

Abstract
Purpose:
Glioblastoma (GBM) is highly resistant to treatment, largely due to disease heterogeneity and resistance mechanisms. We sought to investigate a promising drug that can inhibit multiple aspects of cancer cell survival mechanisms and become an effective therapeutic for GBM patients.
Experimental Design:
To investigate TG02, an agent with known penetration of the blood–brain barrier, we examined the effects as single agent and in combination with temozolomide, a commonly used chemotherapy in GBM. We used human GBM cells and a syngeneic mouse orthotopic GBM model, evaluating survival and the pharmacodynamics of TG02. Mechanistic studies included TG02-induced transcriptional regulation, apoptosis, and RNA sequencing in treated GBM cells as well as the investigation of mitochondrial and glycolytic function assays.
Results:
We demonstrated that TG02 inhibited cell proliferation, induced cell death, and synergized with temozolomide in GBM cells with different genetic background but not in astrocytes. TG02-induced cytotoxicity was blocked by the overexpression of phosphorylated CDK9, suggesting a CDK9-dependent cell killing. TG02 suppressed transcriptional progression of antiapoptotic proteins and induced apoptosis in GBM cells. We further demonstrated that TG02 caused mitochondrial dysfunction and glycolytic suppression and ultimately ATP depletion in GBM. A prolonged survival was observed in GBM mice receiving combined treatment of TG02 and temozolomide. The TG02-induced decrease of CDK9 phosphorylation was confirmed in the brain tumor tissue.
Conclusions:
TG02 inhibits multiple survival mechanisms and synergistically decreases energy production with temozolomide, representing a promising therapeutic strategy in GBM, currently under investigation in an ongoing clinical trial.
Introduction
Glioblastoma (GBM) is the most common primary malignant brain tumor with an annual incidence rate of 27.86 per 100,000 in the United States (1). Despite the statistical improvement of current standard treatment with radiotherapy and temozolomide (TMZ) compared with radiotherapy alone, less than one third of patients survive beyond 2 years and cure is exceedingly rare (2). Therapies targeting particular altered gene products or signal transduction pathways have not provided durable responses in GBM likely due to multiple dynamic and interactive, compensatory, dysregulated pathways (3, 4). Targeting multiple survival mechanisms in tumor can potentially avoid the development of adaptive resistance. In-depth and well-designed preclinical study is the key to identifying such drug candidates to move forward to the clinical trials.
TG02, a pyrimidine-based multikinase inhibitor with good penetration of blood–brain barrier (BBB), has been shown to inhibit multiple cyclin-dependent kinases (CDK) and regulate transcriptional machinery (5, 6). CDK9, the primary target of TG02, is a serine/threonine kinase that is critical for stimulating transcriptional elongation through RNA polymerase II (RNA Pol II; refs. 7–9). CDK7 regulates CDK9 through phosphorylation at Threonine 186 (T186) to facilitate the transcriptional process. CDK9, in turn, controls the RNA Pol II C-terminal domain through phosphorylation of Serine 2 to modulate the transcriptional process (10). When CDK9 activity is inhibited, short-lived proteins are depleted due to their need to be frequently refreshed (11). As a consequence of the transcriptional regulation, TG02 potentially affects multiple essential survival signals of cancers.
TMZ, an orally administered alkylating agent that has proven efficacy in GBM, has a cytotoxic effect associated with induction of cell death (12). However, its treatment effects are limited by multiple resistance mechanisms including, but not limited to, O-6-Methylguanine-DNA Methyltransferase (MGMT) expression (13, 14). We therefore investigated the effects of TG02 in GBM models with both low and high MGMT expression to determine the spectrum of activity of TG02. With a broad spectrum of effects in modulating cell cycle, apoptosis, and survival signaling, TG02 can be a promising therapeutic candidate in high-grade glioma, either as a single agent and/or in combination with TMZ.
Materials and Methods
Reagents and compounds
Antibodies for phospho-CDK9 (#2549), CDK7 (#2916), Survivin (#2808), phospho-H2A.X (Ser139; #9718), Cleaved Caspase-3 (Asp175; #9661), Cleaved PARP (Asp214; #5625), MGMT (#2739), Myeloid Cell Leukemia 1 (Mcl-1) (#5453), HK2 (#2867), Pyruvate kinase isoform M2 (PKM2) (#4053), Lactate dehydrogenase A (LDHA) (#3582), and Stat-3 (#9139) were purchased from Cell Signaling Technology. Anti–b-actin antibody was purchased from Sigma-Aldrich. Antibodies for CDK9 (sc-484), Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (sc-25778), b-actin (sc-47778), and p53 (sc-126) were purchased from Santa Cruz Biotechnology. Anti–x-linked inhibitor of apoptosis (XIAP) (NB100–56183) antibody was purchased from Novus Biologicals. Anti-cytochrome c (ab13575), Phospho-RNA Pol II CTD (ab5095), and mitochondria (ab3298) antibodies were purchased from Abcam. TG02 is licensed and was provided by Tragara Pharmaceuticals. TMZ was purchased from Sigma-Aldrich.
Cell line preparation
The U87, T98G, LN229, HS683, LN18, and Human intestine epithelial cells (HIEC-6, CRL3266) cell lines were purchased from the ATCC. U251 was purchased from Sigma Aldrich. GBM stem cells (GSC) including GSC923 and GSC827 cell lines were obtained from patients’ GBM tissues and generated by the Neuro-Oncology Branch laboratory at National Cancer Institute (NCI) per NCI Institutional Review Board–approved protocol (NCI 02C-0140).
The GSCs were cultured as described previously (15). Briefly, tumor tissues were enzymatically dissociated and grown as spheroids in NBE medium, composed of Neurobasal-A medium, N2 and B27 supplements (Invitrogen) and recombinant human basic fibroblast growth factor (bFGF), EGF, and 1 mmol/L of l-Glutamine, at 37°C with 5% CO2. The U87, U251, LN229, LN18, HS683, T98G, and Mouse glioma GL261 cells were cultured in DMEM medium plus 5% FBS and 1% penicillin/streptomycin. Human astrocytes (HA) were derived from patients undergoing temporal lobe epilepsy surgery, following the informed consent, in accordance with the appropriate Institutional Review Board. Briefly, brain tissues were enzymatically and mechanically dissociated and HA were cultured in DMEM/F12, containing 10% FBS, 8 mg/mL D-glucose, 1% penicillin/streptomycin, and 50 ng/mL amphotericin. All medium constituents were purchased from Life Technologies. Human pulmonary arterial endothelial cells (PAEC) and microvascular endothelial cells (MVEC) were purchased from Lonza. The cells were cultured in endothelial cell growth medium (EBM2-cat# CC-3156, EGM2-cat#-CC-4176, and EGM-2 MV-ca#-CC-4147) from Lonza. HIECs were cultured in OptiMEM1 Reduced Serum Medium with 4% FBS, 20 mmol/L of HEPES, 10 ng/mL of EGF, 10 mmol/L of l-glutamine, and 1% penicillin/streptomycin.
Cell viability assay
The cell viability was determined using a cell counting method. Cells including U87, U251, LN18, LN229, HS683, and T98G were seeded in 12-well cell culture plates at 2 × 104 cells/well. The GSC923 and GSC827 cell lines were seeded at 1 × 105/well. All cells were treated with TG02, TMZ, or a combination of both drugs for 72 hours prior to cell counting using a Beckman Coulter Vi-CELL TM XR cell viability analyzer.
HA and HIEC were seeded at 3.5 × 104/well, and PAEC and MVEC were seeded at 5 × 104/well for overnight culturing. Normal cells were treated with TG02, TMZ, or a combination of both drugs for 72 hours. The cells were replaced to fresh medium without the drugs and were cultured for another 7 days. The cell viability was determined using cell counting method.
Plasmid construction
Expression plasmid for Flag-CDK9 is available through Addgene (plasmid number: 28100).
The expression plasmids of Flag-CDK9 T186E and plasmids of Flag-vector were generated by site-directed mutagenesis according to the manufacturer’s protocol (Invitrogen) as described previously (16). All constructs were verified by DNA sequencing.
Transient transfection
GSC923 and GSC827 cells (5 × 105) were seeded in 12-well plates and transfected with Flag-vector, Flag-CDK9, and Flag-CDK9 T186E expression plasmids using Lipofectamine 2000 reagents (Invitrogen). For U251 cell, the Cellite transfection reagent (Cellomics Technology) was used for cell transfection. After 24-hour transfection, the cells were then treated with 50 nmol/L of TG02 for 24 hours. The cell viability was determined by cell counting method.
Immunoblot analysis
Cell were treated with TG02 (50 nmol/L), TMZ (100 μmol=L), or the combination prior to the cell harvest. The cell lysates were processed with RIPA buffer containing protease inhibitor cocktail (4693132001; Sigma) and phosphatase inhibitor cocktail (4906845001; Sigma) and were collected for Western blotting as describe previously (17).
Colony formation assay
The clonogenic assay was performed by seeding 250 cells per well in 6-well tissue culture dishes. After 24-hour incubation, cells were treated with TG02, TMZ, and TG02/TMZ for 72 hours. Cells were then washed with PBS and incubated in the fresh medium for 14 days. The colonies were stained using 0.5% crystal violet in 25% methanol. Colonies of greater than 50 cells were counted, and the survival fraction was determined by the following equation: survival fraction = numbers of drug treated colonies/numbers of control colonies.
Preparation of subcellular fractions
Cell fractionation was performed as described previously with some modifications (17). Briefly, cells were collected and washed in ice-cold PBS. Cell pellets were resuspended in cytosol extraction buffer with protease inhibitor cocktail on ice followed by centrifugation at 13,000 × g for 30 minutes at 4°C. The supernatant was collected as the cytosolic fraction. The pellet was resuspended in RIPA buffer containing protease inhibitor cocktail on ice for 20 minutes and then centrifuged at 13,400 × g for 20 minutes at 4°C. The supernatant was collected as the particulate fraction.
Raman imaging
GSC923 cells were treated with the indicated drugs for 48 hours prior to the imaging by a DXRxi Raman confocal microscope (Thermo Scientific) using a 780-nm excitation laser, 60X immersed objective, 24 mW of laser power, and 25-µm slit aperture for 1s/pixel/scan integration time. Spectra were collected using the Thermo Scientific OMNIC Software, integrated, and background corrected. OMNICxi profile Chemigram function was fixed between 745 and 755 cm−1 and used for map visualization of Cyt c distribution.
Electron microscopy
Tissue was double-fixed in PBS-buffered glutaraldehyde (2.5%) and osmium tetroxide (0.5%), dehydrated, and embedded into Spurr’s epoxy resin. Ultrathin sections (90 nm) were made and double-stained with uranyl acetate and lead citrate, and viewed in a JEOL JEM 1010 transmission electron microscope (18).
Immunofluorescence microscopy
The cells were treated, collected, washed with PBS, fixed in 4% paraformaldehyde (PFA), and kept in a dark environment for 20 minutes at room temperature. After washing with PBS, the terminal transferase-mediated dUTP-fluorescein nick endlabeling (TUNEL) assay was performed as per the manufacturer’s instructions (Boehringer Mannheim). 4′,6-diamidino-2-phenylindole was applied to the samples after the final wash to visualize cell nuclei. Images were taken under Zeiss LSM710 confocal microscope. TUNEL assay kit was obtained from Roche Diagnostics.
Flow cytometry detection of mitochondrial membrane potential
Mitochondrial membrane potential (∆ym) in live cells was detected by staining the cells with a cell-permeant and lipophilic dye, 3,3′-dihexyloxacarbocyanine Iodide (DiOC6) (D273; Thermo Fisher). Cells were treated with 50 nmol/L of TG02 and/or 100 μmol/L of TMZ for 48 hours before incubation with DiOC6 for 30 minutes. Cellular uptake of DiOC6 was analyzed immediately by flow cytometry on a Beckman Coulter.
ATP determination
In triplicate, 1 × 105 GSC923 cells were collected and lysed after drug treatments, including TG02, TMZ, and TG02/TMZ for 48 hours. The intracellular ATP level was measured in the cell lysate using ATP assay Kit (ab83355) as per the manufacturer’s protocol. Absorbance was measured at OD 584 nm using a Polarstar OPTIMA plate reader.
Measurement of isolated complex I nicotinamide adenine dinucleotide hydride dehydrogenase activity
The nicotinamide adenine dinucleotide hydride (NADH) dehydrogenase activity of complex I was measured using the complex I enzyme activity microplate assay Kit (Abcam). Cells were treated with 50 nmol/L of TG02, 100 µmol/L of TMZ, or combination with both drugs for 48 hours and then were collected and lysed in lysis buffer provided by the manufacturer. A final protein concentration of 5 mg/mL was used for the assays. Absorbance was measured at OD 450 nm using a Polarstar OPTIMA plate reader.
Extracellular acidification rate measurement
Seahorse XF Glycolysis Stress assays were performed as per the manufacturer’s protocol (Seahorse Bioscience). Briefly, 10,000 cells were seeded on XF96 cell culture plates. Cells were treated with TG02 (50 nmol/L), TMZ (100 µmol/L), and both drugs for 48 hours. The extracellular acidification rate (ECAR) was recorded and calculated for basal glycolysis and glycolytic capacity (19). Reagents, including 10 mmol/L Glucose, 2 µmol/L oligomycin, and 50 mmol/L 2-deoxy-glucose (2-DG), were added as per the supplier’s technical specifications.
Cell apoptosis detection
Cells were treated with 50 nmol/L of TG02 and/or 100 µmol=L of TMZ for 48 hours. The apoptosis assay using differential staining of cells with propidium iodide (PI) and Hoechst 33342 (Invitrogen; catalog no. V23201) was performed by flow cytometry analysis based on the manufacturer’s protocol. Nucleated cells were gated on events exhibiting positive Hoechst staining and high forward light scatter properties. Cells that stained PI negative and Hoechst low (due to degraded DNA) were counted as apoptotic cells.
RNA preparation and sequencing
Total RNA was purified using PureLink@RNA Mini Kit as per the manufacturer’s instructions, and the integrity of the RNA was assessed using an RNA600 Pico LabChip kit on a 2100 Bioanalyzer (Agilent Technologies). RNA samples with RNA Integrity Number greater than 9.0 were submitted to ACGT, Inc (http://www.acgtinc.com/) for sequencing, and paired-end sequencing was performed with 125 bp read length. Data are available in the repository Gene Expression Omnibus (GSE107601).
RNA sequencing data analysis
The RNA sequencing data were processed using CCBR Pipeliner-a Next Generation Sequencing data analysis pipeline, developed by CCR Collaborative Bioinformatics Resource (CCBR) (https://github.com/CCBR/Pipeliner), and deployed on the NIH high-performance computing environment (https://helix.nih.gov). Genes with more than 5 counts per million (cpm) in at least two samples were submitted to differential-gene-expression (DEG) analysis. Three DEG algorithms, EdgeR, DEseq2, and voom/Limma, are supported in the RNASeq Pipeline. For each contrast, the genes with an FDR ≤ 0.05 and fold change ≥2 as determined by all three DEG algorithms were considered differentially expressed. Principle component analysis (PCA) and supervised hierarchical clustering (HC) were carried out using Partek Genomics Suite version 6.6. Gene counts as summarized by EdgeR were used in PCA. Mitochondrial complexes used in HC were obtained from QIAGEN’s Ingenuity Pathway Analysis (IPA, QIAGEN Redwood City, www.qiagen.com/ingenuity). Enrichment of curated pathways from IPA’s Knowledge Base was determined using IPA’s significance thresholds.
Intracranial GBM model, bioluminescence image, and treatment
A syngeneic orthotopic GBM mouse model was created for the evaluation of treatment effects of TG02, TMZ, and combined treatment of TG02/TMZ in vivo according to an approved animal study proposal by NCI-Animal Use and Care Committee (ACUC). Mouse glioma GL261 cells that have been stably transduced with the red-shifted firefly luciferase gene (GL261 Red-FLuc) were purchased from PerkinElmer, Inc. Cells were injected stereotactically into the striatum of C57BL/6 albino mice (6–8 weeks old; Charles River Frederick Research Model Facility) using a stereotactic device (coordinates, 2 mm anterior and 2 mm lateral from bregma, and 2.5 mm depth from the dura). Tumor growth was monitored for luciferase expression using the PerkinElmer IVIS Spectrum. Mice were imaged 10 minutes after intraperitoneal injection of luciferin at 150 mg/kg. TG02 administration started on day 5 (p.o. gavage twice a week, 30 mg/kg) and TMZ on day 7 (p.o. gavage daily for 5 days, 5 mg/kg) after GL261 cells intracranial injection. When the animals reached end-points, they were euthanized by perfusion with 4% PFA under anesthesia, and the brains were dissected for histopathologic examination and immunochemistry analysis.
Pharmacodynamics study
An intracranial orthotopic model was utilized for evaluation of pharmacodynamics (PD) of TG02 in vivo, according to an approved animal study proposal by NCI-ACUC. GL261 Red-FLuc were injected stereotactically into the striatum of C57BL/6 albino mice (6–8 weeks old, Charles River Frederick Research Model Facility) using a stereotactic device. Tumor growth was monitored for luciferase expression using the PerkinElmer IVIS Spectrum as previously described. TG02 treatment started on day 5 (p.o. gavage twice a week, 30 mg/kg) after GL261 cell intracranial injection. At day 19, mice were anesthetized and sacrificed. The brain tissues were dissected for histopathologic examination and immunochemistry analysis. The brain tumor samples were obtained and lysed in CelLytic MT Cell Lysis Reagent (Sigma, C3228), and then the samples were homogenized and sonicated. The lysates were centrifuged at 14,000 × g for 20 minutes at 4°C. Supernatants were collected and protein concentration was measured. The protein expressions of CDK9, phopho-CDK9, and GAPDH were determined by Western blotting.
Statistical analysis
Statistical analyses were performed using a GraphPad Prism software (Version 6.05, GraphPad Software, Inc.) and considered significant at *, P < 0.05; **, P < 0.01; ***, P < 0.001, or ****, P < 0.0001 level. Data are shown as mean ± SEM. One-way ANOVA or independent Student t test was used for statistical comparisons. Synergistic effect was analyzed by CompuSyn software (Combo-Syn Inc.; ref. 20).
Results
TG02 inhibits transcriptional progress via CDK9 activity suppression to induce apoptosis
To evaluate the effect of cytotoxicity in TG02-treated GBM, cell viability was tested in four different GBM cell lines, including two primary patient-derived GSC lines from our laboratory (GSC923 and GSC827) and two commercially available GBM cell lines (U251 and LN18). The half maximal effective concentration (EC50) of TG02 is between 36 nmol/L and 58 nmol/L in these cell lines (Fig. 1A). An in vitro assay of the kinase spectrum following TG02 treatment has shown a downregulation in the kinase activity of CDK7 and CDK9 (6). Phosphorylation of CDK9 at T186 positively regulates the transcriptional elongation process through RNA Pol II, a critical mediator in transcription (7, 8). Here, we found that the expression of CDK7 was downregulated significantly in GSC923 and GSC827 and slightly in U251 cells following the treatment with TG02 for 24 hours (Fig. 1B and Supplementary Fig. S1A). Interestingly, TG02-induced downregulation in CDK9 expression was only found in GSC923 and GSC827, but not in U251 cells. This difference may be explained by the changes in expression of the transcription factors that control the promoter regions of CDK9, such as p53 and signal transducer and activator of transcription 3 (STAT3), following TG02 treatment (Supplementary Fig. S2A and S2B). Nevertheless, the phosphorylated CDK9 in all three cell lines was consistently suppressed by treatment with TG02 (Fig. 1B and Supplementary Fig. S1A). As a consequence, the protein expression of phospho-RNA Pol II was decreased following TG02 treatment. We demonstrated that TG02 suppressed the transcriptional process in GBM cells through reducing the CDK9 activity but not necessarily through the decreasing of the protein expression of CDK9.
Figure 1.
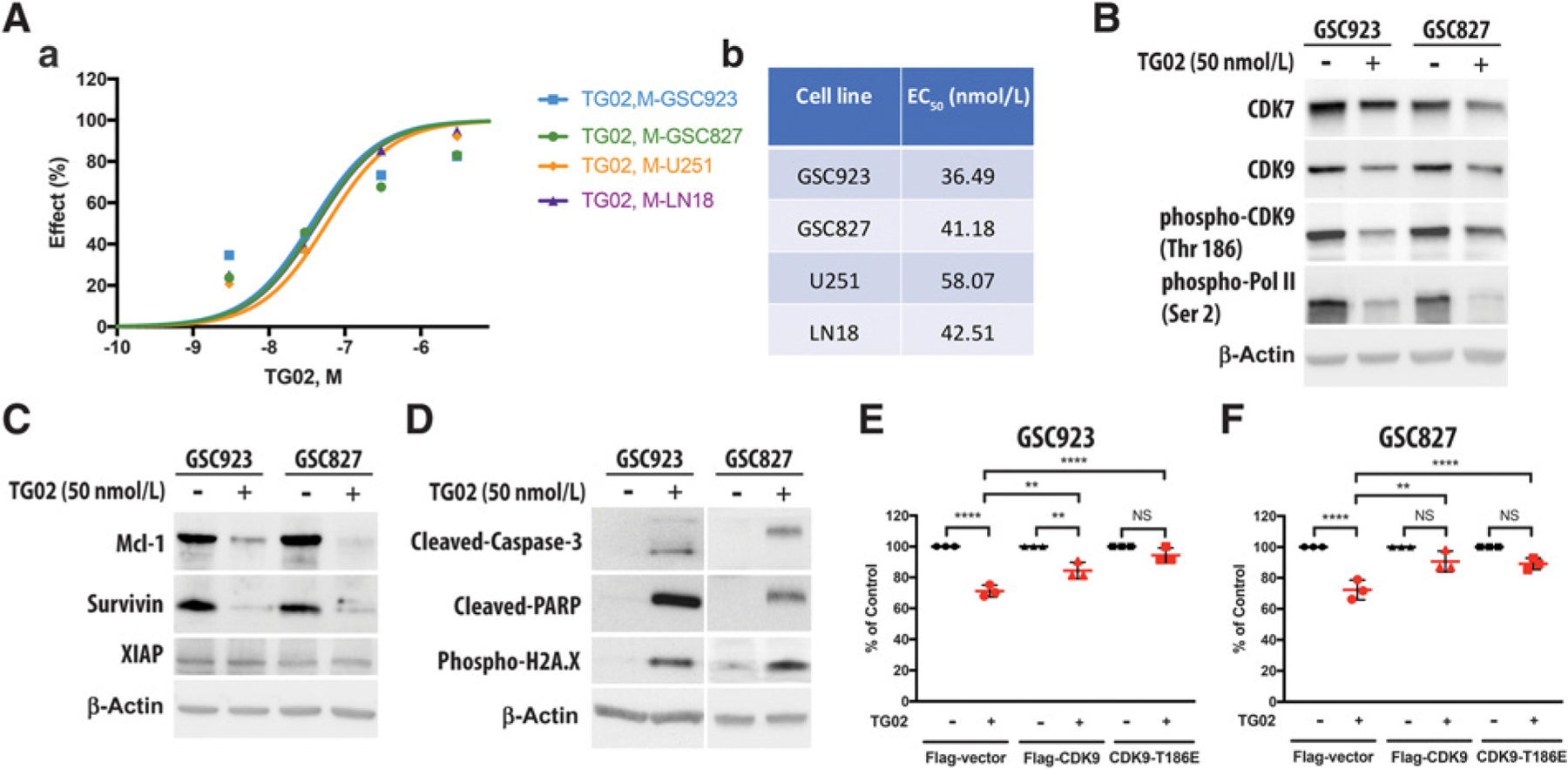
Downregulated phosphorylation of CDK9 is a key factor in TG02-mediated cell death. A, The cytotoxic effect of TG02 in GSC923, GSC823, U251, and LN18 cells. Cells were treated with different dosages by serial dilution of TG02 for 48 hours prior to cell viability testing. The dose-response curve (plot a) and the EC50 of TG02 in GBM cells are shown in a and b, respectively. B, TG02 alters transcriptional regulation by inhibiting phosphorylation of CDK9 and RNA Pol II. The protein expressions of CDK7, CDK9, phosho-CDK9, and phospho-Pol II in GSC923 and GSC827 cells were determined by Western blotting following treatment with TG02 for 24 hours. C, TG02 decreases the expressions of antiapoptotic proteins. The protein expressions of Mcl-1, Survivin, and XIAP in GSC923 and GSC827 were determined by Western blotting after treatment with TG02 for 24 hours. D, TG02 induces cell apoptosis. The protein expressions of cleaved caspase-3, cleaved-PARP, and phospho-H2A.X were detected in GSC923 and GSC827 cells by Western blotting after treating TG02 for 24 hours. E and F, The overexpression phosphorylated mimic form of CDK9 blocks TG02-mediated cell death in both GSC923 (E) and GSC827 cells (F). The cells were transiently transfected with either Flag-vector control, Flag-CDK9, or Flag-CDK9 T186E and then were treated with 50 nmol/L of TG02. The cell viability was determined using cell counting method (n = 3 biological replicates). Cell viability of the nontreated cells was set as 100% of survival for each group, and the cells transfected with Flag-vector, Flag-CDK9, and CDK9-T186E are illustrated by circle, triangle, and square, respectively. **, P < 0.01; ****, P < 0.0001.
Previous studies have shown that inhibition of transcription progression preferentially depletes short-lived proteins, such as Mcl-1 and XIAP, which are essential for cell survival (11). In our studies, we found that TG02 significantly reduced the protein expressions of Mcl-1 and Survivin but not XIAP in GSC923, GSC827, and U251 cells (Fig. 1C and Supplementary Fig. S1B). Furthermore, we confirmed the activation of Caspase-3 and PARP, chromatin condensation and fragmentation in GSC923 and GSC827 following TG02 treatment (Fig. 1D), suggesting that TG02 induces apoptosis through downregulation of short-lived antiapoptotic proteins, including Mcl-1 and Survivin. In order to validate the TG02-induced cytotoxicity is mediated by the suppressed CDK9 kinase activity, we transfected GBM cells with a plasmid expressing CDK9 or point mutation of CDK9 T186E, which mimics CDK9 phosphorylation with sustained kinase activation of CDK9 (21, 22). Overexpression of CDK9 proteins and phosphorylated mimics in GSC923 and GSC827 cells blocked TG02-induced cytotoxicity (Fig. 1E and F). However, in U251 cells, the attenuated TG02-indued cell death was only observed in cells which were overexpressed CDK9 phosphorylated mimic but not in those with overexpressed CDK9 (Supplementary Fig. S1C). These data suggest that TG02-induced cell death is mediated by reducing CDK9 activity and, but not necessarily by decreasing CDK9 expression.
TG02 synergizes with TMZ to inhibit cell growth and induces cell death in GBM
TMZ has proven treatment efficacy but is limited by multiple resistance mechanisms such as MGMT expression. The broad spectrum of the antitumor effects makes TG02 a potential candidate for a combined therapy with TMZ. To test the cytotoxicity of combination of TG02/TMZ in GBM, cell viability was tested in GBM cell lines that have high expression of MGMT (GSC923, LN18, T98G, and HS683) and those have low expression of MGMT (GSC827, U251, LN229, and U87; Supplementary Fig. S3A). A significant decrease in the percentage of viable cells was observed in both groups of cells after TG02 treatment (Fig. 2A and B). A more profound decreasing in cell proliferation was observed following the combined TG02 treatment with TMZ when compared with single treatment of either TG02 or TMZ in some cell lines, such as GSC923, U251, and U87 cells. To further determine the potential combined effect of TG02 and TMZ, we quantitatively measured the dose–effect relationship of each drug alone and their combinations to determine whether a given combination would gain a synergistic effect using COMPUSYN, a computer software, where a combination index (CI) is calculated from the growth inhibition curves (20). The synergistic cytotoxic effect between TG02 and TMZ was observed in GSC923 cells at all tested concentrations of TG02 (12.5–400 nmol/L) and TMZ (25–800 µmol=L), where the CI values were less than 1, indicating a synergistic effect (Fig. 2C–a). A similar synergistic effect between TG02 and TMZ was found in LN18 cells (Supplementary Fig. S3C). In a MGMT low-expressing primary GBM cell line, GSC827 (Fig. 2D), the synergy of combined treatment was observed at all tested concentrations of TG02 and TMZ, but not at lower concentration (6.25 nmol/L of TG02 and 12.5 µmol=L of TMZ). In U251, another MGMT low-expressing cell, the synergistic effect was only observed at lower doses of combination treatment, whereas at higher than 300 nmol/L of TG02 and 300 µmol/L of TMZ (Supplementary Fig. S3B), an antagonistic effect was observed. Overall, these results suggest that TG02 can synergize with TMZ to induce cell death in GBM cells with various genetic backgrounds, more consistently in the MGMT high-expressing cells.
Figure 2.
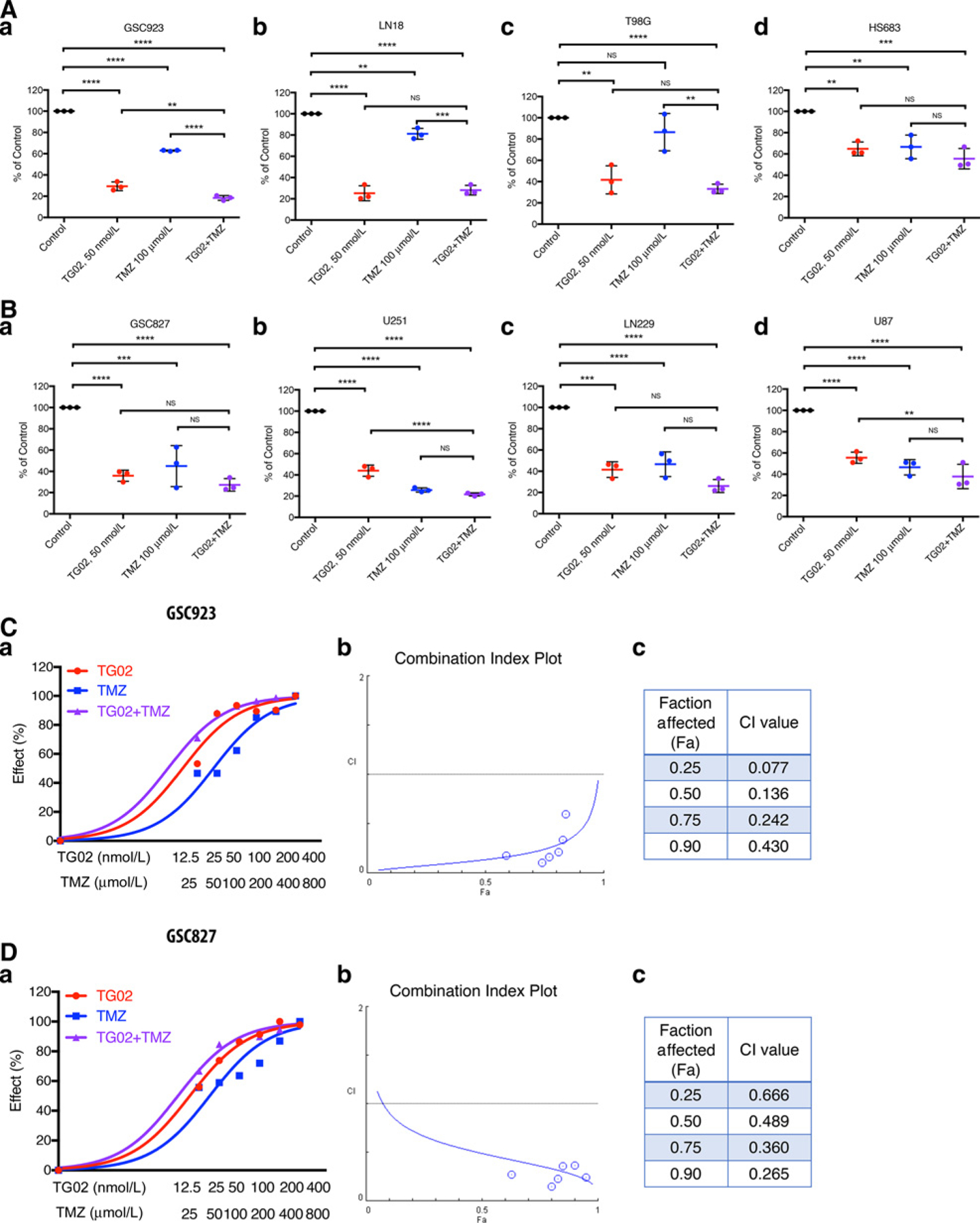
TG02 induces cell death and synergizes with TMZ in GBM. A and B, TG02 inhibits cell growth in both MGMT high-expressing (A, plots a–d) and MGMT low-expressing cells (B, plots a–d). Cell viability was determined after treatment with vehicle, TG02, TMZ, or TG02/TMZ for 72 hours. C and D, Combined TG02/TMZ treatment has a synergistic antiglioma effect in GSC923 (C) and GSC827 cells (D). Dose-response curves (plot a) were plotted from the cell viability following 72 hours of treatment. The synergistic effects of TG02/TMZ in the cells were determined by CI in b. CI values were calculated by COMPUSYN software and are shown in c. CI < 1 is a synergistic, CI = 1 is an additive, and CI > 1 is an antagonistic effect of the two compounds combined (20). **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
TG02 and TMZ induce mitochondrial dysfunction
The constitutive protein expressions of Survivin and Mcl-1 are critical for maintaining mitochondrial integrity and functions in the cells (11, 23, 24). Mcl-1 has been characterized mostly in hematologic malignancies. However, some reports demonstrated aberrant expression of Mcl-1 in some solid tumors, including GBM (25–27). Our findings of TG02-induced reduction of Mcl-1 and Survivin expression suggested a potential mitochondrial dysfunction in TG02-treated GBM. We hypothesized that TG02 induces mitochondrial dysfunction to tumor cells. The RNA sequencing was carried out to analyze the profile of RNA expression following TG02, TMZ, and TG02/TMZ treatment in GSC923 cells. The transcriptomic profiles obtained by CCBR Pipeliner were studied using PCA (Fig. 3A). Expressions of genes of mitochondrial complexes that were derived from the mitochondrial dysfunction pathway in IPA are plotted in a heat map. The supervised HC revealed distinct differences between the TG02 and TG02/TMZ groups compared with the controls and TMZ alone group, suggesting that the changes in mitochondrial function were primarily caused by TG02, but not TMZ. Most of the genes involved in respiratory complexes III to V were downregulated by TG02 (Fig. 3B). Complex I had the most gene expression changes and therefore the overall NADH dehydrogenase activity of complex I was measured. We found that TG02, TMZ, and combination of TG02/TMZ decrease the overall activity of complex I (Fig. 3C). To determine whether the treatments induced mitochondrial damages, mitochondrial membrane integrity was investigated using a fractionation assay followed by Western blotting to check for release of Cyt c in the cytoplasm, an indicator of mitochondrial damage. The ratio of Cyt c/internal control in the mitochondria decreased following TG02, TMZ, and TG02/TMZ treatment. The ratio increased 6.75-, 4.18-, and 9.29-fold in the cytosolic fraction (Fig. 3D), suggesting Cyt c release from mitochondria to cytoplasm following TG02 and TMZ treatment. Raman imaging with Cyt c labeling was performed to explore the intracellular distribution of Cyt c. In the control condition, the labeled Cyt c was locally distributed intracellularly, presumably confined to the mitochondria in the GSC923 cells (Fig. 3E–a). After adding TG02 and TMZ (plots b and c), the labeled Cyt c became more diffusely distributed, suggesting a disruption of the mitochondrial membrane and release of Cyt c. Moreover, the combination of TG02 and TMZ (plot d) resulted in complete disruption of the cell and almost uniform distribution of Cyt c in the entire analyzed area. ∆ym, critical for maintaining the function of mitochondria to produce ATP, was found to be disrupted in TG02 and TG02/TMZ-treated GSC923 (Fig. 3F). Consistent with our previous findings, we observed damaged mitochondria in TG02, TMZ, and TG02/TMZ-treated GSC923 cells using electron microscopy (Fig. 3G). The cell membrane, nuclear envelope, and mitochondria in the untreated tumor cells (plot a) appear intact with well-defined cristae, while chromatin is diffused. Following TG02 or TMZ treatment (plots b and c), an increased number of cytoplasmic vesicles form alongside cristae-damaged mitochondria, indicated by asterisk symbols, suggesting mitochondrial dysfunction. Combined TG02 and TMZ treatment (plot d) led to profound dysmorphic mitochondria, vesicle formation, nuclear envelope separation, condensed nuclei, and profound cell death. We demonstrated that TG02 and TMZ altered mitochondrial membrane potential, disrupted mitochondrial membrane integrity, and released Cyt c from mitochondria. We confirmed that TG02 downregulated CDK9-Pol II kinase activity and decreased the expression of Mcl-1 and Survivin to induce Caspase-3 activation, leading to cell apoptosis. Moreover, we observed similar degrees of downregulation of CDK9-Pol II pathways in combination of TG02/TMZ compared with TG02 alone in GSC923, GSC827, and U251 cells (Supplementary Fig. S4A–S4D). The percentage of apoptotic cells was found to be increased significantly in GSC923 (Fig. 3H and Supplementary Fig. S4E) following TG02 and TG02/TMZ treatment but not in TMZ treatment. Moreover, we did not observe significant increase in apoptotic cells with additional TMZ to TG02, compared with TG02 alone, indicating that the combined treatment of TG02/TMZ-induced cell death is only partially though cell apoptosis. Although TMZ treatment was shown to induce mitochondrial damage, it did not lead to increasing of cell apoptosis.
Figure 3.
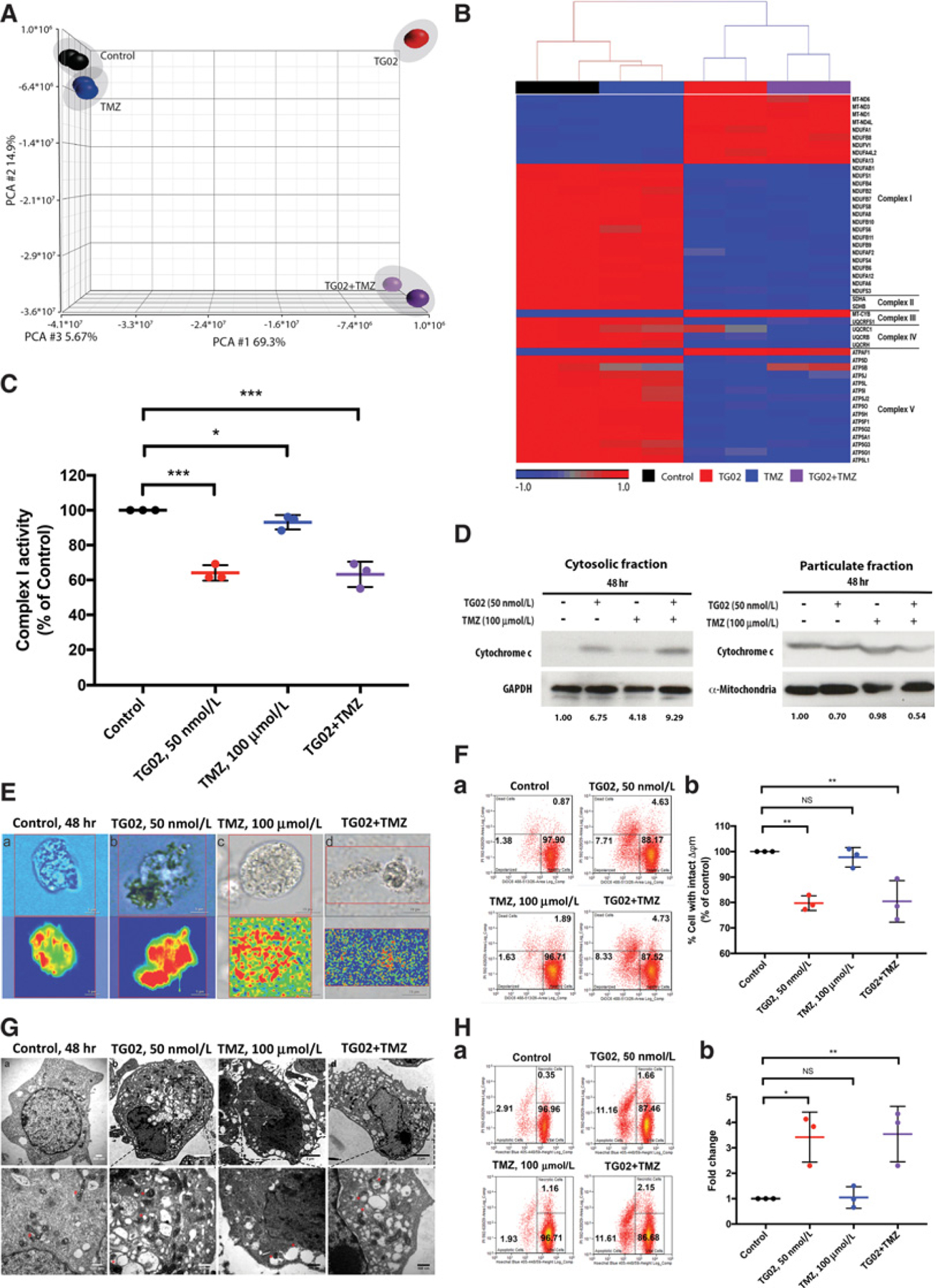
TG02 and TMZ induce mitochondrial dysfunction in GBMs. A, Principal component analysis of the transcriptomic profiles of RNA sequencing of GSC923 that were treated with TG02, TMZ, and TG02/TMZ. B, Supervised HC of mitochondrial complexes I to V. The genes in the heat map were derived from the mitochondrial dysfunction pathway in IPA. C, GSC923 cells were treated with indicated drugs. The NADH dehydrogenase activity was measured using the complex I enzyme activity microplate assay kit (n = 3 biological replicates). D, Western blot of Cyt c in cytosolic and mitochondrial compartments, indicating that Cyt c is released from mitochondria to cytoplasm. GSC923 cells were treated and subjected to cell fractionation separating the cytosolic fraction (left) and particulate fraction (right). We set the intensity of Cyt c/loading control (GAPDH or mitochondria) under no treatment as 1, and the numbers indicate the normalized intensities of Cyt c/loading control. E, The Raman imaging shows the Cyt c location (bottom). Cyt c is shown in red. Top plots are the live images of the cell. F, Flow cytometric analysis of mitochondrial membrane potential changes (∆ym) following TG02, TMZ, and TG02/TMZ treatment in GSC923 cells (a). The statistical analysis was showed in b (n = 3 biological replicates). G, Transmission electron micrographs of TG02-, TMZ-, and TG02/TMZ-treated GSC923 cells. H, TG02 and TG02/TMZ induce cell apoptosis in GSC923. Early apoptosis was determined by staining with Hoechst 33342/PI using flow cytometry (a). The statistical analysis was from three independent experiments (b). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
TG02 synergizes with TMZ in suppression of glycolysis
Reprogramming the energy metabolism toward anaerobic glycolysis is a hallmark of cancer (28). Given the prevalence of glycolysis in tumor cells and the impacts of TG02 on mitochondrial function, we investigated the effect of TG02 on glycolysis in GBM cells. The ECAR was measured as a reflection of glycolytic function in GBM cells following treatments (Fig. 4A). U251, an adherent cell line, rather than GSC line, was chosen for this analysis due to the technique feasibility. Both basal glycolysis and glycolytic capacity were reduced by TMZ, whereas TG02 and TG02/TMZ caused further reduction in the glycolytic capacity (Fig. 4B–a and b). To confirm the glycolytic capability, expressions of several key glycolytic enzymes were evaluated. The protein expression of HK2 was downregulated in both TG02-treated GSC923 and GSC827 cells. PKM2 was decreased in TG02-treated GSC827 but not GSC923 cells. TMZ suppressed the protein expressions of PKM2 and LDHA in GSC923 cells but not GSC827 cells, suggesting that TG02 and TMZ may affect different key steps in glycolysis. More importantly, we found further downregulated protein expressions of HK2, PKM2, and LDHA in both TG02/TMZ-treated GSC923 and GSC827 cells compared with TG02 or TMZ treatment alone (Fig. 4C). Next, we validated the energy productions by measuring the total ATP level in GSC923 cells (29). The cells treated with TG02 showed 30% less ATP compared with control (P < 0.05), whereas 40% less ATP was produced following the combined treatment of TG02/TMZ (P < 0.001; Fig. 4D), suggesting that the TG02-induced reduction of ATP is potentiated by adding TMZ. Our results suggest that the glycolysis suppression is part of the mechanisms for the synergistic effect between TG02 and TMZ. The suppression of the glycolytic pathway is due to the downregulation of the key glycolysis regulators, such as HK2, PKM2, and LDHA. In addition, when treatment-induced mitochondrial dysfunction and glycolysis suppression occur simultaneously, severe cellular energy depletion leads to profound cell death as illustrated in Fig. 4E.
Figure 4.
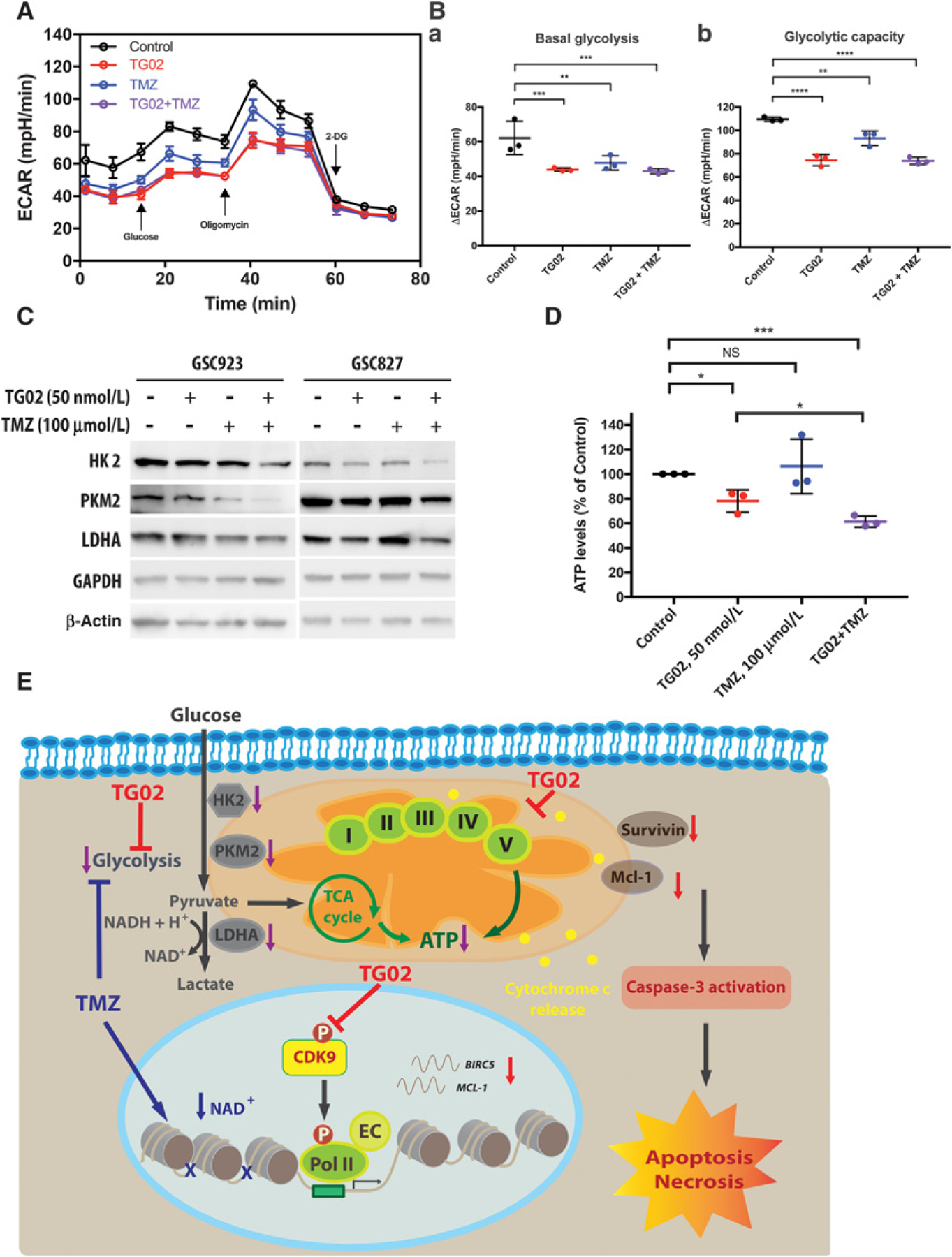
TG02 decreases intracellular ATP levels and suppresses glycolysis as a single agent and in combination with TMZ. A–C, TG02 and TMZ inhibit glycolysis. A, ECAR measurements of U251 cells that were treated with TG02, TMZ, and TG02/TMZ. B, Basal glycolysis (a) and maximal capacity of glycolysis (b) were calculated. C, TG02/TMZ downregulates key enzymes in glycolytic pathway. Western blots of HK2, PKM2, and LDHA in GSC923 and GSC827 cells. D, Intracelluar ATP levels were decreased in TG02 and TG02/TMZ-treated GSC923 cells. ATP levels were determined following TG02, TMZ, and TG02/TMZ treatment. E, Schematic illustrating the mechanisms of the synergism between TG02 and TMZ. TG02 decreased phosphorylation of CDK9 and inhibited RNA Pol II–mediated transcriptional regulation of antiapoptotic proteins, including Mcl-1 and Survivin (also known as BIRC5 gene), leading to release of Cyt c, which induced mitochondrial-mediated apoptosis in GBM cells (shown in red). On the other hand, TG02 also caused disruption of the mitochondrial respiration complexes and inhibition of the glycolysis, resulting in ATP depletion. TMZ induced DNA damage, effectively reducing the level of nicotinamide adenine dinucleotide (NAD+) through PARP activity, and thus suppressed glycolysis to reduce energy production in the cells (shown in blue). The combination of TG02/TMZ treatment downregulated protein expressions of HK2, PKM2, and LDHA to decrease ATP production. The synergism between TG02 and TMZ is partially due to suppressed glycolysis and inhibited ATP generation (shown in purple). As a consequence of disruption of ATP production by combination of TG02/TMZ, the cells undergo apoptosis and necrosis. EC, elongation complex. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
TG02 alone and in combination with TMZ induces cytotoxicity in tumor cells but less in normal cells
A sustained inhibitory effect on cell proliferation and survival was demonstrated by colony formation after GBM cells receiving TG02, TMZ, and TG02/TMZ treatment. Four GBM cell lines including GSC923, GS827, U251, and LN18 were tested and the colonies were counted (Fig. 5A–a–d). We found that none or only few colonies were formed under TG02 treatment in all four GBM cell lines. LN18 and GSC923 cells which express MGMT protein, one of resistant mechanisms to antagonize cytotoxicity of TMZ, were able to form colonies following TMZ treatment (plots a and d). Interestingly, GSC827 cells were capable of forming colonies after TMZ treatment, indicating the cells might have developed resistant mechanisms other than MGMT expression. In all tested cell lines, combination of TG02/TMZ treatment diminished colony forming. We conclude that TG02 and TG02/TMZ treatments have a significant long-term cytotoxic effect on GBMs. The long-term toxicity was also tested in normal cells, including HAs and ECs. In contrast, the TG02-induced cytotoxicity that was found in GBM cells was not observed in HAs and arterial ECs (Fig. 5B–a and b). Microvascular ECs reveal a cytotoxicity from TG02 alone and in combination with TMZ compared with EC from large vessels but considerably less sensitive to the treatments (Fig. 5B–c) compared with GBMs. In addition, we observed that TG02 has more cytotoxic effects on human intestinal epithelial cells, which would explain the gastrointestinal toxicities of TG02 from our in vivo mice studies (Fig. 5B–d) and the clinical experiences with TG02 in cancer patients. To further characterize the possibilities of different sensitivity in response to TG02 between cancer cells and normal cells, we tested the mitochondrial function by measuring the mitochondrial membrane potential changes (∆ym) and cell apoptosis. In normal astrocyte, the treatment with TG02 or TMZ alone did not cause significant mitochondrial dysfunction (Fig. 5C). Although the combined treatment changed mitochondrial membrane potential, neither significant increase in cell apoptosis (Fig. 5D) nor long-term inhibition on cell survival and proliferation (Fig. 5B–a) was observed in normal astrocytes. We previously confirmed that mitochondrial damages and apoptosis were observed after TG02 and TG02/TMZ treatment in GSC923 cells (Fig. 3F and H). These data demonstrate that TG02 alone, and in combination with TMZ, induces mitochondrial dysfunction leading to cell death in GBM but not in normal HAs, suggesting less likelihood of developing irreversible CNS toxicity with the therapeutic dosage in human.
Figure 5.
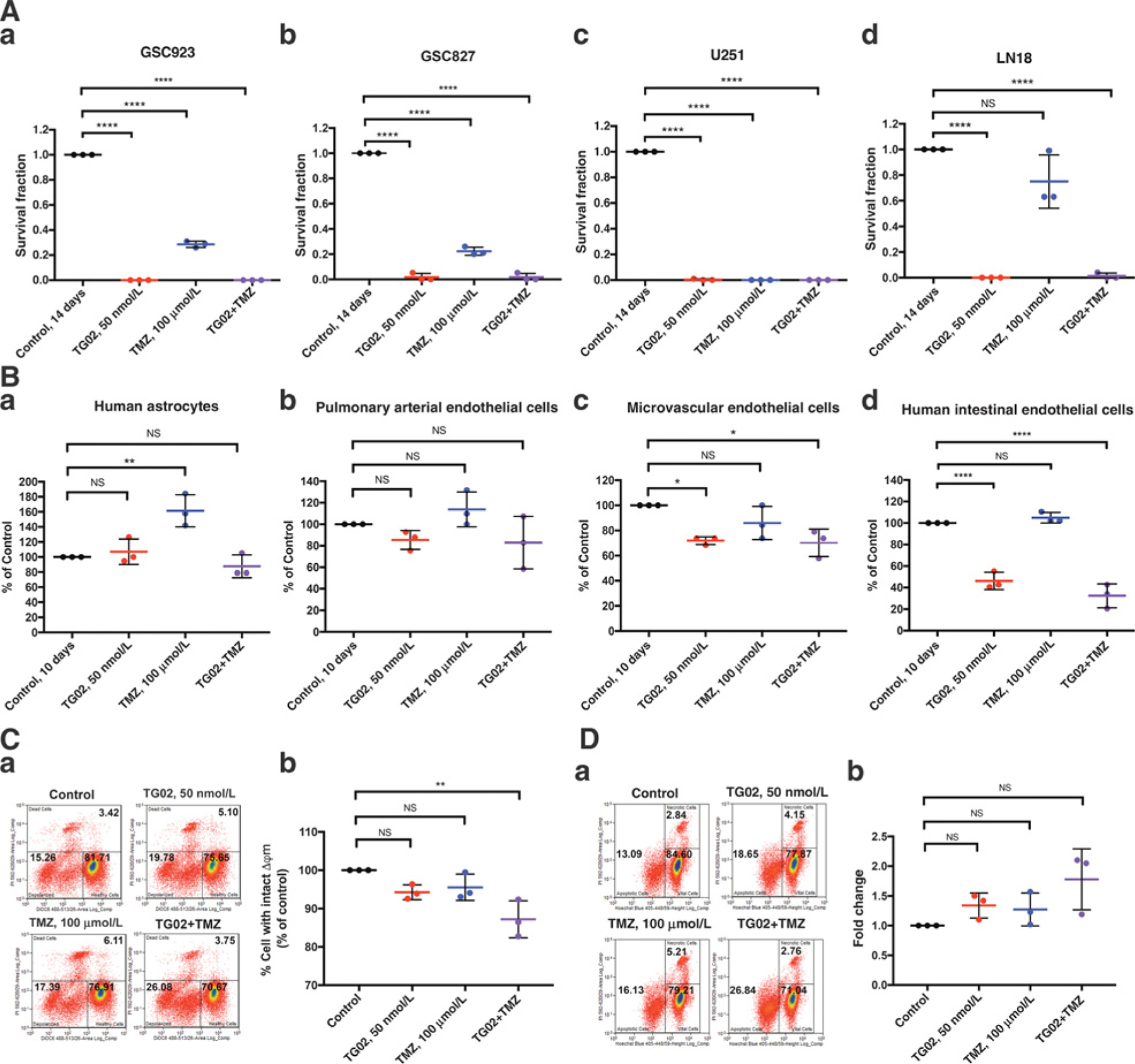
TG02, TMZ, and TG02/TMZ induce cytotoxicity in GBMs but less in normal cells. A, TG02, TMZ, and TG02/TMZ induced cytotoxicity in GSC923, GSC827, U251, and LN18 cells (a–d). Survival fractions are derived from the numbers of colony formation 14 days after 72-hour treatments. The control survival fraction was set as 1, and all other values were normalized to the control. B, HAs (a), normal Human Endothelial Cells (b and c), and normal Epithelial cells (d) are less sensitive to TG02 treatment, reflected by cell viability 7 days after 72-hour treatment. C, Flow cytometric detection of mitochondrial membrane potential changes (∆ym) after TG02, TMZ, and TG02/TMZ treatment in HAs (a). The statistical results from 3 biological replicates are shown in b. D, Flow cytometric detection of apoptosis in HAs following TG02, TMZ, and TG02/TMZ treatment (a). The statistical results from 3 biological replicates are shown in b. *, P < 0.05; **, P < 0.01; ****, P < 0.0001.
Survival benefit from combination treatment of TG02 and TMZ
To avoid the impact of immune deficiency–related drug toxicities on the treatment responses, this syngeneic model was selected. The mouse glioma 261 (GL261) tumor model has been described and utilized for experimental GBM therapy (30). In realizing the cells for in vivo experiments are different from those used for the in vitro experiments, the key cell-based assays were performed using GL261 cells. We investigated the synergistic cytotoxic effect of TMZ and TG02 in GL261 cells. The synergism between both drugs was demonstrated at relatively high affected fractions of inhibition (Fig. 6A–a–c). Mice implanted with orthotopic GL261 allografts were used as the in vivo model in this study. To confirm the PD effect of TG02, 6 mice with GL261 glioma were treated with vehicle or TG02. All mice were sacrificed 20 days after tumor implantation, and the brain tumor tissues were harvested for both histologic and Western blot analyses. In all TG02-treated mice, protein expressions of CDK9 and phospho-CDK9 were decreased in tumor tissue compared with vehicle control (Fig. 6B–a and b). Bioluminescence imaging (BLI) from 2 mice of each cohort showed a decrease in tumor volume by day 19 (Fig. 6B–c). The results suggest that TG02 suppresses CDK9 activity in brain tumor tissue which may confer an antiglioma effect. The correlation between the BLI signals and tumor was confirmed by hematoxylin and eosin staining, where areas of polymorphic tumor cells were demonstrated (Fig. 6B–d). Following this histologic confirmation, the potential survival benefit of TG02, TMZ, and the combination was tested. GL261 mice were randomized into 4 treatment cohorts by tumor size based on the BLI tumor values obtained prior to treatment to minimize selection bias. The dosing schedules are provided in Fig. 6C–a. Median survival times of the mice in the vehicle, TG02, TMZ, and combination treatment were 24, 24.5, 27.5, and 32 days, respectively (Fig. 6C–b). There is no survival benefit in TG02- or TMZ-treated mice compared with the vehicle group. However, we observed significantly prolonged survival in the cohort receiving the combination of TG02 and TMZ (P = 0.0026). Several mice in both TG02 alone and TG02/TMZ combined cohorts died due to gastrointestinal toxicities, such as diarrhea, which could explain the lack of survival benefit of the mice receiving TG02 alone. Animals in the cohort that received combined treatment may better tolerate the toxicity due to the better general condition as a result of better antiglioma effects from the combined treatment. The combination treatment affected tumor growth as tumor measurements in mice that received the combination treatment of TG02 and TMZ were 25% smaller compared with the baseline, whereas no reduction in tumor size was observed in other cohorts (Fig. 6C–c). The BLI of two subjects from each cohort on days 5, 12, and 20 were showed (Fig. 6C–d), which illustrated that the combination treatment of TG02/TMZ decreased tumor burden in the GL261 orthotopic mouse model. Overall, combined treatment with TG02/TMZ prolonged survival in the mouse orthotopic GL261 model, suggesting that the combination of TG02 and TMZ has a synergistic antiglioma effect.
Figure 6.

Treatment of combined TG02 and TMZ has an antiglioma effect in GL261 mouse GBM model. A, Combined TG02/TMZ treatment has a synergistic antiglioma effect in GL261 cells. Cells were treated with various concentrations of TG02, TMZ, and TG02/TMZ for 72 hours, and cell viability was determined by cell counting (a). The synergistic effect of TG02/TMZ was evaluated by CI index in b. CI values were calculated by COMPUSYN software and are shown in c. B, TG02 downregulates CDK9 and phospho-CDK9 in vivo. The protein expressions of CDK9, phospho-CDK9, and GAPDH were determined from GL261 tumor tissues with or without TG02 treatment (a). The bar graph represents the normalized relative intensity quantification of each Western blot band. The intensity of each band was relative to the corresponding band of vehicle 1 and then normalized to the relative GAPDH (b). Imaging tumor size of orthotopic glioblastoma xenografts derived from luciferase-expressing GL261 was tracked by bioluminescence at 12 and 19 days (c). Hematoxylin and eosin stain of mice brain tissues in vehicle and TG02 treatment (d). The scale bar represents 1 mm (top) and 100 µm (bottom). C, Combination treatment of TG02/TMZ prolongs mice survival in the allograft model. a,A schematic illustration of drug administration. b, Kaplan–Meier survival plots of C57BL/6 albino mice (n = 5–7 per cohort) with GL261 allografts showing the median overall survivals of 24, 24.5, 27.5, and 32 days in cohorts which received vehicle, TG02, TMZ, or combination treatment, respectively. The result was analyzed using the Log-rank test for trends in GraphPad Prism software (χ2 = 9.063; df 1; **, P 0.0026). c, The intensity of BLI of tumors in cohorts received vehicle, TG02, TMZ, or combination treatment. The values were calculated and normalized to the initial intensity at day 5. d, Representative BLI of 2 mice from each cohort that were recorded at days 5, 12, and 20.
Discussion
The development of brain tumor treatments remains challenging despite extensive research efforts in the field of neuro-oncology. GBMs are known for its infiltrative behavior, intra- and intertumoral heterogeneity, and the rapid development of resistance to conventional cytotoxic chemotherapies (31, 32). Preclinical studies using in vitro and in vivo testing of conventional tumor models have not consistently correlated with clinical benefit, but most of these studies have had limited interrogation of the treatments’ mechanisms of action.
We performed a series of preclinical studies to investigate a new drug, TG02, with the goal of obtaining extensive preclinical information, improving the development of clinical studies. Although TG02 was touted as a robust CDK9 inhibitor, our studies uncovered several additional distinct mechanisms of actions that are independent to each other, thereby potentially reducing or delaying the development of alternate resistance pathways. We demonstrated that TG02 transcriptionally modulated antiapoptotic proteins, including Mcl-1 and Survivin, via downregulation of CDK9 activity in patient-derived GBM cells. These proteins are essential for maintaining mitochondrial integrity, which is consistent with our findings of mitochondrial dysfunction after TG02 treatment. However, other factors may contribute to the TG02-induced mitochondrial dysfunction, which are worthy of future investigation. We also discovered that TG02 suppressed glycolytic function, unlikely to be related to mitochondrial dysfunction because compensation of energy production should lead to glycolytic upregulation rather than downregulation. In the context of strong inhibition of both energy production pathways, cancer cells are likely to undergo cell death with a lesser chance of developing resistant mechanisms. Importantly, we demonstrated cell growth inhibition, mitochondrial dysfunction, and apoptosis in GBM cells but not in the normal HAs and endothelial cells, suggesting that it will be feasible to target a therapeutic window for TG02 treatment in patients. Finally, prolonged survival and decreased tumor volume in a mouse intracranial xenograft model under combined TG02 and TMZ treatment confirm the in vitro results demonstrating synergy, providing additional support for further testing this combination.
Mitochondria provide most of the energy in all mammalian cells, whereas tumor cells rely more on glycolysis for energy production. The mitochondrial respiratory complexes catalyze the transfer of electrons from NADH to create a proton gradient across the inner membrane and drive ATP production via ATP synthases (33, 34). A serial assay of mitochondrial function was conducted, and mitochondrial damages were confirmed in both TG02 and TMZ treatment in our studies (Fig. 3). Although some evidence suggested that TMZ induces mitochondrial dysfunction, a significant cell apoptosis was not detected (Fig. 3H). This is consistent with the evidence that there is no further increase in apoptosis after combined treatment, compare with TG02 treatment alone (Fig. 3H). Therefore, the drug-induced mitochondrial dysfunction does not fully explain the synergistic cytotoxic effects of TG02/TMZ.
Glycolysis, the major source of energy production in GBM, was found to be suppressed following TMZ, TG02, and TMZ/TG02 treatment (Fig. 4A–D). HK2 and PKM2 are key regulators of glycolysis and promote cancer cell growth and GSC self-renewal (35, 36). Here, we found that glycolytic capacity was downregulated by TG02, TMZ, and TG02/TMZ treatment and that protein expressions of HK2, PKM2, and LDHA were decreased in combination TG02/TMZ treatment. TMZ, an alkylating agent, induces DNA damage leading to PARP activation, a DNA damage repair protein, resulting in cytoplasmic NAD+ depletion, which inhibits glycolysis. We demonstrated that glycolytic capacity was suppressed by TMZ after 48-hour treatment. However, the oxygen consumption rate and the ATP production have been found to be increased by TMZ, suggesting an adaptive response to DNA damage and high energy demand (37). The reduction in energy generation by glycolysis increases cellular reliance on alternative energy sources, particularly oxidative phosphorylation. Therefore, as illustrated in Fig. 4E, the synergistic effects of TG02 and TMZ may in part relate to impairment of both glycolysis and mitochondrial function, further enhanced by the increase in energy requirements for DNA repair, such as Base Excision Repair, from TMZ treatment. However, the drug interaction between TG02 and TMZ on the roles of replication stress is still unclear. The detailed mechanisms of TG02/TMZ-induced suppression of glycolysis are under investigation.
The combined effects of the drugs on tumor cells are complex, but combinatorial regimens are increasingly investigated in refractory disease, such as GBM, because single-agent treatments have been proven ineffective. Targeting multiple components of tumor cell survival and proliferation may both enhance the cytotoxic effect and reduce the potential for development of therapeutic resistance. In the case of TG02, extensive preclinical testing elucidated several complementary mechanisms of action as well as synergy with a commonly used brain tumor chemotherapy agent. Although a precise mechanism for the synergy between TG02 and TMZ remains to be completely determined, these results provide strong support for the further investigation of this novel agent and has led to the implementation of a clinical trial for patients with malignant glioma (NCT02942264).
Supplementary Material
Translational Relevance.
Therapies that target multiple pathways of tumor cell survival mechanisms reduce the potential for development of therapeutic resistance, a challenge in the treatment of glioblastoma. We extensively investigated TG02, a novel agent known to cross the blood–brain barrier. Our investigations uncovered the single-agent effects and several complementary mechanisms of action that underlie the synergy with an oral alkylating agent, temozolomide. We demonstrated that in addition to suppressing transcription through inhibition of cyclin-dependent kinase 9, TG02 also decreases cellular ATP levels by suppression of glycolysis and mitochondrial function, inducing cell death in glioblastoma cells but not in normal astrocytes. Cellular ATP production is further decreased with the combined treatment of TG02 and temozolomide by inhibiting glycolysis, which could partially explain the synergistic effects. The synergy was further characterized by a prolonged survival that was observed in a syngeneic mouse glioblastoma model receiving combined treatment of TG02 and temozolomide. In summary, TG02 targets multiple survival mechanisms and synergistically decreases energy production with temozolomide, representing a promising therapeutic strategy in refractory glioblastoma. These preclinical findings, supporting pharmacologic efficacy of TG02 and temozolomide in GBM, have led to the launching of a phase I/II clinical trial (NCT02942264).
Acknowledgments
The authors thank Dionne L. Davis (NOB/NCI/NIH) for the technical assistance of in vivo studies and Tragara Pharmaceuticals for providing TG02. This research was supported by the Intramural Research Program of the NIH.
Footnotes
Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
T. Estok holds ownership interest (including patents) in Tragara Pharmaceuticals, Inc. No potential conflicts of interest were disclosed by the other authors.
References
- 1.Ostrom QT, Gittleman H, Fulop J, Liu M, Blanda R, Kromer C, et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2008–2012. Neuro Oncol 2015;17Suppl 4:iv1–iv62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005;352:987–96. [DOI] [PubMed] [Google Scholar]
- 3.Huang TT, Sarkaria SM, Cloughesy TF, Mischel PS. Targeted therapy for malignant glioma patients: lessons learned and the road ahead. Neurotherapeutics 2009;6:500–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang H, Xu T, Jiang Y, Xu H, Yan Y, Fu D, et al. The challenges and the promise of molecular targeted therapy in malignant gliomas. Neoplasia 2015;17:239–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 2009;9:153–66. [DOI] [PubMed] [Google Scholar]
- 6.Goh KC, Novotny-Diermayr V, Hart S, Ong LC, Loh YK, Cheong A, et al. TG02, a novel oral multi-kinase inhibitor of CDKs, JAK2 and FLT3 with potent anti-leukemic properties. Leukemia 2012;26:236–43. [DOI] [PubMed] [Google Scholar]
- 7.Loyer P, Trembley JH, Katona R, Kidd VJ, Lahti JM. Role of CDK/cyclin complexes in transcription and RNA splicing. Cell Signal 2005;17: 1033–51. [DOI] [PubMed] [Google Scholar]
- 8.Romano G, Giordano A. Role of the cyclin-dependent kinase 9-related pathway in mammalian gene expression and human diseases. Cell Cycle 2008;7:3664–8. [DOI] [PubMed] [Google Scholar]
- 9.Nechaev S, Adelman K. Pol II waiting in the starting gates: Regulating the transition from transcription initiation into productive elongation. Biochim Biophys Acta 2011;1809:34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Larochelle S, Amat R, Glover-Cutter K, Sanso M, Zhang C, Allen JJ, et al. Cyclin-dependent kinase control of the initiation-to-elongation switch of RNA polymerase II. Nat Struct Mol Biol 2012;19:1108–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lam LT, Pickeral OK, Peng AC, Rosenwald A, Hurt EM, Giltnane JM, et al. Genomic-scale measurement of mRNA turnover and the mechanisms of action of the anti-cancer drug flavopiridol. Genome Biol 2001;2: RESEARCH0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gunther W, Pawlak E, Damasceno R, Arnold H, Terzis AJ. Temozolomide induces apoptosis and senescence in glioma cells cultured as multicellular spheroids. Br J Cancer 2003;88:463–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bocangel DB, Finkelstein S, Schold SC, Bhakat KK, Mitra S, Kokkinakis DM. Multifaceted resistance of gliomas to temozolomide. Clin Cancer Res 2002;8:2725–34. [PubMed] [Google Scholar]
- 14.Hirose Y, Berger MS, Pieper RO. p53 effects both the duration of G2/M arrest and the fate of temozolomide-treated human glioblastoma cells. Cancer Res 2001;61:1957–63. [PubMed] [Google Scholar]
- 15.Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 2006;9:391–403. [DOI] [PubMed] [Google Scholar]
- 16.Su YT, Gao C, Liu Y, Guo S, Wang A, Wang B, et al. Monoubiquitination of filamin B regulates vascular endothelial growth factor-mediated trafficking of histone deacetylase 7. Mol Cell Biol 2013;33:1546–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Su YT, Chang HL, Shyue SK, Hsu SL. Emodin induces apoptosis in human lung adenocarcinoma cells through a reactive oxygen species-dependent mitochondrial signaling pathway. Biochem Pharmacol 2005;70:229–41. [DOI] [PubMed] [Google Scholar]
- 18.Ogilvy AJ, Shen D, Wang Y, Chan CC, Abu-Asab MS. Implications of DNA leakage in eyes of mutant mice. Ultrastruct Pathol 2014;38:335–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu Y, Kwintkiewicz J, Liu Y, Tech K, Frady LN, Su YT, et al. Chemosensitivity of IDH1-mutated gliomas due to an impairment in PARP1-mediated DNA repair. Cancer Res 2017;77:1709–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res 2010;70:440–6. [DOI] [PubMed] [Google Scholar]
- 21.Trent J, Meltzer P, Rosenblum M, Harsh G, Kinzler K, Mashal R, et al. Evidence for rearrangement, amplification, and expression of c-myc in a human glioblastoma. Proc Natl Acad Sci U S A 1986;83:470–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Q, Price JP, Byers SA, Cheng D, Peng J, Price DH. Analysis of the large inactive P-TEFb complex indicates that it contains one 7SK molecule, a dimer of HEXIM1 or HEXIM2, and two P-TEFb molecules containing Cdk9 phosphorylated at threonine 186. J Biol Chem 2005;280:28819–26. [DOI] [PubMed] [Google Scholar]
- 23.Thomas LW, Lam C, Edwards SW. Mcl-1; the molecular regulation of protein function. FEBS Lett 2010;584:2981–9. [DOI] [PubMed] [Google Scholar]
- 24.Hagenbuchner J, Kuznetsov AV, Obexer P, Ausserlechner MJ. BIRC5/Survivin enhances aerobic glycolysis and drug resistance by altered regulation of the mitochondrial fusion/fission machinery. Oncogene 2013;32:4748–57. [DOI] [PubMed] [Google Scholar]
- 25.Gratas C, Sery Q, Rabe M, Oliver L, Vallette FM. Bak and Mcl-1 are essential for Temozolomide induced cell death in human glioma. Oncotarget 2014;5:2428–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zervantonakis IK, Iavarone C, Chen HY, Selfors LM, Palakurthi S, Liu JF, et al. Systems analysis of apoptotic priming in ovarian cancer identifies vulnerabilities and predictors of drug response. Nat Commun 2017;8:365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Warr MR, Shore GC. Unique biology of Mcl-1: therapeutic opportunities in cancer. Curr Mol Med 2008;8:138–47. [DOI] [PubMed] [Google Scholar]
- 28.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–74. [DOI] [PubMed] [Google Scholar]
- 29.Lane N, Martin W. The energetics of genome complexity. Nature 2010;467:929–34. [DOI] [PubMed] [Google Scholar]
- 30.Szatmari T, Lumniczky K, Desaknai S, Trajcevski S, Hidvegi EJ, Hamada H, et al. Detailed characterization of the mouse glioma 261 tumor model for experimental glioblastoma therapy. Cancer Sci 2006;97:546–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013;501:338–45. [DOI] [PubMed] [Google Scholar]
- 32.Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY, et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 2014;343:189–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Labbe K, Murley A, Nunnari J. Determinants and functions of mitochondrial behavior. Annu Rev Cell Dev Biol 2014;30:357–91. [DOI] [PubMed] [Google Scholar]
- 34.Kuhlbrandt W. Structure and function of mitochondrial membrane protein complexes. BMC Biol 2015;13:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wolf A, Agnihotri S, Munoz D, Guha A. Developmental profile and regulation of the glycolytic enzyme hexokinase 2 in normal brain and glioblastoma multiforme. Neurobiol Dis 2011;44:84–91. [DOI] [PubMed] [Google Scholar]
- 36.Dong G, Mao Q, Xia W, Xu Y, Wang J, Xu L, et al. PKM2 and cancer: the function of PKM2 beyond glycolysis. Oncol Lett 2016;11:1980–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oliva CR, Nozell SE, Diers A, McClugage SG 3rd, Sarkaria JN, Markert JM, et al. Acquisition of temozolomide chemoresistance in gliomas leads to remodeling of mitochondrial electron transport chain. J Biol Chem 2010;285:39759–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.