]
- 602.Sun HH, White CB, Dedinas J, Cooper R and Sedlock DM, J. Nat. Prod, 1991, 54, 1440–1443. [DOI] [PubMed] [Google Scholar]
- 603.Jiang W, Zhou W, Uchida H, Kikumori M, Irie K, Watanabe R, Suzuki T, Sakamoto B, Kamio M and Nagai H, Mar. Drugs, 2014, 12, 2748–2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 604.Hagiwara N, Irie K, Funaki A, Koshimizu K, Hayashi H and Arai M, Agric. Biol. Chem, 1988, 52, 641–648. [Google Scholar]
- 605.Ströch K, PhD thesis, University of Göttingen, 2003.
- 606.Nakae K, Hosokawa N, Sawa R, Kubota Y, Masuda T, Ohba S, Igarashi M, Nakagawa N, Nishimura Y and Akamatsu Y, J. Antibiot, 2006, 59, 11–17. [DOI] [PubMed] [Google Scholar]
- 607.Irie K, Hagiwara N, Funaki A, Hayashi H, Arai M and Koshimizu K, Agric. Biol. Chem, 1987, 51, 1733–1735. [Google Scholar]
- 608.Aimi N, Odaka H, Sakai SI, Fujiki H, Suganuma M, Moore RE and Patterson GML, J. Nat. Prod, 1990, 53, 1593–1596. [DOI] [PubMed] [Google Scholar]
- 609.Zhang L, Hoshino S, Awakawa T, Wakimoto T and Abe I, ChemBioChem, 2016, 17, 1407–1411. [DOI] [PubMed] [Google Scholar]
- 610.Jiang W, Tan S, Hanaki Y, Irie K, Uchida H, Watanabe R, Suzuki T, Sakamoto B, Kamio M and Nagai H, Mar. Drugs, 2014, 12, 5788–5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 611.Youssef DTA, Shaala LA, Mohamed GA, Ibrahim SRM, Banjar ZM, Badr JM, McPhail KL, Risinger AL and Mooberry SL, Nat. Prod. Res, 2015, 29, 703–709. [DOI] [PubMed] [Google Scholar]
- 612.Irie K, Hagiwara N, Funaki A, Koshimizu K, Hayashi H, Arai M and Tokuda H, Agric. Biol. Chem, 1988, 52, 3193–3195. [Google Scholar]
- 613.Huang H, Yao Y, He Z, Yang T, Ma J, Tian X, Li Y, Huang C, Chen X, Li W, Zhang S, Zhang C and Ju J, J. Nat. Prod, 2011, 74, 2122–2127. [DOI] [PubMed] [Google Scholar]
- 614.Abe I, J. Antibiot, 2018, 71, 763–768. [DOI] [PubMed] [Google Scholar]
- 615.Awakawa T and Abe I, Org. Biomol. Chem, 2018, 16, 4746–4752. [DOI] [PubMed] [Google Scholar]
- 616.Irie K, Nakagawa Y, Tomimatsu S and Ohigashi H, Tetrahedron Lett, 1998, 39, 7929–7930. [Google Scholar]
- 617.Edwards DJ and Gerwick WH, J. Am. Chem. Soc, 2004, 126, 11432–11433. [DOI] [PubMed] [Google Scholar]
- 618.Read JA and Walsh CT, J. Am. Chem. Soc, 2007, 129, 15762–15763. [DOI] [PubMed] [Google Scholar]
- 619.Huynh MU, Elston MC, Hernandez NM, Ball DB, Kajiyama SI, Irie K, Gerwick WH and Edwards DJ, J. Nat. Prod, 2010, 73, 71–74. [DOI] [PubMed] [Google Scholar]
- 620.He F, Mori T, Morita I, Nakamura H, Alblova M, Hoshino S, Awakawa T and Abe I, Nat. Chem. Biol, 2019, 15, 1206–1213. [DOI] [PubMed] [Google Scholar]
- 621.Awakawa T, Zhang L, Wakimoto T, Hoshino S, Mori T, Ito T, Ishikawa J, Tanner ME and Abe I, J. Am. Chem. Soc, 2014, 136, 9910–9913. [DOI] [PubMed] [Google Scholar]
- 622.Ma J, Zuo D, Song Y, Wang B, Huang H, Yao Y, Li W, Zhang S, Zhang C and Ju J, ChemBioChem, 2012, 13, 547–552. [DOI] [PubMed] [Google Scholar]
- 623.Newton AC, Crit. Rev. Biochem. Mol. Biol, 2018, 53, 208–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 624.Mochly-Rosen D, Das K and Grimes KV, Nat. Rev. Drug Discov, 2012, 11, 937–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 625.Nakayasu M, Fujiki H, Mori M, Sugimura T and Moore RE, Cancer Lett, 1981, 12, 271–277. [DOI] [PubMed] [Google Scholar]
- 626.Fujiki H, Mori M, Nakayasu M, Terada M, Sugimura T and Moore RE, Proc. Natl. Acad. Sci. U. S. A, 1981, 78, 3872–3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 627.Arcoleo JP and Weinstein IB, Carcinogenesis, 1985, 6, 213–217. [DOI] [PubMed] [Google Scholar]
- 628.Basu A, Lazo JS and Kozikowski AP, Biochemistry, 1992, 31, 3824–3830. [DOI] [PubMed] [Google Scholar]
- 629.Stahn S, Thelen L, Albrecht IM, Bitzer J, Henkel T and Teusch NE, Pharmacol. Res. Perspect, 2016, 4, e00230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 630.Nishiwaki S, Fujiki H, Yoshizawa S, Suganuma M, Furuya‐Suguri H, Okabe S, Nakayasu M, Okabe K, Muratake H, Natsume M, Umezawa K, Sakai S. ‐i and Sugimura T, Japanese J. Cancer Res, 1991, 82, 779–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 631.Borgman P, Lopez RD and Lane AL, Org. Biomol. Chem, 2019, 17, 2305–2314. [DOI] [PubMed] [Google Scholar]
- 632.Choules MP, Klein LL, Lankin DC, McAlpine JB, Cho SH, Cheng J, Lee H, Suh JW, Jaki BU, Franzblau SG and Pauli GF, J. Org. Chem, 2018, 83, 6664–6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 633.Tuntiwachwuttikul P, Taechowisan T, Wanbanjob A, Thadaniti S and Taylor WC, Tetrahedron, 2008, 64, 7583–7586. [Google Scholar]
- 634.Raju R, Piggott AM, Huang XC and Capon RJ, Org. Lett, 2011, 13, 2770–2773. [DOI] [PubMed] [Google Scholar]
- 635.Wang M, Feng X, Cai L, Xu Z and Ye T, Chem. Commun, 2012, 48, 4344–4346. [DOI] [PubMed] [Google Scholar]
- 636.Wang H and Reisman SE, Angew. Chemie, Int. Ed, 2014, 53, 6206–6210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 637.Lacey E, Power M, Wu Z, and Rickards RW, US Pat., WO1998009968A1, 1997.
- 638.Che Q, Zhu T, Qi X, Mándi A, Kurtán T, Mo X, Li J, Gu Q and Li D, Org. Lett, 2012, 14, 3438–3441. [DOI] [PubMed] [Google Scholar]
- 639.Che Q, Zhu T, Keyzers RA, Liu X, Li J, Gu Q and Li D, J. Nat. Prod, 2013, 76, 759–763. [DOI] [PubMed] [Google Scholar]
- 640.Che Q, Li J, Li D, Gu Q and Zhu T, J. Antibiot, 2016, 69, 467–469. [DOI] [PubMed] [Google Scholar]
- 641.Alqahtani N, Porwal SK, James ED, Bis DM, Karty JA, Lane AL and Viswanathan R, Org. Biomol. Chem, 2015, 13, 7177–7192. [DOI] [PubMed] [Google Scholar]
- 642.Yao T, Liu J, Liu Z, Li T, Li H, Che Q, Zhu T, Li D, Gu Q and Li W, Nat. Commun, 2018, 9, 4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 643.Fordos MJ, Hebd CR. Seances Acad. Sci, 1863, 56, 1128–1131. [Google Scholar]
- 644.Guttenberger N, Blankenfeldt W and Breinbauer R, Bioorganic Med. Chem, 2017, 25, 6149–6166. [DOI] [PubMed] [Google Scholar]
- 645.Laursen JB and Nielsen J, Chem. Rev, 2004, 104, 1663–1685. [DOI] [PubMed] [Google Scholar]
- 646.Pierson LS and Pierson EA, Appl. Microbiol. Biotechnol, 2010, 86, 1659–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 647.Karnetová J, Tax J, Stajner K, Vaněk Z and Krumphanzl V, Folia Microbiol, 1983, 28, 51–53. [DOI] [PubMed] [Google Scholar]
- 648.Tax J, Sedmera P, Vokoun J, Urban J, Karnetová J, Stajner K, Vaněk Z and Krumphanzl V, Collect. Czechoslov. Chem. Commun, 1983, 48, 527–532. [Google Scholar]
- 649.Krastel P, Zeeck A, Gebhardt K, Fiedler HP and Rheinheimer J, J. Antibiot, 2002, 55, 801–806. [DOI] [PubMed] [Google Scholar]
- 650.Gebhardt K, Schimama J, Krastel P, Dettner K, Rheinheimer J, Zeeck A and Fiedler HP, J. Antibiot, 2002, 55, 794–800. [DOI] [PubMed] [Google Scholar]
- 651.Heine D, Martin K and Hertweck C, J. Nat. Prod, 2014, 77, 1083–1087. [DOI] [PubMed] [Google Scholar]
- 652.Wu C, Van Wezel GP and Hae Choi Y, J. Antibiot, 2015, 68, 445–452. [DOI] [PubMed] [Google Scholar]
- 653.Saleh O, Flinspach K, Westrich L, Kulik A, Gust B, Fiedler HP and Heide L, Beilstein J. Org. Chem, 2012, 8, 501–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 654.Shin-ya K, Furihata K, Hayakawa Y, Seto H, Kato Y and Clardy J, Tetrahedron Lett, 1991, 32, 943–946. [Google Scholar]
- 655.Shin-ya K, Furihata K, Teshima Y, Hayakawa Y and Seto H, J. Org. Chem, 1993, 58, 4170–4172. [Google Scholar]
- 656.Shin-ya K, Hayakawa Y and Seto H, J. Nat. Prod, 1993, 56, 1255–1258. [Google Scholar]
- 657.Zendah I, Riaz N, Nasr H, Frauendorf H, Schüffler A, Raies A and Laatsch H, J. Nat. Prod, 2012, 75, 2–8. [DOI] [PubMed] [Google Scholar]
- 658.Shinichiro K, Kazutoshi S, Yuji Y, Michiko M, Hiroyuki K and Junichiro M, J. Antibiot, 1993, 46, 1485–1493. [Google Scholar]
- 659.Kunigami T, Shin-Ya K, Furihata K, Furihata K, Hayakawa Y and Seto H, J. Antibiot, 1998, 51, 880–882. [DOI] [PubMed] [Google Scholar]
- 660.Khan ST, Izumikawa M, Motohashi K, Mukai A, Takagi M and Shin-ya K, FEMS Microbiol. Lett, 2010, 304, 89–96. [DOI] [PubMed] [Google Scholar]
- 661.Izumikawa M, Tabrez Khan S, Takagi M and Shin-ya K, J. Nat. Prod, 2010, 73, 208–212. [DOI] [PubMed] [Google Scholar]
- 662.Han H, Guo Z-K, Zhang B, Zhang M, Shi J, Li W, Jiao R-H, Tan R-X and Ge H-M, Chin. J. Nat. Med, 2019, 17, 475–480. [DOI] [PubMed] [Google Scholar]
- 663.Ohlendorf B, Schulz D, Erhard A, Nagel K and Imhoff JF, J. Nat. Prod, 2012, 75, 1400–1404. [DOI] [PubMed] [Google Scholar]
- 664.Song Y, Huang H, Chen Y, Ding J, Zhang Y, Sun A, Zhang W and Ju J, J. Nat. Prod, 2013, 76, 2263–2268. [DOI] [PubMed] [Google Scholar]
- 665.Zeyhle P, Bauer JS, Steimle M, Leipoldt F, Rösch M, Kalinowski J, Gross H and Heide L, ChemBioChem, 2014, 15, 2385–2392. [DOI] [PubMed] [Google Scholar]
- 666.de M APD. Espindola, PhD thesis, UC San Diego, 2008. [Google Scholar]
- 667.Imai S, Furihata K, Hayakawa Y, Seto H and Noguchi T, J. Antibiot, 1989, 42, 1196–1198. [DOI] [PubMed] [Google Scholar]
- 668.Ōmura S, Eda S, Funayama S, Komiyama K, Takahashi Y and Woodruff HB, J. Antibiot, 1989, 42, 1037–1042. [DOI] [PubMed] [Google Scholar]
- 669.Funayama S, Eda S, Komiyama K, Omura S and Tokunaga T, Tetrahedron Lett, 1989, 30, 3151–3154. [Google Scholar]
- 670.Nakayama O, Yagi M, Tanaka M, Kiyoto S, Okuhara M and Kohsaka M, J. Antibiot, 1989, 42, 1221–1229. [DOI] [PubMed] [Google Scholar]
- 671.Kondratyuk TP, Park EJ, Yu R, Van Breemen RB, Asolkar RN, Murphy BT, Fenical W and Pezzuto JM, Mar. Drugs, 2012, 10, 451–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 672.Asolkar RN, Singh A, Jensen PR, Aalbersberg W, Carté BK, Feussner KD, Subramani R, DiPasquale A, Rheingold AL and Fenical W, Tetrahedron, 2017, 73, 2234–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 673.Saleh O, Gust B, Boll B, Fiedler HP and Heide L, J. Biol. Chem, 2009, 284, 14439–14447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 674.Haagen Y, Unsöld I, Westrich L, Gust B, Richard SB, Noel JP and Heide L, FEBS Lett, 2007, 581, 2889–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 675.Seeger K, Flinspach K, Haug-Schifferdecker E, Kulik A, Gust B, Fiedler HP and Heide L, Microb. Biotechnol, 2011, 4, 252–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 676.Qin Z, Wang X, Rateb ME, Ass’ad LA, Jaspars M, Deng Z, Yu Y and Deng H, FEMS Microbiol. Lett, 2014, 352, 62–68. [DOI] [PubMed] [Google Scholar]
- 677.Zeyhle P, Bauer JS, Kalinowski J, Shin-ya K, Gross H and Heide L, PLoS One, 2014, 9, e99122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 678.Leipoldt F, Zeyhle P, Kulik A, Kalinowski J, Heide L and Kaysser L, PLoS One, 2015, 10, e0143237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 679.Ozaki T, Zhao P, Shinada T, Nishiyama M and Kuzuyama T, J. Am. Chem. Soc, 2014, 136, 4837–4840. [DOI] [PubMed] [Google Scholar]
- 680.Shin-ya K, Shimizu S, Kunigami T, Hayakawa Y, Seto H and Furihata K, J. Antibiot, 1995, 48, 1378–1381. [DOI] [PubMed] [Google Scholar]
- 681.Conda-Sheridan M, Udumula V, Endres JL, Harper CN, Jaramillo L, Zhong HA and Bayles KW, Eur. J. Med. Chem, 2017, 125, 710–721. [DOI] [PubMed] [Google Scholar]
- 682.Otoguro K, Ishiyama A, Iwatsuki M, Namatame M, Nishihara-Tukashima A, Nakashima T, Shibahara S, Kondo S, Yamada H and Ömura S, J. Antibiot, 2010, 63, 579–581. [DOI] [PubMed] [Google Scholar]
- 683.Cimmino A, Andolfi A and Evidente A, in Microbial Phenazines: Biosynthesis, Agriculture and Health, Springer Berlin Heidelberg, Berlin, Heidelberg, 2013, pp. 217–243. [Google Scholar]
- 684.Matsumoto M and Seto H, J. Antibiot, 1991, 44, 1471–1473. [DOI] [PubMed] [Google Scholar]
- 685.Imai S, Noguchi T and Seto H, J. Antibiot, 1993, 46, 1232–1238. [DOI] [PubMed] [Google Scholar]
- 686.Olsen RK, Daves GD, Moore HW, Folkers K, Smith JL, Parson WW and Rudney H, J. Am. Chem. Soc, 1965, 87, 2298–2300. [DOI] [PubMed] [Google Scholar]
- 687.Olsen RK, Doyle Daves G, Moore HW, Folkers K, Parson WW and Rudney H, J. Am. Chem. Soc, 1966, 88, 5919–5923. [DOI] [PubMed] [Google Scholar]
- 688.Xu MJ, Liu XJ, Zhao YL, Liu D, Xu ZH, Lang XM, Ao P, Lin WH, Yang SL, Zhang ZG and Xu J, Mar. Drugs, 2012, 10, 639–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 689.You ZY, Wang YH, Zhang ZG, Xu MJ, Xie SJ, Han TS, Feng L, Li XG and Xu J, Mar. Drugs, 2013, 11, 4035–4049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 690.Jiao X, Yao Y, Yang B, Liu X, Li X, Yang H, Li L, Xu J, Xu M and Xie P, Org. Biomol. Chem, 2016, 14, 1805–1813. [DOI] [PubMed] [Google Scholar]
- 691.Guo ZK, Wang R, Chen FX, Liu TM and Yang MQ, Phytochem. Lett, 2018, 25, 132–135. [Google Scholar]
- 692.Hu Y, Legako AG, Espindola APDM and MacMillan JB, J. Org. Chem, 2012, 77, 3401–3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 693.Hu Y and MacMillan JB, Org. Lett, 2011, 13, 6580–6583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 694.Saleh H, Petras D, Mainz A, Kerwat D, Nalbantsoy A, Erzurumlu Y and Süssmuth RD, J. Nat. Prod, 2016, 79, 1532–1537. [DOI] [PubMed] [Google Scholar]
- 695.Mukherji R, Zhang S, Chowdhury S and Stallforth P, Angew. Chemie, Int. Ed, 2020, 59, 6192–6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 696.Jaki B, Orjala J and Sticher O, J. Nat. Prod, 1999, 62, 502–503. [DOI] [PubMed] [Google Scholar]
- 697.Jaki B, Heilmann J and Sticher O, J. Nat. Prod, 2000, 63, 1283–1285. [DOI] [PubMed] [Google Scholar]
- 698.Prinsep MR, Thomson RA, West ML and Wylie BL, J. Nat. Prod, 1996, 59, 786–788. [DOI] [PubMed] [Google Scholar]
- 699.Gurr JR, O’Donnell TJ, Luo Y, Yoshida WY, Hall ML, Mayer AMS, Sun R and Williams PG, J. Nat. Prod, 2020, 83, 1691–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 700.Jaki B, Orjala J, Heilmann J, Linden A, Vogler B and Sticher O, J. Nat. Prod, 2000, 63, 339–343. [DOI] [PubMed] [Google Scholar]
- 701.Nakanishi S, Osawa K, Saito Y, Kawamoto I, Kuroda K and Kase H, J. Antibiot, 1992, 45, 341–347. [DOI] [PubMed] [Google Scholar]
- 702.Uchida R, Yokota S and Tomoda H, J. Antibiot, 2014, 67, 783–786. [DOI] [PubMed] [Google Scholar]
- 703.Konya M, Shimoyama K, Arima S, Fukuda T, Uchida R, Tomoda H and Nagamitsu T, Org. Lett, 2020, 22, 5131–5134. [DOI] [PubMed] [Google Scholar]
- 704.Moosmann P, Ueoka R, Grauso L, Mangoni A, Morinaka BI, Gugger M and Piel J, Angew. Chemie, Int. Ed, 2017, 56, 4987–4990. [DOI] [PubMed] [Google Scholar]
- 705.Tsuda M, Nemoto A, Komaki H, Tanaka Y, Yazawa K, Mikami Y and Kobayashi J, J. Nat. Prod, 1999, 62, 1640–1642. [DOI] [PubMed] [Google Scholar]
- 706.Liu D, Yang A, Wu C, Guo P, Proksch P and Lin W, Bioorganic Med. Chem. Lett, 2014, 24, 5288–5293. [DOI] [PubMed] [Google Scholar]
- 707.Yang Y, Fu L, Zhang J, Hu L, Xu M and Xu J, PLoS One, 2014, 9, e99537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 708.He BB, Zhou T, Bu XL, Weng JY, Xu J, Lin S, Zheng JT, Zhao YL and Xu MJ, ACS Catal, 2019, 5391–5399. [Google Scholar]
- 709.Hayashi Y, Onaka H, Itoh N, Seto H and Dairi T, Biosci. Biotechnol. Biochem, 2007, 71, 3072–3081. [DOI] [PubMed] [Google Scholar]
- 710.Ozaki T, Shinde SS, Gao L, Okuizumi R, Liu C, Ogasawara Y, Lei X, Dairi T, Minami A and Oikawa H, Angew. Chemie, Int. Ed, 2018, 57, 6629–6632. [DOI] [PubMed] [Google Scholar]
- 711.Ichimura M, Eiki R, Osawa K, Nakanishi S and Kase H, Biochem. J, 1996, 316, 311–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 712.Ishiyama A, Otoguro K, Namatame M, Nishihara A, Furusawa T, Masuma R, Shiomi K, Takahashi Y, Ichimura M, Yamada H and Omura S, J. Antibiot, 2008, 61, 627–632. [DOI] [PubMed] [Google Scholar]
- 713.Shen B, Curr. Opin. Chem. Biol, 2003, 7, 285–295. [DOI] [PubMed] [Google Scholar]
- 714.Weissman KJ, Philos. Trans. R. Soc. A Math. Phys. Eng. Sci, 2004, 362, 2671–2690. [DOI] [PubMed] [Google Scholar]
- 715.Helfrich EJN and Piel J, Nat. Prod. Rep, 2016, 33, 231–316. [DOI] [PubMed] [Google Scholar]
- 716.Murray LAM, McKinnie SMK, Moore BS and George JH, Nat. Prod. Rep, 2020, DOI: 10.1039/D0NP00018C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 717.Austin MB and Noel JP, Nat. Prod. Rep, 2003, 20, 79–110. [DOI] [PubMed] [Google Scholar]
- 718.Yazaki K, Sasaki K and Tsurumaru Y, Phytochemistry, 2009, 70, 1739–1745. [DOI] [PubMed] [Google Scholar]
- 719.Ding WJ, Zhang SQ, Wang JH, Lin YX, Liang QX, Zhao WJ and Li CY, J. Asian Nat. Prod. Res, 2013, 15, 209–214. [DOI] [PubMed] [Google Scholar]
- 720.Cao DD, Van Trinh TT, Mai HDT, Vu VN, Le HM, Thi QV, Nguyen MA, Duong TT, Tran DT, Chau VM, Ma R, Shetye G, Cho S, Murphy BT and Pham VC, Mar. Drugs, 2019, 17, 529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 721.Cao DD, Do TQ, Doan Thi Mai H, Vu Thi Q, Nguyen MA, Le Thi HM, Tran DT, Chau VM, Cong Thung D and Pham VC, Nat. Prod. Res, 2020, 34, 413–420. [DOI] [PubMed] [Google Scholar]
- 722.Zołnierczyk AK, Mączka WK, Grabarczyk M, Wińska K, Woźniak E and Anioł M, Fitoterapia, 2015, 103, 71–82. [DOI] [PubMed] [Google Scholar]
- 723.Funayama S, Ishibashi M, Anraku Y, Komiyama K and Omura S, Tetrahedron Lett, 1989, 30, 7427–7430. [Google Scholar]
- 724.Ishibashi M, Funayama S, Anraku Y, Komiyama K and Omtura S, J. Antibiot, 1991, 44, 390–395. [DOI] [PubMed] [Google Scholar]
- 725.Dormer PG, Smith AB, Funayama S and Omura S, Tetrahedron Lett, 1992, 33, 1717–1720. [Google Scholar]
- 726.Panthee S, Takahashi S, Takagi H, Nogawa T, Oowada E, Uramoto M and Osada H, J. Antibiot, 2011, 64, 509–513. [DOI] [PubMed] [Google Scholar]
- 727.Kawahara T, Nagai A, Takagi M and Shin-ya K, J. Antibiot, 2012, 65, 579–581. [DOI] [PubMed] [Google Scholar]
- 728.Kagamizono T, Kawashima A, Kishimura Y, Yamagishi M, Tsuchida Y, Kondo H and Hanada K, Biosci. Biotechnol. Biochem, 1993, 57, 766–769. [Google Scholar]
- 729.Sedmera P, Pospíšil S and Novák J, J. Nat. Prod, 1991, 54, 870–872. [Google Scholar]
- 730.Charan RD, Schlingmann G, Bernan VS, Feng X and Carter GT, J. Antibiot, 2005, 58, 271–274. [DOI] [PubMed] [Google Scholar]
- 731.Hardt IH, Jensen PR and Fenical W, Tetrahedron Lett, 2000, 41, 2073–2076. [Google Scholar]
- 732.Kalaitzis JA, Hamano Y, Nilsen G and Moore BS, Org. Lett, 2003, 5, 4449–4452. [DOI] [PubMed] [Google Scholar]
- 733.Peña-López M, Martínez MM, Sarandeses LA and Sestelo JP, Chem. Eur. J, 2009, 15, 910–916. [DOI] [PubMed] [Google Scholar]
- 734.Funayama S, Ishibashi M, Komiyama K and Omura S, J. Org. Chem, 1990, 55, 1132–1133. [Google Scholar]
- 735.Kawasaki T, Hayashi Y, Kuzuyama T, Furihata K, Itoh N, Seto H and Dairi T, J. Bacteriol, 2006, 188, 1236–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 736.Kumano T, Tomita T, Nishiyama M and Kuzuyama T, J. Biol. Chem, 2010, 285, 39663–39671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 737.Isogai S, Nishiyama M and Kuzuyama T, Bioorganic Med. Chem. Lett, 2012, 22, 5823–5826. [DOI] [PubMed] [Google Scholar]
- 738.Komiyama K, Funayama S, Anraku Y, Ishibashi M, Takahashi Y and Ōmura S, J. Antibiot, 1990, 43, 247–252. [DOI] [PubMed] [Google Scholar]
- 739.Carretero-Molina D, Ortiz-López FJ, Martín J, Oves-Costales D, Díaz C, De La Cruz M, Cautain B, Vicente F, Genilloud O and Reyes F, Mar. Drugs, 2020, 18, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 740.Shiomi K, Nakamura H, Iinuma H, Naganawa H, Isshiki K, Takeuchi T, Umezawa H and Iitaka Y, J. Antibiot, 1986, 39, 494–501. [DOI] [PubMed] [Google Scholar]
- 741.Gomi S, Ohuchi S, Sasaki T, Itoh J and Sezaki M, J. Antibiot, 1987, 40, 740–749. [DOI] [PubMed] [Google Scholar]
- 742.Fukuda DS, Mynderse JS, Baker PJ, Berry DM, Boeck LD, Yao RC, Mertz FP, Nakatsukasa WM, Mabe J, Ott J, Counter FT, Ensminger PW, Allen NE, Alborn WE and Hobbs JN, J. Antibiot, 1990, 43, 623–633. [DOI] [PubMed] [Google Scholar]
- 743.Henkel T and Zeeck A, J. Antibiot, 1991, 44, 665–669. [DOI] [PubMed] [Google Scholar]
- 744.Kagamizono T, Hamaguchi T, Ando T, Sugawara K, Adachi T and Osada H, J. Antibiot, 1999, 52, 75–80. [DOI] [PubMed] [Google Scholar]
- 745.Kuroda M, Ogita T, Enokida R, Okazaki H, and Kinoshita T, JP Pat., 7010857.A, 1995.
- 746.Kuroda M, Ogita T, Enokida R, Okazaki H, and Kinoshita T, JP Pat., 1134009522.A, 1999.
- 747.Shiomi K, Nakamura H, Iinuma H, Naganawm H, Takeuchi T, Umezawa H and Iitaka Y, J. Antibiot, 1987, 40, 1213–1219. [DOI] [PubMed] [Google Scholar]
- 748.Motohashi K, Sue M, Furihata K, Ito S and Seto H, J. Nat. Prod, 2008, 71, 595–601. [DOI] [PubMed] [Google Scholar]
- 749.Wu Z, Li S, Li J, Chen Y, Saurav K, Zhang Q, Zhang H, Zhang W, Zhang W, Zhang S and Zhang C, Mar. Drugs, 2013, 11, 2113–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 750.Farnaes L, Coufal NG, Kauffman CA, Rheingold AL, Dipasquale AG, Jensen PR and Fenical W, J. Nat. Prod, 2014, 77, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 751.Soria-Mercado IE, Jensen PR, Fenical W, Kasse S and Golen J, Acta Crystallogr. Sect. E Struct. Reports Online, 2004, 60, o1627–o1629. [Google Scholar]
- 752.Soria-Mercado IE, Prieto-Davo A, Jensen PR and Fenical W, J. Nat. Prod, 2005, 68, 904–910. [DOI] [PubMed] [Google Scholar]
- 753.Bin Cheng Y, Jensen PR and Fenical W, European J. Org. Chem, 2013, 2013, 3751–3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 754.Lacret R, Pérez-Victoria I, Oves-Costales D, De La Cruz M, Domingo E, Martín J, Díaz C, Vicente F, Genilloud O and Reyes F, Mar. Drugs, 2016, 14, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 755.Winter JM, Moffitt MC, Zazopoulos E, McAlpine JB, Dorrestein PC and Moore BS, J. Biol. Chem, 2007, 282, 16362–16368. [DOI] [PubMed] [Google Scholar]
- 756.Cho JY, Kwon HC, Williams PG, Jensen PR and Fenical W, Org. Lett, 2006, 8, 2471–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 757.Shiomi K, Iinuma H, Naganawa H, Isshiki K, Takeuchi T and Umezawa H, J. Antibiot, 1987, 40, 1740–1745. [DOI] [PubMed] [Google Scholar]
- 758.Moore BS, Synlett, 2018, 29, 401–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 759.Winter JM and Moore BS, J. Biol. Chem, 2009, 284, 18577–18581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 760.Miles ZD, Diethelm S, Pepper HP, Huang DM, George JH and Moore BS, Nat. Chem, 2017, 9, 1235–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 761.McKinnie SMK, Miles ZD, Jordan PA, Awakawa T, Pepper HP, Murray LAM, George JH and Moore BS, J. Am. Chem. Soc, 2018, 140, 17840–17845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 762.Bernhardt P, Okino T, Winter JM, Miyanaga A and Moore BS, J. Am. Chem. Soc, 2011, 133, 4268–4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 763.Winter JM, Jansma AL, Handel TM and Moore BS, Angew. Chemie, Int. Ed, 2009, 48, 767–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 764.Shiomi K, Iinuma H, Hamada M, Naganawa H, Manabe M, Matsuki C, Takeuchi T and Umezawa H, J. Antibiot, 1986, 39, 487–493. [DOI] [PubMed] [Google Scholar]
- 765.Shomura T, Gomi S, Ito M, Yoshida J, Tanaka E, Amano S, Watabe H-O, Ohuchi S, Itoh J, Sezaki M, Takebe H and Uotani K, J. Antibiot, 1987, 40, 732–739. [DOI] [PubMed] [Google Scholar]
- 766.Farnaes L, La Clair JJ and Fenical W, Org. Biomol. Chem, 2014, 12, 418–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 767.Dantzig AH, Minor PL, Garrigus JL, Fukuda DS and Mynderse JS, Biochem. Pharmacol, 1991, 42, 2019–2026. [DOI] [PubMed] [Google Scholar]
- 768.Hori Y, Abe Y, Shigematsu N, Goto T, Okuhara M and Kohsaka M, J. Antibiot, 1993, 46, 1890–1893. [DOI] [PubMed] [Google Scholar]
- 769.Hwang JS, Kim GJ, Choi HG, Kim MC, Hahn D, Nam JW, Nam SJ, Kwon HC, Chin J, Cho SJ, Hwang H and Choi H, J. Nat. Prod, 2017, 80, 2269–2275. [DOI] [PubMed] [Google Scholar]
- 770.Shin-ya K, Imai S, Furihata K, Hayakawa Y, Kato Y, Vanduyne GD, Clardy J and Seto H, J. Antibiot, 1990, 43, 444–447. [DOI] [PubMed] [Google Scholar]
- 771.Takagi H, Motohashi K, Miyamoto T, Shin-ya K, Furihata K and Seto H, J. Antibiot, 2005, 58, 275–278. [DOI] [PubMed] [Google Scholar]
- 772.Park JS and Kwon HC, Mar. Drugs, 2018, 16, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 773.Wessels P, Gohrt A, Zeeck A, Drautz H and Zahner H, J. Antibiot, 1991, 44, 1013–1018. [DOI] [PubMed] [Google Scholar]
- 774.Shin-ya K, Shimazu A, Hayakawa Y and Seto H, J. Antibiot, 1992, 45, 124–125. [DOI] [PubMed] [Google Scholar]
- 775.Volkmann C, Hartjen U, Zeeck A and Fiedler HP, J. Antibiot, 1995, 48, 522–524. [DOI] [PubMed] [Google Scholar]
- 776.Umezawa K, Masuoka S, Ohse T, Naganawa H, Kondo S, Ikeda Y, Kinoshita N, Hamada M, Sawa T and Takeuchi T, J. Antibiot, 1995, 48, 604–607. [DOI] [PubMed] [Google Scholar]
- 777.Martucci H, Campit SE, Gee SR, Bray WM, Gokey T, Cada AK, Yen TY, Minoura K, Guliaev AB, Lokey RS and Amagata T, J. Nat. Prod, 2017, 80, 684–691. [DOI] [PubMed] [Google Scholar]
- 778.Izumikawa M, Nagai A, Hashimoto J, Takagi M and Shin-ya K, J. Antibiot, 2010, 63, 729–731. [DOI] [PubMed] [Google Scholar]
- 779.Pathirana C, Jensen PR and Fenical W, Tetrahedron Lett, 1993, 33, 7663–7666. [Google Scholar]
- 780.Murray LAM, McKinnie SMK, Pepper HP, Erni R, Miles ZD, Cruickshank MC, López-Pérez B, Moore BS and George JH, Angew. Chemie, Int. Ed, 2018, 57, 11009–11014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 781.Shin-ya K, Furihata K, Hayakawa Y and Seto H, Tetrahedron Lett, 1990, 31, 6025–6026. [Google Scholar]
- 782.Gallagher KA and Jensen PR, BMC Genomics, 2015, 16, 960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 783.Sakoulas G, Nam SJ, Loesgen S, Fenical W, Jensen PR, Nizet V and Hensler M, PLoS One, 2012, 7, e29439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 784.Kaysser L, Bernhardt P, Nam SJ, Loesgen S, Ruby JG, Skewes-Cox P, Jensen PR, Fenical W and Moore BS, J. Am. Chem. Soc, 2012, 134, 11988–11991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 785.Ryu MJ, Hwang S, Kim S, Yang I, Oh DC, Nam SJ and Fenical W, Org. Lett, 2019, 21, 5779–5783. [DOI] [PubMed] [Google Scholar]
- 786.Gao J, Ko TP, Chen L, Malwal SR, Zhang J, Hu X, Qu F, Liu W, Huang JW, Cheng YS, Chen CC, Yang Y, Zhang Y, Oldfield E and Guo RT, Angew. Chemie, Int. Ed, 2018, 57, 683–687. [DOI] [PubMed] [Google Scholar]
- 787.Teufel R, Kaysser L, Villaume MT, Diethelm S, Carbullido MK, Baran PS and Moore BS, Angew. Chemie, Int. Ed, 2014, 53, 11019–11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 788.Diethelm S, Teufel R, Kaysser L and Moore BS, Angew. Chemie, Int. Ed, 2014, 53, 11023–11026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 789.Hayakawa Y, Yamazaki Y, Kurita M, Kawasaki T, Takagi M and Shin-ya K, J. Antibiot, 2010, 63, 379–380. [DOI] [PubMed] [Google Scholar]
- 790.Wong WR, Oliver AG and Linington RG, Chem. Biol, 2012, 19, 1483–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 791.Nam SJ, Kauffman CA, Paul LA, Jensen PR and Fenical W, Org. Lett, 2013, 15, 5400–5403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 792.Guo YA, Zhao M, Xu Z and Ye T, Chem. Eur. J, 2017, 23, 3572–3576. [DOI] [PubMed] [Google Scholar]
- 793.Guo CJ, Knox BP, Chiang YM, Lo HC, Sanchez JF, Lee KH, Oakley BR, Bruno KS and Wang CCC, Org. Lett, 2012, 14, 5684–5687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 794.Hamed A, Abdel-Razek AS, Frese M, Stammler HG, El-Haddad AF, Ibrahim TMA, Sewald N and Shaaban M, Molecules, 2018, 23, 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 795.Itoh T, Tokunaga K, Radhakrishnan EK, Fujii I, Abe I, Ebizuka Y and Kushiro T, ChemBioChem, 2012, 13, 1132–1135. [DOI] [PubMed] [Google Scholar]
- 796.Itoh T, Tokunaga K, Matsuda Y, Fujii I, Abe I, Ebizuka Y and Kushiro T, Nat. Chem, 2010, 2, 858–864. [DOI] [PubMed] [Google Scholar]
- 797.Walsh CT, Garneau-Tsodikova S and Howard-Jones AR, Nat. Prod. Rep, 2006, 23, 517–531. [DOI] [PubMed] [Google Scholar]
- 798.Shinichiro K, Kazutoshi S, Hiroyuki K, Atsuo O, Michiko M and Junichiro M, J. Antibiot, 1993, 46, 892–899. [Google Scholar]
- 799.Han X, Liu Z, Zhang Z, Zhang X, Zhu T, Gu Q, Li W, Che Q and Li D, J. Nat. Prod, 2017, 80, 1684–1687. [DOI] [PubMed] [Google Scholar]
- 800.Macherla VR, Liu J, Bellows C, Teisan S, Nicholson B, Lam KS and Potts BCM, J. Nat. Prod, 2005, 68, 780–783. [DOI] [PubMed] [Google Scholar]
- 801.Riclea R and Dickschat JS, Chem. Eur. J, 2011, 17, 11930–11934. [DOI] [PubMed] [Google Scholar]
- 802.Liu D-Z and Liang B-W, Nat. Prod. Commun, 2014, 9, 1934578X1400900. [PubMed] [Google Scholar]
- 803.Liu DZ and Liang BW, J. Antibiot, 2014, 67, 415–417. [DOI] [PubMed] [Google Scholar]
- 804.Liu DZ and Liang BW, Magn. Reson. Chem, 2013, 52, 57–59. [DOI] [PubMed] [Google Scholar]
- 805.Liu DZ, Liang BW and Li XF, Chem. Biodivers, 2015, 12, 153–156. [DOI] [PubMed] [Google Scholar]
- 806.Kwon HC, Espindola APDM, Park JS, Prieto-Davó A, Rose M, Jensen PR and Fenical W, J. Nat. Prod, 2010, 73, 2047–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 807.Fenical W and Jensen PR, Nat. Chem. Biol, 2006, 2, 666–673. [DOI] [PubMed] [Google Scholar]
- 808.Gallagher KA, Rauscher K, Ioca LP and Jensen PR, Appl. Environ. Microbiol, 2013, 79, 6894–6902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 809.Raju R, Piggott AM, Barrientos Diaz LX, Khalil Z and Capon RJ, Org. Lett, 2010, 12, 5158–5161. [DOI] [PubMed] [Google Scholar]
- 810.Schmidt J, Khalil Z, Capon RJ and Stark CBW, Beilstein J. Org. Chem, 2014, 10, 1228–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 811.Shang XF, Morris-Natschke SL, Liu YQ, Guo X, Xu XS, Goto M, Li JC, Yang GZ and Lee KH, Med. Res. Rev, 2018, 38, 775–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 812.Shang XF, Morris-Natschke SL, Yang GZ, Liu YQ, Guo X, Xu XS, Goto M, Li JC, Zhang JY and Lee KH, Med. Res. Rev, 2018, 38, 1614–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 813.Augustiniak H, Gerth K, Höfle G, Irschik H, Jansen R, Kunze B, Reichenbach H, Steinmetz H, and Trowitzsch-Kienast W, Ger. Offen. 42, 3520229 A1, 1986. [Google Scholar]
- 814.Kunze B, Hofle G, Reichenbach H and Hofle G, J. Antibiot, 1987, 40, 258–265. [DOI] [PubMed] [Google Scholar]
- 815.Höfle G, Böhlendorf B, Fecker T, Sasse F and Kunze B, J. Nat. Prod, 2008, 71, 1967–1969. [DOI] [PubMed] [Google Scholar]
- 816.Kitagawa W and Tamura T, J. Antibiot, 2008, 61, 680–682. [DOI] [PubMed] [Google Scholar]
- 817.Nachtigall J, Schneider K, Nicholson G, Goodfellow M, Zinecker H, Imhoff JF, Süssmuth RD and Fiedler HP, J. Antibiot, 2010, 63, 567–569. [DOI] [PubMed] [Google Scholar]
- 818.Zhang M, Yang CL, Xiao YS, Zhang B, Deng XZ, Yang L, Shi J, Wang YS, Li W, Jiao RH, Tan RX and Ge HM, J. Antibiot, 2017, 70, 853–855. [DOI] [PubMed] [Google Scholar]
- 819.Höfle G and Irschik H, J. Nat. Prod, 2008, 71, 1946–1948. [DOI] [PubMed] [Google Scholar]
- 820.Dekker KA, Inagaki T, Gootz TD, Huang LH, Kojima Y, Kohlbrenner WE, Matsunaga Y, McGuirk PR, Nomura E, Sakakibara T, Sakemi S, Suzuki Y, Yamauchi Y and Kojima N, J. Antibiot, 1998, 51, 145–152. [DOI] [PubMed] [Google Scholar]
- 821.Kawada M, Inoue H, Ohba SI, Hatano M, Amemiya M, Hayashi C, Usami I, Abe H, Watanabe T, Kinoshita N, Igarashi M, Masuda T, Ikeda D and Nomoto A, J. Antibiot, 2013, 66, 543–548. [DOI] [PubMed] [Google Scholar]
- 822.Le T, Lee E, Lee J, Hong A, Yim C-Y, Yang I, Choi H, Chin J, Cho S, Ko J, Hwang H, Nam S-J and Fenical W, Mar. Drugs, 2019, 17, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 823.Matsuo Y, Imagawa H, Nishizawa M and Shizuri Y, Science, 2005, 307, 1598. [DOI] [PubMed] [Google Scholar]
- 824.Gao X, Matsuo Y and Snider BB, Org. Lett, 2006, 8, 2123–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 825.Wang XJ, Gong DL, Wang JD, Zhang J, Liu CX and Xiang WS, Bioorganic Med. Chem. Lett, 2011, 21, 2313–2315. [DOI] [PubMed] [Google Scholar]
- 826.Sandmann A, Dickschat J, Jenke-Kodama H, Kunze B, Dittmann E and Müller R, Angew. Chemie, Int. Ed, 2007, 46, 2712–2716. [DOI] [PubMed] [Google Scholar]
- 827.Pistorius D, Li Y, Mann S and Müller R, J. Am. Chem. Soc, 2011, 133, 12362–12365. [DOI] [PubMed] [Google Scholar]
- 828.Höfle G and Kunze B, J. Nat. Prod, 2008, 71, 1843–1849. [DOI] [PubMed] [Google Scholar]
- 829.Stec E, Pistorius D, Müller R and Li SM, ChemBioChem, 2011, 12, 1724–1730. [DOI] [PubMed] [Google Scholar]
- 830.Pistorius D, Li Y, Sandmann A and Müller R, Mol. Biosyst, 2011, 7, 3308–3315. [DOI] [PubMed] [Google Scholar]
- 831.Kitagawa W, Ozaki T, Nishioka T, Yasutake Y, Hata M, Nishiyama M, Kuzuyama T and Tamura T, ChemBioChem, 2013, 14, 1085–1093. [DOI] [PubMed] [Google Scholar]
- 832.Oettmeier W, Dostatni R, Majewski C, Höfle G, Fecker T, Kunze B and Reichenbach H, Zeitschrift fur Naturforsch. - Sect. C J. Biosci, 1990, 45, 322–328. [Google Scholar]
- 833.Meunier B, Madgwick SA, Reil E, Oettmeier W and Rich PR, Biochemistry, 1995, 34, 1076–1083. [DOI] [PubMed] [Google Scholar]
- 834.Aussel L, Pierrel F, Loiseau L, Lombard M, Fontecave M and Barras F, Biochim. Biophys. Acta - Bioenerg, 2014, 1837, 1004–1011. [DOI] [PubMed] [Google Scholar]
- 835.Collins MD and Jones D, Microbiol. Rev, 1981, 45, 316–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 836.Hiraishi A, J. Biosci. Bioeng, 1999, 88, 449–460. [DOI] [PubMed] [Google Scholar]
- 837.Søballe B and Poole RK, Microbiology, 1999, 145, 1817–1830. [DOI] [PubMed] [Google Scholar]
- 838.Nowicka B and Kruk J, Biochim. Biophys. Acta - Bioenerg, 2010, 1797, 1587–1605. [DOI] [PubMed] [Google Scholar]
- 839.Morton RA, Nature, 1958, 182, 1764–1767. [DOI] [PubMed] [Google Scholar]
- 840.Chen YH, Lu MC, Chang YC, Hwang TL, Wang WH, Weng CF, Kuo J and Sung PJ, Tetrahedron Lett, 2012, 53, 1675–1677. [Google Scholar]
- 841.Sassa T and Yoshkoshi H, Agric. Biol. Chem, 1983, 47, 187–189. [Google Scholar]
- 842.Torigoe K, Wakasugi N, Sakaizumi N, Ikejima T, Suzuki H, Kojiri K and Suda H, J. Antibiot, 1996, 49, 314–317. [DOI] [PubMed] [Google Scholar]
- 843.Ishii S, Fujii M and Akita H, Chem. Pharm. Bull, 2009, 57, 1103–1106. [DOI] [PubMed] [Google Scholar]
- 844.Abby SS, Kazemzadeh K, Vragniau C, Pelosi L and Pierrel F, arXiv, 2020, preprint, arXiv:2005.00394. [DOI] [PubMed] [Google Scholar]
- 845.Li W, Trends Biochem. Sci, 2016, 41, 356–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 846.Payne KAP, White MD, Fisher K, Khara B, Bailey SS, Parker D, Rattray NJW, Trivedi DK, Goodacre R, Beveridge R, Barran P, Rigby SEJ, Scrutton NS, Hay S and Leys D, Nature, 2015, 522, 497–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 847.White MD, Payne KAP, Fisher K, Marshall SA, Parker D, Rattray NJW, Trivedi DK, Goodacre R, Rigby SEJ, Scrutton NS, Hay S and Leys D, Nature, 2015, 522, 502–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 848.Ferguson KL, Eschweiler JD, Ruotolo BT and Marsh ENG, J. Am. Chem. Soc, 2017, 139, 10972–10975. [DOI] [PubMed] [Google Scholar]
- 849.Frydman B and Rapoport H, J. Am. Chem. Soc, 1963, 85, 823–825. [Google Scholar]
- 850.Powls R, Redfearn E and Trippett S, Biochem. Biophys. Res. Commun, 1968, 33, 408–411. [DOI] [PubMed] [Google Scholar]
- 851.Ishii M, Kawasumi T, Igarashi Y, Kodama T and Minoda Y, Agric. Biol. Chem, 1983, 47, 167–169. [Google Scholar]
- 852.Ishii M, Kawasumi T and Igarashi Y, J. Bacteriol, 1987, 169, 2380–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 853.Holsclaw CM, Sogi KM, Gilmore SA, Schelle MW, Leavell MD, Bertozzi CR and Leary JA, ACS Chem. Biol, 2008, 3, 619–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 854.Howarth OW, Grund E, Kroppenstedt RM and Collins MD, Biochem. Biophys. Res. Commun, 1986, 140, 916–923. [DOI] [PubMed] [Google Scholar]
- 855.Dairi T, Methods Enzymol, 2012, 515, 107–122. [DOI] [PubMed] [Google Scholar]
- 856.Suvarna K, Stevenson D, Meganathan R and Hudspeth MES, J. Bacteriol, 1998, 180, 2782–2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 857.Hiratsuka T, Furihata K, Ishikawa J, Yamashita H, Itoh N, Seto H and Dairi T, Science, 2008, 321, 1670–1673. [DOI] [PubMed] [Google Scholar]
- 858.Joshi S, Fedoseyenko D, Mahanta N, Manion H, Naseem S, Dairi T and Begley TP, Curr. Opin. Chem. Biol, 2018, 47, 134–141. [DOI] [PubMed] [Google Scholar]
- 859.Cotrim CA, Weidner A, Strehmel N, Bisol TB, Meyer D, Brandt W, Wessjohann LA and Stubbs MT, ChemistrySelect, 2017, 2, 9319–9325. [Google Scholar]
- 860.Hein S, Klimmek O, Polly M, Kern M and Simon J, Mol. Microbiol, 2017, 104, 449–462. [DOI] [PubMed] [Google Scholar]
- 861.Sogi KM, Holsclaw CM, Fragiadakis GK, Nomura DK, Leary JA and Bertozzi CR, ACS Infect. Dis, 2016, 2, 800–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 862.Mougous JD, Senaratne RH, Petzold CJ, Jain M, Lee DH, Schelle MW, Leavell MD, Cox JS, Leary JA, Riley LW and Bertozzi CR, Proc. Natl. Acad. Sci. U. S. A, 2006, 103, 4258–4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 863.Charan RD, Schlingmann G, Janso J, Bernan V, Feng X and Carter GT, J. Nat. Prod, 2004, 67, 1431–1433. [DOI] [PubMed] [Google Scholar]
- 864.McAlpine JB, Banskota AH, Charan RD, Schlingmann G, Zazopoulos E, Piraee M, Janso J, Bernan VS, Aouidate M, Farnet CM, Feng X, Zhao Z and Carter GT, J. Nat. Prod, 2008, 71, 1585–1590. [DOI] [PubMed] [Google Scholar]
- 865.Bonitz T, Zubeil F, Grond S and Heide L, PLoS One, 2013, 8, e85707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 866.Ratnayake AS, Janso JE, Feng X, Schlingmann G, Goljer I and Carter GT, J. Nat. Prod, 2009, 72, 496–499. [DOI] [PubMed] [Google Scholar]
- 867.Boufaied N, Wioland MA, Falardeau P and Gourdeau H, Anticancer. Drugs, 2010, 21, 543–552. [DOI] [PubMed] [Google Scholar]
- 868.Mason WP, Belanger K, Nicholas G, Vallières I, Mathieu D, Kavan P, Desjardins A, Omuro A and Reymond D, J. Neurooncol, 2012, 107, 343–349. [DOI] [PubMed] [Google Scholar]
- 869.Kolevzon A, Clinical Trial, NCT03493607, 2018. https://clinicaltrials.gov/ct2/show/NCT03493607?term=NCT03493607&draw=2&rank=1 (accessed September 2, 2020).
- 870.Leys D, Curr. Opin. Chem. Biol, 2018, 47, 117–125. [DOI] [PubMed] [Google Scholar]
- 871.Annaval T, Han L, Rudolf JD, Xie G, Yang D, Chang CY, Ma M, Crnovcic I, Miller MD, Soman J, Xu W, Phillips GN and Shen B, ACS Chem. Biol, 2018, 13, 2728–2738. [DOI] [PubMed] [Google Scholar]
- 872.Hu Y, Wang K and MacMillan JB, Org. Lett, 2013, 15, 390–393. [DOI] [PubMed] [Google Scholar]
- 873.Marshall SA, Payne KAP, Fisher K, White MD, Ní Cheallaigh A, Balaikaite A, Rigby SEJ and Leys D, Nat. Commun, 2019, 10, 2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 874.Van Heijenoort Y, Leduc M, Singer H and Van Heijenoort J, J. Gen. Microbiol, 1987, 133, 667–674. [DOI] [PubMed] [Google Scholar]
- 875.Thorne KJ and Kodicek E, Biochem. J, 1966, 99, 123–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 876.Higashi Y, Strominger JL and Sweeley CC, J. Biol. Chem, 1970, 245, 3697–3702. [PubMed] [Google Scholar]
- 877.Huang LY, Wang SC, Cheng TJR and Wong CH, Biochemistry, 2017, 56, 5417–5427. [DOI] [PubMed] [Google Scholar]
- 878.Wang X, Ribeiro AA, Guan Z and Raetz CRH, Biochemistry, 2009, 48, 1162–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 879.Wallhausser KH, Nesemann G, Prave P and Steigler A, Antimicrob. Agents Chemother, 1965, 5, 734–736. [PubMed] [Google Scholar]
- 880.Huber G, Schacht U, Weidenmüller HL, Schmidt-Thomé J, Duphorn J and Tschesche R, Antimicrob. Agents Chemother, 1965, 5, 737–742. [PubMed] [Google Scholar]
- 881.Ostash B and Walker S, Nat. Prod. Rep, 2010, 27, 1594–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 882.Welzel P, Witteler F‐J, Müller D and Riemer W, Angew. Chemie, Int. Ed, 1981, 20, 121–123. [Google Scholar]
- 883.Fehlhaber HW, Girg M, Seibert G, Hobert K, Welzel P, Van Heijenoort Y and Van Heijenoort J, Tetrahedron, 1990, 46, 1557–1568. [Google Scholar]
- 884.Slusarchyk WA and Weisenborn FL, Tetrahedron Lett, 1969, 10, 659–662. [DOI] [PubMed] [Google Scholar]
- 885.Weisenborn FL, Bouchard JL, Smith D, Pansy F, Maestrone G, Miraglia G and Meyers E, Nature, 1967, 213, 1092–1094. [DOI] [PubMed] [Google Scholar]
- 886.Meyers E, Miraglia GJ, Smith DA, Basch HI, Pansy FE, Trejo WH and Donovick R, Appl. Microbiol, 1968, 16, 603–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 887.Slusarchyk WA, Osband JA and Weisenborn FL, Tetrahedron, 1973, 29, 1465–1472. [Google Scholar]
- 888.Subramaniam-Niehaus B, Schneider T, Metzger JW and Wohlleben W, Zeitschrift fur Naturforsch. Sect. C - J. Biosci, 1997, 52, 217–226. [DOI] [PubMed] [Google Scholar]
- 889.Uchida R, Iwatsuki M, Kim YP, Ohte S, Mura S and Tomoda H, J. Antibiot, 2010, 63, 151–155. [DOI] [PubMed] [Google Scholar]
- 890.Uchida R, Iwatsuki M, Kim YP, Ömura S and Tomoda H, J. Antibiot, 2010, 63, 157–163. [DOI] [PubMed] [Google Scholar]
- 891.Arai M, Torikata A, Enokita R, Fukatsu H, Nakayama R and Yoshida K, J. Antibiot, 1977, 30, 1049–1054. [DOI] [PubMed] [Google Scholar]
- 892.Arai M, Nakayama R, Yoshida K, Takeuchi M, Teramoto S and Torikata A, J. Antibiot, 1977, 30, 1055–1059. [DOI] [PubMed] [Google Scholar]
- 893.Takahashi S, Serita K, Arai M, Seto H, Furihata K and Otake N, Tetrahedron Lett, 1983, 24, 499–502. [Google Scholar]
- 894.He H, Shen B, Korshalla J, Siegel MM and Carter GT, J. Antibiot, 2000, 53, 191–195. [DOI] [PubMed] [Google Scholar]
- 895.Koyama N, Tokura Y, Takahashi Y and Tomoda H, Bioorganic Med. Chem. Lett, 2013, 23, 860–863. [DOI] [PubMed] [Google Scholar]
- 896.Ostash B, Saghatelian A and Walker S, Chem. Biol, 2007, 14, 257–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 897.Schuricht U, Hennig L, Findeisen M, Welzel P and Arigoni D, Tetrahedron Lett, 2001, 42, 3835–3837. [Google Scholar]
- 898.Ren F, Ko TP, Feng X, Huang CH, Chan HC, Hu Y, Wang K, Ma Y, Liang PH, Wang AHJ, Oldfield E and Guo RT, Angew. Chemie, Int. Ed, 2012, 51, 4157–4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 899.Zhang L, Chen CC, Ko TP, Huang JW, Zheng Y, Liu W, Wang I, Malwal SR, Feng X, Wang K, Huang CH, Hsu STD, Wang AHJ, Oldfield E and Guo RT, Angew. Chemie, Int. Ed, 2016, 55, 4716–4720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 900.Torikata A, Yoshikawa H, Katayama T, Arai M, Nakahara M and Kitano N, J. Antibiot, 1977, 30, 1060–1063. [DOI] [PubMed] [Google Scholar]
- 901.Halliday J, McKeveney D, Muldoon C, Rajaratnam P and Meutermans W, Biochem. Pharmacol, 2006, 71, 957–967. [DOI] [PubMed] [Google Scholar]
- 902.Wada SI, Ohba SI, Someno T, Hatano M and Nomoto A, J. Antibiot, 2014, 67, 319–322. [DOI] [PubMed] [Google Scholar]
- 903.Zhang M, Wada SI, Amemiya F, Watanabe T and Shibasaki M, Chem. Pharm. Bull, 2015, 63, 961–966. [DOI] [PubMed] [Google Scholar]
- 904.Wada SI, Kubota Y, Sawa R, Umekita M, Hatano M, Ohba SI, Hayashi C and Igarashi M, J. Antibiot, 2015, 68, 521–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 905.Ohtake S and Wang YJ, J. Pharm. Sci, 2011, 100, 2020–2053. [DOI] [PubMed] [Google Scholar]
- 906.Scheer H, in Chlorophylls and Bacteriochlorophylls, 2007, pp. 1–26. [Google Scholar]
- 907.Qiu NW, Jiang DC, Wang XS, Wang BS and Zhou F, Photosynthetica, 2019, 57, 974–984. [Google Scholar]
- 908.Fujita Y and Yamakawa H, in Modern Topics in the Phototrophic Prokaryotes: Metabolism, Bioenergetics, and Omics, Springer International Publishing, Cham, 2017, pp. 67–122. [Google Scholar]
- 909.Senge MO and Smith KM, in Anoxygenic Photosynthetic Bacteria, Kluwer Academic Publishers, Dordrecht, 2006, pp. 137–151. [Google Scholar]
- 910.Senge MO, Ryan AA, Letchford KA, MacGowan SA and Mielke T, Symmetry, 2014, 6, 781–843. [Google Scholar]
- 911.Walter E, Schreiber J, Zass E and Eschenmoser A, Helv. Chim. Acta, 1979, 62, 899–920. [Google Scholar]
- 912.Scheer H, Svec WA, Cope BT, Studier MH, Scott RG and Katz JJ, J. Am. Chem. Soc, 1974, 96, 3714–3716. [Google Scholar]
- 913.Holt AS, Purdie JW and Wasley JWF, Can. J. Chem, 1966, 44, 88–93. [Google Scholar]
- 914.Caple MB, Chow H and Strouse CE, J. Biol. Chem, 1978, 253, 6730–6737. [PubMed] [Google Scholar]
- 915.Purdie JW and Holt AS, Can. J. Chem, 1965, 43, 3347–3353. [Google Scholar]
- 916.Gloe A, Pfennig N, Brockmann H and Trowitzsch W, Arch. Microbiol, 1975, 102, 103–109. [DOI] [PubMed] [Google Scholar]
- 917.Mizoguchi T, Oh-oka H and Tamiaki H, Photochem. Photobiol, 2007, 81, 666–673. [DOI] [PubMed] [Google Scholar]
- 918.Brockmann H, Knobloch G, Schweer I and Trowitzsch W, Arch. Mikrobiol, 1973, 90, 161–164. [PubMed] [Google Scholar]
- 919.Kobayashi M, Oh-Oka H, Akutsu S, Akiyama M, Tominaga K, Kise H, Nishida F, Watanabe T, Amesz J, Koizumi M, Ishida N and Kano H, Photosynth. Res, 2000, 63, 269–280. [DOI] [PubMed] [Google Scholar]
- 920.Brockmann H and Lipinski A, Arch. Microbiol, 1983, 136, 17–19. [Google Scholar]
- 921.Mizoguchi T, Harada J and Tamiaki H, FEBS Lett, 2006, 580, 6644–6648. [DOI] [PubMed] [Google Scholar]
- 922.Chew AGM and Bryant DA, Annu. Rev. Microbiol, 2007, 61, 113–129. [DOI] [PubMed] [Google Scholar]
- 923.Zappa S, Li K and Bauer CE, Adv. Exp. Med. Biol, 2010, 675, 229–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 924.Thweatt JL, Canniffe DP and Bryant DA, in Advances in Botanical Research, 2019, vol. 90, pp. 35–89. [Google Scholar]
- 925.Oster U, Bauer CE and Rüdiger W, J. Biol. Chem, 1997, 272, 9671–9676. [DOI] [PubMed] [Google Scholar]
- 926.Frigaard NU, Voigt GD and Bryant DA, J. Bacteriol, 2002, 184, 3368–3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 927.Garcia-Gil LJ, Gich FB and Fuentes-Garcia X, Arch. Microbiol, 2003, 179, 108–115. [DOI] [PubMed] [Google Scholar]
- 928.Addlesee HA and Hunter CN, J. Bacteriol, 1999, 181, 7248–7255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 929.Mayfield JA, Dehner CA and DuBois JL, Curr. Opin. Chem. Biol, 2011, 15, 260–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 930.Poulos TL, Chem. Rev, 2014, 114, 3919–3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 931.Swenson SA, Moore CM, Marcero JR, Medlock AE, Reddi AR and Khalimonchuk O, Cells, 2020, 9, 579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 932.Ward B, in Molecular Medical Microbiology: Second Ed., Elsevier, 2014, vol. 1–3, pp. 201–233. [Google Scholar]
- 933.Mogi T, Saiki K and Anraku Y, Mol. Microbiol, 1994, 14, 391–398. [DOI] [PubMed] [Google Scholar]
- 934.Puustinen A and Wikstrom M, Proc. Natl. Acad. Sci. U. S. A, 1991, 88, 6122–6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 935.Steffens GCM, Biewald R and Buse G, Eur. J. Biochem, 1987, 164, 295–300. [DOI] [PubMed] [Google Scholar]
- 936.Hosler JP, Fetter J, Tecklenburg MMJ, Espe M, Lerma C and Ferguson- Miller S, J. Biol. Chem, 1992, 267, 24264–24272. [PubMed] [Google Scholar]
- 937.Dailey HA, Dailey TA, Gerdes S, Jahn D, Jahn M, O’Brian MR and Warren MJ, Microbiol. Mol. Biol. Rev, 2017, 81, e00048–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 938.Saiki K, Mogi T, Ogura K and Anraku Y, J. Biol. Chem, 1993, 268, 26041–26045. [PubMed] [Google Scholar]
- 939.Saiki K, Mogi T and Anraku Y, Biochem. Biophys. Res. Commun, 1992, 189, 1491–1497. [DOI] [PubMed] [Google Scholar]
- 940.Brown KR, Allan BM, Do P and Hegg EL, Biochemistry, 2002, 41, 10906–10913. [DOI] [PubMed] [Google Scholar]
- 941.Brown BM, Wang Z, Brown KR, Cricco JA and Hegg EL, Biochemistry, 2004, 43, 13541–13548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 942.Zhang X, King-Smith E, Dong L-B, Yang LC, Rudolf JD, Shen B and Renata H, Science, 2020, 369, 799–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.