

Abstract
Background
Surrogate endpoints (i.e., intermediate endpoints intended to predict for patient-centered outcomes) are increasingly common. However, little is known about how surrogate evidence is handled in the context of health technology assessment (HTA).
Objectives
1) To map methodologies for the validation of surrogate endpoints and 2) to determine their impact on acceptability of surrogates and coverage decisions made by HTA agencies.
Methods
We sought HTA reports where evaluation relied on a surrogate from 8 HTA agencies. We extracted data on the methods applied for surrogate validation. We assessed the level of agreement between agencies and fitted mixed-effects logistic regression models to test the impact of validation approaches on the agency’s acceptability of the surrogate endpoint and their coverage recommendation.
Results
Of the 124 included reports, 61 (49%) discussed the level of evidence to support the relationship between the surrogate and the patient-centered endpoint, 27 (22%) reported a correlation coefficient/association measure, and 40 (32%) quantified the expected effect on the patient-centered outcome. Overall, the surrogate endpoint was deemed acceptable in 49 (40%) reports (k-coefficient 0.10, P = 0.004). Any consideration of the level of evidence was associated with accepting the surrogate endpoint as valid (odds ratio [OR], 4.60; 95% confidence interval [CI], 1.60–13.18, P = 0.005). However, we did not find strong evidence of an association between accepting the surrogate endpoint and agency coverage recommendation (OR, 0.71; 95% CI, 0.23–2.20; P = 0.55).
Conclusions
Handling of surrogate endpoint evidence in reports varied greatly across HTA agencies, with inconsistent consideration of the level of evidence and statistical validation. Our findings call for careful reconsideration of the issue of surrogacy and the need for harmonization of practices across international HTA agencies.
Keywords: health technology assessment, outcomes research, surrogate, validation
Background
In recent years, regulatory agencies, including the European Medicines Agency (EMA) and the Food and Drug Administration (FDA) in the United States, have increasingly approved drugs and biologics on the basis of surrogate endpoints.1 A surrogate endpoint is defined as a biomarker or physiological measure, laboratory test result, imaging result, or another replacement endpoint that is thought to capture the causal pathway through which the disease process affects the patient-centered outcomes.2
When used as primary outcomes, surrogate endpoints enable clinical trials of smaller sample size, shorter duration, and lower cost than trials with a patient-centered primary endpoint.3 The uptake of surrogate endpoints in pivotal trials is typically associated with expedited drug review and accelerated approval programs, resulting in market authorization based on less rigorous evidence (i.e., fewer and smaller studies) without an appropriate comparator or single-arm studies.4 However, once licensed, patient access to these products typically depends on assessment by a health technology assessment (HTA) agency that informs a country’s or region’s coverage of reimbursement decision.5 While regulatory bodies are primarily concerned with the efficacy-safety, HTA agencies seek to assess the long-term comparative effectiveness and economic consequences of health technologies, alongside other considerations such as equity, severity of disease, or unmet need. Recent research has shown that the methodological guidelines of HTA agencies often take a conservative approach to the use of surrogate endpoints to support their coverage recommendations, for example, by 1) expressing a preference for patient-relevant outcomes (such as mortality), 2) recommending that surrogate endpoints should only be used in situations where patient-relevant outcomes are not available or their evidence is limited, or 3) limiting use of surrogate outcomes to validated measures.6,7
Four previous studies have investigated the impact of surrogate endpoints on HTA decisions. Two studies focused on cancer drugs,8,9 and 2 considered the range of technology appraisals undertaken by either the National Institute of Health and Care Excellence (NICE) in the United Kingdom or the Canadian Common Drug Review.10,11 However, these previous studies did not assess HTA agencies’ approach to validation of the surrogate endpoints or how this related to their coverage recommendation.
The objectives of this study were 1) to map the methodological approaches for the validation of surrogate endpoints applied in reports across a sample of international HTA agencies and 2) to assess how the consideration of the validity of the surrogate endpoints influences the coverage or reimbursement decisions made by these agencies.
Methods
Selection of HTA Reports
We applied a 2-step approach to the selection and inclusion of HTA reports in this study. First, we sought to identify health technologies and related HTA reports that involved the use of surrogate endpoints. We used the surrogate endpoint definition of the US National Institutes of Health, that is, a biomarker (or intermediate endpoint) intended to substitute for a clinical endpoint.12 We screened the guidance published by NICE between May 2013 and June 2018. All technology appraisal guidance, medical technologies guidance, and diagnostics guidance reports published in this timeframe were screened by one of the research team (BG) for inclusion on the basis that they included discussion of a surrogate endpoint.
Second, based on a selected list of NICE evaluations (and reports), we then identified HTA evaluation reports for the same health technology and clinical indication from a further sample of 6 HTA agencies. These agencies included Health Improvement Scotland (HIS)/Scottish Medicines Consortium (SMC) in Scotland, Haute Autorité de Santé (HAS) in France, Pharmaceutical Benefits Advisory Committee (PBAC) and Medical Services Advisory Committee (MSAC) in Australia, Canadian Agency for Drugs and Technologies in Health (CADTH) in Canada, Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG)/Gemeinsame Bundesausschuss (G-BA) in Germany, Zorginstituut Nederland (ZiN) in the Netherlands, and Országos Gyógyszerészeti és Élelmezés-egészségügyi Intézet (NIPN) in Hungary. These agencies span different geographical areas, include some of the most prominent HTA organizations worldwide, and are known to follow methodological guidelines that include consideration of surrogate endpoints with different levels of detail.7 Between August and September 2018, we sought all relevant reports from these agencies, irrespective of language and publication date.
Framework for Assessment and Validation of Surrogate Endpoints
In the biostatistics literature, several approaches have been discussed that would identify when a biomarker is “likely to predict” a patient-centered endpoint of interest.13 Most common methods are framed within the causal inference and meta-analytic paradigms.14,15 The 2-stage meta-analytic approach developed by Burzykowski et al.15 requires demonstration of strong correlation between the surrogate and definitive endpoints (individual-level surrogacy) as well as correlation of treatment effects on both endpoints (trial-level surrogacy). Meta-analysis of individual patient data (IPD) remains the optimal approach because it enables the standardization of methods across IPD sets and robust analysis at both the patient and trial levels. However, because IPD meta-analyses are time and resource intensive, meta-analyses of outcome correlation or trial-level associations using aggregate data are more often reported. Bayesian multivariate meta-analytic methods of estimation are increasingly used, as they take into account the correlation between the treatment effects on the surrogate and patient-centered outcomes in addition to the uncertainty in the surrogate relationship.16
A recent overview of HTA guidelines identified that only 5 HTA agencies provide detailed advice on the statistical methods that should be used for the validation of surrogate endpoints.7 These guidelines note the current lack of consensus on the minimum criteria to establish the validity of surrogates.7 Numerical values discussed as thresholds for acceptable surrogacy include a coefficient of determination R2≥ 0.6 or 0.717,18 or a coefficient of correlation R≥ 0.85.19
In 2017, Ciani et al.20 proposed a methodological framework for the incorporation and reporting of the use of surrogate endpoints in HTA. A 3-step approach was recommended: 1) to establish the level of evidence available (i.e., whether the relationship between the putative surrogate endpoint and patient-centered endpoint of interest is supported by clinical plausibility, observational data, or meta-analyses of multiple randomized controlled trials [RCTs]); 2) to assess the strength of the association between the surrogate and patient-centered outcomes: observational association or treatment effect assessment (e.g., correlation coefficient at the individual and at the trial level); and 3) to quantify the expected effect on the patient-centered outcome given the observed effect on the surrogate endpoint. Table 1 elaborates this 3-stage methodological framework, illustrated with examples of good practice.
Table 1.
Methods for the Validation of Surrogate Endpoints: 3-Stage Framework
| Level of Evidence | Strength of the Association | Quantification of the Expected Effect on the Patient-Centered Outcome | |
|---|---|---|---|
| Level 1: Randomized controlled trials showing that treatment changes in the surrogate are associated with treatment changes in the final outcomeLevel 2: Epidemiological/observational studies showing consistent association between surrogate and final outcomeLevel 3: Pathophysiological studies and understanding of the disease process demonstrating the biological plausibility of relation between surrogate and final outcome |
For trial-level surrogacy Meta-analysis of individual patient data/aggregate data from randomized controlled trials that have assessed both the surrogate and patient-centered endpoints With trial/country/center as the analysis unit Preferably within the same indication and treatment class For individual-level surrogacy As above or even single large randomized controlled trials/observational studies that have assessed both the surrogate and patient-centered endpoints |
For trial-level surrogacy Coefficient of correlation (Kendall’s τ, Spearman’s ρ, Pearson within-study correlations from multivariate meta-analyses) Coefficient of determination from weighted/unweighted adjusted/unadjusted linear regression of treatment effects on endpoints/copula models For individual-level surrogacy Coefficient of correlation (Kendall’s τ, Spearman’s ρ, Pearson) Coefficient of determination from weighted/unweighted adjusted/unadjusted linear regression of treatment effects on endpoints/copula models Hazard ratio from Cox regressions/Bayesian hierarchical analysis |
For trial-level surrogacy Prediction based on the estimated regression equation for the trial-level surrogacy and observed effect on the surrogate endpoint Intercept, slope, and conditional variance of the linear model of the relationship between the treatment effects on the surrogate endpoint and the effects on the final outcome based on aggregate data Bayesian multivariate meta-analyses Surrogate threshold effect, the minimum treatment effect on the surrogate necessary to predict a nonzero effect on the patient-centered outcomes using the 95% prediction limits of the regression line |
Data Extraction from Reports
We developed a structured extraction form for included HTA reports based on the above framework, previous studies,21 and the Consolidated Health Economic Evaluation Reporting Standards (CHEERS) checklist.22 We considered the following categories of information: general characteristics of the evaluation/report, characteristics of the health technology, and orphan status designation. Orphan designation is attributed to medicines that are intended to treat, prevent, or diagnose a rare disease (usually no more than 5 in 10,000 in the relevant jurisdiction) that is life-threatening or chronically debilitating; that are unlikely to generate sufficient returns to justify the investment needed for the medicines’ development; and that provide a significant benefit in relation to the efficacy or safety of the treatment, prevention, or diagnosis of the same condition.23–25
We analyzed characteristics of the included surrogate endpoint (i.e., source of evidence, justification for use, methods for validation, how surrogate endpoint was incorporated in economic modeling [if undertaken], and other considerations), how uncertainty was dealt with in relation to consideration of the surrogate endpoint (including restricted coverage or price discounts), and final coverage/reimbursement recommendation. Following the 3-step validation framework described above,20 we assessed 1) the level of evidence available to support the surrogate–to–final outcome relationship (e.g., an individual patient data meta-analysis of RCTs would represent the highest level of evidence), 2) whether the report discussed the association between surrogate and final outcome with a related metric (e.g., Spearman’s ρ) given, and 3) whether the report discussed quantification of the expected treatment effect on the patient-centered endpoint based on the observed effect on the surrogate endpoint, either from previous evidence or based on the decision model in the report (Table 1). In addition, we assessed the level of acceptability of the surrogate endpoint. For example, “increase in total kidney volume correlates to growth in cyst volume and was considered to be an appropriate surrogate for disease progression” would be a statement that indicates acceptability of total kidney volume as a surrogate by the appraisal committee. Finally, we investigated how the surrogate endpoint was used in the development of the cost-effectiveness model and the reimbursement/coverage recommendation made. We recorded if finance-based (e.g., “Patient Access Schemes” in the United Kingdom intended to provide the National Health Service with access to the technology based on confidential discount from list price) or performance-based risk-sharing arrangements (e.g., plans to track the performance of the product over a specified period of time to inform the amount or level of reimbursement based on the health outcomes achieved) were agreed with the manufacturer.26
The data extraction form was piloted on 3 HTA reports (by OC, BG, RST). Following this pilot, information was extracted from each HTA report by one of the authors. Non-English reports were data extracted by coauthors who were native or proficient speakers and translated into English. A random sample of the reports (n = 36) was checked for accuracy of data extraction by another member of the team (OC, BG, or RST).
Data Analysis and Synthesis
We used tables and descriptive statistics to summarize extracted data and enable comparison of information across agencies (for a given health technology) and within agencies (across HTA reports). Two key areas of results presentation are 1) the methodological handling of surrogate endpoints in HTA reports and how this influences the acceptability of surrogate endpoints and 2) how surrogate endpoint validity influences the final reimbursement/coverage recommendation made by HTA agencies. In case of multiple evaluations made by an agency for the same technology, we considered the latest evaluation. Given that clinical evidence often accumulates after marketing authorization, we considered this to be a conservative approach (i.e., looking at the highest evidence base for surrogate validation).
We determined the level of agreement between agencies in terms of acceptability of surrogate endpoint and final recommendations made using a generalization of the κ coefficient for binary observations and multiple observers. We interpreted κ values as follows: values ≤0 as indicating no agreement and 0.01 to 0.20 as none to slight, 0.21 to 0.40 as fair, 0.41 to 0.60 as moderate, 0.61 to 0.80 as substantial, and 0.81 to 1.00 as almost perfect agreement.27
We collapsed categorical variables into binary responses (acceptable surrogate v. no/unclear; approved technology v. rejected/restricted), and we fitted univariable and multivariable mixed-effects logistic regression models to test 1) the impact of level of evidence, reporting a metric of association, and quantifying the expected effect on the patient-centered outcome and orphan status on the HTA agency’s acceptability of the surrogate endpoint and (2) the impact of the acceptability of the surrogate endpoint (and previous variables) on the final coverage recommendations given by the HTA agency. We applied the standard 2-tailed P < 0.05 threshold for the interpretation of statistical significance of regression coefficients. We conducted all statistical analyses in Stata/SE 16.1 (StataCorp, College Station, TX).
Results
Description of Health Technologies under Assessment and Included Reports
We screened a total of 291 HTA reports from NICE, of which 23 (8%) were included in the analysis. Among the 23 technologies assessed, 21 (91%) were pharmaceuticals and 2 (9%) were medical devices. Twelve (52%) technologies were used for an oncology indication, 3 (13%) for a cardiovascular indication, 2 (9%) for either an endocrinology or a nephrology indication, and the remainder spread across a variety of conditions (i.e., chronic hepatitis C, biliary cholangitis, vitreomacular traction, pulmonary fibrosis). A summary of the technologies included is available in Table 2.
Table 2.
Summary of Characteristics of HTA Reports
| Characteristic | Total No. (%) of HTA Reports (N = 124) |
|---|---|
| Drugs | 122 (98) |
| Medical device | 2 (2) |
| HTA agencies | |
| NICE | 23 (19) |
| HIS/SMC | 20 (16) |
| HAS | 20 (16) |
| PBAC/MSAC | 15 (12) |
| CADTH | 13 (10) |
| IQWiG/G-BA | 13 (10) |
| ZiN | 9 (7) |
| NIPN | 11 (9) |
| Disease area | |
| Cancer | 65 (52) |
| Cardiovascular | 17 (14) |
| Pulmonology | 8 (6) |
| Nephrology | 8 (6) |
| Endocrinology | 7 (6) |
| Infectious disease | 7 (6) |
| Ophthalmology | 6 (5) |
| Gastroenterology | 6 (5) |
| Orphan status | 8 (6) |
| Surrogate validation | |
| Surrogate accepted (yes) | 49 (40) |
| Level of evidence assessed (yes) | 61 (49) |
| Strength of association provided (yes) | 27 (22) |
| Quantification of effect provided (yes) | 40 (32) |
| Final recommendation given | |
| Approved | 32 (26) |
| Restricted | 61 (49) |
| Rejected | 20 (16) |
| No recommendation | 11 (9) |
CADTH, Canadian Agency for Drugs and Technologies in Health; HAS, Haute Autorité de Santé; HIS/SMC, Health Improvement Scotland/Scottish Medicines Consortium; HTA, health technology assessment; IQWiG/G-BA, Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen/Gemeinsame Bundesausschuss; NICE, National Institute for Health and Care Excellence; NIPN, Országos Gyógyszerészeti és Élelmezés-egészségügyi Intézet; PBAC/MSAC, Pharmaceutical Benefits Advisory Committee/Medical Services Advisory Committee; ZiN, Zorginstituut Nederland.
The most frequently considered surrogate endpoint, progression-free survival, was used in the evaluation of 7 (30%) technologies (axitinib, 2 indications of bortezomib, brentuximab, cobimetinib, pertuzumab, ribociclib), all intended for oncology indications. Major/complete cytogenetic response was used in 4 (17%) oncologic evaluations (bosutinib, dasatinib first and second line, pertuzumab). Changes in low-density lipoprotein cholesterol levels were used in 2 (9%) technologies intended for dyslipidemia (alirocumab, evolocumab). Other surrogate endpoints were biomarkers (parathyroid hormone, testosterone level, prostate-specific antigen, alkaline phosphatase, bilirubin, glycated hemoglobin, sustained virologic response), functional measurements (forced vital capacity, venous blood flow, change in total kidney volume), or measure of clinical response (e.g., proportion of patients with nonsurgical resolution of focal vitreomacular traction).
We identified a total of 124 reports across all 8 HTA agencies matching these NICE appraisals (Figure 1). These reports included a total of 341 archived documents (including the reports, associated recommendations, appendices, and responses to consultation) that were obtained and screened for data extraction (see Supplementary Material). Four technologies (alirocumab, evolocumab, pirfenidone, ribociclib) were evaluated across all 8 agencies. One technology (geko device; FirstKind Ltd High Wycombe, UK was only evaluated by NICE. The median number of evaluations per technology was 5.
Figure 1.
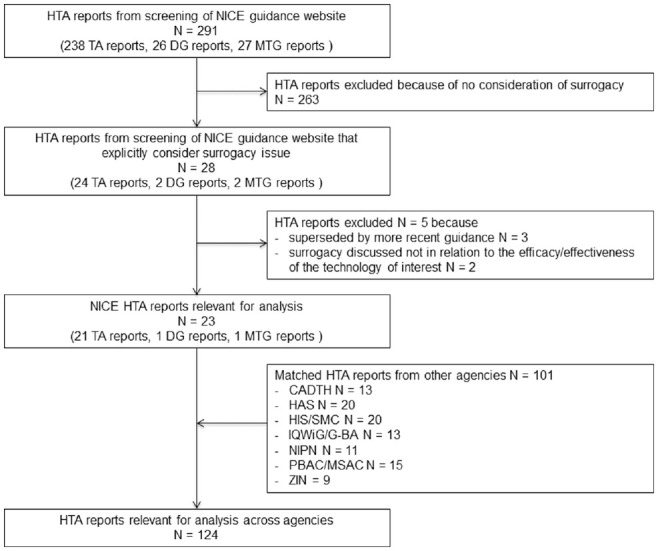
Flow diagram of health technology assessment report selection. CADTH, Canadian Agency for Drugs and Technologies in Health; DG, diagnostic guidance; HAS, Haute Autorité de Santé; HIS/SMC, Health Improvement Scotland/Scottish Medicines Consortium; IQWiG/G-BA, Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen/Gemeinsame Bundesausschuss; MTG, medical technologies guidance; NIPN, Országos Gyógyszerészeti és Élelmezés-egészségügyi Intézet; PBAC/MSAC, Pharmaceutical Benefits Advisory Committee/Medical Services Advisory Committee; TA, technology appraisal; ZIN, Zorginstituut Nederland.
How validation of surrogate endpoints is empirically addressed in HTA reports
To investigate how the validation of putative surrogate endpoints was addressed in practice, each of the 124 unique reports was considered as a separate observation (Table 3).
Table 3.
Characteristics of Health Technologies and Related HTA Evaluationa
| No. | Technology | Indication | Clinical Area | Main Surrogate Endpoint(s) [Patient-Centered Endpoint Substituted for] | NICE | HIS/SMC | HAS | PBAC/MSAC | CADTH | IQWiG/G-BA | ZiN | NIPN | HTA across Agencies |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|

| |||||||||||||
CADTH, Canadian Agency for Drugs and Technologies in Health; HAS, Haute Autorité de Santé; HER2, human epidermal growth factor receptor 2; HIS/SMC, Health Improvement Scotland/Scottish Medicines Consortium; HTA, health technology assessment; IQWiG/G-BA, Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen/Gemeinsame Bundesausschuss; NICE, National Institute for Health and Care Excellence; NIPN, Országos Gyógyszerészeti és Élelmezés-egészségügyi Intézet; PBAC/MSAC, Pharmaceutical Benefits Advisory Committee/Medical Services Advisory Committee; ZiN, Zorginstituut Nederland; —, Not assessed.
 , approved for reimbursement;
, approved for reimbursement;  , restricted reimbursement (either restricted prescription or subject to a price change); \color{75}\blacksquare, rejected.
, restricted reimbursement (either restricted prescription or subject to a price change); \color{75}\blacksquare, rejected.
Multiple evaluations available.
One European Network of HTA (EUnetHTA) report identified (https://www.eunethta.eu/the-joint-assessment-on-continuous-glucose-monitoring-cgm-real-time-and-flash-glucose-monitoring-fgm-as-personal-standalone-systems-in-patients-with-diabetes-mellitus-treated-with-insuli/).
Reports sought from MSAC.
The level of evidence to establish the validity of the surrogate was clearly assessed in 61 (49%) evaluations and not assessed in 57 (46%). In the other 6 reports, this information was unclear (5%). Only 27 reports (22%) reported a measure of strength of association between the putative surrogate endpoint and the patient-relevant endpoint of interest, and in the majority of the evaluations (97, 78%), there was no correlation metric reported. Forty (32%) evaluations quantified the predicted effect of the surrogate endpoint on the patient-centered outcome; the majority of reports did not (72, 58%) or failed to provide enough information (12, 10%) for us to judge whether this was actually done. The surrogate endpoints were overall deemed “acceptable” in 49 reports (40%), “unacceptable” in 23 (18%), and with no clear statement on acceptability provided in the remaining 52 (42%) evaluations (Suppl. Table S1).
Variation between agencies
The level of depth and scrutiny applied by different agencies in relation to the validation of surrogate endpoints varied (Figure 2). NICE was the agency most likely to report on the level of evidence (22/23), strength of association (7/23), and quantification of effect (17/23) related to the validation of a putative surrogate endpoint. In contrast, HAS and NIPN were the agencies with the least level of information reported in terms of validation.
Figure 2.

Steps of the validation of surrogate endpoints performed by health technology assessment agencies. CADTH, Canadian Agency for Drugs and Technologies in Health; DG, diagnostic guidance; HAS, Haute Autorité de Santé; HIS/SMC, Health Improvement Scotland/Scottish Medicines Consortium; IQWiG/G-BA, Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen/Gemeinsame Bundesausschuss; MTG, medical technologies guidance; NIPN, Országos Gyógyszerészeti és Élelmezés-egészségügyi Intézet; PBAC/MSAC, Pharmaceutical Benefits Advisory Committee/Medical Services Advisory Committee; TA, technology appraisal; ZIN, Zorginstituut Nederland.
IQWiG appeared to apply a particularly strict approach with respect to the acceptability of surrogate endpoints, with no surrogate outcome explicitly deemed valid. Pairwise κ coefficients revealed moderate to substantial (>0.40) agreement on the acceptability of the surrogate endpoint between NICE and SMC, as well as between PBAC and NIPN HTA. Overall, there was very low level of agreement across the 8 agencies (0.10; P = 0.04) (Suppl. Table S2).
Variation between health technologies
High consistency in acceptability was seen for cholesterol level used in the assessment of alirocumab in hypercholesterolemia (only IQWiG did not accept the validity of this putative surrogate endpoint28) (Suppl. Figure S1). Total kidney volume used in the assessment of tolvaptan in autosomal dominant polycystic kidney disease was accepted in 5 of 6 assessments (CADTH stated that the relationship between total kidney volume and clinically important endpoints “remains to be elucidated”29). For other health technologies, conclusions about the validity of the surrogate endpoints were conflicting. For example, alkaline phosphatase and bilirubin were deemed valid in the assessment of obeticholic acid for primary biliary cholangitis by 3 agencies (NICE,30 SMC,31 CADTH32) and invalid by 3 agencies (HAS,33 IQWiG/G-BA,34 ZIN35).
Level of evidence
The acceptability of the putative surrogate measure should be based on the related level of evidence (see Table 1). This can be as low as expert opinion, as in the NICE HTA assessment of progression-free survival (PFS) of brentuximab vedotin in CD30-positive Hodgkin lymphoma,36 or as high as individual patient data meta-analyses of RCTs, as seen in the evaluation of pathological complete response of pertuzumab in human epidermal growth factor receptor 2–positive breast cancer.37 However, a higher level of evidence did not always result in a positive opinion expressed by the committee in relation to the acceptability of the surrogate. For example, based on the CollaborativeTrials in Neoadjuvant Breast Cancer pooled individual patient data meta-analysis, CADTH concluded that there is insufficient evidence to support the validity of pathological complete response as a surrogate for long-term outcomes in breast cancer.38 In contrast, informed by clinicians’ opinion, NICE accepted PFS for brentuximab vedotin in CD30-positive Hodgkin lymphoma.36
Strength of association
Reports often discussed the concept of association or correlation between the 2 endpoints of interest but rarely reported an actual metric (e.g., R2, Spearman’s ρ correlation coefficient). For example, the pirfenidone in idiopathic pulmonary fibrosis appraisal by NICE39 cited 1 study showing that there is a moderate correlation between changes in percent predicted forced vital capacity and changes in a disease-specific health-related quality-of-life measure (i.e., Spearman’s ρ correlation coefficient of –0.32). Lack of reporting of correlation metrics may reflect the difficult interpretation of these values, limited methods guidance, or presumed confidence in the validity of the surrogate.
Quantification of effect on patient-relevant outcomes
Quantification of the expected treatment effect on the patient-centered outcome based on the observed effect on the surrogate endpoint was rarely reported. In some cases, this quantification was a risk equation based on previous longitudinal studies or registries in the same (or similar) therapy area. In the appraisal of evolocumab in primary hypercholesterolemia/mixed dyslipidemia, treatment effects were modeled with published risk equations from the Framingham Heart Study and the UK REACH registry for cardiovascular disease patients.40 The surrogate threshold effect (STE) has been proposed as key metric to identify the minimum level of observed effect on the surrogate endpoint in order to predict a significant effect on the patient-centered outcome.41 However, STE was only included in the IQWiG report on ribociclib in locally advanced or metastatic breast cancer.42
Use of surrogate endpoint evidence in cost-effectiveness models
For those reports that included a cost-effectiveness analysis, surrogate endpoints were usually a key input in the decision model. For example, annual change in total kidney volume was used as an intermediate step to model change in estimated glomerular filtration rate (eGFR) in the cost-effectiveness model of tolvaptan in autosomal dominant polycystic kidney disease.43 While quantification of the treatment effect on the final outcome based on the surrogate could be an output of the decision model, we did not find any examples of this across reports in this study. Despite a pivotal trial powered for a surrogate primary endpoint, the available cost-effectiveness models were developed using immature survival data from short-term studies extrapolated to obtain estimates of the full survival benefit.44,45 Evidence around the validation of the primary surrogate endpoint could inform the choice of the methods for performing the extrapolation in economic models (e.g., how plausible the extrapolated portions are),46 but we never encountered this across our sample of HTA reports.
While surrogate endpoints are generally assumed to replace patient-relevant outcomes, such as overall survival, in cost-effectiveness models, they may also be used to predict health-related quality of life. For example, a key utility value was an assumed 0.04 increase in health-related quality of life for patients experiencing a sustained virologic response with the use of the ledipasvir-sofosbuvir drug combination in chronic hepatitis C evaluation.47 They may also be used to predict health care resource consumption/costs (e.g., PFS as a proxy for time on treatment with biologic cobimetinib for the management of unresectable or metastatic melanoma).48
Multivariable regression analysis showed that reporting about the level of evidence supporting the relationship between the putative surrogate and the patient-centered endpoint of interest increased the probability of accepting the validity of the surrogate endpoints (odds ratio [OR], 4.60; 95% confidence interval [CI], 1.60–13.18; P = 0.005), regardless of whether this evidence is biological, plausibility anecdotal, observational, or experimental (Table 4). That these other elements are statistically significant in univariate regressions suggests that they are correlated with reporting of evidence.
Table 4.
Factors Associated with Surrogate Acceptability and Recommendation Given
| Factor | Multivariate Regression Analysis,a OR (95% CI) [P Value] | Univariate Regression Analysis,a OR (95% CI) [P Value] |
|---|---|---|
| Factors associated with acceptability of surrogate endpoint | ||
| Level of evidence assessed | 4.60 (1.60–13.18) [0.005] | 5.51 (2.42–12.55) [<0.001] |
| Strength of association provided | 1.23 (0.40–3.74) [0.72] | 2.69 (1.04–6.97) [0.041] |
| Quantification of effect provided | 1.17 (0.38–3.61) [0.78] | 3.52 (1.43–8.65) [0.006] |
| Orphan status | 0.52 (0.81–3.39) [0.50] | 0.38 (0.06–2.36) [0.30] |
| Factors associated with positive recommendation | ||
| Acceptability of surrogate endpoint | 0.71 (0.23–2.20) [0.55] | 0.52 (0.19–1.46) [0.21] |
| Level of evidence assessed | 0.32 (0.07–1.37) [0.12] | 0.40 (0.15–1.09) [0.07] |
| Strength of association provided | 2.30 (0.51–10.45) [0.28] | 1.42 (0.43–4.66) [0.57] |
| Quantification of effect provided | 1.12 (0.27–4.74) [0.87] | 0.57 (0.20–1.63) [0.29] |
| Orphan status | 8.61 (1.03–72.94) [0.047] | 11.38 (1.55–83.58) [0.02] |
CI, confidence interval; OR, odds ratio.
From mixed-effect logistic regression with clustering at the level of the health technology. OR >1 indicates higher odds of the surrogate deemed acceptable or technology receiving positive recommendation.
What impact does use of surrogate endpoints have on the recommendations given?
We were able to examine the recommendations based on 113 assessments (11 [9%] HTA recommendations given by NIPH were not publicly accessible) (Table 2). Pairwise κ coefficients show at least modest (>0.20) agreement on the final recommendation given by IQWiG/G-BA and SMC and substantial (>0.60) agreement on the final recommendation given by IQWiG/G-BA and HAS. Overall, the level of agreement across the 8 agencies was relatively low (0.18; P = 0.004) (Suppl. Table S3).
For 8 (6%) of the recommendations, orphan drug designation was associated with either full approval (n = 6) or restricted approval (n = 2). A patient access scheme was mandated in 19 (16%) of the restricted recommendations by NICE and SMC, with risk-sharing agreements being required in 3 (2%) of these restricted recommendations. In 10 (8%) of the restricted recommendations, a price reduction was required. Lack of benefit, high uncertainty on outcomes, or insufficient evidence on the relationship between the surrogate and patient-relevant outcomes was explicitly cited in 13 (11%) rejections. In contrast, 6 (5%) approval recommendations were made despite stated uncertainty in clinical or cost-effectiveness evidence (Suppl. Table S4).
With the exception of orphan status (OR, 8.61; 95% CI, 1.03–72.94; P = 0.047), none of the other factors were predictive of the final coverage recommendation (Table 4).
Discussion
In this study, we mapped the methods used in 124 surrogate endpoint-based HTA evaluations/reports on 23 different health technologies across 8 HTA agencies. Based on a previously proposed 3-step framework for the validation of surrogate outcomes,20 we found that 61 (49%) reports discussed the level of evidence to support the relationship between the surrogate endpoint and the patient-centered outcome based on IPD meta-analyses of RCTs in the relevant indication. Only 27 (22%) evaluations reported a correlation coefficient or other association measure. When available, these associations were usually below recommended thresholds for acceptability of surrogate (i.e., the lower limit of the 95% CI for R≥ 0.85 recommended by IQWiG).49 Forty (32%) reports quantified the expected effect on the patient-centered outcome given the observed effect on the surrogate outcome. A clear statement around the acceptability of the surrogate endpoint was provided in 49 (40%) reports, while 23 (19%) rejected the validity of the proposed surrogate endpoint. Our regression models showed that searching for evidence of the relationship between the surrogate and patient-centered outcome was a predictor of the HTA agency’s acceptance of the surrogate endpoint but did not show any significant effect for the other steps in the validation process.
Among the 113 assessments with a policy recommendation, 32 (28%) technologies were fully approved, 20 (18%) were rejected, and 61 (54%) received restricted approval. To handle the decision uncertainty as the result of the use surrogate endpoints, HTA agencies often used conditional approval based on price discount agreements (including patient access and risk-sharing schemes with evidence development), had restricted indications, or applied more permissive evaluation frameworks (such as orphan technology designation, end-of-life treatment, or specialist coverage programs, such as the Cancer Drugs Fund in the United Kingdom). For example, when evaluating bosutinib, all HTA agencies had access to results of the main study that reported major cytogenetic response and immature overall survival data. IQWiG approved bosutinib as an orphan medicine despite concluding that major cytogenetic response and overall survival were limited. The Scottish Medicine Consortium also found “high uncertainty around the survival estimate” but still approved bosutinib as part of the ultra-orphan process. While the reimbursement of drugs authorized with orphan designation may vary across Europe, orphan status is usually a policy imperative that commits HTA agencies to recommend even without evidence of additional benefit.50 We found weak evidence that the acceptability of the surrogate endpoint was associated with the final coverage decision made by HTA agencies.
We found considerable variability in the level of scrutiny applied with respect to the surrogacy issue across HTA agencies. This variability is in part explained by differences in the methodological guidelines followed by the HTA agencies.7 Different expertise available to the committee, different level of reporting, or different interpretations of the definition of surrogate endpoints may also play a role. Some surrogate endpoints, especially so-called intermediate endpoints (e.g., progression-free survival, disease-free survival, event-free survival), may be considered not to require validation by HTA agencies as they have been already accepted by a regulatory body for marketing authorization. In several cases, HTA agencies quoted EMA or FDA approval documents to support their acceptance of the validity of a surrogate endpoint. However, it is important to recognize that the mandate of regulators is not the same as HTA organizations.11 The underlying evidence for the accepted surrogate endpoints for regulatory review may be weak or missing.51,52 As a life-cycle evaluation to health care technologies has become more widespread, regulatory agencies have gained statutory authority to order postmarketing studies, typically in the case of approvals based on uncertain evidence. However, only 1 in 10 new drug indications approved by the US FDA on the basis of surrogate endpoints has been shown to have at least 1 postapproval trial validating the use of the surrogate or demonstrating improved overall survival.53–55
Surrogate endpoint evidence affects the assessment of clinical and cost-effectiveness of a health technology.7 However, we found limited consideration in the economic elements of the HTA reports included in this study. For example, some cost-effectiveness models were based on extrapolations of immature survival data from short-term studies rather than use of validated primary surrogate endpoint data. Furthermore, there was little use of biomarkers or intermediate endpoints as replacements for either health-related quality of life or health care resource consumption/costs.
Limitations
Our analyses were limited to consideration of publicly available information, and reporting details varied greatly between agencies. As we based our initial selection of technologies on a text search for surrogacy terms of NICE reports, we may have excluded reports/technologies using surrogate endpoint evidence. We cannot exclude the possibility that consideration of surrogacy issues occurred during HTA committee meetings but that these observations were not reported in public documents. Some of the non-English reports were not double screened due to lack of language expertise across the coauthors. Although we identified only 2 nondrug technologies in our sample, we believe that the findings of the report apply equally to such technologies, including medical devices.
Conclusions
We found that the handling of surrogate endpoint evidence varied greatly across HTA reports and agencies, with inconsistent consideration of level of evidence and statistical validation. Consideration of the level of evidence supporting the relationship between the surrogate endpoint and patient-centered outcome increased the likelihood of acceptability of a surrogate endpoint. However, we did not find strong evidence supporting an association between accepting the surrogate and the coverage recommendation made about the treatment. Claims of surrogate validity need to be considered contextually, given that the relationship between surrogate endpoint and patient-relevant outcome is typically treatment and indication specific.
HTA evaluation reports often refer to regulatory (FDA or EMA) statements about the acceptability of surrogate endpoints. However, regulators are more focused on safety and shorter-term efficacy, and registration trials are often specifically designed to answer these questions. Given that HTA agencies focus on a longer-term perspective and seek to assess clinical effectiveness and cost-effectiveness, their considerations on the acceptability of surrogate endpoints may differ from those of regulators.56
Our findings demonstrate the need for further consideration of the issue of surrogacy and for harmonization of practices between regulatory and HTA agencies and across international jurisdictions.
Supplemental Material
Supplemental material, sj-doc-1-mdm-10.1177_0272989X21994553 for Validity of Surrogate Endpoints and Their Impact on Coverage Recommendations: A Retrospective Analysis across International Health Technology Assessment Agencies by Oriana Ciani, Bogdan Grigore, Hedwig Blommestein, Saskia de Groot, Meilin Möllenkamp, Stefan Rabbe, Rita Daubner-Bendes and Rod S. Taylor in Medical Decision Making
Footnotes
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Financial support for this study was provided entirely by the European Union’s Horizon 2020 research and innovation program under grant 779306 (COMED—Pushing the Boundaries of Cost and Outcome Analysis of Medical Technologies). The funding agreement ensured the authors’ independence in designing the study, interpreting the data, writing, and publishing the report. The results only reflect the authors’ views, and the European Union is not responsible for any use that may be made of the information it contains. None of the authors are employed by the health technology assessment agencies included in this study or were involved as appraisal committee members in the included evaluations. OC completed this manuscript during her Fulbright Visiting Scholarship at Yale School of Public Health.
ORCID iDs: Oriana Ciani  https://orcid.org/0000-0002-3607-0508
https://orcid.org/0000-0002-3607-0508
Bogdan Grigore https://orcid.org/0000-0003-4241-7595
Supplemental Material: Supplementary material for this article is available on the Medical Decision Making website at http://journals.sagepub.com/home/mdm.
Contributor Information
Oriana Ciani, Centre for Research on Health and Social Care Management, SDA Bocconi, Milan, Lombardia, Italy; Evidence Synthesis & Modelling for Health Improvement, University of Exeter Medical School, Exeter, Devon, UK.
Bogdan Grigore, Evidence Synthesis & Modelling for Health Improvement, University of Exeter Medical School, Exeter, Devon, UK.
Hedwig Blommestein, Institute for Medical Technology Assessment, Erasmus School of Health Policy & Management, Erasmus University Rotterdam, Rotterdam, The Netherlands.
Saskia de Groot, Institute for Medical Technology Assessment, Erasmus School of Health Policy & Management, Erasmus University Rotterdam, Rotterdam, The Netherlands.
Meilin Möllenkamp, Hamburg Center for Health Economics, Universität Hamburg, Hamburg, Germany.
Stefan Rabbe, Hamburg Center for Health Economics, Universität Hamburg, Hamburg, Germany.
Rita Daubner-Bendes, Syreon Research Institute, Budapest, Hungary; MRC/CSO Social and Public Health Sciences Unit & Robertson Centre for Biostatistics, Institute of Health and Well Being, University of Glasgow, Glasgow, Scotland, UK.
Rod S. Taylor, Evidence Synthesis & Modelling for Health Improvement, University of Exeter Medical School, Exeter, Devon, UK MRC/CSO Social and Public Health Sciences Unit & Robertson Centre for Biostatistics, Institute of Health and Well Being, University of Glasgow, Glasgow, Scotland, UK.
References
- 1. Darrow JJ, Avorn J, Kesselheim AS. FDA approval and regulation of pharmaceuticals, 1983. –2018. JAMA. 2020;323(2):164–176. [DOI] [PubMed] [Google Scholar]
- 2. DeMets DL, Psaty BM, Fleming TR. When can intermediate outcomes be used as surrogate outcomes? JAMA. 2020;323(12):1184–5. [DOI] [PubMed] [Google Scholar]
- 3. Wittes J, Lakatos E, Probstfield J. Surrogate endpoints in clinical trials: cardiovascular diseases. Stat Med. 1989;8:415–25. [DOI] [PubMed] [Google Scholar]
- 4. Naci H, Smalley KR, Kesselheim AS. Characteristics of preapproval and postapproval studies for drugs granted accelerated approval by the US Food and Drug Administration. JAMA. 2017;318(7):626–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Torbica A. HTA around the world: broadening our understanding of cross-country differences. Value Health. 2020;23(1):1–2. [DOI] [PubMed] [Google Scholar]
- 6. Velasco Garrido M, Mangiapane S. Surrogate outcomes in health technology assessment: an international comparison. Int J Technol Assess Health Care. 2009;25:315–22. [DOI] [PubMed] [Google Scholar]
- 7. Grigore B, Ciani O, Dams F, et al. Surrogate endpoints in health technology assessment: an international review of methodological guidelines. Pharmacoeconomics. 2020;38(10):1055–70. [DOI] [PubMed] [Google Scholar]
- 8. Pinto A, Naci H, Neez E, et al. Association between the use of surrogate measures in pivotal trials and health technology assessment decisions: a retrospective analysis of NICE and CADTH reviews of cancer drugs. Value Health. In press. [DOI] [PubMed] [Google Scholar]
- 9. Kleijnen S, Lipska I, Leonardo Alves T, et al. Relative effectiveness assessments of oncology medicines for pricing and reimbursement decisions in European countries. Ann Oncol. 2016;27(9):1768–75. [DOI] [PubMed] [Google Scholar]
- 10. Elston J, Taylor RS. Use of surrogate outcomes in cost-effectiveness models: a review of United Kingdom health technology assessment reports. Int J Technol Assess Health Care. 2009;25:6–13. [DOI] [PubMed] [Google Scholar]
- 11. Rocchi A, Khoudigian S, Hopkins R, et al. Surrogate outcomes: experiences at the Common Drug Review. Cost Eff Resour Alloc. 2013;11:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. De Gruttola VG, Clax P, DeMets DL, et al. Considerations in the evaluation of surrogate endpoints in clinical trials: summary of a National Institutes of Health workshop. Control Clin Trials. 2001;22:485–502. [DOI] [PubMed] [Google Scholar]
- 13. Weir CJ, Walley RJ. Statistical evaluation of biomarkers as surrogate endpoints: a literature review. Stat Med. 2006;25:183–203. [DOI] [PubMed] [Google Scholar]
- 14. Van der Elst W, Molenberghs G, Alonso A. Exploring the relationship between the causal-inference and meta-analytic paradigms for the evaluation of surrogate endpoints. Stat Med. 2016;35(8):1281–98. [DOI] [PubMed] [Google Scholar]
- 15. Burzykowski T, Molenberghs G, Buyse M. The Evaluation of Surrogate Endpoints. New York: Springer Science + Business Media; 2005. [Google Scholar]
- 16. Bujkiewicz S, Achana F, Papanikos T, Riley RD, Abrams KR. NICE DSU Technical Support Document 20: multivariate meta-analysis of summary data for combining treatment effects on correlated outcomes and evaluating surrogate endpoints. 2019. Available from: http://www.nicedsu.org.uk
- 17. Belin L, Tan A, De Rycke Y, Dechartres A. Progression-free survival as a surrogate for overall survival in oncology trials: a methodological systematic review. Br J Cancer. 2020;122(11):1707–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xie W, Halabi S, Tierney JF, et al. A systematic review and recommendation for reporting of surrogate endpoint evaluation using meta-analyses. JNCI Cancer Spectr. 2019;3(1):pkz002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Institut fuär Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG). Aussagekraft von surrogatendpunkten in der onkologie [Validity of surrogate parameters in oncology]. IQWiG-Berichte 80; 2011. Available from: https://www.iqwig.de/download/a10-05_rapid_report_surrogatendpunkte_in_der_onkologie.pdf?rev=117386 [Google Scholar]
- 20. Ciani O, Buyse M, Drummond M, Rasi G, Saad ED, Taylor RS. Time to review the role of surrogate end points in health policy: state of the art and the way forward. Value Health. 2017;20(3):487–495. [DOI] [PubMed] [Google Scholar]
- 21. Taylor RS, Elston J. The use of surrogate outcomes in modelbased cost-effectiveness analyses: a survey of UK Health Technology Assessment reports. Health Technol Assess. 2009;13(8): iii, ix–xi, 1–50. [DOI] [PubMed] [Google Scholar]
- 22. Husereau D, Drummond M, Petrou S, et al. Consolidated Health Economic Evaluation Reporting Standards (CHEERS) statement. Cost Effectiveness Resource Allocation. 2013;11:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. European Medicines Agency. Orphan designation: overview. Available from: https://www.ema.europa.eu/en/human-regulatory/overview/orphan-designation-overview
- 24. Australian Government, Therapeutic Goods Administration. Orphan drug designation eligibility criteria. Available from: https://www.tga.gov.au/publication/orphan-drug-designation-eligibility-criteria
- 25. CADTH. Drugs for rare diseases: a review of national and international health technology assessment agencies and public payers’ decision-making processes. 2018. Available from: https://www.cadth.ca/sites/default/files/pdf/es0326_drugs_for_rare_diseases.pdf
- 26. Garrison LP, Jr, Towse A, Briggs A, et al. Performance-based risk-sharing arrangements—good practices for design, implementation, and evaluation: report of the ISPOR good practices for performance-based risk-sharing arrangements task force. Value Health. 2013;16(5):703–19. [DOI] [PubMed] [Google Scholar]
- 27. McHugh ML. Interrater reliability: the kappa statistic. Biochem Med. 2012;22:276–82. [PMC free article] [PubMed] [Google Scholar]
- 28. Gemeinsame Bundesausschuss. Nutzenbewertungsverfahren zum Wirkstoff Alirocumab (Hypercholesterinämie oder gemischte Dyslipidämie) 2016. Available from: https://www.g-ba.de/bewertungsverfahren/nutzenbewertung/407/ [Google Scholar]
- 29. CADTH Common Drug Review. Tolvaptan. 2018. Available from: https://cadth.ca/sites/default/files/cdr/pharmacoeconomic/SR0435_Jinarc_PE_Report.pdf [Google Scholar]
- 30. National Institute for Health and Care Excellence. Obeticholic acid for treating primary biliary cholangitis [TA443]. 2017. Available from: https://www.nice.org.uk/guidance/ta443 [Google Scholar]
- 31. Scottish Medicines Consortium. Obeticholic acid, 5mg and 10mg film-coated tablets (Ocaliva®) SMC No (1232/17). 2017. Available from: https://www.scottishmedicines.org.uk/media/2055/obeticholic_acid_ocaliva_final_may_2017_amended_170517_for_website.pdf [Google Scholar]
- 32. CADTH Common Drug Review. Obeticholic acid. 2017. Available from: https://www.cadth.ca/sites/default/files/cdr/ [Google Scholar]
- 33. Haute Authorite de Sante. OCALIVA (obeticholic acid), bile acid. 2017. Available from: https://www.has-sante.fr/jcms/c_2773278/en/ocaliva-obeticholic-acid-bile-acid [Google Scholar]
- 34. Gemeinsame Bundesausschuss. Nutzenbewertungsverfahren zum Wirkstoff Obeticholsäure. 2017. Available from: https://www.gba.de/bewertungsverfahren/nutzenbewertung/276/ [Google Scholar]
- 35. Zorginstituut Nederland. DOOR HET ZORGINSTITUUT AANGEPASTE VERSIE VAN HET EVALUATIERAPPORT DAG 60 OCALIVA. 2017. Available from: https://www.zorginstituutnederland.nl/publicaties/adviezen/2018/07/18/gvs-advies-obeticholzuur-ocaliva-voorde-behandeling-van-primaire-biliaire-cholangitis-pbc [Google Scholar]
- 36. National Institute for Health and Care Excellence. Brentuximab vedotin for treating CD30-positive Hodgkin lymphoma [TA524]. 2018. Available from: https://www.nice.org.uk/guidance/ta524 [Google Scholar]
- 37. Cortazar P, Zhang L, Untch M, et al. Pathological complete response and long-term clinical benefit in breast cancer: the CTNeoBC pooled analysis. Lancet. 2014;384(9938):164–72. [DOI] [PubMed] [Google Scholar]
- 38. CADTH. Pan-Canadian Oncology Drug Review. Perjeta or Perjeta-herceptin combo pack for neoadjuvant breast cancer—details. 2015. Available from: https://www.cadth.ca/perjeta-orperjeta-herceptin-combo-pack-neoadjuvant-breast-cancerdetails [Google Scholar]
- 39. National Institute for Health and Care Excellence. Pirfenidone for treating idiopathic pulmonary fibrosis [TA504]. 2018. Available from: https://www.nice.org.uk/guidance/ta504/resources/pirfenidone-for-treating-idiopathic-pulmonary-fibrosispdf-82606719541957 [Google Scholar]
- 40. National Institute for Health and Care Excellence. Evolocumab for treating primary hypercholesterolaemia and mixed dyslipidaemia [TA394]. 2016. Available from: https://www.nice.org.uk/guidance/ta394/chapter/1-recommendations [Google Scholar]
- 41. Burzykowski T, Buyse M. Surrogate threshold effect: an alternative measure for meta-analytic surrogate endpoint validation. Pharm Stat. 2006;5:173–86. [DOI] [PubMed] [Google Scholar]
- 42. Gemeinsame Bundesausschuss. Nutzenbewertungsverfahren zum Wirkstoff Ribociclib (Mammakarzinom, HR+, HER2–, postmenopausale Frauen, Kombination mit Aromatasehemmer). 2018. Available from: https://www.g-ba.de/bewertungsverfahren/nutzenbewertung/311/ [Google Scholar]
- 43. National Institute for Health and Care Excellence. Tolvaptan for treating autosomal dominant polycystic kidney disease [TA358]. 2015. Available from: https://www.nice.org.uk/guidance/ta358 [Google Scholar]
- 44. Scottish Medicines Consortium. Axitinib, 1mg and 5mg, film-coated tablets (Inlyta®) SMC No. (855/13) 2013. Available from: https://www.scottishmedicines.org.uk/media/1284/axitinib_inlyta_resubmission_final_october_2013_amended_011113.pdf [Google Scholar]
- 45. National Institute for Health and Care Excellence. Axitinib for treating advanced renal cell carcinoma after failure of prior systemic treatment [TA333]. 2015. https://www.nice.org.uk/guidance/ta333 [DOI] [PubMed] [Google Scholar]
- 46. Latimer N. NICE DSU Technical Support Document 14: undertaking survival analysis for economic evaluations alongside clinical trials—extrapolation with patient-level data. 2011. Available from: http://www.nicedsu.org.uk [PubMed]
- 47. SMC. Ledipasvir/sofosbuvir, 90mg/400mg, film-coated tablet (Harvoni®). Available from: https://www.scottishmedicines.org.uk/media/1905/ledipasvir_sofosbuvir_harvoni_final_february_2015_for_website.pdf
- 48. CADTH. Cotellic for metastatic melanoma. Available from: https://www.cadth.ca/cotellic-metastatic-melanoma-details
- 49. Ciani O, Davis S, Tappenden P, et al. Validation of surrogate endpoints in advanced solid tumours: systematic review of statistical methods, results, and implications for policy makers. Int J Technol Assess Health Care. 2014;30:13. [DOI] [PubMed] [Google Scholar]
- 50. Malinowski KP, Kawalec P, Trabka W, et al. Reimbursement of orphan drugs in Europe in relation to the type of authorization by the European Medicines Agency and the decision making based on health technology assessment. Front Pharmacol. 2018;9:1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vreman RA, Naci H, Goettsch WG, et al. Decision making under uncertainty: comparing regulatory and health technology assessment reviews of medicines in the United States and Europe. Clin Pharmacol Ther. 2020;108(2):350–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gyawali B, Hey SP, Kesselheim AS. Evaluating the evidence behind the surrogate measures included in the FDA’s table of surrogate endpoints as supporting approval of cancer drugs. EClinicalMedicine. 2020;21:100332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pease AM, Krumholz HM, Downing NS, Aminawung JA, Shah ND, Ross JS. Postapproval studies of drugs initially approved by the FDA on the basis of limited evidence: systematic review. BMJ. 2017;357:j1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wallach JD, Ciani O, Pease AM, et al. Comparison of treatment effect sizes from pivotal and postapproval trials of novel therapeutics approved by the FDA based on surrogate markers of disease: a meta-epidemiological study. BMC Med. 2018;16(1):45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kim C, Prasad V. Cancer drugs approved on the basis of a surrogate end point and subsequent overall survival: an analysis of 5 years of US Food and Drug Administration approvals. JAMA Intern Med. 2015;175(12):1992–4. [DOI] [PubMed] [Google Scholar]
- 56. Ruof J, Knoerzer D, Dünne A-A, et al. Analysis of endpoints used in marketing authorisations versus value assessments of oncology medicines in Germany. Health Policy. 2014;118:242–54. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, sj-doc-1-mdm-10.1177_0272989X21994553 for Validity of Surrogate Endpoints and Their Impact on Coverage Recommendations: A Retrospective Analysis across International Health Technology Assessment Agencies by Oriana Ciani, Bogdan Grigore, Hedwig Blommestein, Saskia de Groot, Meilin Möllenkamp, Stefan Rabbe, Rita Daubner-Bendes and Rod S. Taylor in Medical Decision Making