

Abstract
Electrophoresis or electrochromatography carried out in nanometer columns (width and depth) offers some attractive benefits compared to microscale columns. These advantages include unique separation mechanisms that are scale dependent, fast separation times, and simpler workflow due to the lack of a need for column packing and/or wall coatings to create a stationary phase. We report the use of thermoplastics, in this case PMMA, as the substrate for separating single-stranded DNAs (ssDNAs). Electrophoresis nanochannels were created in PMMA using nanoimprint lithography (NIL), which can produce devices at lower cost and in a higher production mode compared to the fabrication techniques required for glass devices. The nanochannel column in PMMA was successful in separating ssDNAs in free solution that was not possible using microchip electrophoresis in PMMA. The separation could be performed in <1 s with resolution >1.5 when carried out using at an electric field strength of 280 V/cm and an effective column length of 60 μm (100 nm × 100 nm, depth and width). The ssDNAs transport through the PMMA column was driven electrokinetically under the influence of an EOF. The results indicated that the separation was dominated by chromatographic effects using an open tubular nanoelectrochromatography (OT-NEC) mode of separation. Interesting to these separations was that no column packing was required nor a wall coating to create the stationary phase; the separation was affected using the native polymer that was UV/O3 activated and an aqueous buffer mobile phase.
Keywords: DNA, Electrochromatography, Nanofluidics, Thermoplastics
1. Introduction
Development of DNA separations for clinical applications are trending toward miniaturization and methods that can support point-of-care testing (POCT) [1,2]. With advances in the design and fabrication of microfluidic “lab-on-a-chip” (LOC) devices over the last two decades, there have been numerous reports on microscale electrophoretic separations of DNA [3]. However, the majority of these methods still require a gel-based matrix for the separation due to the length independent mobility of DNA for both double stranded and single stranded forms in free solution [4,5]. Employing automated chip-based gel electrophoresis systems for DNA separations still remains challenging due to the limitations imposed by the requirement of gel loading and replacement in microchip electrophoresis as well as wall-coatings to suppress the EOF [6–9]. Due to the high EOF in glass-based devices induced by the high surface charge density, wall-coatings are required such as linear polyacrylamides that are covalently anchored to the wall of the microchannel or dynamic coatings [10,11]. If the separation could be performed in free solution, challenges associated with the need for gel matrices would be eliminated. Also, new materials that have an intrinsically lower EOF compared to fused silica would also negate the need for dynamic or permanent wall coatings.
Nanofluidic separations have garnered attention due to unique nanoscale phenomena, such as electric double layer (EDL) overlap and transverse electromigration leading to unique separation modalities [12–14]. In addition, advantages of nanofluidic separations include low sample and reagent consumption, ultrafast separations, and ease of integration to LOC devices to allow for sample preprocessing before the separation. The increased surface area-to-volume ratio in nanofluidic channels also allows for solute/wall interactions that can influence the electrokinetic transport of analytes through nanochannels. Field-dependent mobilities are observed as well due to intermittent motion of solutes through nanochannels when surface roughness is comparable to the critical dimension of the nanochannel [15].
Moreover, adsorption–desorption events between solutes and the column wall can be prominent in nanochannels compared to microchannels due to scaling effects. Kitamori et al. [16] utilized adsorption/desorption effects in nanochannels to demonstrate open tubular chromatographic separations, which he coined “extended nanochromatography” [17]. They performed a wide range of chromatographic separations by using pressure driven flow in glass nanofluidic devices with channel dimensions of approximately 250 nm in depth and 1000 nm in width and lengths >5 cm. Reverse phase chromatographic separation of amino acids was performed by chemically modifying the nanochannel surface with a C18 stationary phase [18]. Moreover, in a recent review on nanoscale electrophoresis by Santiago et al. [19], he reported the effects of wall interactions, which can dominate compared to electrophoretic effects indicating that separations in nanoscale columns can be considered to be more chromatographic in nature than electrophoretic.
Recent publications of DNA separations have demonstrated unique nanoscale phenomena for free-solution electrokinetic length-based separations of double-stranded (ds) DNA in glass and silicon nanofluidic devices. Cross et al. [20] showed length-dependent mobilities of DNAs (2–10 kb) electrically driven through nanoslits (19 and 70 nm depths; only one dimension <100 nm). Peterson et al. [21] reported that oligonucleotides could be separated via nanochannel electrophoresis and proposed steric effects alone contributed to the residence time distribution of 100–1000 base pair (bp) oligonucleotides within the nanochannel. Pennathur et al. [22] showed the free solution electrokinetic separation of dsDNAs in a glass nanoslit device (40, 100, and 1560 nm depths). In this work, the authors investigated ionic strength and channel dimension effects on the separation of dsDNAs from 10 to 100 bp in length. They suggested that separation efficiency was dependent on the finite EDL thickness with respect to the nanoslit dimensions. The best separation for these fragments were achieved in 100 nm deep nanoslits using sodium borate buffers at 1 to 10 mM; ratio of the channel critical dimension to thickness of EDL (d/λd) was 33.
Unfortunately, the utilization of glass-based nanochannel devices (nanoslit possess one dimension ≤100 nm while nanochannels have two dimensions ≤100 nm) is challenged by the fact that they require sophisticated device fabrication techniques that are high in cost and require specially trained personnel to generate the prerequisite devices, which can impede their use in research, clinical, or forensic applications. In addition, the surface chemistry is restricted to the silanol groups and requires modifications to change the surface chemistry.
Recently, thermoplastics have been demonstrated as viable substrates for nanofluidics due to their biocompatibility, optical properties comparable to glass, and their ability to be manufactured at large scale and low-cost using replication techniques such as nanoimprint lithography, NIL [15,23–25]. Another benefit of thermoplastics for nanofluidics is their diverse surface chemistries, which are determined by the monomers comprising the polymer chains. For example, PMMA possesses methyl esters, while cyclic olefin copolymer (COC) contains ethylene and norbornene units. Therefore, both would result in different adsorption/desorption effects with solutes, and surface charge densities different that would affect the magnitude of the EOF. In addition, a diverse range of surface activation methods that can alter the surface chemistry of the polymer, for example, changing its wettability and/or altering the EOF [25,26]. UV/O3 or O2 plasma are methods that have been reported to generate oxygen-containing species for many plastics.
There have been a limited number of studies on nanoscale separations using thermoplastic devices [27,28], Weerakoon-Ratnayake et al. [27] showed the electrokinetic transport of silver nanoparticles (AgNPs) using PMMA nanoslits. The authors were able to demonstrate size-dependent mobilities of AgNPs in free solution, which was not possible using microscale columns. Differences in the mobilities were observed for 60 and 100 nm AgNPs at high electric field strengths. Most recently, O’Neil et al. [28–30] showed the separation of deoxynucleotide monophosphates (dNMPs) in free solution using thermoplastic nanochannel devices fabricated in PMMA substrates with COC cover plates. High electric field strengths provided better separations. In addition, the mobility order of the dNMPs was closely related to the hydrophobicity of each dNMP. However, the separation was dominated by electrophoresis compared to chromatographic mechanisms.
Most of the nanoscale separations noted above were performed on dsDNA. Compared with dsDNA, ssDNA is highly flexible, thermodynamically less stable, and more hydrophobic. Published literature associated with ssDNA reveals a large range of persistence lengths spanning from 1 to 6 nm under a variety of conditions [31–33]. The base-to-base spacing for ssDNA is 0.43 nm compared to 0.34 nm for dsDNA. ssDNA is also amphophilic, where the nucleobases are hydrophobic, and the phosphodiester backbone is hydrophilic. However, both ssDNAs and dsDNAs migrate as free draining coils in free solution and as such, their electrophoretic mobility is independent of the number of nucleotides the DNA possesses. Therefore, in both cases seizing matrices are required to sort ssDNAs and dsDNAs. Hydrogen bonding, hydrophobic interactions, and van der Waals interactions are the largest contributors to DNA-surface interactions [34]. Also, ssDNAs have the ability to expose either nucleobases or the phosphodiester backbone to a surface depending on the chemical composition of the surface [35].
In this work, we report the use of thermoplastic nanochannels (100 nm × 100 nm in depth and width) fabricated in PMMA for the free-solution electrokinetically driven separation of ssDNAs. To track the motion of ssDNAs, they were fluorescently tagged with their motion monitored using laser-induced fluorescence microscopy. We found that we could separate ssDNAs in free solution with an effective migration time that was inversely related to the length of the ssDNA indicating that the separation was affected by solute/wall interactions. Also, when using microscale columns where infrequent wall interactions would be expected to occur, the ssDNA fragments comigrated. Unique to these separations is that no monolayer coating of the polymer surface was required and also, the channel critical dimension was larger than the EDL thickness. Therefore, we define our separation as open tubular nanoelectrochromatography (OT-NEC). We will present results on the use of different electrochromatographic parameters and their effects on the resolution of the separation including electric field strength, column length, and type of substrate material used.
2. Materials and methods
2.1. Reagent and materials
Silicon (100) (Si) wafers were purchased from University Wafers (Boston, MA). Impact modified (IM) and non-impact modified (NIM) PMMA substrates were purchased from ePlastics (San Diego, CA) and Good Fellow (Berwyn, PA), respectively. Cyclic olefin copolymer (COC) 8007 was purchased from TOPAS Advanced Polymers (Florence, KY). COC 6015 was purchased from Knightsbridge Plastics, Inc. (Fremont, CA). UV curable polyurethane (PUA) resin was secured from Chansung Sheet Co. Ltd. (Chuncheognam-do, Korea). ATTO 532 end-labeled ssDNAs with three different lengths (Oligo35, Oligo50, Oligo70) were purchased from Integrated DNA technologies (San Jose, CA) and consisted of a random sequence. Ultra-pure TRIS-borate EDTA buffer 10× (TBE) and molecular biology grade water was obtained from Thermo Scientific (Waltham, MA).
2.2. Fabrication of microchannel devices
T-shaped (50 μm depth × 100 μm width and 5 cm long electrophoresis column) microfluidic devices (Fig. 1) were produced using a hot embossing machine (Wabash P3H-15-CLX, IN, USA) into PMMA. The upper platen was kept at 155°C and lower platen at 80°C with 1250 psi pressure applied for 3 min. Embossed devices were diced using a bandsaw and reservoirs were mechanically drilled. The devices were subsequently cleaned with 5% Micro-90, IPA, and nanopure water. Following embossing and hole drilling, the substrates were UV/O3 treated for 16 min at 22 mW/cm2 and sealed with a 150 μm thick PMMA sheet by using thermal fusion bonding at 105°C for 1 h. Microchannel dimensions were measured before and after bonding using a rapid laser-scanning confocal microscope (VK-X250, Keyence, IL, USA).
Figure 1.
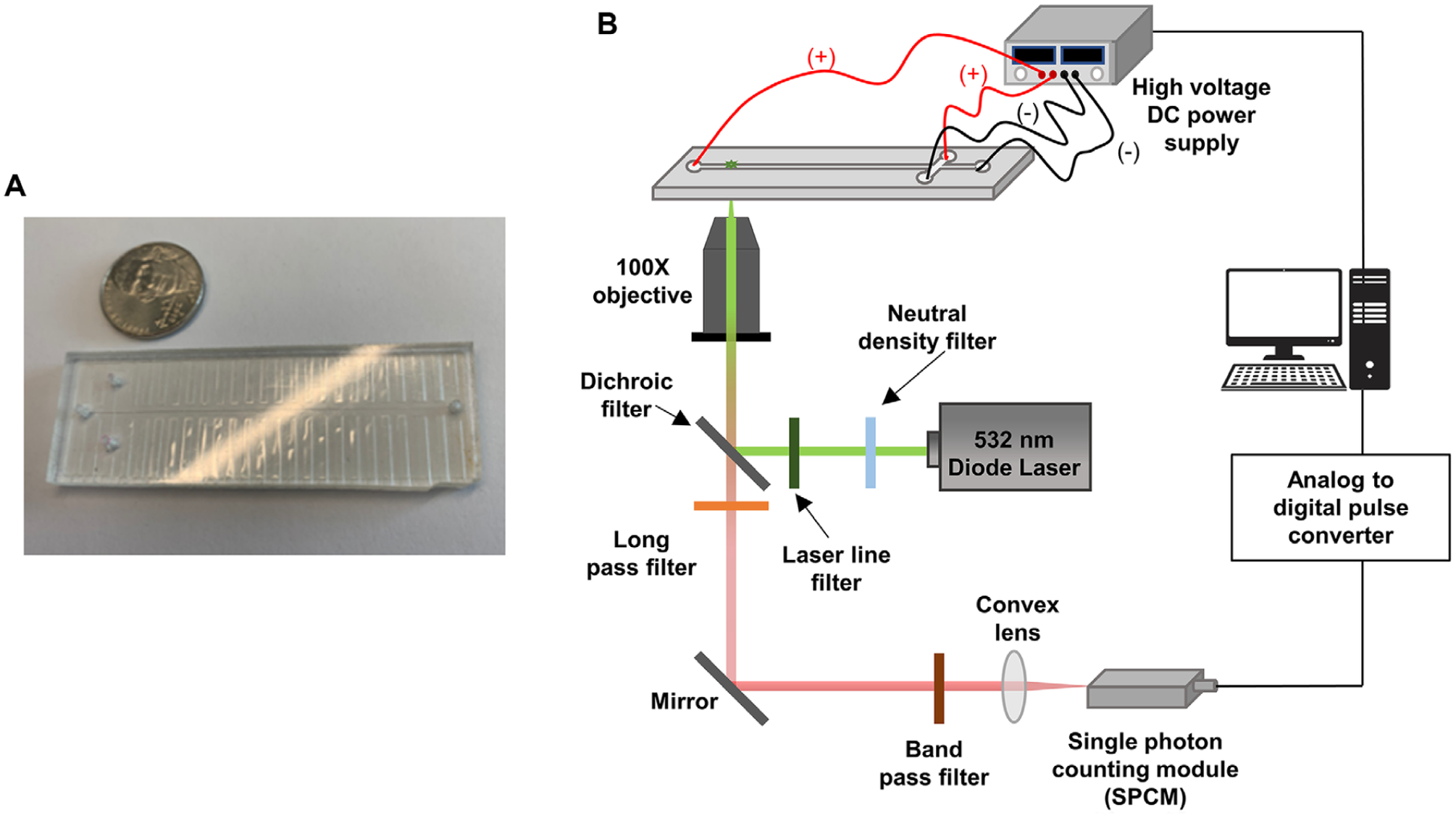
(A) An image of the T-chip used for the microscale electrophoresis. The chip was made via hot embossing into PMMA. (B) Schematic diagram of the in-house built microchip electrophoresis laser-induced fluorescence detector that utilized a 20 mW, 532 nm excitation laser with edge filter. The detector contained a 560 nm long pass filter, 532 nm dichroic filter and SPCM-AQR single photon counting module within the optical train. A 100× high numerical aperture (NA = 1.3) microscope objective was used to focus the laser beam onto the microchannel and collect the fluorescence.
2.3. Free solution separation of ssDNAs by microchip electrophoresis
The microfluidic device was primed with 50% methanol–water mixture for 5 min. Then, it was filled with TBE buffer at pH 8.3. Separation was carried out using the injection end set as the anode, and the detection end as the cathode. Injection was initiated by applying a positive voltage to the sample reservoir and grounding the sample waste reservoir for the amount of time required to completely fill the cross channel. The remaining reservoirs were floating during injection. After injection, a positive voltage was applied to the electrophoresis buffer reservoir and the electrophoresis waste reservoir was grounded. Detection was done 4.0 cm from the injection cross using a home-built laser-induced fluorescence detector equipped with a single photon avalanche diode counting module.
The laser-induced fluorescence (LIF) detector illustrated in Fig. 1 was configured in an epillumination format having a 532 nm, 20 mW excitation laser (LaserGlow Technologies, Toronto, Ontario, Canada), XF 3085 edge filter (Horiba Scientific, Middlesex, UK), 560 nm long pass filter (Omega Optical, Brattleboro, VT), a 532 nm dichroic filter (Omega Optical Brattleboro, VT), and a SPCM-AQR single photon counting module (Perkin Elmer Optoelectronics, Waltham, MA). A 100× high numerical aperture (NA = 1.3) microscope objective from Nikon (Natick, MA) was used to focus the laser beam onto the microchip and collect the fluorescence.
2.4. Fabrication of nanochannel devices
Fabrication of nanochannel devices were accomplished following a procedure previously reported by our group with slight modifications [15,36]. Access microchannels were fabricated in a Si wafer through photolithography followed by wet Si etching. Then, nanochannels were fabricated by focused ion beam milling into the Si wafer. Resin stamps were produced from the Si master by UV curing of a PUA resin for 3 min that was situated on a COC plate coated with a NOA72 adhesive. Next, the structures on the resin stamp were thermally imprinted into PMMA substrates using NIL with a Nanonex 2500 nanoimprinting machine that consisted of air cushion thermal imprinting. Unlike the pressing method for imprinting where solid parallel plates are used, air cushion press methods have improved uniformity in pressure and imprinting speed. The conditions used for imprinting are given in Table 1 for different PMMA substrates used in these experiments. Metrology of the devices were done by SEM imaging (see Fig. 2). The depths of the nanochannels imprinted in different PMMAs were measured using a scanning probe microscope, SPM (SPM-9700HT, SHIMADZU, Japan), and are given in the Supporting Information Table S1.
Table 1.
Calculated radius of gyration (Rg) of ssDNA using the equation, , where σ is the persistence length (5 nm) [55] of the ssDNA and L is the contour length [L = number of nucleotides × length per (0.43 nm)]
| Radius of gyration (Rg)/nm | |
|---|---|
| Oligo 35 | 5.0 |
| Oligo 50 | 5.9 |
| Oligo 70 | 7.1 |
Figure 2.

SEM images of the Si master, resin stamp, and imprinted device. (A) SEM of the Si master at 500X magnification. (B) Enlarged area of the entrance funnel formed in the Si master. (C) UV-imprinted nanochannel device to form the resin stamp with PUA. (D) Enlarged nanochannel area in the resin stamp. (E) Nanochannels thermally imprinted into a PMMA substrate. (F) Enlarged area of nanochannel imprinted into PMMA.
After imprinting the nanochannel device, sealing was done using a COC cover plate (100 μm thickness) using the conditions delineated in Supporting Information Table S2. The bond strength between the cover plate and substrate (IM-PMMA or NIM-PMMA) was evaluated using a crack opening method [37,38]. The bond strength was determined by placing a razor blade of known thickness (tb) between the thermally bonded substrate and cover plate to induce an inter-facial fracture with a length of L from the edge of the razor blade. If the elastic moduli of the cover plate and substrate are Ep and Es, respectively, the bond strength is determined using equation (1), where ts and tp are the thickness of the cover plate and substrate, respectively.
| (1) |
The elastic moduli of IM-PMMA was 1.6 GPa and for NIM-PMMA, it was 3.3 GPa that were obtained experimentally using a Q800 dynamic mechanical analyzer (TA instruments, New Castle, DE). The elastic moduli of the COC 8007 cover plate was 3.0 GPa, which was provided by the manufacturer. All measurements were performed five times and the average bond strength is reported along with the standard deviation in the measurements. The NIM-PMMA devices had a bond strength of 0.086 ± 0.014 mJ/cm2 while for the IM-PMMA devices was 0.084 ± 0.018 mJ/cm2. According to this data, there was no significant difference between the bond strength of IM-PMMA/COC devices and NIM-PMMA/COC devices.
2.5. Surface roughness measurements by AFM
To determine the surface roughness of IM-PMMA and NIM-PMMA, an AFM analysis was conducted. The tip used for imaging was operated at a frequency of 300 kHz and a radius <15 nm. Tapping mode was used with a scanning frequency of 1 Hz. First, IM-PMMA and NIM-PMMA surfaces (1 cm × 1 cm) were imaged by AFM to measure roughness without exposing to O2 plasma. Then, both PMMA surfaces were exposed to O2 plasma at 50 mW for 1 min and surface roughness was measured by AFM and RMS roughness was reported for each of these different surfaces.
2.6. EOF measurements
The current monitoring method described by Huang et al. [39] was used to measure the EOF in nanochannel devices. A nanochannel device possessing a single nanochannel (100 μm long, and approximately 100 nm × 100 nm, width × depth) connecting two access microchannels was fabricated as described previously. The chip was first primed with 50% v/v water–methanol, drained, and flushed with nuclease free water. Then, the device was filled with 45.0 mM TBE buffer and allowed to equilibrate for 4 min under a 1 V DC bias. After achieving a constant current trace, one reservoir was replaced with 48.0 mM TBE. Pt electrodes were placed in the reservoirs at each end of the nanochannel under a 1 V DC bias. Signals were acquired using pClamp10 software and Digidata 1440B low noise digitizer set at a rate of 10 kHz sampling frequency.
2.7. Detection system for the nanoelectrochromatography
The imaging system for monitoring the transport of ssDNAs through the thermoplastic nanochannel used a fluorescence imaging system we have reported [28]. More information can be found in Supporting Information Fig. S1.
2.8. Nanoelectrochromatographic separation of ssDNAs
Assembled nanochannel devices were primed with 50% methanol–water mixture for 5 min. Then, using a vacuum pump methanol–water mixture was removed from the nanofluidic device. Following priming, it was filled with 45 mM TBE buffer (pH 8.3) and allowed to equilibrate for 10 min. ATTO 532-labeled ssDNAs were prepared in 45 mM TBE. Next, carrier electrolyte in one of the reservoirs connecting microchannels was replaced with the ssDNA solution. Afterward, the carrier electrolyte in the opposite side of the same microchannel was removed and vacuum was applied to make sure it was completely filled. Once the microchannel was filled, all of the other reservoirs were filled with the same volume of carrier electrolyte. Finally, a square voltage (Vpp) was applied using an ATTEN ATF20B function waveform generator for a period of 10 s to allow for multiple injections of the dye labeled ssDNAs into the nanochannels. Events were recorded for 18 000 frames allowing multiple events to be analyzed. All solutions that were used for the nanofluidic device experiments were filtered through 0.2 μm filters to remove large particulates that may cause device clogging.
2.9. Data analysis
Determination of the migration time for fluorescently labeled ssDNAs were determined using a particular nanochannel column length and determining the time for the ssDNAs to travel through the nanochannel column. For more information on determining the effective mobilities of the dye-labeled ssDNAs, see the Supporting Information (Fig. S2 and S3 as well as equation S1).
3. Results and discussion
3.1. Microscale separation of ssDNAs
To understand ssDNA electrokinetically driven separation scaling effects in terms of channel dimensions (width, depth, and length), we first investigated the free solution electrokinetic separation of dye-labeled ssDNAs of different lengths (35mer, 50mer, 70mer) in 45 mM TBE (pH 8.3) using a PMMA microchannel device (column length = 5 cm, width = 100 μm, and depth = 50 μm) with laser-induced fluorescence detection (see Fig. 3A). The ssDNAs migrated from anode to cathode in the same direction as the EOF. As seen in Fig. 3B, all three ssDNAs comigrated with an apparent mobility of 1.80 × 10−5 cm2/Vs. Here, the free draining behavior of the ssDNA resulted in the electrophoretic mobilities to be independent of ssDNA length. For ssDNAs, it has been shown that the electrophoretic mobility increases with increasing nucleotides to ~10 nucleotides and then the mobility becomes independent of DNA nucleotide length in free solution [40]. However, we should note that previously we demonstrated the ability to electrokinetically separate dsDNAs using a PMMA device surface modified with a C18 monolayer and including an ion-pairing agent in the carrier electrolyte, 25 mM TFAA, but required the use of a low electric field strength (67 V/cm) to observe any type of separation for a series of dsDNA fragments of different lengths [41].
Figure 3.
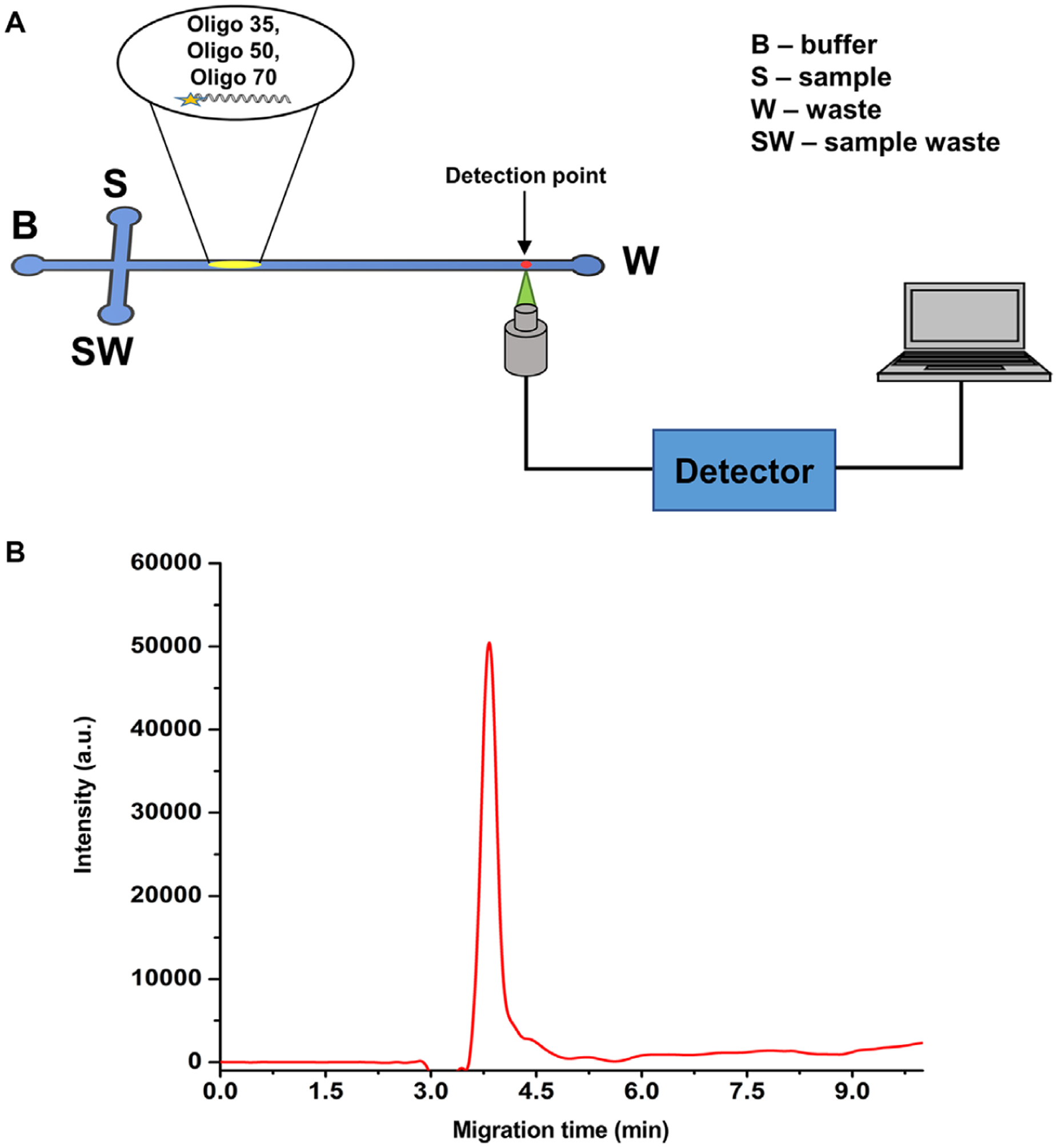
(A) Schematic diagram of experimental set up used for the microscale electrophoresis, where a T-shaped microchip was used. A sample plug was electrokinetically introduced into the separation channel by applying a potential across the S and SW reservoirs. A photon avalanche detector was used to capture the fluorescence signal at the detection point. (B) Electropherogram obtained for the free solution microchip (PMMA) electrophoresis of ssDNAs with microchannel dimensions of 50 μm × 100 μm (depth and width, respectively) with a length of 5.0 cm. The applied voltage for the electrophoresis was 5 kV (1000 V/cm) with a 45 mM TBE buffer (pH 8.3) used as the background electrolyte. The ssDNAs (5 μM) were injected electrokinetically into the separation channel.
3.2. PMMA material effects on the electroosmotic flow (EOF)
Nanochannels were fabricated using two different types of PMMA substrates to understand material effects on the nanoscale separation of ssDNAs. In all cases, no monolayer coating of the PMMA was undertaken. PMMA is a brittle thermoplastic with excellent optical properties. Two types of PMMA were evaluated here including IM-PMMA and NIM-PMMA. NIM-PMMA has a low impact strength meaning that it has a low capacity to absorb energy before fracture. To improve the impact strength of PMMA, acrylic modifiers are blended with the polymer matrix without degrading its optical transparency; this is called IM-PMMA [42,43]. These two materials were tested because each PMMA plastic can be activated using O2 plasma or UV/O3 irradiation, which can alter the surface charge through photo-oxidation reactions creating carboxylic acids groups as we have shown in our previous work [15,44]. Because the extent of surface carboxylic acids formed as well as the distribution of −COOH groups on the surface is dependent on the material type, it was hypothesized that the presence of the acrylic modifiers in IM-PMMA could affect the separation performance using nanoscale electrophoresis, especially in the case of wall interactions that may affect the separation [44].
Surface roughness in nanofluidic devices may occur either during replication due to imperfections in the molding tool [45] or following activation with an O2 plasma or UV/O3 irradiation [15]. Molecular dynamic (MD) simulations have shown that surface roughness has a large influence on the EOF and surface wettability depends on the magnitude of the roughness height (hr). The EOF can be significantly different for a situation where λd/hr <<1 compared to a situation where λd/hr ~1. Also, when surface roughness is comparable to the thickness of the EDL, effects on the EOF [46] and streaming potential can be observed [47]. However, the effect becomes insignificant when λd > hr [48]. According to Zhang et al. [49], the fluid flow experiences greater resistance on rough surfaces causing molecules to adsorb onto the surface of the nanochannel. In addition, their simulations indicated a decrease in the zeta potential and EOF with increasing roughness, which agreed with other MD simulation studies [50]. We hypothesized that the presence of the acrylic modifiers in IM-PMMA may also affect the surface roughness compared to PMMA without the acrylic modifiers following O2 plasma activation, which would affect the EOF.
We first measured the surface roughness of sheet IM-PMMA and NIM-PMMA before and after O2 plasma activation to serve as a model for our nanochannel devices. Surface topographical profiling was done using sheet PMMAs due to the difficulties associated with using AFM profiling of nanochannels arising from tip effects. Figure 4A and B shows AFM images of IM-PMMA and NIM-PMMA without O2 plasma activation, and Fig. 4C and D shows AFM images of IM-PMMA and NIM-PMMA after O2 plasma activation, respectively. After exposing to O2 plasma, the RMS roughness of the surface increased for both materials (see Fig. 4E). O2 plasma not only modified the surface chemically, but also created sub-nanometer and nanometer surface roughness features through slight etching of the surface during the activation process [45]. The surface roughness of IM-PMMA prior to and after O2 plasma activation was much higher than that of NIM-PMMA. Higher RMS roughness for IM-PMMA may be due to the acrylic modifiers present in IM-PMMA; difference in etching rates for the additives compared to PMMA in IM-PMMA will enhance its roughness with respect to NIM-PMMA. Thus, the surface of NIM-PMMA nanochannel devices would be expected to be less rough compared to IM-PMMA nanochannels following O2 plasma treatment.
Figure 4.
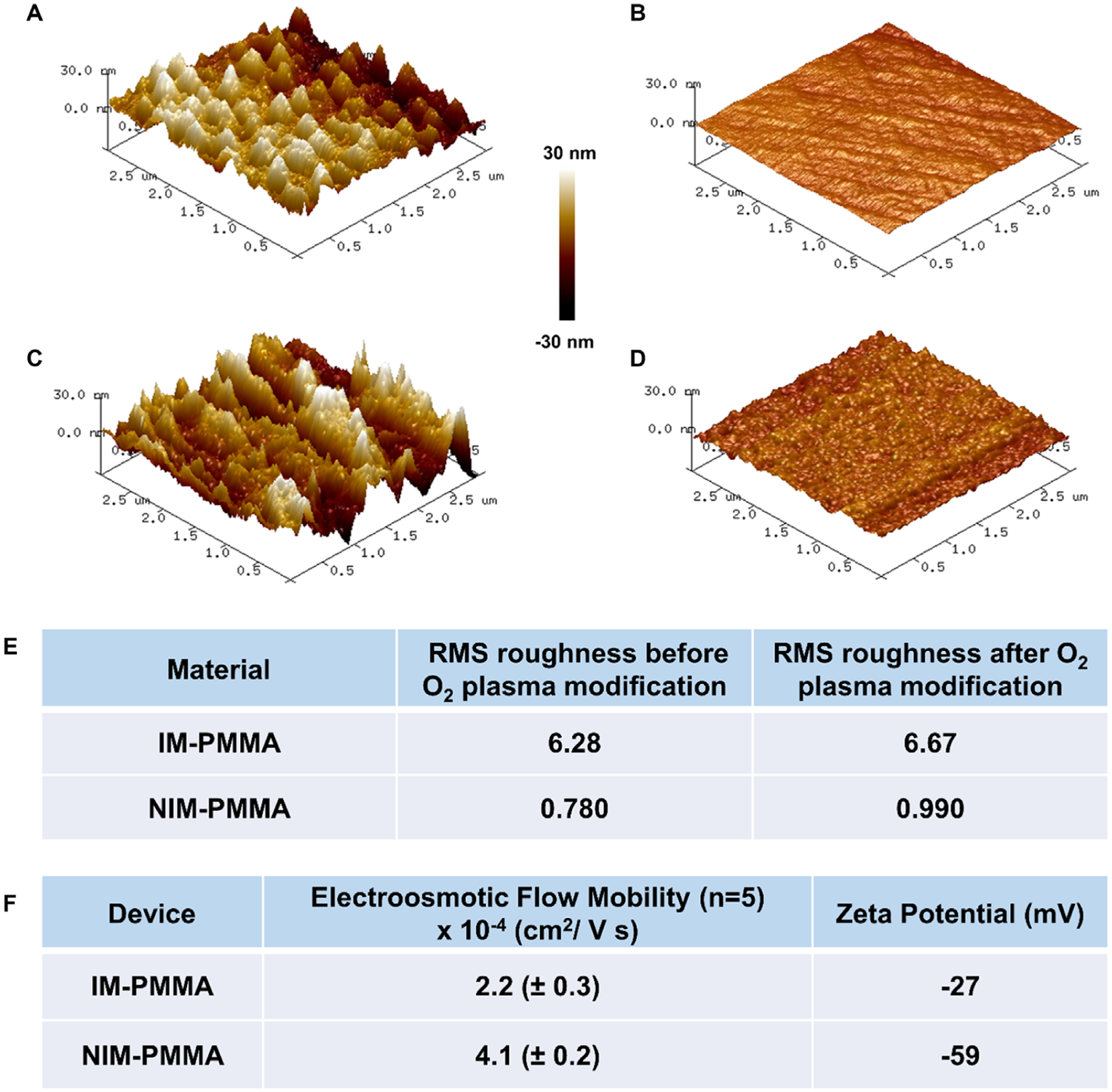
Characterization of surface roughness by AFM for IM-PMMA and NIM-PMMA surfaces before and after O2 plasma activation. Shown are AFM images of: (A) IM-PMMA; (B) NIM-PMMA; (C) O2 plasma treated IM-PMMA; and (D) O2 plasma treated NIM PMMA. Plasma activation was done at 50 mW for 1 min. These images were taken by scanning an area of 3 μm × 3 μm. (E) Comparison of the measured root mean square (RMS) roughness of both PMMA types before and after O2 plasma activation. (F) The measured EOF for NIM-PMMA and IM-PMMA nanochannel devices as well as the zeta potential following O2 plasma activation (EOF measured at pH = 8.3).
The EOF of thermoplastic nanochannel devices was measured by the buffer replacement method [39]. For these experiments, we used O2 plasma activated nanochannel devices made from both IM-PMMA and NIM-PMMA substrates. According to our previous studies, the majority of surface functionalities following O2 plasma activation of PMMA are carboxylic acid groups, which provide a negatively charged surface at pH 8.3 due to deprotonation of the carboxylic acid groups [15,51]. The thickness of the EDL in these experiments was estimated to be ~1.3 nm at the TBE buffer concentrations used herein. The zeta potential (ζ) was calculated using equation (2), where ε0 is the permittivity of vacuum, εr is the dielectric constant of the buffer (80.1), and η is the viscosity of the buffer. The viscosity of the buffer was assumed to be the same as the viscosity of pure water (8.9 × 10−4 Pa/s) [15].
| (2) |
As seen in Fig. 4F, the EOF and zeta potential were smaller for IM-PMMA compared to that of NIM-PMMA. These observations are in agreement with the aforementioned molecular dynamic simulation results, which indicated increased surface roughness typically reduces the EOF. In addition, IM-PMMA also showed lower a zeta potential compared to NIM-PMMA, which could mean that the acrylic modifiers are less prone to undergo surface photo-oxidation during O2 plasma activation. The EOF for NIM-PMMA (4.1 × 10−4 cm2/Vs) and IM-PMMA (2.2 × 10−4 cm2/Vs) are higher than the measured EOF reported by Uba et al. [15] (1.02 × 10−4 cm2/Vs), but in close agreement with the EOF (4.74 × 10−4 cm2/Vs) calculated using surface charge data [15].
3.3. Open-tubular nanoelectrochromatography (OTNEC) separation of ssDNAs
Because the free solution electrokinetic separation of the ssDNAs was not observed using PMMA-based microchip devices, we sought to determine if reducing the size of the channel could result in the ability to electrokinetically sort ssDNAs in free solution using thermoplastic nanochannels (dimensions were 100 nm × 100 nm in depth and width, respectively). Figure 5A and B shows the effective mobilities of the ssDNAs used herein at varying electric field strengths for both NIM-PMMA and IM-PMMA, respectively. The background electrolyte used here was 45 mM TBE resulting in λd = 1.3 nm. Therefore, d/λd was ~80, which would suggest plug-like flow indicating that transverse electromigration would not be in effect here [52]. As can be seen in Fig. 5A, B, each of the ssDNAs had different μeff indicating that they could be separated using nanocolumns in both IM-PMMA and NIM-PMMA at all electric field strengths investigated, even near 1000 V/cm that resulted in no separation of these ssDNAs using microelectrophoresis (see Fig. 3B).
Figure 5.
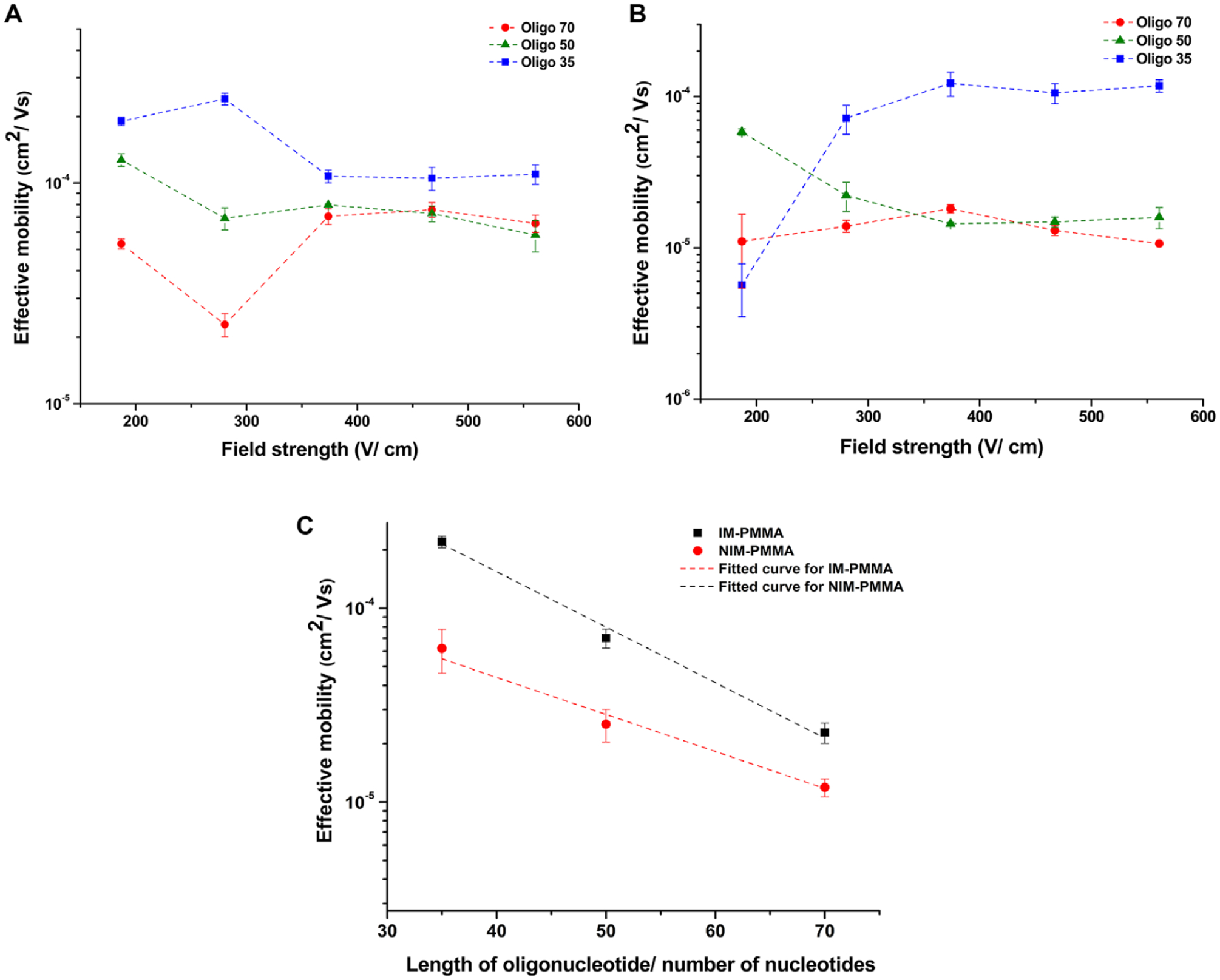
(A) Effective mobility of oligonucleotides versus the field strength in NIM-PMMA (substrate) – COC 8007 (cover plate) nanochannels (100 nm depth × 100 nm width and 60 μm effective length). (B) Effective mobility of ssDNAs versus electric field strength in IM-PMMA (substrate)-COC 8007 (cover plate) nanochannels (100 nm depth × 100 nm width and 60 nm effective length). Note: the lines in (A) and (B) are shown following trends and are not intended to indicate a functional fit to the data point. (C) Semi log plot of effective mobility versus length of ssDNAs in NIM-PMMA and IM-PMMA at 280 V/cm.
So why is not the same type of separation observed in microchip electrophoresis (see Fig. 3B)? Because the nanochannel dimensions (depth and width) are much smaller than in the case of the microchannel, which results in a significantly higher surface area-to-volume ratio in the nanochannel (~1000-fold higher), the nanochannel allows for more frequent solute–wall interactions and thus, the separation can be influenced by potential chromatographic effects. In contrast, the microchannel, due to its larger size, limits the number of solute–wall interactions. Because significantly fewer wall interactions would be expected in the microchannel, the electrokinetic transport of the ssDNAs are dominated by electrophoretic effects, which result in no observable separation due to the free draining behavior of DNAs in free solution.
Therefore, we define the nanoscale separation observed here to be open tubular nanoelectrochromatography (OT-NEC) and use the term effective mobility (μeff) typically used in conventional capillary electrochromatography (CEC), where analytes are separated according to their partitioning between both the liquid and stationary phases as well as any differences in their apparent electrophoretic mobilities [53,54]. The flat flow profile of the EOF provides better efficiencies than that associated with the parabolic flow profiles obtained using pressure-driven flows due to reduced Taylor dispersion. In our experiment, the dimensions of the separation column are in the nanoscale and the background carrier electrolyte acts as the mobile phase, which is driven through the nanochannel by the EOF. Instead of a conventionally sized capillary column (microscale domain) in which the wall is coated with a stationary phase or packed with silica particles coated with a stationary phase, we utilized here an open nanochannel with the surface carboxyl groups formed during O2 plasma activation as well as the native polymer of the thermoplastic serving as the stationary phase. As such, for thermoplastic OT-NEC, we can forgo the chemical steps associated with forming the stationary phase or packing the column to affect separations using electrochromatography.
The velocity (v) of an analyte moving through the nanochannel is defined by equation (3), where k´ is the capacity factor [53];
| (3) |
For a given solute, vapp (apparent velocity) is the sum of the EOF velocity and the electrophoretic velocity of a particular solute. The μeff for a solute migrating through the thermoplastic nanochannel can be deduced by dividing equation (3) by the electric field strength (E; V/cm) and is given by equation (4). In OT-NEC, solutes with similar apparent mobilities (μapp, see equation 5) can have different μeff depending on the degree of wall interactions they experience within the nanochannel, which is determined by k′.
| (4) |
| (5) |
3.4. Effect of electric field strength on μeff
The radius of gyration (Rg) of each ssDNA was calculated by [40], where σ is the persistence length (5 nm) [55] of ssDNA and L is the contour length (L = number of nucleotides × distance between two adjacent nucleotides, which is 0.43 nm). The values of Rg for each of the ssDNAs used in these studies are given in Table 1. According to the calculated Rg values for these ssDNAs, Rg < channel dimension (h) in all cases and the persistence length of ssDNA << channel depth and width. Thus, we can assume that these ssDNA migrate through the nanochannel as randomly coiled molecules.
As evident from Fig. 5A and B, the effective mobility of each ssDNA does not depend on the electric field strength in both materials except for electric field strengths ≤280 V/cm. At low electric field strengths, the molecules migrate slower than at higher electric field strengths and thus, have more time to interact with the nanochannel wall. We believe similar effects are responsible for the electric field dependent mobility observed for the ssDNA at electric field strengths ≤280 V/cm. In addition to the field-independent mobility observed at electric fields >280 V/cm, the effective mobility difference among different length ssDNAs is reduced. For example, the effective mobility of Oligo50 and Oligo70 approach each other in both PMMA substrates. This may have resulted from reduced solute/wall interactions of the ssDNAs due to higher EOF velocities at high electric field strengths (veof μeof E).
In Fig. 5C, we present the relationship between the effective mobility and the length of the ssDNA at 280 V/cm for both IM-PMMA and NIM-PMMA using a semi-logarithmic scale. The effective mobility and the length of the ssDNA are related through an exponential function, y = 0.002e−(0.064x) for NIM-PMMA, where y is the effective mobility and x is the number of nucleotides in the ssDNA molecule. In the case of IM-PMMA, this relationship was y = 0.003e(−0.047x). The smallest distinguishable nucleotide length difference we can discern is approximately seven nucleotides based on mobility differences for NIM-PMMA at three standard deviation units (99.7%). Therefore, for just the range of sizes investigated herein (35–70 nucleotides), the OT-NEC can effectively identify five different ssDNAs via mobility matching at the 99.7% confidence level. However, the size range of ssDNAs that can be used in the separation can be increased as well. For example, if we used as an upper limit, a ssDNA with 200 nucleotides, the multiplexing power of OT-NEC becomes ~24.
3.5. Separation of oligonucleotides in IM-PMMA and NIM-PMMA
Figure 6A and B shows histograms of the effective migration time of the model ssDNAs in IM-PMMA and NIM-PMMA nanochannel devices, respectively, at an electric field strength of 280 V/cm. This field strength was selected because it provided the best separation in terms of resolution for all ssDNAs in both nanochannel devices. The migration order of the ssDNAs in both materials was the same; Oligo35 < Oligo50 < Oligo70, where the number indicates the number of nucleotides in the ssDNA. The migration time increased with increasing chain length of the ssDNAs. These thermoplastic nanochannels were activated with O2 plasma to generate surface-confined carboxylic acid groups. At pH 8.3, these groups are expected to be deprotonated and produce an EOF that travels from anode to cathode.
Figure 6.
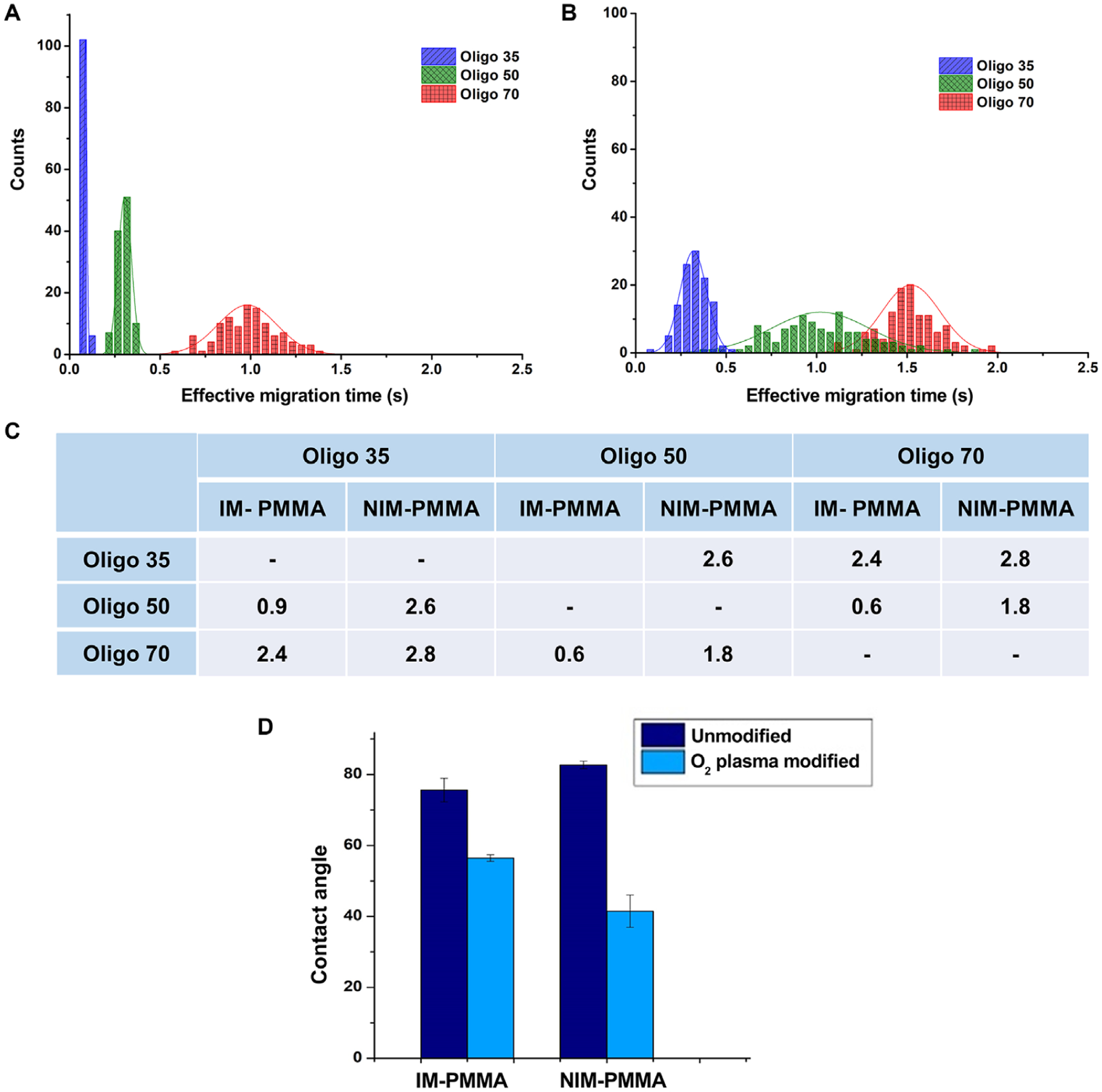
(A) Histogram of effective migration time of Oligo 35, Oligo 50 and Oligo 70 at an electric field strength of 280 V/cm that were separated in NIM-PMMA-COC8007 device. (B) Histogram of effective migration time of Oligo35, Oligo50 and Oligo70 at a field strength of 280 V/cm that were separated in IM-PMMA-COC 8007 device. Histograms were fitted in to a Gaussian distribution and each bin represent 0.05 s for n = 100 events. (C) Calculated separation resolution between the 3 ssDNA. (D) Contact angle of IM-PMMA and NIM-PMMA before and after oxygen plasma modification.
ssDNAs are amphiphilic and because of their single strand character, they have the ability to expose either their phosphodiester backbone or nucleobases to the surface. Therefore, the ssDNAs can interact with the surface through electrostatic interactions, van der Waals interactions, hydrogen bonding, and/or hydrophobic interactions [56]. Because the nanochannel surface is negatively charged, the question arises: Can the ssDNAs overcome electrostatic effects arising from the channel surface to allow sufficient interactions with the wall either through hydrogen bonding and/or hydrophobic interactions? We suspect that solute/wall interactions must be the dominate separation mode because the electrophoresis did not result in separation as indicated in the microchip results where solute/wall interactions would be minimal (see Fig. 3B).
Previous studies of DNA adsorption to silica surfaces have shown that even though electrostatic repulsion exists between the negatively charged DNA and the negatively charged silica surface, when the solution pH was greater than the pKa of the surface silanol groups, the phosphate/silanol and hydrophobic interactions are sufficiently strong to overcome electrostatic repulsions [56]. In addition to the aforementioned interactions, several investigators have proposed that ssDNA adsorption to silica surfaces can occur through hydrogen bonding via the exposed nucleobases to the silica surface. In light of these observations, we believe that ssDNA when moving electrokinetically through thermoplastic nanochannels will overcome electrostatic repulsions arising from the negatively charged carboxylate surface groups and interact with the native plastic via hydrogen bonding and/or hydrophobic interactions. If this is indeed the case, then the longer ssDNAs should show a longer effective migration time than the shorter ssDNAs, exactly what our data indicated (see Fig. 5C). Previous research has shown that DNA can be separated in free solution using glass-based devices when d/λd is close to unity due to non-plug like flow [20]. In our case, d/λd ~80 so that plug flow would be expected.
The resolution of adjacent peaks was calculated using R = 1.18(tm2 − tm1)/ (w0.51 + w0.52.), where tm1 and tm2 are migration times of the peak pair and w0.51, w0.52 are the full width at half maximum for each Gaussian peak. The separation resolution between the three ssDNAs in NIM-PMMA ranged from 1.8 to 2.6 (Fig. 6C), which exceeded baseline resolution (R = 1.5). The separation resolution between the three ssDNA in IM-PMMA ranged from 0.6 to 2.4 (Fig. 6C). Only the separation resolution between Oligo35 and Oligo70 (R = 2.4) exceeded baseline resolution in IM-PMMA. A recent study by O’Neil et al. [44], which mapped surface charge of IM-PMMA via super resolution fluorescence imaging, showed that there is a heterogeneous distribution of surface charge for IM-PMMA after O2 plasma activation. This heterogeneous surface charge distribution for IM-PMMA created “patches” of unmodified polymer on the surface. These hydrophobic “patches” on the surface of IM-PMMA can generate hydrophobic interactions with the exposed nucleobases in ssDNA in addition to hydrogen bonding and van der Waals interactions (exposed nucleobases in ssDNA are hydrophobic. As seen in Fig. 6D, the contact angle of IM-PMMA after O2 plasma modification is higher than that of the NIM-PMMA, which suggests that IM-PMMA is more hydrophobic compared to NIM-PMMA most likely due to the presence of the acrylic modifiers. This could increase the number of interactions of the ssDNA with the channel wall through hydrophobic interactions for IM-PMMA. Indeed, inspection of the migration times for IM-PMMA were slightly longer than those for NIM-PMMA. Also, the broader width of the migration time histograms for IM-PMMA would seem to indicate more solute/wall interactions that are kinetically slower in terms of the adsorption/desorption kinetics compared to the NIM-PMMA case, which would result in higher peak dispersion. As a note, peak dispersion is defined here as the standard deviations in the histograms shown in Fig. 6A and B, which are built from migration times for many OT-NEC runs. Finally, the presence of the additives found in IM-PMMA, which has different chemistry, can give rise to mixed retention mechanisms that are absent in the case of NIM-PMMA. However, in both cases (IM- and NIM-PMMA), length-based separations of ssDNAs were observed with the shorter DNAs moving faster through the column.
The retention factor (kι) is another parameter that measures partitioning of a solute with the stationary phase, which in this case is the PMMA nanochannel wall that has been O2 plasma activated. In the absence of wall interactions, all ssDNAs migrate with the same electrophoretic mobility due to the free draining behavior of ssDNAs as shown by Fig. 3B. Therefore, is constant for all ssDNAs. Hence, kι can be calculated using equation (6), where teff is the effective migration time of a charged analyte, teof is the effective migration time of a non-retained analyte, which in this case is proportional to the EOF. Calculated retention factors for the three ssDNAs using equation (6) are given in Table 2.
Table 2.
Retention factors of oligonucleotides in IM-PMMA and NIM-PMMA
| Retention factor (k′) | ||
|---|---|---|
| IM-PMMA | NIM-PMMA | |
| Oligo35 | 2.1 | 0.7 |
| Oligo50 | 9 | 5 |
| Oligo70 | 14 | 18 |
| (6) |
The retention factors increased with increasing length of the ssDNA in both materials. For IM-PMMA, retention factors ranged from 2.1 to 14 and NIM-PMMA retention factors ranged from 0.7 to 18. Oligo35 had the lowest retention factor in both materials while Oligo70 showed the highest retention factor. These results are in agreement with molecular dynamic simulations performed by Monserud et al. [57], where the residence time of DNAs in a nanoslit increased with increasing length of DNA due to increased interactions with the surface. One can anticipate that larger molecules will have greater stationary phase interactions. Regardless of whether the molecular conformation is extended or randomly coiled, the interaction strength between a ssDNA and a nanochannel surface with the appropriate chemistry will increase with the number of nucleotides due to increasing van der Waals contacts or other interactions.
Effect of column length on separation performance
Because we observed better separation performance using NIM-PMMA, the remainder of the experiments performed used this material as the substrate. We investigated the separation performance of the ssDNAs at different effective column lengths. Thus, we compared separation column lengths of 30 μm (see Fig. 7) and 60 μm effective column lengths. Histograms of the effective migration times were all taken at a field strength of 280 V/cm. Theoretical plates per meter were calculated using [5.54(teff/w0.5)] × [100/column length (cm)]. The separation resolution was determined by the number of theoretical plates, retention factor, and the selectivity of a particular solute as expressed by equation (7) (Purnell’s equation) [58].
Figure 7.
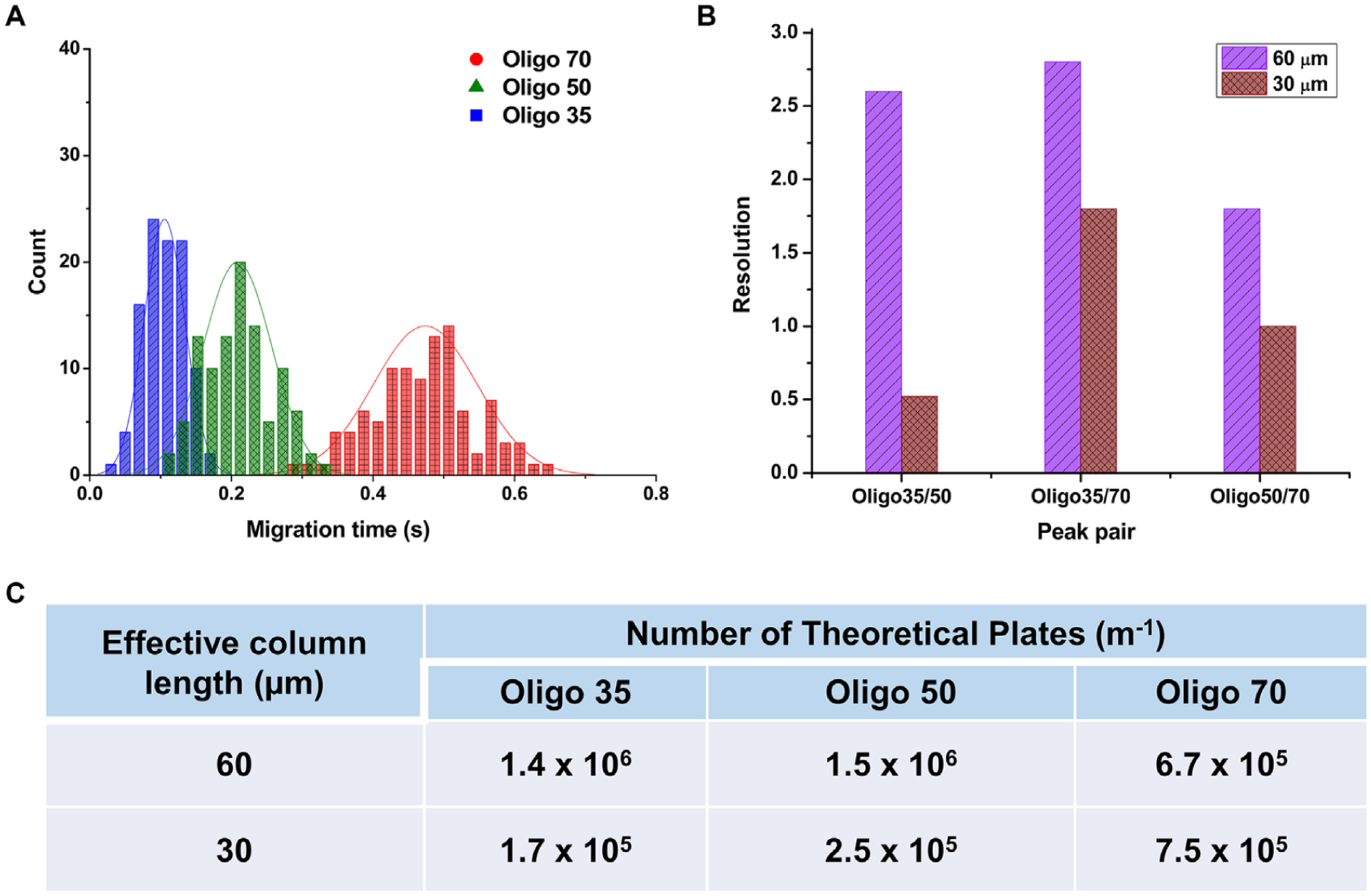
(A) Histogram of migration time for ssDNAs at an effective column length of 30 μm through a 100 nm × 100 nm NIM-PMMA nanochannel at a field strength of 280 V/cm. Histograms were fit to a Gaussian and each histogram represents 100 events. (B) Comparison of peak resolution at an effective column length of 60 μm and 30 μm. (C) Calculated theoretical plates for ssDNAs at effective column lengths of 30 μm and 60 μm. N = 5.54 (tm/w0.5)2 was used to calculate the number of theoretical plates and to obtained theoretical plates in units of m−1.
| (7) |
According to Fig. 7A and B, the resolution for the 30 μm length column decreased compared to the 60 μm column length for all three ssDNAs (resolution at 30 μm length ranged from 0.5 to 1.8). In contrast to electrophoresis where the resolution is not affected by column length, in electrochromatography the efficiency decreases with decreasing column length as the number of theoretical plates are directly proportional to column length (N = L/H, where L is column length and H is the height equivalent to theoretical plate). The number of theoretical plates at 30 μm effective column length was lower for Oligo35 and Oligo50 as compared to that for the 60 μm effective column length as shown in Fig. 7. However, for Oligo70 we noticed a slight increase in the number of theoretical plates at a shorter column length.
4. Concluding remarks
We were able to demonstrate in this study the ability to separate ssDNAs in thermoplastic nanochannels even when the EDL thickness was significantly less than the critical channel dimension and thus, plug-like flow was present and as such, transverse electromigration was not in effect here (d/λd ~ 80). Due to chromatographic effects for these electrokinetically driven separations, the column diameter could be reduced to improve separation resolution due to potential increased wall interactions. However, when d/λd approaches 1, significant double layer overlap will induce more of a parabolic flow profile that can give rise to Taylor dispersion that will degrade separation efficiency.
Microchip electrophoresis using a PMMA chip indicated no separation was present for the ssDNAs, which seemed to indicate minimal surface interactions and thus, electrophoretic effects dominate. Due to the free draining behavior of ssDNAs, their electrophoretic mobility is independent of length. However, for the nanoscale column separation of the model ssDNAs, it was observed using electrokinetic transport that due to scaling effects, surface interactions by the solute resulted in separation at all electric field strengths due to OT-NEC. We were able to achieve separation resolutions >1.5 for Oligo35, Oligo50, and Oligo70 using a NIM-PMMA device. Unique to these results is that no column packing was required as typically done for CEC and also, no surface monolayer of a hydrophobic coating was necessary, most likely due to the chemical nature of the thermoplastic that served as the stationary phase. ssDNA length-dependent surface interactions played a major role in determining the migration time of each ssDNA through thermoplastic nanochannels with longer ssDNAs showing longer migration times. IM-PMMA devices showed higher dispersion in their migration times than NIM-PMMA devices, which could have arisen from mixed retention mechanisms due to the acrylic modifiers used in IM-PMMA.
The thermoplastic nanochannel devices used in these experiments had 4 channels per device and a success rate of making devices >90%. For each device, >100 separations could be performed and we did not observe any clogging of the nanochannel. However, all solutions were filtered using a 200 nm filter to potentially mitigate this issue.
Our data showed a significant influence of surface roughness on the EOF and zeta potential as well with the EOF decreasing for highly rough surfaces. The degree of surface roughness was dependent on the nature of the PMMA material used. For example, IM-PMMA showed higher surface roughness compared to NIM-PMMA and as such, showed a lower EOF as well as zeta potential. In any case, both devices could be operated in a normal mode in which the EOF was sufficient to drive all of the ssDNAs in the same direction irrespective of length.
Separation efficiency was affected by column length with longer columns providing better separation resolution. This is consistent with a chromatographic mode of separation dominating as the resolution is dependent on column length and this is not the case when electrophoresis is the dominate separation mechanism. Thus, while the separation resolution provided 7 base resolution, this may be improved using longer nano-columns. However, even with 7 base resolution, there are a number of applications that can be realized, such as detecting single nucleotide polymorphisms when using a ligase detection reaction (LDR) as we have shown [59], the detection of microsatellite instability, or short tandem repeats for human identification.
While nanoscale electrophoresis has been reported for a variety of analytes, the major substrate consisted of glass or quartz. However, glass is challenged on two fronts: (1) The fabrication techniques for making nanochannels is complicated by the need for sophisticated equipment to make the necessary channels that lowers production rate and requires increased cost of producing each device. This significantly reduces accessibility of nanoscale electrophoresis to the research community as well as the commercial sector. On the other hand, thermoplastics are adaptable to injection molding, even on the nanometer scale, to allow production of devices in a high throughput mode and at low cost [60]. (2) To provide hydrophobic stationary phases for reverse-phase chromatography, appending monolayers to the channel wall are required. This was not required in our thermoplastic (PMMA) devices. All that was required to affect the separations observed here was activation of the plastic surface using an O2 plasma.
Supplementary Material
Acknowledgments
The authors would like to thank the NIH for funding of this work (NIBIB: P41 EB020594; NCI: P30 CA168524; P20 GM130423, P20 GM103638). The authors would also like to thank the National Science Foundation for partial support of this work as well (1507577). We would like to thank Dr. Cindy L. Berrie and Dr. Nian J.B. Kamathewatta for their support on AFM imaging. Finally, the authors would like to thank the Kansas University Nanofabrication Facility for assistance in making the nanofluidic devices used in this work. We would also like to acknowledge Dr. Mateusz Hupert for assistance in collecting the microchip electrophoresis data and Drs. Malgorzata Witek and Kumuditha Ratnayake for helpful discussions on the data interpretation.
Abbreviations:
- AgNP
silver nanoparticle
- COC
cyclic olefin copolymer
- dNMP
deoxynucleotide monophosphate
- LOC
lab-on-a-chip
- NIL
nanoimprint lithography
- OT-NEC
open-tubular nanoelectrochromatography
- POCT
point-of-care testing
Footnotes
Additional supporting information may be found online in the Supporting Information section at the end of the article.
The authors have declared no conflict of interest.
5 References
- [1].Roda A, Michelini E, Caliceti C, Guardigli M, Mirasoli M, Simoni P, Anal. Bioanal. Chem 2018, 410, 669–677. [DOI] [PubMed] [Google Scholar]
- [2].Sonker M, Kim D, Egatz-Gomez A, Ros A, Annu. Rev. Anal. Chem 2019, 12, 475–500. [DOI] [PubMed] [Google Scholar]
- [3].Khandurina J, Guttman A, Curr. Opin. Chem. Biol 2003, 7, 595–602. [DOI] [PubMed] [Google Scholar]
- [4].Mayer P, Slater GW, Drouin G, Anal. Chem 1994, 66, 1777–1780. [Google Scholar]
- [5].Grossman PD, Colburn JC, Capillary Electrophoresis: Theory and Practice, Academic Press, San Diego: 1992. [Google Scholar]
- [6].Bae YC, Soane D, J. Chromatogr. A 1993, 652, 17–22. [DOI] [PubMed] [Google Scholar]
- [7].Barron AE, Soane DS, Blanch HW, J. Chromatogr. A 1993, 652, 3–16. [DOI] [PubMed] [Google Scholar]
- [8].Fang Y, Zhang JZ, Hou JY, Lu H, Dovichi NJ, Electrophoresis 1996, 17, 1436–1442. [DOI] [PubMed] [Google Scholar]
- [9].Viovy J-L, Rev. Mod. Phys 2000, 72, 813. [Google Scholar]
- [10].Doherty EA, Meagher RJ, Albarghouthi MN, Barron AE, Electrophoresis 2003, 24, 34–54. [DOI] [PubMed] [Google Scholar]
- [11].Dolník V, Electrophoresis 2004, 25, 3589–3601. [DOI] [PubMed] [Google Scholar]
- [12].Napoli M, Eijkel JC, Pennathur S, Lab Chip 2010, 10, 957–985. [DOI] [PubMed] [Google Scholar]
- [13].Pennathur S, Santiago JG, Anal. Chem 2005, 77, 6782–6789. [DOI] [PubMed] [Google Scholar]
- [14].Pennathur S, Santiago JG, Anal. Chem 2005, 77, 6772–6781. [DOI] [PubMed] [Google Scholar]
- [15].Uba FI, Pullagurla SR, Sirasunthorn N, Wu J, Park S, Chantiwas R, Cho Y-K, Shin H, Soper SA, Analyst 2015, 140, 113–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mawatari K, Kazoe Y, Shimizu H, Pihosh Y, Kitamori T, Anal. Chem 2014, 86, 4068–4077 [DOI] [PubMed] [Google Scholar]
- [17].Shimizu H, Toyoda K, Mawatari K, Terabe S, Kitamori T, Anal. Chem 2019, 91, 3009–3014. [DOI] [PubMed] [Google Scholar]
- [18].Smirnova A, Shimizu H, Mawatari K, Kitamori T, J. Chromatogr. A 2015, 1418, 224–227. [DOI] [PubMed] [Google Scholar]
- [19].Baldessari F, Santiago JG, J. Nanobiotechnol 2006, 4, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Cross JD, Strychalski EA, Craighead H, J. Appl. Phys 2007, 102, 024701. [Google Scholar]
- [21].Svec F, Fréchet J, Hilder E, Peterson D, Rohr T, Photopolymerized and Photografted Porous Polymer Monoliths for Fabrication of Microfluidic Analytical Systems, Springer, Dordrecht: 2002. [Google Scholar]
- [22].Pennathur S, Baldessari F, Santiago JG, Kattah MG, Steinman JB, Utz PJ, Anal. Chem 2007, 79, 8316–8322. [DOI] [PubMed] [Google Scholar]
- [23].Chantiwas R, Hupert ML, Pullagurla SR, Balamurugan S, Tamarit-López J, Park S, Datta P, Goettert J, Cho Y-K, Soper SA, Lab Chip 2010, 10, 3255–3264. [DOI] [PubMed] [Google Scholar]
- [24].Chantiwas R, Park S, Soper SA, Kim BC, Takayama S, Sunkara V, Hwang H, Cho Y-K, Chem. Soc. Rev 2011, 40, 3677–3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Weerakoon-Ratnayake KM, O’Neil CE, Uba FI, Soper SA, Lab Chip 2017, 17, 362–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Chai J, Lu F, Li B, Kwok DY, Langmuir 2004, 20, 10919–10927. [DOI] [PubMed] [Google Scholar]
- [27].Weerakoon-Ratnayake KM, Uba FI, Oliver-Calixte NJ, Soper SA, Anal. Chem 2016, 88, 3569–3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].O’Neil C, Amarasekara CA, Weerakoon-Ratnayake KM, Gross B, Jia Z, Singh V, Park S, Soper SA, Anal. Chim. Acta 2018, 1027, 67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Arumay P, Yaakov L, PLoS Comput. Biol 2019, 15, e1006768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Liautard JP, Chromatogr J. A 1984, 285, 221–225. [DOI] [PubMed] [Google Scholar]
- [31].Chen H, Meisburger SP, Pabit SA, Sutton JL, Webb WW, Pollack L, Proc. Natl. Acad. Sci. USA 2012, 109, 799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Mills JB, Vacano E, Hagerman PJ, J. Mol. Biol 1999, 285, 245–257. [DOI] [PubMed] [Google Scholar]
- [33].Tinland B, Pluen A, Sturm J, Weill G, Macromolecules 1997, 30, 5763–5765. [Google Scholar]
- [34].Melzak KA, Sherwood CS, Turner RF, Haynes CA, J. Colloid Interface Sci 1996, 181, 635–644. [Google Scholar]
- [35].Maheswari PU, Rajendiran V, Palaniandavar M, Thomas R, Kulkarni G, Inorg. Chim. Acta 2006, 359, 4601–4612. [Google Scholar]
- [36].Uba FI, Hu B, Weerakoon-Ratnayake K, Oliver-Calixte N, Soper SA, Lab Chip 2015, 15, 1038–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ramm P, Lu JJ-Q, Taklo MM, Handbook of Wafer Bonding, John Wiley & Sons, Weinheim: 2011. [Google Scholar]
- [38].Tsao C, Hromada L, Liu J, Kumar P, DeVoe D, Lab Chip 2007, 7, 499–505. [DOI] [PubMed] [Google Scholar]
- [39].Huang X, Gordon MJ, Zare RN, Anal. Chem 1988, 60, 1837–1838. [Google Scholar]
- [40].Stellwagen E, Lu Y, Stellwagen NC, Biochemistry 2003, 42, 11745–11750. [DOI] [PubMed] [Google Scholar]
- [41].Galloway M, Soper SA, Electrophoresis 2002, 23, 3760–3768. [DOI] [PubMed] [Google Scholar]
- [42].Biwa S, Ito N, Ohno N, Mech. Mater 2001, 33, 717–728. [Google Scholar]
- [43].Cho K, Yang J, Park CE, Polymer 1998, 39, 3073–3081. [Google Scholar]
- [44].Oneil CE, Jackson JM, Shim S-H, Soper SA, Anal. Chem 2016, 88, 3686–3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hu Y, Werner C, Li D, Anal. Chem 2003, 75, 5747–5758. [DOI] [PubMed] [Google Scholar]
- [46].Ziarani A, Mohamad A, Nanoscale Microscale Thermophys. Eng 2008, 12, 154–169. [Google Scholar]
- [47].Park H, Lee H, Int. J. Heat Mass Transfer 2012, 55, 3295–3306. [Google Scholar]
- [48].Qiao R, Microfluid Nanofluidics 2007, 3, 33–38. [Google Scholar]
- [49].Zhang C, Lu P, Chen Y, Int. Commun. Heat Mass Transfer 2014, 59, 101–105. [Google Scholar]
- [50].Qiao R, Aluru NR, Colloids Surf. A Physicochem. Eng. Asp 2005, 267, 103–109. [Google Scholar]
- [51].Henry AC, Tutt TJ, Galloway M, Davidson YY, McWhorter CS, Soper SA, McCarley RL, Anal. Chem 2000, 72, 5331–5337. [DOI] [PubMed] [Google Scholar]
- [52].Rogers B, Adams J, Pennathur S, Nanotechnology: understanding small systems, CRC Press, Boca Raton: 2014. [Google Scholar]
- [53].Lelièvre F, Yan C, Zare RN, Gareil P, J. Chromatogr. A 1996, 723, 145–156. [Google Scholar]
- [54].Xiang R, Horváth C, Anal. Chem 2002, 74, 762–770. [DOI] [PubMed] [Google Scholar]
- [55].Qian W, Doi K, Kawano S, Biophys. J 2017, 112, 838–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Wu J, Wang H, Zhu A, Long F, ACS Omega 2018, 3, 5605–5614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Monserud JH, Schwartz DK, Biomacromolecules 2012, 13, 4002–4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Kapnissi-Christodoulou CP, Nicolaou AG, Stavrou IJ, J. Chromatogr. A 2016, 1467, 145–154. [DOI] [PubMed] [Google Scholar]
- [59].Sinville R, Coyne J, Meagher RJ, Cheng YW, Barany F, Barron A, Soper SA, Electrophoresis 2008, 29, 4751–4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Becker H, Lab Chip 2009, 9, 2759–2762. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.