

Abstract
The frontonasal dysplasias are a group of craniofacial phenotypes characterized by hypertelorism, nasal clefting, frontal bossing, and abnormal hairline. These conditions are caused by recessive mutations in members of the aristaless gene family, resulting in abnormal cranial neural crest migration and differentiation. We report a family with a dominantly inherited craniofacial phenotype comprised of frontal bossing with high hairline, ptosis, hypertelorism, broad nasal tip, large anterior fontanelle, cranial base anomalies, and sagittal synostosis. Chromosomal microarray identified a heterozygous 108.3 kilobase deletion of chromosome 2p21 segregating with phenotype and limited to the sine oculis homeobox gene SIX2 and surrounding noncoding DNA. Similar to the human SIX2 deletion phenotype, one mouse model of frontonasal dysplasia, brachyrrhine, exhibits dominant inheritance and impaired cranial base chondrogenesis associated with reduced Six2 expression. We report the first human autosomal dominant frontonasal dysplasia syndrome associated with SIX2 deletion and with phenotypic similarities to murine models of Six2 Loss-of-function.
Keywords: frontonasal dysplasia, Brachyrrhine, craniosynostosis, ptosis, hypertelorism
INTRODUCTION
Frontonasal dysplasia (FND) syndromes include a wide phenotypic spectrum comprised of ocular hypertelorism, broad or cleft nose with abnormal nasal tip, frontal bossing, and hairline anomalies. FND type 1 involves the nose, philtrum, and eyelids (ptosis); type 2 includes alopecia, enlarged parietal foramina, and may be associated with genital abnormalities; type 3 has the most severe facial features and includes ocular abnormalities (microphthalmia or anophthalmia) and pinna malformations. Classical FND types 1–3 are caused by recessive mutations in the Drosophila aristaless-like homeobox genes (ALX3, ALX4, and ALX1, respectively) [Kayserili et al., 2009, 2012; Twigg et al., 2009; Uz et al., 2010; Kariminejad et al., 2014]. These transcription factors are expressed in neural-crest derived mesenchyme and lateral plate mesoderm and regulate cell fate determination and differentiation [ten Berge et al., 1998; Beverdam et al., 2001; Ettensohn et al., 2003; Kayserili et al., 2009, 2012; Kariminejad et al., 2014]. FND associated with craniosynostosis is seen in craniofrontonasal syndrome caused by mutation in EFNB1 [Twigg et al., 2004; Wieland et al., 2004]. Functional alterations of these proteins result in abnormal development of midline craniofacial neural crest derivatives.
Here, we present the genetic analysis of a family with autosomal dominant FND segregating with chromosome 2p21 deletion encompassing the SIX2 gene. This phenotype is consistent with a mouse model of Six2 Loss-of function, brachyrrhine. [Lozanoff, 1993; Ma and Lozanoff, 1993, 1996; Lozanoff et al., 1994, 2001]. Together, these findings support that SIX2 haploinsufficiency confers an autosomal dominantly transmitted FND in humans with phenotype similar to analogous craniofacial phenotypes of Six2 and brachyrrhine (Br) mouse models.
CLINICAL REPORT
The proband was referred to Genetics Clinic at 22 months of age for evaluation of craniosynostosis and dysmorphic features. She was the 2,756 g (10th centile) and 48.3 cm (25th centile) product of a full term vaginal delivery to a 37 year old gravida 2 para 2 woman after a pregnancy complicated by maternal hypothyroidism treated with levothyroxine. She was noted to have dysmorphic features including bilateral ptosis and epicanthus inversus at birth. Her primary care physician ordered a chromosome analysis which showed a normal female karyotype of 46,XX. Psychomotor development was normal and she reached normal milestones at the appropriate ages. Because she had a large anterior fontanelle, a 3D cranial computed tomography (CT) scan was performed documenting synostosis of the posterior sagittal suture. Physical examination at 22 months showed a healthy dysmorphic girl whose weight was 11.1 kg (35th centile), length 83 cm (35th centile), head circumference 50 cm (95th centile). The head was large with frontal bossing, large anterior fontanelle measuring 6.4 by 4.0 cm, flat nasal bridge, and wide nasal tip. She had ptosis and epicanthus inversus. She was hypeteloric with inner canthal distance 3.4 cm and outer canthal distance of 9.1 cm (both > 95th centile). There was bilateral ptosis with epicanthus inversus. The palate was intact but the lateral palatine ridges were wide. Dentition was normal. Ears were normally shaped but posteriorly rotated (see Fig. 1A,B). The remainder of the examination was normal, including the musculoskeletal and limb examination.
FIG. 1.

Frontonasal dysplasia phenotype. A–B: Facial features of the proband at 21 months of age, including high hairline, frontal bossing, hypertelorism, ptosis, and broad nasal tip. C–D: Facial features of the proband’s mother as an adult, including high frontal hairline, frontal bossing, hypertelorism, unilateral ptosis, broad nasal tip, and prognathia. E–H: Cranial CT 3D reconstruction for the proband at 31 months of age demonstrating wide anterior fontanelle (E, arrow), metopic synostosis (E, arrowhead), parietal foramina (F, arrowheads), sagittal synostosis (F, arrow), persistent craniopharyngeal canal (G and H, arrow), and premature lateral spheno-occipital synchondrosis (H, arrowheads). [Color figure can be seen in the online version of this article, available at http://wileyonlinelibrary.com/journal/ajmga]
Her mother had a strikingly similar appearance to the proband, having a high broad forehead, flat nasal bridge, wide nasal tip, epicanthus inversus, and ptosis (Fig. 1C,D). Her mother’s head circumference was 54.5 cm (50th centile) whereas her father’s head circumference was 61 cm (>98th centile). Maternal grandparents and other relatives were not available for examination. Per maternal report, the proband’s maternal uncle has very similar facial features, including hypertelorism, broad nasal bridge, and broad forehead. There was no known family history of hypertension or renal disease.
Review of the head CT with 3D reconstruction of the cranium confirmed large anterior fontanelle (Fig. 1E, arrow), persistent metopic suture (Fig. 1E, arrowhead), bilateral parietal foramina (Fig. 1F, arrowheads), and complete sagittal synostosis by 31 months (Fig. 1F, arrow). Subtle cranial base anomalies were present, including premature lateral spheno-occipital synchondrosis (Fig. 1H inset, arrowheads) and persistent craniopharyngeal canal (Fig. 1G, arrow, and Fig. 1H inset, arrow). Macrocephaly developed over that period to 53.3 cm (+2.8 SD). Renal ultrasound was normal, as were renal function testing for creatinine and electrolytes including calcium and phosphorous. Thyroid testing was normal. Her neurocognitive development has continued to be normal for age.
Sequence analysis of genes associated with parietal foramina (ALX4, MSX2) and craniosynostosis (FGFR1, FGFR2, FGFR3, TWIST) did not reveal any pathogenic variants. Single nucleotide polymorphism (SNP)-based microarray (Human610-Quad Bead-Chip, Illumina) was performed and identified a 108.3 kilobase (kb) deletion of chromosome 2p21, with decreased log ratio and absence of heterozygosity across 39 markers (rs4953156 to rs17319875; chr2:45,189,763–45,298,085; GRCh37/hg19) (Fig. 2B). This included a complete and isolated deletion of the SIX2 gene (chr2:45,232,324–45,236,542) and surrounding noncoding DNA (Fig. 2A). Parental testing confirmed that the SIX2 deletion was present in the proband’s mother and absent in her father. Sanger sequencing of SIX2 exons in proband and mother revealed no pathogenic variants on the remaining allele. The SIX3 gene (chr2:45,169,037–45,173,216) proximal to the deletion was also sequenced in proband and mother, and no pathogenic variants were detected.
FIG. 2.
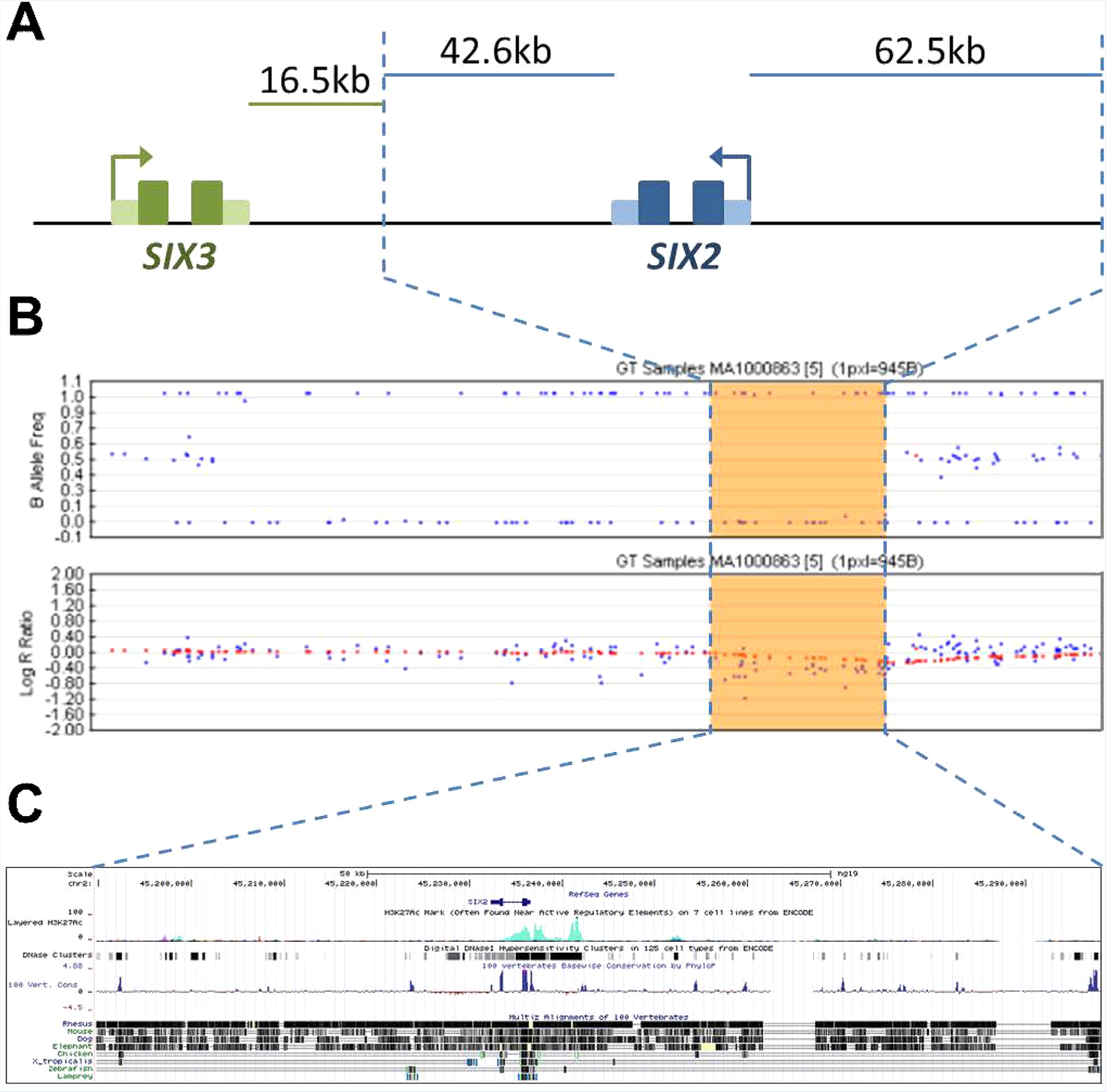
2p21 deletion including SIX2. A: Schematization of the 108.3 kb chromosome 2p21 deletion (blue dashed lines) encompassing the SIX2 gene (blue) and surrounding noncoding DNA. The SIX3 gene (green) is proximal to and not included in the deletion. B: Log ratio and B-allele frequencies depicted across the deletion. C: Regulatory elements within the deletion (chr2:45,189,763–45,298,085; hg19) depicted using UCSC genome browser (genome.ucsc.edu), including ENCODE regulatory elements predicted by H3K27Ac marks, DNAse hypersensitivity, and conservation in mammalian, vertebrate, and invertebrate species. [Color figure can be seen in the online version of this article, available at http://wileyonlinelibrary.com/journal/ajmga]
DISCUSSION
This case characterizes a novel dominantly inherited frontonasal dysplasia, similar to FND type 1 and associated with heterozygous deletion of the SIX2 gene. The orthologues Drosophila sine oculis and murine Six2 are critical for cell fate determination [Serikaku and O’Tousa, 1994]. In mouse, Six2 is critical for cranial base formation, and loss-of-function models result in frontonasal dysplasia. Notably, FND types 1–3 are caused by recessive mutations in the Drosophila aristaless homeobox transcription factors (ALX1, ALX3, and ALX4) that are critical for differentiation of neural crest cells into midline midface craniofacial structures and the branchial arches [Mavrogiannis et al., 2001; Twigg et al., 2009; Uz et al., 2010]. Neither biochemical nor epistatic association of aristaless/ALX and sine oculis/SIX has been described. Together, this dominantly inherited FND phenotype implicates an additional family of homeobox transcription factors as candidate causes for FND.
Genes of the SIX family encode homeodomain transcription factors with high sequence similarity to the Drosophila gene sine oculis and are associated with branchial arch, ear, and renal malformations. Mutations in SIX1 and SIX5 cause branchio-oto-renal syndrome, characterized by cervical fistulae, ear anomalies, and renal hypodysplasia [Ruf et al., 2004; Hoskins et al., 2007]. Like other members of this family, the SIX2 protein contains a DNA-binding homeodomain and an upstream SIX domain that functions in binding site specificity and protein-protein interactions [Boucher et al., 2000]. Missense dominant mutations in the human SIX2 gene have been associated with isolated renal hypodysplasia without craniofacial involvement [Weber et al., 2008]. However, these demonstrate incomplete penetrance or multifactorial etiology, as parents of multiple patients carrying these mutations have normal kidney function. In our case, neither the proband nor her mother has a history of hypertension or other signs of renal dysfunction or dysplasia. One possibility is that whole gene deletion results in haploin-sufficiency, whereas missense mutations produce an abnormal protein with altered function, either gain- or partial loss-of-function.
Targeted deletion of the murine Six2 gene results in craniofacial defects, renal hypodysplasia, and neonatal lethality in homozygous animals [Self et al., 2006]. Six2 mutant mice have cranial base deficits resulting in a compressed cranial appearance, with no cartilage between the basioccipital, basisphenoid, and presphenoid bones [He et al., 2010]. Additionally, there is absent intrasphenoidal and reduced spheno-occipital synchondrosis. This reduction of cartilage and impaired synchondrosis is likely due to premature chondrocyte maturation and decreased proliferation, thereby prematurely depleting the pool of cartilagenous progenitor cells [He et al., 2010]. No craniofacial or renal phenotype has been reported for heterozygous Six2 mutant mice. On the other hand, the radiation-induced mouse mutant brachyrrhine (Br) carries a semi-dominant mutation resulting in frontonasal dysplasia (FND) and renal hypoplasia [McBratney et al., 2003; Fogelgren et al., 2008]. The heterozygous mutant (Br/+) survives to adulthood, while homozygous mutants (Br/Br) die at birth. Homozygous brachyrrhine mice (Br/Br) have severe frontonasal dysplasia, anterior cranial base malformations including unfused trabecular cartilage and absent presphenoid bone, midfacial retrusion with resulting retrognathia, and renal hypoplasia resulting in intrauterine growth restriction and embryonic lethality [Lozanoff, 1993; Ma and Lozanoff, 1993, 1996; Lozanoff et al., 1994, 2001; Fogelgren et al., 2009]. Similar to Six2 mutant mice, chondrogenesis is impaired.
The brachyrrhine heterozygous mouse (Br/+) has an intermediate phenotype including midface hypoplasia, hypertelorism, and sphenoidal abnormalities. Frontonasal dysplasia and anterior cranial base malformations in heterozygous mice (Br/+) include fused trabecular cartilage lacking lateral chondrification and absent orbital cartilages. The Br/+ craniofacial phenotype is most similar to the human SIX2 heterozygous deletion phenotype in terms of viability, frontonasal dysplasia, and cranial base abnormalities, particularly of the sphenoid and spheno-occipital synchondrosis. Linkage and expression analyzes link the brachyrrhine mouse phenotype to a critical region containing the Six2 gene and highly overlapping the homologous human 2p21 deletion. Notably, in the brachyrrhine mouse no mutation was found in the Six2 coding sequence or 1.5 kb upstream of the promoter region. However, the brachyrrhine mouse has a dose dependent decrease in Six2 expression, suggesting existence of a novel distal cis-regulatory element [Fogelgren et al., 2008]. Therefore, deletion of a cis-regulatory element of SIX2 may contribute to the frontonasal dysplasia observed in our patients (Fig. 2C).
Molecular mechanisms governing SIX2 expression and function appear to play orthologous roles in human and mouse craniofacial development. Six2 expression in the craniofacial mesenchyme is regulated by Hox family member Hoxa2, which regulates the development of second branchial arch derivatives that comprise the stapes, styloid, and lesser hyoid through inhibition of Six2 expression [Kutejova et al., 2005]. Hoxa2 overexpression in chicken or mouse results in abnormalities of the facial skeleton and decreased levels of Six2 in the cranial mesenchyme [Kanzler et al., 1998; Creuzet et al., 2002]. Other studies indicate that, in the second branchial arch, Six2 regulates Igf1 and Igfbp5 in bone development, and dysregulation of this pathway results in bone overgrowth in Hoxa2 mutants [Kutejova et al., 2005, 2008]. Noncoding Six2 regulatory elements (Fig. 2C) are likely pleiotropic, as Six2 has shown to be repressed by Hoxa2 and activated by Hox11 at a single enhancer [Yallowitz et al., 2009].
In conclusion, SIX2 haploinsufficiency results in frontonasal dysplasia likely due to altered neural crest cell fate specification and differentiation in the branchial arches. Contributions of other genes regulating SIX2 expression, including HOX genes, and of factors downstream of SIX2 function, including IGF genes, likely constitute a significant pathway in craniofacial development. This pathway may intersect with the functions of the ALX gene family. Therefore, SIX2 and other genes within this pathway should be considered in patients exhibiting similar frontonasal dysplasia phenotypes.
ACKNOWLEDGMENTS
We thank the family for their participation in this study. This study was sponsored in part by the National Institute on Deafness and Other Communication Disorders (NIDCD/NIH) research grants R01 DC012564 to Z.M.A and internal departmental funds to R.B.H.
Grant sponsor: National Institute on Deafness and Other Communication Disorders (NIDCD/NIH); Grant number: R01 DC012564; Grant sponsor: Internal Departmental Funds.
Footnotes
Conflict of interest: none.
REFERENCES
- Beverdam A, Brouwer A, Reijnen M, Korving J, Meijlink F. 2001. Severe nasal clefting and abnormal embryonic apoptosis in Alx3/Alx4 double mutant mice. Development 128:3975–3986. [DOI] [PubMed] [Google Scholar]
- Boucher CA, Winchester CL, Hamilton GM, Winter AD, Johnson KJ, Bailey ME. 2000. Structure, mapping and expression of the human gene encoding the homeodomain protein, SIX2. Gene 247:145–151. [DOI] [PubMed] [Google Scholar]
- Creuzet S, Couly G, Vincent C, Le Douarin NM. 2002. Negative effect of Hox gene expression on the development of the neural crest-derived facial skeleton. Development 129:4301–4313. [DOI] [PubMed] [Google Scholar]
- Ettensohn CA, Illies MR, Oliveri P, De Jong DL. 2003. Alx1, a member of the Cart1/Alx3/Alx4 subfamily of Paired-class homeodomain proteins, is an essential component of the gene network controlling skeletogenic fate specification in the sea urchin embryo. Development 130:2917–2928. [DOI] [PubMed] [Google Scholar]
- Fogelgren B, Kuroyama MC, McBratney-Owen B, Spence AA, Malahn LE, Anawati MK, Cabatbat C, Alarcon VB, Marikawa Y, Lozanoff S. 2008. Misexpression of Six2 is associated with heritable frontonasal dysplasia and renal hypoplasia in 3H1 Br mice. Dev Dyn 237:1767–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogelgren B, Yang S, Sharp IC, Huckstep OJ, Ma W, Somponpun SJ, Carlson EC, Uyehara CF, Lozanoff S. 2009. Deficiency in Six2 during prenatal development is associated with reduced nephron number, chronic renal failure, and hypertension in Br/+ adult mice. Am J Physiol Renal Physiol 296:F1166–F1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He G, Tavella S, Hanley KP, Self M, Oliver G, Grifone R, Hanley N, Ward C, Bobola N. 2010. Inactivation of Six2 in mouse identifies a novel genetic mechanism controlling development and growth of the cranial base. Dev Biol 344:720–730. [DOI] [PubMed] [Google Scholar]
- Hoskins BE, Cramer CH, Silvius D, Zou D, Raymond RM, Orten DJ, Kimberling WJ, Smith RJ, Weil D, Petit C, Otto EA, Xu PX, Hildebrandt F. 2007. Transcription factor SIX5 is mutated in patients with branchio-oto-renal syndrome. Am J Hum Genet 80:800–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanzler B, Kuschert SJ, Liu YH, Mallo M. 1998. Hoxa-2 restricts the chondrogenic domain and inhibits bone formation during development of the branchial area. Development 125:2587–2597. [DOI] [PubMed] [Google Scholar]
- Kariminejad A, Bozorgmehr B, Alizadeh H, Ghaderi-Sohi S, Toksoy G, Uyguner ZO, Kayserili H. 2014. Skull defects, alopecia, hypertelorism, and notched alae nasi caused by homozygous ALX4 gene mutation. Am J Med Genet Part A 164A:1322–1327. [DOI] [PubMed] [Google Scholar]
- Kayserili H, Altunoglu U, Ozgur H, Basaran S, Uyguner ZO. 2012. Mild nasal malformations and parietal foramina caused by homozygous ALX4 mutations. Am J Med Genet Part A 158A:236–244. [DOI] [PubMed] [Google Scholar]
- Kayserili H, Uz E, Niessen C, Vargel I, Alanay Y, Tuncbilek G, Yigit G, Uyguner O, Candan S, Okur H, Kaygin S, Balci S, Mavili E, Alikasifoglu M, Haase I, Wollnik B, Akarsu NA. 2009. ALX4 dysfunction disrupts craniofacial and epidermal development. Hum Mol Genet 18:4357–4366. [DOI] [PubMed] [Google Scholar]
- Kutejova E, Engist B, Mallo M, Kanzler B, Bobola N. 2005. Hoxa2 downregulates Six2 in the neural crest-derived mesenchyme. Development 132:469–478. [DOI] [PubMed] [Google Scholar]
- Kutejova E, Engist B, Self M, Oliver G, Kirilenko P, Bobola N. 2008. Six2 functions redundantly immediately downstream of Hoxa2. Development 135:1463–1470. [DOI] [PubMed] [Google Scholar]
- Lozanoff S 1993. Midfacial retrusion in adult brachyrrhine mice. Acta Anat (Basel) 147:125–132. [DOI] [PubMed] [Google Scholar]
- Lozanoff S, Johnston J, Ma W, Jourdan-Le Saux C. 2001. Immunohistochemical localization of Pax2 and associated proteins in the developing kidney of mice with renal hypoplasia. J Histochem Cytochem 49:1081–1097. [DOI] [PubMed] [Google Scholar]
- Lozanoff S, Jureczek S, Feng T, Padwal R. 1994. Anterior cranial base morphology in mice with midfacial retrusion. Cleft Palate Craniofac J 31:417–428. [DOI] [PubMed] [Google Scholar]
- Ma W, Lozanoff S. 1993. External craniofacial features, body size, and renal morphology in prenatal brachyrrhine mice. Teratology 47:321–332. [DOI] [PubMed] [Google Scholar]
- Ma W, Lozanoff S. 1996. Morphological deficiency in the prenatal anterior cranial base of midfacially retrognathic mice. J Anat 188:547–555. [PMC free article] [PubMed] [Google Scholar]
- Mavrogiannis LA, Antonopoulou I, Baxova A, Kutilek S, Kim CA, Sugayama SM, Salamanca A, Wall SA, Morriss-Kay GM, Wilkie AO. 2001. Haploinsufficiency of the human homeobox gene ALX4 causes skull ossification defects. Nat Genet 27:17–18. [DOI] [PubMed] [Google Scholar]
- McBratney BM, Margaryan E, Ma W, Urban Z, Lozanoff S. 2003. Frontonasal dysplasia in 3H1 Br/Br mice. Anat Rec A Discov Mol Cell Evol Biol 271:291–302. [DOI] [PubMed] [Google Scholar]
- Ruf RG, Xu PX, Silvius D, Otto EA, Beekmann F, Muerb UT, Kumar S, Neuhaus TJ, Kemper MJ, Raymond RM Jr, Brophy PD, Berkman J, Gattas M, Hyland V, Ruf EM, Schwartz C, Chang EH, Smith RJ, Stratakis CA, Weil D, Petit C, Hildebrandt F. 2004. SIX1 mutations cause branchio-oto-renal syndrome by disruption of EYA1-SIX1-DNA complexes. Proc Natl Acad Sci USA 101:8090–8095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Self M, Lagutin OV, Bowling B, Hendrix J, Cai Y, Dressler GR, Oliver G. 2006. Six2 is required for suppression of nephrogenesis and progenitor renewal in the developing kidney. EMBO J 25:5214–5228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serikaku MA, O’Tousa JE. 1994. Sine oculis is a homeobox gene required for Drosophila visual system development. Genetics 138:1137–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Berge D, Brouwer A, el Bahi S, Guenet JL, Robert B, Meijlink F. 1998. Mouse Alx3: An aristaless-like homeobox gene expressed during embryogenesis in ectomesenchyme and lateral plate mesoderm. Dev Biol 199:11–25. [DOI] [PubMed] [Google Scholar]
- Twigg SR, Kan R, Babbs C, Bochukova EG, Robertson SP, Wall SA, Morriss-Kay GM, Wilkie AO. 2004. Mutations of ephrin-B1 (EFNB1), a marker of tissue boundary formation, cause craniofrontonasal syndrome. Proc Natl Acad Sci USA 101:8652–8657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twigg SR, Versnel SL, Nurnberg G, Lees MM, Bhat M, Hammond P, Hennekam RC, Hoogeboom AJ, Hurst JA, Johnson D, Robinson AA, Scambler PJ, Gerrelli D, Nurnberg P, Mathijssen IM, Wilkie AO. 2009. Frontorhiny, a distinctive presentation of frontonasal dysplasia caused by recessive mutations in the ALX3 homeobox gene. Am J Hum Genet 84:698–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uz E, Alanay Y, Aktas D, Vargel I, Gucer S, Tuncbilek G, von Eggeling F, Yilmaz E, Deren O, Posorski N, Ozdag H, Liehr T, Balci S, Alikasifoglu M, Wollnik B, Akarsu NA. 2010. Disruption of ALX1 causes extreme microphthalmia and severe facial clefting: Expanding the spectrum of autosomal-recessive ALX-related frontonasal dysplasia. Am J Hum Genet 86:789–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber S, Taylor JC, Winyard P, Baker KF, Sullivan-Brown J, Schild R, Knuppel T, Zurowska AM, Caldas-Alfonso A, Litwin M, Emre S, Ghiggeri GM, Bakkaloglu A, Mehls O, Antignac C, Network E, Schaefer F, Burdine RD. 2008. SIX2 and BMP4 mutations associate with anomalous kidney development. J Am Soc Nephrol 19:891–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieland I, Jakubiczka S, Muschke P, Cohen M, Thiele H, Gerlach KL, Adams RH, Wieacker P. 2004. Mutations of the ephrin-B1 gene cause craniofrontonasal syndrome. Am J Hum Genet 74:1209–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yallowitz AR, Gong KQ, Swinehart IT, Nelson LT, Wellik DM. 2009. Non-homeodomain regions of Hox proteins mediate activation versus repression of Six2 via a single enhancer site in vivo. Dev Biol 335:156–165. [DOI] [PMC free article] [PubMed] [Google Scholar]