

Abstract
Gaucher disease (GD) is the most common lysosomal storage disease caused by deficiency of beta-glucocerebrosidase (GCase) resulting in lysosomal accumulation of its glycolipid substrate glucosylceramide. The activity of GCase depends on many factors such as proper folding and lysosomal localization, which are influenced by mutations in GCase encoding gene, and regulated by various GCase-binding partners including Saposin C, progranulin and heat shock proteins. In addition, proinflammatory molecules also contribute to pathogenicity of GD. In this review, we summarize the molecules that are known to be important for the pathogenesis of GD, particularly those modulating GCase lysosomal appearance and activity. In addition, small molecules that inhibit inflammatory mediators, calcium ion channels and other factors associated with GD are also described. Discovery and characterization of novel molecules that impact GD is not only important for deciphering the pathogenic mechanisms of the disease, but it also provides new targets for drug development to treat the disease.
Keywords: Gaucher Disease, beta-glucocerebrosidase, LIMP-2, Saposin C, progranulin, heat shock proteins
1. INTRODUCTION
Gaucher Disease (GD) is the most prevalent autosomal recessive lysosome storage disease (LSD). GD is caused by the loss-of-function of lysosomal hydrolase enzyme, beta-glucocerebrosidase (GCase). The protein sequence and domain structure of GCase are displayed in Figure 1. Across populations, approximately 70–98% of GD cases are accounted for by five relatively common mutations in GBA1, the gene encoding GCase1: p.N370S, p.L444P, c.84GGIns, IVS2+1G>A and RecNcil [1–4]. GD is classified into three subtypes according to clinical manifestations (Table 1). In Type I, pathology is confined to the reticuloendothelial and skeletal systems with no neuropathic symptoms and the clinical manifestations include hepatosplenomegaly, splenomegaly, and bone disease [5, 6]. The incidence of non-neuronopathic GD is about 1 in 60,000 globally, and the highest incidence occurs in Ashkenazi Jewish community, ranging from 1 in 800 to 1 in 950[7–9]. Types II and III are neuoronopathic GD (nGD) and involve accumulation of GlcCer in brain resulting in neurological damage. Type II GD, also known as acute neuronopathic Gaucher disease, accounts for 5–20% of cases and develops during infancy, usually by 3 to 6 months of age; symptoms include increased tone, seizures, rigidity of the neck and trunk, swallowing disorders and oculomotor paralysis [10]. Type III GD, or chronic neuronopathic GD, accounts for less than 10% cases and involves organomegaly, bone disease and neurological malfunctions [6]. The severity of GD is reported to correlate with the extent of endoplasmic reticulum (ER) retention and proteasomal degradation of GCase, which are affected by its mutations [11].
Fig. 1. Sequence and structure of human acid-β-glucosidase.
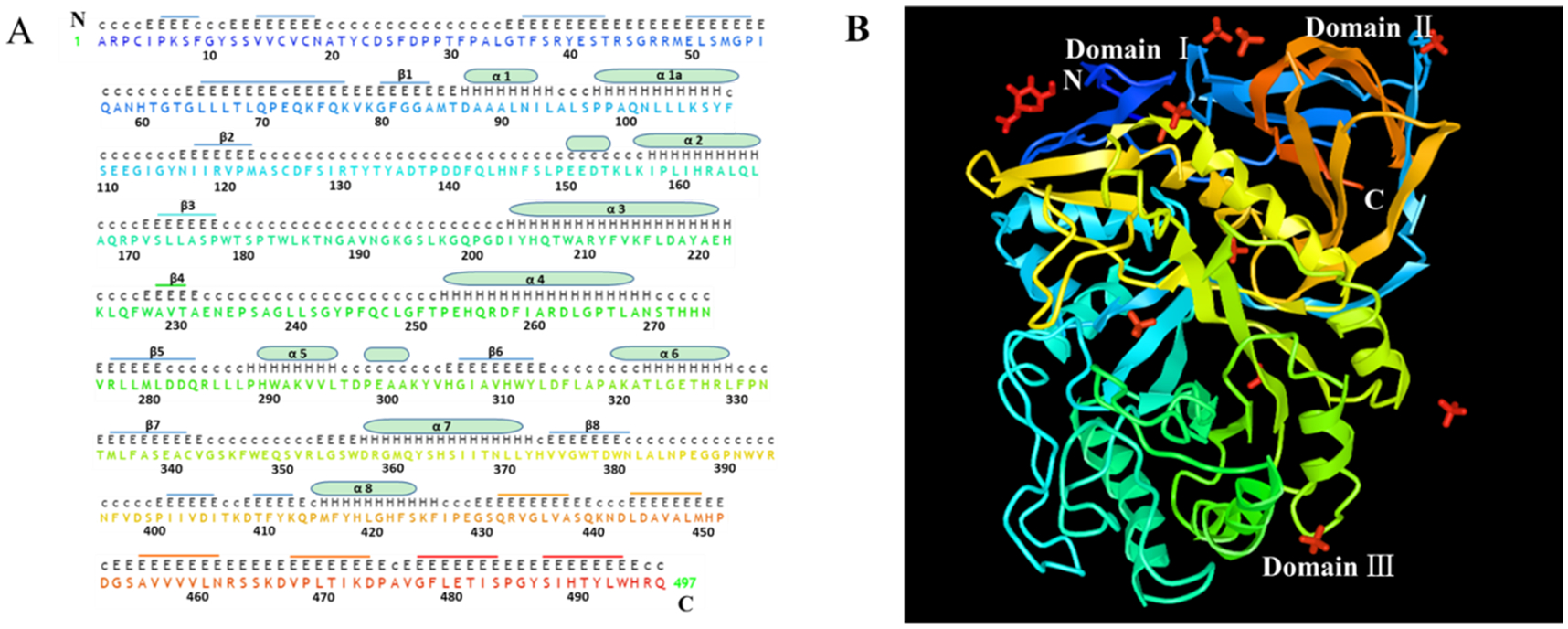
A. The sequence of human acid-β-glucosidase has a total of 497 amino acid residues in which there are eight α helices displayed with cylinders and eight β-strands indicated by stubs. The sequences and stubs in discrepant colors are correspondently presented in X-ray structure in panel B. The upper upper-case letters are the nonstandard residues with coordinates and the upper lower-case letters are the nonstandard residues missing coordinates. The lower upper-case letters indicate the standard residues and the numbers are for amino acid sequence location. B. X-ray structure of α helices and β-strands of human acid-β-glucosidase (from NCBI structure database PDB ID 10GS viewed by iCn3D, https://www.ncbi.nlm.nih.gov/Structure/icn3d/full.html?showseq=1&mmdbid=23543&buidx=1). Acid-β-glucosidase has one N-acetyl-d-glucosamine (top left corner in red color), ten sulfate ions (four limbs in red color), and three domains: domain Ⅰ(aa1-aa29 and aa384–414), domain Ⅱ (an immunoglobulin-like domain consists of aa30-aa76 and aa429-aa497) and domain Ⅲ (a catalytic domain, which is a TIM barrel and contains aa77-aa383 and aa415-aa428).
Table 1.
Clinical characteristics of Gaucher Disease
| Gaucher Disease | Type I | Type II | Type III |
|---|---|---|---|
| Alternative name | nonneuronopathic | acute neuronopathic | chronic neuronopathic |
| Disease onset | childhood or adulthood | 3–6 months old infants | childhood or adolescence |
| Life expectancy | childhood or adulthood | dies in infancy (median 9 months) | childhood or early adulthood |
| Occupying of all GD | 90%, the most common type | 5–20% | less than 10% |
| Prevalence | 1 in 100,000 in general population | less than 1 in 100,000 live births | less than 1 in 100,000 live births |
| Special ethnicity | the highest incidence in Ashkenazi Jewish community, ranging from 1 in 800 to 1 in 950 | no ethnic difference | Norrbottnian region of Sweden (Norrbottnian Gaucher disease), 1 in 50,000 prevalence |
| GCase mutant | N370S | Various | L444P |
| Residual GCase activity | around 15% of control | 1.75% of control | nearly absent |
| Disease course | progressive | rapidly progressive | progressive |
| Involvement | confined to the reticuloendothelial and skeletal systems with no neuropathic symptoms | accumulation of glucosylceramide in brain, without bone involvement | organomegaly, bone disease and neurological malfunctions |
| Clinical manifestations | hepatosplenomegaly, anemia, thrombocytopenia and bone disease | early nervous system signs, increased tone, seizures, rigidity of the neck and trunk, swallowing disorders and oculomotor paralysis, cerebellar signs | late onset nervous problems, abnormal horizontal saccades, oculomotor apraxia, myoclonus, seizures, dementia (late stage), cerebellar signs, extrapyramidal finding |
| Treatment | enzyme replacement therapy, substrate reduction therapy | hematopoietic stem cell transplantation, pharmacological chaperones, gene therapy | hematopoietic stem cell transplantation, pharmacological chaperones, gene therapy |
Residual activity of GCase in GD patients has been characterized; in Type I GD patients with the genotypes c.1226 A﹥G (p.N370S)/c.1226 A﹥G (p.N370S), c.1226 A﹥G (p.N370S)/c.508 C﹥T (p.R131C), c.259 C﹥T (p.R48W)/c.1448 T﹥C (p.L444P), c.259 C﹥T (p.R48W)/c.1448 T﹥C (p.L444P), the residual activity of GCase in macrophages was around 15% of control. In Type II patients, the genotypes c.508 C﹥T (p.R131C)/c.508 C﹥T (p.R131C) were associated with extremely low residual activity at 1.75% of control. Finally, the residual activity of GCase in immortalized lymphocytes was nearly absent in GD3 patients with genotypes c.1342G﹥C (p.D409H)/c.971G﹥C (p.R285P), c.882T>G;c.1342G﹥C (p.H255Q;D409H)/ c.754T﹥A (p.F213I), c.1448 T﹥C (p.L444P)/c.1448 T﹥C (p.L444P) [12].
There are several methods that can be used to treat peripheral symptomology of GD. Historically, standard care has been largely limited to enzyme replacement therapies (ERT), like alglucerase and imiglucerase, and substrate reduction therapies (SRT), including miglustat and eliglustat. Treatment using hematopoietic stem cell transplantation is not routinely used owing to the problems of graft rejection and the shortage of available donors. However, emergent treatments using small molecules including pharmacological chaperones (i.e., ambroxo and isofagomine), proteasome inhibitors like MG-132, proteostasis regulators (i.e., celastrol and MG-132) and endoplasmic reticulum (ER)-associated degradation (ERAD) inhibitors (i.e., kifunensine and eeyarestatin I), possess enormous therapeutic potential [6, 13–20], and have potential benefit against neuronopathic forms of the disease. Synergistic effects may be obtainable using both a proteostasis regulator and a pharmacologic chaperone to restore the function of misfolded protein [14].
Despite the development of multiple strategies for non-neuronopathic GD, there is no effective treatment available for the neurological manifestations of Types II and III disease. The continued study of the molecular mechanisms underlying nGD is essential for improved application of existing therapies and identification of new therapies. During the past decade, the roles of numerous molecules involved in the pathogenesis of GD and regulating GCase activity have been uncovered (summarized in Table 2 and Table 3). Membrane proteins, including LIMP-2 and saposins, are involved in the disease and their expressions are important for GCase activity [21]. Additional molecules, such as progranulin (PGRN), HSP70, phosphatidylinositol 4-kinases, and Saposin-C are required for lysosomal trafficking of GCase. Inflammatory molecules such as TNFα, IL-1β, RipK3, type I IFN response proteins, macrophage colony-stimulating factor (MCSF), and complement cascade proteins trigger inflammation in GD and thus enhance the pathogenicity of GD. TMEM106B and gpNMB cause lysosomal dysfunction associated with GD. Proteins such as heat shock proteins, PGRN, FKBP10, calnexin also act as molecular chaperones and mediate mutant enzyme degradation. Ca2+ channel RyaR mediates calcium release and modulate GD. Herein, we give an overview about the roles of these molecules crucial in the GD pathogenesis, which are also briefly summarized in Table 2 and Figure 2.
Table 2.
Molecules known to play roles in Gaucher Disease
| Molecules regulating GD | Functions |
|---|---|
| LIMP-2 | LIMP-2 is a binding receptor for GCase and targets it to the lysosome [22] |
| Prosaposins | Prosaposin is a precursor protein which is cleaved into glycoproteins saposin (Sap) A-D, of which, Saposin C functions as activator of GCase [27] |
| HSP27 | HSP27 participates in the proteosomal degradation of mutant GCase[66] |
| HSP70 | HSP70 is involved in the lysosomal localization of GCase and is recruited by PGRN to GCase/ LIMP2 upon stress[61] |
| HSP90 | HSP90 binds mutated GCase and directs its towards endoplasmic reticulum associated degradation (ERAD) and proteasome degradation pathway[70] |
| ERdj3 | ERdj3 is an ER localized Hsp40 which interacts with GCase and promotes mutant GCase degradation[73] |
| PGRN | PGRN is required for lysosomal appearance of GCase and a cochaperone of HSP70, and affects intracellular sublocation of GCase[60, 61] |
| CHI3L1 | The expression of CHI3L1, a downstream mediator of PGRN, correlates with GD phenotype[63]. |
| TNFα | TNFα induces bone manifestations, neuroinflammation and perturbs myelin and its levels are increased in GD [79, 80, 96] |
| IL-1β | IL-1β levels are elevated in serum of Gaucher patients and was detected in the fetal brains of Gaucher mice [79, 96] |
| M-CSF | M-CSF is elevated in serum of Type1 GD[97]. Its mRNA is increased in neuronopathic GD mouse model and contributes to neuroinflammation[75] |
| p38 | p38 is a proinflammatory kinase and is activated in Gaucher disease due to absence of ceramide mediated suppression of p38[100] |
| TCP1 | Interaction between TCP1 and mutant GCase mediates the degradation of mutant GCase[64] |
| c-Cbl | The interaction of c-Cbl with GCase is increased in GD, which results in increased proteasome mediated degradation of GCase [64] |
| Type I IFN response associated molecules | Accumulated GlcCer triggers activation of IFN response in nGD and is associated with neuroinflammation[102] |
| Ripk3 | Ripk3 plays a key role in necroptosis and neuroinflammation in nGD and its expression is elevated in nGD brains[103]. |
| C1q | The complement protein C1q is upregulated following GCase inhibition and is associated with neuroinflammation [105]. |
| C5a and C5aR1 | GCase deficiency stimulates C5a generation and activates C5aR1. C5a/C5aR1 worsens GD by upregulating glucosylceramide synthase [106, 107]. |
| TMEM106B | TMEM106B regulates lysosomal function by controlling lysosome size, number, and trafficking [112] |
| FKBP10 | FKBP10 is a component of GCase proteostasis network and binds mutant GCase and causes proteasome mediated degradation of the enzyme[25] |
| RyaR | Ca2+ release from ER via the ryanodine receptor RyaR is linked with GlcCer accumulation and correlates with severity of GD[123, 124] |
| OPN | OPN modulates gene expression of cytokines, macrophage differentiation and migration and its levels are elevated in plasma in Type I GD[127] |
| gpNMB | gpNMB is involved in lysosomal stress, has anti-inflammatory actions in macrophages and its expression is upregulated in serum of GD patients[128] |
| Neopterin | Neopterin is derived from GTP, produced by immune cells such as macrophages, and shown to act as potential biomarker for GD[129, 130] |
| α-Synuclein | GD patients are vulnerable to develop Parkinson disease and exhibit features of PD such as α-synuclein accumulation [118, 121] |
Table 3.
Approaches/molecules that enhance the GCase activity
Fig. 2. Summary and illustration of the molecules known to be involved in the regulation of Gaucher Disease.
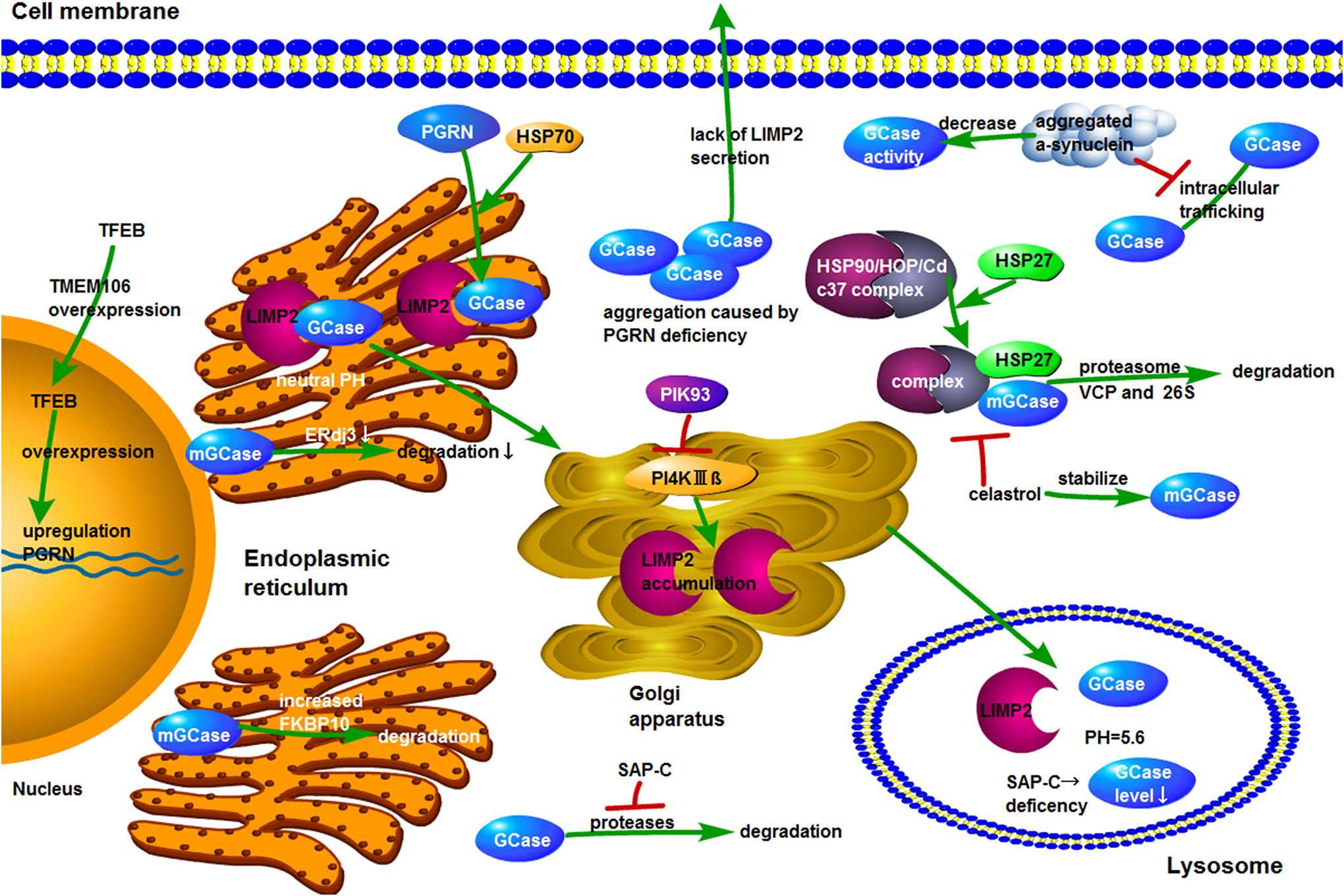
The overexpression of TMEM106 leads to the translocation of TFEB from cytoplasm to nucleus. GRN encoding PGRN is the target of TFEB and overexpression of TFEB results in upregulation of PGRN. GCase binds to LIMP2 at neutral pH in endoplasmic reticulum and then traffics to lysosome where GCase is separated from LIMP2 at acid condition. When LIMP2 is deficient, delivery of GCase to lysosome is reduced and GCase secretion is increased. Inhibition of PI4KIIIβ promotes LIMP-2 accumulation in Golgi. PGRN deficiency causes the aggregation of GCase in cytoplasm. HSP70 is recruited by PGRN to the GCase/LIMP2 complex. During degradation of mutant GCase, HSP90/HOP/Cdc37 chaperone complex first identifies the mutant GCase and then recruits HSP27, which leads to degradation of GCase mutants by VCP and 26S proteasome. Celastrol interferes with HSP90 chaperone function by hindering the assembly of chaperone complex required for proteasomal degradation of mutant GCase, thereby increasing the amount and catalytic activity of mutant GCase. The absence of SAP-C results in lower levels of GCase in acidic structures such as mature lysosomes, whereas application of SAP-C protects GCase from the degradation by proteases. A decrease in the concentration of ERdj3 reduces the rate of mutant GCase degradation. Elevated levels of FKBP10 promotes the degradation of mutant GCase. The aggregated α-synuclein reduces GCase activity and impedes the intracellular trafficking of GCase.
2. LIMP-2 and Gaucher Disease
Lysosomal integral membrane protein type-2 (LIMP-2), also known as SCARB2, is a binding partner of GCase, which is mainly expressed on the lysosomal membrane and plays a crucial role in regulating the transport of GCase to lysosome by a mannose 6-phosphate-independent trafficking [22, 23]. Binding between GCase and LIMP-2 is acutely influenced by key amino acids in the highly conserved region of GCase; alterations to aspartic acid 399 or the di-isoleucines, isoleucine 402 or isoleucine 403, diminish the binding of GCase to LIMP2, disrupt the pH-dependent binding, decrease the delivery of GCase to lysosome and dramatically elevated GCase secretion [24]. LIMP-2 is recognized as an important component of the GCase proteostasis involved in ER-to-Golgi-to-lysosome GCase trafficking. Knocking down the expression of LIMP-2 by siRNA results in reduced GCase activity, with GCase activity diminished by 3.3-fold in wild-type (WT) fibroblasts, by 2.2-fold in N370S fibroblasts and by 2.2-fold in L444P fibroblasts [25]. By contrast, overexpression of LIMP-2 in L444P GCase fibroblasts enhanced the endoglycosidase H resistant post-ER GCase glycoform and lysosomal delivery of GCase [25].
The lysosomal localization of GCase is also influenced by Phosphatidylinositol 4-kinases (PI4Ks), which regulate intracellular trafficking of its receptor, LIMP-2 [26]. During its trafficking to lysosomes, GCase interacts with LIMP-2 in the ER, which augments its trafficking to Golgi and finally to lysosomes. Inhibition of PI4KIIIβ using PIK93 promotes LIMP-2 accumulation in Golgi, indicating that the exit of LIMP-2 from Golgi is facilitated by PI4KIIIβ. Reduction in PI4KIIα levels inhibits post-Golgi trafficking of LIMP-2, leading to build up of LIMP-2 in enlarged endosomal vesicles. Overall, these findings implicate that PI4Ks might play critical role in GD owing to its ability to control LIMP-2 dependent GCase localization to lysosomes.
3. Prosaposins and Gaucher Disease
Prosaposin (PSAP) is a glycoprotein encoded by PSAP gene and acts as precursor of the sphingolipid activator proteins, known as Saposins (SAP) A, B, C, and D, which are 8–11 kDa amphipathic glycoproteins involved in sphingolipid degradation [27, 28]. Of these, SAP-C activates GCase hydrolysis of GlcCer in a dose-dependent manner. In addition, the degradation of GlcCer triggered by SAP-C is pH dependent and is optimum in the presence of both SAP-C and bis-(monoacylglycero)-phosphate [29]. However, the lack of either PSAP or SAP-C can be compensated for by the presence of the other in proper proportions. Patients with PSAP deficiency display manifestations of Type II GD and deficiency of SAP-C is related to Type I or Type Ⅲ GD phenotypes [27, 28, 30, 31]. Importantly, SAP-C is an essential activator in the GCase-catalyzed hydrolysis of GlcCer. The three spatially closed regions (TIM barrel-helix 6 and helix 7, and the Ig-like domain) of the GCase surface are involved in the binding to SAP-C[32]. Defects in SAP-C lead to a lethal GlcCer storage and exposure to functional SAP-C can elevate the lysosomal GCase activity in a dose-dependent manner.
SAP-C level determined by its stability and degradation affects the GCase sub-location. SAP-C’s disulfide bridges provide resistance to degradation and alteration of the protein’s structure resultant of mutation contributes to reduced stability [33]. Additionally, a GD-associated mutation in a cysteine residue of SAP-C disrupts the protein structure resulting in reduced half-life and accelerated autophagy mediated degradation of the mutant SAP-C [33, 34]. The absence of SAP-C results in lower levels of GCase in acidic structures such as mature lysosomes but higher levels in LysoTracker negative vesicles in SAP-C deficient fibroblasts, indicating that SAP-C impacts the GCase intracellular localization[35].
SAP-C level is regulated by CTSB (cathepsin B) and CTSD (cathepsin D) activity which involves in the regulation of autolysosome. The accumulated autophagosomes caused by SAP-C insufficiency in SAP-C deficient fibroblasts is due to suspended autolysosome breakdown, which to some extent, is an outcome of decreased levels and enzymatic activity of CTSB and CTSD [36]. In SAP-C deficient fibroblasts, the expressions of autophagy associated proteins such as Beclin 1, Atg5 and Atg7 are not affected, and increasing the expression of CTSB and CTSD effectively rescues autolysosome degradation [36]. Two compounds, BCM-95 and 2-hydroxypropyl-β-cyclodextrin (HP-β-CD), boost lysosome function by stimulating autophagy, lysosomal cholesterol and ceramide clearance, and enhancing expression and activity of CTSB and CTSD in SAP-C deficient fibroblasts[37].
SAP-C also plays an important role in the central nervous system (CNS) and maintenance of axonal integrity. The role of SAP-C in the CNS and the relevance of this functionality to GD is demonstrated by several murine models. In SAP-C knockout (KO) mice, the GlcCer, lactosylceramide and their deacylated analogues are accumulated, and neuromotor activity is decreased, hippocampal long-term potentiation is impaired, and weak hind limbs and progressive ataxia are presented [38]. Further, the GD mouse models, 4L/PS-NA and 9H/PS-NA, were derived from the backcross of prosaposin and saposin deficient mice (PS-NA) to the mice with point genetic mutants, V394L/V394L or D409H/D409H, of GCase. These mice display GlcCer accumulation in the liver, lung and spleen and CNS [39]. The mouse model 4L;C*, which displays a type III GD phenotype, was created by the cross of SAP-C knockout mice (C−/−) to the mice with point mutated GCase (V394L/V394L) [40]. 4L;C* mice exhibit decreased activity and expression of GCase and accumulation of GlcCer with onset of central nervous system symptoms at 30 days followed by death at 48 days resultant of neurological abnormalities. Overall, this model is considered appropriate to study the neuronal form of GD [40].
Importantly, some lines of evidence suggest that SAP-C may have potential as a therapeutic target in particular cases of GD. The activity of mutant GCase can be increased 2–3 folds at pH 4.05 in GD patient spleen homogenates by following addition combination and stimulation with healthy controls; an effect partially dependent on the SAP-C from healthy control tissues [41]. Further, application of chemically synthesized SAP-C at 1 μM can improve the GCase activity by 14 to 22 times and can also shield GCase from the degradation by proteases[42, 43].
Cumulatively, these findings highlight the role of SAP-C as an optimizer of GCase activity with important functionality in glycosphingolipid degradation and particular importance to maintenance of cellular metabolism and protection against axonal dysfunction and retrograde degeneration in the CNS.
4. Progranulin and Gaucher Disease
Progranulin (PGRN) is a highly conserved cysteine rich glycoprotein which regulates wide array of biological functions including cell growth, cell survival, tumorigenesis, wound repair, immunomodulation and inflammation [44–50]. PGRN is highly expressed in many cell types including neurons, astrocytes, chondrocytes, microglia, epithelial, myeloid and immune cells [51–53]. The important roles of PGRN in the lysosome are attested through multiple lines of evidence. PGRN is reported to be associated with the master regulator of lysosome biogenesis, TFEB factor, under the abnormal lysosomal storage conditions [54, 55]. GRN, encoding PGRN, is the target of TFEB and overexpression of TFEB results in increased PGRN expression [56]. In PGRN KO mice, increased lysosome biogenesis occurs in brain concomitant with increases in immunoreactivity of LAMP1 and gene expressions of lysosomal enzyme CTSD, protein ATP6V0D2, and the master regulator TFEB [57]. Homozygous mutations in GRN are known to cause neuronal ceroid lipofuscinosis (NCL), a rare lysosomal storage disease, which is characterized by aggregation of lipopigments in lysosomes and neurodegenerative impact on cognition and sensorimotor ability [58, 59].
Strikingly, ovabumin (OVA)-challenged and aged PGRN KO mice also develop a GD phenotype, including typical Gaucher cells, β-glucocerebroside accumulation, and classical tubular like-structural transformation of lysosomes [60]. In accordance with the findings from mouse models, GD patients displayed reduced serum levels of PGRN in association with two GRN gene variants (rs4792937 and rs5848) exhibiting increased rates of occurrence in GD patients [60].
PGRN is required for the trafficking of GCase to lysosome and is involved in GD by affecting the intracellular localization of GCase rather than influencing the enzymatic activity or expression. PGRN deficiency causes the aggregation of GCase in cytoplasm [60, 61]. Recombinant PGRN (rPGRN) has positive treatment effects on GD both in vitro and in vivo [60]. rPGRN ameliorates accumulation of GlcCer in fibroblasts originated from Type I and Type II GD patients and in the PGRN KO bone marrow derived macrophages (BMDM). In the PGRN KO OVA-induced GD mouse model, rPGRN inhibits the formation of Gaucher-like cells. Similarly, in the well-established D409V/− GD mice model, the use of rPGRN alleviates the severity of disease phenotype, decreases GlcCer storage and also decreases number and size of Gaucher cells [60]. A PGRN-derived protein, termed Pcgin, composed of C-terminal 96 amino acids of PGRN, is also therapeutic for GD as it functions by decreasing size and number of Gaucher - like cells in lungs and reduces accumulation of GlcCer in GD mouse models [61]. Pcgin also enhances lysosomal localization of mutant GCase and reduces GlcCer accumulation in GD fibroblasts [61]. In contrast to PGRN, Pcgin does not have the oncogenic activity of PGRN and is a potentially safe therapeutic molecule.
Chitinase-3-like protein 1 (CHI3L1), also known as YKL-40, is a glycoprotein secreted by several cell types including monocytes/macrophages, chondrocytes and is associated with inflammation, extracellular tissue remodeling in many diseases such as rheumatoid arthritis, fibrosis, type II diabetes and cancers [62]. CHI3L1 levels are higher in PGRN KO mice GD model and also in GD patients compared to healthy controls. Treatment with Pcgin or imiglucerase significantly decreases its levels in both PGRN KO mice and the fibroblasts from GD patients [63]. CHI3L1 is a downstream mediator of PGRN and a potential diagnostic biomarker of GD [63].
5. Heat shock proteins and Gaucher Disease
Under physiological conditions, nascent GCase undergoes cleavage and glycosylation and translocates from ER to Golgi for further modifications and finally traffics to lysosomes [64]. The folding and maturation of GCase inside cells is assisted by many chaperones, co-chaperones, and folding enzymes which constitute the proteostasis network. Mutated and improperly folded GCase is degraded by the ubiquitin–proteasome pathway. Heat shock proteins (HSP) are molecular chaperones which bind GCase and regulate its degradation. HSP 27, known to bind to polyubiquitin chains and the 26S proteasome and mediate degradation of the ubiquitinated proteins by the 26S proteasome under stressful conditions, interacts with mutant GCase [65]. During degradation of mutant GCase, HSP90/HOP/Cdc37 chaperone complex first identifies the mutant GCase and then recruits HSP27, which leads to degradation of GCase mutants by valosin-containing protein (VCP) and 26S proteasome. Suppression of HSP27 elevates both the amount and enzyme activity of GCase, and may represent a potential treatment strategy for GD [66].
HSP70 is a highly conserved chaperone which regulates the proper folding and unlocks disaggregation of many proteins and HSP70 deficiency leads to the GCase accumulation [67]. In GD, HSP70 is recruited by PGRN to the GCase/LIMP2 complex and HSP70 deficiency lead to reduced GCase detection [61]. Specifically, PGRN functions as a co-chaperone molecule of HSP70 and recruits HSP70 to form a ternary complex required for lysosomal appearance of GCase [61]. The C-terminal granulin E domain of PGRN is necessary for the association between PGRN and GCase as revealed by a series of C-terminal and N-terminal deletion mutants [61]. The finding that PGRN acts as a lysosomal enzyme chaperone [60, 61] was also extended by a recent article in which PGRN was demonstrated to act as a chaperone molecule of another lysosomal enzyme, cathepsin D (CSTD); an interaction also mediated through the granulin E domain [68]. It is conceivable that PGRN could have a more general function, beyond its associations with GCase and CSTD, as a lysosomal protein chaperone. Demonstration of associations of PGRN with additional lysosome enzymes forthcoming, application of this chaperone molecule or its derivatives may lead to innovative therapeutics for a host of lysosome storage diseases and neurodegenerative disorders.
HSP90, an important molecular chaperone involved in protein folding, is also reported to be crucial to target the mutant GCase for proteasomal degradation [69, 70]. The misfolded GCase is recognized and bound by HSP90, which leads to the degradation of GCase through endoplasmic reticulum associated degradation (ERAD) and VCP/protein 97 (p97)/proteasome degradation pathway[70, 71]. The deacetylation (K286R) of HSP90 mediates recognition and degradation of mutant GCase and acetylation (K286Q and K286A) of HSP90 decreases the ubiquitination of GCase mutants and therefore limits degradation, which may form the basis of a possible GD treatment strategy. Indeed, histone deacetylase (HDAC) inhibitors, like LB205 and SAHA, can elevate the quantity and increase the activity of misfolded GCase protein by increasing the acetylated HSP90 [69, 70]. Recognition of the critical role of HSPs in modulating GCase activity has generated interest in HSP90 inhibitor-based therapeutics have gained importance. Celastrol, derived from the root of Tripterygium Wilfordii (Thunder of God Vine) and Celastrus Regelii, have been implicated in some lysosomal storage diseases because it can modulate chaperone functions [72]. Celastrol interferes with HSP90 chaperone function by hindering the assembly of chaperone complex required for proteasomal degradation of mutant GCase in GD, thereby increasing the amount and catalytic activity of mutant GCase. It also induces expression of BAG family molecular chaperone regulator 3 (BAG3) which stabilizes mutant GCase and increases catalytic activity of BAG3. The possibility of “off-target” effects suggests that further studies are required prior to its clinical trials in GD. Overall, despite the possibility of having side effects, HDAC inhibitors and celastrol may possess the potential to serve as a substitute for enzyme replacement therapy for nGD.
ERdj3, the DnaJ homolog subfamily B member 11 (DNAJB11), is an ER associated HSP40 which can interact with both WT and mutant GCase. A decrease in the concentration of ERdj3 reduces the rate of mutant GCase degradation and favors the folding and trafficking of mutant enzyme via pro-folding ER calnexin pathway in patient-derived fibroblasts [73]. The reduced binding between mutant GCase and TCP1, a subunit of the TCP1 ring complex (TRiC) chaperonin complex and enhanced interaction with c-Cbl, a member of the family of E3 ubiquitin ligases, propels the mutant GCase towards the proteasomal degradation pathway [64]. Inhibition of c-Cbl results in increased GCase activity in GD and normal fibroblasts.
6. Inflammatory mediators and Gaucher Disease
In a GD mouse model with a L444P point mutation in the GBA gene, multisystem inflammation is demonstrated, where the macrophages, lymphocytes, and neutrophils are clustered, and liver TNF-α mRNA is about threefold higher than in controls [74]. It is believed that inflammation in the brain also plays a role in neuronal cell death in the nGD. The accumulation of GlcCer in neurons can activate the microglia, which promotes the release of inflammatory cytokines, reactive oxygen and nitrogen species to amplify the inflammatory response. In grey matter, the mRNA levels of proinflammatory cytokines such as IL-1β, TNFα, and M-CSF increase alongside disease severity [75]. In the mouse model of nGD, neuroinflammation including microglial activation and astrogliosis are associated with selective neuron loss [76].
TNFα level is increased in GD, may induce clinical signs, like bone manifestations, and displays a positive correlation to disease severity that the highest level is detected in the most advanced nGD type [77–79]. Moreover, a polymorphism in the TNFα promoter has been demonstrated to correlate with serum levels of TNFα and disease severity. Higher TNFα levels and incidence of nGD were observed in patients heterozygous for the polymorphism[78]. TNFα is suggested to disrupt myelin by altering the ionic channel expression and membrane potential of oligodendrocytes, and causes damage of oligodendrocytes [80, 81]. Inhibition of inflammation via targeting TNFα, the top cytokine in the inflammatory cascade, is believed to hold potential benefits for GD patients. PGRN and its derivatives are particularly promising, because extracellular PGRN and its derivatives exerts potent anti-inflammatory actions via directly binding to TNF receptors and antagonizing TNFα inflammatory activity [82–94], whereas intracellular PGRN and its derivatives functions as the chaperons of GCase and enhances the lysosomal appearance of GCase [60, 61, 95].
Serum level of interleukin-1 beta (IL-1β) was also increased and related to disease severity in GD patients[79]. In type I GD patients, those with more obvious clinical manifestations had higher IL-1β level than the patients having milder manifestation. In the GD mouse established by double GBA gene KO (C57BL/6J-Gbatm1Nsb), the level of IL-1β in fetal brain was significantly higher than in the WT mouse[96].
In Type I GD patients, increased levels of several cytokines including M-CSF (2–8 fold), sCD14 (2–5 fold) and IL-8 (2–20 fold) have been detected and a positive correlation between disease severity and the cytokine levels has been observed [97]. In both GD patients and murine disease models, elevated serum level of IL-6 and increased activation of p38 are observed [98, 99]. Knockdown of GCase in human breast cancer MCF-7 cells results in increased p38 activation and subsequently increased production of IL-6 upon exposure to phorbol 12-myristate 13-acetate (PMA). In the presence of normal GCase, GlcCer is cleaved to ceramide which is reported to have anti-inflammatory effect by inhibiting p38 via activation of ceramide-activated protein phosphatases. The decreased ceramide formation caused by GCase mutation may therefore enhance p38 activation and drive inflammatory response in GD [100].
The type I IFN response, usually activated in response to viral and bacterial infection [101], also contributes to the pathology of initial stages of nGD [102]. A recent report of microarray data from the Gbaflox/flox; nestin-Cre mouse model, with GBA1 deficiency confined to cells of neuronal lineage, i.e., neuron and macroglia, illustrates activation of the type I IFN response during neuroinflammation made evident by massive induction of type I IFN-stimulated genes (ISGs), including pathogen recognition receptors (PRRs) such as Toll-like receptors (TLR), C-type lectin receptors, CLEC7A (Dectin-1), CLEC5A (MDL-1) [24], the scavenger receptors CD36, and MSR1 (SR-A1), interferon regulatory factor (IRF) such as IRF7, IRF8, IRF9, IRF1, IFNα and IFNβ, and antiviral genes. The proposed mechanism of activation of IFN in nGD suggests that accumulated GlcCer activate PRR, which triggers activation of the antiviral response and production of IFNα and IFNβ in neurons. The IFN then activates the surrounding microglia by binding to their IFNAR, thus triggering the IFN signaling pathways. Deficiency of IFN-I receptor results in inhibition of neuroinflammation in nGD [102]. However, the exact role of IFN response in nGD is not yet clear and further studies are required to clarify whether it is useful or harmful for the disease.
Recently the protein serine-threonine kinase, receptor-interacting protein kinase-3 (Ripk3) has been implicated in necroptosis and neuroinflammation in nGD brains. Ripk1 and Ripk3 levels are increased in mouse nGD brain. Modulating Ripk3 has been shown to be beneficial for GD [103]. In this study, injection of conduritol B-epoxide (CBE) resulted in GD manifestation in Ripk3+/− mice whereas Rip3−/− mice displayed inhibition of the disease in liver and brain, had enhanced survival and motor coordination, suggesting the potential of RIPK3 as therapeutic target for nGD and type I GD. However, the inhibitors of RIPK3 are yet to be identified.
7. Complement and Gaucher Disease
Proteomics analysis aiming to identify the diagnostic serum markers and protein signatures for GD patients who were ongoing ERT has shown that the complement cascade proteins (i.e., C3, C4d region, C5, C8 gamma chain and alpha chain) have been shown to change more than 30% before and after therapy, which indicates that complement may play a role in the pathology of GD[104]. The expression of complement protein, C1q, which is a part of the C1 complex of the complement system, is induced in striatum, substantia nigra and motor cortex by suppression of lysosomal GCase using a selective GCase irreversible inhibitor CBE for 28 days (100mg/kg/day through intraperitoneal injection) and this causes extensive neuroinflammatory reaction [105]. Accumulation of GlcCer resulting from deficiency or inhibition of GCase triggers production of complement-activating GC-specific IgG autoantibodies, which stimulate C5a production and C5aR1 activation, that subsequently increase the expression of UDP-glucose ceramide glucosyltransferase, an enzyme that synthesizes GlcCer [106]. Overall, a series of events, consisting of GlcCer accumulation, recruitment of immune cells, inflammatory cascade activation, are precipitated by complement activation and favor GD progression. Deficiency of C5aR1 protects mice from CBE induced GD. However, daily administration of CBE over a 29–35 day course results in serious signs of GD and even fatality in WT and C5aR2-deficient mice. The application of C5aR1 antagonist A8(Δ 71− 73) to block the receptor decreases the GlcCer accumulation and inflammation in mice, and thus may serve as a potential treatment strategy for GD patients. In summary, C5a-C5aR1 axis plays a crucial role in GD pathology and is an attractive therapeutic target for GD [106, 107].
8. TMEM106B and Gaucher Disease
Transmembrane protein 106B (TMEM106B) is a cytoplasmic/lysosomal protein that is expressed on the membranes of endosomes and lysosomes in neurons, glial and endothelial cells [108, 109]. TMEM106B was first identified as one of the genetic risk factors for frontotemporal lobar degeneration (FTLD) [110]. TMEM106B overexpression results in the enlargement of LAMP1- and TMEM106B-positive organelles, improper lysosome acidification, decrease in lysosome number, and enhances lysosomal stress, which causes endosomal-lysosome dysfunction [109, 111, 112]. Overexpression of TMEM106B stimulates the translocation of the mTOR-sensitive transcription factor, TFEB, which is also a marker of lysosomal stress, to neuronal nuclei from cytoplasm and thus regulates lysosomal stress [112].
Intriguingly, GRN and TMEM106b genes exert opposite effects on lysosome function by regulating lysosomal enzyme expression in an opposite manner. TMEM106b deficient 5-month old mice display a significant reduction in dipeptidyl-peptidase 2 (DPPII) and CTSB levels whereas GRN deficient brains exhibit a significant increase in levels of these proteins [113]. Knocking out both GRN and TMEM106b normalizes the levels of these proteins to that of WT. At 5-months age, LAMP1 is not significantly altered in GRN deficient mice, but is decreased in TMEM106b deficient samples and in the double KO brain, and LAMP1 levels are normalized to WT levels, demonstrating that interaction between TMEM106B and PGRN regulates LAMP1 levels. Although TMEM106B deficiency normalizes the higher CTSB and DPPII enzyme activities in 7-month-old GRN KO brain lysate, it does not significantly change Hex A/B/S activity, indicating that some lysosomal enzyme activities can be selectively rescued by TMEM106B deficiency. TMEM106b deficiency significantly downregulates vacuolar-ATPase (V-ATPase) V0 domain subunits V0a1, V0c, and V0d1 and accessary protein 1 (AP1) and thus hampers lysosomal acidification, which then reduces lysosomal enzyme activity and proteolysis and thus normalizes lysosomal protein levels in GRN deficient neurons [113].
9. FKBP10 and Gaucher Disease
The ER localized molecular chaperone FK506 binding protein 10 (FKBP10) belongs to the FKBP-type peptidyl-prolyl cis/trans isomerase family and was identified as a crucial GCase proteostasis network component by comparative proteomic analysis of patient-derived LSD fibroblasts treated with proteostasis regulators (MG-132 or diltiazem) [25]. In ER, elevated levels of FKBP10 promotes the degradation of mutant GCase whereas reduced concentration of FKBP10 increase the folding and activity of GCase, which alleviates LSD [25]. FKBP10 influences enzyme degradation and folding decision in ER of LSD. The fate of the newly synthesized unfolded mutant lysosomal enzyme to undergo either degradation or folding depends on whether it binds with either FKBP10 or calnexin in the ER, respectively. After treating with diltiazem or MG-132, FKBP10 levels in L444P GCase fibroblasts decreased 2-fold. By knocking down the expression of FKBP10, the GCase activity is enhanced by 1.4-fold in L444P GCase fibroblasts (that is about 18% of WT GCase activity) and by 2.0-fold in Gaucher’s G202R mutant fibroblasts. In contrast, overexpression of FKBP10 in L444P GCase fibroblasts reduces roughly 20% of GCase activity [25]. In brief, silencing FKBP10 can potentially increase lysosomal mutant enzyme function and mitigate LSD.
10. Parkinson’s disease and Gaucher Disease
GD is also associated with Parkinson’s disease (PD), a neurodegenerative disorder portrayed by aggregation of soluble synaptic protein α-synuclein (α-syn) into insoluble amyloid fibrils in Lewy body inclusions [114]. GD patients often present symptoms of PD and α-synuclein-positive Lewy bodies, suggesting co-occurrence of PD; in fact, PD is highly prevalent in GD subjects carrying mutations in the GBA gene [115, 116], indicating that GBA mutations increase susceptibility to PD. Several studies demonstrate that accumulation of α-syn is linked with reduction in GCase activity [105, 117]. Attenuation of lysosomal degradation pathway by accumulation of GlcCer in neurons results in occurrence of high α-syn protein levels, which is associated with neurotoxicity caused by the inherent ability of α-syn to produce amyloid fibrils [118]. High levels of toxic α-syn impede the intracellular trafficking of GCase, which inhibits lysosomal function of GCase, thus causing further accumulation of GlcCer. This type of positive feedback loop favors piling up of oligomeric forms of α-syn, which ultimately leads to neurodegenerative disease. A non-inhibitory small molecule modulator of GCase, NCGC00188758 (N-(4-Ethynylphenyl)-5,7-dimethylpyrazolo[1,5-a]pyrimidine-3-carboxamide), can increase GCase activity, decrease pathological α-syn aggregates and repair lysosomal function in human midbrain synucleinopathy models and is thus advantageous for PD[119]. Overexpression of GBA1 via AAV-GBA1 intra-cerebral gene delivery attenuated accumulation of α-syn and shielded the midbrain dopamine neurons from toxic effects of α-syn in rodent models by modulating autophagy [120]. Promoting differentiation of iPSCs to dopaminergic neurons and macrophages in GD patients is in favor of the a noninhibitory small molecule treatment, NCGC607 (2-[2-(4-Iodoanilino)-2-oxoethoxy]-N-[2-(N-methylanilino)-2-oxoethyl]benzamide) to repair the GCase activity and diminish substrate storage [121]. This molecule also lowered expression of α-syn in Type I GD – PD and Type II GD dopaminergic neurons. These findings indicate that therapies aimed at increasing GCase function to treat GD also reduce the accumulation of α-syn associated with PD [118, 122].
11. Summary and Perspectives
GD pathogenicity is a complex phenomenon and can be influenced by molecules that regulate GCase trafficking (LIMP-2, PGRN, HSPs), inflammatory mediators (IL-1β, TNFα, RipK3, PGRN), lysosomal stress mediators such as TMEM106B and PGRN, molecular chaperones such as FKBP10, and heat shock proteins such as HSP90, HSP70. Moreover, additional molecules are also reported to contribute to GD. Calcium ions play an important role in the protein folding in the ER and malfunction of calcium homeostasis acts as an important mediator of neuropathophysiology in typeⅡGD [123, 124]. The major Ca2+ channel ryanodine receptor (RyaR), which regulates calcium efflux from ER to cytoplasm, has been implicated in pathophysiology of nGD. RyaRs have recently gained importance as therapeutic target in nGD. Antagonization of RyaRs using dantrolene abrogates intracellular calcium release via RyaRs and results in neuroprotective effects and impedes disease progression in nGD [125]. Lacidipine is a selective L-type Ca2+ channel inhibitor which increases the L444P GCase mutant enzyme activity by enhancing its folding in ER via altering calcium homeostasis and enhancing expression of ER chaperone binding immunoglobulin protein (BiP) [126].
Osteopontin (OPN), also called secreted phosphoprotein 1 (SPP 1), is found in cancer, bone cells and many types of immune cells, including T-cells and macrophages [127]. High levels of OPN in the plasma of type I GD patients suggest its potential as biomarker of GD. Glycoprotein, non-Metastatic Melanoma B (gpNMB), which is involved in osteoblastogenesis and osteoclast mediated bone resorption, lysosomal stress, an anti-inflammatory role in macrophage, is upregulated in serum of GD patients and its levels correlate with accumulation of glucosylsphingosine and well known GD biomarkers, including chitotriosidase and CCL18, which suggests its potential as marker of GD progression or response to therapy [128]. Neopterin, a pteridine catabolic product of GTP, is synthesized by activated macrophages and dendritic cells in response to interferon-γ and TNF-α, has recently been accepted as reliable marker of activation of immune system. Elevated serum levels of neopterin were observed in Type I GD patients [129]. Plasma concentration of neopterin mirrors the overall activation and accumulation of Gaucher cells and correlates with the degree of immune activation in GD. Another study demonstrated the significance of neopterin as a biomarker particularly in chitotriosidase-deficient patients [130].
In brief, identification and characterization of molecules associated with GD is of utmost importance for unraveling the molecular mechanisms underlying disease pathogenesis. Therapies based on molecules which impact GD by regulating GCase folding, trafficking, or stability, inflammatory pathways or other processes linked with GD can be designed, in order to overcome the drawbacks of currently available therapeutic options for GD. Moreover, because of link between GD and PD, treatment options for GD may also prove to be beneficial for PD.
Acknowledgments
We apologize to the colleagues whose publications are not included due to the space limitation. This work was supported by NIH research grants R01AR062207, R01AR061484, 1R01NS103931, and a DOD research grant W81XWH-16-1-0482. Yuehong Chen was funded by China Scholarship Council (grant number 201606240021).
Footnotes
Conflict of interest
We herein declare that we have no conflict of interest.
References
- [1].Barkhuizen M, Anderson DG and Grobler AF, Advances in GBA-associated Parkinson’s disease--Pathology, presentation and therapies, Neurochem Int. 93 (2016) 6–25. [DOI] [PubMed] [Google Scholar]
- [2].Beutler E, Gelbart T and Scott CR, Hematologically important mutations: Gaucher disease, Blood Cells Mol Dis. 35 (2005) 355–64. [DOI] [PubMed] [Google Scholar]
- [3].Grabowski GA and Horowitz M, Gaucher’s disease: molecular, genetic and enzymological aspects, Baillieres Clin Haematol. 10 (1997) 635–56. [DOI] [PubMed] [Google Scholar]
- [4].Koprivica V, Stone DL, Park JK, Callahan M, Frisch A, Cohen IJ, et al. , Analysis and classification of 304 mutant alleles in patients with type 1 and type 3 Gaucher disease, Am J Hum Genet. 66 (2000) 1777–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Adar T, Ilan Y, Elstein D and Zimran A, Liver involvement in Gaucher disease - Review and clinical approach, Blood Cells Mol Dis. 68 (2018) 66–73. [DOI] [PubMed] [Google Scholar]
- [6].Cox TM, Gaucher disease: clinical profile and therapeutic developments, Biologics. 4 (2010) 299–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Cassinerio E, Graziadei G and Poggiali E, Gaucher disease: a diagnostic challenge for internists, Eur J Intern Med. 25 (2014) 117–24. [DOI] [PubMed] [Google Scholar]
- [8].Beutler E and Grabowski GA, Glucosylceramide lipidosis–Gaucher disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The metabolic and molecular bases of inherited diseases. 8th edn., New York: McGraw-Hill. 36 (2001) 35–68. [Google Scholar]
- [9].Mignot C, Gelot A, Bessieres B, Daffos F, Voyer M, Menez F, et al. , Perinatal-lethal Gaucher disease, Am J Med Genet A. 120a (2003) 338–44. [DOI] [PubMed] [Google Scholar]
- [10].Stirnemann J, Belmatoug N, Camou F, Serratrice C, Froissart R, Caillaud C, et al. , A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments, Int J Mol Sci. 18 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ron I and Horowitz M, ER retention and degradation as the molecular basis underlying Gaucher disease heterogeneity, Hum Mol Genet. 14 (2005) 2387–98. [DOI] [PubMed] [Google Scholar]
- [12].Malini E, Zampieri S, Deganuto M, Romanello M, Sechi A, Bembi B, et al. , Role of LIMP-2 in the intracellular trafficking of beta-glucosidase in different human cellular models, Faseb j. 29 (2015) 3839–52. [DOI] [PubMed] [Google Scholar]
- [13].Sawkar AR, D’Haeze W and Kelly JW, Therapeutic strategies to ameliorate lysosomal storage disorders--a focus on Gaucher disease, Cell Mol Life Sci. 63 (2006) 1179–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Mu TW, Ong DS, Wang YJ, Balch WE, Yates JR 3rd, Segatori L, et al. , Chemical and biological approaches synergize to ameliorate protein-folding diseases, Cell. 134 (2008) 769–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Deegan P, Fernandez-Sasso D, Giraldo P, Lau H, Panahloo Z and Zimran A, Treatment patterns from 647 patients with Gaucher disease: An analysis from the Gaucher Outcome Survey, Blood Cells Mol Dis. (2016) [DOI] [PubMed] [Google Scholar]
- [16].Babajani G and Kermode AR, Alteration of the proteostasis network of plant cells promotes the post-endoplasmic reticulum trafficking of recombinant mutant (L444P) human beta-glucocerebrosidase, Plant Signal Behav. 9 (2014) e28714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wang F, Song W, Brancati G and Segatori L, Inhibition of endoplasmic reticulum-associated degradation rescues native folding in loss of function protein misfolding diseases, J Biol Chem. 286 (2011) 43454–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Giugliani R, Vairo F, Kubaski F, Poswar F, Riegel M, Baldo G, et al. , Neurological manifestations of lysosomal disorders and emerging therapies targeting the CNS, The Lancet Child & Adolescent Health. (2017) [DOI] [PubMed] [Google Scholar]
- [19].Deegan P, Fernandez-Sasso D, Giraldo P, Lau H, Panahloo Z and Zimran A, Treatment patterns from 647 patients with Gaucher disease: An analysis from the Gaucher Outcome Survey, Blood Cells Mol Dis. 68 (2018) 218–225. [DOI] [PubMed] [Google Scholar]
- [20].Platt FM, Emptying the stores: lysosomal diseases and therapeutic strategies, Nat Rev Drug Discov. 17 (2018) 133–150. [DOI] [PubMed] [Google Scholar]
- [21].zimmer k.p., coutre p.l., aerts h.m.f.g., harzer k., fukuda m., o’brien j., et al. , INTRACELLULAR TRANSPORT OF ACID β-GLUCOSIDASE AND LYSOSOME-ASSOCIATED MEMBRANE PROTEINS IS AFFECTED IN GAUCHER’S DISEASE (G202R MUTATION), journal of pathology. 188 (1999) 407–414. [DOI] [PubMed] [Google Scholar]
- [22].Tabuchi N, Akasaki K, Sasaki T, Kanda N and Tsuji H, Identification and characterization of a major lysosomal membrane glycoprotein, LGP85/LIMP II in mouse liver, J Biochem. 122 (1997) 756–63. [DOI] [PubMed] [Google Scholar]
- [23].Blanz J, Zunke F, Markmann S, Damme M, Braulke T, Saftig P, et al. , Mannose 6-phosphate-independent Lysosomal Sorting of LIMP-2, Traffic. 16 (2015) 1127–36. [DOI] [PubMed] [Google Scholar]
- [24].Liou B, Haffey WD, Greis KD and Grabowski GA, The LIMP-2/SCARB2 binding motif on acid beta-glucosidase: basic and applied implications for Gaucher disease and associated neurodegenerative diseases, J Biol Chem. 289 (2014) 30063–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ong DS, Wang YJ, Tan YL, Yates JR 3rd, Mu TW and Kelly JW, FKBP10 depletion enhances glucocerebrosidase proteostasis in Gaucher disease fibroblasts, Chem Biol. 20 (2013) 403–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Jovic M, Kean MJ, Szentpetery Z, Polevoy G, Gingras AC, Brill JA, et al. , Two phosphatidylinositol 4-kinases control lysosomal delivery of the Gaucher disease enzyme, beta-glucocerebrosidase, Mol Biol Cell. 23 (2012) 1533–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Schulze H, Kolter T and Sandhoff K, Principles of lysosomal membrane degradation: Cellular topology and biochemistry of lysosomal lipid degradation, Biochim Biophys Acta. 1793 (2009) 674–83. [DOI] [PubMed] [Google Scholar]
- [28].Yoneshige A, Suzuki K, Suzuki K and Matsuda J, A mutation in the saposin C domain of the sphingolipid activator protein (Prosaposin) gene causes neurodegenerative disease in mice, J Neurosci Res. 88 (2010) 2118–34. [DOI] [PubMed] [Google Scholar]
- [29].Abdul-Hammed M, Breiden B, Schwarzmann G and Sandhoff K, Lipids regulate the hydrolysis of membrane bound glucosylceramide by lysosomal beta-glucocerebrosidase, J Lipid Res. 58 (2017) 563–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Qi X, Leonova T and Grabowski GA, Functional human saposins expressed in Escherichia coli. Evidence for binding and activation properties of saposins C with acid beta-glucosidase, J Biol Chem. 269 (1994) 16746–53. [PubMed] [Google Scholar]
- [31].Tylki-Szymanska A, Czartoryska B, Vanier MT, Poorthuis BJ, Groener JA, Lugowska A, et al. , Non-neuronopathic Gaucher disease due to saposin C deficiency, Clin Genet. 72 (2007) 538–42. [DOI] [PubMed] [Google Scholar]
- [32].Atrian S, Lopez-Vinas E, Gomez-Puertas P, Chabas A, Vilageliu L and Grinberg D, An evolutionary and structure-based docking model for glucocerebrosidase-saposin C and glucocerebrosidase-substrate interactions - relevance for Gaucher disease, Proteins. 70 (2008) 882–91. [DOI] [PubMed] [Google Scholar]
- [33].Motta M, Camerini S, Tatti M, Casella M, Torreri P, Crescenzi M, et al. , Gaucher disease due to saposin C deficiency is an inherited lysosomal disease caused by rapidly degraded mutant proteins, Hum Mol Genet. 23 (2014) 5814–26. [DOI] [PubMed] [Google Scholar]
- [34].Tatti M, Motta M and Salvioli R, Autophagy in Gaucher disease due to saposin C deficiency, Autophagy. 7 (2011) 94–5. [DOI] [PubMed] [Google Scholar]
- [35].Vaccaro AM, Motta M, Tatti M, Scarpa S, Masuelli L, Bhat M, et al. , Saposin C mutations in Gaucher disease patients resulting in lysosomal lipid accumulation, saposin C deficiency, but normal prosaposin processing and sorting, Hum Mol Genet. 19 (2010) 2987–97. [DOI] [PubMed] [Google Scholar]
- [36].Tatti M, Motta M, Di Bartolomeo S, Scarpa S, Cianfanelli V, Cecconi F, et al. , Reduced cathepsins B and D cause impaired autophagic degradation that can be almost completely restored by overexpression of these two proteases in Sap C-deficient fibroblasts, Hum Mol Genet. 21 (2012) 5159–73. [DOI] [PubMed] [Google Scholar]
- [37].Tatti M, Motta M, Scarpa S, Di Bartolomeo S, Cianfanelli V, Tartaglia M, et al. , BCM-95 and (2-hydroxypropyl)-beta-cyclodextrin reverse autophagy dysfunction and deplete stored lipids in Sap C-deficient fibroblasts, Hum Mol Genet. 24 (2015) 4198–211. [DOI] [PubMed] [Google Scholar]
- [38].Sun Y, Ran H, Zamzow M, Kitatani K, Skelton MR, Williams MT, et al. , Specific saposin C deficiency: CNS impairment and acid beta-glucosidase effects in the mouse, Hum Mol Genet. 19 (2010) 634–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Sun Y, Quinn B, Witte DP and Grabowski GA, Gaucher disease mouse models: point mutations at the acid beta-glucosidase locus combined with low-level prosaposin expression lead to disease variants, J Lipid Res. 46 (2005) 2102–13. [DOI] [PubMed] [Google Scholar]
- [40].Sun Y, Liou B, Ran H, Skelton MR, Williams MT, Vorhees CV, et al. , Neuronopathic Gaucher disease in the mouse: viable combined selective saposin C deficiency and mutant glucocerebrosidase (V394L) mice with glucosylsphingosine and glucosylceramide accumulation and progressive neurological deficits, Hum Mol Genet. 19 (2010) 1088–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ho MW and O’Brien JS, Gaucher’s disease: deficiency of ‘acid’ -glucosidase and reconstitution of enzyme activity in vitro, Proc Natl Acad Sci U S A. 68 (1971) 2810–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Yoneshige A, Muto M, Watanabe T, Hojo H and Matsuda J, The effects of chemically synthesized saposin C on glucosylceramide-beta-glucosidase, Clin Biochem. 48 (2015) 1177–80. [DOI] [PubMed] [Google Scholar]
- [43].Sun Y, Qi X and Grabowski GA, Saposin C is required for normal resistance of acid beta-glucosidase to proteolytic degradation, J Biol Chem. 278 (2003) 31918–23. [DOI] [PubMed] [Google Scholar]
- [44].Jian J, Konopka J and Liu C, Insights into the role of progranulin in immunity, infection, and inflammation, Journal of leukocyte biology. (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Jian J, Konopka J and Liu C, Insights into the role of progranulin in immunity, infection, and inflammation, J Leukoc Biol. 93 (2013) 199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Jian J, Li G, Hettinghouse A and Liu C, Progranulin: A key player in autoimmune diseases, Cytokine. (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Liu CJ, Progranulin: a promising therapeutic target for rheumatoid arthritis, FEBS Lett. 585 (2011) 3675–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Liu CJ and Bosch X, Progranulin: A growth factor, a novel TNFR ligand and a drug target, Pharmacol Ther. 133 (2012) 124–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Wei J, Hettinghouse A and Liu C, The role of progranulin in arthritis, Ann N Y Acad Sci. (2016) [DOI] [PubMed] [Google Scholar]
- [50].He Z and Bateman A, Progranulin (granulin-epithelin precursor, PC-cell-derived growth factor, acrogranin) mediates tissue repair and tumorigenesis, J Mol Med (Berl). 81 (2003) 600–12. [DOI] [PubMed] [Google Scholar]
- [51].Toh H, Chitramuthu BP, Bennett HP and Bateman A, Structure, function, and mechanism of progranulin; the brain and beyond, J Mol Neurosci. 45 (2011) 538–48. [DOI] [PubMed] [Google Scholar]
- [52].Xu K, Zhang Y, Ilalov K, Carlson CS, Feng JQ, Di Cesare PE, et al. , Cartilage oligomeric matrix protein associates with granulin-epithelin precursor (GEP) and potentiates GEP-stimulated chondrocyte proliferation, J Biol Chem. 282 (2007) 11347–55. [DOI] [PubMed] [Google Scholar]
- [53].Feng JQ, Guo FJ, Jiang BC, Zhang Y, Frenkel S, Wang DW, et al. , Granulin epithelin precursor: a bone morphogenic protein 2-inducible growth factor that activates Erk1/2 signaling and JunB transcription factor in chondrogenesis, Faseb j. 24 (2010) 1879–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, et al. , A gene network regulating lysosomal biogenesis and function, Science. 325 (2009) 473–7. [DOI] [PubMed] [Google Scholar]
- [55].Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, et al. , TFEB links autophagy to lysosomal biogenesis, Science. 332 (2011) 1429–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Kao AW, McKay A, Singh PP, Brunet A and Huang EJ, Progranulin, lysosomal regulation and neurodegenerative disease, Nat Rev Neurosci. 18 (2017) 325–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Tanaka Y, Chambers JK, Matsuwaki T, Yamanouchi K and Nishihara M, Possible involvement of lysosomal dysfunction in pathological changes of the brain in aged progranulin-deficient mice, Acta Neuropathol Commun. 2 (2014) 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Smith KR, Damiano J, Franceschetti S, Carpenter S, Canafoglia L, Morbin M, et al. , Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage, Am J Hum Genet. 90 (2012) 1102–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Gotzl JK, Mori K, Damme M, Fellerer K, Tahirovic S, Kleinberger G, et al. , Common pathobiochemical hallmarks of progranulin-associated frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis, Acta neuropathologica. (2014) [DOI] [PubMed] [Google Scholar]
- [60].Jian J, Zhao S, Tian QY, Liu H, Zhao Y, Chen WC, et al. , Association Between Progranulin and Gaucher Disease, EBioMedicine. 11 (2016) 127–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Jian J, Tian QY, Hettinghouse A, Zhao S, Liu H, Wei J, et al. , Progranulin Recruits HSP70 to beta-Glucocerebrosidase and Is Therapeutic Against Gaucher Disease, EBioMedicine. 13 (2016) 212–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Di Rosa M, Distefano G, Zorena K and Malaguarnera L, Chitinases and immunity: Ancestral molecules with new functions, Immunobiology. 221 (2016) 399–411. [DOI] [PubMed] [Google Scholar]
- [63].Jian J, Chen Y, Liberti R, Fu W, Hu W, Saunders-Pullman R, et al. , Chitinase-3-like Protein 1: A Progranulin Downstream Molecule and Potential Biomarker for Gaucher Disease, EBioMedicine. 28 (2018) 251–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Lu J, Chiang J, Iyer RR, Thompson E, Kaneski CR, Xu DS, et al. , Decreased glucocerebrosidase activity in Gaucher disease parallels quantitative enzyme loss due to abnormal interaction with TCP1 and c-Cbl, Proc Natl Acad Sci U S A. 107 (2010) 21665–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Parcellier A, Schmitt E, Gurbuxani S, Seigneurin-Berny D, Pance A, Chantome A, et al. , HSP27 is a ubiquitin-binding protein involved in I-kappaBalpha proteasomal degradation, Mol Cell Biol. 23 (2003) 5790–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Yang C, Wang H, Zhu D, Hong CS, Dmitriev P, Zhang C, et al. , Mutant glucocerebrosidase in Gaucher disease recruits Hsp27 to the Hsp90 chaperone complex for proteasomal degradation, Proc Natl Acad Sci U S A. 112 (2015) 1137–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Nillegoda NB, Kirstein J, Szlachcic A, Berynskyy M, Stank A, Stengel F, et al. , Crucial HSP70 co-chaperone complex unlocks metazoan protein disaggregation, Nature. 524 (2015) 247–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Beel S, Moisse M, Damme M, De Muynck L, Robberecht W, Van Den Bosch L, et al. , Progranulin functions as a cathepsin D chaperone to stimulate axonal outgrowth in vivo, Hum Mol Genet. (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Lu J, Yang C, Chen M, Ye DY, Lonser RR, Brady RO, et al. , Histone deacetylase inhibitors prevent the degradation and restore the activity of glucocerebrosidase in Gaucher disease, Proc Natl Acad Sci U S A. 108 (2011) 21200–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Yang C, Rahimpour S, Lu J, Pacak K, Ikejiri B, Brady RO, et al. , Histone deacetylase inhibitors increase glucocerebrosidase activity in Gaucher disease by modulation of molecular chaperones, Proc Natl Acad Sci U S A. 110 (2013) 966–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Bendikov-Bar I, Ron I, Filocamo M and Horowitz M, Characterization of the ERAD process of the L444P mutant glucocerebrosidase variant, Blood Cells Mol Dis. 46 (2011) 4–10. [DOI] [PubMed] [Google Scholar]
- [72].Yang C, Swallows CL, Zhang C, Lu J, Xiao H, Brady RO, et al. , Celastrol increases glucocerebrosidase activity in Gaucher disease by modulating molecular chaperones, Proc Natl Acad Sci U S A. 111 (2014) 249–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Tan YL, Genereux JC, Pankow S, Aerts JM, Yates JR 3rd and Kelly JW, ERdj3 is an endoplasmic reticulum degradation factor for mutant glucocerebrosidase variants linked to Gaucher’s disease, Chem Biol. 21 (2014) 967–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Mizukami H, Mi Y, Wada R, Kono M, Yamashita T, Liu Y, et al. , Systemic inflammation in glucocerebrosidase-deficient mice with minimal glucosylceramide storage, Journal of Clinical Investigation. 109 (2002) 1215–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Vitner EB, Farfel-Becker T, Eilam R, Biton I and Futerman AH, Contribution of brain inflammation to neuronal cell death in neuronopathic forms of Gaucher’s disease, Brain. 135 (2012) 1724–35. [DOI] [PubMed] [Google Scholar]
- [76].Farfel-Becker T, Vitner EB, Pressey SN, Eilam R, Cooper JD and Futerman AH, Spatial and temporal correlation between neuron loss and neuroinflammation in a mouse model of neuronopathic Gaucher disease, Hum Mol Genet. 20 (2011) 1375–86. [DOI] [PubMed] [Google Scholar]
- [77].Michelakakis H, Spanou C, Kondyli A, Dimitriou E, Van Weely S, Hollak CE, et al. , Plasma tumor necrosis factor-a (TNF-a) levels in Gaucher disease, Biochim Biophys Acta. 1317 (1996) 219–22. [DOI] [PubMed] [Google Scholar]
- [78].Altarescu G, Zimran A, Michelakakis H and Elstein D, TNF-alpha levels and TNF-alpha gene polymorphism in type I Gaucher disease, Cytokine. 31 (2005) 149–52. [DOI] [PubMed] [Google Scholar]
- [79].Barak V, Acker M, Nisman B, Kalickman I, Abrahamov A, Zimran A, et al. , Cytokines in Gaucher’s disease, Eur Cytokine Netw. 10 (1999) 205–10. [PubMed] [Google Scholar]
- [80].Soliven B, Szuchet S and Nelson DJ, Tumor necrosis factor inhibits K+ current expression in cultured oligodendrocytes, J Membr Biol. 124 (1991) 127–37. [DOI] [PubMed] [Google Scholar]
- [81].Selmaj KW and Raine CS, Tumor necrosis factor mediates myelin and oligodendrocyte damage in vitro, Ann Neurol. 23 (1988) 339–46. [DOI] [PubMed] [Google Scholar]
- [82].Fu W, Hu W, Shi L, Mundra JJ, Xiao G, Dustin ML, et al. , Foxo4- and Stat3-dependent IL-10 production by progranulin in regulatory T cells restrains inflammatory arthritis, FASEB J. 31 (2017) 1354–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Liu C, Li XX, Gao W, Liu W and Liu DS, Progranulin-derived Atsttrin directly binds to TNFRSF25 (DR3) and inhibits TNF-like ligand 1A (TL1A) activity, PLoS One. 9 (2014) e92743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Mundra JJ, Jian J, Bhagat P and Liu CJ, Progranulin inhibits expression and release of chemokines CXCL9 and CXCL10 in a TNFR1 dependent manner, Sci Rep. 6 (2016) 21115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Tang W, Lu Y, Tian QY, Zhang Y, Guo FJ, Liu GY, et al. , The Growth Factor Progranulin Binds to TNF Receptors and Is Therapeutic Against Inflammatory Arthritis in Mice, Science. 332 (2011) 478–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Tian Q, Zhao S and Liu C, A solid-phase assay for studying direct binding of progranulin to TNFR and progranulin antagonism of TNF/TNFR interactions, Methods Mol Biol. 1155 (2014) 163–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Wei F, Zhang Y, Zhao W, Yu X and Liu CJ, Progranulin Facilitates Conversion and Function of Regulatory T Cells under Inflammatory Conditions, PLoS One. 9 (2014) e112110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Williams A, Wang EC, Thurner L and Liu CJ, Review: Novel Insights Into Tumor Necrosis Factor Receptor, Death Receptor 3, and Progranulin Pathways in Arthritis and Bone Remodeling, Arthritis Rheumatol. 68 (2016) 2845–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Zhao Y, Liu B and Liu CJ, Establishment of a surgically-induced model in mice to investigate the protective role of progranulin in osteoarthritis, J Vis Exp. (2014) e50924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Zhao YP, Liu B, Tian QY, Wei JL, Richbourgh B and Liu CJ, Progranulin protects against osteoarthritis through interacting with TNF-alpha and beta-Catenin signalling, Ann Rheum Dis. 74 (2015) 2244–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Zhao YP, Tian QY, Frenkel S and Liu CJ, The promotion of bone healing by progranulin, a downstream molecule of BMP-2, through interacting with TNF/TNFR signaling, Biomaterials. 34 (2013) 6412–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Zhao YP, Tian QY, Liu B, Cuellar J, Richbourgh B, Jia TH, et al. , Progranulin knockout accelerates intervertebral disc degeneration in aging mice, Sci Rep. 5 (2015) 9102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Zhao YP, Tian QY and Liu CJ, Progranulin deficiency exaggerates, whereas progranulin-derived Atsttrin attenuates, severity of dermatitis in mice, FEBS Lett. 587 (2013) 1805–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Zhao YP, Wei JL, Tian QY, Liu AT, Yi YS, Einhorn TA, et al. , Progranulin suppresses titanium particle induced inflammatory osteolysis by targeting TNFalpha signaling, Sci Rep. 6 (2016) 20909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Jian J, Hettinghouse A and Liu CJ, Progranulin acts as a shared chaperone and regulates multiple lysosomal enzymes, Genes Dis. 4 (2017) 125–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Hong YB, Kim EY and Jung SC, Upregulation of proinflammatory cytokines in the fetal brain of the Gaucher mouse, J Korean Med Sci. 21 (2006) 733–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Hollak CE, Evers L, Aerts JM and van Oers MH, Elevated levels of M-CSF, sCD14 and IL8 in type 1 Gaucher disease, Blood Cells Mol Dis. 23 (1997) 201–12. [DOI] [PubMed] [Google Scholar]
- [98].Kitatani K, Wada M, Perry D, Usui T, Sun Y, Obeid LM, et al. , Activation of p38 Mitogen-Activated Protein Kinase in Gaucher’s Disease, PLoS One. 10 (2015) e0136633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Allen MJ, Myer BJ, Khokher AM, Rushton N and Cox TM, Pro-inflammatory cytokines and the pathogenesis of Gaucher’s disease: increased release of interleukin-6 and interleukin-10, Qjm. 90 (1997) 19–25. [DOI] [PubMed] [Google Scholar]
- [100].Kitatani K, Sheldon K, Anelli V, Jenkins RW, Sun Y, Grabowski GA, et al. , Acid beta-glucosidase 1 counteracts p38delta-dependent induction of interleukin-6: possible role for ceramide as an anti-inflammatory lipid, J Biol Chem. 284 (2009) 12979–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Gonzalez-Navajas JM, Lee J, David M and Raz E, Immunomodulatory functions of type I interferons, Nat Rev Immunol. 12 (2012) 125–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Vitner EB, Farfel-Becker T, Ferreira NS, Leshkowitz D, Sharma P, Lang KS, et al. , Induction of the type I interferon response in neurological forms of Gaucher disease, J Neuroinflammation. 13 (2016) 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Vitner EB, Salomon R, Farfel-Becker T, Meshcheriakova A, Ali M, Klein AD, et al. , RIPK3 as a potential therapeutic target for Gaucher’s disease, Nat Med. 20 (2014) 204–8. [DOI] [PubMed] [Google Scholar]
- [104].Vissers JP, Langridge JI and Aerts JM, Analysis and quantification of diagnostic serum markers and protein signatures for Gaucher disease, Mol Cell Proteomics. 6 (2007) 755–66. [DOI] [PubMed] [Google Scholar]
- [105].Rocha EM, Smith GA, Park E, Cao H, Graham AR, Brown E, et al. , Sustained Systemic Glucocerebrosidase Inhibition Induces Brain alpha-Synuclein Aggregation, Microglia and Complement C1q Activation in Mice, Antioxid Redox Signal. 23 (2015) 550–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Pandey MK, Burrow TA, Rani R, Martin LJ, Witte D, Setchell KD, et al. , Complement drives glucosylceramide accumulation and tissue inflammation in Gaucher disease, Nature. 543 (2017) 108–112. [DOI] [PubMed] [Google Scholar]
- [107].Pandey MK, Grabowski GA and Kohl J, An unexpected player in Gaucher disease: The multiple roles of complement in disease development, Semin Immunol. (2018) [DOI] [PubMed] [Google Scholar]
- [108].Busch JI, Martinez-Lage M, Ashbridge E, Grossman M, Van Deerlin VM, Hu F, et al. , Expression of TMEM106B, the frontotemporal lobar degeneration-associated protein, in normal and diseased human brain, Acta Neuropathol Commun. 1 (2013) 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Nicholson AM and Rademakers R, What we know about TMEM106B in neurodegeneration, Acta Neuropathol. 132 (2016) 639–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Van Deerlin VM, Sleiman PM, Martinez-Lage M, Chen-Plotkin A, Wang LS, Graff-Radford NR, et al. , Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions, Nat Genet. 42 (2010) 234–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Chen-Plotkin AS, Unger TL, Gallagher MD, Bill E, Kwong LK, Volpicelli-Daley L, et al. , TMEM106B, the risk gene for frontotemporal dementia, is regulated by the microRNA-132/212 cluster and affects progranulin pathways, J Neurosci. 32 (2012) 11213–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Stagi M, Klein ZA, Gould TJ, Bewersdorf J and Strittmatter SM, Lysosome size, motility and stress response regulated by fronto-temporal dementia modifier TMEM106B, Mol Cell Neurosci. 61 (2014) 226–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Klein ZA, Takahashi H, Ma M, Stagi M, Zhou M, Lam TT, et al. , Loss of TMEM106B Ameliorates Lysosomal and Frontotemporal Dementia-Related Phenotypes in Progranulin-Deficient Mice, Neuron. 95 (2017) 281–296 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Neudorfer O, Giladi N, Elstein D, Abrahamov A, Turezkite T, Aghai E, et al. , Occurrence of Parkinson’s syndrome in type I Gaucher disease, Qjm. 89 (1996) 691–4. [DOI] [PubMed] [Google Scholar]
- [115].Lwin A, Orvisky E, Goker-Alpan O, LaMarca ME and Sidransky E, Glucocerebrosidase mutations in subjects with parkinsonism, Mol Genet Metab. 81 (2004) 70–3. [DOI] [PubMed] [Google Scholar]
- [116].Yun SP, Kim D, Kim S, Kim S, Karuppagounder SS, Kwon SH, et al. , alpha-Synuclein accumulation and GBA deficiency due to L444P GBA mutation contributes to MPTP-induced parkinsonism, Mol Neurodegener. 13 (2018) 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Xu YH, Sun Y, Ran H, Quinn B, Witte D and Grabowski GA, Accumulation and distribution of alpha-synuclein and ubiquitin in the CNS of Gaucher disease mouse models, Mol Genet Metab. 102 (2011) 436–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, et al. , Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies, Cell. 146 (2011) 37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Mazzulli JR, Zunke F and Tsunemi T, Activation of beta-Glucocerebrosidase Reduces Pathological alpha-Synuclein and Restores Lysosomal Function in Parkinson’s Patient Midbrain Neurons, 36 (2016) 7693–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Rocha EM, Smith GA, Park E, Cao H, Brown E, Hayes MA, et al. , Glucocerebrosidase gene therapy prevents alpha-synucleinopathy of midbrain dopamine neurons, Neurobiol Dis. 82 (2015) 495–503. [DOI] [PubMed] [Google Scholar]
- [121].Aflaki E, Borger DK, Moaven N and Stubblefield BK, A New Glucocerebrosidase Chaperone Reduces alpha-Synuclein and Glycolipid Levels in iPSC-Derived Dopaminergic Neurons from Patients with Gaucher Disease and Parkinsonism, 36 (2016) 7441–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Wong YC and Krainc D, Lysosomal trafficking defects link Parkinson’s disease with Gaucher’s disease, Mov Disord. 31 (2016) 1610–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Korkotian E, Schwarz A, Pelled D, Schwarzmann G, Segal M and Futerman AH, Elevation of intracellular glucosylceramide levels results in an increase in endoplasmic reticulum density and in functional calcium stores in cultured neurons, J Biol Chem. 274 (1999) 21673–8. [DOI] [PubMed] [Google Scholar]
- [124].Pelled D, Trajkovic-Bodennec S, Lloyd-Evans E, Sidransky E, Schiffmann R and Futerman AH, Enhanced calcium release in the acute neuronopathic form of Gaucher disease, Neurobiol Dis. 18 (2005) 83–8. [DOI] [PubMed] [Google Scholar]
- [125].Liou B, Peng Y, Li R, Inskeep V, Zhang W, Quinn B, et al. , Modulating ryanodine receptors with dantrolene attenuates neuronopathic phenotype in Gaucher disease mice, Hum Mol Genet. 25 (2016) 5126–5141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Wang F, Chou A and Segatori L, Lacidipine remodels protein folding and Ca 2+ homeostasis in Gaucher’s disease fibroblasts: a mechanism to rescue mutant glucocerebrosidase, Chem Biol. 18 (2011) 766–76. [DOI] [PubMed] [Google Scholar]
- [127].Vairo F, Sperb-Ludwig F, Wilke M, Michellin-Tirelli K, Netto C, Neto EC, et al. , Osteopontin: a potential biomarker of Gaucher disease, Ann Hematol. 94 (2015) 1119–25. [DOI] [PubMed] [Google Scholar]
- [128].Murugesan V, Liu J, Yang R, Lin H, Lischuk A, Pastores G, et al. , Validating glycoprotein non-metastatic melanoma B (gpNMB, osteoactivin), a new biomarker of Gaucher disease, Blood Cells Mol Dis. 68 (2018) 47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Casal JA, Lacerda L, Perez LF, Pinto RA, Clara Sa Miranda M and Carlos Tutor J, Relationships between serum markers of monocyte/macrophage activation in type 1 Gaucher’s disease, Clin Chem Lab Med. 40 (2002) 52–5. [DOI] [PubMed] [Google Scholar]
- [130].Drugan C, Drugan TC, Miron N, Grigorescu-Sido P, Nascu I and Catana C, Evaluation of neopterin as a biomarker for the monitoring of Gaucher disease patients, Hematology. 21 (2016) 379–86. [DOI] [PubMed] [Google Scholar]