

Abstract
Background
Targeted agents have improved the outcome of a subset of non‐small cell lung cancer (NSCLC). Molecular profiling by next‐generation sequencing (NGS) allows screening for multiple genetic alterations both in tissue and in plasma, but limited data are available concerning its feasibility and impact in real‐world clinical practice.
Methods
Patients with advanced NSCLC consecutively referring to our Institution for potential eligibility to VISION trial (NCT02864992) were prospectively enrolled. They were already screened with standard method, and EGFR/ALK/ROS‐1 positive cases were excluded. NGS was performed in plasma and tissue using the Guardant360 test covering 73 genes and the Oncomine Focus Assay covering 59 genes, respectively.
Results
The study included 235 patients. NGS was performed in plasma in 209 (88.9%) cases; 78 of these (37.3%) were evaluated also in tissue; tissue only was analyzed in 26 cases (11.1%). Half of the tissue samples were deemed not evaluable. Druggable alterations were detected in 13 (25%) out of 52 evaluable samples and 31 of 209 (14.8%) of plasma samples. Improved outcome was observed for patients with druggable alterations if treated with matched targeted agents: they had a longer median overall survival (not reached) compared with the ones who did not start any targeted therapy (9.1 months; 95% confidence interval, 4.6–13.6; p = .046). The results of NGS testing potentially also affected the outcome of patients treated with immunotherapy.
Conclusion
Systematic real‐life NGS testing showed the limit of tissue analysis in NSCLC and highlighted the potentiality of genetic characterization in plasma in increasing the number of patients who may benefit from NGS screening, both influencing the clinical decision‐making process and affecting treatment outcome.
Implications for Practice
Genetic characterization of cancer has become more important with time, having had positive implications for treatment specificity and efficacy. Such analyses changed the natural history of advanced non‐small cell lung cancer (aNSCLC) with the introduction of drugs targeted to specific gene alterations (e.g., EGFR mutations, ALK and ROS‐1 rearrangements). In the field of cancer molecular characterization, the applicability of the analysis of a wide panel of genes using a high‐throughput sequencing approach, such as next‐generation sequencing (NGS), is still a matter of research. This study used NGS in a real‐world setting to systematically and prospectively profile patients with aNSCLC. The aim was to evaluate its feasibility and reliability, as well as consequent access to targeted agents and impact on clinical outcome whenever a druggable alteration was detected either in tumor tissue samples or through liquid biopsy.
Keywords: Oncogene addiction, Actionable genetic alterations, Genetic characterization, Next‐generation sequencing, Liquid biopsy
Short abstract
The availability of targeted therapies has profoundly changed the diagnostic and therapeutic pathways in solid tumors, in particular in advanced non‐small cell lung cancer; however, limited data are available concerning the real impact of next‐generation sequencing plasma characterization in clinical practice. This article reports a real‐world experience, aiming to analyze actual effect on clinical outcome.
Introduction
The availability of targeted therapies has profoundly changed the diagnostic and therapeutic pathways in solid tumors, especially in advanced non‐small cell lung cancer (aNSCLC). In several instances, therapeutic algorithms based on genetic characterization have led to the use of on‐label targeted agents in a subset of patients with oncogene‐addicted tumors. Until recently most molecular information used for clinical decision has stemmed from single genetic alteration testing [1, 2, 3, 4, 5, 6, 7], whereas the potentialities of wide genetic screening in clinical practice are now emerging [8, 9, 10].
In aNSCLC, according to the European Society for Medical Oncology clinical practice guidelines, testing for EGFR mutations and rearrangements involving ALK and ROS‐1 genes are considered mandatory, and BRAF V600E mutations are rapidly approaching this status, as first‐line BRAF/MEK inhibitors have been approved by some regulatory agencies. HER2, MET exon 14 mutations, and gene fusions involving RET and NTRK‐1 are rapidly gaining the role of potentially interesting targets [11]. The molecular testing guidelines of the American Society of Clinical Oncology also highlight the importance of BRAF testing in patients with advanced lung adenocarcinoma (LADC) [12, 13] as well as testing RET, HER2, KRAS, and MET as part of larger panels.
Next‐generation sequencing (NGS) allows screening for multiple genetic alterations and can be performed both in tissue and in blood [14]. The real clinical impact of systematic NGS analysis in aNSCLC compared with standard methods is still under investigation.
Tissue biopsies represent the current standard for molecular testing, but availability of adequate material to perform a wide genetic characterization is challenging in non‐small cell lung cancer (NSCLC). Even considering standard molecular testing, tissue biopsies are not diagnostic in 20% to 30% of patients, highlighting the need for alternative sources of tumor DNA [14, 15].
Circulating tumor DNA (ctDNA) is a subset of cell‐free DNA (cfDNA) that can be found in plasma and represents genetic material both from the primary tumor and from metastases [16, 17]. Plasma ctDNA harboring specific somatic mutations is relatively specific for cancer; it might be usefully representative of tumor burden and mirror intratumor heterogeneity [17, 18]. Thus, cfDNA analysis holds considerable promise as a surrogate marker in multiple contexts: monitoring response to local or systemic therapies, definition of minimal residual disease and risk of relapse, following radical‐intent treatments, and identifying of acquired drug‐resistance mechanisms [19, 20, 21].
However, limited data are available concerning the real impact of NGS plasma characterization in clinical practice and the correct application of NGS results for the management of patients with NSCLC [22, 23].
The present article reports a real‐world prospective experience of systematic NGS analysis in patients with aNSCLC with the aim of analyzing feasibility, reliability, access to targeted agents, and impact on clinical outcome.
Materials and Methods
Patients and Molecular Analyses
From August 2017 to September 2019, patients with stage IIIB/IV NSCLC treated at Rete Oncologica Veneta and referring to our institution for potential eligibility for the VISION trial (NCT02864992) were prospectively screened, and clinical data were collected. VISION is a single‐arm, open‐label, phase II trial, aimed to assess activity and tolerability of tepotinib, a highly selective small molecule inhibitor of c‐Met in patients with aNSCLC harboring MET exon 14 skipping alterations or MET amplification.
Patients’ data recorded at baseline included patient demographics, Eastern Cooperative Oncology Group performance status (ECOG PS) at time of first‐line systemic treatment start, smoking history, and weight loss of more than 5% during the 6 months before cancer diagnosis [24]. Tumor data collected included histology; EGFR, ALK, and ROS‐1 status as they determined for LADCs; programmed death ligand 1 (PD‐L1) status, when available; and radiological staging. Information about treatments undergone during the course of the disease and their response were collected.
The ethics committee of our institution approved the study on February 20, 2017. The latest version of the protocol was approved by the ethics committee of our institution on the December 6, 2018.
Written informed consent was signed before any trial‐related activities were carried out. Prescreening informed consent was obtained prior to NGS testing in tumor tissue and/or plasma. The prescreening informed consent approved by our ethics committee was version 3.0. The study was performed in accordance with the Declaration of Helsinki.
Tumor tissue for NGS testing was obtained from archived samples or from freshly obtained formalin‐fixed paraffin‐embedded (FFPE) tumor tissue. ctDNA was isolated and tested from freshly collected plasma samples.
MET alterations were searched in either plasma samples or tissue tumor samples, using the Guardant360 test (Guardant Health, Redwood City, CA), covering 73 genes and all somatic alterations recognized as potential targets by National Comprehensive Cancer Network, and Oncomine Focus Assay (Thermo Fisher Scientific, Waltham, MA; testing done by MolecularMD Inc., Portland, OR) covering 59 genes, respectively.
Additional details about inclusion criteria, study procedures, and protocol are provided in the supplemental online material.
Statistical Analysis
The primary aim of the study was to describe the feasibility of routine use of multiple genetic testing by NGS in aNSCLC and its impact on clinical management in real‐world clinical practice.
After assessing feasibility, we analyzed the impact of the presence of potentially druggable alterations (not previously found with standard methods) on outcome according to the access to matched targeted drugs.
The secondary aims of the study were to explore the association of the presence of druggable alterations with outcome in the whole study population and among patients treated with immunotherapy.
Because of the observational exploratory nature of the work, a sample size was not calculated.
Druggable alterations according to NGS results were defined as EGFR sensitizing mutations or deletions, ALK rearrangements (that were not identified with standard diagnostics), RET fusions, ROS‐1 translocations, MET exon 14 skipping mutations, MET amplifications, HER2 exon 20 mutations, HER2 amplifications, and BRAF V600E mutations [25].
Progression‐free survival (PFS) was calculated from the first day of systemic treatment to the first radiological or clinical disease progression (PD) or death from any cause. Overall survival (OS) was calculated from the first day of any systemic treatment to death from any cause. Radiological response (RR) was assessed according to RECIST version 1.1; disease control rate was defined as complete response plus partial response (PR) plus stable disease (SD).
Variables were presented by using median value for continuous variables and percentages (numbers) for categorical variables and their relationship with the presence of target alteration was assessed using the Mann‐Whitney test, Kruskal‐Wallis test, and the chi‐squared test as appropriate.
Univariate logistic regression models and results were reported using odds ratios (ORs) with their 95% confidence intervals (CIs). Median PFS and OS were estimated by using Kaplan‐Meier methods, and a log‐rank test was used to compare survival between groups. Hazard ratios (HRs) and their 95% CIs were calculated with the Cox regression method. Statistical significance level was set at p < .05 for all tests. All statistical analyses were performed with SPSS version 20.0 software (SPSS Inc., Chicago, IL).
Results
Patients and Treatments
A total of 235 patients were included: 130 (55.3%) men and 105 (44.7%) women. Clinical‐pathological features of the study population are summarized in Table 1. Median follow‐up time was 11.7 (range, 1.6–26.6) months; 54.9% of enrolled patients was still alive at the time of this analysis.
Table 1.
Patients’ clinical features
| Variable | Patients’ population (n = 235), n (%) |
|---|---|
| Gender | |
| Male | 130 (55.3) |
| Female | 105 (44.7) |
| Age at diagnosis of aNSCLC, median (range), years | 68.5 (31.8–88.2) |
| Performance status | |
| 0 | 78 (33.2) |
| ≥1 | 157 (66.8) |
| Weight loss | 40 (17.0) |
| Smoking status | |
| Never smoker | 63 (26.8) |
| Active smoker | 54 (23.0) |
| Former smoker | 118 (50.2) |
| Cancer histology | |
| Adenocarcinoma | 204 (86.8) |
| Squamous cell carcinoma | 12 (5.1) |
| Not otherwise specified carcinoma | 12 (5.1) |
| Sarcomatoid carcinoma | 3 (1.3) |
| Large cell carcinoma | 2 (0.9) |
| Adenosquamous carcinoma | 2 (0.9) |
| Number of metastatic sites | |
| 1 | 100 (42.6) |
| 2 | 81 (34.5) |
| 3 | 38 (16.2) |
| 4 | 11 (4.7) |
| Extrathoracic metastases | 118 (50.2) |
| Liver metastasis | 26 (11.1) |
| Bone metastasis | 64 (27.2) |
| PD‐L1 testing | |
| Not done | 34 (14.5) |
| Done | 201 (85.5) |
| PD‐L1 status cutoff 1% | |
| <1% | 104 (51.7) |
| ≥1% | 97 (48.3) |
| PD‐L1 status cutoff 50% | |
| <50% | 152 (75.6) |
| ≥50% | 49 (24.4) |
| Total number of systemic treatments | |
| 0 | 20 (8.5) |
| 1 | 86 (36.6) |
| 2 | 94 (40.0) |
| 3 | 20 (8.5) |
| 4 | 10 (4.3) |
| 5 | 4 (1.7) |
| 6 | 1 (0.4) |
Abbreviations: aNSCLC, advanced non‐small cell lung cancer; PD‐L1, programmed death ligand 1.
First‐line systemic therapy was started in 215 cases (91.5%): 165 (76.7%) received a platinum‐based doublet, 35 (16.3%) were treated with pembrolizumab, 9 (4.2%) were eligible only for single‐agent chemotherapy because of comorbidities, and 2 (0.9%) started targeted treatment after the results of the NGS testing (supplemental online Table 1).
Among the 20 patients who did not undergo any systemic treatment, 6 patients (2.6%) received first‐line locoregional treatments for oligometastatic disease, and 14 (5.9%) patients experienced a fast decline of performance status, preventing the chance of any kind of active treatment.
Seventy‐one of 129 (55.0%), 10 of 36 (27.8%), and 2 of 15 (13.3%) patients were treated with immune‐checkpoint inhibitors (ICIs) in the second, third, and fourth line of systemic treatment, respectively. Further details on the administered therapies and patients’ responses are shown in supplemental online Table 1.
NGS Analyses
Molecular analyses were performed exclusively in tumor tissue in 26 cases (11.1%), in plasma in 131 cases (55.7%), and both in tissue and plasma in 78 cases (33.2%).
Overall, tumor tissue was analyzed in 104 cases: 18 samples (17.3%) were obtained from archived surgical tissue, whereas 86 (82.7%) were obtained from FFPE biopsy tissue. Fifty‐two samples (50.0%) were defined as not evaluable by central pathology review because of insufficient tumor tissue available. Almost all not‐evaluable samples were obtained from nonsurgical biopsies (49 of 52; 94.2%), whereas 17 of 52 (32.7%) of evaluable samples were surgical samples. Median time needed to receive the report of druggable MET alterations and to have the complete NGS testing report was 8 (range, 1–29) and 11 (range, 1–31) working days, respectively.
Plasma samples of 209 patients were analyzed, and all samples were evaluable. Twenty‐nine samples were found negative for the presence of any genetic alterations: among these, eight patients underwent liquid biopsy before any systemic treatment. Seven patients were tested twice, at the time of PD to sequential lines of systemic treatment. The first assessment for five of these cases showed no detectable tumor‐related somatic alterations; three of five turned out positive for somatic genetic alterations at subsequent plasma testing performed after PD. Median time needed to have the complete NGS testing report was 10 working days (range, 4–29).
Seventy‐eight patients were tested both in tissue and in plasma: 43 tissue tumor samples were not suitable for NGS testing, and 4 showed no evidence of oncogenic alterations; 14 plasma samples showed no detectable alterations. Among the remaining 28 cases, 20 genetic alterations were detected in tissue and 14 confirmed in plasma. Four of eight druggable alterations were found both in tissue and in plasma.
Thirty‐two of 209 patients (15.3%) tested in plasma were found positive for potentially druggable alterations; nine patients had also tissue evaluable, and the alterations were confirmed in four cases.
Details about genetic alterations found in tissue and plasma are summarized in supplemental online Figure 1.
Druggable Alterations: Tissue Analyses
A druggable oncogenic alteration was found in 13 patients (24.5% of evaluable samples) in tumor. We found four KIF5B‐RET translocations, three BRAF V600E mutations, one MET exon 14 fusion skipping alteration, one case of MET exon 14 fusion skipping alteration and MET gene amplification, one common sensitizing HER2 exon 20 insertion (G776delinsVC), and one HER2 amplification, later confirmed using fluorescence in situ hybridization (FISH) test. Besides these 11 cases, an EZR–ROS‐1 translocation was detected with NGS in a former smoker (16 pack‐years) male patient with 3+ for ROS‐1 at immunohistochemistry; confirmative FISH diagnostic for this patient turned out not evaluable. An EGFR sensitizing mutation on exon 21 was also found: this patient's tissue had not been routinely tested for EGFR before because the patient had a light smoking habit and a squamous cell carcinoma.
Druggable Alterations: Plasma Analyses
Plasma NGS allowed detection of druggable oncogenic driver alterations in 32 of 209 patients (15.3%).
EGFR sensitizing alterations were found in four cases. Two patients were diagnosed with squamous cell carcinoma, had previous smoking habits, and had not been tested on tissue for EGFR; one did not have enough tissue available to perform the test. These three patients were found positive by real‐time polymerase chain reaction (RT‐PCR) testing after additional confirmatory liquid biopsy. Another patient carried an uncommon exon 20 mutation (A767_V769dup), which was not confirmed either in tissue, using RT‐PCR, or in liquid biopsy, also by using RT‐PCR.
ALK translocations were observed in three cases, previously negative at immunohistochemistry. In two of them, subsequent FISH testing for ALK rearrangement was deemed not evaluable. In the other, material was insufficient to perform FISH testing, and the patient underwent rebiopsy: the sample was evaluable and positive for ALK rearrangement.
KIF5B‐RET translocation was detected in two cases: one was also analyzed and confirmed in tissue.
Among four patients positive for BRAF V600E mutation in plasma, one had also tissue tested, and the alteration was confirmed.
Fourteen patients carried MET alterations: seven patients had MET exon 14 fusion skipping alteration, five had MET gene amplification, and two had both these alterations (one also positive in tissue).
Four patients carried common sensitizing exon 20 insertion involving HER2 gene (G776delinsVC); one of them had the same mutation in tissue.
CD74–ROS‐1 gene fusion was detected in one patient, who was diagnosed of LADC, immunohistochemically negative for ROS‐1 protein. Subsequent FISH testing was deemed not evaluable.
Clinical and Pathological Features of Patients Carrying a Potentially Druggable Genetic Alteration
In the whole study population, being a current or a former smoker was associated with lower chance of carrying druggable molecular alterations (OR, 0.21; 95% CI, 0.10–0.42; p < .001). No other clinical features, such as gender or age, showed such a correlation (OR, 1.22; 95% CI, 0.62–2.41; p = .557 for female; OR, 1.00; 95% CI, 0.97–1.03; p = .979 for older age; supplemental online Table 2).
The distribution of PD‐L1 tumor proportion score values showed higher values for the subgroup of patients carrying druggable alterations (p = .037, Mann‐Whitney test).
Druggable Alterations and Targeted Treatments
Five patients were positive for RET rearrangement; three of them received off‐label cabozantinib, experiencing SD as best RR. One of them is still on treatment after 76 weeks. The remaining two patients died because of rapid progression while on ICI treatment and never received targeted therapy.
BRAF V600E mutation was found in six cases: two died before starting any treatment, one died after first‐line chemotherapy, one is receiving second‐line ICI treatment, and two started off‐label dabrafenib and trametinib. These two patients experienced a good disease control, still ongoing after 37 and 64 weeks, respectively.
Druggable MET alterations were detected in 15 cases. Four patients received tepotinib as part of the VISION clinical trial, and RR cannot be disclosed yet. Five patients are undergoing first‐line therapy, and six either died or experienced a fast decline of performance status that prevented any other active treatment.
Somatic gene alterations involving HER2 gene were found in five patients: three died after completion of first‐line chemotherapy, and two are receiving second‐line standard systemic treatment.
Two patients tested positive for ROS‐1 gene rearrangement; one is still receiving maintenance pemetrexed, whereas the other had clinical benefit from crizotinib for 38 weeks.
Among the five patients with EGFR alterations, four began EGFR tyrosine kinase inhibition: two achieved SD and two PR as best RR. The one with the uncommon exon 20 insertion is still receiving first‐line chemotherapy.
Three patients were positive for ALK rearrangement; two are currently receiving alectinib after 15 and 5 weeks, respectively, whereas one is still on treatment with first‐line chemotherapy.
Details about response to targeted treatments and duration of clinical benefit in patients with a potentially druggable alteration are summarized in Figure 1.
Figure 1.
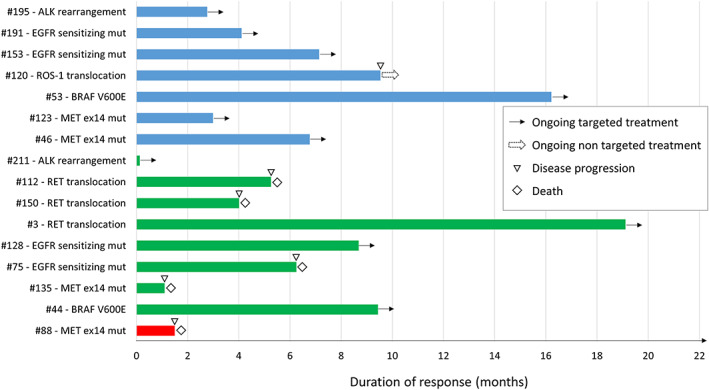
Response to targeted agents among patients found positive for potentially druggable alterations after next‐generation sequencing analysis in tissue or in plasma (previously screened for EGFR/ALK/ROS‐1 with standard method and found negative or not evaluable). In blue, patients who achieved partial response as best response, in green patients with stable disease as best response and in red patients who experienced disease progression as best response. Abbreviations: ex14, exon 14; mut, mutation.
Druggable Alterations and Immune‐Checkpoint Inhibitors
Four of five patients carrying RET rearrangement received prior treatment with ICIs, achieving one PR and four PD as best RR.
Among patients carrying BRAF V600E mutation, one has just started second‐line treatment with pembrolizumab, whereas another patient was treated with nivolumab prior to targeted therapy, obtaining SD for over 9 months.
ICIs were part of prior oncologic treatment for 10 patients with MET alterations, and best responses recorded were four PR, four SD, and two PD.
No patients with either somatic gene alterations involving HER2 gene or ROS‐1 gene rearrangement ever received ICIs. Conversely, immunotherapy was administered in one of three patients with ALK translocation, having PD as best response, and in three of five patients carrying EGFR sensitizing alterations, with their best response being one stability and two PD.
Survival Analyses
Median OS of the study population was 21.8 (95% CI, 17.9–25.7) months (Fig. 2).
Figure 2.
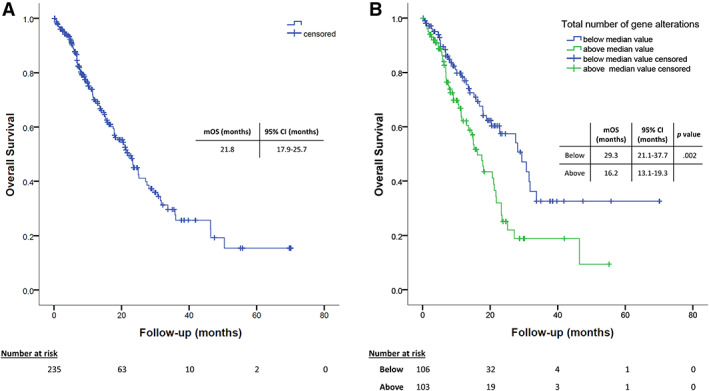
Survival analyses. (A): Estimation of mOS of the whole study population. (B): Estimation of mOS according to the number of genetic alterations found in plasma, dichotomized according to the median number of alterations (median value, 2). Abbreviations: CI, confidence interval; mOS, median overall survival.
Impact on OS of gender, age at diagnosis, smoking status, ECOG PS, weight loss before cancer diagnosis, histology, presence of druggable target alterations, and number and subtype of metastatic sites were evaluated in univariate analysis (supplemental online Table 3). ECOG PS at diagnosis of stage IV disease (HR, 1.93; 95% CI, 1.28–2.93; p = .002), weight loss (HR, 2.43; 95% CI, 1.54–3.93; p < .001), number of metastatic sites (HR, 1.41; 95% CI, 1.14–1.74; p = .002), and having bone (HR, 2.26; 95% CI, 1.49–3.42; p < .001) or liver (HR, 1.79; 95% CI, 1.01–3.17; p = .047) or extrathoracic metastasis (HR, 1.53; 95% CI, 1.03–2.28; p = .038) had a significant impact on OS. ECOG PS, weight loss, and bone metastasis were confirmed to independently affect OS in multivariate analysis (HR, 1.56; 95% CI, 1.04–2.36; p = .033; HR, 2.46; 95% CI, 1.53–3.96; p < .001 and HR, 2.22; 95% CI, 1.30–3.78; p = .0013, respectively).
The same analyses were performed in the 209 patients with plasma samples tested, confirming the results of the whole population (supplemental online Table 4).
The presence of potentially druggable alterations had no significant impact on outcome: median OS was 22.8 (95% CI, 19.1–26.5) months in the absence of druggable alterations versus 18 (95% CI, 3.84–32.2) months among positive patients (p = .179; Fig. 3A). Of interest, patients carrying druggable alterations who started a matched targeted agent had longer median OS (not reached), compared with the ones who did not (9.1 months; 95% CI, 4.6–13.6; p = .046, log‐rank test; Fig. 3B). Although the difference was not statistically significant, patients with druggable alterations who started a matched targeted agent showed a numerically longer median OS (not reached) in comparison with the rest of the study population (21.7 months; 95% CI, 17.4–25.9; p = .173, log‐rank test).
Figure 3.
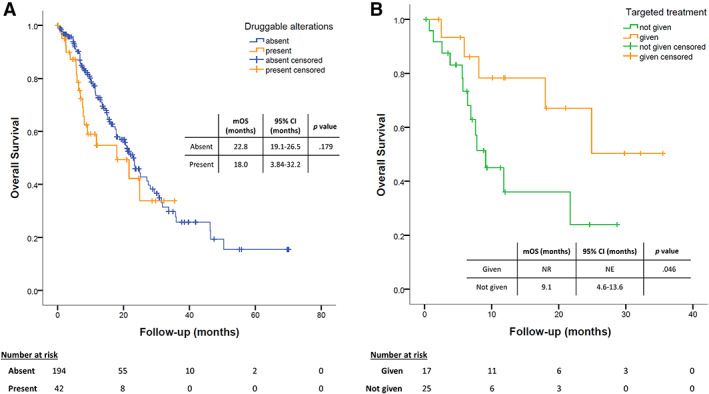
Survival analyses. (A): Estimation of overall survival according to the presence of a potentially druggable genetic alteration found by next‐generation sequencing analyses. (B): Kaplan‐Meier comparison of patients who were found positive for a potentially druggable alteration and received targeted agents accordingly versus patients who did not. Abbreviations: CI, confidence interval; mOS, median overall survival; NE, not evaluable; NR, not reached.
In the subgroup of patients who received immunotherapy and who had their plasma tested before ICI (n = 69), those who carried a druggable molecular alteration had a median PFS comparable to the others (6.5 months; 95% CI, 0.3–12.8 vs. 5.8 months; 95% CI, 3.1–8.5; p = .316) but showed a trend for a shorter median OS: 8.1 (95% CI, 9.8–9.4) versus 22.8 months (95% CI, 17.6–27.9; p = .133, log‐rank test; HR, 2.01; 95% CI, 0.78–5.68; p = .143, Cox regression test; Fig. 4), although it was not statistically significant. On the other side, different patterns of response have been shown according to the kind of potentially druggable alteration found, as described above (Fig. 5).
Figure 4.
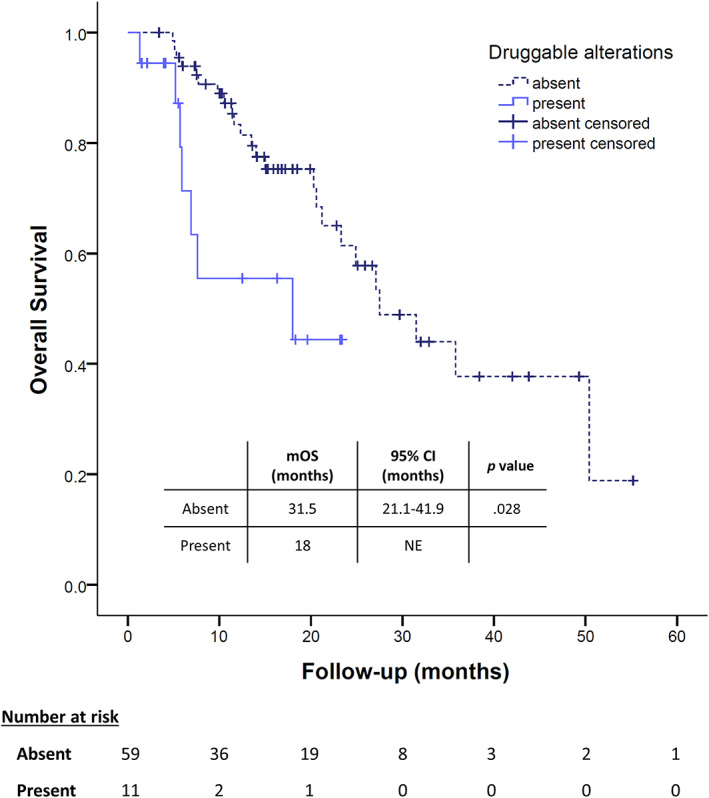
Overall survival analyses among patients who received immunotherapy during the course of the disease according to the presence of a potentially druggable alteration. Abbreviations: CI, confidence interval; mOS, median overall survival; NE, not evaluable.
Figure 5.
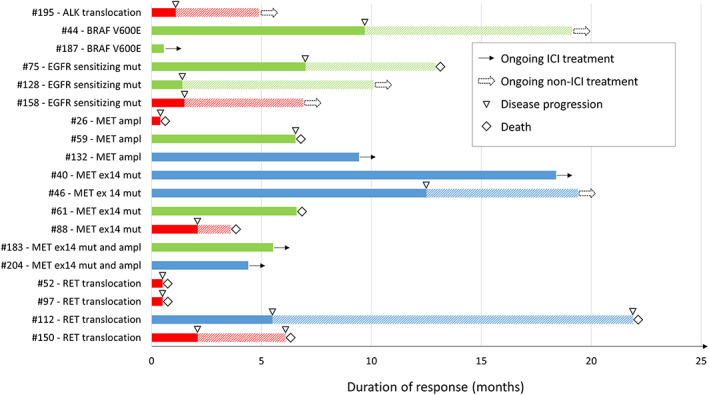
Response to ICI agents among patients found positive for potentially druggable alterations after next‐generation sequencing analysis in tissue or in plasma (previously screened for EGFR/ALK/ROS‐1 with standard method and found negative or not evaluable). In blue, patients who achieved partial response as best response, in green patients with stable disease as best response and in red patients who experienced disease progression as best response. Abbreviations: ampl, amplification; ex14, exon 14; ICI, immune‐checkpoint inhibitor; mut, mutation.
Discussion
Molecular characterization of aNSCLC has led to considerable clinical benefit for patients carrying molecular alterations for which targeted agents are available [11, 12, 13, 26]. Indeed, as an example, patients with ROS‐1 rearrangement treated with crizotinib reached a median OS of 51 months [27], a result considered unreachable before the introduction of targeted agents.
Routine molecular testing in lung cancer is performed in tissue, and analysis includes molecular alterations with matched drugs available in clinical practice. In parallel, less invasive diagnostic procedures to obtain tumor samples are increasingly used in the context of a disease for which surgical approach is rare [28, 29], whereas extensive molecular testing for potentially druggable alterations [30, 31] might also allow the development of more personalized treatments also for patients with wild type EGFR/ALK/ROS‐1. This promising approach can be further fostered by the use of NGS analysis in plasma, which can overcome the limitation of tissue availability, also allowing for dynamic evaluation during treatment [21, 32].
Outside of limited reports, evidence that this approach can have clinical impact on real‐world patients is still debatable [20, 33, 34]. In 2014 a pivotal work showed the benefit of multiple molecular testing for LADC in clinical practice and the impact of targeted agents in this setting [35]. However, it must be stressed that among the molecular alterations tested, Kris and colleagues included EGFR and ALK sensitizing alterations, which are now routinely tested in current clinical practice, and other driver mutations, such as AKT1, MEK1, or NRAS, that are currently orphans of a targeted treatment [35]. On the other hand, more recent studies including different solid tumors showed that increasing genetic information and offering off‐label drugs might not be translated into clinical benefit for patients with cancer [36, 37].
Our study is based on real‐world data collection aiming to describe the impact of genetic characterization through NGS performed in plasma and in tissue in clinical practice. Real‐world evidence, although characterized by several methodological limitations, can provide useful information about reliability and transferability of molecular diagnostic in clinical practice, thus helping in planning new organizational models to better apply NGS screening in clinical practice [38].
In our study, we first tested the feasibility of the analysis in clinical practice, showing that tissue genetic characterization was feasible in only 50% of samples. Moreover, 32.7% of these evaluable cases came from surgical samples, underlining the difficulties of obtaining material suitable for NGS analysis from biopsies in real‐life patients. On the contrary, liquid biopsy was always evaluable; however, 29 samples were found negative for the presence of any genetic alterations. This potential limit can be related to tumor burden and tumor shedding [39, 40]. This consideration is supported by our finding that genetic alterations were found by repeating liquid biopsy at the moment of progression in three of five negative cases. These observations confirm that plasma NGS is feasible in clinical practice, opening the opportunity to apply this technique for disease monitoring evaluation of tumor genetic heterogeneity and early detection of acquired treatment resistance mechanisms [32, 41, 42]. The feasibility of NGS analysis in plasma in NSCLC was also recently evaluated in other series including a comparable number of patients [22, 43, 44].
In our series, we also highlighted another potential limitation of performing genetic screening only in plasma. Tumor heterogeneity and disease burden, in addition to potential technical issues, might account for potential discrepancy between tissue and plasma results. The number of patients having both tissue and plasma available was not sufficient to draw any conclusions on the topic. Overall, whenever feasible, both tissue and plasma NGS should still be performed.
After assessing feasibility and reliability, our data allowed us to estimate the clinical impact of multigene screening in a real‐world setting. First of all, we observed that even in a population already tested for the presence of EGFR, ALK, and ROS‐1 sensitizing alterations with standard techniques, NGS was able to find about 18% of patients carrying potentially druggable alterations. The identification of a potentially druggable alteration and the availability of a matched agent can have a significant impact on prognosis (Fig. 3). Indeed, in the presence of druggable alterations, only patients treated with targeted agents had improved outcome.
These observations are potentially relevant for clinical practice, even though the analysis is not able to fully exclude intrinsic biological differences related to different potentially druggable alterations. Even selecting genetic alterations with matched targeted drugs potentially available, we could not establish the potential influence of intrinsic prognostic value of each alteration.
Unfortunately, in our experience, a matched targeted treatment was available only for a limited subset of the study population: 39% of the patients carrying a potentially druggable alteration and 6.8% of the overall study population (including also the not evaluable samples, which were 50% of the tissue samples). This low rate is due to many factors, mirroring real‐world clinical potential limitations to this approach. First of all, patients have been screened but started systemic treatment according to clinical practice, and several patients carrying potentially druggable alterations are still on standard treatment and might be subsequently eligible. Some patients also experienced a rapid decline of clinical conditions preventing any further treatments. Limited access to matched targeted drugs is also due to limitations for the off‐label use of targeted agents and limited availability of clinical trials for rare molecular alterations. Notably, however, three patients who were classified with standard methods as having wild‐type tumors turned to be positive for EGFR/ALK/ROS‐1 alterations and received the matched approved drug after NGS testing.
In addition to the potential for matched targeted agents, plasma NGS may allow the identification of patients at different prognosis or might influence sensitivity to other treatments, thus potentially impacting on clinical decision‐making process. In our experience, a higher number of genetic alterations has been associated with worse prognosis (Fig. 2B). Moreover, the presence of a potentially druggable alteration has been associated with higher level of PD‐L1 expression and with a trend for shorter survival in patients treated with ICIs, even though heterogeneity of response and duration was found (Fig. 5). These observations are in line with prior reports and highlight the potentialities of liquid biopsy in clinical practice in evaluating indication and timing for immunotherapy [45, 46].
Our data also highlight the importance of formally established molecular tumor boards in cancer institutes and networks of community hospitals to define criteria for patient selection, material to be tested and tests to be used, interpretation of results, access to matched therapies, and clinical outcomes [47, 48]. This need stems from the complexity of analyzing and interpreting the results of multigene NGS analysis in clinical practice, the issues related to access to targeted drugs, and the importance of integrating molecular information in diagnostic‐therapeutic pathways of patients with aNSCLC.
Conclusion
We describe a real‐world experience with NGS analysis systematically and prospectively performed in patients with aNSCLC, providing insight into its feasibility and its potential clinical implications, including the effective use of matched targeted agents, but also its potential limitations in clinical practice of the near future.
Author Contributions
Conception/design: Laura Bonanno, PierFranco Conte
Provision of study material or patients: Laura Bonanno, Lorenzo Calvetti, Stefano Frega, Giulia Pasello
Collection and/or assembly of data: Laura Bonanno, Alberto Pavan, Alessandra Ferro, Lorenzo Calvetti
Data analysis and interpretation: Laura Bonanno, Alberto Pavan
Manuscript writing: Laura Bonanno, Alberto Pavan, Alessandra Ferro
Final approval of manuscript: Laura Bonanno, Alberto Pavan, Alessandra Ferro, Lorenzo Calvetti, Stefano Frega, Giulia Pasello, Giuseppe Aprile, Valentina Guarneri, PierFranco Conte
Disclosures
The authors indicated no financial relationships.
Supporting information
See http://www.TheOncologist.com for supplemental material available online.
Appendix S1: Supplementary Information
Acknowledgments
The molecular analyses were performed for the VISION trial, and they were all provided by Merck. The consent for publication of the molecular data and correlation with clinical features and outcome has been provided by Merck Medical Direction. Merck was allowed to review the manuscript but was not involved in this project more than providing the molecular analyses as part of the screening for the VISION trial.
Disclosures of potential conflicts of interest may be found at the end of this article.
No part of this article may be reproduced, stored, or transmitted in any form or for any means without the prior permission in writing from the copyright holder. For information on purchasing reprints contact Commercialreprints@wiley.com. For permission information contact permissions@wiley.com.
References
- 1. Yates LR, Seoane J, Le Tourneau C et al. The European Society for Medical Oncology (ESMO) Precision Medicine Glossary. Ann Oncol 2018;29:30–35. [DOI] [PubMed] [Google Scholar]
- 2. Lindeman NI, Cagle PT, Aisner DL et al. Updated molecular testing guideline for the selection of lung cancer patients for treatment with targeted tyrosine kinase inhibitors: Guideline from the College of American Pathologists, the International Association for the Study of Lung Cancer, and the Association for Molecular Pathology. Arch Pathol Lab Med 2018;142:321–346. [DOI] [PubMed] [Google Scholar]
- 3. Bang YJ, Van Cutsem E, Feyereislova A et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2‐positive advanced gastric or gastro‐oesophageal junction cancer (ToGA): A phase 3, open‐label, randomised controlled trial. Lancet 2010;376:687–697. [DOI] [PubMed] [Google Scholar]
- 4. Demetri GD, von Mehren M, Blanke CD et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med 2002;347:472–480. [DOI] [PubMed] [Google Scholar]
- 5. Hauschild A, Grob JJ, Demidov L V et al. Dabrafenib in BRAF‐mutated metastatic melanoma: A multicentre, open‐label, phase 3 randomised controlled trial. Lancet 2012;380:358–365. [DOI] [PubMed] [Google Scholar]
- 6. Robson M, Im SA, Senkus E et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med 2017;377:523–533. [DOI] [PubMed] [Google Scholar]
- 7. Kaufman B, Shapira‐Frommer R, Schmutzler RK et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol 2015;33:244–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schwaederle M, Zhao M, Lee JJ et al. Association of biomarker‐based treatment strategies with response rates and progression‐free survival in refractory malignant neoplasms. JAMA Oncol 2016;2:1452. [DOI] [PubMed] [Google Scholar]
- 9. Haslem DS, Van Norman SB, Fulde G et al. A Retrospective analysis of precision medicine outcomes in patients with advanced cancer reveals improved progression‐free survival without increased health care costs. J Oncol Pract 2017;13:e108–e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sicklick JK, Kato S, Okamura R et al. Molecular profiling of cancer patients enables personalized combination therapy: The I‐PREDICT study. Nat. Med. 2019;25:744–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Planchard D, Popat S, Kerr K et al. Metastatic non‐small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow‐up. Ann Oncol 2018;29:iv192–iv237. [DOI] [PubMed] [Google Scholar]
- 12. Kalemkerian GP, Narula N, Kennedy EB et al. Molecular testing guideline for the selection of patients with lung cancer for treatment with targeted tyrosine kinase inhibitors: American Society of Clinical Oncology endorsement of the College of American Pathologists/International Association for the Study of Lung Cancer/Association for Molecular Pathology clinical practice guideline update. J Clin Oncol 2018;36:911–919. [DOI] [PubMed] [Google Scholar]
- 13. Lindeman NI, Cagle PT, Beasley MB et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: Guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathogy. Arch Pathol Lab Med 2013;137:828–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ahlborn LB, Rohrberg KS, Gabrielaite M et al. Application of cell‐free DNA for genomic tumor profiling: A feasibility study. Oncotarget 2019;10:1388–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. VanderLaan PA, Yamaguchi N, Folch E et al. Success and failure rates of tumor genotyping techniques in routine pathological samples with non‐small‐cell lung cancer. Lung Cancer 2014;84:39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Diaz LA, Bardelli A. Liquid biopsies: Genotyping circulating tumor DNA. J Clin Oncol 2014;32:579–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bettegowda C, Sausen M, Leary RJ et al. Detection of circulating tumor DNA in early‐ and late‐stage human malignancies. Sci Transl Med 2014;6:224ra24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Diehl F, Schmidt K, Choti MA et al. Circulating mutant DNA to assess tumor dynamics. Nat Med 2008;14:985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bronkhorst AJ, Ungerer V, Holdenrieder S. The emerging role of cell‐free DNA as a molecular marker for cancer management. Biomol Detect Quantif 2019;17:100087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Miller TE, Yang M, Bajor D et al. Clinical utility of reflex testing using focused next‐generation sequencing for management of patients with advanced lung adenocarcinoma. J Clin Pathol 2018;71:1108–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rolfo C, Mack PC, Scagliotti GV et al. Liquid biopsy for advanced non‐small cell lung cancer (NSCLC): A statement paper from the IASLC. J Thorac Oncol 2018;13:1248–1268. [DOI] [PubMed] [Google Scholar]
- 22. Sabari JK, Offin M, Stephens D et al. A prospective study of circulating tumor DNA to guide matched targeted therapy in lung cancers. J Natl Cancer Inst 2019;111:575–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Drilon A, Wang L, Arcila ME et al. Broad, hybrid capture‐based next‐generation sequencing identifies actionable genomic alterations in lung adenocarcinomas otherwise negative for such alterations by other genomic testing approaches. Clin Cancer Res 2015;21:3631–3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fearon K, Strasser F, Anker SD et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol 2011;12:489–495. [DOI] [PubMed] [Google Scholar]
- 25. Wood DE, Chair V, Aggarwal C et al. NCCN Clinical Practice Guidelines in Oncology: Non‐Small Cell Lung Cancer. Version 7.2019. Plymouth Meeting, PA: National Comprehensive Cancer Network, 2019. [Google Scholar]
- 26. Lim C, Tsao MS, Le LW et al. Biomarker testing and time to treatment decision in patients with advanced nonsmall‐cell lung cancer. Ann Oncol 2015;26:1415–1421. [DOI] [PubMed] [Google Scholar]
- 27. Shaw AT, Riely GJ, Bang YJ et al. Crizotinib in ROS1‐rearranged advanced non‐small‐cell lung cancer (NSCLC): Updated results, including overall survival, from PROFILE 1001. Ann Oncol 2019;30:1121–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Roy‐Chowdhuri S, Chen H, Singh RR et al. Concurrent fine needle aspirations and core needle biopsies: A comparative study of substrates for next‐generation sequencing in solid organ malignancies. Mod Pathol 2017;30:499–508. [DOI] [PubMed] [Google Scholar]
- 29. VanderLaan PA, Wang HH, Majid A et al. Endobronchial ultrasound‐guided transbronchial needle aspiration (EBUS‐TBNA): An overview and update for the cytopathologist. Cancer Cytopathol 2014;122:561–576. [DOI] [PubMed] [Google Scholar]
- 30. Wu Z, Yang Z, Dai Y et al. Update on liquid biopsy in clinical management of non‐small cell lung cancer. Onco Targets Ther 2019;12:5097–5109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fancello L, Gandini S, Pelicci PG et al. Tumor mutational burden quantification from targeted gene panels: Major advancements and challenges. J Immunother Cancer 2019;7:183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zulato E, Attili I, Pavan A et al. Early assessment of KRAS mutation in cfDNA correlates with risk of progression and death in advanced non‐small‐cell lung cancer. Br J Cancer 2020; May 7 [E pub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen M, Zhao H. Next‐generation sequencing in liquid biopsy: Cancer screening and early detection. Hum Genomics 2019;13:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bonanno L, Calabrese F, Nardo G et al. Morphological and genetic heterogeneity in multifocal lung adenocarcinoma: The case of a never‐smoker woman. Lung Cancer 2016;96:52–55. [DOI] [PubMed] [Google Scholar]
- 35. Kris MG, Johnson BE, Berry LD et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 2014;311:1998–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Le Tourneau C, Delord JP, Gonçalves A et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): A multicentre, open‐label, proof‐of‐concept, randomised, controlled phase 2 trial. Lancet Oncol 2015;16:1324–1334. [DOI] [PubMed] [Google Scholar]
- 37. Belin L, Kamal M, Mauborgne C et al. Randomized phase II trial comparing molecularly targeted therapy based on tumor molecular profiling versus conventional therapy in patients with refractory cancer: Cross‐over analysis from the SHIVA trial. Ann Oncol 2017;28:590–596. [DOI] [PubMed] [Google Scholar]
- 38. Di Maio M, Perrone F, Conte P. Real‐world evidence in oncology: Opportunities and limitations. The Oncologist 2020;25:e746–e752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sacher AG, Paweletz C, Dahlberg SE et al. Prospective validation of rapid plasma genotyping for the detection of EGFR and KRAS mutations in advanced lung cancer. JAMA Oncol 2016;2:1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sacher AG, Komatsubara KM, Oxnard GR. Application of plasma genotyping technologies in non–small cell lung cancer: A practical review. J Thorac Oncol 2017;12:1344–1356. [DOI] [PubMed] [Google Scholar]
- 41. Pasini L, Ulivi P. Liquid biopsy for the detection of resistance mechanisms in NSCLC: Comparison of different blood biomarkers. J Clin Med 2019;8:998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lim M, Kim CJ, Sunkara V et al. Liquid biopsy in lung cancer: Clinical applications of circulating biomarkers (CTCs and ctDNA). Micromachines 2018;9:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li BT, Janku F, Jung B et al. Ultra‐deep next‐generation sequencing of plasma cell‐free DNA in patients with advanced lung cancers: Results from the Actionable Genome Consortium. Ann Oncol 2019;30:597–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Aggarwal C, Thompson JC, Black TA et al. Clinical implications of plasma‐based genotyping with the delivery of personalized therapy in metastatic non–small cell lung cancer. JAMA Oncol 2019;5:173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sabari JK, Montecalvo J, Chen R et al. PD‐L1 expression and response to immunotherapy in patients with MET exon 14‐altered non‐small cell lung cancers (NSCLC). J Clin Oncol 2017;35:8512–8512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mazieres J, Drilon A, Lusque A et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: Results from the IMMUNOTARGET registry. Ann Oncol 2019;30:1321–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. van der Velden DL, van Herpen CML, van Laarhoven HWM et al. Molecular tumor boards: Current practice and future needs. Ann Oncol 2017;28:3070–3075. [DOI] [PubMed] [Google Scholar]
- 48. Le‐Rademacher J, Dahlberg S, Lee JJ et al. Biomarker clinical trials in lung cancer: Design, logistics, challenges, and practical considerations. J Thorac Oncol 2018;13:1625–1637. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
See http://www.TheOncologist.com for supplemental material available online.
Appendix S1: Supplementary Information