

TO THE EDITOR:
The striking advances in sequencing technology have led to an era in which a wide variety of specific genetic mutations in clinically similar syndromes can be identified. Thus, many entities have been recently described and are being studied to identify their clinical manifestations.
Activated phosphoinositide 3-kinase δ syndrome (APDS) is a gain-of-function mutation that manifests by immune dysregulation.1
It is a newly defined primary immunodeficiency syndrome that was previously considered part of common variable immune deficiency (CVID). However, next-generation sequencing defined APDS as a distinct disease in which the mechanism of hyperactive signaling and disturbed immunological response was first described in 2013.2
The PI3K that is responsible for the clinical syndrome of APDS is a lipid kinase heterodimer that is a combination of a catalytic subunit p110 and a regulatory subunit p85. These 2 subunits compose the PI3K complex that is involved in the signaling pathway of both B-and T-cell receptors.2
The liver is the site where the systemic immune system meets translocated bacterial products and intestinally derived inflammatory markers borne by the portal circulation and therefore might be expected to be affected by immune dysregulation. Matsuda et al3 have shown that deregulation of the PI3K/AKT pathway can lead to metabolic dysfunction including nonalcoholic fatty liver disease (NAFLD).3 Liver involvement leading to nodular regenerative hyperplasia (NRH) was previously described in patients with CVID4,5; however, not much is known about liver involvement in APDS. In a recent study by Coulter et al6 describing 53 patients with APDS, 3 patients had cirrhosis, of whom 1 also had sclerosing cholangitis from previous Cryptosporidium infection. The aim of our study was to evaluate the hepatic manifestations of APDS and to describe the range of findings in a relatively large cohort of patients with this rare disease. As a newly described disease with a newly ascribed etiology, it is of value to identify the clinical features of the disease to allow clinicians to anticipate and screen for different disease manifestations.
In this retrospective study, genetically sequenced APDS patients followed at the National Institutes of Health laboratory of clinical infectious diseases were identified. Detailed review of the patients’ medical and laboratory records was performed.
Because of the longitudinal collection of patients’ data, and the fluctuating nature of liver enzyme abnormalities, patients were categorized per the pattern of the elevated transaminases into 2 groups. Transaminitis that lasted for more than 6 months was considered persistent while disturbance lasting for less than 6 months was considered transient. Then the elevation was categorized in relationship to normal values into 3 grades: Mild: an elevation of up to 2 times the upper limit of normal ALT (ULN); Moderate: 2 to 5 times the ULN; and Severe: higher than 5 times the ULN. The age of initial transaminase elevation was also recorded.
Cross-sectional abdominal imaging performed on first evaluation at the National Institutes of Health was reviewed by a single radiologist with attention to liver and spleen sizes as well as for findings suggestive of portal hypertension.
Clinically indicated liver biopsies, when performed, were reviewed by a single hepatopathologist (D.E.K.).
A total of 33 patients with APDS from 28 families were followed between January 2007 and April 2017 with an average follow-up of 37.07 ± 30.9 months (Table I). The average age of patients was 20.36 ± 17.2 years, with 19 (57%) males. We identified 9 (27%) patients (5 males), average age 17.1 years, with elevation of transaminases. Five of these patients showed persistent duration, while 4 showed intermittent elevation; as for the magnitude of the transaminitis, 2 were categorized as grade A, 6 as grade B, and 1 as grade C. In 8 patients, viral hepatitis (B and C) were tested and found to be negative. Five of the patients with elevated transaminases had PI3KCD mutations, whereas 4 had mutations in PI3KR1. In 7, alanine aminotransferase (ALT) level was more than aspartate aminotransferase (AST) level (1 with alkaline phosphatase [ALP] elevation as well), 1 showed a higher AST level compared with the ALT level, and 1 had a fluctuating pattern. Five patients normalized liver enzymes during follow-up (Figure 1). The elevated transaminases were first noticed at a mean age of 17.2 ± 6.3 years. Patients with R1 mutation had significantly higher occurrence of transaminitis (66.6% vs 18.5%; P = .03).
TABLE I.
Patient characteristics
| Characteristic | Patients with elevated liver enzymes (n = 9) | Patients without liver enzyme elevation (n = 24) |
|---|---|---|
| Age (y), mean | 17.1 | 21.5 |
| Sex (males), n (%) | 5 (55) | 14 (58) |
| Mutation type, n | ||
| R1 P-85 | 4 | 2 |
| P-110 | 5 | 22 |
| Highest ALT* | 65 U/L (62–132) | 22 U/L (15–34) |
| ALP (median)* | 175 U/L (99–226) | 95 U/L (68–154) |
| Platelets, K/uL (median)* | 248 (164–354) | 192 (145–234) |
| Spleen size corrected to height† | 0.94 (n = 7) | 0.81 (n = 16) |
ALP, Alkaline phosphatase; ALT, alanine aminotransferase.
Expressed as median (interquartile range).
Measured in centimeters.
FIGURE 1.
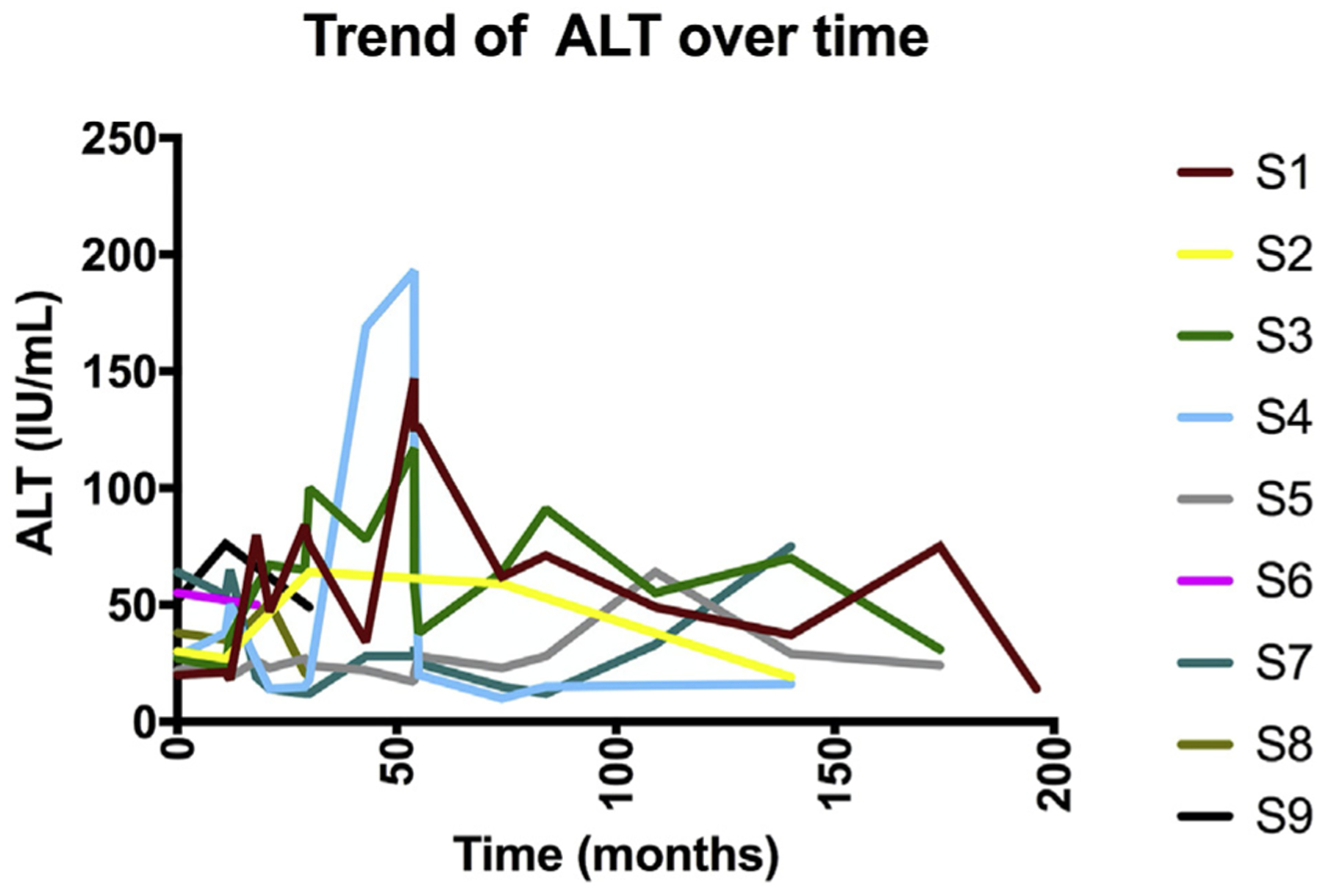
Trends in ALT for patients with transaminitis during the follow-up period. ALT, Alanine aminotransferase.
Five patients had clinically indicated liver biopsies, 1 of them with no previous elevation of liver enzymes. Four of the biopsies showed histology significant for NRH (Figure 2), including the patient with no disturbance in transaminases; the fifth biopsy was positive for steatosis. Hepatic venous portal gradient was measured in 4 patients and was mildly elevated in all of them (5.5 mm Hg average). Clinical manifestations of portal hypertension were seen in only 1 patient (mild ascites).
FIGURE 2.
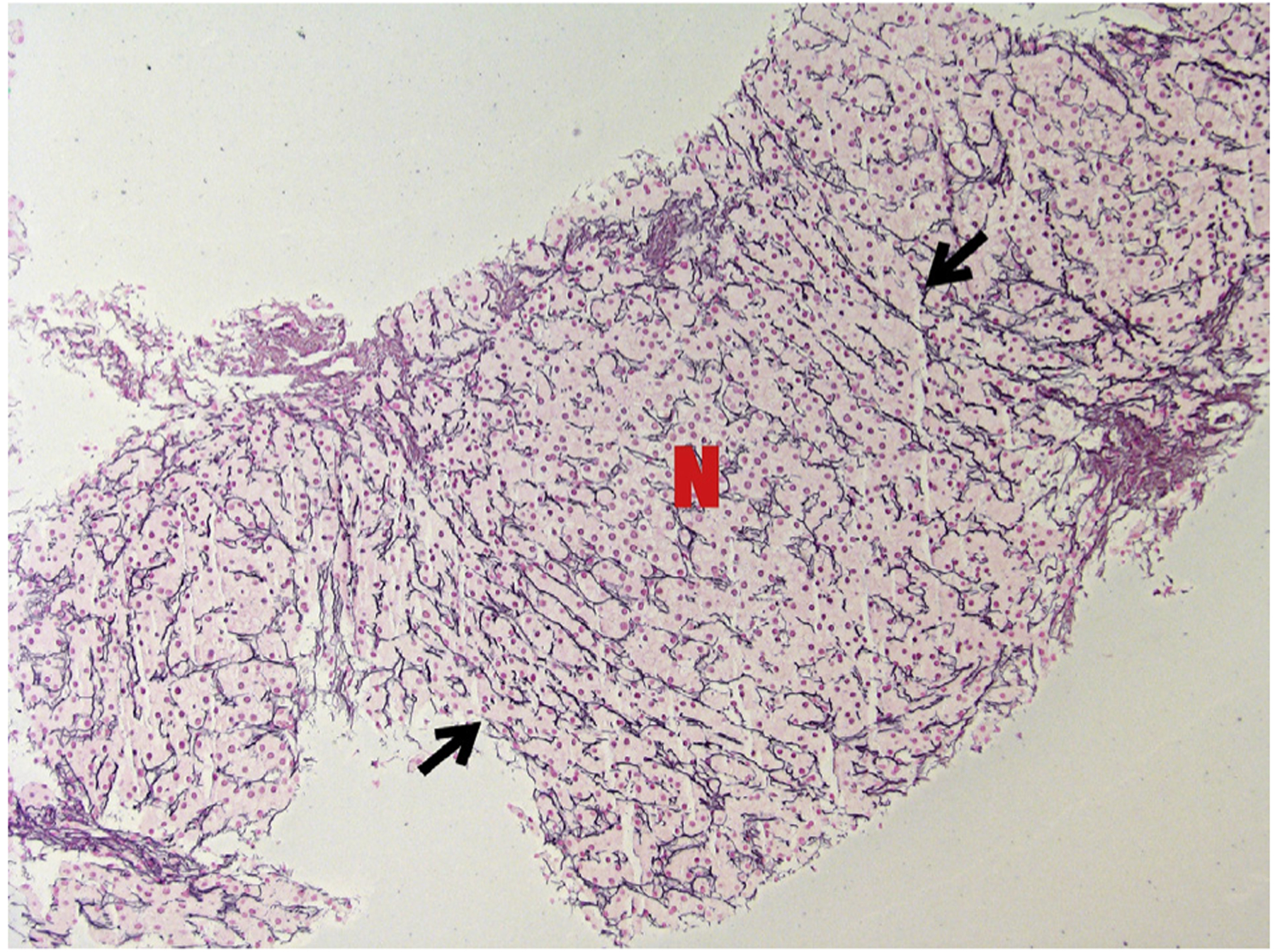
Reticulin stain of PI-3K patient’s liver biopsy showing a nodule where hepatocytes are arranged in thick plates (marked with N). At the borders of the nodule, compressed plates of hepatocytes marked with black arrows (magnification × 100).
None of the patients received any treatment on the basis of the presence of liver disease, nor were their intravenous immunoglobulin doses modified.
In this report of liver manifestations in a cohort of a rare, newly described disease, we found that 30% of the patients (10 of 33) had some form of liver disturbance. In most patients, this disturbance was first noticed within the second decade of life, and resolved spontaneously. Of the patients who were biopsied, all but 1 showed disturbed hepatic architecture suggestive of NRH with mild elevation of portal pressures in all patients. There was no evidence of active liver infection during the episode of liver enzyme elevation.
We report a higher prevalence of liver disease in our cohort compared with that reported previously.6 This might be because we had longitudinal follow-up of our cohort, allowing us to track fluctuations in liver enzymes. In addition, we had a higher number of liver biopsies and greater sensitivity for the detection of NRH, which is something not commonly pursued in normal evaluation of liver biopsies (reticulin stain).
Genetic sequencing of the broader pool that was once considered CVID enabled the identification of this unique cohort of patients with APDS. With improvements in genetic sequencing techniques and increased availability, there is likely to be an increased number of patients diagnosed with APDS. APDS is an important example of a disease in which an understanding of the etiology will lead to accurate diagnoses and better treatments, thus leading to improved survival, giving time for organ-specific complications to occur and influence outcomes. This has been described in other settings7,8 and in CVID in general.9
At present, the only curative approach to APDS is a stem cell transplant. In this cohort, the predominant hepatic manifestation documented was NRH, which is known to lead to poor outcomes after stem cell transplant, and if diagnosed before transplant may influence the choice of myeloablative preconditioning. With the anticipation of a larger number of patients getting diagnosed with APDS in the future, understanding the underlying liver disease will not only lead to clinical implications but also influence their treatment options.
Clinical Implications.
This first report of hepatic manifestations of activated phosphoinositide 3-kinase δ syndrome shows nodular regenerative hyperplasia as a predominant finding, which has implications for the development of portal hypertension, therapies, and for stem cell transplantation in these patients.
Acknowledgments
This work was supported by the intramural research programs of the National Institute of Diabetes and Digestive and Kidney Diseases, National Institute of Allergy and Infectious Diseases, and National Cancer Institute, National Institutes of Health.
Footnotes
Conflicts of interest: The authors declare that they have no relevant conflicts of interest.
REFERENCES
- 1.Lucas CL, Chandra A, Nejentsev S, Condliffe AM, Okkenhaug K. PI3Kδ and primary immunodeficiencies. Nat Publ Gr 2016;4:702–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Elgizouli M, Lowe DM, Speckmann C, Schubert D, Hülsdünker J, Eskandarian Z, et al. Activating PI3Kδ mutations in a cohort of 669 patients with primary immunodeficiency. Clin Exp Immunol 2016;183:221–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matsuda S, Kobayashi M, Kitagishi Y. Roles for PI3K/AKT/PTEN pathway in cell signaling of nonalcoholic fatty liver disease. ISRN Endocrinol 2013;2013(Figure 1): 472432. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3570922&tool=pmcentrez&rendertype=abstract. Accessed December 11,2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Malamut G, Ziol M, Suarez F, Beaugrand M, Viallard JF, Lascaux AS, et al. Nodular regenerative hyperplasia: the main liver disease in patients with primary hypogammaglobulinemia and hepatic abnormalities. J Hepatol 2008;48: 74–82. [DOI] [PubMed] [Google Scholar]
- 5.Ward C, Lucas M, Piris J, Collier J, Chapel H. Abnormal liver function in common variable immunodeficiency disorders due to nodular regenerative hyperplasia. Clin Exp Immunol 2008;153:331–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coulter TI, Chandra A, Bacon CM, Babar J, Curtis J, Screaton N, et al. Clinical spectrum and features of activated phosphoinositide 3-kinase δ syndrome: a large patient cohort study. J Allergy Clin Immunol 2017;139:597–606.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hussain N, Feld JJ, Kleiner DE, Hoofnagle JH, Garcia-eulate R, Ahlawat S, et al. Hepatic abnormalities in patients with chronic granulomatous disease. Hepatology 2007;45:675–83. [DOI] [PubMed] [Google Scholar]
- 8.O’Brien K, Hussain N, Warady BA, Kleiner DE, Kleta R, Bernardini I, et al. Nodular regenerative hyperplasia and severe portal hypertension in cystinosis. Clin Gastroenterol Hepatol 2006;4:387–94. [DOI] [PubMed] [Google Scholar]
- 9.Fuss IJ, Friend J, Yang Z, He JP, Hooda L, Boyer J, et al. Nodular regenerative hyperplasia in common variable immunodeficiency. J Clin Immunol 2013;33: 748–58. [DOI] [PMC free article] [PubMed] [Google Scholar]