

Abstract
Background
Liver metastasis of colorectal cancer (CRC) is an important cause of death from CRC, but its molecular mechanism is still unclear. In recent years, whole-exome sequencing has played an increasingly important role in the study of the occurrence and development of diseases, especially malignant tumors. Its high throughput and low cost advantages enable researchers to explore the pathogenic genes of diseases, and screen potential molecular markers and therapeutic targets from the level of genomics.
Methods
This study collected the primary tumor tissues, matched paracancerous, normal tissues, and liver metastases of 4 CRC patients admitted to the Department of General Surgery of the First Affiliated Hospital of Soochow University, and performed high-depth whole-exome sequencing, with the sequencing depth of each sample reaching 123× on average, then filtered the sequencing data, compared them, and analyzed the bioinformatics data.
Results
we found 8,565 single nucleotide variants (SNV) and 429 insertions/deletions (InDel) in the primary and hepatic lesion tissues, and the genes with the highest mutation frequency were titin (TTN), obscurin (OBSCN), and homeodomain-interacting protein kinase 2 (HIPK2). The Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of the mutant genes was conducted, and it was found that the mutant genes were mainly concentrated in the cells, cell parts, and cellular process of GO. The results of KEGG pathway analysis showed that mutations were mainly distributed in circadian entrainment, insulin secretion, and glutamatergic synapse. Further, we identified 723 SNV and Indel genes with high frequency mutations including TTN, OBSCN, and hydrocephalus-inducing protein homolog (HYDIN) across all tissues of liver metastases. The GO analysis showed that the mutated genes in liver metastatic tissues were mainly concentrated in cell, cell part, and cellular process. The KEGG pathway analysis showed that high frequency mutation genes were focused on gastric acid secretion, bile secretion, and melanogenesis.
Conclusions
This study found some candidate genes related to the occurrence of CRC and liver metastasis through whole-exome sequencing of relevant tissues in CRC patients with liver metastasis, which is expected to provide new markers and therapeutic targets for such patients.
Keywords: Whole-exome sequencing, colorectal cancer (CRC), liver metastasis
Introduction
Colorectal cancer (CRC) is one of the most common digestive system malignancies in the world. In China, nearly one fifth of CRC patients have been found to have liver metastasis (LM) at their first diagnosis, and half of CRC patients have LM during the disease progression. The involvement of LM is a very important factor in the prognosis of CRC (1,2), it is caused by the interaction of multiple factors, and its clinical phenotype, characteristics, and molecular mechanism of metastasis are also currently hot topics in both clinical and basic research. The variation of tumor cell genome is an important factor for the occurrence and metastasis of tumors (3-5). In recent years, whole-exome sequencing has been widely used in the study of various diseases, especially in oncology research. Several functional genes associated with lung cancer targeting or immunotherapy have been identified in non-small cell lung carcinoma (NSCLC) (6,7). It has also been repeatedly confirmed by whole-exome sequencing that protein 53 (P53), Kirsten rat sarcoma viral oncogene homolog (KRAS), adenomatous polyposis coli (APC), and other genes play an important role in the occurrence and progression of CRC (8,9). However, the genomic characteristics of LM from CRC are still unclear. Though there are many studies on the NGS of colorectal cancer, but relatively few studies on the gene detection of colorectal cancer with liver metastasis, and the data of exome sequencing of colorectal cancer with liver metastasis are even less in China.
In this study, primary tumor tissues, matched normal tissues, and liver metastasis specimens of 4 CRC patients with LM from the General Surgery Department of the First Affiliated Hospital of Soochow University were collected, and high-depth complete exome sequencing was performed to analyze the mutated genes, tumor mutation burden (TMB), molecular functions, and signal pathways related to CRC with LM. It is hoped that this study can deepen the understanding of CRC with LM and provide potential marker molecules and therapeutic targets for the diagnosis and treatment of CRC with LM. We present the following article in accordance with the MDAR reporting checklist (available at http://dx.doi.org/10.21037/jgo-21-9).
Methods
Participants and samples
CRC patient sample acquisition
We collected primary CRC tumor tissues, matched normal tissues, and liver metastatic tumor tissues of 4 patients from the Department of General Surgery of the First Affiliated Hospital of Soochow University from September 2019 to April 2020. Informed written consent was provided by all patients before inclusion in this study. Respective tumor tissue samples from a histologically confirmed adenocarcinoma by two molecular pathologists were matched with the inclusion criteria. This study was conducted in accordance with the Declaration of Helsinki (as revised in 2013) and was approved by the Ethics Committee of the First Affiliated Hospital of Soochow University (No. 2021-023).
DNA extraction
The DNA was extracted from primary tumor tissues, liver metastatic tissues, and matched normal tissues using E.Z.N.A. Tissue DNA kit (Omega Bio-Tek, Norcross, GA, USA) and QIAamp DNA FFPE Tissue kit (Qiagen Sciences, Venlo, Netherlands). The DNA extracted from normal tissues were used as germline DNA control. The samples with cancer cell populations were estimated by pathologists to ensure more than 75% of cells were cancer cells. A bioanalyzer (Agilent, Palo Alto, CA, USA) was used to assess DNA quantity.
Whole-exome sequencing and data processing
Sample genome DNA was fragmented using NEBNext dsDNA Fragmentase (NEB, Ipswich, MA, USA) followed by DNA ends repairing. End-repaired DNA fragments were detailed and ligated with the NEBNext adaptor (NEB, Ipswich, MA, USA). Biotinylated RNA library baits and magnetic beads were mixed with the barcoded library for selection of targeted regions using the SureSelect Human All Exon V6 Kit (Agilent Technologies, Palo Alto, CA, USA). The captured sequences were further amplified for 150 bp paired-end sequencing in Illumina X-ten system (Illumina, San Diego, CA, USA).
Somatic single nucleotide variant and insertion/deletion identification
To identify somatic single nucleotide variants (SNVs) and insertion/deletions (InDels), the Burrows-Wheeler Aligner (BWA) (10) was used to align the clean reads from each sample against the human reference genome (GRCh38). The picard tool (http://picard.sourceforge.net/index.shtml) removed the read duplicates derived from library polymerase chain reaction (PCR). Somatic SNV and InDel calling was processed for multi-sample by MuTect (https://software.broadinstitute.org/cancer/cga/mutect) (11). Tumor mutation burden (TMB) is calculated as the number of somatic mutations per million bases (MB), including somatic single nucleotide variants (SNVs) and insertion/deletions (InDels).
SNV
The gene sets screened were used for functional annotation analysis using in-house script, which consisted of the Kyoto Encyclopedia of Genes and Genomes (KEGG) (12) and Gene Ontology (GO) (13) database. The significance of gene group enrichment was defined by a modified Fisher’s exact test and a P value <0.05 was considered to indicate a statistically significant difference.
Statistical analyses
The software SPSS (IBM Corp., Armonk, NY, USA) (14) was used to analyze all the correlated biological and clinical variables.
Results
Participant characteristics
The mean age of the 4 CRC patients with LM was 64.5 years (range, 46–77 years), and included 3 males and 1 female. All 4 were diagnosed with adenocarcinoma, and did not receive adjuvant chemoradiotherapy before Surgery. The tumor stage of 4 participants were stage IV. Half (n=2) of them were histological grade 2 and another 2 were histological grade 3. All 4 participants had lymphatic invasion and perineural invasion. The carcinoembryonic antigen (CEA) was increased in all 4 participants (Table 1).
Table 1. Clinicopathological participant characteristics.
| Characteristics | No. of cases [%] |
|---|---|
| Total number | |
| Age, years, mean [range] | 64.5 [46–77] |
| Gender | |
| Male | 3 [75] |
| Female | 1 [25] |
| Pathological diagnosis | |
| Adenocarcinoma | 4 [100] |
| Tumor stage at diagnosis | |
| III | 0 |
| IV | 4 [100] |
| Histological grade | |
| Grade 2 | 2 [50] |
| Grade 3 | 2 [50] |
| Vascular invasion | |
| Yes | 2 [50] |
| No | 2 [50] |
| Lymphatic invasion | |
| Yes | 4 [100] |
| No | 0 |
| Perineural invasion | |
| Yes | 2 [50] |
| No | 2 [50] |
| CEA value at diagnosis | |
| Normal | 0 |
| Increased | 4 [100] |
Whole-exome sequencing
We collected the primary tumor tissues, matched paracancerous normal tissues, and LMs of 4 patients admitted to the Department of General Surgery of the First Affiliated Hospital of Soochow University, and performed high-depth whole-exome sequencing, with the sequencing depth of each sample reaching 123× on average (Table S1).
Identification of somatic single nucleotide polymorphisms and InDels
The normal tissue samples data of all participants matched the original site of cancer tissue samples data. To compare the LMs sample data analysis, we took the first participants’ normal tissue and named it CRC01C (CRCO1 CONTROL), the original cancer organization was named CRC01CvsT (CRCO1 CONTROL vs. tumor), LMs sample was named CRC01CvsLM (CRCO1 CONTROL vs. liver metastasis), and then named the rest of the participant samples in the same way. By filtering the sequencing comparing and analyzing the data, 8,565 SNVs and 429 Indels were detected across all samples; CRC04CvsT had the largest number of SNVs (5,940), followed by CRC01CvsLM (1,094) (Table S2). Significantly mutated genes (SMG) are those genes whose mutation frequency is significantly higher than those in the background. Generally, differences of somatic SNV and InDel are analyzed comprehensively. MuSiC (The Genome Institute, Washington University, USA) software (15) was used to search for genes with higher mutation frequency in tumor samples (compared with control samples). A significantly mutated gene (SMG) test was performed for each type of mutation, whose test method was convolution test (CT). We found that the genes with the highest frequency of mutation were titin (TTN), obscurin (OBSCN), and homeodomain-interacting protein kinase 2 (HIPK2). We identified the 30 genes with the highest frequency of mutations. We also calculated the TMB of each sample respectively on the average TMB over 16 mutations per MB (5–60 mutations/MB) (Figure 1).
Figure 1.

Landscape of somatic mutations in all samples. Figure left: proportion of the number of gene mutations. Figure right: significance of high-frequency mutations. Up: Sample mutation frequency. The types of mutations are shown, including (I) non-synonymous mutations (green), (II) shear site mutations (yellow), (III) causes code shift insertion (purple), and (IV) causes code shift deletion (red).
KEGG and GO analysis
We conducted GO and KEGG pathway analysis of the mutant genes, revealing that the mutant genes were mainly concentrated in the cells, cell parts, and cellular process of GO (Figure 2). Results of KEGG pathway analysis showed that mutations were mainly distributed in circadian entrainment, insulin secretion, and glutamatergic synapse (Figure 3).
Figure 2.

GO analysis histogram of the mutated genes of all samples. GO, Gene Ontology.
Figure 3.
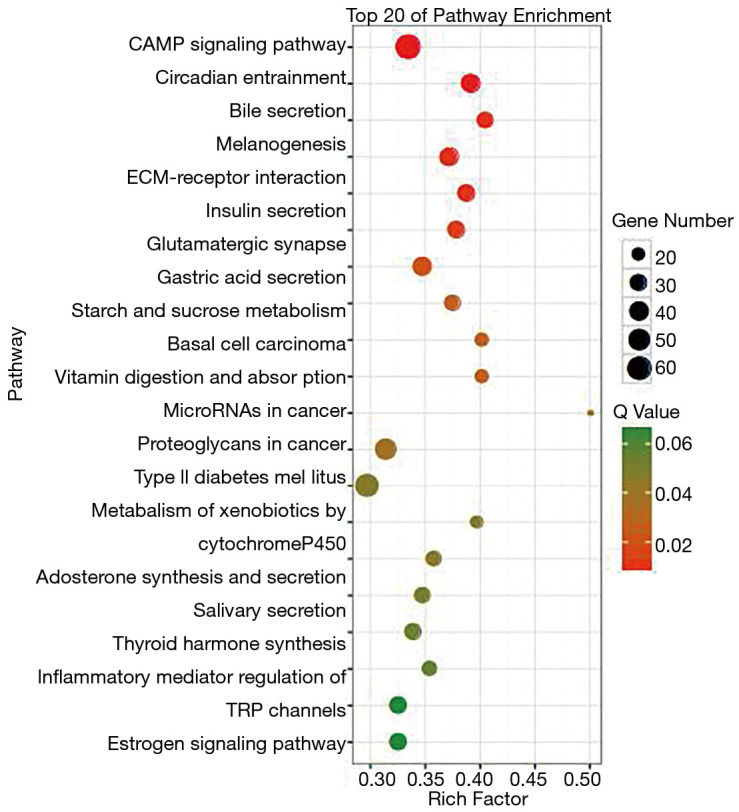
KEGG bubble of mutant genes of all samples. KEGG, Kyoto Encyclopedia of Genes and Genomes.
Gene mutation and KEGG and GO analysis in LM samples
We further analyzed the high frequency mutated genes of LMs, which were TTN, OBSCN and hydrocephalus-inducing protein homolog (HYDIN) (Figure 4). The GO analysis showed that the mutated genes in LM tissues were mainly concentrated in the cell, cell parts, and cellular process (Figure 5). The KEGG pathway analysis showed that high frequency mutation genes were focused on gastric acid secretion, bile secretion, and melanogenesis (Figure 6).
Figure 4.

Landscape of somatic mutations in CRCLM samples. Figure left: proportion of gene mutations. Figure right: significance of highfrequencymutations. Figure right: sample mutation frequency. The types of mutations are shown, including (I) non-synonymous mutations (green), (II) shear site mutations (yellow), (III) causes code shift insertion (purple), and (IV) causes code shift deletion (red). CVRCLM, colorectal cancer liver metastasis.
Figure 5.

GO analysis histogram of mutant genes in colorectal cancer liver metastasis samples. GO, Gene Ontology.
Figure 6.
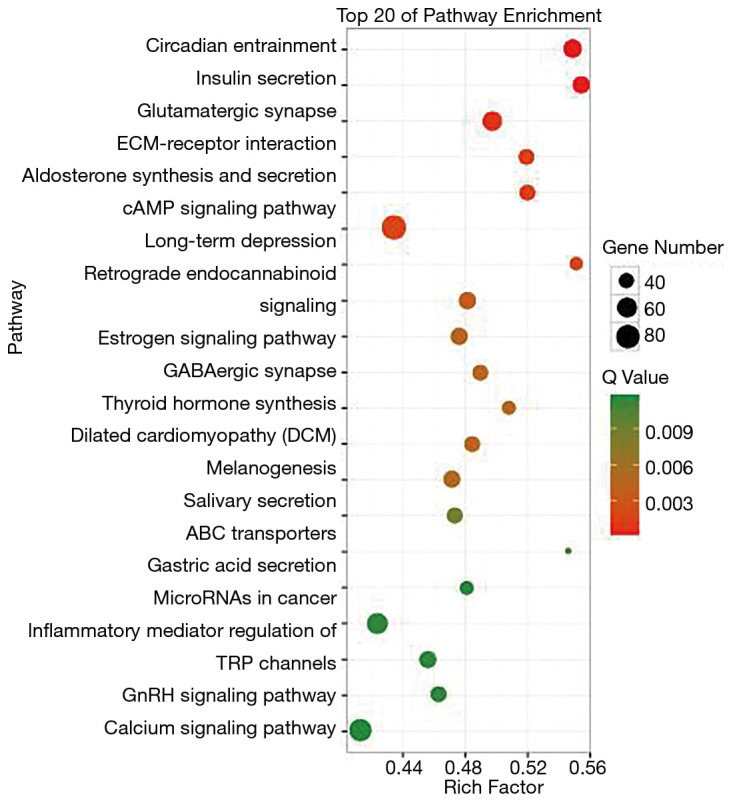
KEGG bubble diagram of liver metastases from colorectal cancer. KEGG, Kyoto Encyclopedia of Genes and Genomes.
Discussion
Tumor metastasis is an important factor in tumor progression, and distant metastasis is often the main reason for tumor treatment failure. The process of LM of CRC is extremely complex, involving many tumor-related molecules, which are induced and regulated by their respective specific signaling pathways. With the continuous progress of all kinds of research techniques in recent years, a growing body of evidence has shown that tumor development and metastasis are related to the cancer genome (16),transcriptome (17), epigenetics (18), proteome (19),metabolome (20), and tumor microenvironment (21) interaction cross-regulation of a dynamic complex process. This study collected the primary tumor tissues, matched paracancerous normal tissues, and metastatic tissues of 4 patients admitted to the General Surgery Department of the First Affiliated Hospital of Soochow University, and conducted high-depth whole-exome sequencing, in an attempt to explore the molecular mechanism of CRC LM from the genomic level. We performed an average sequencing depth of 123× per sample. By filtering, comparing, and analyzing the data, we identified 8,565 SNVs and 429 Indels in primary and hepatic metastases, and we found that the genes with the highest frequency of mutation were TTN, OBSCN, HIPK2, and HYDIN. The TTN gene was the most frequently mutated gene and had mutations in all 4 participants with LM. The TTN gene is known to encode rhabdomyin, and mutations in this gene are also associated with familial hypertrophic cardiomyopathy (HCM) 9 (22). It has also been identified as a chromosomal structural protein, and TTN has been found to promote bone metastasis in breast cancer (23). We speculated that variation of TTN might lead to chromosome instability and thus promote the occurrence and metastasis of tumors. The OBSCN gene is over 150 kb long and contains over 80 exons encoding a protein of approximately 720 kDa that belongs to the sarcomeric signaling protein family (24,25). It is an important signaling protein that is involved in the modulation of multiple cellular signals. We hypothesized that OBSCN mutation might lead to abnormal intracellular signaling pathway transduction that catalyzed changes in cell function.
Due to the small number of patients with liver metastasis of colorectal cancer, it is difficult to obtain appropriate tissue samples of liver metastasis of colorectal cancer for sequencing. We will also increase the number of samples of liver metastasis of colorectal cancer for total exon sequencing in the future work to obtain more data. And due to financial constraints, this study failed to combine with other omics studies such as transcriptome and proteomics. We will try to address these issues in the future to further explore the molecular mechanism of CRC LM.
Conclusions
In this study, we found some candidate genes related to the occurrence of CRC and LM through whole-exome sequencing of relevant tissues in CRC patients with LM. These findings are expected to provide us with new markers and therapeutic targets for CRC patients with LM.
Acknowledgments
Funding: This work was supported by Project of Nature Science Foundation of China (81672348, 81702048), National Science Foundation of Jiangsu Province of China (BK20191172), Program of Postgraduate Research Innovation in University of Jiangsu Province (KYCX19_1993), Project of Key laboratory of Clinical pharmacy of Jiangsu Province of China (XZSYSKF2020027), Project of Science and Technology Development Plan of Suzhou City of China (SYS2019007), Project of Gusu Medical Key Talent of Suzhou City of China (GSWS2020005), Project of Medical Science and Technology Development Foundation of Suzhou City of China (SYS201734), The Science and Technology Project Foundation of Suzhou (SS201852, SS202093), the Science and Education for Health Foundation of Suzhou for Youth (KJXW2019074) and Project of Medical Specialty and Community of Baoshan District of Shanghai City of China (BSZK-2018-A08).
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. This study was conducted in accordance with the Declaration of Helsinki (as revised in 2013) and was approved by the Ethics Committee of the First Affiliated Hospital of Soochow University (No. 2021-023). Informed written consent was provided by all patients before inclusion in this study.
Open Access Statement: This is an Open Access article distributed in accordance with the Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License (CC BY-NC-ND 4.0), which permits the non-commercial replication and distribution of the article with the strict proviso that no changes or edits are made and the original work is properly cited (including links to both the formal publication through the relevant DOI and the license). See: https://creativecommons.org/licenses/by-nc-nd/4.0/.
Reporting Checklist: The authors have completed the MDAR checklist. Available at http://dx.doi.org/10.21037/jgo-21-9
Data Sharing Statement: Available at http://dx.doi.org/10.21037/jgo-21-9
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at http://dx.doi.org/10.21037/jgo-21-9). The authors have no conflicts of interest to declare.
(English Language Editor: J. Jones)
References
- 1.Weitz J, Koch M, Debus J, et al. Colorectal cancer. Lancet 2005;365:153-65. 10.1016/S0140-6736(05)17706-X [DOI] [PubMed] [Google Scholar]
- 2.Engstrand J, Nilsson H, Strömberg C, et al. Colorectal cancer liver metastases - a population-based study on incidence, management and survival. BMC Cancer 2018;18:78. 10.1186/s12885-017-3925-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bhullar DS, Barriuso J, Mullamitha S, et al. Biomarker concordance between primary colorectal cancer and its metastases. EBioMedicine 2019;40:363-74. 10.1016/j.ebiom.2019.01.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Huizen NA, Coebergh van den Braak RRJ, Doukas M, et al. Up-regulation of collagen proteins in colorectal liver metastasis compared with normal liver tissue. J Biol Chem 2019;294:281-9. 10.1074/jbc.RA118.005087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alves JM, Prado-López S, Cameselle-Teijeiro JM, et al. Rapid evolution and biogeographic spread in a colorectal cancer. Nat Commun 2019;10:5139. 10.1038/s41467-019-12926-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haratani K, Hayashi H, Tanaka T, et al. Tumor immune microenvironment and nivolumab efficacy in EGFR mutation-positive non-small-cell lung cancer based on T790M status after disease progression during EGFR-TKI treatment. Ann Oncol 2017;28:1532-9. 10.1093/annonc/mdx183 [DOI] [PubMed] [Google Scholar]
- 7.Lusk CM, Watza D, Dyson G, et al. Profiling the Mutational Landscape in Known Driver Genes and Novel Genes in African American Non-Small Cell Lung Cancer Patients. Clin Cancer Res 2019;25:4300-8. 10.1158/1078-0432.CCR-18-2439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boutin AT, Liao WT, Wang M, et al. Oncogenic Kras drives invasion and maintains metastases in colorectal cancer. Genes Dev 2017;31:370-82. 10.1101/gad.293449.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang J, Lin Y, Huang Y, et al. Genome landscapes of rectal cancer before and after preoperative chemoradiotherapy. Theranostics 2019;9:6856-66. 10.7150/thno.37794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010;26:589-95. 10.1093/bioinformatics/btp698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cibulskis K, Lawrence MS, Carter SL, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 2013;31:213-9. 10.1038/nbt.2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 2000;28:27-30. 10.1093/nar/28.1.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000;25:25-9. 10.1038/75556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weaver B, Wuensch KL. SPSS and SAS programs for comparing Pearson correlations and OLS regression coefficients. Behav Res Methods 2013;45:880-95. 10.3758/s13428-012-0289-7 [DOI] [PubMed] [Google Scholar]
- 15.Dees ND, Zhang Q, Kandoth C, et al. MuSiC: identifying mutational significance in cancer genomes. Genome Res 2012;22:1589-98. 10.1101/gr.134635.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Birkbak NJ, McGranahan N. Cancer Genome Evolutionary Trajectories in Metastasis. Cancer Cell 2020;37:8-19. 10.1016/j.ccell.2019.12.004 [DOI] [PubMed] [Google Scholar]
- 17.Berglund E, Maaskola J, Schultz N, et al. Spatial maps of prostate cancer transcriptomes reveal an unexplored landscape of heterogeneity. Nat Commun 2018;9:2419. 10.1038/s41467-018-04724-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nebbioso A, Tambaro FP, Dell'Aversana C, et al. Cancer epigenetics: Moving forward. PLoS Genet 2018;14:e1007362. 10.1371/journal.pgen.1007362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Álvarez-Chaver P, De Chiara L, Martínez-Zorzano VS. Proteomic Profiling for Colorectal Cancer Biomarker Discovery. Methods Mol Biol 2018;1765:241-69. 10.1007/978-1-4939-7765-9_16 [DOI] [PubMed] [Google Scholar]
- 20.Kumar A, Misra BB. Challenges and Opportunities in Cancer Metabolomics. Proteomics 2019;19:e1900042. 10.1002/pmic.201900042 [DOI] [PubMed] [Google Scholar]
- 21.Vitale I, Manic G, Coussens LM, et al. Macrophages and Metabolism in the Tumor Microenvironment. Cell Metab 2019;30:36-50. 10.1016/j.cmet.2019.06.001 [DOI] [PubMed] [Google Scholar]
- 22.Neidhardt J, Fehr S, Kutsche M, et al. Tenascin-N: characterization of a novel member of the tenascin family that mediates neurite repulsion from hippocampal explants. Mol Cell Neurosci 2003;23:193-209. 10.1016/S1044-7431(03)00012-5 [DOI] [PubMed] [Google Scholar]
- 23.Chiovaro F, Martina E, Bottos A, et al. Transcriptional regulation of tenascin-W by TGF-beta signaling in the bone metastatic niche of breast cancer cells. Int J Cancer 2015;137:1842-54. 10.1002/ijc.29565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Russell MW, Raeker MO, Korytkowski KA, et al. Identification, tissue expression and chromosomal localization of human Obscurin-MLCK, a member of the titin and Dbl families of myosin light chain kinases. Gene 2002;282:237-46. 10.1016/S0378-1119(01)00795-8 [DOI] [PubMed] [Google Scholar]
- 25.Carlsson L, Yu JG, Thornell LE. New aspects of obscurin in human striated muscles. Histochem Cell Biol 2008;130:91-103. 10.1007/s00418-008-0413-z [DOI] [PubMed] [Google Scholar]