

Abstract
Lessons Learned
A biweekly TAS‐102 plus BEV schedule in patients with heavily pretreated mCRC showed equivalent efficacy with less toxicity compared with the current schedule of TAS‐102 plus BEV combination.
Biweekly TAS‐102 plus BEV combination could reduce unnecessary dose reduction of TAS‐102, maintain higher doses, and possibly be effective even in cases without chemotherapy‐induced neutropenia (CIN).
The prespecified subgroup analysis of this study showed an obvious association between CIN within the first two cycles and prognosis of biweekly TAS‐102 plus BEV.
Background
TAS‐102 (trifluridine/tipiracil) plus bevacizumab (BEV) combination therapy has shown promising activity in patients with metastatic colorectal cancer (mCRC). However, the previously reported dose and schedule for the TAS‐102 (70 mg/m2/day on days 1–5 and 8–12, every 4 weeks) plus BEV (5 mg/kg on day 1, every 2 weeks) regimen is complicated by severe hematological toxicities and difficult administration schedules. Here, we evaluated the efficacy and safety of a more convenient biweekly TAS‐102 plus BEV combination.
Methods
Patients with mCRC who were refractory or intolerant to standard chemotherapies were enrolled. Patients received biweekly TAS‐102 (twice daily on days 1–5, every 2 weeks) with BEV (5mg/kg on day 1, every 2 weeks). The primary endpoint was progression‐free survival rate at 16 weeks (16‐w PFS rate).
Results
From October 2017 to January 2018, 46 patients were enrolled. The recommended phase II dose was determined to be TAS‐102 (70 mg/m2/day). Of the 44 eligible patients, the 16‐w PFS rate was 40.9% (95% confidence interval, 26.3%–56.8%), and the null hypothesis was rejected (p < .0001). Median progression‐free survival (PFS) and overall survival were 4.29 months and 10.86 months, respectively. Disease control rate was 59.1%. Common grade 3 or higher adverse events were hypertension (40.9%), neutropenia (15.9%), and leucopenia (15.9%).
Conclusion
Biweekly TAS‐102 plus BEV showed promising antitumor activity with safety.
Discussion
The combination of TAS‐102 (trifluridine/tipiracil hydrochloride, also known as Lonsurf) plus BEV has shown promising antitumor activity in mCRC [1, 2, 3, 4, 5]. However, TAS‐102 plus BEV combination therapy was accompanied by an increase in toxicity. Furthermore, scheduling of the standard schedule for TAS‐102 plus BEV combination therapy is somewhat complicated, with TAS‐102 given by oral administration on days 1–5 and 8–12 in a 4‐week cycle and BEV by single intravenous administration every 2 weeks [4, 5]. Here, therefore, we planned this phase Ib/II study of biweekly TAS‐102 in combination with BEV with the expectation of equivalent efficacy but with less toxicity and a simpler regimen schedule. This multicenter prospective trial of biweekly TAS‐102 plus BEV—the BiTS study—met its primary endpoint, with PFS at 16 weeks exceeding the prespecified threshold.
In previous trials [4, 6], CIN related to TAS‐102 was associated with better prognosis, suggesting that avoiding a dose reduction of TAS‐102 caused by hematological toxicities and maintaining a higher dose could be beneficial to patients. We therefore conducted a prespecified analysis to evaluate the relationship between CIN and antitumor activity in this study. Prespecified subgroup analyses and the multivariable Cox regression analyses to evaluate factors affecting PFS and overall survival (OS) showed that age and Eastern Cooperative Oncology Group performance status (ECOG PS) affected PFS and that CIN within the first two cycles affected OS (Figs. 1 and 2).
Figure 1.
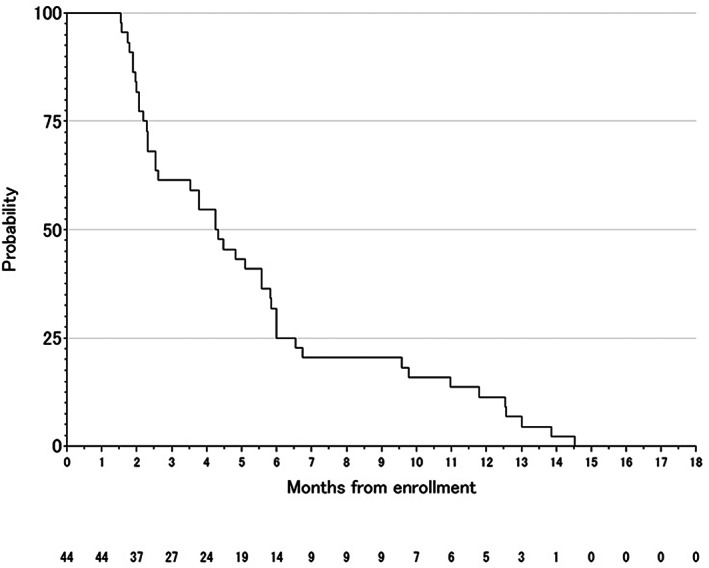
Progression‐free survival.
Figure 2.
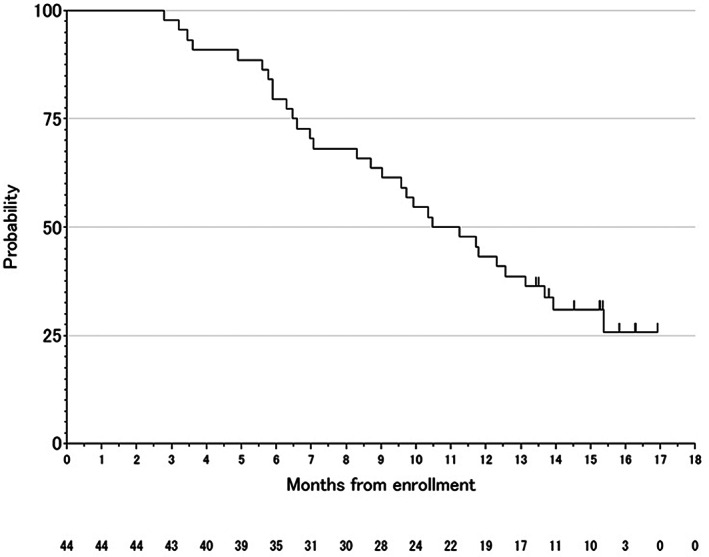
Overall survival.
Biweekly TAS‐102 (days 1–5, every 2 weeks) plus BEV combination showed equivalent efficacy with less toxicity compared with the current schedule of TAS‐102 (days 1–5 and 8–12 in a 4‐week cycle) plus BEV combination [4, 5]. Allowing that it is difficult to draw conclusions from cross‐trial comparisons because of differences in study design and patient characteristics, the biweekly TAS‐102 plus BEV combination might be considered an alternative option with a simple schedule.
Trial Information
| Disease | Colorectal cancer; advanced cancer |
| Stage of Disease/Treatment | Metastatic/advanced |
| Prior Therapy | More than two prior regimens |
| Type of Study | Phase II, single arm |
| Primary Endpoint | Progression‐free survival rate at 16 weeks |
| Secondary Endpoints | Overall survival, progression‐free survival, overall response rate, safety |
| Additional Details of Endpoints or Study Design | |
| Study design: From October 2017 to January 2018, 46 patients were enrolled (6 patients in phase Ib and 40 in phase II). Of these 46 patients, two were excluded from the study assessment because they did not start the protocol treatment. In the phase Ib component, no dose‐limiting toxicity (DLT) was observed in the first six patients at dose level 1. Accordingly, the recommended phase II dose (RP2D) was determined to be 5 mg/kg for BEV and 70 mg/m2/day for TAS‐102. All patients had received fluoropyrimidine, oxaliplatin, irinotecan, and anti–vascular endothelial growth factor (VEGF) antibody as prior chemotherapies before enrollment to this trial. All RAS wild‐type patients received anti–epidermal growth factor receptor (EGFR) antibody as a prior chemotherapy before enrollment. Nine patients (20.5%) received regorafenib before enrollment. | |
| This was a prospective, investigator‐initiated, open‐label, single‐arm, multicenter, phase Ib/II study conducted in 15 Japanese hospitals of the combination of biweekly TAS‐102 with BEV in patients with unresectable mCRC who were refractory or intolerant to standard chemotherapies. | |
| The study was conducted in two components. The initial phase Ib dose de‐escalation component based on the rolling six design aimed to determine the RP2D of TAS‐102 (35 mg/m2 by twice‐daily oral administration on days 1–5 for 2 weeks, dose level 1) with biweekly BEV (5 mg/kg). A total of six patients were enrolled concomitantly at dose level 1. If two or fewer DLTs occurred at dose level 1, TAS‐102 (35 mg/m2 given orally twice daily on days 1–5 for 2 weeks) with biweekly BEV would be the RP2D in this study. De‐escalation occurred when three or more DLTs occurred at dose level 1, and another six patients were included at dose level 0 (TAS‐102: 30 mg/m2 given orally twice daily on days 1–5 for 2 weeks). | |
| DLT was defined as a grade 3 or higher nonhematological toxicity, excluding controllable nausea, vomiting, hypertension, and transient electrolyte abnormalities; grade 4 neutropenia lasting 7 days or more; grade 3 or higher febrile neutropenia; grade 4 thrombocytopenia; or unresolved toxicities causing a delay of 2 weeks or longer in initiation of the next cycle. | |
| In the phase II component, all patients received the RP2D until disease progression, unacceptable toxicity, withdrawal of consent, or changes meeting the criteria for protocol discontinuation. This trial was carried out in accordance with the Helsinki Declaration and Ethical Guidelines for Clinical Studies and was approved by the institutional review boards of all participating institutions. All participating patients were required to give written informed consent before entering the study. | |
| Patients: Inclusion criteria were age ≥ 20 years; histologically confirmed unresectable metastatic colorectal adenocarcinoma; refractory or intolerant to fluoropyrimidine, irinotecan, oxaliplatin, anti‐VEGF therapy, and anti‐EGFR therapy (for tumors with wild‐type RAS); able to take oral drugs; ECOG PS 0 or 1; evaluable lesion according to RECIST version 1.1; and adequate organ function within 7 days before enrollment (hemoglobin ≥8.0 g/dL, neutrophil count ≥1,500/mm3, platelet count ≥75,000/mm3, total bilirubin ≤1.5 mg/dL, serum transaminases ≤100 IU/L, serum creatinine ≤1.5 mg/dL, and proteinuria ≤1+). | |
| Endpoints: The primary endpoint in the phase II component was investigator‐assessed 16‐w PFS rate. Major secondary endpoints included PFS, time to treatment failure (TTF), OS, objective response rate (ORR), disease control rate (DCR), and safety. Enhanced computed tomography (CT) or magnetic resonance imaging of the chest, abdomen, and pelvis was implemented every 8 weeks during the first year after initiation of the treatment and every 12 weeks thereafter. Tumor response was assessed by each institution using RECIST version 1.1 criteria. | |
| Study landmarks were defined as follows: PFS was defined as the time from study enrollment to the first disease progression or death, whichever occurred first; TTF as the time from study enrollment to the date of first disease progression, protocol discontinuation for any reason, or death, whichever occurred first; OS as the time from study enrollment to the date of death from any cause; ORR as the percentage of patients relative to the total enrolled subjects who achieved a complete (CR) or partial response (PR) based on CT scan images; and DCR as the percentage of patients relative to the total enrolled subjects who achieved a CR or PR plus stable disease based on CT scan images. Furthermore, right‐sided primary was defined as cancer located in the cecum, ascending colon, or transverse colon, and left‐sided primary as cancer in the descending colon, sigmoid colon, or rectum. | |
| Patients were examined and tested every 2 weeks by the local laboratory in each participating center. In these 2‐weekly assessments, patients were monitored for hematology, serum chemistry, and urinalysis. Adverse events were reported every 2 weeks and assessed according to the Common Terminology Criteria for Adverse Events, version 4.0. | |
| Statistical analysis: We set the threshold 16‐w PFS rate at 15% and the expected 16‐w PFS rate at 38.7% on the basis of the results of a previous study [7] in the phase II part. Given a one‐sided alpha of .025 and statistical power of 90% with about 10% ineligible or dropout patients, we set 45 patients as the target sample size in this study. | |
| The analytical population for efficacy was defined as all enrolled eligible patients who received RP2D at least once and were assessed for efficacy endpoint once. Primary analysis was to estimate the 16‐w PFS rate with two‐sided 95% exact confidence interval (CI). A one‐side binomial exact test was also conducted against the null hypothesis (threshold 16‐w PFS rate, 15%). For the secondary analysis, the Kaplan‐Meier method was used to estimated PFS, TTF, and OS. ORR and DCR were analyzed in the same way as in the primary analysis. | |
| We prespecified factors that would likely affect PFS or OS, namely, age, body mass index, ECOG PS, tumor location, clinical stage, RAS status, prior regorafenib administration, and neutropenia of grade 2 or higher within the first two cycles. These prognostic factors were analyzed by multivariable Cox regression analysis for PFS and OS. Backward variable selection (threshold exclusion criteria for p = .20) was also adopted to select variables that affected PFS or OS. Adjusted hazard ratio was estimated in the multivariable model. Descriptive analysis was shown for the safety outcomes. | |
| All analyses were done with SAS version 9.4 (SAS Institute, Inc., Cary, NC). | |
| Investigator's Analysis | Active and should be pursued further |
Drug Information
| Drug 1 | |
| Generic/Working Name | TAS‐102 |
| Trade Name | Lonsurf |
| Company Name | Taiho Pharmaceutical Co., Ltd. |
| Dose | Milligrams (mg) per squared meter (m2) |
| Route | Oral (p.o.) |
| Schedule of Administration | 35 mg/m2 twice daily on days 1–5, every 2 weeks |
| Drug 2 | |
| Generic/Working Name | Bevacizumab |
| Trade Name | Avastin |
| Company Name | Chugai Pharmaceutical Co., Ltd. |
| Drug Type | Antibody |
| Drug Class | VEGF |
| Dose | 5 milligrams (mg) per kilogram (kg) |
| Route | IV |
| Schedule of Administration | Day 1, every 2 weeks |
Patient Characteristics
| Number of Patients, Male | 24 |
| Number of Patients, Female | 20 |
| TNM Stage | |
| IIIA | 3 (6.8%) |
| IIIB | 2 (4.5%) |
| IVA | 14 (31.8%) |
| IVB | 25 (56.8%) |
| TNM classification (Union for International Cancer Control 7th edition). | |
| Age | Median (range): 69 years (33–82 years) |
| Number of Prior Systemic Therapies | Median (range): 3 (1–5) |
| Performance Status: ECOG |
0 — 25 1 — 19 2 — 3 — Unknown — |
| Detailed Patient Characteristics | |
| Age, years | |
| <65 | 8 (18.2%) |
| ≥65 | 36 (81.8%) |
| Body mass index | |
| <25 | 38 (86.4%) |
| ≥25 | 6 (13.6%) |
| RAS status | |
| Wild‐type | 25 (56.8%) |
| Mutant | 19 (43.2%) |
| Prior therapy: Yes | 44 (100%) |
| Fluoropyrimidine | 44 (100%) |
| Oxaliplatin | 44 (100%) |
| Irinotecan | 44 (100%) |
| Anti‐VEGF inhibitor | 44 (100%) |
| Anti‐EGFR antibody | 25 (56.8%) |
| Regorafenib | 9 (20.5%) |
| Prior number of regimens | |
| 1 | 1 (2.3%) |
| 2 | 18 (40.9%) |
| 3 | 13 (29.5%) |
| ≥4 | 12 (27.3%) |
| Diagnosis | |
| Colon | 27 (61.4%) |
| Rectum | 17 (38.6%) |
| Primary tumor locationa | |
| Left‐sided | 31 (70.5%) |
| Right‐sided | 13 (29.5%) |
| Cancer Types or Histologic Subtypes | Well‐differentiated tubular adenocarcinoma (tub1), 16; moderately differentiated tubular adenocarcinoma (tub2), 26; other, 2 |
| Abbreviations: EGFR, epidermal growth factor receptor; VEGF, vascular endothelial growth factor. | |
| aRight‐sided was defined as cancer that is located in the cecum, ascending colon, or transverse colon, and left‐sided as cancer located in the descending colon, sigmoid colon, or rectum. | |
Primary Assessment Method
| Title | 16‐w PFS Rate |
| Number of Patients Enrolled | 46 |
| Number of Patients Evaluable for Toxicity | 44 |
| Number of Patients Evaluated for Efficacy | 44 |
| Evaluation Method | RECIST version 1.1 |
| (Median) Duration Assessments PFS | 4.29 months |
| Outcome Notes | |
| Of the 44 enrolled patients, 24 (54.5%) received subsequent chemotherapy, mainly with regorafenib (n = 12). Prespecified subgroup analyses and the multivariable Cox regression analyses to evaluate factors affecting PFS and OS are summarized in Figures 3 and 4, respectively. These analyses showed that age and ECOG PS affected the PFS and that chemotherapy‐induced neutropenia within the first two cycles affected OS. No clear association was seen between RAS status, primary tumor location, prior regorafenib administration, and the efficacy of biweekly FTD/TPI plus BEV combination. | |
Figure 3.
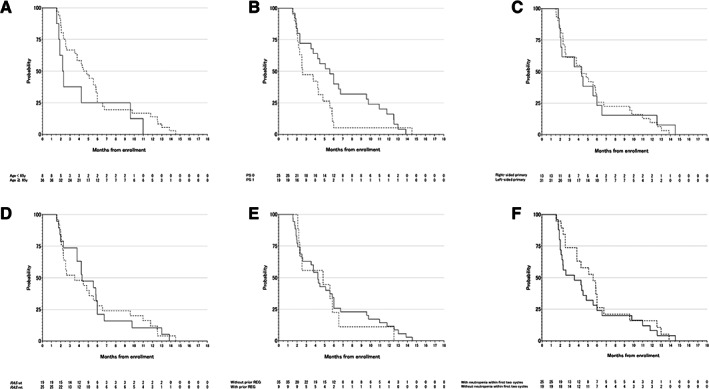
Prespecified subgroup analysis for progression‐free survival (PFS). (A): PFS by age. Straight line, age < 65 years; dotted line, age ≥ 65 years. (B): PFS by Eastern Cooperative Oncology Group (ECOG) PS. Straight line, ECOG PS 0; dotted line, ECOG PS 1. (C): PFS by primary location. Straight line, right‐sided primary; dotted line, left‐sided primary. (D): PFS by RAS status. Straight line, RAS wild‐type; dotted line, RAS mutant‐type. (E): PFS by prior regorafenib administration. Straight line, without prior regorafenib; dotted line, with prior regorafenib. (F): PFS by chemotherapy‐induced neutropenia within the first two cycles. Straight line, without neutropenia within the first two cycles; dotted line, with neutropenia within the first two cycles. Abbreviations: mt, mutant; PS, performance status; REG, regorafenib; wt, wild type.
Figure 4.
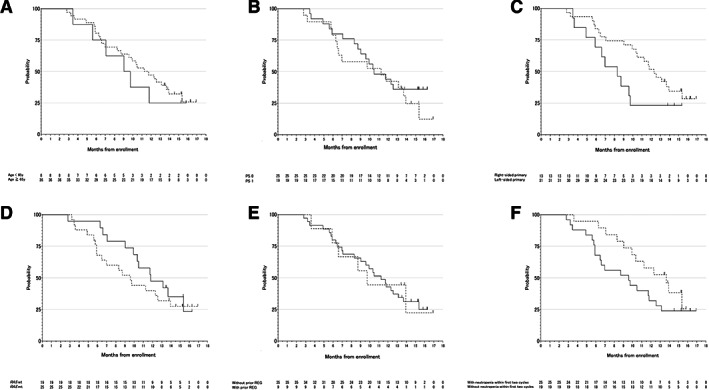
Prespecified subgroup analysis for overall survival (OS). (A): OS by age. Straight line, age < 65 years; dotted line, age ≥ 65 years. (B): OS by Eastern Cooperative Oncology Group (ECOG) PS. Straight line, ECOG PS 0; dotted line, ECOG PS 1. (C): OS by primary location. Straight line, right‐sided primary; dotted line, left‐sided primary. (D): OS by RAS status. Straight line, RAS wild‐type; dotted line, RAS mutant‐type. (E): OS by prior regorafenib administration. Straight line, without prior regorafenib; dotted line, with prior regorafenib. (F): OS by chemotherapy‐induced neutropenia within the first two cycles. Straight line, without neutropenia within the first two cycles; dotted line, with neutropenia within the first two cycles. Abbreviations: mt, mutant; PS, performance status; REG, regorafenib; wt, wild type.
Secondary Assessment Method
| Title | OS, TTF, ORR |
| Number of Patients Enrolled | 46 |
| Number of Patients Evaluable for Toxicity | 44 |
| Number of Patients Evaluated for Efficacy | 44 |
| Response Assessment CR | n = 0 (0%) |
| Response Assessment PR | n = 0 (0%) |
| Response Assessment SD | n = 26 (59.1%) |
| Response Assessment PD | n = 17 (38.6%) |
| Response Assessment OTHER | n = 1 (2.3%) |
| (Median) Duration Assessments OS | 10.86 months |
| Outcome Notes | |
| Of the 44 eligible patients, 16‐w PFS rate was 40.9% (95% CI, 26.3%–56.8%), and the null hypothesis of a 16‐w PFS rate ≤ 15% was rejected (p < .0001). With a median follow‐up of 15.36 months (range, 2.79–16.93), median PFS was 4.29 months (95% CI, 2.54–5.83), median TTF was 4.16 months (95% CI, 2.39–5.82), and median OS was 10.86 months (95% CI, 8.32–13.68). | |
| DCR was 59.1% (95% CI, 43.3%–73.7%). | |
Adverse Events
| All Cycles Name | NC/NA, % | Grade 1, % | Grade 2, % | Grade 3, % | Grade 4, % | Grade 5, % | All grades, % |
|---|---|---|---|---|---|---|---|
| White blood cell decreased | 32 | 16 | 36 | 16 | 0 | 0 | 68 |
| Neutrophil count decreased | 36 | 7 | 41 | 16 | 0 | 0 | 64 |
| Anemia | 11 | 41 | 39 | 9 | 0 | 0 | 89 |
| Platelet count decreased | 52 | 36 | 7 | 5 | 0 | 0 | 48 |
| Alanine aminotransferase increased | 78 | 20 | 0 | 2 | 0 | 0 | 22 |
| Aspartate aminotransferase increased | 55 | 43 | 2 | 0 | 0 | 0 | 45 |
| Blood bilirubin increased | 73 | 18 | 2 | 7 | 0 | 0 | 27 |
| Creatinine increased | 57 | 43 | 0 | 0 | 0 | 0 | 43 |
| Febrile neutropenia | 100 | 0 | 0 | 0 | 0 | 0 | 0 |
| Hypertension | 9 | 14 | 36 | 41 | 0 | 0 | 91 |
| Anorexia | 32 | 36 | 23 | 9 | 0 | 0 | 68 |
| Proteinuria | 31 | 32 | 30 | 7 | 0 | 0 | 69 |
| Fatigue | 36 | 32 | 32 | 0 | 0 | 0 | 64 |
| Nausea | 40 | 39 | 14 | 7 | 0 | 0 | 60 |
| Peripheral sensory neuropathy | 48 | 48 | 2 | 2 | 0 | 0 | 52 |
| Mucositis oral | 70 | 23 | 7 | 0 | 0 | 0 | 30 |
| Vomiting | 77 | 16 | 7 | 0 | 0 | 0 | 23 |
| Palmar‐plantar erythrodysesthesia syndrome | 80 | 20 | 0 | 0 | 0 | 0 | 20 |
| Diarrhea | 81 | 14 | 0 | 5 | 0 | 0 | 19 |
| Fever | 82 | 9 | 7 | 2 | 0 | 0 | 18 |
| Weight loss | 82 | 7 | 9 | 2 | 0 | 0 | 18 |
| Dry skin | 84 | 11 | 5 | 0 | 0 | 0 | 16 |
| Alopecia | 86 | 7 | 7 | 0 | 0 | 0 | 14 |
| Dysgeusia | 87 | 11 | 2 | 0 | 0 | 0 | 13 |
| Epistaxis | 89 | 11 | 0 | 0 | 0 | 0 | 11 |
| Rash acneiform | 89 | 9 | 2 | 0 | 0 | 0 | 11 |
| Thromboembolic event | 96 | 0 | 0 | 2 | 2 | 0 | 4 |
Adverse Events Legend
Abbreviation: NC/NA, no change from baseline/no adverse event.
The content of the worst adverse event observed over the entire cycle in each case is shown above. Fourteen (30.4%) patients required at least one dose reduction of TAS‐102, primarily owing to anorexia. Twenty‐five (54.3%) patients required dose delays during the treatment period, predominantly owing to neutropenia. The median treatment interruption was 8 days (range, 1–28). No patient suffered from febrile neutropenia. Finally, no treatment‐related deaths were observed. Patients received the study treatment for a median of 6.5 cycles (range, 1–24 cycles). Discontinuation of protocol treatment was mainly due to disease progression (n = 39, 88.6%), and the remaining five cases were due to adverse events (11.4%). The median relative dose intensity of TAS‐102 and BEV was 80.9% (range, 44.0%–100%) and 81.5% (range, 50.0%–100%), respectively.
Assessment, Analysis, and Discussion
| Completion | Study completed |
| Investigator's Assessment | Active and should be pursued further |
The prespecified analysis to evaluate the relationship between chemotherapy‐induced neutropenia (CIN) and antitumor activity in this study discovered an obvious association between CIN within the first two cycles and prognosis of biweekly TAS‐102 plus bevacizumab (BEV). In particular, overall survival (OS) was prolonged in the group with CIN compared with that without CIN (hazard ratio, 0.44; 95% confidence interval, 0.2–0.95; p = .036). However, survival with biweekly TAS‐102 plus BEV combination was better than with TAS‐102 alone (median OS, 6.7–7.1 months) [3, 5], even in the group without CIN (median OS, 9.5 months; Fig. 4F). One therapeutic effect of anti–vascular endothelial growth factor antibodies represented by BEV is an increase in drug delivery to tumor because of normalization of the tumor vasculature. A preclinical study showed that the combination of TAS‐102 plus BEV significantly suppresses tumor growth compared with TAS‐102 alone in colorectal cancer xenograft models and that the concentration of phosphorylated trifluridine in tumors was higher with TAS‐102 plus BEV than with TAS‐102 monotherapy [8]. Considering that the therapeutic effect is promising despite mild hematological toxicities in the biweekly TAS‐102 plus BEV combination, the current schedule of TAS‐102 might need to be reconsidered when combined with BEV. The TAS‐102 dosing schedule was initially considered to be continuous daily dosing, but the current schedule, on days 1–5 and 8–12 in a 4‐week cycle, was eventually determined after a phase I study [9, 10]. To date, however, no study has investigated a biweekly schedule. Based on our present results, the biweekly TAS‐102 plus BEV combination could reduce unnecessary dose reduction of TAS‐102, maintain higher doses, and possibly be effective even in cases without CIN.
Although several guidelines recommend TAS‐102 monotherapy as late‐line systemic chemotherapy for metastatic colorectal cancer (mCRC) [11, 12, 13], a recent prospective trial showed the efficacy of TAS‐102 plus BEV combination in the first‐line setting for mCRC [14]. In general, TAS‐102 plus BEV combination therapy does not show tumor shrinkage in the late‐line setting. In contrast to late‐line use, however, TAS‐102 plus BEV combination is expected to induce tumor shrinkage when administered in the first‐line setting (objective response rate 33.8% in TASCO1 trial [14], 40.5% in KSCC1602 trial [15]). These findings suggest that TAS‐102 plus BEV may be introduced in first‐line treatment in patients for whom intensive chemotherapy is not indicated. The safety results of our study also suggest that this biweekly TAS‐102 plus BEV regimen is beneficial for patients with mCRC who are not eligible for intensive therapy.
Several limitations of this study warrant mention. First, it was conducted under a nonrandomized design with a relatively small sample size. Second, the recommended dose for TAS‐102 when administered biweekly in combination with BEV may be undetermined because of the phase Ib dose de‐escalation design. Third, the impact of biweekly TAS‐102 with BEV on quality of life was not evaluated. Nevertheless, toxicity appeared to be lower than in other studies. The simple schedule is considered preferable for both patients and health care professionals.
In conclusion, biweekly TAS‐102 plus BEV in combination for patients with mCRC shows promising antitumor efficacy and acceptable toxicity and might represent a treatment option for patients with heavily treated mCRC.
Disclosures
Hironaga Satake: Ono Pharmaceutical, Taiho Pharmaceutical, Takeda Pharmaceutical (RF), Bayer, Bristol‐Myers Squibb, Chugai Pharmaceutical, Daiichi Sankyo, Eli Lilly Japan, Merck Bio Pharma, Merck Sharp & Dohme, Ono Pharmaceutical, Sanofi, Taiho Pharmaceutical, Takeda, Yakult Honsha, (H); Takeshi Kato: Bayer, Chugai Pharmaceutical, Eli‐Lilly Japan, Merck Biopharma, Taiho Pharmaceutical, Takeda Pharmaceutical (RF, H); Koji Oba: Chugai Pharmaceutical, Daiichi Sankyo, Eisai, Ono Pharmaceutical, Takeda Pharmaceutical (H); Masahito Kotaka: Chugai Pharmaceutical, Taiho Pharma (H); Yoshinori Kagawa: Chugai Pharmaceutical, Taiho Pharma (H); Toshihiko Matsumoto: Sanofi Pharma (RF); Akitaka Makiyama: Eli Lilly, Chugai Pharmaceutical, Takeda Pharmaceutical (H); Akihito Tsuji: Bayer, Bristol‐Myers Squibb Japan, Chugai Pharmaceutical, Daiichi Sankyo, Eli‐Lilly, Merck Biopharma, Takeda Pharmaceutical, Taiho Pharmaceutical (H); Yoshito Komatsu: Merck Sharp & Dohme K.K., Taiho Pharmaceutical, Yakult Honsha, Bayer, Daiichi Sankyo, ONO Pharmaceutical, NanoCarrier, Eisai, Sanofi‐Aventis S.A., Sysmex Corporation, Shionogi, IQVIA Services Japan K.K., Parexel International Corporation, Astellas Pharma Inc., Mediscience Planning Inc., Sumitomo Dainippon Pharma, A2 Healthcare Corporation (RF), Eli Lilly Japan K.K., Taiho Pharmaceutical, Takeda Pharmaceutical, Bayer Yakuhin, Bristol‐Myers Squibb Co., Sanofi‐Aventis S.A., Merck Biopharma, Yakult Honsha, Ono Pharmaceutical, Nipro Corporation, MOROO, Asahi Kasei Pharma Cooperation, Mitsubishi Tanabe Pharma Corporation, Otsuka Pharmaceutical, Medical Review, Shiseido (H); Takayuki Yoshino: Novartis Pharma K.K., Merck Sharp & Dohme K.K., Sumitomo Dainippon Pharma, Chugai Pharmaceutical, Sanofi K.K., Daiichi Sankyo, PAREXEL International Inc., Ono Pharmaceutical, GlaxoSmithKline K.K. (RF); Kentaro Yamazaki: Chugai Pharma, Daiichi Sankyo, Yakult Honsha, Takeda, Bayer, Merck Biofarma, Eli Lilly, Sanofi, Ono Pharmaceutical, Merck Sharp & Dohme, Bristol‐Myers Squibb, Taiho Pharmaceutical (H); Eiji Oki: Taiho Pharmaceutical, Takeda Pharmaceutical, Bayer, Eli Lilly Japan (H); Junichi Sakamoto: Takeda Pharmaceutical (C/A), Chugai Pharmaceutical (H). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
Figures
Acknowledgments
The authors thank the patients and families who participated in this study, as well as the members of the Data and Safety Monitoring Committee (Yoshihiro Kakeji, Keiji Hirata, and Kei Muro). The study content was presented in part at the European Society for Medical Oncology Asia 2018 Congress, Singapore, November 23–25, 2018; the 2019 Gastrointestinal Cancers Symposium, San Francisco, CA, January 17–19, 2019; and the 2020 Gastrointestinal Cancers Symposium, San Francisco, CA, January 23–25, 2020. This study was supported in part by the nonprofit organization Epidemiological and Clinical tResearch Information Network. Editorial assistance and English editing were provided by Guy Harris of DMC Corp.
Footnotes
- ClinicalTrials.gov Identifier: UMIN000029198
- Sponsor: Nonprofit organization (NPO) Epidemiological Clinical Research Information Network, under a funding contract with Taiho Pharmaceutical Co. Ltd., Japan
- Principal Investigator: Hironaga Satake
- IRB Approved: Yes
References
- 1. Yoshino T, Mizunuma N, Yamazaki K et al. TAS‐102 monotherapy for pretreated metastatic colorectal cancer: A double‐blind, randomised, placebo‐controlled phase 2 trial. Lancet Oncol 2012;13:993–1001. [DOI] [PubMed] [Google Scholar]
- 2. Bendell JC, Rosen LS, Mayer RJ et al. Phase 1 study of oral TAS‐102 in patients with refractory metastatic colorectal cancer. Cancer Chemother Pharmacol 2015;76:925–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mayer RJ, Van Cutsem E, Falcone A et al. Randomized trial of TAS‐102 for refractory metastatic colorectal cancer. N Engl J Med 2015;372:1909–1919. [DOI] [PubMed] [Google Scholar]
- 4. Kuboki Y, Nishina T, Shinozaki E et al. TAS‐102 plus bevacizumab for patients with metastatic colorectal cancer refractory to standard therapies (C‐TASK FORCE): An investigator‐initiated, open‐label, single‐arm, multicentre, phase 1/2 study. Lancet Oncol 2017;18:1172–1181. [DOI] [PubMed] [Google Scholar]
- 5. Pfeiffer P, Yilmaz M, Moller S et al. TAS‐102 with or without bevacizumab in patients with chemorefractory metastatic colorectal cancer: An investigator‐initiated, open‐label, randomised, phase 2 trial. Lancet Oncol 2020;21:412–420. [DOI] [PubMed] [Google Scholar]
- 6. Yoshino T, Cleary JM, Van Cutsem E et al. Neutropenia and survival outcomes in metastatic colorectal cancer patients treated with trifluridine/tipiracil in the RECOURSE and J003 trials. Ann Oncol 2020;31:88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Skolnik JM, Barrett JS, Jayaraman B et al. Shortening the timeline of pediatric phase I trials: The rolling six design. J Clin Oncol 2008;26:190–195. [DOI] [PubMed] [Google Scholar]
- 8. Tsukihara H, Nakagawa F, Sakamoto K et al. Efficacy of combination chemotherapy using a novel oral chemotherapeutic agent, TAS‐102, together with bevacizumab, cetuximab, or panitumumab on human colorectal cancer xenografts. Oncol Rep 2015;33:2135–2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hong DS, Abbruzzese JL, Bogaard K et al. Phase I study to determine the safety and pharmacokinetics of oral administration of TAS‐102 in patients with solid tumors. Cancer 2006;107:1383–1390. [DOI] [PubMed] [Google Scholar]
- 10. Overman MJ, Varadhachary G, Kopetz S et al. Phase 1 study of TAS‐102 administered once daily on a 5‐day‐per‐week schedule in patients with solid tumors. Invest New Drugs 2008;26:445–454. [DOI] [PubMed] [Google Scholar]
- 11. Van Cutsem E, Cervantes A, Adam R et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol 2016;27:1386–1422. [DOI] [PubMed] [Google Scholar]
- 12. Yoshino T, Arnold D, Taniguchi H et al. Pan‐Asian adapted ESMO consensus guidelines for the management of patients with metastatic colorectal cancer: A JSMO‐ESMO initiative endorsed by CSCO, KACO, MOS, SSO and TOS. Ann Oncol 2018;29:44–70. [DOI] [PubMed] [Google Scholar]
- 13.NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines): Colon Cancer. Version 2.2020. Plymouth Meeting, PA: National Comprehensive Cancer Network 2020.
- 14. Lesniewski‐Kmak K, Moiseyenko V, Saunders M et al. O‐022 phase II study evaluating trifluridine/tipiracil + bevacizumab and capecitabine + bevacizumab in first‐line unresectable metastatic colorectal cancer (mCRC) patients who are non‐eligible for intensive therapy (TASCO1): Results of the primary analysis. Ann Oncol 2018;29(suppl 5):O‐022a. [Google Scholar]
- 15. Oki E, Makiyama A, Miyamoto Y et al. Trifluridine/tipiracil (FTD/TPI) plus bevacizumab in elderly patients with previously untreated metastatic colorectal cancer (KSCC1602): A multicenter, phase II clinical trial. J Clin Oncol 2019;37(suppl 15):3548a. [Google Scholar]