

Abstract
Amyotrophic lateral sclerosis (ALS) causes rapidly progressive paralysis and death within 5 years from diagnosis due to degeneration of the motor circuits. However, a significant population of ALS patients also shows cognitive impairments and progressive hippocampal pathology. Likewise, the mutant SOD1(G93A) mouse model of ALS (mSOD1), in addition to loss of spinal motor neurons, displays altered spatial behavior and hippocampal abnormalities including loss of parvalbumin-positive interneurons (PVi) and enhanced long-term potentiation (LTP). However, the cellular and molecular mechanisms underlying these morpho-functional features are not well understood. Since removal of TrkB.T1, a receptor isoform of the brain-derived neurotrophic factor, can partially rescue the phenotype of the mSOD1 mice, here we tested whether removal of TrkB.T1 can normalize the number of PVi and the LTP in this model. Stereological analysis of hippocampal PVi in control, TrkB.T1−/−, mSOD1, and mSOD1 mice deficient for TrkB.T1 (mSOD1/T1−/−) showed that deletion of TrkB.T1 restored the number of PVi to physiological level in the mSOD1 hippocampus. The rescue of PVi neuron number is paralleled by a normalization of high-frequency stimulation-induced LTP in the pre-symptomatic mSOD1/T1−/− mice. Our experiments identified TrkB.T1 as a cellular player involved in the homeostasis of parvalbumin expressing interneurons and, in the context of murine ALS, show that TrkB.T1 is involved in the mechanism underlying structural and functional hippocampal degeneration. These findings have potential implications for hippocampal degeneration and cognitive impairments reported in ALS patients at early stages of the disease.
Keywords: Amyotrophic lateral sclerosis, SOD1(G93A) mouse, Parvalbumin-positive interneurons, Hippocampus, TrkB, Long-term potentiation
1. Introduction
Amyotrophic lateral sclerosis (ALS) leads to paralysis and death. As such, studies have been mainly focusing on circuits in the spinal cord and motor cortex. While the central and peripheral motor circuits are possibly at the epicenter of characteristic ALS symptoms, alterations of circuits including those of the hippocampus - far away from sensory inputs and motor outputs - are also present in ALS patients and in the mutant SOD1(G93A) mouse (mSOD1) model of ALS (Abdulla et al., 2014; Phukan et al., 2007; Quarta et al., 2015; Takeda et al., 2009). These changes may account for the cognitive deficits observed in about half of the ALS patients (Abrahams et al., 2005; Lillo and Hodges, 2010; Ringholz et al., 2005).
A pivotal component of hippocampal circuits is represented by inhibitory GABAergic, fast-spiking parvalbumin-positive interneurons (PVi; (Bartos et al., 2002; Baude et al., 2006; Kawaguchi et al., 1987). Hippocampal PVi comprise a subset of axo-axonic, basket, and bistratified cells. Functionally, these cells grant synchronized oscillatory inputs to pyramidal neurons (Klausberger, 2005). At the behavioral level, hippocampal PVi are relevant for hippocampus-dependent performance, including spatial learning and memory (Murray et al., 2011).
Recently, we have reported a reduction in this population of PVi in pre-symptomatic mSOD1 mice concurrently with increased anxiety-like behavior and impaired spatial navigation behavior (Quarta et al., 2015). However, the cellular and molecular reasons underlying these latter pathological features are not well understood.
The development and function of selected neural cells, including GABAergic interneurons, is under tight control by brain derived neurotrophic factor (BDNF), and its cognate receptor tropomyosin receptor kinase B (TrkB; Angelov and Angelova, 2017; Barde et al., 1982; Klein et al., 1989; Waterhouse et al., 2012; Zheng et al., 2011). Alternative splicing generates a catalytic form of TrkB (full length TrkB, TrkB.FL) and truncated isoforms lacking the intracellular kinase domain (TrkB.T1, TrkB.T2, and TrkB.T-Shc; Klein et al., 1990; Wong and Garner, 2012).
TrkB·FL signaling induces, via the PI3K/AKT, RAS/MAPK, PLC/PKC, AMPK/ACC and JAK/STAT pathways, proliferation, survival, and differentiation effects and contributes to the synaptic plasticity maintenance (Huang and Reichardt, 2003). TrkB.T1 (T1) is the predominant TrkB isoform in the mature brain (Dorsey et al., 2006) and has a more elusive and less studied signaling (Fenner, 2012; Ferrer et al., 1999; Fryer et al., 1996; Silhol et al., 2005). A canonical function of T1 is to act as the dominant-negative receptor to inhibit TrkB·FL signaling to decrease the pro-survival BDNF effects on neuronal survival, differentiation, and plasticity (Fig. 1). However, cells from both CNS (Rose et al., 2003) and peripheral organs such as the heart respond directly to BDNF through TrkB.T1 with release of calcium from internal stores (Fulgenzi et al., 2015). Furthermore, deletion of the T1 isoform induces alterations in the CNS (Carim-Todd et al., 2009), skeletal (Dorsey et al., 2012) and cardiac muscle (Fulgenzi et al., 2015).
Fig. 1.
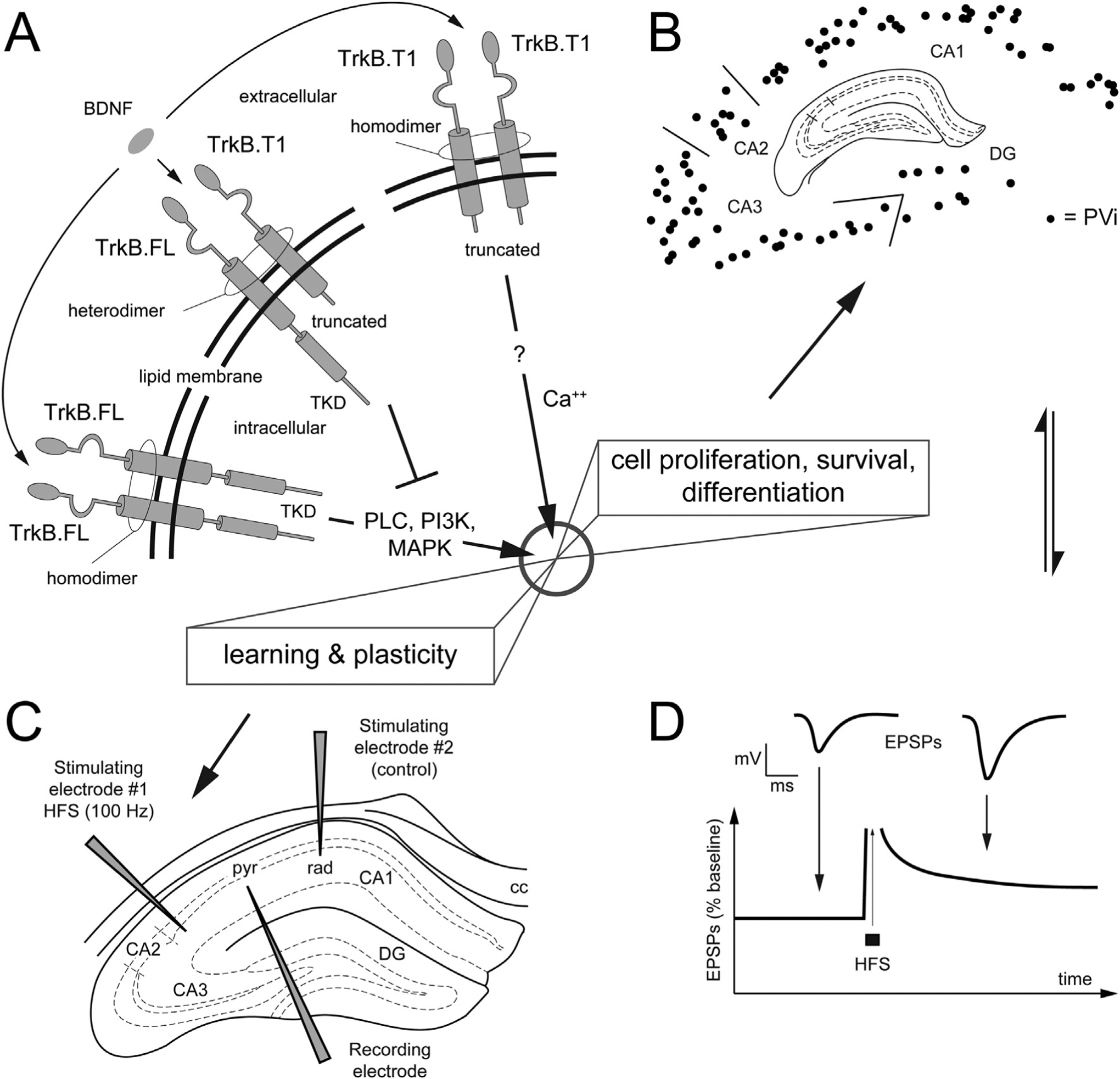
Molecular structure of TrkB isoforms and morphofunctional components investigated in the present work.
A. Schematic representation of the molecular structure of TrkB isoforms in the lipid membrane (full length = FL, truncated isoform T1). Extracellularly, the TrkB receptors are formed by a combination of Cysteine Rich Repeats, Leucine Rich Repeats and Immunoglobulin domains that is identical across isoforms. Intracellularly, the tyrosine kinase domain (TKD) is unique to the FL isoform. In the presence of brain-derived neurotrophic factor (BDNF), TrkB·FL homodimerization leads to receptor autophosphorylation at the TKD and downstream signaling (e.g., via the phosphorylation of PLC, PI3K, MAPK) to induce cell proliferation, survival/death, differentiation as well as plasticity. The TrkB.T1 isoform contains a short intracellular domain and lacks the TKD. In the presence of BDNF, T1 forms a heterotimer with FL, to exert its dominant negative inhibition (interrupted line in the figure) of FL-mediated signaling. In astrocytes, T1 is the only expressed isoform, where it controls the sequestration of BDNF. Note that while FL and T1 are the main isoforms, other isoforms exist, i.e., T2 e SHC (not shown).? = unknown intracellular signaling.
B, C, D. Morphofunctional components investigated in the present study: we focus our neuroanatomical analysis on a subpopulation of hippocampal inhibitory neurons that express parvalbumin (PV), the PV+ interneurons (PVi). B. Schematics of the charts PVi; our structural approach on PVi is complemented with electrophysiological investigation (C, D) aimed at comparing synaptic plasticity at the Schaffer’s collateral, via classic LTP experiments.
It is currently not known whether endogenous T1 is involved in the maintenance of the hippocampal PVi. In the present study we first investigated this aspect, by performing a stereological quantification of the hippocampal PVi population in TrkB.T1 knockout mice.
Alterations in BDNF/TrkB signaling have been associated with neurological disorders, including ALS. Hence, the study of the organization and targeting BDNF/TrkB signaling holds great promise to treat neurodegenerative conditions (Angelov and Angelova, 2017; Angelova and Angelov, 2017; Angelova et al., 2013; Geral et al., 2013; Guerzoni et al., 2017). However, in ALS exogenous BDNF delivery does not protect against neurodegeneration and, surprisingly, endogenous BDNF expression is increased in this clinical population, while phosphorylation levels of TrkB are reduced (Kust et al., 2002; Mutoh et al., 2000).
In the mSOD1 mouse model of ALS, T1 limits the pro-survival effects of BDNF on motoneurons (Yanpallewar et al., 2012a). From this observation, we therefore theorized that the dominant negative receptor T1, by negatively regulating the BDNF/TrkB pro-survival signaling, could be involved in the loss of hippocampal PVi observed in the mSOD1 model. To test this hypothesis, we evaluate the impact of T1 deletion on the PVi population in mSOD1 mice.
BDNF-TrkB signaling and PVi are implicated in the regulation of long-term potentiation (LTP), a cellular model of learning (Kumar, 2011; Leal et al., 2015; Levine et al., 1995), which was shown to be abnormally increased in the mSOD1 mouse (Spalloni et al., 2006). In an attempt to determine the relevance of T1 in these measures of synaptic strength, we performed experiments to evaluate the impact of T1 deletion on LTP in mSOD1 mice.
2. Materials and methods
SOD1(G93A) mice [B6SJL-TGN(SOD1-G93A)1GUR/J; 002726] were acquired from The Jackson Laboratory (Bar Harbor, ME) and crossed to T1 knockout mice backcrossed on C57/B16J background for at least 10 generations (Yanpallewar et al., 2012a). Experimental groups were mSOD1, wild-type littermates (wt), T1 knockout (T1−/−) and mSOD1/T1−/− mice. All mice were 8 weeks old.
Animals were kept under controlled environmental parameters and veterinarian assistance. All animals were infection-free, were kept under constant light/dark (12/12 h) cycle and fed a diet of rodent chow and water ad libitum. Care was taken to avoid animal stress and discomfort during handling and surgery. All experiments were conducted with the experimenter blind to the genotype of the animals. Protocols followed the National Institutes of Health Guidelines for animal care and use of laboratory animals, and were approved by the NCI-Frederick ACUC committee.
2.1. Immunohistochemistry
The immunohistochemical procedure for the visualization of PVi has been previously described (Quarta et al., 2015). Briefly, animals were deeply anesthetized (Avertin 250 mg/kg i.p.) and perfused transcardially with PBS, followed by 4% paraformaldehyde in 0.1 M PBS, pH 7.4. Brains were dissected out, soaked overnight in 30% buffered sucrose at 4 °C, and cut on a cryostat microtome into 40 μm thick coronal sections. The dorsal hippocampus was investigated between −1.46/−1.58 mm and −2.70/−2.80 mm from bregma (anterior and posterior limit, respectively; (Paxinos and Franklin, 2013). Every fifth section containing the hippocampus was collected for the immunohistochemical study of PVi. Some sections were collected separately for thionin staining in order to determine hippocampal cytoarchitecture (0.25% thionin solution; (Armed Forces Institute of Pathology (U.S.) and Luna, 1968). Sections processed for PV immunohistochemistry were incubated in 3% normal goat serum for 1 h and then in monoclonal mouse anti-PV antibodies (Sigma-Aldrich, mouse monoclonal, PARV-19, #P3088, RRID:AB_477329, 1:10000), in PBS/0.3% Triton X-100/3% normal goat serum, for 36 h at 4 °C. All sections were then incubated in goat anti-mouse biotinylated secondary antibodies (Sigma-Aldrich Co., Cat# B7264, RRID:AB_258607, dilution 1:200) in PBS/0.3% Triton X-100 for 1 h, followed by the avidin–biotin procedure (Pierce, Rockford, IL, USA), and reacted with 3,3′-diaminobenzidine dihydrochloride (Sigma-Aldrich). Sections were then mounted on gelatin-coated slides, dehydrated, and coverslipped (Eukitt, Kindler, Germany).
Digital images were acquired with CCD video camera module (20× HI PLAN Leica objective). The entire coronal section of dorsal hippocampus was reconstructed by joining the acquired images and then analyzed with the public domain ImageJ program (Schneider et al., 2012). All cell profiles were counted for each acquired hippocampal section. As in our previous investigations (Del Tongo et al., 2009; Quarta et al., 2015) estimates were performed on the hippocampus of both hemispheres and are reported here as PVi per hippocampus. Quantitative evaluations were performed by using the optical dissector method on counts of cell profiles (Schmitz and Hof, 2005; Sterio, 1984). Image reconstructions were printed out and we analyzed, at the microscope stage under high magnification (40× HI PLAN Leica objective), the same sections with the focus on the upper surface (i.e., the look-up section). The somatic profiles in the focus, through the observation of the look-up section, were marked on the printouts, counted, and subtracted from the total cell counts of the image reconstructions (Jinno and Kosaka, 2002, 2009) to obtain the value N, which expresses the estimated number of PVi per section.
The number of PVi counted per dorsal hippocampus was calculated as follows:
where s represents the single section and n is the last section counted (typically, 6 sections were counted per animal).
2.2. Electrophysiology
Electrical stimulation of Schaffer collaterals generates excitatory postsynaptic potentials (EPSP) in the postsynaptic CA1 neuron. Brief, high-frequency trains of stimuli (HFS) to the Schaffer collaterals cause LTP, which is quantified as the normalized field EPSP (fEPSP) response at 55–60 min after tetanus stimulation (Bliss and Lømo, 1973; Lomo, 1966).
Mice were placed under deep anaesthesia (Avertin, 250 mg/kg i.p.) and transcardially perfused with 25–30 ml of room temperature carbogenated N-methyl-D-glucamine artificial cerebrospinal fluid (NDMG-ACSF composition in mM: N-methyl-D-glucamine 93, HCl 93, KCl 2.5, NaH2PO4 1.2, NaHCO3 30, HEPES 20, Sodium ascorbate 5, Thiourea 2, Sodium pyruvate 3, Glucose 25, MgSO4 10, CaCl2 0.5, N-acetyl-l-Cysteine 12, pH 7.4, 300–310 mOsm), which has been shown to facilitate the preservation of the GABAergic interneurons activity, including that of PVi (Pan et al., 2015; Ting et al., 2014). Brains were gently extracted from the skull within 1 min, sectioned at 300 μm with a vibroslicer (Leica) incubated for further 10 min in the same NMDG-ACSF at 37 °C and then transferred in the holding solution (Composition in mM: NaCl 92, KCl 2.5, NaH2PO4 1.2, NaHCO3 30, Hepes 20, Glucose 25, Sodium ascorbate 5, Thiourea 2, Sodium pyruvate 3, MgSO4 2, CaCl2 2, N-acetyl-l-Cysteine 12, pH 7.4, 300–310 mOsm) until used for recording. Samples (wt, n = 9 slices; mSOD1, n = 8 slices; mSOD1/T1−/−, n = 8 slices) were used within 10 h after sectioning. Slices were perfused with ACSF (Composition in mM: NaCl 124, KCl 2.5, NaH2PO4 1.2, NaHCO3 24, Hepes 5, Glucose 12.5, MgSO4 2, CaCl2 2, pH 7.4, 300–310 mOsm) at 28 °C at the rate of 2 ml/min. No other drugs were used during the recording. For the recordings, two Teflon-coated concentric platinum–iridium electrodes were placed in the stratum radiatum in the CA1 subfield of the dorsal hippocampus, 300–400 μm apart. Borosilicate glass recording electrodes were pulled (Sutter Instruments P90), ACSF filled to get 4–7 MΩ resistance, and placed in the apical dendritic region of CA1 pyramidal neurons evenly spaced with respect to the stimulating electrode. Field potentials were obtained by alternate stimulation of the two electrodes by activation of the Schaffer collaterals. One of the electrodes was used as a control electrode, whereas the other was used to deliver the conditioning protocol. Stimulation intensity for the two channels was set so that the response was 50% of the maximal response. Input/Output analysis was performed at the begin of each recording for all slices. Stimulus intensity was increased in discrete step from 0 until maximal response was achieved. 10 fEPSP were recorded and averaged for each level. Fiber Volley amplitude was measured with respect to baseline level (i.e. average of the signal on the 15 ms preceding the stimulus) and plotted against the initial fEPSP slope.
Baseline recording was obtained by stimulating the slice every 20 s for at least 45 min. Once the baseline was stabilized to obtain LTP, two 100 Hz trains (lasting 1 s each) every 20 s were delivered to the stimulating electrode. Baseline recording was then resumed and followed for 1 h. Field potential was recorded (Multiclamp 700b; Axon Instruments), digitized (10 kHz Digidata 1324), low-pass filtered (3 kHz, eight-pole Bessel), and stored (Clampex 9.2; Axon Instruments). Signals were analyzed off line (Clampfit 9.2; Axon Instruments), and the size of the fEPSP was evaluated by measuring the initial slope of the signal expressed as percentage of the variation from the baseline value (average of 5 min before the conditioning protocol; Carim-Todd et al., 2009; Yanpallewar et al., 2012b). Slices that showed > 5% difference from baseline to the end of recording measured at the control electrode where discarded. Paired Pulse was obtained for delays of 20 ms (with 20 ms increment every 20 s up to 400 ms) using the control stimulating electrode and calculating the ratio of the rising slope (10% to 60%) of the first to the second fEPSP.
2.3. Statistical analysis
Immunohistochemical data are shown per group as mean ± standard deviation. Data of wt, mSOD1, T1−/− and mSOD1/T1−/− mice are compared with one-way or two-way ANOVA, and Bonferroni’s post hoc test. Electrophysiological results are presented as percentage of the baseline slope of fEPSP ± standard error of the mean and followed by ANOVA and Bonferroni’s post hoc test, when necessary.
3. Results
3.1. Hippocampal parvalbumin-positive interneurons
Neurons across genotypes displayed gross morphological similarity and comparable spatial distribution. Most of the PVi were present in the stratum pyramidale of CA1, CA2, and CA3, and in the granular layer and hilus of DG (Fig. 2A–D). The majority of PVi were small- to medium-sized fusiform cells, and triangular neurons (Fig. 2, insets A1–D2).
Fig. 2.
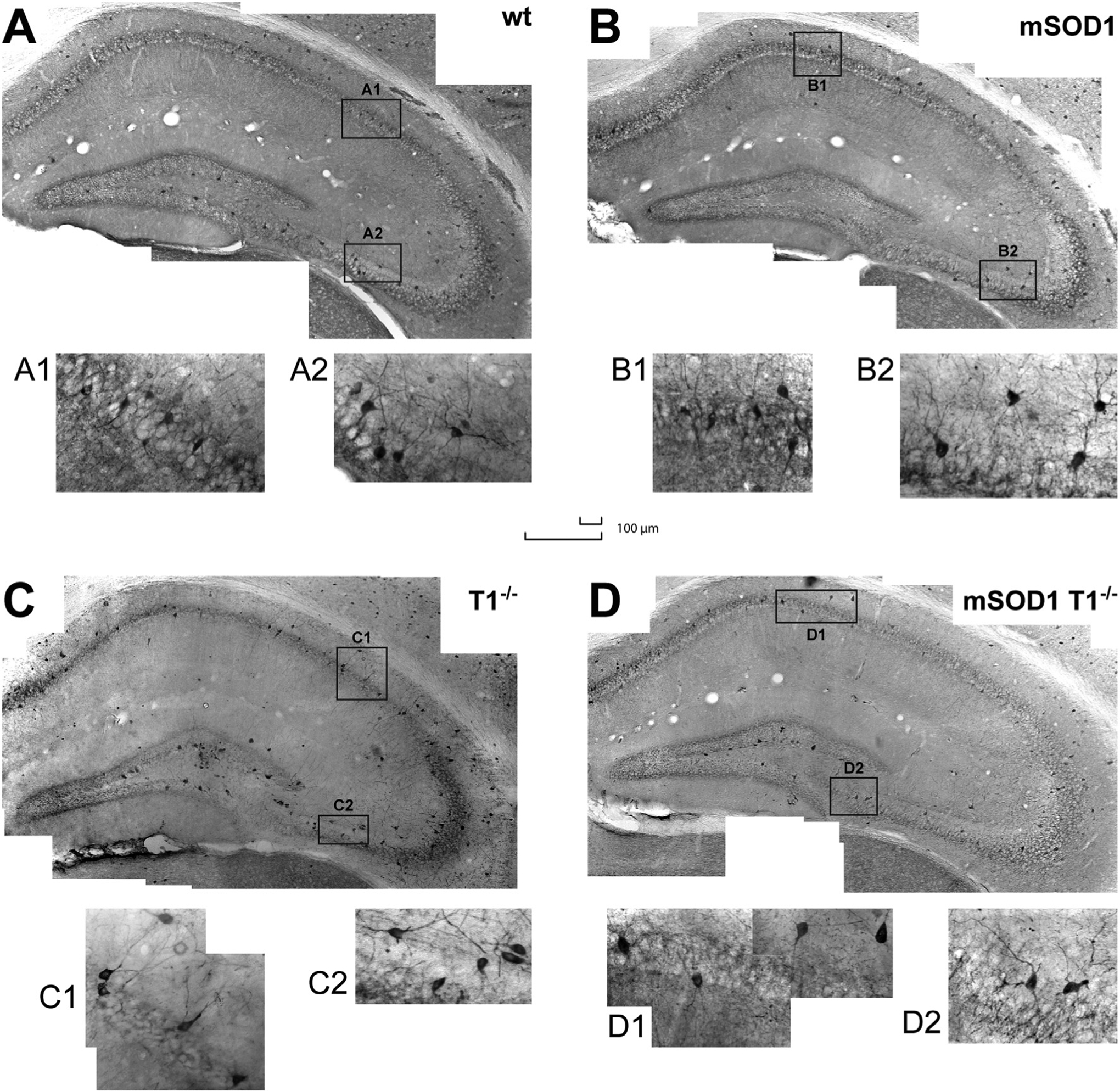
The topographic distribution and gross morphology of hippocampal PVi in wt and mSOD1 mice are not influenced by TrkB.T1.
Histological sections of the hippocampus illustrate the topology of PVi (20×) for representative wt (A), mSOD1 (B), T1−/− (C), mSOD1/T1−/− mice (D). Distribution of PVi in the different subfield is preserved across genotypes. Two insets, one for CA1 and one for CA3 (40×), display morphological features of PVi for wt (A1–A2), mSOD1 (B1–B2), T1−/− (C1–C2), mSOD1/T1−/− mice (D1–D2). Note how the neuronal morphology (insets) appears comparable among genotypes. In the CA1–3 subfields, PVi are mostly distributed in the stratum pyramidale, while in the DG they are found mainly in the granular layer. Both scale bars: 100 μm (large scale for insets).
We have previously reported that the mSOD1 mouse displays a reduced number of PVi in the hippocampus already at a pre-symptomatic phase (Quarta et al., 2015). To assess whether T1 is involved in the maintenance of hippocampal PVi population we performed unbiased stereological estimations of these neurons, separately for the four genotypes. The analysis revealed quantitative differences in the hippocampal PVi populations among the different genotypes (two-way ANOVA, genotype effect: F(3,167) = 50.85, p < 0.01).
When analyzed separately for the four hippocampal subfields, posthoc tests showed that these findings arise from subfield-specific differences among genotypes. The number of PVi in DG and CA2 was comparable among the experimental groups, albeit trend towards numerical increase (for T1) and decrease (for mSOD1) compared to wt were observed. In particular, the estimations of PVi in the DG were 106.33 ± 26.54 in wt, 75.36 ± 19.94 in mSOD1, 130.39 ± 30.96 in T1−/−, and 106.25 ± 17.27 in mSOD1/T1−/− mice (Fig. 3 DG). Analogous results were obtained for the PVi populations in CA2 (wt: 95.28 ± 14.49, mSOD1: 61.11 ± 11.37; T1−/−: 97.82 ± 9.47, mSOD1/T1−/−: 87.29 ± 19.08; Fig. 3 CA2).
Fig. 3.
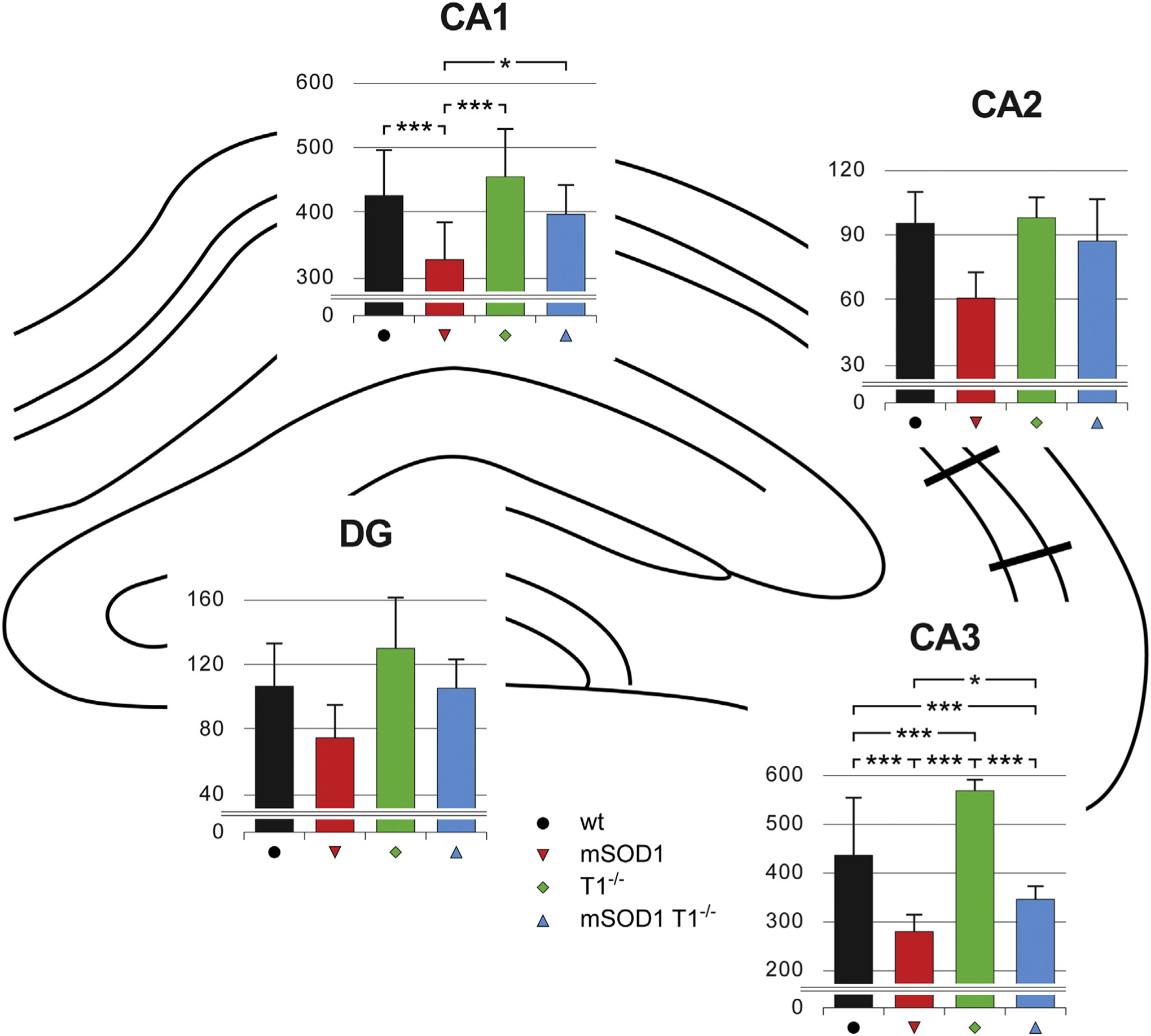
TrkB.T1 shrinks the population of hippocampal PVi in wt and in mSOD1 mice.
Stereological estimations of hippocampal PVi populations are shown, for each subfield, in graphs for wt (n = 6), mSOD1(n = 5), T1−/− (n = 6), mSOD1/T1−/− (n = 6) mice. For each graph, the ordinate indicates the PVi number. The number of PVi is comparable among groups in DG and CA2. T1 deletion results in an increase of PVi number in CA3. In mSOD1 mice both the PVi populations of CA1 and CA3 are found to be reduced compared to wt animals. Knocking out T1 in mSOD1 mice completely restores PVi population in CA1; in CA3, PVi number increases with respect to mSOD1 mice, albeit below the level of controls. Asterisks denote statistical significant differences (* = p < 0.05; *** = p < 0.01).
In line with our previous study (Quarta et al., 2015), the PVi population was found reduced in mSOD1 mice compared to wt animals, for both CA1 (wt: 422.56 ± 70.63, mSOD1: 323.12 ± 55.57, p < 0.001; Fig. 3 CA1), and CA3 (wt: 433.86 ± 118.50, mSOD1: 279.20 ± 42.95, p < 0.001; Fig. 3 CA3). The estimate of hippocampal PVi in mice deficient for this receptor isoform indicated that the PVi population in CA1 did not differ between T1 deficient mice and control animals (wt: 422.56 ± 70.63, T1−/−: 451.45 ± 72.76; Fig. 3 CA1). At variance with results obtained in CA1, the deletion of this receptor led to an increase in the number of PVi that was observed only in CA3 (wt: 433.86 ± 118.50, T1−/−: 565.98 ± 22.27, p < 0.001; Fig. 3 CA3). To our knowledge, these data provide the first evidence for a role of T1 in the maintenance of hippocampal interneurons.
To assess whether T1 is involved in the loss of hippocampal PVi occurring in mSOD1 mice, we next compared the stereological estimates of these interneurons in mSOD1/T1−/− mice with the quantifications of PVi number obtained from wt and mSOD1 mice. The number of PVi in mSOD1/T1−/− was comparable to that of wt mice in CA1 (wt: 422.56 ± 70.63, mSOD1/T1−/−: 395.83 ± 44.66; Fig. 3 CA1), which is indicative for a complete rescue of PVi in this subfield. In the CA3 there was a significant increase in the population of PVi in mSOD1/T1−/− mice compared to mSOD1 animals (mSOD1: 279.20 ± 42.95, mSOD1/T1−/−: 344.79 ± 27.13; p < 0.001). However, this numerical increase did not fully compensate the PVi loss, as the population of these interneurons was lower in mSOD1/T1−/− mice compared to wt animals (wt: 433.86 ± 118.50, mSOD1/T1−/−: 344.79 ± 27.13; p < 0.001; Fig. 3 CA3). Together, these findings indicate that T1 takes part in the loss of hippocampal PVi occurring in pre-symptomatic mSOD1 mice.
3.2. Synaptic plasticity
To investigate whether the rescue of PVi observed in mSOD1 mice knocked-out for T1 also affect the efficiency of synaptic communication, we assessed LTP at the Schaffer collaterals in mice of the three genotypes (wt, mSOD1, and mSOD1/T1−/−, Fig. 3). In the past, we reported the recordings for T1−/− mice, showing that endogenous expression of this receptor is not necessary for LTP elicited by HFS (Carim-Todd et al., 2009).
Acute hippocampal slices from wt, mSOD1, and mSOD1/T1−/− animals were stimulated in CA3 and recordings of fEPSP were obtained in CA1 (Fig. 4A–C).
Fig. 4.
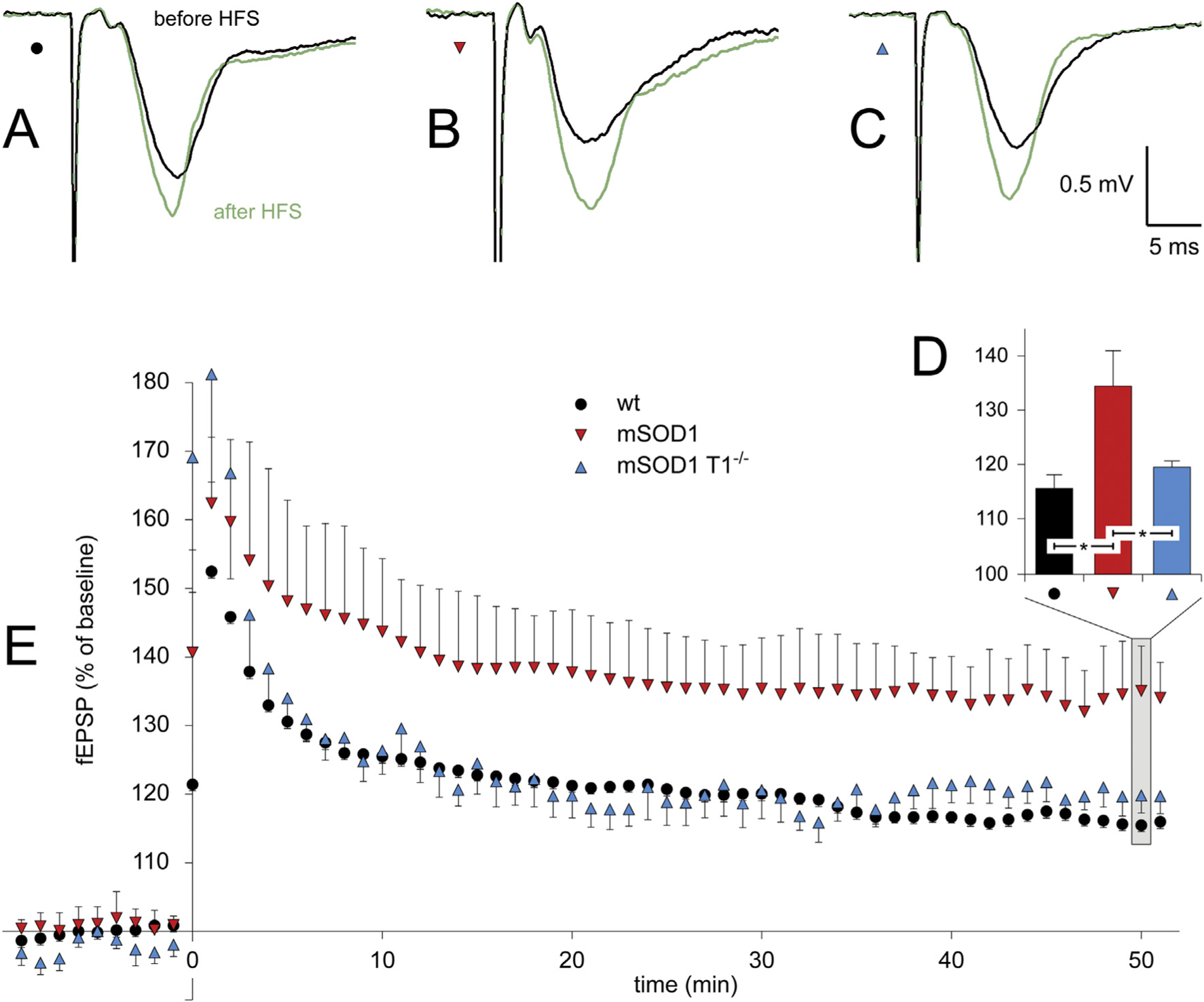
TrkB.T1 enhances LTP in presymptomatic mSOD1 mice.
Representative fEPSP recordings from wt (A), mSOD (B) and mSOD1/T1−/− (C) animals. For each case, the traces are obtained before (gray) and 50 min (green) after the high-frequency stimulation (HFS) conditioning protocol (2 times at 100 ms, 100 Hz trains). The mSOD1 mice (n = 8 slices) exhibit enhanced levels - with respect to wt mice (n = 9 slices) - of long-term potentiation (LTP) 50 min following HFS; at the same time point, mSOD1/T1−/− mice (n = 8 slices) display levels of LTP resembling those of wt mice (D). Following HFS, the potentiation of fEPSP is higher in mSOD1 mice compared to wt mice; the post-HFS fEPSP is comparable between wt mice and mSOD1/T1−/− mice (E). Asterisks denote statistical significant differences (* = p < 0.05). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
No differences between genotypes were observed in terms of basal synaptic communication, as assessed by the input–output curves from the CA3–CA1 subfields (Supplementary Fig. 1, A). Moreover, in the context of the mSOD1 mouse, we found that TrkB.T1 does not modulate paired pulse facilitation (Supplementary Fig. 1, B–C).
We compared LTP in CA1 of the dorsal hippocampus 50 min post-HFS (Fig. 4 A–D). At this time point, we observed differences between genotypes (one-way ANOVA; wt: 15.73 ± 2.5; mSOD1: 34.5 ± 6.47; mSOD1/T1−/−: 19.63 ± 1.12, genotype effect: F(2,24) = 6.26, p < 0.01). The values for mSOD1 mice were higher compared to wt mice (pairwise comparison: p < 0.01).
A significant difference was observed when comparing mSOD1 with mSOD1/T1−/− (pairwise comparison: p = 0.0336). Importantly, the values of mSOD1/T1−/− and wt mice were similar, indicating that the LTP levels were normalized in mSOD1 deficient for T1.
4. Discussion
We reported here experiments carried out to assess the influence of the BDNF receptor TrkB.T1 on hippocampal physiopathology in murine ALS. We found that this receptor i) takes part in the maintenance of the PVi population in specific hippocampal subfields and ii) is involved in two morpho-functional features of the hippocampal phenotype observed in the mSOD1 mouse, i.e., the loss of PVi and the increased LTP.
While the spinal pathology has been well characterized in ALS, only in recent years the potential contribution of cortical pathology has begun to be investigated. Currently, there is increasing interest in identifying cortical components that may regulate motor neuron survival and function, and recent reports have uncovered an interneuron pathology in motor cortex as well as non-cortical regions in ALS (Clark et al., 2017; Minciacchi et al., 2009).
4.1. TrkB.T1 influence on PVi maintenance and rescue of PVi loss by TrkB.T1 deletion in the mSOD1 mouse
Our results unveil for the first time that the PVi population in the hippocampus is increased in mice lacking this receptor isoform. Results indicate that the numerical increase of PVi is significant for CA3. This finding may be suggestive for a subfield specific action of T1; however, considering that CA3 is the largest subfield in the hippocampus, it remains possible that the differences observed in this subfield may more easily reach statistical significance compared to CA1, CA2, and DG, where we nonetheless observed a trend towards increase of PVi number. While it remains to be determined if T1 exert a subfield specific influence or a generalized action on these neurons, our results strongly point to this receptor as an important intracellular player involved in PVi maintenance, which could in turn influence hippocampal information processing.
Abnormal T1 levels have been documented in other neurodegenerative disorders, such as Alzheimer’s disease (Ferrer et al., 1999; Silhol et al., 2005) and Down syndrome, a disorder also associated with cognitive deficits (Dorsey et al., 2002; Hernandez and Fisher, 1996). The accelerated death of hippocampal neurons in a mouse model for Down syndrome, is not rescued by exogenous BDNF delivery (Dorsey et al., 2002) but instead by restoring the physiological levels of T1 (Dorsey et al., 2006). It is currently unknown if the expression levels of T1 are increased in the hippocampus or region-specific PVi selectively overexpress TrkB.T1 in mSOD1 mouse model of ALS. It should be noted that expression of T1, indeed, has been reported in PVi in the brain (Bracken and Turrigiano, 2009; Ohira and Hayashi, 2009). Regardless, the current findings point to a significant contribution of T1 to the hippocampal PVi neurodegeneration occurring in the mSOD1 mouse. Previously, we also reported that T1 participates in the early loss of α-motorneurons in this ALS model (Yanpallewar et al., 2012a). Here, we provide evidence indicating that other types of neurons, in another CNS region, i.e. PVi in the hippocampus, are also susceptible to the regulation by T1 in mSOD1 mice. We speculate that this receptor could be an active cellular player mediating the effect of the mutant protein or it blunts the trophic effects of the TrkB·FL in the PVi. In the light of present results, it is possible that, in ALS, antagonizing T1 action could result in reduced hippocampal interneuron degeneration.
4.2. Autocrine and/or paracrine influence of TrkB.T1 on PVI in mSOD1 mice
The genetic deletion of T1 likely bears at least two neural outcomes that affect intracellular signaling and possibly mediate the effects observed herein: from one side, the loss of dominant-negative inhibition in neurons, on the other side the lack of TrkB.T1-induced signaling in astrocytes.
While the co-localization of T1 and PVi has been reported in the visual cortex (Bracken and Turrigiano, 2009; Ohira and Hayashi, 2009), it is unknown if the same expression pattern is present in the hippocampus. If expressed in PVi, T1 could limit pro-survival effects of BDNF signaling. Interestingly, deletion of TrkB results in PVi loss (Zheng et al., 2011), supporting the hypothesis that PVi are dependent on TrkB signaling. Pyramidal neurons, known to express T1, were shown to be sensitive to T1 manipulation (Michaelsen et al., 2010) and could influence the homeostasis of PVi.
Additionally or alternatively, T1 could indirectly influence hippocampal PVi via astrocytes; T1 is the only TrkB isoform expressed in these glia cells, where it regulates calcium signaling (Matyas et al., 2017; Rose et al., 2003). In the future it will be important to address whether astrocytes, through BDNF-T1 mediated calcium signaling, regulate the GABAergic neurons survival and function and if T1 deletion has any relevance on the astrogliosis known to occur in the mSOD1 mouse.
GABAergic synaptic efficacy can be regulated by removal of GABA and this effect is enhanced in astrocytes upon BDNF stimulation (Vaz et al., 2011). Furthermore, astrocytes modulate inhibitory synapse formation via distinct presynaptic and postsynaptic mechanisms involving BDNF (Elmariah et al., 2005). It is therefore conceivable that astrocytic BDNF-T1 signaling could affect the homeostasis of hippocampal GABAergic neurons and the excitatory neurotransmission (Boddum et al., 2016; Losi et al., 2014).
While the cellular mechanisms mediating the results presented herein remain to be discovered, our work encourage future enquiries, including conditional knock-out of T1 in PVi, in other neuron types, or in astrocytes, to determine the cell type-specific roles of T1 on interneuron and LTP homeostasis.
4.3. A link between PVi, BDNF signaling and LTP in SOD1 mutant mice?
Our results extend the knowledge on electrophysiological abnormalities in the hippocampus of the mSOD1 mouse (Spalloni et al., 2006), indicating that LTP enhancement occurs in an early pre-symptomatic phase. As blocking GABA-mediated transmission facilitates LTP induction at excitatory synapses (Meredith et al., 2003; Wigström and Gustafsson, 1983), we suspect that a decrease in the PVi population may lead to a reduced inhibitory input, which would at least partially account for the increased LTP observed in the mSOD1 mouse. Accordingly, LTP increases have also been reported in concomitance with decreases of hippocampal PVi number in other neurodegenerative conditions (Auffret et al., 2010; Levenga et al., 2013; McEachern and Shaw, 1999; Nisticò et al., 2013). If an inverse relation links the GABAergic PVi population and LTP, hyperexcitability induced by PVi loss should facilitate LTP, as it is indeed the case (Casasola et al., 2004). Consistently, in ALS patients, cortical hyperexcitability appears to be closely related to the altered interplay between excitatory cortico-motoneurons and inhibitory interneurons (Bae et al., 2013). Our results in the mSOD1 model extend this notion to hippocampal circuits and encourage future enquiry in ALS patients. Since it has been reported that increased hippocampal activation is a dysfunctional condition and that targeting excess hippocampal activity has therapeutic potential (Bakker et al., 2012), counterbalancing for the hippocampal hyperactivity may be beneficial for the cognitive profile of ALS patients.
Previous report indicate that BDNF enhances synaptic transmission in hippocampal neurons via postsynaptic TrkB receptors (Levine et al., 1995). The post-synaptic argument would fit well with our results indicating a complete rescue of PVi in CA1 of mSOD1 mice deficient for T1. However, since the PVi population is partially restored in CA3, we cannot rule out that presynaptic neurotransmitter release could also be altered by an increase in the number of PVi neurons leading to enhanced GABA transmission. PVi-specific genetic deletion of TrkB cause a 20% loss of these neurons and alters hippocampal gamma (30 to 90 Hz range) oscillations (Buzsáki and Wang, 2012; Zheng et al., 2011), advocating for a pivotal role of TrkB in PVi survival and function and - more specifically - in network communication. Changes in hippocampal oscillations has been associated to changes of LTP (Bikbaev, 2007; Kalweit et al., 2017).
LTP is normally related with animal learning. Therefore, the increased LTP level observed in the pre-symptomatic mSOD1 mouse apparently stride with the abnormal spatial behavior observed in our previous work (Quarta et al., 2015). Intriguingly, increased LTP has also been associated with impaired learning (Migaud et al., 1998) and it has been suggested that saturation of LTP may reduce information-storage capacity, consistent with theories proposing that plasticity may be detrimental to network efficiency (Cohen et al., 2015, 2017; Roth-Alpermann et al., 2006; Woollett and Maguire, 2009).
In the context of the mSOD1 mouse, our results suggest that T1 deletion act as “LTP-desaturation” strategy since T1 does not alter LTP in physiological conditions (Carim-Todd et al., 2009). It is important to underline that the implications of our electrophysiological findings must be contextualized to the HFS protocol employed. For example, theta-burst stimulation (TBS) elicits LTP via intracellular pathways that are partially different from those involved in HFS (Zhu et al., 2015). As such, the relative influence of T1 for induction and maintenance of LTP could vary depending on to the stimulation paradigm. In the future investigations, testing other LTP-inducing paradigms such as those based on BDNF might unravel novel roles for T1 in synaptic function.
Future studies will determine whether T1 modulates the performance in spatial navigation tasks. T1 has been shown to regulate anxiety-like behavior (Carim-Todd et al., 2009) and sleep (Watson et al., 2015). Given the robust interplay between spatial navigation and sleep (Guan et al., 2004; Nguyen et al., 2013; Ravassard et al., 2016), and between anxiety and spatial navigation (Hund and Minarik, 2006), at least subtle aspects of this latter behavior may be affected in the T1−/− mouse, especially during adulthood, when T1 becomes the predominant TrkB isoform (Fenner, 2012; Silhol et al., 2005). Considering that manipulating the expression of T1 or TrkB·FL alters the spatial behavior impairments in a mouse model of Alzheimer’s disease (Kemppainen et al., 2012), an important question that remains open is whether T1 deletion may be a viable strategy to rescue, at least in part the cognitive abnormalities of the mSOD1 mouse.
In conclusion, this study provides the first evidence for a role of T1 in the survival of hippocampal PVi, and suggest that antagonizing the activity of this receptor may help attenuating hippocampal GABAergic dysfunctions and normalize the synaptic plasticity in mSOD1 mouse.
Supplementary Material
Acknowledgments
Grants
Grant sponsor: Pegaso PhD Fellowship founded by the Region of Tuscany to EQ. GF, SY and LT were supported by the Intramural Research Program of the NCI, Center for Cancer Research, NIH.
Abbreviations:
- ALS
amyotrophic lateral sclerosis
- BDNF
brain-derived neurotrophic factor
- CA1
Cornu Ammonis 1
- CA2
Cornu Ammonis 2
- CA3
Cornu Ammonis 3
- DG
dentate gyrus
- fEPSP
field excitatory postsynaptic potentials
- FTD
fronto-temporal dementia
- HFS
high frequency stimulation
- LTP
long-term potentiation
- mSOD1
mutant SOD1(G93A)
- PBS
phosphate-buffered saline
- PVi
parvalbumin-positive interneurons
- SOD1
superoxide dismutase 1
- SOD1(G93A)
B6SJL-TgN(SOD1-G93A)1Gur
- TrkB
tropomyosin receptor kinase B
- TrkB·FL
full length TrkB
- TrkB.T1
truncated TrkB
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.mcn.2018.03.010.
Disclosures
No conflicts of interest, financial or otherwise, are declared by the authors.
References
- Abdulla S, Machts J, Kaufmann J, Patrick K, Kollewe K, Dengler R, Heinze HJ, Petri S, Vielhaber S, Nestor PJ, 2014. Hippocampal degeneration in patients with amyotrophic lateral sclerosis. Neurobiol. Aging 35, 2639–2645. [DOI] [PubMed] [Google Scholar]
- Abrahams S, Leigh PN, Goldstein LH, 2005. Cognitive change in ALS: a prospective study. Neurology 64, 1222–1226. [DOI] [PubMed] [Google Scholar]
- Angelov B, Angelova A, 2017. Nanoscale clustering of the neurotrophin receptor TrkB revealed by super-resolution STED microscopy. Nano 9, 9797–9804. [DOI] [PubMed] [Google Scholar]
- Angelova A, Angelov B, 2017. Dual and multi-drug delivery nanoparticles towards neuronal survival and synaptic repair. Neural Regen. Res 12, 886–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelova A, Angelov B, Drechsler M, Lesieur S, 2013. Neurotrophin delivery using nanotechnology. Drug Discov. Today 18, 1263–1271. [DOI] [PubMed] [Google Scholar]
- Armed Forces Institute of Pathology (U.S.), Luna LG, 1968. Manual of Histologic Staining methods: of the Armed Forces Institute of Pathology Blakiston Division, New York. [Google Scholar]
- Auffret A, Gautheron V, Mattson MP, Mariani J, Rovira C, 2010. Progressive age-related impairment of the late long-term potentiation in Alzheimer’s disease Presenilin-1 mutant knock-in mice. J. Alzheimers Dis 19, 1021–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae JS, Simon NG, Menon P, Vucic S, Kiernan MC, 2013. The puzzling case of hyperexcitability in amyotrophic lateral sclerosis. J. Clin. Neurol 9, 65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker A, Krauss GL, Albert MS, Speck CL, Jones LR, Stark CE, Yassa MA, Bassett SS, Shelton AL, Gallagher M, 2012. Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron 74, 467–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barde YA, Edgar D, Thoenen H, 1982. Purification of a new neurotrophic factor from mammalian brain. EMBO J 1, 549–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartos M, Vida I, Frotscher M, Meyer A, Monyer H, Geiger JRP, Jonas P, 2002. Fast synaptic inhibition promotes synchronized gamma oscillations in hippocampal interneuron networks. Proc. Natl. Acad. Sci 99, 13222–13227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baude A, Bleasdale C, Dalezios Y, Somogyi P, Klausberger T, 2006. Immunoreactivity for the GABAA receptor 1 subunit, somatostatin and Connexin36 distinguishes axoaxonic, basket, and bistratified interneurons of the rat hippocampus. Cereb. Cortex 17, 2094–2107. [DOI] [PubMed] [Google Scholar]
- Bikbaev A, 2007. Hippocampal network activity is transiently altered by induction of long-term potentiation in the dentate gyrus of freely behaving rats. Front. Behav. Neurosci 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TVP, Lømo T, 1973. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J. Physiol 232, 331–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boddum K, Jensen TP, Magloire V, Kristiansen U, Rusakov DA, Pavlov I, Walker MC, 2016. Astrocytic GABA transporter activity modulates excitatory neurotransmission. Nat. Commun 7, 13572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracken BK, Turrigiano GG, 2009. Experience-dependent regulation of TrkB isoforms in rodent visual cortex. Dev. Neurobiol 69, 267–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsáki G, Wang X-J, 2012. Mechanisms of gamma oscillations. Annu. Rev. Neurosci 35, 203–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carim-Todd L, Bath KG, Fulgenzi G, Yanpallewar S, Jing D, Barrick CA, Becker J, Buckley H, Dorsey SG, Lee FS, et al. , 2009. Endogenous truncated TrkB.T1 receptor regulates neuronal complexity and TrkB kinase receptor function in vivo. J. Neurosci 29, 678–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casasola C, Montiel T, Calixto E, Brailowsky S, 2004. Hyperexcitability induced by GABA withdrawal facilitates hippocampal long-term potentiation. Neuroscience 126, 163–171. [DOI] [PubMed] [Google Scholar]
- Clark RM, Blizzard CA, Young KM, King AE, Dickson TC, 2017. Calretinin and neuropeptide Y interneurons are differentially altered in the motor cortex of the SOD1G93A mouse model of ALS. Sci. Rep 7, 44461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen EJ, Quarta E, Fulgenzi G, Minciacchi D, 2015. Acetylcholine, GABA and neuronal networks: a working hypothesis for compensations in the dystrophic brain. Brain Res. Bull 110, 1–13. [DOI] [PubMed] [Google Scholar]
- Cohen EJ, Quarta E, Bravi R, Granato A, Minciacchi D, 2017. Neural plasticity and network remodeling: from concepts to pathology. Neuroscience 344, 326–345. [DOI] [PubMed] [Google Scholar]
- Del Tongo C, Carretta D, Fulgenzi G, Catini C, Minciacchi D, 2009. Parvalbumin-positive GABAergic interneurons are increased in the dorsal hippocampus of the dystrophic mdx mouse. Acta Neuropathol 118, 803–812. [DOI] [PubMed] [Google Scholar]
- Dorsey SG, Bambrick LL, Balice-Gordon RJ, Krueger BK, 2002. Failure of brain-derived neurotrophic factor-dependent neuron survival in mouse trisomy 16. J. Neurosci 22, 2571–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsey SG, Renn CL, Carim-Todd L, Barrick CA, Bambrick L, Krueger BK, Ward CW, Tessarollo L, 2006. In vivo restoration of physiological levels of truncated TrkB.T1 receptor rescues neuronal cell death in a trisomic mouse model. Neuron 51, 21–28. [DOI] [PubMed] [Google Scholar]
- Dorsey SG, Lovering RM, Renn CL, Leitch CC, Liu X, Tallon LJ, Sadzewicz LD, Pratap A, Ott S, Sengamalay N, et al. , 2012. Genetic deletion of trkB.T1 increases neuromuscular function. Am. J. Phys. Cell Physiol 302, C141–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmariah SB, Oh EJ, Hughes EG, Balice-Gordon RJ, 2005. Astrocytes regulate inhibitory synapse formation via Trk-mediated modulation of postsynaptic GABAA receptors. J. Neurosci 25, 3638–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenner BM, 2012. Truncated TrkB: beyond a dominant negative receptor. Cytokine Growth Factor Rev 23, 15–24. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Marín C, Rey MJ, Ribalta T, Goutan E, Blanco R, Tolosa E, Martí E, 1999. BDNF and full-length and truncated TrkB expression in Alzheimer disease. Implications in therapeutic strategies. J. Neuropathol. Exp. Neurol 58, 729–739. [DOI] [PubMed] [Google Scholar]
- Fryer RH, Kaplan DR, Feinstein SC, Radeke MJ, Grayson DR, Kromer LF, 1996. Developmental and mature expression of full-length and truncated TrkB receptors in the rat forebrain. J. Comp. Neurol 374, 21–40. [DOI] [PubMed] [Google Scholar]
- Fulgenzi G, Tomassoni-Ardori F, Babini L, Becker J, Barrick C, Puverel S, Tessarollo L, 2015. BDNF modulates heart contraction force and long-term homeostasis through truncated TrkB.T1 receptor activation. J. Cell Biol 210, 1003–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geral C, Angelova A, Lesieur S, 2013. From molecular to nanotechnology strategies for delivery of neurotrophins: emphasis on brain-derived neurotrophic factor (BDNF). Pharmaceutics 5, 127–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Z, Peng X, Fang J, 2004. Sleep deprivation impairs spatial memory and decreases extracellular signal-regulated kinase phosphorylation in the hippocampus. Brain Res 1018, 38–47. [DOI] [PubMed] [Google Scholar]
- Guerzoni LPB, Nicolas V, Angelova A, 2017. In vitro modulation of TrkB receptor signaling upon sequential delivery of curcumin-DHA loaded carriers towards promoting neuronal survival. Pharm. Res 34, 492–505. [DOI] [PubMed] [Google Scholar]
- Hernandez D, Fisher EMC, 1996. Down syndrome genetics: unravelling a multifactorial disorder. Hum. Mol. Genet 5, 1411–1416. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF, 2003. Trk receptors: roles in neuronal signal transduction. Annu. Rev. Biochem 72, 609–642. [DOI] [PubMed] [Google Scholar]
- Hund AM, Minarik JL, 2006. Getting from here to there: spatial anxiety, wayfinding strategies, direction type, and wayfinding efficiency. Spat. Cogn. Comput 6, 179–201. [Google Scholar]
- Jinno S, Kosaka T, 2002. Patterns of expression of calcium binding proteins and neuronal nitric oxide synthase in different populations of hippocampal GABAergic neurons in mice. J. Comp. Neurol 449, 1–25. [DOI] [PubMed] [Google Scholar]
- Jinno S, Kosaka T, 2009. Stereological Estimation of Numerical Densities of Glutamatergic Principal Neurons in the Mouse Hippocampus. Hippocampus NA–NA. [DOI] [PubMed] [Google Scholar]
- Kalweit AN, Amanpour-Gharaei B, Colitti-Klausnitzer J, Manahan-Vaughan D, 2017. Changes in neuronal oscillations accompany the loss of hippocampal LTP that occurs in an animal model of psychosis. Front. Behav. Neurosci 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y, Katsumaru H, Kosaka T, Heizmann CW, Hama K, 1987. Fast spiking cells in rat hippocampus (CA1 region) contain the calcium-binding protein parvalbumin. Brain Res 416, 369–374. [DOI] [PubMed] [Google Scholar]
- Kemppainen S, Rantamaki T, Jeronimo-Santos A, Lavasseur G, Autio H, Karpova N, Karkkainen E, Staven S, Vicente Miranda H, Outeiro TF, et al. , 2012. Impaired TrkB receptor signaling contributes to memory impairment in APP/PS1 mice. Neurobiol. Aging 33 (1122), e23–39. [DOI] [PubMed] [Google Scholar]
- Klausberger T, 2005. Complementary roles of cholecystokinin- and parvalbumin-expressing GABAergic neurons in hippocampal network oscillations. J. Neurosci 25, 9782–9793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein R, Parada LF, Coulier F, Barbacid M, 1989. trkB, a novel tyrosine protein kinase receptor expressed during mouse neural development. EMBO J 8, 3701–3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein R, Conway D, Parada LF, Barbacid M, 1990. The trkB tyrosine protein kinase gene codes for a second neurogenic receptor that lacks the catalytic kinase domain. Cell 61, 647–656. [DOI] [PubMed] [Google Scholar]
- Kumar A, 2011. Long-term potentiation at CA3–CA1 hippocampal synapses with special emphasis on aging, disease, and stress. Front. Aging Neurosci 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kust BM, Copray JCVM, Brouwer N, Troost D, Boddeke HWGM, 2002. Elevated levels of neurotrophins in human biceps brachii tissue of amyotrophic lateral sclerosis. Exp. Neurol 177, 419–427. [DOI] [PubMed] [Google Scholar]
- Leal G, Afonso PM, Salazar IL, Duarte CB, 2015. Regulation of hippocampal synaptic plasticity by BDNF. Brain Res 1621, 82–101. [DOI] [PubMed] [Google Scholar]
- Levenga J, Krishnamurthy P, Rajamohamedsait H, Wong H, Franke TF, Cain P, Sigurdsson EM, Hoeffer CA, 2013. Tau pathology induces loss of GABAergic interneurons leading to altered synaptic plasticity and behavioral impairments. Acta Neuropathol. Commun 1, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine ES, Dreyfus CF, Black IB, Plummer MR, 1995. Brain-derived neurotrophic factor rapidly enhances synaptic transmission in hippocampal neurons via post-synaptic tyrosine kinase receptors. Proc. Natl. Acad. Sci 92, 8074–8077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillo P, Hodges JR, 2010. Cognition and behaviour in motor neurone disease. Curr. Opin. Neurol 23, 638–642. [DOI] [PubMed] [Google Scholar]
- Lomo T, 1966. Frequency potentiation of excitatory synaptic activity in the dentate area of the hippocampal formation. Acta Physiol. Scand 68, 28. [Google Scholar]
- Losi G, Mariotti L, Carmignoto G, 2014. GABAergic interneuron to astrocyte signalling: a neglected form of cell communication in the brain. Philos. Trans. R. Soc. B 369, 20130609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matyas JJ, O’Driscoll CM, Yu L, Coll-Miro M, Daugherty S, Renn CL, Faden AI, Dorsey SG, Wu J, 2017. Truncated TrkB.T1-mediated astrocyte dysfunction contributes to impaired motor function and neuropathic pain after spinal cord injury. J. Neurosci 37, 3956–3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEachern JC, Shaw CA, 1999. The plasticity–pathology continuum: defining a role for the LTP phenomenon. J. Neurosci. Res 58, 42–61. [PubMed] [Google Scholar]
- Meredith RM, Floyer-Lea AM, Paulsen O, 2003. Maturation of long-term potentiation induction rules in rodent hippocampus: role of GABAergic inhibition. J. Neurosci 23, 11142–11146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelsen K, Zagrebelsky M, Berndt-Huch J, Polack M, Buschler A, Sendtner M, Korte M, 2010. Neurotrophin receptors TrkB.T1 and p75NTR cooperate in modulating both functional and structural plasticity in mature hippocampal neurons. Eur. J. Neurosci 32, 1854–1865. [DOI] [PubMed] [Google Scholar]
- Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RG, Morrison JH, et al. , 1998. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature 396, 433–439. [DOI] [PubMed] [Google Scholar]
- Minciacchi D, Kassa RM, Del Tongo C, Mariotti R, Bentivoglio M, 2009. Voronoi-based spatial analysis reveals selective interneuron changes in the cortex of FALS mice. Exp. Neurol 215, 77–86. [DOI] [PubMed] [Google Scholar]
- Murray AJ, Sauer J-F, Riedel G, McClure C, Ansel L, Cheyne L, Bartos M, Wisden W, Wulff P, 2011. Parvalbumin-positive CA1 interneurons are required for spatial working but not for reference memory. Nat. Neurosci 14, 297–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutoh T, Sobue G, Hamano T, Kuriyama M, Hirayama M, Yamamoto M, Mitsuma T, 2000. Decreased phosphorylation levels of TrkB neurotrophin receptor in the spinal cords from patients with amyotrophic lateral sclerosis. Neurochem. Res 25, 239–245. [DOI] [PubMed] [Google Scholar]
- Nguyen ND, Tucker MA, Stickgold R, Wamsley EJ, 2013. Overnight sleep enhances Hippocampus-dependent aspects of spatial memory. Sleep 36, 1051–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisticò R, Mango D, Mandolesi G, Piccinin S, Berretta N, Pignatelli M, Feligioni M, Musella A, Gentile A, Mori F, et al. , 2013. Inflammation subverts hippocampal synaptic plasticity in experimental multiple sclerosis. PLoS One 8, e54666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohira K, Hayashi M, 2009. A new aspect of the TrkB signaling pathway in neural plasticity. Curr. Neuropharmacol 7, 276–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan G, Li Y, Geng H-Y, Yang J-M, Li K-X, Li X-M, 2015. Preserving GABAergic interneurons in acute brain slices of mice using the N-methyl-D-glucamine-based artificial cerebrospinal fluid method. Neurosci. Bull 31, 265–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ, 2013. Paxinos and Franklin’s the Mouse Brain in Stereotaxic Coordinates Elsevier Academic Press, Amsterdam; Boston. [Google Scholar]
- Phukan J, Pender NP, Hardiman O, 2007. Cognitive impairment in amyotrophic lateral sclerosis. Lancet Neurol 6, 994–1003. [DOI] [PubMed] [Google Scholar]
- Quarta E, Bravi R, Scambi I, Mariotti R, Minciacchi D, 2015. Increased anxiety-like behavior and selective learning impairments are concomitant to loss of hippocampal interneurons in the presymptomatic SOD1(G93A) ALS mouse model. J. Comp. Neurol 523, 1622–1638. [DOI] [PubMed] [Google Scholar]
- Ravassard P, Hamieh AM, Joseph MA, Fraize N, Libourel P-A, Lebarillier L, Arthaud S, Meissirel C, Touret M, Malleret G, et al. , 2016. REM sleep-dependent bidirectional regulation of hippocampal-based emotional memory and LTP. Cereb. Cortex 1991 (26), 1488–1500. [DOI] [PubMed] [Google Scholar]
- Ringholz GM, Appel SH, Bradshaw M, Cooke NA, Mosnik DM, Schulz PE, 2005. Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology 65, 586–590. [DOI] [PubMed] [Google Scholar]
- Rose CR, Blum R, Pichler B, Lepier A, Kafitz KW, Konnerth A, 2003. Truncated TrkB-T1 mediates neurotrophin-evoked calcium signalling in glia cells. Nature 426, 74–78. [DOI] [PubMed] [Google Scholar]
- Roth-Alpermann C, Morris RGM, Korte M, Bonhoeffer T, 2006. Homeostatic shutdown of long-term potentiation in the adult hippocampus. Proc. Natl. Acad. Sci 103, 11039–11044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz C, Hof PR, 2005. Design-based stereology in neuroscience. Neuroscience 130, 813–831. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW, 2012. NIH image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silhol M, Bonnichon V, Rage F, Tapia-Arancibia L, 2005. Age-related changes in brain-derived neurotrophic factor and tyrosine kinase receptor isoforms in the hippocampus and hypothalamus in male rats. Neuroscience 132, 613–624. [DOI] [PubMed] [Google Scholar]
- Spalloni A, Geracitano R, Berretta N, Sgobio C, Bernardi G, Mercuri NB, Longone P, Ammassari-Teule M, 2006. Molecular and synaptic changes in the hippocampus underlying superior spatial abilities in pre-symptomatic G93A+/+ mice over-expressing the human Cu/Zn superoxide dismutase (Gly93 → ALA) mutation. Exp. Neurol 197, 505–514. [DOI] [PubMed] [Google Scholar]
- Sterio DC, 1984. The unbiased estimation of number and sizes of arbitrary particles using the disector. J. Microsc 134, 127–136. [DOI] [PubMed] [Google Scholar]
- Takeda T, Uchihara T, Arai N, Mizutani T, Iwata M, 2009. Progression of hippocampal degeneration in amyotrophic lateral sclerosis with or without memory impairment: distinction from Alzheimer disease. Acta Neuropathol 117, 35–44. [DOI] [PubMed] [Google Scholar]
- Ting JT, Daigle TL, Chen Q, Feng G, 2014. Acute brain slice methods for adult and aging animals: application of targeted patch clamp analysis and optogenetics. Methods Mol. Biol 221–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaz SH, Jørgensen TN, Cristóvão-Ferreira S, Duflot S, Ribeiro JA, Gether U, Sebastião AM, 2011. Brain-derived Neurotrophic factor (BDNF) enhances GABA transport by modulating the trafficking of GABA Transporter-1 (GAT-1) from the plasma membrane of rat cortical astrocytes. J. Biol. Chem 286, 40464–40476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse EG, An JJ, Orefice LL, Baydyuk M, Liao G-Y, Zheng K, Lu B, Xu B, 2012. BDNF promotes differentiation and maturation of adult-born neurons through GABAergic transmission. J. Neurosci 32, 14318–14330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson AJ, Henson K, Dorsey SG, Frank MG, 2015. The truncated TrkB receptor influences mammalian sleep. Am. J. Phys. Regul. Integr. Comp. Phys 308, R199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigström H, Gustafsson B, 1983. Facilitated induction of hippocampal long-lasting potentiation during blockade of inhibition. Nature 301, 603–604. [DOI] [PubMed] [Google Scholar]
- Wong J, Garner B, 2012. Evidence that truncated TrkB isoform, TrkB-Shc can regulate phosphorylated TrkB protein levels. Biochem. Biophys. Res. Commun 420, 331–335. [DOI] [PubMed] [Google Scholar]
- Woollett K, Maguire EA, 2009. Navigational expertise may compromise anterograde associative memory. Neuropsychologia 47, 1088–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanpallewar SU, Barrick CA, Buckley H, Becker J, Tessarollo L, 2012a. Deletion of the BDNF truncated receptor TrkB.T1 delays disease onset in a mouse model of amyotrophic lateral sclerosis. PLoS One 7, e39946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanpallewar SU, Barrick CA, Palko ME, Fulgenzi G, Tessarollo L, 2012b. Tamalin is a critical mediator of electroconvulsive shock-induced adult neuroplasticity. J. Neurosci 32, 2252–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng K, An JJ, Yang F, Xu W, Xu Z-QD, Wu J, Hökfelt TGM, Fisahn A, Xu B, Lu B, 2011. TrkB signaling in parvalbumin-positive interneurons is critical for gamma-band network synchronization in hippocampus. Proc. Natl. Acad. Sci. U. S. A 108, 17201–17206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu G, Liu Y, Wang Y, Bi X, Baudry M, 2015. Different patterns of electrical activity lead to long-term potentiation by activating different intracellular pathways. J. Neurosci 35, 621–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.