

Abstract
Pin1 is a unique isomerase that regulates protein conformation and function after phosphorylation. Pin1 aberration contributes to some neurological diseases, notably Alzheimer’s disease, but its role in epilepsy is not fully understood. We found that Pin1-deficient mice had significantly increased seizure susceptibility in multiple chemical inducing models and developed age-dependent spontaneous epilepsy. Electrophysiologically, Pin1 ablation enhanced excitatory synaptic transmission to prefrontal cortex (PFC) pyramidal neurons without affecting their intrinsic excitability. Biochemically, Pin1 ablation upregulated AMPA receptors and GluA1 phosphorylation by acting on phosphorylated CaMKII. Clinically, Pin1 was decreased significantly, whereas phosphorylated CaMKII and GluA1 were increased in the neocortex of patients with epilepsy. Moreover, Pin1 expression restoration in the PFC of Pin1-deficient mice using viral gene transfer significantly reduced phosphorylated CaMKII and GluA1 and effectively suppressed their seizure susceptibility. Thus, Pin1-CaMKII-AMPA receptors are a novel axis controlling epileptic susceptibility, highlighting attractive new therapeutic strategies.
Keywords: AMPA receptor, CaMKII, epilepsy, Pin1, prefrontal cortex
Introduction
Epilepsy is a devastating disorder characterized by increasing excitability of brain (Staley 2015; Li et al. 2017). The key to preventing and controlling epilepsy lies in understanding epileptogenesis. Epileptogenesis is triggered by the imbalance between excitatory and inhibitory pathways that result in progressive changes in brain structure and function, leading to recurrent seizures. Some factors might serve as suppressors or protectors during epileptogenesis. Identifying these factors is necessary and urgent for developing more effective and safe treatments.
The peptidyl-prolyl isomerase Pin1 is a unique enzyme that isomerizes substrate proteins between cis and trans conformations after they are phosphorylated on specific Ser or Thr residues preceding a Pro residue (pSer/Thr-Pro) (Lu et al. 1996; Kozono et al. 2018; Nechama et al. 2018; Chen et al. 2019). Pin1 is now known to have a profound impact on many key proteins involved in diverse cellular processes (Lu 2004; Lu and Zhou 2007; Lee et al. 2011; Liou et al. 2011). Pin1 aberrations contribute to many diseases, notably Alzheimer’s disease (AD), and thus, Pin1 has become an attractive novel drug target for controlling the deregulation of protein conformations (Lu 2004; Driver et al. 2015). Interestingly, recent studies have shown that Pin1 specifically catalyzes phosphorylated tau from the pathogenic “cis” back into the physiologic “trans” conformation (Lu et al. 2007), which can now be detected by cis and trans conformation-specific antibodies (Lu et al. 2016). Recently, it was reported that the expression of Pin1 is downregulated in the brain tissue of patients with temporal lobe epilepsy (Tang et al. 2017). However, the role and mechanisms of Pin1 in epileptogenesis are still unknown.
It is well known that the central nervous system networks of synaptically connected excitatory glutamatergic neurons are critical in generating epileptic synchronization. The fast synaptic excitation predominantly mediated by AMPA receptors is especially important. AMPA receptor antagonists markedly reduce or abolish epileptiform activity in a broad range of animal seizure models, which suggests that AMPA receptors are crucial to epileptic discharges (Rogawski 2013). More recently, evidence from clinical trials that perampanel has acceptable safety and significant efficacy in the treatment of human partial-onset seizures validates the AMPA receptor as a novel target for epilepsy therapy (French et al. 2013). A key mediator of AMPA receptors is calcium/calmodulin-dependent protein kinase II (CaMKII), which is strongly expressed at excitatory synapses. CaMKII activity can drive AMPA receptors to synapses by a mechanism that requires their GluA1 subunit (Hayashi et al. 2000). GluA1 is phosphorylated by CaMKII at Ser831, and such phosphorylation may enhance the conductance of AMPA receptors (Derkach et al. 1999; Kristensen et al. 2011). A recent study found that Pin1 bound to phosphorylated CaMKII and weakened its activity (Shimizu et al. 2018). Therefore, we speculate that Pin1 might interact with CaMKII to affect AMPA receptors associated with epilepsy.
Here, we used multiple chemical inducing models of epilepsy to investigate seizure susceptibility in Pin1−/− (knockout, KO) mice. We found that Pin1 KO mice were more susceptible to not only in pilocarpine-induced status epilepticus (SE) but also in pentylenetetrazole (PTZ)-kindled chronic epilepsy. Moreover, these mice also developed age-dependent spontaneous epilepsy. Although the morphology and the intrinsic excitability of pyramidal neurons in the prefrontal cortex (PFC) were unaltered, the excitatory synaptic transmission mediated by AMPA receptors was enhanced in these neurons in Pin1 KO mice. This enhancement was associated with the increased level of total AMPA receptors and phosphorylated GluA1 Ser831. Pin1 acted on the phosphorylated Thr176-Pro motif in CaMKII to suppress CaMKII Thr286 and formed a complex with CaMKII and GluA1 in the PFC, which may be one of the pathways by which AMPA receptor function is regulated. The clinical significance of these findings lies in that Pin1 was decreased, whereas phosphorylated GluA1 Ser831 and CaMKII Thr286 were increased in the neocortex of patients with epilepsy. Moreover, viral Pin1 re-expression in the PFC of Pin1 KO mice reduced the level of phosphorylated CaMKII Thr286 and GluA1 Ser831 and partially restored the epilepsy-related electrophysiological and behavioral impairments in these mice. Thus, Pin1 acts on the phosphorylated Thr176-Pro motif in CaMKII to suppress GluA1 Ser831 and excitatory synaptic transmission through AMPA receptors, uncovering a novel pathway underlying the pathophysiology of epilepsy and offering attractive new therapeutic strategies.
Materials and Methods
Animals and Reagents
Experiments were performed in Pin1 KO and WT mice. Pin1 KO mice were described previously (Kondo et al. 2017). As Pin1 KO mice are infertile, Pin1 KO and WT littermates were obtained by mating with heterozygous mice. Mice ~3 weeks old were used in the electrophysiology experiments. Mice were raised under a 12-h light/12-h dark cycle with food and water provided ad libitum. All experiments complied with the guidelines of the Animal Advisory Committee at the Fujian Medical University and the US National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. Picrotoxin (PTX) was purchased from Tocris Bioscience. Other chemicals were purchased from Sigma–Aldrich.
Seizure induction and behavioral observation
Male mice (at least 2 months old) were used for induction. Intraperitoneal injection (i.p.) of pilocarpine at a single dose (100 mg/kg) was given to the experimental animals to induce SE. To reduce peripheral cholinergic effects, methylscopolamine (1 mg/kg i.p.) was administered 30 min prior. Behavior was observed for a minimum of 2 h after pilocarpine injection. For induction of chronic epilepsy, mice were treated with a single i.p. dose of 20 mg/kg PTZ for 7 days.
Seizure Assessment
SE onset is typically characterized by continuous limbic seizure activity, which is interrupted by generalized convulsive seizures (Bankstahl et al. 2012). Seizures were assessed according to a modified version of the Racine scale (Racine 1972). This scale categorizes five stages of intensity: facial twitching (stage 1); tail stiffening and head nodding (stage 2); forelimb myoclonus (stage 3); loss of postural control (stage 4); and loss of righting reflexes, falling, and bouncing (stage 5).
Animal Surgery and EEG Recording
Male mice were anesthetized with 1% isoflurane and placed in a stereotaxic frame. Five stainless-steel screws were implanted into the skull to provide EEG contacts (two prefrontal sites: M/L, R ± 1.5 mm, A/P + 2 mm), ground (cerebellar), and mechanical support (Ni et al. 2016). EEGs were recorded and analyzed using the BL-420F Data Acquisition & Analysis System (Chengdu TME Technology). Signals were amplified and filtered between 1 and 300 Hz and referenced to the cerebellar ground screw.
Virus Injection
AAV-Ctrl (AAV2/9-GFP, 1.6 × 1012 vector genomes per mL) and AAV-Pin1 (AAV2/9-m-Pin1-3xFlag-GFP, 1.2 × 1012 vector genomes per mL) were made by Hanbio Biotechnology. Four-week-old mice were anesthetized with 2% isoflurane and placed in a stereotaxic frame. AAV vectors (2 μL) carrying either Pin1-3xFlag-GFP or GFP only were bilaterally injected into the PFC with the following coordinates: M/L, R ± 1.5 mm, A/P + 2 mm, D/V − 1 mm, relative to the bregma. The virus injection rate was 200 nL/min. Animals were used at least 21 days after virus injection.
Slice Preparation
Mice at the age of 21–28 days were used. Coronal brain slices (300 μm) through the PFC were prepared with a Vibroslice (VT 1000S, Leica) in ice-cold artificial cerebrospinal fluid (ACSF) containing the following (in mM): 126 NaCl, 2.5 KCl, 1.25 NaH2PO4, 2 MgSO4, 2 CaCl2, 26 NaHCO3, and 10 glucose. Slices were allowed to recover for at least 2 h in ACSF (1 h at 34 °C followed by 1 h at 22 °C). Then, slices were transferred to the recording chamber and superfused (3 mL/min) with ACSF at 32–33 °C. All solutions were saturated with 95% O2 and 5% CO2.
Electrophysiology
Neurons were visualized by an infrared-sensitive CCD camera with a 40× water-immersion lens (Olympus). For whole-cell recording, glass pipettes (3–4 MΩ) were filled with a solution containing (in mM) 130 K-gluconate, 20 KCl, 10 HEPES buffer, 4 Mg-ATP, 0.3 Na-GTP, 10 disodium phosphocreatine and 0.2 EGTA, with the pH adjusted to 7.25 with KOH and 290 mOsm. For spontaneous inhibitory postsynaptic current (IPSC) recordings, we used a pipette solution containing the following (in mM): 130 CsCl, 4 NaCl, 10 tetraethylammonium (TEA), 10 HEPES, 2 Na2-ATP, 0.5 Na3-GTP, and 0.2 EGTA, with the pH adjusted to 7.25 with CsOH. Recordings were started 2–3 min after a stable whole-cell configuration was obtained. Access resistance (Ra, <20 MΩ) was not compensated and was continually monitored throughout each experiment. Recordings were terminated whenever the input resistance increased by 30% or the access resistance exceeded 20 MΩ. Signals were acquired using a MultiClamp 700B amplifier controlled by Clampex 10.2 software via a Digidata 1440A interface (Molecular Devices) and filtered at 2 kHz, digitized at 10 kHz. The resting membrane potential (RMP) was the average membrane potential within 2 min after obtaining the whole-cell configuration. The input resistance (Rin) was calculated from small voltage deflections induced by rectangular hyperpolarizing current injections (−50 pA). We used the first spike evoked by the minimum current needed to elicit an action potential (AP) applied from −70 mV to quantify spike properties. AP amplitude was calculated as the voltage difference between the AP threshold and the AP peak. AP duration was measured as the duration at half-maximal amplitude. AHP amplitude was defined as the voltage difference between the AP threshold and the depth of the AHP. The root-mean-square noise level was 3–5 pA, and a threshold of 10–15 pA was used to detect and measure excitatory postsynaptic currents (EPSCs) and IPSCs. Ten to Twenty individual events of each neuron were taken from recordings every 30 s to 1 min. Evoked EPSCs were generated with a two-concentric bipolar stimulating electrode (FHC) positioned 100 μm from the neuron under recording. Paired pulses of 0.1 ms were delivered at 25-ms intervals and synchronized using a Mater-8 stimulator (A.M.P.I). The holding potential was −70 mV. All collected events were averaged and analyzed using Clampfit 10.2 (Molecular Devices) and Mini Analysis 6.0 software (Synaptosoft).
Immunohistochemistry
After inducing deep anesthesia, mice were perfused with ice-cold saline followed by 4% paraformaldehyde (PFA) in 0.1 M PBS, pH 7.4. The brains were removed, postfixed overnight in 4% PFA at 4 °C, and transferred to 30% sucrose in PBS. Coronal sections (40 μm) were cut on a cryostat (CM3050 S, Leica). After washing three times in 0.5% Triton X-100 in PBS (PBST), the sections were incubated in blocking buffer containing 3% bovine serum albumin and 5% normal goat serum in PBST for 1 h at RT and then with primary antibodies in blocking buffer overnight at 4 °C (mouse anti-Pin1, 1:400, from Kun Ping Lu; rabbit anti-NeuN, 1:1000, Abcam; mouse anti-NeuN, 1:1000, Abcam). After washing three times with PBST, the sections were incubated with Alexa Fluor 488- or Alexa Fluor 555-conjugated secondary antibodies at RT for 1 h. The sections were washed another three times in PBST and then incubated with DAPI (0.1 μg/mL) at RT for 5 min. After another three wash cycles in PBST, sections were mounted with Pro-long anti-fade medium (Invitrogen). All images were acquired on Zeiss Axio Observer A1. Quantitative analysis was performed with NIH ImageJ software.
Western Blot Analysis
Protein samples were run on 10% SDS-polyacrylamide gels and then transferred to polyvinylidene difluoride membranes. After blocking in 5% nonfat milk for 1 h at room temperature (RT), membranes were incubated with the respective primary antibodies at 4 °C overnight (mouse anti-Pin1, 1:3000, from Kun Ping Lu; rabbit anti-GluA1/2/3/4, 1:200, Santa Cruz Biotechnology; rabbit anti-p-Ser831 GluA1, 1:1000, Abcam; rabbit anti-p-Ser845 GluA1, 1:1000, Abcam; rabbit anti-GluN2A, 1:1000, Abcam; rabbit anti-GluN2B, 1:1000, Abcam; goat anti-vGAT, 1:200, Santa Cruz Biotechnology; mouse anti-CaMKII 1:200, Santa Cruz Biotechnology; rabbit anti-p-CaMKII Thr286, 1:1000, Abcam; rabbit anti-PKC, 1:1000, Abcam; rabbit anti-PKA, 1:1000, Abcam; mouse anti-actin, 1:3000, TransGen Biotech; rabbit anti-Flag, 1:1000, CST). After washing three times in 0.1% Tween 20 in TBS, the membranes were incubated with horseradish peroxidase-conjugated secondary antibodies in 3% nonfat milk for 1 h at RT and visualized with an electrochemiluminescence kit (Thermo Scientific).
Coimmunoprecipitation
Relevant proteins were obtained from WT mouse PFC homogenates and HEK293T cells transfected with CaMKIIα-Flag or GluA1-Flag (Sino Biological) in lysis buffer containing 20 mM Tris (pH 7.5), 150 mM NaCl, 1% Triton X-100, 10 mM EDTA, and protease and phosphatase inhibitor mixture. Extracts were incubated with rabbit anti-Pin1 antibodies (Abcam) for 1 h at 4 °C, and 15 μL of protein A/G PLUS-Agarose (Santa Cruz) was then added, followed by further incubation for 2 h at 4 °C. The precipitated proteins were washed 4–6 times in the same buffer and subjected to western analysis. Control aliquots were incubated with rabbitIgGs.
Plasmid Constructs and GST Pulldown Assays
CaMKIIα-Flag was purchased from (Sino Biological). Specific point mutations were introduced using the Fast Mutagenesis System (TransGen Biotech). All mutations were fully sequenced to exclude the possibility of other site mutations. CaMKIIα mutant proteins were expressed in HEK293T cells by transient transfection using TurboFect (Thermo Scientific). Extracts were incubated with 1 μM GST or GST-Pin1 for 1 h at 4 °C in lysis buffer containing 20 mM Tris (pH 7.5), 150 mM NaCl, 1% Triton X-100, 10 mM EDTA, and 0.1% BSA, and 15 μL of glutathione agarose beads (GE Healthcare) were then added, followed by further incubation for 1 h at 4 °C. The precipitated proteins were washed 4–6 times in the same buffer and subjected to immunoprecipitation.
Human Brain Tissues
Cortical tissues were obtained from 12 patients with intractable epilepsy, and control samples were obtained from six patients with brain trauma. Epilepsy was diagnosed according to criteria established by the International League Against Epilepsy (Kwan et al. 2010). The patients with intractable epilepsy conformed to the following criteria: typical clinical symptoms and electroencephalographic features, three or more antiepileptic drugs taken for at least 2 years at the maximal doses. The control patients had no history of epilepsy or other neurological diseases. Formal consent for the use of data and brain tissues was signed by the patients and their legal guardians. This research was approved by the Ethics Committee of Fujian Medical University. A portion of the resected brain tissues was stored in liquid nitrogen until being used for western blot. The remaining samples were fixed in 4% paraformaldehyde and then sectioned for immunofluorescence analysis.
Statistical Analysis
Experiments were routinely repeated at least three times, and the repeat number was increased according to effect size or sample variation. We estimated the sample size considering the variation and mean of the samples. No statistical method was used to predetermine sample size. No animals or samples were excluded from any analysis. Animals were randomly assigned groups for in vivo studies. All data are presented as the mean ± standard error of the mean (SEM) (n = number of individual samples), followed by determining significant differences using the two tailed Student’s t-test or analysis of variance (ANOVA) test, where *P < 0.05, **P < 0.01, and ***P < 0.001.
Data Availability
The authors declare that the main data supporting the findings of this study are within the article and its Supplementary Information files. Extra data are obtained from the corresponding authors upon request.
Results
Pin1 KO Increased Epileptic Susceptibility in Mice
To assess the pathophysiological consequences of Pin1 dysfunction, we measured seizure susceptibility in Pin1 KO mice. First, the genotypes of mice were confirmed by PCR (Fig. 1A), which was further confirmed by lack of Pin1 protein in the PFC of Pin1 KO mice by immunoblotting analysis (Fig. 1B) and immunostaining analysis (Fig. 1C). We used pilocarpine to induce robust SE (a state of continuous seizure activity), and most of the animals developed SE, as expected (Cavalheiro et al. 1996). However, the latency to SE was shorter in Pin1 KO mice than in Pin1+/+ (wild-type, WT) mice (WT, 36.8 ± 2.2 min vs. KO, 19.7 ± 2.6 min, P < 0.001; Fig. 1D), and Pin1 KO mice showed a higher SE stage (WT, 2.5 ± 0.2 vs. KO, 3.8 ± 0.2, P < 0.001; Fig. 1D). Moreover, the stage 5 seizure duration within the entire course of SE was longer in Pin1 KO mice, suggesting more severe epilepsy (WT, 14.8 ± 5.5 s vs. KO, 128.8 ± 12.3 s, P < 0.001; Fig. 1D). We also analyzed the electroencephalograms (EEGs) of mice after pilocarpine injection (Fig. 1E) and found that Pin1 KO mice showed higher amplitude (WT, 45.9 ± 4.9 μV vs. KO, 184.6 ± 7.4 μV, P < 0.001; Fig. 1E) of electrographic seizure (ES) and much more prolongation of electrographic seizure duration (ESD) (WT, 9.9 ± 0.3 min vs. KO, 28.6 ± 2.0 min, P < 0.001; Fig. 1E) in 1-h monitoring. These results indicate that pilocarpine can induce a more severe SE state in Pin1 KOmice.
Figure 1.
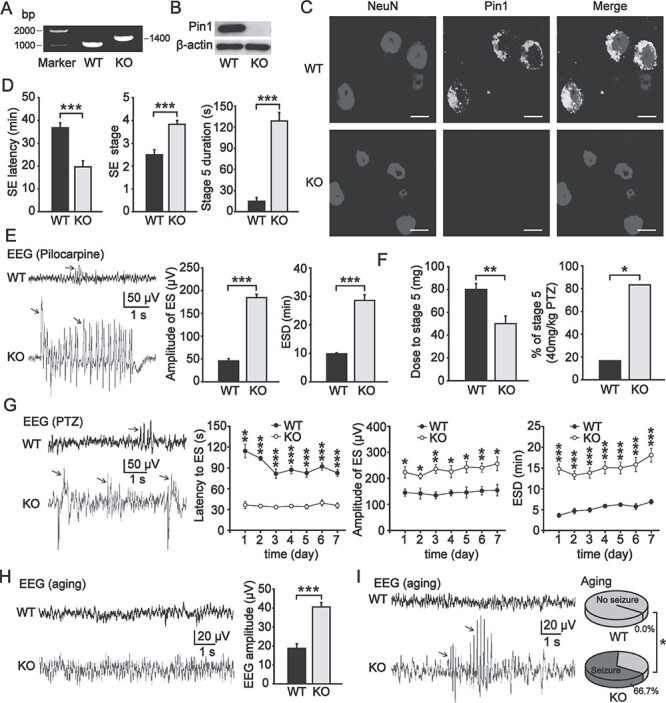
Seizure susceptibility increased in Pin1 KO mice. (A) Genotyping of Pin1 KO mice. (B) Western blot analysis indicated that Pin1 was deleted in Pin1 KO mice. (C) Pin1 was detectable in PFC pyramidal neurons in WT mice and was abolished in Pin1 KO mice. Scale bar, 10 μm. (D) Pilocarpine induced more severe SE in Pin1 KO mice (2–4 months). Left, the latency to SE was shorter in Pin1 KO mice (n = 6; ***P < 0.001). Middle, a higher SE stage was induced in Pin1 KO mice (n = 6; ***P < 0.001). Right, stage 5 seizure duration of SE was longer in Pin1 KO mice (n = 6; ***P < 0.001). (E) Left, representative EEG traces of WT mice (black) and Pin1 KO mice (gray) show ES (arrows) induced by pilocarpine. Middle, the amplitude of ES was higher in Pin1 KO mice (n = 6; ***P < 0.001). Right, the ESD was longer in Pin1 KO mice (n = 6; ***P < 0.001). (F) PTZ induced epileptogenesis more easily in Pin1 KO mice. Left, the total dose of PTZ needed to induce stage 5 seizures was lower in Pin1 KO mice (n = 6; **P < 0.01). Right, more Pin1 KO mice developed stage 5 seizures at 40 mg/kg PTZ (n = 6; *P < 0.05). (G) Left panel, representative EEG traces of WT mice (black) and Pin1 KO mice (gray) showing the ES (arrows) induced by PTZ. Right panel, chronic epilepsy induced by the administration of 20 mg/kg PTZ for 7 days showed shorter latency to ES, higher amplitude of ES, and longer ESD in Pin1 KO mice (n = 5; ***P < 0.001, *P < 0.05, **P < 0.01). (H) Pin1 KO mice (6–8 months) developed age-dependent seizure susceptibility. Representative EEG traces of aged WT mice (black) and Pin1 KO mice (gray) showed that the amplitude of EEG increased in Pin1 KO mice (n = 5; ***P < 0.001). (I) Left, representative EEG traces of aged WT mice (black) and Pin1 KO mice (gray) showed paroxysmal ES (arrows) in Pin1 KO mice. Right, spontaneous epilepsy emerged in more than half of the aged Pin1 KO mice but in none of the aged WT mice (n = 6; *P < 0.05).
To confirm these results, we applied another common chemical kindling model of epilepsy induced by PTZ, a GABA receptor antagonist, which has been widely used as a convulsant agent for the kindling model of epilepsy (Sood et al. 2008). Mice were repeatedly intraperitoneally injected a subconvulsant dose of 20 mg/kg PTZ. We found that the total dose of PTZ needed for mice to achieve stage 5 seizures was lower in the Pin1 KO group (WT, 80.0 ± 5.2 mg vs. KO, 50.0 ± 6.8 mg, P < 0.01; Fig. 1F). Moreover, for two doses of 20 mg/kg PTZ, more Pin1 KO mice developed stage 5 seizures (WT, 16.7% vs. KO, 83.3%, P < 0.05; Fig. 1F). We further induced chronic epilepsy in mice by administering a single dose of 20 mg/kg PTZ every day for 7 days and recorded EEGs (Fig. 1G) for 1-h each day. We observed shorter latency to ES, higher amplitude of ES and longer ESD (Fig. 1G) in Pin1 KO mice. These data suggest that Pin1 can inhibit epileptogenesis in the progression of epilepsy.
The above results demonstrate that Pin1 KO mice are more susceptible to chemically induced epilepsy. To address whether Pin1 KO mice develop spontaneous epilepsy without any chemical inducer, we compared both young and relatively older Pin1 KO mice with their WT littermates. We did not observe any spontaneous epilepsy in young Pin1 KO mice (2–4 months). We then compared the EEG recordings of relatively older Pin1 KO mice in relation to their Pin1 WT littermates (6–8 months). Notably, the amplitude of EEG increased in these Pin1 KO mice (WT, 18.7 ± 2.5 μV vs. KO, 40.5 ± 2.4 μV, P < 0.001; Fig. 1H). More importantly, these mice showed a few paroxysmal ESDs during EEG monitoring (Fig. 1I). Meanwhile, spontaneous stage 1–2 seizures occasionally emerged in more than half of these mice (66.7%), but none of the WT mice exhibited any seizure onsets over the long-term behavioral observation (P < 0.05; Fig. 1I). These data indicate that Pin1 deficiency may lead to increased seizure susceptibility in an age-dependent manner, which is consistent with the observation that Pin1 KO mice appear normal until they develop age-dependent neuronal degeneration (Liou et al. 2003; Pastorino et al. 2006) and precious studies that some genetic models exhibit adult-onset epilepsy (Lapray et al. 2010; Ketzef and Gitler 2014).
Pin1 KO Does Not Significantly Alter Intrinsic Excitability in Pyramidal Neurons in the PFC
To investigate the mechanisms underlying seizure susceptibility, we examined the properties of pyramidal neurons in the PFC of Pin1 KO mice. First, Pin1 KO mice showed a normal development of cortical layers, and the laminar distribution of NeuN-positive neurons was similar (Fig. 2A). Further detailed quantification of the laminar distribution (Fig. 2A) indicated no significant change between the genotypes. We also compared the pyramidal neuron soma size, and no difference was found in Pin1 KO mice (Fig. 2A). These data suggest that Pin1 dysfunction has no effect on some morphologic characters of pyramidal neurons in the PFC, which is consistent with previous study (Liou et al. 2003).
Figure 2.
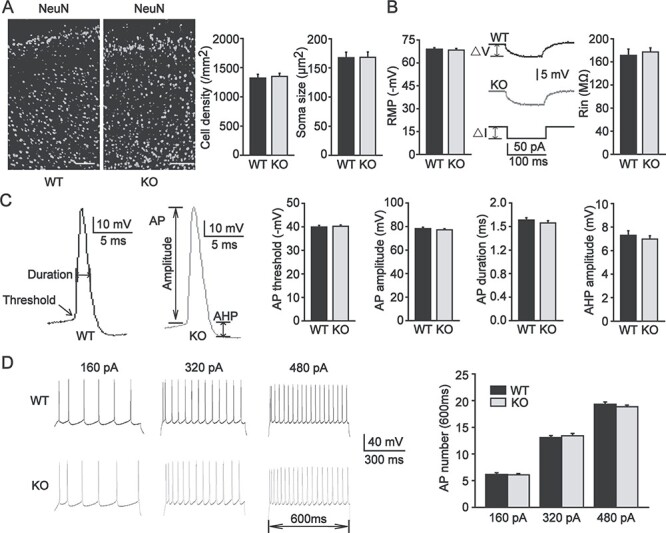
The morphology and intrinsic excitability of pyramidal neurons in the PFC were unchanged in Pin1 KO mice. (A) Left panel, coronal brain sections immunostained with NeuN in the PFC of WT and Pin1 KO mice. Scale bar, 100 μm. Right panel, quantification of the laminar distribution of pyramidal neurons (left) did not change in Pin1 KO mice (n = 18 sections, from 6 mice of each group). The soma size of PFC pyramidal neurons (right) was similar between WT and Pin1 KO mice (n = 48 neurons, from 6 mice of each group). (B) Left panel, the RMP of PFC pyramidal neurons did not change in Pin1 KO mice. Right panel, Rin was calculated from △V/△I and was similar in both genotypes. (C) Representative AP traces of WT mice (black) and Pin1 KO mice (gray) showed that AP threshold, amplitude, and duration and AHP amplitude were also similar. (D) Representative AP firing of WT mice (black) and Pin1 KO mice (gray). PFC pyramidal neurons were injected with a series of 600-ms step depolarizing current pulses and showed no difference in the number of APs generated at the 160, 320, and 480 pA levels (n = 36 neurons, from 6 mice of each group).
Next, we explored the electrophysiological characteristics of pyramidal neurons in the PFC without Pin1. Whole-cell recordings of pyramidal neurons in acute PFC slices obtained from Pin1 KO mice and WT littermates were performed in current-clamp mode. We first measured the intrinsic excitability of pyramidal neurons in the PFC. These neurons presented similar RMP and input resistance (Rin) values in both genotypes (Fig. 2B), thus indicating that Pin1 had no effect on the passive membrane properties of pyramidal neurons. Then, we analyzed and compared several firing parameters of pyramidal neurons between WT and Pin1 KO mice. The voltage threshold for AP generation was indistinguishable, and AP amplitude and AP duration were similar. We observed no change in afterhyperpolarization (AHP) amplitude (Fig. 2C). These data also indicated that the active membrane characteristics of pyramidal neurons did not change. We further examined the relationship between somatic current injection and AP firing in pyramidal neurons from WT and Pin1 KO mice. Pyramidal neurons were stimulated with a series of 600-ms step depolarizing current pulses (holding potential, −70 mV; increment, 40 pA). There was no difference in the number of APs at any of the depolarizing levels of 160, 320 or 480 pA (Fig. 2D). These results suggest no obvious alteration in the intrinsic excitability of pyramidal neurons in Pin1-deficientPFC.
Pin1 KO Enhances Excitatory Synaptic Transmission Mediated by AMPA Receptors
To determine whether Pin1 regulates the activity of PFC pyramidal neurons, spontaneous AP firing was recorded in whole-cell configurations. For bath application of 10 mM KCl, the firing rate increased in WT mice to 71 ± 8/min and in Pin1 KO mice to 146 ± 10/min (P < 0.001; Fig. 3A), suggesting enhanced activity-dependent excitability. Because the intrinsic properties of pyramidal neurons shown above did not change, we next determined whether Pin1 affects synaptic transmission in the PFC. First, we examined glutamatergic transmission to pyramidal neurons. Spontaneous AMPA-mediated EPSCs were recorded in voltage-clamp mode (holding potential, −70 mV) from both genotypes in the presence of picrotoxin (PTX, 100 μM) to block GABAA-mediated IPSCs. EPSCs recorded from Pin1 KO mice exhibited higher amplitude than those recorded from WT control littermates (WT, 53.8 ± 6.3 pA vs. KO, 96.6 ± 4.0 pA, P < 0.001), but no significant difference was found in frequency (WT, 1.3 ± 0.2 Hz vs. KO, 1.4 ± 0.2 Hz; Fig. 3B). The observed effects were selective for EPSCs since no significant changes were detected in the amplitude or frequency (Fig. 3C) of IPSCs recorded in the presence of kynurenic acid (2 mM, to block the glutamatergic transmissions). Next, to distinguish presynaptic or postsynaptic effects on the increased amplitude of EPSCs, we characterized the paired-pulse ratios (PPRs) of evoked EPSCs in response to a pair of stimulations between the two genotypes. At synapses, a second stimulation may generate a smaller response because of the depletion of vesicles in the releasable pool by the first stimulation. Notably, the PPRs of EPSCs induced by paired stimuli at 25-ms intervals were similar (Fig. 3D), indicating that there is no difference in the presynaptic release of glutamate. These results show that Pin1 KO enhances excitatory synaptic transmission of PFC pyramidal neurons through postsynaptic AMPA receptors.
Figure 3.
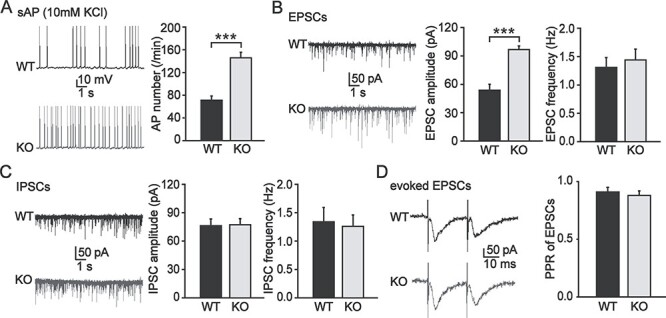
Excitatory synaptic transmission of PFC pyramidal neurons mediated by AMPA receptors was enhanced in Pin1 KO mice. (A) Representative spontaneous AP firing traces of WT mice (black) and Pin1 KO mice (gray) showed enhanced activity-dependent excitability in Pin1 KO mice (n = 24 neurons, from 6 mice of each group; ***P < 0.001). (B) Representative EPSC traces of WT mice (black) and Pin1 KO mice (gray) revealed that EPSC amplitude but not frequency increased significantly (n = 32 neurons, from 8 mice of each group; ***P < 0.001). (C) Representative IPSC traces of WT mice (black) and Pin1 KO mice (gray) showed no difference both in amplitude or frequency (n = 32 neurons, from 8 mice of each group). (D) Representative paired EPSC traces of WT mice (black) and Pin1 KO mice (gray) showed similar PPRs, indicating no difference in the presynaptic release of glutamate (n = 16 neurons, from 4 mice of each group).
Pin1 KO Increases Total AMPA Receptors and GluA1 Phosphorylated at Ser831
To examine the biochemical basis for Pin1 KO to enhance excitatory synaptic transmission mediated by AMPA receptors, we assessed AMPA receptor expression levels in the PFC by western blot. The total level of AMPA receptors was significantly increased in Pin1 KO mice (231 ± 38% of WT, P < 0.01). As it has been reported that GluA1 phosphorylation at Ser831 potentiates AMPA currents (Barria et al. 1997), we further found that GluA1 phosphorylated at Ser831 of AMPA receptors was increased in PFC slices without Pin1 (202 ± 26% of WT, P < 0.01). This effect appeared to be specific because we did not find any change in the expression of another phosphorylated GluA1 Ser845 (Fig. 4A,B) or in the protein expression of NMDA receptor subunits (Fig. 4C,D) between Pin1 KO and WT mice. Moreover, vGAT, a marker of GABAergic inhibitory synapses, was similar in both genotypes (Fig. 4C,D). These results indicate that the increased expression of AMPA receptors, especially GluA1 phosphorylated at Ser831, may lead to higher excitement of PFC pyramidal neurons through the potentiation of excitatory synaptic transmission, which may contribute to epileptic susceptibility in Pin1 KOmice.
Figure 4.
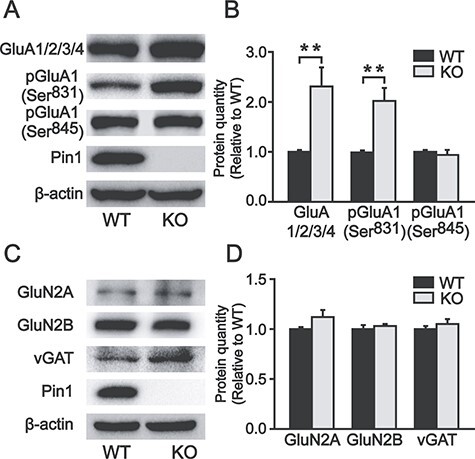
Expression of total AMPA receptors and phosphorylated GluA1 Ser831 increased in the PFC of Pin1 KO mice. (A, B) The total AMPA receptors and the phosphorylated GluA1 Ser831 of AMPA receptors increased in PFC slices of Pin1 KO mice (n = 6; **P < 0.01). However, the phosphorylated GluA1 Ser845 of AMPA receptors showed no difference (n = 6). (C, D) NMDA receptor subunits and vGAT showed no change (n = 6).
Pin1 Affects GluA1 Ser831 Phosphorylation by Acting on CaMKII
Pin1 is an isomerase specifically targeting Pro-directed Ser/Thr phosphorylation (Yaffe et al. 1997), and Ser831 in GluA1 is not Pro-directed phosphorylation, making it unlikely that Pin1 directly acts on GluA1 phosphorylated Ser831. However, GluA1 Ser831 has been demonstrated to be directly phosphorylated by CaMKII, and Pin1 can interact with CaMKII in a phosphorylation-specific manner (Mammen et al. 1997; Tatara et al. 2010). Therefore, we hypothesized that Pin1 might act on CaMKII to regulate GluA1 phosphorylation in the PFC. To test this hypothesis, we examined CaMKII and its activation of phosphorylation at Thr286. Although the total level of CaMKII was unaltered, CaMKII phosphorylation at Thr286 was significantly increased in Pin1 KO mice (334 ± 48% of WT, P < 0.001; Fig. 5A,B). GluA1 Ser831 can also be directly phosphorylated by PKC, while GluA1 Ser845 is phosphorylated specifically by PKA (Roche et al. 1996). Therefore, we examined the expression of these two kinases and found no change in Pin1 KO mice (Fig. 5C,D). These data suggest that Pin1 may regulate the activity of CaMKII phosphorylated Thr286, leading to an increase in GluA1 phosphorylated at Ser831. To examine whether Pin1 interacts with CaMKII and GluA1, endogenous Pin1 was immunoprecipitated from mouse PFC homogenates using an anti-Pin1 antibody. The coprecipitated CaMKII and GluA1 demonstrated a complex formation of these proteins with Pin1 (Fig. 5E). To examine the interaction between Pin1 and these proteins, we expressed CaMKIIα-Flag or GluA1-Flag in HEK293T cells. Cell lysates were immunoprecipitated with anti-Pin1 antibody, and bound protein complexes were analyzed by western blotting using anti-Flag antibodies for CaMKIIα and GluA1 detection, respectively. Pin1 coimmunoprecipitated with CaMKIIα but not GluA1 (Fig. 5F), indicating an interaction between Pin1 and CaMKII but not GluA1. CaMKII contains four potential Pro-directed Ser/Thr phosphorylation sites, which may be putative Pin1 binding sites: Thr176, Ser181, Ser234, and Thr241. To identify which site is involved in the interaction of Pin1 and CaMKII, we implemented a CaMKII point mutation from Thr (T) or Ser (S) to Ala (A) and performed GST-Pin1 pulldown assays in vitro. HEK293T cells transfected with different CaMKII mutants were incubated with GST-Pin1 or with GST alone as a negative control. As shown in Fig. 5G, only the T176A mutation, but not the other mutations in CaMKII, abolished the interaction of CaMKII with Pin1, indicating that Pin1 regulates CaMKII activity via acting on the phosphorylated Thr176-Pro motif. To verify whether CaMKII-Thr176 is the Pin1 binding site, we created a phosphorylation mimic form of T176 (T176D/S181A/S234A/T241A). The coimmunoprecipitation test revealed that the T176D of CaMKII binds to Pin1, indicating that Pin1-CamKII binding is phosphorylation dependent manner (Supplementary Fig. 1A). These results suggest that Pin1 may affect the function of AMPA receptors by regulating phosphorylated CaMKII.
Figure 5.
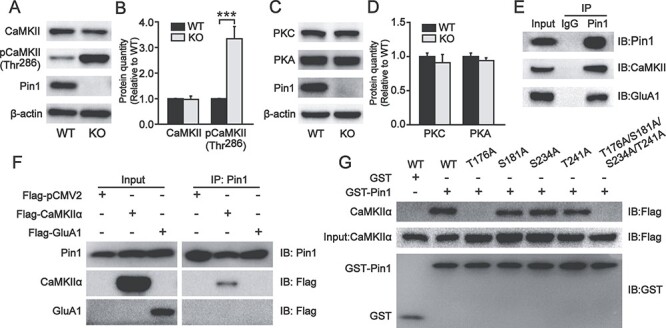
The expression of phosphorylated CaMKII Thr286 increased in the PFC of Pin1 KO mice, and Pin1 could interact with CaMKII and GluA1. (A, B) The expression of total CaMKII was unaltered in PFC slices of Pin1 KO mice (n = 6). However, phosphorylated CaMKII Thr286, which is a specific kinase for GluA1 Ser831, increased significantly (n = 6; ***P < 0.001). (C, D) These two kinases for GluA1, PKC, and PKA showed no change in Pin1 KO mice (n = 6). (E) Coimmunoprecipitation of endogenous Pin1 complexes from WT mouse PFC homogenates. Western blots were performed with anti-CaMKII and anti-GluA1 antibodies. Rabbit IgGs were used as a negative control. CaMKII and GluA1 can both be coprecipitated by endogenous Pin1 (n = 4). (F) Samples of HEK293T cells expressing CaMKIIα-Flag or GluA1-Flag were immunoprecipitated with anti-Pin1 antibody. Western blots were performed with anti-Flag antibody for CaMKIIα and GluA1 detection, showing that only CaMKIIα can be detected (n = 4). (G) GST-Pin1 pulldown from samples of HEK293T cells expressing CaMKIIα-Flag WT or mutants: 4 putative Pin1 binding sites Thr or Ser were mutated to Ala (T176A, S181A, S234A, T241A, and T176A/S181A/S234A/T241A). GST was used as a negative control. Pulled down CaMKIIα-Flag mutants were detected using an anti-Flag antibody. CaMKIIα, including the T176A mutant, could not be detected in GST-Pin1 pulldown assays (n = 4).
The Pin1-CaMKII-AMPA Receptor Axis Is Deregulated in the Brains of Epilepsy Patients
To examine the clinical relevance of the above biochemical changes in Pin1-CaMKII-AMPA receptors in human epilepsy, we examined the levels of Pin1, AMPA receptors, GluA1 phosphorylation at Ser831 and CaMKII phosphorylation at Thr286 in brain tissues of 12 epilepsy patients and six age-matched controls (Supplementary Table 1). Pin1 expression assayed by immunofluorescent staining (Fig. 6A) and immunoblotting (Fig. 6B,C) was significantly reduced in the patients with epilepsy (Tang et al. 2017). More importantly, we found that GluA1 phosphorylated at Ser831 and CaMKII phosphorylated at Thr286 were both increased in the human neocortex tissues of patients with epilepsy, as we observed in Pin1 KO mice. However, the total level of AMPA receptors did not change significantly (Fig. 6B,C). These results show the deregulation of the Pin1-CaMKII-AMPA receptor axis in epilepsy patients.
Figure 6.
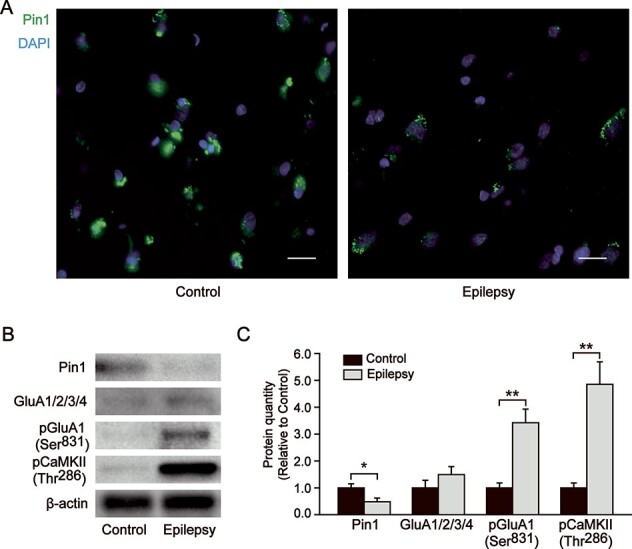
Pin1 decreased but phosphorylated GluA1 Ser831 and CaMKII Thr286 increased in the human brain tissues of patients with epilepsy. (A) Pin1 immunostaining decreased in the human epileptic brain sections. Scale bar, 50 μm. (B, C) Western blots also confirmed the decreased expression of Pin1 in the human brain tissues of patients with epilepsy (control, n = 6; epilepsy, n = 12; *P < 0.05). Although the total AMPA receptors did not change significantly, phosphorylated GluA1 Ser831 and CaMKII Thr286 both increased in the human neocortex tissues and in Pin1 KO mice (control, n = 6; epilepsy, n = 12; **P < 0.01).
Restoring Pin1 Expression in Pin1 KO Mice Rescues Biochemical Defects in Phosphorylated CaMKII and GluA1 but also Recovers Functional Impairments in Seizure Susceptibility
The above results demonstrate that Pin1-deficiency leads to increases in phosphorylated CaMKII and GluA1 and seizure susceptibility in mice and that Pin1 is reduced accompanied by increased phosphorylated CaMKII and GluA1 in human epilepsy patients, suggesting a critical role of the Pin1-CaMKII-AMPA receptor axis in epileptogenesis. To further support this hypothesis, we examined whether the deregulation of the Pin1-CaMKII-AMPA receptor axis and epileptic phenotypes can be reversed by restoring Pin1 in the PFC of Pin1 KO mice. To this end, AAV viruses expressing either both Pin1 and the marker GFP (AAV-Pin1) or GFP only (AAV controls, AAV-Ctrl) were bilaterally injected into the PFC of Pin1 KO mice. The expression of Pin1 in these areas was confirmed by immunofluorescence and western blot analysis (Supplementary Fig. 2A,B). More importantly, in the SE model induced by pilocarpine at a single dose (100 mg/kg), we found that the amplitude of ES was lower (AAV-Ctrl, 127.4 ± 5.2 μV vs. AAV-Pin1, 92.48 ± 6.7 μV, P < 0.01), the latency to ES was longer (AAV-Ctrl, 10.5 ± 1.3 min vs. AAV-Pin1, 14.7 ± 1.0 min, P < 0.05), the SE stage was lower (AAV-Ctrl, 2.31 ± 0.04 vs. AAV-Pin1, 1.35 ± 0.09, P < 0.001), and stage 3–5 seizure duration was shorter (AAV-Ctrl, 47.5 ± 3.8 s vs. AAV-Pin1, 22.2 ± 3.2 s, P < 0.001) in the AAV-Pin1 group than in the AAV-Ctrl group (Fig. 7A,B; Supplementary Video 1,2). These epilepsy-related functional outcomes were further supported by biochemical changes, as shown by reduced CaMKII phosphorylation at Thr286 (71.8 ± 2.95% of AAV-Ctrl, P < 0.001) and GluA1 phosphorylation at Ser831 (68.9 ± 2.31% of AAV-Ctrl, P < 0.001) in the PFC in the AAV-Pin1 group compared with the AAV-Ctrl group (Fig. 7C,D). Thus, restoring Pin1 expression in the PFC of Pin1-deficient mice not only rescues biochemical defects in phosphorylated CaMKII and GluA1 but also recovers functional impairments in seizure susceptibility.
Figure 7.
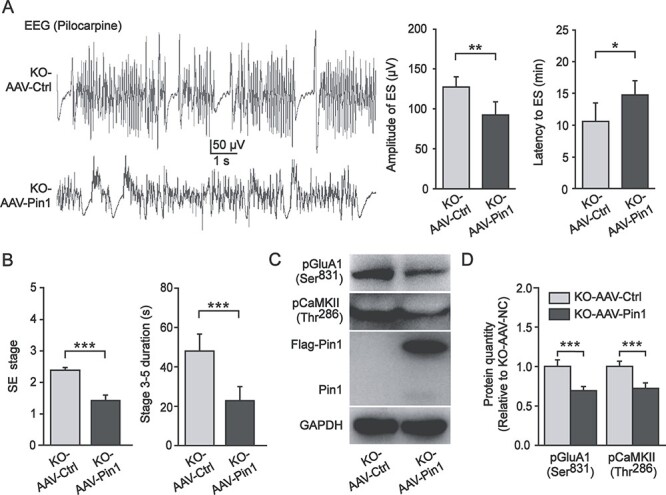
AAV-Pin1 expression in the PFC of Pin1 KO mice partially rescued the impairments in these mice. (A) Left, representative EEG traces of the AAV-Pin1 group and the AAV-Ctrl group show ES induced by pilocarpine. Right, the amplitude of ES was lower in the AAV-Pin1 group (n = 6; **P < 0.01), and the latency to ES was higher in the AAV-Pin1 group (n = 6; *P < 0.05). (B) Behavioral observation showed lower SE stage and shorter stage 3–5 seizure duration in the AAV-Pin1 group (n = 6; ***P < 0.001). (C, D) Phosphorylated CaMKII Thr286 and GluA1 Ser831 decreased in the PFC of Pin1 KO mice after AAV-Pin1 injection (n = 6; ***P < 0.001).
Discussion
Although there is evidence for a role of Pin1 in epilepsy, the underlying mechanisms and overall impact on epilepsy are still unknown. In the present study, we showed for the first time that Pin1 is a critical regulator of seizure activity in epilepsy in vitro and in vivo. First, Pin1-deficient mice showed increased severity of SE in epilepsy induced by pilocarpine or PTZ and displayed spontaneous epilepsy that increased in an age-dependent manner. Second, the morphology and the intrinsic excitability of pyramidal neurons in the PFC were similar between Pin1 KO and WT mice. Third, the excitatory synaptic transmission mediated by AMPA receptors in the PFC was enhanced with the ablation of Pin1. Fourth, Pin1 was found to be associated with phosphorylated CaMKII Thr176 in vitro and in vivo. Fifth, this binding decreased the levels of AMPA receptors and phosphorylated GluA1 Ser831. Sixth, Pin1 levels were inversely correlated with phosphorylated GluA1 Ser831 and CaMKII Thr286 in human brain tissues of patients with epilepsy. Finally, AAV-mediated Pin1 re-expression in the PFC of Pin1 KO mice reduced the level of phosphorylated CaMKII Thr286 and GluA1 Ser831 and seizure susceptibility.
Pin1 is an intracellular signaling molecule that plays a critical role in many neurological disorders, such as AD (Butterfield et al. 2006; Albayram et al. 2016), traumatic brain injury (Albayram et al. 2016), chronic traumatic encephalopathy (Albayram et al. 2016), Parkinson disease (Ryo et al. 2006; Ghosh et al. 2013), amyotrophic lateral sclerosis (Kesavapany et al. 2007), and Huntington’s disease (Grison et al. 2011). Pin1 is inactivated in the hippocampus of patients with mild cognitive impairment and AD (Butterfield et al. 2006; Sultana et al. 2006). Pin1 knockout causes mice to develop diffuse phenotypes of premature aging similar to AD in humans (Lee et al. 2009; Driver et al. 2014). Transgenic overexpression of Pin1 in postnatal neurons in mice is able to protect against age-dependent neurodegeneration (Lim et al. 2008). A recent study in an epileptic mouse model reported evidence that seizure activity downregulates Pin1 expression levels (Tang et al. 2017). In the present study, Pin1 KO mice displayed severe induced seizures and age-dependent spontaneous epilepsy, indicating that endogenous Pin1 acts as an inhibitor during epileptogenesis. All of these studies suggest that Pin1 has strong neuroprotective effects.
The expression levels of Pin1 vary widely in different tissues. Pin1 is downregulated in cells that are not proliferating. In contrast, Pin1 is highly expressed in neurons from the beginning of neuronal differentiation (Becker and Bonni 2006; Hamdane et al. 2006). Pin1 immunoreactivity is mainly distributed in hippocampal and cortical pyramidal neurons in human brains (Dakson et al. 2011). In agreement with these studies, we observed Pin1 immunofluorescence in PFC pyramidal neurons according to colocalization with NeuN, a neuronal marker. Although Pin1 is expressed in these neurons, its physiological function remains unclear. Therefore, we explored the cellular mechanisms underlying epileptic susceptibility in Pin1 KO mice. We found that PFC pyramidal neurons showed activity-dependent hyperexcitability but normal morphology. Meanwhile, the unchanged intrinsic excitability indicated that the neuronal excitability was not regulated by ion channels on these neurons.
The function of Pin1 at synapses is still poorly understood. Electrophysiological experiments have demonstrated that the deletion of Pin1 specifically affects GABAergic inhibitory transmission to postsynaptic pyramidal neurons in the hippocampus (Antonelli et al. 2014). Moreover, Pin1 contributes to synaptic plasticity at excitatory synapses (Westmark et al. 2010). The interaction of Pin1 and metabotropic glutamate receptors potentiates dopamine-dependent synaptic plasticity of striatal medium-spiny neurons (Park et al. 2013). It has been recently reported that Pin1 negatively modulates NMDA-mediated synaptic signaling in hippocampal principal neurons (Antonelli et al. 2016). In contrast, we showed that Pin1 affected excitatory synaptic transmission mediated by AMPA receptors in the PFC pyramidal neurons, suggesting that Pin1 might play a different role in diverse neurons of various brain regions. Our work further emphasized the critical role of Pin1 in synaptic transmission. Inhibition and excitatory synaptic transmission are important in epilepsy (Van Liefferinge et al. 2013). In this study, we found that the enhancement of AMPA-mediated excitatory synaptic transmission may cause hyperexcitability of pyramidal neurons and higher amplitude of EEG in the PFC, leading to seizure susceptibility in Pin1-deficientmice.
In the brain, Pin1 regulates many important proteins and protects them from dysfunctional postphosphorylation structural changes (Pastorino et al. 2012; Driver et al. 2015). The best characterized neuronal substrates of Pin1 are cytoskeletal proteins such as tau, amyloid precursor protein, and neurofilaments (Lu et al. 1999; Pastorino et al. 2006; Rudrabhatla and Pant 2010). Pin1 can restore misfolded proteins to their normal physiological condition, promote degradation, and avoid the accumulation of insoluble proteins (Lu et al. 1999; Ma et al. 2012). Recently, Pin1 has been reported as a key modulator of GABAA and NMDA receptors in the hippocampus (Antonelli et al. 2014; Antonelli et al. 2016). This study revealed that the total level of AMPA receptors increased in the PFC without Pin1. However, NMDA receptor subunits and vGAT at GABAergic inhibitory synapses had no change in the PFC. Furthermore, phosphorylated GluA1 Ser831 of AMPA receptors was also increased in Pin1 KO mice. AMPA receptors are distributed widely in all brain areas relevant to epilepsy and have been identified as suitable targets of antiepileptic therapy. Therefore, the increased expression of total AMPA receptors and phosphorylated GluA1 Ser831 might be an essential molecular mechanism underlying epileptic susceptibility. Because GluA1 Ser831 is specifically phosphorylated by CaMKII and CaMKII phosphorylation potentiates AMPA receptor function (Yakel et al. 1995; Mammen et al. 1997), we found that phosphorylated CaMKII Thr286 was significantly increased in Pin1 KO mice and that Pin1 could form a complex with CaMKII and GluA1 by directly binding to phosphorylated CaMKII. The kinase domain of CaMKII is followed by auto-inhibitory and Ca2+/CaM binding domain, which regulates CaMKII activity (Myers et al. 2017). Pin1 binding to phosphorylated CaMKII Thr176 in the kinase domain may promote the conformational change of CaMKII and be required for the activation of CaMKII by auto-phosphorylation of Thr286. Further studies are required how Pin1 regulates CaMKII activity through Thr176 phosphorylation.
A recent study reported that CaMKII could bind with and regulate Pin1 in retinal neurons after glutamate injury (Wang et al. 2019). Interestingly, we also showed this glutamate-mediated excitotoxicity in the opposite direction. Most importantly, reduced Pin1 and enhanced GluA1 Ser831 and CaMKII Thr286 phosphorylation were validated in human brain tissues of patients with epilepsy. Our studies further showed that AAV-mediated Pin1 expression in the PFC of Pin1 KO mice could partially rescue seizure susceptibility in these mice, presumably reducing the expression of phosphorylated CaMKII Thr286 and GluA1 Ser831.
In summary, we discovered that the Pin1-CaMKII-AMPA receptor axis plays an important role in epileptic inhibition. Pin1 directly bound to CaMKII at the Thr176 site. In the PFC glutamatergic excitatory synapses of Pin1 KO mice, the expression of phosphorylated CaMKII Thr286 significantly increased, leading to an increase in phosphorylated GluA1 Ser831 in this model (Fig. 8) and resulting in the potentiation of AMPA currents. Taken together, our findings reveal a novel Pin1 signaling pathway during epileptogenesis, strongly suggesting the potential utility of Pin1 as a target for seizure protection.
Figure 8.
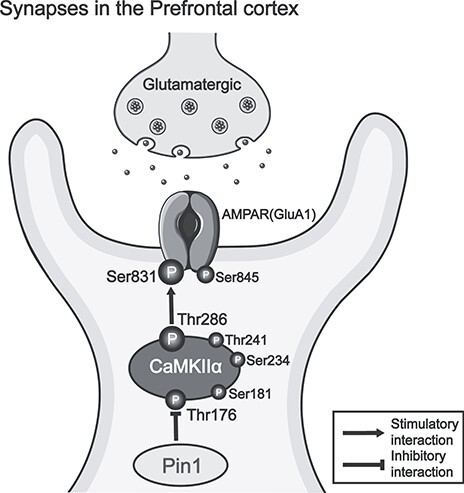
A model to illustrate the role of the Pin1-CaMKII-AMPA receptor axis in PFC glutamatergic excitatory synapses. Pin1 directly interacts with CaMKII by binding its Thr176 site, regulating the expression of phosphorylated CaMKII Thr286 and leading to a change in phosphorylated GluA1 Ser831.
Funding
Natural Science Foundation of Fujian Province (2020J01602); Innovation of Science and Technology, Fujian Province (2017Y9054); Collaborative Innovation Center for Stem Cells Translational Medicine (Fujian 2011 Program); National Institutes of Health (R01CA167677).
Notes
We are grateful for the support of the First Affiliated Hospital of Fujian Medical University, which supplied patient brain tissues. We also thank all the patients and their families for their participation in this study. The study has approval of the Ethics Committee of Fujian Medical University. Conflict of Interest: None declared.
Author Contributions
H.L. and X.H. designed the experiments; K.P.L. and X.Z.Z. supervised the plan and provided Pin1 KO mice; H.L., X.H., F.Y., A.L., D.Z., and N.M. carried out the experiments and analyzed the data; F.Y., A.L., and D.Z. raised the mice; X.H. and H.L. wrote the paper; H.L., K.P.L., and X.Z.Z. developed the concepts and prepared and revised the manuscriptz prior to submission; Y.L. and L.C. provided human specimens; T.H.L,S.L., L.W., X.Y., and M.Z. provided advice and technical assistance.
Supplementary Material
Contributor Information
Xiaojun Hou, Fujian Key Laboratory for Translational Research in Cancer and Neurodegenerative Diseases, Institute for Translational Medicine, The School of Basic Medical Sciences, Fujian Medical University, Fuzhou, Fujian, 350108, China; Fuzhou Children’s Hospital of Fujian Medical University, Fuzhou, Fujian, 350005, China.
Fan Yang, Fujian Key Laboratory for Translational Research in Cancer and Neurodegenerative Diseases, Institute for Translational Medicine, The School of Basic Medical Sciences, Fujian Medical University, Fuzhou, Fujian, 350108, China.
Angcheng Li, Fujian Key Laboratory for Translational Research in Cancer and Neurodegenerative Diseases, Institute for Translational Medicine, The School of Basic Medical Sciences, Fujian Medical University, Fuzhou, Fujian, 350108, China.
Debao Zhao, Fujian Key Laboratory for Translational Research in Cancer and Neurodegenerative Diseases, Institute for Translational Medicine, The School of Basic Medical Sciences, Fujian Medical University, Fuzhou, Fujian, 350108, China.
Nengjun Ma, Fujian Key Laboratory for Translational Research in Cancer and Neurodegenerative Diseases, Institute for Translational Medicine, The School of Basic Medical Sciences, Fujian Medical University, Fuzhou, Fujian, 350108, China.
Linying Chen, Fujian Key Laboratory for Translational Research in Cancer and Neurodegenerative Diseases, Institute for Translational Medicine, The School of Basic Medical Sciences, Fujian Medical University, Fuzhou, Fujian, 350108, China; The First Affiliated Hospital of Fujian Medical University, Fuzhou, Fujian, 350009, China.
Suijin Lin, Fujian Key Laboratory for Translational Research in Cancer and Neurodegenerative Diseases, Institute for Translational Medicine, The School of Basic Medical Sciences, Fujian Medical University, Fuzhou, Fujian, 350108, China.
Yuanxiang Lin, The First Affiliated Hospital of Fujian Medical University, Fuzhou, Fujian, 350009, China.
Long Wang, Fujian Key Laboratory for Translational Research in Cancer and Neurodegenerative Diseases, Institute for Translational Medicine, The School of Basic Medical Sciences, Fujian Medical University, Fuzhou, Fujian, 350108, China.
Xingxue Yan, Fujian Key Laboratory for Translational Research in Cancer and Neurodegenerative Diseases, Institute for Translational Medicine, The School of Basic Medical Sciences, Fujian Medical University, Fuzhou, Fujian, 350108, China.
Min Zheng, Fujian Key Laboratory for Translational Research in Cancer and Neurodegenerative Diseases, Institute for Translational Medicine, The School of Basic Medical Sciences, Fujian Medical University, Fuzhou, Fujian, 350108, China.
Tae Ho Lee, Fujian Key Laboratory for Translational Research in Cancer and Neurodegenerative Diseases, Institute for Translational Medicine, The School of Basic Medical Sciences, Fujian Medical University, Fuzhou, Fujian, 350108, China.
Xiao Zhen Zhou, Division of Translational Therapeutics, Department of Medicine and Cancer Research Institute, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA, 02115, USA.
Kun Ping Lu, Division of Translational Therapeutics, Department of Medicine and Cancer Research Institute, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA, 02115, USA; Broad Institute of MIT and Harvard, Cambridge, MA, 02142, USA.
Hekun Liu, Fujian Key Laboratory for Translational Research in Cancer and Neurodegenerative Diseases, Institute for Translational Medicine, The School of Basic Medical Sciences, Fujian Medical University, Fuzhou, Fujian, 350108, China.
References
- Albayram O, Herbert MK, Kondo A, Tsai C-Y, Baxley S, Lian X, Hansen M, Zhou XZ, Lu KP. 2016. Function and regulation of tau conformations in the development and treatment of traumatic brain injury and neurodegeneration. Cell Biosci. 6:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonelli R, De Filippo R, Middei S, Stancheva S, Pastore B, Ammassari-Teule M, Barberis A, Cherubini E, Zacchi P. 2016. Pin1 modulates the synaptic content of NMDA receptors via prolyl-isomerization of PSD-95. J Neurosci. 36:5437–5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonelli R, Pizzarelli R, Pedroni A, Fritschy JM, Del Sal G, Cherubini E, Zacchi P. 2014. Pin1-dependent signalling negatively affects GABAergic transmission by modulating neuroligin2/gephyrin interaction. Nat Commun. 5:5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankstahl M, Muller CJ, Wilk E, Schughart K, Loscher W. 2012. Generation and characterization of pilocarpine-sensitive C57BL/6 mice as a model of temporal lobe epilepsy. Behav Brain Res. 230:182–191. [DOI] [PubMed] [Google Scholar]
- Barria A, Derkach V, Soderling T. 1997. Identification of the Ca2+/calmodulin-dependent protein kinase II regulatory phosphorylation site in the alpha-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate-type glutamate receptor. J Biol Chem. 272:32727–32730. [DOI] [PubMed] [Google Scholar]
- Becker EB, Bonni A. 2006. Pin1 mediates neural-specific activation of the mitochondrial apoptotic machinery. Neuron. 49:655–662. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Abdul HM, Opii W, Newman SF, Joshi G, Ansari MA, Sultana R. 2006. Pin1 in Alzheimer's disease. J Neurochem. 98:1697–1706. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Poon HF, St Clair D, Keller JN, Pierce WM, Klein JB, Markesbery WR. 2006. Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: insights into the development of Alzheimer's disease. Neurobiol Dis. 22:223–232. [DOI] [PubMed] [Google Scholar]
- Cavalheiro EA, Santos NF, Priel MR. 1996. The pilocarpine model of epilepsy in mice. Epilepsia. 37:1015–1019. [DOI] [PubMed] [Google Scholar]
- Chen L, Xu X, Wen X, Xu S, Wang L, Lu W, Jiang M, Huang J, Yang D, Wang J, et al. 2019. Targeting PIN1 exerts potent antitumor activity in pancreatic ductal carcinoma via inhibiting tumor metastasis. Cancer Sci. 110:2442–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dakson A, Yokota O, Esiri M, Bigio EH, Horan M, Pendleton N, Richardson A, Neary D, Snowden JS, Robinson A, et al. 2011. Granular expression of prolyl-peptidyl isomerase PIN1 is a constant and specific feature of Alzheimer's disease pathology and is independent of tau, Abeta and TDP-43 pathology. Acta Neuropathol. 121:635–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derkach V, Barria A, Soderling TR. 1999. Ca2+/calmodulin-kinase II enhances channel conductance of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate type glutamate receptors. Proc Natl Acad Sci USA. 96:3269–3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driver JA, Zhou XZ, Lu KP. 2014. Regulation of protein conformation by Pin1 offers novel disease mechanisms and therapeutic approaches in Alzheimer's disease. Discov Med. 17:93–99. [PMC free article] [PubMed] [Google Scholar]
- Driver JA, Zhou XZ, Lu KP. 2015. Pin1 dysregulation helps to explain the inverse association between cancer and Alzheimer's disease. Biochim Biophys Acta. 1850:2069–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French JA, Krauss GL, Steinhoff BJ, Squillacote D, Yang H, Kumar D, Laurenza A. 2013. Evaluation of adjunctive perampanel in patients with refractory partial-onset seizures: results of randomized global phase III study 305. Epilepsia. 54:117–125. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Saminathan H, Kanthasamy A, Anantharam V, Jin H, Sondarva G, Harischandra DS, Qian Z, Rana A, Kanthasamy AG. 2013. The peptidyl-prolyl isomerase Pin1 up-regulation and proapoptotic function in dopaminergic neurons: relevance to the pathogenesis of Parkinson disease. J Biol Chem. 288:21955–21971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grison A, Mantovani F, Comel A, Agostoni E, Gustincich S, Persichetti F, Del Sal G. 2011. Ser46 phosphorylation and prolyl-isomerase Pin1-mediated isomerization of p53 are key events in p53-dependent apoptosis induced by mutant huntingtin. Proc Natl Acad Sci USA. 108:17979–17984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdane M, Dourlen P, Bretteville A, Sambo AV, Ferreira S, Ando K, Kerdraon O, Begard S, Geay L, Lippens G, et al. 2006. Pin1 allows for differential Tau dephosphorylation in neuronal cells. Mol Cell Neurosci. 32:155–160. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R. 2000. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 287:2262–2267. [DOI] [PubMed] [Google Scholar]
- Kesavapany S, Patel V, Zheng YL, Pareek TK, Bjelogrlic M, Albers W, Amin N, Jaffe H, Gutkind JS, Strong MJ, et al. 2007. Inhibition of Pin1 reduces glutamate-induced perikaryal accumulation of phosphorylated neurofilament-H in neurons. Mol Biol Cell. 18:3645–3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketzef M, Gitler D. 2014. Epileptic synapsin triple knockout mice exhibit progressive long-term aberrant plasticity in the entorhinal cortex. Cereb Cortex. 24:996–1008. [DOI] [PubMed] [Google Scholar]
- Kondo A, Albayram O, Zhou XZ, Lu KP. 2017. Pin1 knockout mice: a model for the study of tau pathology in Alzheimer's disease. Methods Mol Biol. 1523:415–425. [DOI] [PubMed] [Google Scholar]
- Kozono S, Lin YM, Seo HS, Pinch B, Lian X, Qiu C, Herbert MK, Chen CH, Tan L, Gao ZJ, et al. 2018. Arsenic targets Pin1 and cooperates with retinoic acid to inhibit cancer-driving pathways and tumor-initiating cells. Nat Commun. 9:3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen AS, Jenkins MA, Banke TG, Schousboe A, Makino Y, Johnson RC, Huganir R, Traynelis SF. 2011. Mechanism of Ca2+/calmodulin-dependent kinase II regulation of AMPA receptor gating. Nat Neurosci. 14:727–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan P, Arzimanoglou A, Berg AT, Brodie MJ, Allen Hauser W, Mathern G, Moshe SL, Perucca E, Wiebe S, French J. 2010. Definition of drug resistant epilepsy: consensus proposal by the ad hoc task force of the ILAE commission on therapeutic strategies. Epilepsia. 51:1069–1077. [DOI] [PubMed] [Google Scholar]
- Lapray D, Popova IY, Kindler J, Jorquera I, Becq H, Manent JB, Luhmann HJ, Represa A. 2010. Spontaneous epileptic manifestations in a DCX knockdown model of human double cortex. Cereb Cortex. 20:2694–2701. [DOI] [PubMed] [Google Scholar]
- Lee TH, Chen CH, Suizu F, Huang P, Schiene-Fischer C, Daum S, Zhang YJ, Goate A, Chen RH, Zhou XZ, et al. 2011. Death-associated protein kinase 1 phosphorylates Pin1 and inhibits its prolyl isomerase activity and cellular function. Mol Cell. 42:147–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TH, Tun-Kyi A, Shi R, Lim J, Soohoo C, Finn G, Balastik M, Pastorino L, Wulf G, Zhou XZ, et al. 2009. Essential role of Pin1 in the regulation of TRF1 stability and telomere maintenance. Nat Cell Biol. 11:97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Fu X, Smith NA, Ziobro J, Curiel J, Tenga MJ, Martin B, Freedman S, Cea-Del Rio CA, Oboti L, et al. 2017. Loss of CLOCK results in dysfunction of brain circuits underlying focal epilepsy. Neuron. 96:387–401 e386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J, Balastik M, Lee TH, Nakamura K, Liou YC, Sun A, Finn G, Pastorino L, Lee VM, Lu KP. 2008. Pin1 has opposite effects on wild-type and P301L tau stability and tauopathy. J Clin Invest. 118:1877–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou YC, Sun A, Ryo A, Zhou XZ, Yu ZX, Huang HK, Uchida T, Bronson R, Bing G, Li X, et al. 2003. Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration. Nature. 424:556–561. [DOI] [PubMed] [Google Scholar]
- Liou YC, Zhou XZ, Lu KP. 2011. Prolyl isomerase Pin1 as a molecular switch to determine the fate of phosphoproteins. Trends Biochem Sci. 36:501–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu KP. 2004. Pinning down cell signaling, cancer and Alzheimer's disease. Trends Biochem Sci. 29:200–209. [DOI] [PubMed] [Google Scholar]
- Lu KP, Finn G, Lee TH, Nicholson LK. 2007. Prolyl cis-trans isomerization as a molecular timer. Nat Chem Biol. 3:619–629. [DOI] [PubMed] [Google Scholar]
- Lu KP, Hanes SD, Hunter T. 1996. A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature. 380:544–547. [DOI] [PubMed] [Google Scholar]
- Lu KP, Kondo A, Albayram O, Herbert MK, Liu H, Zhou XZ. 2016. Potential of the antibody against cis-phosphorylated tau in the early diagnosis, treatment, and prevention of Alzheimer disease and brain injury. JAMA Neurol. 73:1356–1362. [DOI] [PubMed] [Google Scholar]
- Lu KP, Zhou XZ. 2007. The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat Rev Mol Cell Biol. 8:904–916. [DOI] [PubMed] [Google Scholar]
- Lu PJ, Wulf G, Zhou XZ, Davies P, Lu KP. 1999. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature. 399:784–788. [DOI] [PubMed] [Google Scholar]
- Ma SL, Pastorino L, Zhou XZ, Lu KP. 2012. Prolyl isomerase Pin1 promotes amyloid precursor protein (APP) turnover by inhibiting glycogen synthase kinase-3beta (GSK3beta) activity: novel mechanism for Pin1 to protect against Alzheimer disease. J Biol Chem. 287:6969–6973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammen AL, Kameyama K, Roche KW, Huganir RL. 1997. Phosphorylation of the alpha-amino-3-hydroxy-5-methylisoxazole4-propionic acid receptor GluR1 subunit by calcium/calmodulin-dependent kinase II. J Biol Chem. 272:32528–32533. [DOI] [PubMed] [Google Scholar]
- Myers JB, Zaegel V, Coultrap SJ, Miller AP, Bayer KU, Reichow SL. 2017. The CaMKII holoenzyme structure in activation-competent conformations. Nat Commun. 8:15742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nechama M, Kwon J, Wei S, Kyi AT, Welner RS, Ben-Dov IZ, Arredouani MS, Asara JM, Chen CH, Tsai CY, et al. 2018. The IL-33-PIN1-IRAK-M axis is critical for type 2 immunity in IL-33-induced allergic airway inflammation. Nat Commun. 9:1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni KM, Hou XJ, Yang CH, Dong P, Li Y, Zhang Y, Jiang P, Berg DK, Duan S, Li XM. 2016. Selectively driving cholinergic fibers optically in the thalamic reticular nucleus promotes sleep. Elife. 5:e10382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JM, Hu JH, Milshteyn A, Zhang PW, Moore CG, Park S, Datko MC, Domingo RD, Reyes CM, Wang XJ, et al. 2013. A prolyl-isomerase mediates dopamine-dependent plasticity and cocaine motor sensitization. Cell. 154:637–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastorino L, Ma SL, Balastik M, Huang P, Pandya D, Nicholson L, Lu KP. 2012. Alzheimer's disease-related loss of Pin1 function influences the intracellular localization and the processing of AbetaPP. J Alzheimers Dis. 30:277–297. [DOI] [PubMed] [Google Scholar]
- Pastorino L, Sun A, Lu PJ, Zhou XZ, Balastik M, Finn G, Wulf G, Lim J, Li SH, Li X, et al. 2006. The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-beta production. Nature. 440:528–534. [DOI] [PubMed] [Google Scholar]
- Racine RJ. 1972. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 32:281–294. [DOI] [PubMed] [Google Scholar]
- Roche KW, O'Brien RJ, Mammen AL, Bernhardt J, Huganir RL. 1996. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron. 16:1179–1188. [DOI] [PubMed] [Google Scholar]
- Rogawski MA. 2013. AMPA receptors as a molecular target in epilepsy therapy. Acta Neurol Scand Suppl. 197:9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudrabhatla P, Pant HC. 2010. Phosphorylation-specific peptidyl-prolyl isomerization of neuronal cytoskeletal proteins by Pin1: implications for therapeutics in neurodegeneration. J Alzheimers Dis. 19:389–403. [DOI] [PubMed] [Google Scholar]
- Ryo A, Togo T, Nakai T, Hirai A, Nishi M, Yamaguchi A, Suzuki K, Hirayasu Y, Kobayashi H, Perrem K, et al. 2006. Prolyl-isomerase Pin1 accumulates in lewy bodies of parkinson disease and facilitates formation of alpha-synuclein inclusions. J Biol Chem. 281:4117–4125. [DOI] [PubMed] [Google Scholar]
- Shimizu T, Kanai K, Sugawara Y, Uchida C, Uchida T. 2018. Prolyl Isomerase Pin1 directly regulates calcium/calmodulin-dependent protein kinase II activity in mouse brains. Front Pharmacol. 9:1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sood N, Sahai AK, Medhi B, Chakrabarti A. 2008. Dose-finding study with nicotine as a proconvulsant agent in PTZ-induced seizure model in mice. J Biomed Sci. 15:755–765. [DOI] [PubMed] [Google Scholar]
- Staley K. 2015. Molecular mechanisms of epilepsy. Nat Neurosci. 18:367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultana R, Boyd-Kimball D, Poon HF, Cai J, Pierce WM, Klein JB, Markesbery WR, Zhou XZ, Lu KP, Butterfield DA. 2006. Oxidative modification and down-regulation of Pin1 in Alzheimer's disease hippocampus: a redox proteomics analysis. Neurobiol Aging. 27:918–925. [DOI] [PubMed] [Google Scholar]
- Tang L, Zhang Y, Chen G, Xiong Y, Wang X, Zhu B. 2017. Down-regulation of Pin1 in temporal lobe epilepsy patients and mouse model. Neurochem Res. 42:1211–1218. [DOI] [PubMed] [Google Scholar]
- Tatara Y, Terakawa T, Uchida T. 2010. Identification of Pin1-binding phosphorylated proteins in the mouse brain. Biosci Biotechnol Biochem. 74:2480–2483. [DOI] [PubMed] [Google Scholar]
- Van Liefferinge J, Massie A, Portelli J, Di Giovanni G, Smolders I. 2013. Are vesicular neurotransmitter transporters potential treatment targets for temporal lobe epilepsy? Front Cell Neurosci. 7:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Liao L, Huang Y, Wang M, Zhou H, Chen D, Liu F, Ji D, Xia X, Jiang B, et al. 2019. Pin1 is regulated by CaMKII activation in glutamate-induced retinal neuronal regulated necrosis. Front Cell Neurosci. 13:276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westmark PR, Westmark CJ, Wang S, Levenson J, O'Riordan KJ, Burger C, Malter JS. 2010. Pin1 and PKMzeta sequentially control dendritic protein synthesis. Sci Signal. 3:ra18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe MB, Schutkowski M, Shen M, Zhou XZ, Stukenberg PT, Rahfeld JU, Xu J, Kuang J, Kirschner MW, Fischer G, et al. 1997. Sequence-specific and phosphorylation-dependent proline isomerization: a potential mitotic regulatory mechanism. Science. 278:1957–1960. [DOI] [PubMed] [Google Scholar]
- Yakel JL, Vissavajjhala P, Derkach VA, Brickey DA, Soderling TR. 1995. Identification of a Ca2+/calmodulin-dependent protein kinase II regulatory phosphorylation site in non-N-methyl-d-aspartate glutamate receptors. Proc Natl Acad Sci USA. 92:1376–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that the main data supporting the findings of this study are within the article and its Supplementary Information files. Extra data are obtained from the corresponding authors upon request.