

Abstract
In light of the worldwide epidemic of obesity, and in recognition of hypertension as a major factor in the cardiovascular morbidity and mortality associated with obesity, The Obesity Society and the American Society of Hypertension agreed to jointly sponsor a position paper on obesity‐related hypertension to be published jointly in the journals of each society. The purpose is to inform the members of both societies, as well as practicing clinicians, with a timely review of the association between obesity and high blood pressure, the risk that this association entails, and the options for rational, evidenced‐based treatment. The position paper is divided into six sections plus a summary as follows: pathophysiology, epidemiology and cardiovascular risk, the metabolic syndrome, lifestyle management in prevention and treatment, pharmacologic treatment of hypertension in the obese, and the medical and surgical treatment of obesity in obese hypertensive patients.
The United States is currently facing a very real obesity epidemic. The most recent National Health and Nutrition Examination Survey indicates that approximately two thirds of US adults are presently classified as overweight or obese (body mass index [BMI] ≥25) and one third as obese (BMI ≥30). 1 , 2 While the numbers alone are formidable, they leave unaddressed the medical costs associated with obesity and obesity‐related comorbidities, not the least of which is obesity‐related hypertension. Given the frequent concurrence of obesity and hypertension, it is no coincidence that as the rate of obesity continues to rise, so too does the rate of hypertension. It is estimated that at least 75% of the incidence of hypertension is related directly to obesity. 1 It is essential, therefore, to develop treatment strategies for the management of obesity in order to reduce the development of obesity‐related hypertension as well as to effectively manage high blood pressure (BP) in the obese.
Recent publications have estimated that the annual medical burden of obesity and obesity‐related diseases in the United States totaled roughly $147 billion in 2008, 1 and that projected obesity‐related medical expenses will more than double by 2018, topping $344 billion, or about 21% of total healthcare spending. 1 Although lifestyle changes aimed at prevention, especially in childhood, are the ultimate solution to the societal problem of obesity and its complications, the scope of illness caused by obesity demands immediate attention and therapeutic intervention in the obese population. Given the important role that obesity plays in the pathogenesis of hypertension, the leadership of both The Obesity Society and The American Society of Hypertension have commissioned this position paper for the purpose of providing the membership of both societies, as well as the community of clinicians in practice, with a current and timely summary on the relationship between weight and BP, on the cardiovascular (CV) risk imposed and on the management of obesity‐related hypertension.
The Pathophysiology of Obesity‐Related Hypertension
The association of obesity and hypertension has been recognized since the beginning of the 20th century, when BP was first measured in populations (Table I). 3 This relationship between body weight and BP was demonstrated prospectively in the Framingham Heart Study in the 1960s. 4 The nature of the linkage between BP and body weight remained obscure until the mid‐1980s when basic clinical and population‐based research significantly clarified many aspects of the relationship between these two common and complex regulatory disturbances. Appreciation of the clinical significance of obesity‐related hypertension has grown substantially over this same time period, to the point where obesity is recognized as a major cause of high BP, and the combination of obesity and hypertension is recognized as a pre‐eminent cause of CV risk.
Table I.
Pathogenesis of Obesity‐Related Hypertension
| Central (abdominal) obesity |
| Insulin resistance (hyperinsulinemia) |
| ↑ Leptin levels |
| ↑ Sympathetic nervous system (SNS) activity |
| Insulin |
| Leptin |
| ↑ Renin‐angiotensin‐aldosterone system activity |
| SNS stimulation of renin release |
| ↑ Angiotensinogen from intra‐abdominal adipocytes |
| ↑ Aldosterone production (in excess of angiotensin stimulation) |
| Salt sensitivity (↑ renal sodium reabsorption) |
| SNS |
| Insulin |
| Angiotensin (all) |
| Aldosterone |
| Intra‐renal blood flow redistribution |
Until the 1980s there was no cogent explanation for the documented association between weight and BP. Trivial attributions, such as small cuff/large arm artifact, or excessive salt intake, have been excluded as a cause for this association. Similarly, a purely hemodynamic explanation, based on increased plasma volume and increased cardiac output, are not sufficient explanations since the latter do not account for the increase in peripheral resistance noted in obese hypertensive patients when compared with normotensive obese patients. Clues to the basic mechanisms involved in the link between obesity and hypertension first appeared in the 1940s and 1950s with the important observations by Vague. 5 Based on observations made in his own obesity practice, Vague noted that the CV and metabolic complications of obesity were more common in patients with the upper body obesity phenotype, which he called “android,” as compared with lower body obesity, which he referred to as “gynoid.” These prescient observations attracted little attention until the 1980s when population‐based studies in Scandinavia, using waist to hip ratio as a quantifiable surrogate for the upper body phenotype, demonstrated significant CV risk (hypertension, myocardial infarction, and type 2 diabetes mellitus) in association with a high waist to hip ratio. 6 , 7 , 8 Parallel lines of research showed that insulin resistance was also associated with the upper body phenotype, 9 , 10 , 11 and many subsequent clinical and population‐based studies showed an association of insulin levels and/or insulin resistance with hypertension in both obese and nonobese people. 12 , 13 Thus, insulin, hypertension and the android or central obesity phenotype tracked together in population‐based and clinical studies. These observations formed the basis for our current understanding of the pathophysiology of obesity‐related hypertension.14 Pathogenetic factors are reviewed here since they provide the basis for a rational therapeutic strategy.
Insulin and Sympathetic Activity
The relationship of insulin to BP, although controversial at first, has a plausible explanation, and insulin is now generally acknowledged to play a role in the pathophysiology of obesity‐related hypertension. 14 Since insulin stimulates the sympathetic nervous system (SNS), 16 , 17 and since obese patients have increased SNS activity, 18 , 19 , 20 a role for insulin‐mediated SNS stimulation seems a likely factor in the pathogenesis of high BP in the setting of central obesity. This is supported by studies demonstrating concomitant decreases in BP and SNS activity when insulin is lowered by low energy diets in obese patients. 21 Insulin also has a direct action on the kidney to stimulate sodium retention. 22
Leptin and the SNS
Leptin, the polypeptide product of the ob/ob gene, is produced in adipocytes and secreted into plasma where the circulating concentration reflects the fat mass of the individual. 23 Leptin is a potent appetite suppressant and, like insulin, stimulates the SNS. 24 , 25 Although leptin deficiency causes severe obesity in animals and humans, all but a vanishingly small minority of obese humans have elevated leptin levels. 23 , 26 When infused into animals, leptin raises the BP, 27 stimulates the SNS, 28 and has been shown to correlate with BP, in at least some human populations. 29 , 30 Both insulin‐mediated and leptin‐mediated SNS activation may be viewed as mechanisms recruited in obese patients to stabilize weight and restore energy balance by stimulating sympathetically mediated thermogenesis with consequent enhancement of energy output. 14 , 31
Renin‐Angiotensin‐Aldosterone System
The renin‐angiotensin‐aldosterone system (RAAS) is activated in obesity. 32 , 33 , 34 , 35 , 36 Aldosterone levels may be increased out of proportion to the increase in renin activity. 33 Several mechanisms have been thought to underlie RAAS activation, including SNS stimulation of renin release 33 with the generation of angiotensin II; angiotensinogen production in adipose tissue, especially intraabdominal adipocytes 34 , 35 with the generation of angiotensin II and aldosterone; and effects of free fatty acids, along with other poorly defined factors, on aldosterone production and release. 33
Sodium Excretion, Pressure Natriuresis, and Salt Sensitivity
Obesity predisposes the kidney to reabsorb sodium by neural (SNS), hormonal (aldosterone and insulin), and renovascular (angiotensin II) mechanisms. 37 This enhanced sodium avidity shifts the pressure natriuresis curve to the right, 38 thereby necessitating higher arterial pressure to excrete the day’s salt intake and maintain sodium balance and volume homeostasis. This is the basis for the documented salt sensitivity of obesity‐related hypertension 39 and underlines the need for diuretics in the therapeutic regimen.
Other Potential Mechanisms
Other factors that may be implicated in the pathophysiology of obesity‐related hypertension include a decrease in natriuretic peptides 33 , 34 with consequent impairment in salt excretion; a decrease in adiponectin; 40 obstructive sleep apnea, which stimulates the SNS; 41 , 43 , 44 low birth weight, which is associated with excessive weight gain in childhood and adolescence, along with increased risk of hypertension and stimulation of the SNS 45 , 46 , 47 in adulthood; and endothelial dysfunction 48 with consequent blunting of physiological vasodilation. 49
Implications for Therapy
The goal of therapy is to address the CV risk factors attendant upon obesity‐related hypertension. This involves lifestyle changes, pharmaceutical agents, and sometimes surgery. Treatment should address the underlying pathophysiology and modify the surrogate markers and risk factors for CV disease (CVD) toward desirable levels in the apparently justifiable belief that this will translate into decreased CV morbidity and mortality.
Epidemiology of Obesity‐Related Hypertension and CV Risk
Data from recent US National Health and Nutrition Examination Surveys (NHANES) from 2005 to 2008 indicate that the prevalence of hypertension among adults 18 years older in the United States was 30.9%, or nearly 1 in 3 adults. In the context of the entire population, more than 76 million US adults are estimated to have hypertension. 50 At the same time, nearly 70% of American adults are overweight or obese. 51 Given the important pathophysiologic links between weight and BP described above, we can expect a significant increase in the prevalence of hypertension in coming years if trends of increasing weight in the population are not stabilized and reversed.
Epidemiological data unequivocally support the link between body weight and BP. Indeed, greater body weight is one of the major risk factors for high BP. Recent data from NHANES indicate that the prevalence of hypertension among obese individuals, with a BMI ≥30 kg/m2, is 42.5% compared with 27.8% for overweight individuals (BMI 25.0–29.9 kg/m2) and 15.3% for those with BMI <25 kg/m2. 52
Likewise, higher BMI is also associated with increased risk for development of hypertension over time. Data from the long‐standing Framingham Heart Study revealed that compared with normal weight adult men and women, the multivariable‐adjusted relative risks for development of hypertension in long‐term follow‐up were 1.48 and 1.70 for overweight men and women and 2.23 and 2.63 for obese men and women, respectively. 53 With the current obesity epidemic extending into its third decade, prevalence rates of hypertension, which had been falling in the 1970s and 1980s, are again rising.
Numerous studies have also demonstrated the important role of weight gain in BP elevation and of weight reduction in BP lowering. As a general rule, in Western societies, systolic BP (SBP) and diastolic BP (DBP) tend to rise with age beginning at around age 25 in most adults. 54 , 55 This is a “normative” process of aging, but it may not be inevitable or “normal.” In fact, recent data suggest that these “age‐related” increases in SBP and DBP may be avoided in young adults who maintain stable BMI during long‐term follow‐up into middle age. In the Coronary Artery Risk Development in Young Adults (CARDIA) study, young adults (mean age 25 years at baseline) who maintained a stable BMI (within 2 kg/m2 of baseline) at 6 examinations during 15 years had no significant changes in SBP or DBP, whereas those who had an increase in their BMI ≥2 kg/m2 had substantial increases in BP. 56 For example, women who maintained stable BMI in this study had nonsignificant declines in SBP, whereas women whose BMI increased had statistically significant average increases in SBP of 9.8 mm Hg to 12.5 mm Hg. Of note, this weight gain was more important than the baseline weight, since the same patterns were observed for those who were at normal weight or overweight at baseline. Hence, age‐related changes in BP may not be inevitable, and may be caused more by age‐related weight gain than aging per se. These data have important implications for healthcare and public health, since weight maintenance may be a strategy that is easier to achieve than substantial weight loss for preventing or controlling hypertension. 56
The influence of weight gain on BP and the benefits of maintaining stable weight or losing weight extend down even to young children. One large birth cohort study of children examined BMI at ages 5 and 14 and the association with SBP and DBP at age 14. Children who were overweight at age 5 but had normal BMI at age 14 had similar mean SBP and DBP to those who had a normal BMI at both time points. Conversely, children who were overweight at both ages or who had a normal BMI at age 5 and were overweight at age 14 had higher SBP and DBP at age 14 than those who had a normal BMI at both ages, even after adjustment for potential confounders. 57
Risks Associated With Hypertension
Hypertension is a complex phenotype that arises from numerous genetic, environmental, behavioral, and even social origins. As discussed above, obesity is one of the most prevalent risk factors for its development. Regardless of its etiology, however, hypertension is a highly prevalent and highly significant risk factor for the development of all manifestations of CVD, including coronary heart disease (CHD), stroke, heart failure (HF), aortic and peripheral arterial disease, and valvular heart disease.
CVD Risk Across the Spectrum of BP
Individuals with hypertension, currently defined as untreated SBP ≥140 mm Hg or DBP ≥90 mm Hg or use of therapy for elevated BP, have a 2‐ to 3‐fold increased risk for all CVD events combined compared with nonhypertensive individuals. When CVD endpoints are considered individually, relative risks with hypertension are greatest for stroke and HF, and somewhat lower for CHD. Nonetheless, because overall absolute CHD incidence is greater than incidence of stroke and HF, the absolute impact of hypertension on CHD is greater than for other manifestations of CVD. Although much of the focus on CVD risk has been on frank hypertension, it is clear that risks for CVD increase at higher BP levels, even within the so‐called normal range. A recent epidemiologic pooling study of nearly 1 million men and women, which included data on more than 56,000 decedents, revealed that risk for CVD death increases in a continuous fashion at SBP levels starting as low as 115 mm Hg and DBP 75 mm Hg, as discussed in detail below. 58
Other studies provide confirmation of these findings. Data from more than 347,000 middle‐aged men (35 to 57 years) screened for the Multiple Risk Factor Intervention Trial (MRFIT) provide precise estimates of incremental risks with higher BP levels. There is a continuous, graded effect of BP on the multivariable‐adjusted relative risk for CHD mortality beginning at pressures well below 140 mm Hg. 59 , 60 Although relative risks were clearly highest for men with SBP ≥180 mm Hg, these data help make an important point about BP levels in the population of which the majority of CVD events are occurring. Because the majority of the population had lower BP levels, this is where most of the events occurred. Thus, nearly two thirds of excess CHD deaths occurred in men with baseline SBP between 130 and 159 mm Hg, relatively “mild” levels of elevated BP.
For many CVD endpoints, there is effect‐modification by sex, with hypertensive men being at higher absolute risk for CVD events than hypertensive women. HF is a notable exception to this generalization. There is also substantial effect‐modification by age, with older hypertensive patients being at similar or higher relative risk but much greater absolute risk than younger patients. 61 Hypertension rarely occurs in isolation, 62 and it confers increased risk for CVD across the spectrum of overall risk factor burden, with increasing importance in the setting of other risk factors. As shown in Figure 1, absolute levels of predicted hard CHD risk (including CHD death or nonfatal myocardial infarction) increase substantially with increasing risk factor burden, and risk is augmented still further by increases in BP levels from optimal to marked hypertension in men and similarly for women. 63 Thus, BP levels, and the risk they confer, must always be considered in the context of other risk factors and the patient’s global risk for CVD.
Figure 1.
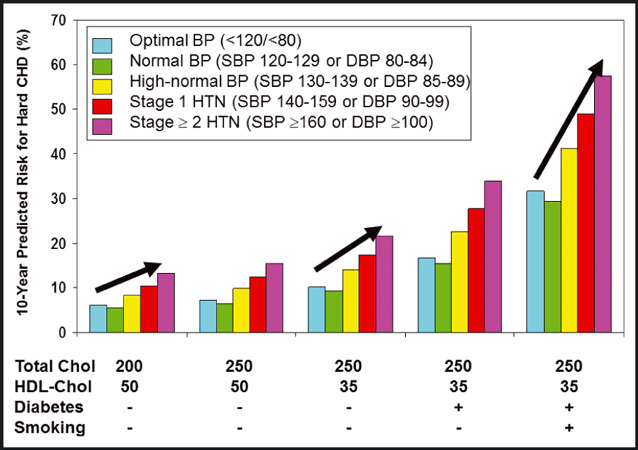
Predicted 10‐year risk for hard coronary heart disease events for a 50‐year‐old man with selected levels of risk factors and blood pressure (BP) stages. Note that risk increases dramatically with greater risk factor burden and with higher BP level at any given risk factor burden. The increase in risk across BP strata is more pronounced when other risk factors are at more adverse levels. Data calculated from reference. 63
BP and CHD
Elevated BP is one of the major risk factors contributing to the estimated 1.4 million CHD events that occur in the United States annually. 51 Numerous studies have documented the graded and continuous risk for CHD endpoints associated with higher BP levels. Risk for CHD mortality is not limited to patients with frank hypertension, however. There is a linear, graded risk for CHD death that extends down even to optimal levels of BP. The Prospective Studies Collaboration observed that beginning at 115 mm Hg, the risk for CHD death doubles for each increase of 20 mm Hg in SBP. Similarly, CHD death risk doubles for each increase of 10 mm Hg in DBP beginning at 75 mm Hg. 58
BP and Stroke
Hypertension is well established as the dominant risk factor in contributing to the 700,000 strokes that occur each year in the United States, 51 even when considered in the context of other known risk factors, such as cigarette smoking, atrial fibrillation, myocardial infarction, and diabetes. Hypertension confers a threefold relative risk for stroke compared with BP levels <140/<90 mm Hg, and approximately 80% of individuals who experience a stroke have antecedent hypertension. The population‐attributable risk (PAR) for stroke associated with hypertension varies between 33% and 53% in different age groups, suggesting that this proportion of strokes would be avoided if hypertension were not present. 64 In adults aged 55 in the Framingham Heart Study, the lifetime risk for stroke was >1 in 6, but it was twice as high among those with hypertension compared with those who had BP <120/<80 mm Hg. 65
As is true for CHD risk prediction, a risk prediction algorithm has been developed to estimate the absolute 10‐year risk of an atherothrombotic brain infarct using standard CVD risk factors plus the presence of atrial fibrillation, HF, and coronary disease. In these equations, hypertension represents the predominant risk factor for stroke, but the risk in people with elevated BP varies over as much as a 10‐fold range depending on the degree of exposure to the other risk factors. 66
Just as with CHD, risk for stroke is not limited to patients with frank hypertension. There is a linear, graded risk for stroke that extends down even to optimal levels of BP. Beginning at 115 mm Hg, the risk for stroke mortality doubles for each increase of 20 mm Hg in SBP. Likewise, stroke mortality risk doubles for each increase of 10 mm Hg in DBP beginning at 75 mm Hg. 58 Thus, at any given age, an individual with SBP of 135 mm Hg is at approximately twice the risk and one with SBP of 155 mm Hg is at four times the risk as someone at the same age with an SBP of 115 mm Hg.
BP and HF
With the aging of the population, the concomitant increase in the prevalence of hypertension, and improved survival after myocardial infarction, HF has become an emerging public health concern. National surveillance data indicate that approximately 5.7 million Americans are living with HF currently, with 670,000 incident cases each year. HF is the leading cause of hospitalization for people 65 years and older in the United States, with 990,000 hospital discharges in 2007. 51
As with stroke, hypertension is also the dominant risk factor for HF. The overall remaining lifetime risk for HF is 1 in 5 for men and women 40 years and older, but at every age there is a stepwise increase in lifetime risk for HF with increasing BP, with approximately a twofold higher risk in patients with stage 2 or treated hypertension compared with those with BP <140/<90 mm Hg. 67 From 75% to 91% of individuals who develop HF have antecedent hypertension. 51 , 68 The PAR for hypertension in HF, ie, the fraction of HF in this population that would not occur in the absence of hypertension, was 59% in women and 39% in men. By contrast, the PARs for myocardial infarction were 13% and 34% for women and men, respectively. 68
BP and Renal Disease
Hypertension is also a major risk factor for the development of renal disease. African Americans have approximately four times the risk of whites of developing end‐stage renal disease (ESRD), in part due to their significantly higher prevalence of hypertension. 51 Data from a large insured population of 316,675 adults with a normal glomerular filtration rate (>60 mL/min/1.73 m2) and no evidence of proteinuria or hematuria at baseline, risks for ESRD increased dramatically with higher baseline BP level. Individuals with hypertension were 2.5 to more than 4 times as likely to develop ESRD, depending on the severity of their hypertension, even after adjustment for other factors. 69 In addition to its contribution to ESRD, elevated BP also occurs in and exacerbates milder forms of chronic kidney disease and worsens proteinuria. Data from the same population indicate that, compared with patients with lean body mass, risk for ESRD was higher with greater baseline weight, with adjusted relative risks of 1.87, 3.57, 6.12, and 7.07 for those with BMI in the overweight, class I (BMI 30–34.9), class II (BMI 35–39.9), and class III (BMI >40) obesity ranges, respectively. 70 Thus, greater baseline weight is also associated with a markedly increased risk for incident ESRD, both through its association with hypertension and independent of this association.
Joint Effects of Obesity and Hypertension on CVD Risk
The association of hypertension with CV risk in the short‐ and long‐term is thus unequivocally established. The association of obesity with short‐term CVD event rates (eg, in the next 10 years) is more difficult to establish, largely because the major effects of obesity appear to act through more proximal risk factors such as diabetes, dyslipidemia, and hypertension. However, longer‐term studies of obesity and CVD do indicate risk for CVD associated with obesity independent of these other risk factors. In addition, several lines of evidence data suggest that obesity and hypertension may have additive effects in increasing risk for CVD over long‐term follow‐up.
Data from the long‐standing Chicago Heart Association Detection Project in Industry, 71 which enrolled more than 38,000 individuals from 1967 to 1973, serve as an example to highlight these joint risks. As shown in Figure 2, 32‐year CVD death rates were higher (and increased in a stepwise fashion) for patients with higher BMI at baseline and no hypertension. For those with hypertension at baseline, CVD death rates were substantially higher overall, and increased in a stepwise fashion for patients with higher baseline BMI levels. A similar pattern of results was observed for individual outcomes of CHD death and stroke death rates, as well as for hospitalizations for CHD, stroke, and HF during follow‐up using Medicare data.
Figure 2.
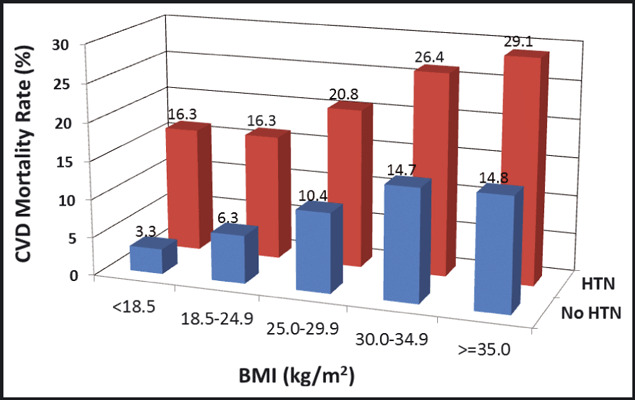
Thirty‐two year rates of death due to cardiovascular disease in participants of the Chicago Heart Association Detection Project in Industry cohort, stratified by baseline body mass index (BMI) and hypertension (HTN) status. CVD indicates cardiovascular disease.
Clinically Relevant Risk Assessment
Given the above data on the prevalence, risks, and sequelae of hypertension and the likelihood that hypertension will be clustered with obesity and other major CVD risk factors (the metabolic syndrome), it is imperative that patients with hypertension be identified and appropriately managed. Risk assessment for individuals with hypertension should take into account global CVD risk based on the presence and severity of these other risk factors. Widely available multivariable risk equations, such as the Framingham CVD risk score or the Reynolds risk score, are means for assessing such risk and identifying individuals who may benefit from more intensive preventive therapy and efforts at lifestyle modification. Adjunctive therapy beyond antihypertensive therapy must be considered, especially in light of clinical trial results indicating significant and substantial reductions in risk for CVD events in treated hypertensive patients with fairly average levels of low‐density lipoprotein (LDL) cholesterol who were randomized to low‐dose statin medication. 72 Likewise, the benefits of adjunctive low‐dose aspirin therapy for CVD risk reduction clearly appear to outweigh the potential risks when specifically targeted to hypertensive patients. 73
Impact of Obesity on Diabetes and CV Risk
An additional important factor in the CV risk associated with obesity and hypertension is the role played by obesity in the development of type 2 diabetes. Obesity, and particularly central adiposity, is the dominant risk factor for the development of type 2 diabetes, which routinely clusters with hypertension because of common underlying pathophysiology. Diabetes exerts a substantial independent and amplifying effect on the CV risks associated with both obesity and hypertension. 74 , 75 Efforts aimed at diminishing the incidence and impact of diabetes, therefore, including both lifestyle changes and the appropriate use of antihypertensive and anti‐obesity therapies, are an essential part of the overall therapeutic plan.
The Metabolic Syndrome
Obesity‐related hypertension frequently occurs in association with other CV risk factors, forming a constellation referred to as the metabolic syndrome (Table II). 76 Although the concept of a metabolic syndrome has achieved widespread acceptance over the past 2 decades, no consensus has developed over the precise definition of the syndrome, nor over the criteria required to establish the diagnosis. No less than five sets of diagnostic criteria have been proposed by different international and national panels including the International Diabetes Federation (IDF), the World Health Organization (WHO), and the US National Cholesterol Education Program Adult Treatment Panel III (NCEP‐ATP III), among others. 77 The differences in criteria, although small and overlapping, point to the imprecision in defining the metabolic syndrome, and to the differences in the perceived importance of the various manifestations. Using the NCEP‐ATP III criteria and the NHANES III survey it has been estimated that about 30% of the US population has the metabolic syndrome. 78 The prevalence increases with age so that by 60 years, more than 40% of people meet criteria for the diagnosis.
Table II.
Metabolic Syndrome
| Many sets of diagnostic criteria |
| Critical components |
| Hypertension |
| Central obesity |
| Insulin resistance (hyperinsulinemia) |
| Characteristic dyslipidemia |
| High triglycerides |
| Low high‐density lipoprotein cholesterol |
| Other associations |
| Impaired glucose tolerance; type 2 diabetes mellitus |
| Microalbuminuria; chronic renal disease |
| Prothrombotic diathesis |
| Small dense low‐density lipoprotein |
| ↑ Inflammatory markers |
Critical Components
The four fundamental components of the metabolic syndrome are central obesity, insulin resistance, hypertension, and a characteristic dyslipidemia (high triglycerides and low high‐density lipoprotein cholesterol). Reaven has identified the importance of insulin resistance as a critical component of the syndrome, which he originally designated “syndrome x” and which was also called the “insulin resistance syndrome,” although these earlier designations have given way to the term metabolic syndrome. 77 Central or android obesity is the usual although not the exclusive cause of the insulin resistance. Insulin resistance and the consequent hyperinsulinemia drive the hypertension (as described above) and the dyslipidemia (stimulation of hepatic very LDL production).
Other Associated Features
In addition to the four principal components, a variety of other abnormalities have been associated with the metabolic syndrome, including type 2 diabetes mellitus, impaired glucose tolerance, renal functional impairment and microalbuminuria, hyperuricemia, prothrombotic coagulation diatheses, small dense LDL cholesterol, and markers of inflammation. 79 All of these abnormalities have been associated with increased CV risk.
Additional Pathogenetic Factors
Although abdominal obesity and insulin resistance are the major threads that connect the various features of the metabolic syndrome, two other factors of potential importance should be mentioned: dietary fructose and disordered sleep. The consumption of high fructose corn syrup has increased dramatically in the past 3 decades, paralleling the increase in obesity and hypertension. Sweetened beverages, such as non‐diet soda, account for 70% of the intake of high fructose corn syrup, 80 , 81 and recent evidence suggests a link between sweetened sodas, hyperuricemia, and the manifestations of the metabolic syndrome. 82 , 83
Another recently described factor that may contribute to the development of the metabolic syndrome is shortened or interrupted sleep. Obstructive sleep apnea, a well‐recognized complication of obesity, is associated with increased SNS activity, which persists during daytime wakefulness. 41 , 43 The SNS overactivity is associated with hypertension. Both the SNS stimulation and the hypertension are reversed with effective treatment of the sleep apnea. Sleep debt, a consequence of shortened or disordered sleep, or night shift work, is also associated with obesity, insulin resistance, and hypertension, raising the possibility that disturbances of normal sleep patterns may play a role in the pathogenesis of the metabolic syndrome. 44 , 84 , 85 , 86 Insufficient sleep may also antagonize weight loss in response to caloric restriction. 87
Is the Metabolic Syndrome a Distinct Entity?
Considerable debate has centered on whether the metabolic syndrome is in fact a discrete entity. Although the abnormalities characteristic of the metabolic syndrome are relatively common in the population at large, there is no doubt that these traits occur together much more commonly than predicted by chance alone. This, however, may reflect the central role of insulin resistance and hyperinsulinemia in the pathogenesis of the different manifestations, rather than a direct linkage of the associated traits. The argument has been advanced, with some merit, that the concept of a metabolic syndrome may alert clinicians to look for associated manifestations when obesity and hypertension coexist and to recognize the CV risk associated with this constellation.
Lifestyle in the Prevention and Management of Obesity and Hypertension
The importance of lifestyle management in the treatment of patients with obesity‐related hypertension cannot be overemphasized (Table III). Adoption of a healthy lifestyle facilitates weight loss, increases responsiveness to antihypertensive medications, and produces independent beneficial effects on cardiac risk factors.
Table III.
Lifestyle Management of Obesity‐Related Hypertension
| Weight loss |
| Dietary Approaches to Stop Hypertension (DASH) diet |
| Salt restriction |
| ↑ Physical activity; exercise |
| Alcohol moderation |
| Behavioral modification |
Developmental Origins of Obesity‐Related Hypertension
The childhood origins of obesity‐related hypertension are well illustrated in a study of 260,000 overweight and obese children in Germany and Switzerland, in which 35% had hypertension with increased ventricular mass or arterial stiffness. 88 Studies of the Bogalusa childhood cohort who were prehypertensive or mildly hypertensive in adulthood showed that when they were tracked back to as young as 4 to 8 years old, they had higher BPs and were heavier and more insulin resistant than their normotensive counterparts. 89 This is in accord with prospective studies showing tracking of adiposity, obesity, and BPs from childhood into adult life. In a recent analysis of four cohort studies followed for a mean of 23 years, overweight or obese children who remained obese as adults had substantially increased risk of hypertension, diabetes, dyslipidemia, and carotid atherosclerosis. 90 Importantly, patterns of food consumption and physical activity also track from childhood to adulthood. 91
Stemming from Barker’s work, there is substantial evidence for effects of intrauterine growth and early postnatal weight gain on adiposity and high BP. 92 These effects are not restricted to low birth weight infants, as shown by the clustering of adiposity and BP along with impaired glucose tolerance and dyslipidemia in 8‐ and 14‐year‐olds born from the lowest and highest birth weight quintiles. 93 , 94 Excessive postnatal weight gain in early childhood dominated over effects of birth weight, 94 especially in children of mothers who smoked in pregnancy or did not breastfeed. 94 In the same Australian cohort, the trajectories for adolescent obesity were well established by the age of 5. 95 For all of these reasons, a consensus is developing that it may be necessary to tackle lifestyle‐induced obesity‐related hypertension at its source: in infancy and early childhood and in the parents.
Evidence that Childhood Obesity can be Prevented or Modified at a Population Level
There is an increasing number of randomized controlled trials attempting to modify childhood obesity in populations rather than in clinic settings. Most of these have been school‐based. Few have also examined effects of the programs on BP. An extensive Cochrane review of lifestyle interventions to prevent obesity in childhood included 55 studies. A meta‐analysis of 37 of these involved 27,946 children, of whom the majority were aged 6 to 12 years. 96 The authors concluded that the “programs overall were effective at reducing adiposity, although not all individual interventions were effective, and there was a high level of observed heterogeneity and possible bias.” Moreover, the effect was relatively small with children in the intervention group “showing a small standardized mean difference in adiposity (measured as BMI or zBMI) of −0.15 kg/m.” Given the unexplained heterogeneity and the likelihood of small study bias, however, these findings must be interpreted cautiously. The authors were unable to distinguish which of the program components contributed to the beneficial effects and suggested that “childhood obesity prevention research must now move towards identifying how effective intervention components can be embedded within health, education and care systems and achieve long term sustainable impacts.”
None of the above studies reported effects on BP or CV risk phenotypes other than obesity. However, two large randomized controlled trials of effects of home‐ and school‐based nutrition and physical activity programs on CV risk factors have been reported from Australia. The first 1147 10‐ to 12‐year‐olds from 30 schools found improved BP, a reduction in fatness, and improved physical fitness. 97 Decline in SBP was significantly greater with a fitness intervention for the boys and with a home nutrition intervention for the girls. The greatest improvements overall were with the combined fitness and home nutrition program.
The second study used cluster analysis to identify 29% of 800 11‐year‐old children at increased CV/metabolic risk. The children in both high‐ and low‐risk clusters were then randomized to two semesters of family nutrition and school‐based physical activity programs. 98 High‐risk children responded better in terms of fatness, fitness, nutrition, and blood cholesterol than did low‐risk children. Boys responded better than the girls during the program but effects were more sustained after a further 6 months in girls.
The lifestyle factors contributing to the rising epidemic of obesity, and hence obesity‐related hypertension, are embedded in changes in society worldwide: increased sedentariness from the car, television, and computers; parental protectiveness in seemingly hostile urban environments; and increased consumption of calorie‐rich foods in the form of soft drinks, fast foods, and sugar‐enriched low‐fat dairy products. Clustering of unhealthy behaviors in obese children, such as poor nutritional habits, high salt consumption, low levels of physical activity, and smoking and alcohol consumption (by adolescence) 99 dictate the need for multifaceted national‐, school‐, and family‐based programs to tackle global CV risk at an early stage. Many such programs are underway worldwide, and are addressing issues relating to infancy, 100 childhood, 101 parents 102 and the community, 103 and through international networks. 104
Prevention of Weight Gain and Hypertension
Lifestyle changes tend not to occur in isolation. Those who are obese tend to show clustering of behaviors predisposing to higher BP including not only disturbed energy balance but less healthy diets with higher salt intake, less fruit and vegetable intake, less low‐fat dairy products and increased saturated fat intake, 99 , 105 sedentary behaviors, and in many communities high alcohol consumption. These pro‐hypertensive behaviors add to the effects of obesity per se. In industrialized communities, low socioeconomic status is a further factor predisposing to obesity, 106 while in developing nations, rising urbanization and westernization with fast‐food patterns and decreased physical activity create obesogenic environments. 107
Long‐term weight gain is insidious, arising from the cumulative effect of excess intake, which may be as little as 50 to 100 kcal/d. 108 In a longitudinal analysis, 105 weight gain in US adults was positively associated with small changes that included increased consumption of sugars, starches, refined grains, and processed foods as well as increased alcohol intake, time spent watching television, and decreased physical activity; weight change related inversely to consumption of fruit and vegetables, whole grains, nuts, and yogurt. These authors suggest that the small daily changes associated with weight gain could be prevented by small changes in lifestyle adhered to in the long‐term. Long‐term behavior change, however, will need recognition of effective strategies from population studies and clinical trials as well as the cooperation of governments and industry. 109 Effects of low socioeconomic status and ethnic differences, with greater predisposition to obesity and hypertension in blacks and obesity in Hispanics, will need particular attention. 106 , 110
Lifestyle Changes in the Management of Obesity‐Related Hypertension
Weight Loss
Systematic reviews consistently report a decrease in SBP of about 1 mm Hg per kg of weight loss with follow‐up of 2 to 3 years. 111 , 112 , 113 , 114 There is attenuation in the longer‐term, with a decrease of about 6 mm Hg in SBP per 10 kg of weight loss. 112 Intervention programs appropriate for obesity‐hypertension combine diet, physical activity, and behavioral modification and aim to achieve long‐term change in health‐related behaviors.
Choice of Weight‐Reducing Diet
In the short‐term, many variations on reduced‐energy diets can achieve weight loss. Diets include very low calorie, balanced deficit (reduction in protein, fat, and carbohydrates), changes in a specific nutrient (low fat, low carbohydrate, low glycemic index, high protein), and those popularized through publications or commercial weight‐reduction plans. 115 , 116 Meta‐analyses and systematic reviews that compare these various dietary approaches do not favor a specific diet for weight reduction. 117 , 118
The DASH Diet
In management of obesity‐related hypertension, a palatable diet rich in components that may lower BP and low in salt is supported by clinical trials. 119 Such information has been incorporated in the Dietary Approaches to Stop Hypertension (DASH) diet 120 for management of BP, endorsed by the Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation and Treatment of High Blood Pressure (JNC 7). 121 This approach focuses on a “prudent diet” rather than the effects of specific nutrients. Appropriate diets for the management of obesity‐related hypertension are rich in potassium, calcium and magnesium, and fiber and low in salt and saturated fat. In terms of foods, these diets promote consumption of vegetables, fruits, low‐fat dairy products, whole grains, nuts, poultry, and fish and discourage salt, red meats, sweet foods, and sugary drinks. Mediterranean 122 and lactoovovegetarian 123 diets are also associated with benefits in relation to CV risk, weight control, and BP, but vegetarian diets are not widely acceptable.
In US adults, long‐term weight gain was related to foods that are discouraged in a prudent diet, while foods promoted in a DASH‐type diet were associated with better weight control. 105 It is important, then, to consider diets for weight management in obesity‐hypertension in a wider context than energy restriction to ensure an adequate content of foods that may ameliorate BP. 119
Several trials have shown early but not sustained benefits of combining DASH‐type diets with other modification of lifestyle. 124 , 125 , 126 , 127 , 128 One study 129 found significant differences between white, African American, Chinese, and Hispanic groups in conforming to a DASH diet assessed in the Multi‐Ethnic Study of Atherosclerosis (MESA), indicating that responses may improve if ethnicity is taken into account in delivery of lifestyle programs that include DASH guidelines.
The Arthritis, Diet, and Activity Promotion Trial (ADAPT) 130 included a 3‐year follow‐up evaluation of a behaviorally based, multifactorial lifestyle program compared with usual care in overweight or obese individuals being treated with not more than two antihypertensive drugs. The 4‐month intervention promoted weight loss using a low‐sodium DASH‐style diet 131 with the inclusion of at least four fish meals weekly, moderate‐intensity physical activity with increased incidental activity, not more than two standard alcoholic drinks daily, and quitting smoking. In the intervention group, weight loss and reduction in waist girth were significantly greater with the intervention after 4 months and 1 year, but decrease in BP was greater only at 4 months. Improvements in diet, notably in fat, sodium, fish, and vegetables, persisted to 1 year in the intervention group. Two years later, physical activity was greater in the intervention group and some dietary improvements were maintained, but with no significant between‐group difference in weight change or BP.
Low‐Salt Diets
Salt sensitivity is commonly associated with obesity. 132 Salt restriction decreases the risk of hypertension with or without weight loss as well as reducing the incidence of CV events. 133 In the Hypertension Prevention Trial, 134 participants with DBP 78 to 89 mm Hg were followed for 3 years after being randomized to one of five groups: control, decreased energy intake, decreased sodium intake, decreased sodium and energy intake, or decreased sodium and increased potassium intake. BP decreased in all groups, with the greatest decrease in patients assigned to reduced energy only. The groups with reduced sodium intake had a significantly lower rate of hypertension.
In the Trials of Hypertension Prevention (TOHP) phase I study, 135 participants with high normal BP were randomized to one of four groups for 18 months: control, weight loss, sodium restriction, or stress management. In the weight reduction group, weight decreased by 3.9 kg and BP by 2.9/2.3 mm Hg. Sodium restriction resulted in a decrease in BP of 1.7/2.9 mm Hg. Seven years later, the odds ratio for hypertension among 181 participants was lower by 77% with weight loss and 35% with sodium restriction.
Phase II of TOHP examined the effects of weight loss, sodium restriction, or both on BP and the incidence of hypertension. 136 , 137 At 6, 18, and 36 months, weight loss favored the weight reduction intervention over usual care, although weight loss was attenuated over time, and change in BP showed a similar pattern. In the sodium reduction group, a decrease in BP was greater at each time point and also became attenuated with time, from 5.1/4.4 mm Hg at 6 months to 0.7/3.0 mm Hg at 3 years.
The Trial of Nonpharmacologic Interventions in the Elderly (TONE) study 138 investigated weight loss and salt restriction and the need for antihypertensive drugs in treated hypertensive patients during follow‐up to a median of 29 months. Weight reduction, sodium restriction, and the combination of both were compared with usual care in obese participants. The relative hazard ratio was 0.60 for reduced sodium alone, 0.64 for weight loss alone, and 0.47 for the combined intervention. The within‐groups rate of adverse events was similar.
Physical Activity
Aerobic exercise can reduce weight and BP, but when exercise is the only intervention, weight losses are small, with an estimated change of 1.6 kg in moderate‐intensity programs continued for 6 to 12 months. 124 , 139 , 140 In a meta‐analysis that included assessment of ambulatory BP 141 it was reported that in studies lasting 4 to 52 weeks, with physical activity as the only intervention, aerobic exercise reduced BP by 3/2.4 mm Hg. The change affected daytime (3.3/3.5 mm Hg) but not nighttime (0.6/1.0 mm Hg) BP. The effect on BP was independent of the estimated weight loss of 1.2 kg. However, when aerobic exercise is combined with calorie restriction for weight control, the effects on ambulatory BP can be substantial. 142
A few studies 141 also examined the effects of resistance training on BP. The estimated decrease in BP (3.2/3.5 mm Hg) was similar to the effects of aerobic exercise, although not statistically significant for SBP and without statistically significant weight change. A more recent meta‐analysis 143 found that resistance training of at least 4 weeks resulted in an estimated decrease of 3.9/3.9 mm Hg in normotensive or prehypertensive individuals, but a decrease of 4.1/1.5 mm Hg in hypertensive patients was not statistically significant. The place of resistance training in programs for management of hypertension is not established.
Comparison of high‐ and low‐intensity exercise programs with 3.5‐ to 12‐month follow‐up favored the higher‐intensity programs with a difference in weight loss of about 1.5 kg. 144 Higher levels of activity may be difficult to maintain in the long‐term, 145 although 146 the maintenance of 10% weight loss for 2 years in women whose activity increased by 275 minutes per week from baseline values has been reported. Physical activity encourages maintenance of weight loss and offers additional benefits in improving CV risk factors. 146
A systematic review of longitudinal studies of sedentary behaviors in adults found insufficient evidence for an association between sedentary behaviors and measures of adiposity or CV risk factors, including self‐reported hypertension, 147 but there was moderate evidence for an association with type 2 diabetes and strong evidence for an association with all‐cause and CVD mortality. However, longitudinal analysis of data from US adults 105 showed that an increase in time spent in watching television predicted weight gain, possibly mediated by lack of physical activity, adverse food choices, and eating snacks while watching TV. While there is some variation in the relationships identified, evidence points to the need to incorporate measures to modify sedentary behavior in lifestyle intervention programs.
Alcohol
The pressor effect of alcohol has been established in clinical trials, with an estimated increase in SBP of 1 mm per 10 g of alcohol. 148 Paradoxically, drinking alcohol at low to moderate levels is associated with lower risk of atherosclerotic disease. 149 Alcohol provides 29 kJ/g and, although weight gain from excess intake might be expected, meta‐analysis has not shown a consistent relationship between alcohol and weight gain. 150 In US adults, however, increased alcohol intake was associated with greater long‐term weight gain. 105 Moderation of heavier daily alcohol intake to no more than one standard drink in women and two standard drinks in men appears prudent, 119 with potential benefits for both weight gain and BP. In a factorial trial of independent and combined effects of alcohol moderation and weight reduction in overweight and obese hypertensive drinkers, effects on BP were additive over a 3‐month period, with the combined modalities achieving a 14/9 mm Hg BP reduction compared with controls who maintained usual weight and drinking habits. 151
Smoking
Although smokers tend to have lower body weight, they may gain weight because of clustering of adverse health behaviors. 152 Smoking increases BP acutely, with an associated rise in arterial stiffness that lasts longer in hypertensive men. 153 There is an important window of opportunity for lifestyle programs to prevent the weight gain (and BP rise) often seen with smoking cessation. 105
Behavioral Modification Techniques
Behavioral modification techniques are considered an essential part of programs to achieve and maintain weight loss. 154 Social and professional support, goal‐setting, self‐monitoring, stimulus control, changing the environment and problem solving, daily self‐weighing, and prevention of relapse are strategies that have had some success in improving adherence to weight loss programs. 154 , 155 , 156 In the Weight Loss Maintenance Randomized Controlled Trial, 157 improvement in maintenance at 30 months was associated with monthly personal contact with the program staff and regular use of an internet‐based intervention. 158 Contact by telephone, including text messaging, mail, or e‐mail, have been used to maintain contact with staff, 155 but rapid changes in technology with availability of, for example, social networking and applications for mobile phones offer new opportunities. At this time, there is no consensus about the most effective behavioral strategies for lifestyle modification, particularly in the long‐term. Trials that are in progress 154 may clarify the best options for encouraging the maintenance of lifestyle change that is so critical to weight control.
Long‐Term Effects of Lifestyle Interventions on Outcomes
Although limited data are available, a few interventions and meta‐analyses report long‐term benefits associated with improvements in lifestyle, especially in at‐risk populations. In 10‐ to 15‐year follow‐up of prehypertensive adults who took part in the TONE studies, the risk of a CV event was lower by 25% to 30% in those who had been assigned to the salt‐restricted group. 133 In adults at risk for diabetes, a program of diet and physical activity reduced the risk of diabetes by 58%, with size of the decrease related to the extent of change in lifestyle. 159 In adults with impaired glucose tolerance, a program of diet and physical activity was more effective for the prevention of diabetes than either metformin or usual care, with respective incidence rates per 100 person‐years of 4.8, 7.8, and 11. 160
Among the more than 100,000 nonsmokers who completed the Cancer Prevention Study II (CPS‐II) Nutrition Cohort lifestyle questionnaire in 1992–1993, 161 responses were scored with reference to the American Cancer Society lifestyle guidelines for weight control, physical activity, diet, and alcohol. In a 14‐year follow‐up, individuals with the highest scores had a significantly lower risk for mortality from all causes, CVD, and cancer than those with low scores.
Meta‐analyses have shown reduced morbidity and mortality associated with lifestyle interventions used for secondary prevention. 162 , 163 However, authors stress the need for more high‐quality studies of adequate duration. 162 , 163 , 164
Treatment of Hypertension in the Obese
BP Thresholds and Targets
According to JNC 7, normal BP is considered a reading of <120/80 mm Hg averaged over two or more seated BP readings completed during two or more office visits (Table IV). As hypertension progresses, the risk of death from ischemic heart disease or stroke (beginning with a normal BP of 115/75 mm Hg) doubles with each increment of 20 mm Hg in SBP or 10 mm Hg in DBP across the entire BP range from 115/75 mm Hg to 185/115 mm Hg. Individuals in the general population with a SBP of 120 to 139 mm Hg or a DBP of 80 to 89 mm Hg are considered prehypertensive and require lifestyle modification to prevent CVD. Patients with prehypertension are at twice the risk of developing hypertension as those with normal BP. Stage 1 hypertension is defined as SBP between 140 and 159 mm Hg or DBP between 90 and 99 mm Hg, and stage 2 hypertension is defined as SBP ≥160 mm Hg or DBP ≥100 mm Hg. To reduce the incidence of CV and renal complications, target BPs for the general population should be <140/90 mm Hg and for patients with established diabetes or chronic kidney disease <130/80 mm Hg, 121 but there is limited evidence in support of this lower threshold. 165
Table IV.
Antihypertensive Agents
| Target: ≤140/90 |
| Renin‐angiotensin‐aldosterone system inhibition |
| Angiotensin‐converting enzyme inhibitors; angiotensin receptor blockers |
| Diuretics |
| Low‐dose thiazide or thiazide‐like agent |
| Loop diuretics (if required) |
| Potassium–sparing agents |
| Avoid β‐blockers except for specific cardiac indication |
| All agents potentiated by weight loss |
As in individuals with diabetes and chronic kidney disease, many authorities have recommended lower target BPs for obese individuals. This recommendation is partially due to the constellation of risk factors associated with obesity and the metabolic syndrome, and is also attributed to the fact that hypertension in obese patients has proven more difficult to control than hypertension in the nonobese population. In fact, even modest weight loss increases the likelihood of achieving goal BPs. 166 Although logical, there is no strong evidence to support lowering BP much beyond the defined 140/90 mm Hg threshold. 165
Antihypertensive Agents: RAAS Inhibitors
More than 100 medications are available for the direct treatment of hypertension, acting on a variety of systems throughout the body. Although there is no evidence based on longitudinal outcome studies in obese patients, recommendations for the use of specific antihypertensive agents in an obese population have emerged. Angiotensin‐converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), β‐blockers, calcium channel blockers (CCBs), and thiazide diuretics are all effective in lowering BP in most obese patients. 167 Although lowering BP is of paramount importance, studies suggest that antagonizing the RAAS system has special significance in obese patients. 168 , 169 As noted above, angiotensin is overexpressed in obesity, directly contributing to obesity‐related hypertension, making the case to consider ACE inhibitors/ARBs as first‐line agents. In comparison to ARBs or ACE inhibitors, β‐blocker‐ and thiazide‐based regimens increase insulin resistance and are associated with an increase in new cases of diabetes. In contrast, regimens based on RAAS inhibition are associated with significantly fewer cases of new diabetes. 168 , 170 , 171 This is of particular importance in the obese population, a group at heightened risk for the development of type 2 diabetes. 172 In addition, ACE inhibitors and ARBs have not been associated with weight gain or insulin resistance and provide renal protection in diabetes, a highly prevalent disease among obese persons. The Second Australian National Blood Pressure (ANBP2) trial 173 reported slightly better outcomes in hypertensive white men (average BMI 27.4) treated with a regimen that began with an ACE inhibitor compared with a regimen that began with a diuretic. Calcium channel blockers are also effective in the treatment of obesity‐related hypertension and have not been associated with weight gain or adverse changes in lipids. 174 , 175 , 176
Diuretics and β‐Blockers
Although thiazide diuretics are often recommended as first‐line agents for the treatment of hypertension, 177 their known dose‐related side effects, which include dyslipidemia and insulin resistance, are undesirable in obese populations prone to the metabolic syndrome and type 2 diabetes. This causes a therapeutic dilemma since obesity‐related hypertension is salt‐sensitive and diuretics will be required to control BP in most cases. Many experts recommend low‐dose thiazides (12.5 to 25 mg of hydrochlorothiazide or equivalent agent) along with close lipid and glucose monitoring. If greater diuretic effect is required to control BP, the use of loop diuretics and/or the addition of potassium‐sparing agents such as spironolactone, eplerenone, or amiloride should be considered, given the importance of aldosterone in obesity‐related hypertension.
β‐Blockers have been shown to cause insulin resistance and have been closely associated with weight gain and higher body weights, as well as decreased diet‐induced thermogenesis, fat oxidation rate, and weekly habitual activity. 178 , 179 , 180 The use of β‐blockers should be limited to obese patients with specific CV indications such as post–myocardial infarction and HF. When β‐blockers are indicated, agents with a vasodilating component such as carvedilol and nebivolol appear to have less weight gain potential and less of an impact on carbohydrate and lipid metabolism. 179 , 181 Although appropriate in HF, the protective effect of these agents with vasodilator effects post–myocardial infarction has not been definitively established.
Medical Treatment of Obesity in Hypertensive Patients
Lifestyle management is a critical component of the treatment regimen for all obese hypertensive patients (Table V). According to National Institutes of Health guidelines, pharmacotherapy for obesity can be considered if a patient has a BMI ≥30, or a BMI ≥27 if weight‐related comorbidities, including hypertension, type 2 diabetes, dyslipidemia, and/or obstructive sleep apnea, are present. 182 Many therapeutic targets have been recently identified, on both the intake and the expenditure side of the energy balance equation, thereby providing hope that new agents, with limited off‐target effects, may become available in the future.
Table V.
Bariatric Agents
| Approved medications |
| Orlistat (Alli): causes fat malabsorption |
| Acarbose: causes sucrose malabsorption |
| Phenteramine (approved for short‐term use only) |
| ↓ Appetite |
| ↑ Energy expenditure |
| Lorcaserin |
| ↓ Apetite |
| Medications for diabetes that cause weight loss |
| Metformin (insulin sensitizer that ↓ hepatic glucose output and ↓ glucose absorption) |
| Incretin‐based agents that ↑ insulin and ↓ glucagon secretion |
| GLP1 agonists: exenatide, liguratide |
| DPP‐4 inhibitors: sitagliptin; saxagliptin; linagliptin |
| Pramlinatide (amytin congener) |
| Suppresses appetite: delays gastric emptying; increases satiety |
| Combination agents |
| Phentermine/topiramate (Qnexa, Qsymia) |
| Naltrexone/bupropion (investigational) |
Inhibition of Nutrient Absorption
On the intake side, targets include nutrient absorption and appetite suppression. Orlistat induces fat malabsorption by inhibiting pancreatic lipase. Acarbose, both an intestinal alpha‐glucosidase and pancreatic alpha‐amylase inhibitor, blocks the enzymes required to digest complex sugars into absorbable glucose and other monosaccharides. Acarbose has limited use in the United States and Europe in the treatment of diabetes and only a small impact on weight.
Appetite Suppression
Appetite is a critical target for obesity control. Research on the regulation of appetite has identified endogenous systems that both stimulate (orexigenic) and suppress (anorexigenic) food intake, yielding a variety of attractive druggable targets. 183 , 184 Endogenous ligands, such as leptin and serotonin (5HT), work by simultaneously inhibiting the orexigenic centers in the brain and stimulating the anorexigenic pathways. Several appetite suppressants have been associated with significant weight loss in humans but off‐target effects have resulted in the withdrawal of these agents. Fenfluramine, a congener of norepinephrine and amphetamine, releases serotonin from neurons in the hypothalamus and was an effective weight loss agent when combined with phentermine (Fen‐Phen), but the association with pulmonary hypertension, cardiac valvular lesions, and cardiac fibrosis resembling the fibroelastosis seen in the carcinoid syndrome led to its withdrawal from the market in 1997. Sibutramine, a centrally acting uptake inhibitor of norepinephrine and serotonin, was removed from the market in 2010 because of an increase in CV events. The endocannabinoid antagonist rimonabant, which diminished the intake of high fat and sweet foods, was withdrawn because of an increase in depressive symptoms and suicidal ideation. Accordingly, the search for more effective and safer anorexigenic agents is ongoing. The Food and Drug Administration (FDA) has recently approved lorcaserin (Belviq), a selective 5HT2c receptor agonist, which promotes weight loss through satiety.
Increasing Energy Output
In addition to physical activity, stimulating energy output by increasing thermogenesis has emerged as a potential critical target of anti‐obesity therapy. During therapeutic dieting, resting metabolic rate, and therefore energy expenditure, decreases significantly and antagonizes weight loss. Reversing this decrease in metabolic rate would have a substantial effect on weight loss. Recent studies utilizing fluoro‐deoxy‐flucose positron emission tomographic scans have resuscitated brown adipose tissue (BAT) as a druggable therapeutic target for stimulating energy expenditure by increasing metabolic rate. 185 BAT, via SNS activation and a β‐receptor cyclase–linked mechanism, uncouples fatty acid oxidation from ATP formation generating heat rather than stored forms of energy. The demonstration of functional BAT in adult humans raises the possibility that β3‐adrenergic agonists might stimulate energy output; however, none are currently available. Another area of interest is the possibility that white adipose tissue might be converted to BAT, thereby replacing a fuel storage organ with an energy producing one. 186 , 187 While the feasibility of this has been suggested on the basis of animal experiments, proof of principle in humans has not yet been established. Another potential target involves interference with adipose tissue blood flow; however, these studies are in their infancy.
Orlistat (Xenical/Alli)
Orlistat is an inhibitor of gastrointestinal lipases, thereby decreasing the absorption of dietary fat. Roughly 30% of ingested lipids are not absorbed in the presence of orlistat 120 mg, resulting in a caloric deficit that has been shown to produce statistically significant weight loss. Due to its mechanism of action, orlistat induces gastrointestinal side effects if too much dietary fat is consumed. Many long‐term studies have assessed the efficacy of orlistat. In one, 188 the mean placebo‐corrected weight loss in the orlistat group was 2.8 kg while in combination with a hypocaloric diet and lifestyle modification. Statistically significant but small placebo‐corrected reductions in BP, and LDL cholesterol were noted.
A half‐dose (60 mg) formulation of orlistat is now commercially available over the counter and marketed as Alli. 189 This dose has been shown to reduce absorption of ingested fat by approximately 25% as compared with roughly 30% with the 120‐mg dose. 190 The lower dose is only slightly less effective. 191 Nearly two thirds of weight lost with this agent was maintained at the end of 2 years, and the losses remained significantly greater than with placebo. After 2 years of treatment, SBP was significantly reduced only with the higher dose. 192 Orlistat, therefore, induces modest weight loss, along with improved lipid parameters, but has only minimal effects on BP.
Phentermine
Phentermine, an adrenergic agonist, is thought to promote weight loss by activation of the central nervous system and SNS, with a subsequent decrease in food intake and increased resting energy expenditure. 193 Phentermine is FDA approved for the short‐term treatment of obesity (up to 3 months). Given the increased release of norepinephrine promoted by phentermine, it has the potential to raise BP and heart rate. 194 , 195 A meta‐analysis 196 evaluated nine randomized controlled trials of phentermine for weight loss in obese patients. The pooled analysis showed that those treated with phentermine along with diet and lifestyle lost 3.6 kg more than patients treated with diet and lifestyle alone.
Another recent study 197 examined the impact of treatment with phentermine and a low‐carbohydrate diet in obese patients. Weight loss was significantly greater in the phentermine group compared with the placebo group. Additionally, SBP and DBP decreased from baseline in both groups and heart rate did not differ.
Therapies for Comorbid Diseases
Many of the medications available to treat type 2 diabetes are associated with weight gain and increased fat deposition. For overweight and obese patients with diabetes, however, there are FDA‐approved medications that have been associated with weight loss and reduced BP. The effects of these agents on weight, and especially BP, are rather modest but stand in contrast to the gain in weight frequently seen with insulin, thiazolidinediones, and insulin secretagogues.
Metformin
Metformin is an oral antihyperglycemic medication approved as first‐line therapy in the management of type 2 diabetes. It has been shown to promote mild weight loss by decreasing hepatic glucose production, decreasing intestinal absorption of glucose, and improving insulin sensitivity by increasing peripheral glucose uptake and utilization. 198 In the Diabetes Prevention Program trial, 3234 overweight and prediabetic patients were treated with either lifestyle intervention, metformin 850 mg twice a day, or placebo and followed for an average of 2.8 years. During this time, the mean weight loss was 0.1, 2.1, and 5.6 kg in the placebo, metformin, and lifestyle‐intervention groups, respectively. BP decreased in the lifestyle intervention group but not in the metformin group. 160 , 199 , 200
Incretin Therapy (GLP‐1 Agonists and DPP‐4 Inhibitors)
Incretins play an important role in glucose homeostasis by increasing insulin release from pancreatic β‐cells and suppressing the release of glucagon from pancreatic α‐cells. Glucagon‐like peptide 1 (GLP‐1) is a major endogenous incretin, but it is not useful therapeutically because it is rapidly metabolized by dipeptidyl peptidase‐4 (DPP‐4). Exenatide (Byetta) along with its extended‐release form Bydureon liraglutide (Victoza) are injectable GLP‐1 agonists that mimic endogenous GLP‐1 but have a longer duration of action, making them suitable therapies for type 2 diabetes. GLP‐1 agonists enhance glucose‐dependent insulin secretion, suppress inappropriate glucagon secretion (leading to decreased hepatic glucose output and decreased insulin demand), and slow gastric emptying. In addition to improving glycemic control, these GLP‐1 agonists have been shown to decrease food intake and enhance satiety, suggesting a possible role in the treatment of obesity. 201 Three inhibitors of the metabolizing enzyme DPP‐4 that prolong the half‐life of endogenous GLP‐1 are approved for use in the United States: sitagliptin (Januvia), saxagliptin (Onglyza), and linagliptin (Trajenta). These are given orally once per day.
Multiple trials assessing the efficacy of exenatide have demonstrated statistically significant reductions in weight and BP 202 , 203 , 204 , 205 over a prolonged period but the changes in BP were small. 206 , 207 In a comparison of therapy for type 2 diabetes 208 involving exenatide, sitagliptin, and insulin in a large retrospective study, the incretin‐based treatments were associated with weight loss in comparison to small weight gains in the insulin‐treated group. Weight loss was significantly associated with small reductions in both SBP and DBP in all treatment groups. Similar findings were noted with liraglutide, a long‐acting once‐daily GLP‐1 analogue, in comparison with a sulfonylurea or insulin.
Pramlintide
Amylin is a β‐cell polypeptide co‐stored with insulin and co‐released with insulin in response to meals. In diabetic patients, amylin, as well as insulin secretion, is deficient. Pramlintide is a synthetic injectable congener that mimics the actions of amylin. Pramlintide promotes weight loss by a variety of effects that include slowing gastric emptying, increasing satiety, decreasing postprandial glucagon secretion, and centrally decreasing appetite and total caloric intake. 209 Approved doses vary for type 1 diabetes (pramlintide dose: 30–60 μg subcutaneously before meals) and type 2 diabetes (pramlintide dose: 60–120 μg subcutaneously before meals). In type 1 diabetic patients, pramlintide fostered modest weight loss when added to an insulin regimen and was without effect on BP. 210 , 211 Somewhat greater weight loss was noted in type 2 diabetic patients treated with pramlintide. 212
Studies aimed at assessing the weight loss potential of pramlintide in obese patients without type 2 diabetes demonstrated reductions in BP that were directly associated with weight loss. 213 , 214 In one study, 215 placebo‐corrected weight loss at month 12 for the pramlintide 120 μg three times daily group averaged 6.1 kg, with higher doses providing little additional benefit. Furthermore, the pramlintide 120 μg three times daily group demonstrated a significant placebo‐corrected 4.6‐mm Hg reduction in SBP and a trend toward improvement with a placebo‐corrected 2.6‐mm Hg decrease in DBP.
Combination Therapies
Low‐dose, controlled‐release phentermine plus topiramate (Qynexa, Qysmia) has recently been approved for the treatment of obesity. Phentermine has been described above, and topiramate as monotherapy is FDA approved as an anti‐epileptic and for migraine prophylaxis. In the 56‐week phase 3 trial, overweight or obese adults with two or more comorbidities (including hypertension, dyslipidemia, prediabetes, diabetes, and/or abdominal obesity) were randomly assigned to one of two possible doses of Qysmia or placebo. Mean body weight decreased 1.4, 8.1, and 10.2 kg in the placebo, low‐dose, and high‐dose treatment groups, respectively. Moderate decreases in BP accompanied weight loss. 216 This combination was recently approved by the FDA.
A sustained‐release (once‐daily) combination of naltrexone and bupropion (NB) is currently under investigation as a new weight loss therapy. Naltrexone is an opioid antagonist FDA approved for the treatment of opioid addiction and alcohol dependence. Bupropion is a norepinephrine and dopamine reuptake inhibitor FDA approved as an antidepressant (Wellbutrin), and to assist in smoking cessation (Zyban). NB was studied in a 56‐week phase 3 trial in individuals with either uncomplicated obesity (BMI 30–45), or BMI ≥27 to 45 with comorbidities, including hypertension or dyslipidemia. Participants were assigned to receive two possible doses of once‐daily NB or placebo. Mean weight loss was 1.3%, 5%, and 6.1% in the placebo, low‐dose, and high‐dose NB treatment groups, respectively. A small increase in pulse rate and a transient increase in BP was noted in the NB treatment groups. 217
Bariatric Surgery
Bariatric surgeries affect or restrict the flow of food through the gastrointestinal tract. Restrictive surgical procedures, such as laparoscopic‐adjustable gastric banding (LAGB), induce earlier satiety by decreasing the volume of the stomach. The Roux‐en‐Y gastric bypass (RYGB) involves both restriction of the stomach and bypass of the small bowel. The sleeve gastrectomy, in which the fundus of the stomach is removed, is becoming increasingly popular. However, RYGB and LAGB remain the most broadly used surgical treatments for morbid obesity and associated conditions, including obesity‐related hypertension. 218
In an extensive meta‐analysis of 136 studies, Buchwald and colleagues 219 evaluated the impact of bariatric surgery on weight loss and obesity‐related comorbidities including diabetes and hypertension. The mean percentage of excess weight loss ((Initial Weight − Ideal Weight/Ideal Weight) was 61.2% for all patients. Diabetes resolved in 76.8% of patients and resolved or improved in 86.0%, whereas hypertension resolved in 61.7% of patients and resolved or improved in 78.5%.
The Swedish Obese Subjects Study aimed to assess the long‐term benefits of bariatric surgery. The study evaluated obese patients who underwent bariatric surgery at least 2 years (4047 patients) or 10 years (1703 patients) before data analysis as compared with contemporaneously matched, conventionally treated obese control patients. After 2 years, patients’ weight had increased by 0.1% in the control group and had decreased by 23.4% in the bariatric surgery group, whereas after 10 years, weight had increased by 1.6% in the control group and decreased by 16.1% in the bariatric surgery group. At 2 years, the surgery group demonstrated significant decreases in SBP (−4.4 mm Hg) and DBP (−5.2 mm Hg) as compared with increases in SBP (+0.5 mm Hg) and DBP (+0.3 mm Hg) seen in the control group. At 10 years, SBP in the surgery group had increased 0.5 mm Hg from initial baseline, as compared with 4.4 mm Hg in the control group, while DBP remained 2.6 mm Hg below baseline, as compared with 2.0 mm Hg below baseline in the control group. Although recovery from hypertension was more common in the surgery group at both 2 years (21% control vs 34% surgery) and 10 years (11% control vs 19% surgery), no change in hypertension incidence rate was noted between groups at the 2‐ and 10‐year analysis. 220
Subgroup analysis of patients with ischemic heart disease demonstrated both sustained weight loss (surgery: −26.3 kg after 2 years and −17.3 kg after 10 years; control: −2.3 kg after 2 years and −4.3 kg after 10 years) and decreased incidence of hypertension (surgery: −15% after 2 years and −23.1% after 10 years; control: +21.2% after 2 years and 0.0% after 10 years). 221 Thus, bariatric surgery demonstrated a beneficial effect on weight and hypertension in both the short‐ and long‐term.
Summary and Conclusions
Obesity‐related hypertension is an important public health issue. As the prevalence of obesity increases, the prevalence of hypertension with its associated CV risk will increase as well. While primary and even primordial prevention is the long‐term goal for diminishing the prevalence of obesity, control of both obesity and hypertension in the population at risk is the overriding current challenge. Treating hypertension in the obese requires addressing the obesity as part of the therapeutic plan. Lifestyle management is required in every case, with a focus on weight loss and risk reduction. Some have likened the treatment of obesity with caloric restriction alone to the treatment of hypertension with sodium restriction: it works if extreme enough, but it is not a feasible long‐term strategy. In most patients, additional therapies including medications, aggressive diet counseling and behavioral techniques, and sometimes bariatric surgery will be required.
Lessons From the Treatment of Hypertension
Since redundancy is the hallmark of physiological regulation, complex regulatory disturbances such as obesity and hypertension cannot usually be addressed by a single agent. In the treatment of hypertension, agents that block the RAAS, the SNS and renal sodium excretion are frequently required. The need to address multiple targets in the treatment of obesity will likely be required as well. This means agents that reduce appetite as well as agents that increase metabolism will be needed in most patients. In the treatment of hypertension, agents that operate in the periphery, outside the central nervous system, provide BP control with a minimum of off‐target side effects. In the treatment of obesity, peripherally acting agents (β3‐agonists, for example) may be useful at increasing energy expenditure. Drugs that regulate appetite will likely involve the central nervous system. The challenge here will involve precise target localization to avoid more general and unacceptable effects on the brain.
Lifestyle Management
Preventing and managing obesity‐related hypertension requires multiple, parallel efforts with involvement of government, industry, health professionals, and individual self‐care. The importance of increasing physician effectiveness through advocacy has been appropriately emphasized: 222 “Environmental interventions are among the most effective for improving public health. In addition to addressing lifestyle issues in the office, physicians should advocate for environmental approaches at institutional, local, state, and federal levels through speaking, writing, and collaborating with others. In the United States, the timing is right to synergize with efforts such as the White House Task Force on Childhood Obesity and the Surgeon General’s emphasis on changing the national conversation from a negative one about obesity and illness to a positive one about health and fitness.”
Although more research needs to be done to find the most effective ways of helping obese hypertensive patients to sustain lifestyle changes, the first step is for health professionals to understand the options available and to provide clear advice on achievable goals. This needs to be backed up by behavioral modification techniques to assist individuals to sustain weight control and increase fitness. The end objectives are to substantially reduce the risk of CV and obesity‐related metabolic disease, to minimize the need for medications, and to render the required pharmacologic therapy more effective.
Disclosures: This joint position paper also published in Obesity. (Landsberg L, Aronne L, Beilin L, Burke V, Igel L, Lloyd‐Jones D, Sowers, J. Obesity Related Hypertension: Pathogenesis, Cardiovascular Risk, and Treatment. Obesity. 2012;doi:10.1038/oby.20181).
Conflict of Interest: Dr. Landsberg discloses no relevant or financial conflicts of interest. Dr. Sowers discloses participation in 2 NIH grants and is an advisory board member for Merck. Dr. Beilin discloses no relevant or financial conflict of interest. Dr. Burke discloses no relevant or financial conflicts of interest. Dr. Igel discloses no relevant or financial conflicts of interest. Dr. Lloyd‐Jones discloses no relevant or financial conflicts of interest. Dr. Aronne discloses participation in contracted research with Amylin Pharmaceuticals Inc, High Point Pharmaceuticals LLC, Medical University of South Carolina and Novo Nordisk. Dr. Aronne is a consultant or on the Advisory Boards for the following: Amylin Pharmaceuticals Inc, Ethicon Endo‐Surgery Inc, GlaxoSmithKline Consumer Healthcare LP, Novo Nordisk, Orexigen Therapeutics Inc, VIVUS Inc, Takeda Pharmacueticals and Zafgen Inc. Dr. Aronne has ownership interest in Cardiometabolic Support Network LLC and Myos Corporation. No funding or editorial support was provided for this article.
References
- 1. American Heart Association, Overweight and Obesity Statistics – 2009 Update. 2009.
- 2. Flegal KM, Carroll MD, Ogden CL, Curtin LR. Prevalence and trends in obesity among US adults, 1999–2008. JAMA. 2010;303:235–241. [DOI] [PubMed] [Google Scholar]
- 3. Pickering G. High Blood Pressure. New York, NY: Grune and Stratton, Inc; 1968. [Google Scholar]
- 4. Kannel WB, Brand N, Skinner JJ Jr, et al. The relation of adiposity to blood pressure and development of hypertension. The Framingham study. Ann Intern Med. 1967;67:48–59. [DOI] [PubMed] [Google Scholar]
- 5. Vague J. The degree of masculine differentiation of obesities: a factor determining predisposition to diabetes, atherosclerosis, gout, and uric calculous disease. Am J Clin Nutr. 1956;4:20–34. [DOI] [PubMed] [Google Scholar]
- 6. Lapidus L, Bengtsson C, Larsson B, et al. Distribution of adipose tissue and risk of cardiovascular disease and death: a 12 year follow up of participants in the population study of women in Gothenburg, Sweden. Br Med J (Clin Res Ed). 1984;289:1257–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Larsson B, Svardsudd K, Welin L, et al. Abdominal adipose tissue distribution, obesity, and risk of cardiovascular disease and death: 13 year follow up of participants in the study of men born in 1913. Br Med J (Clin Res Ed). 1984;288:1401–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cassano PA, Segal MR, Vokonas PS, Weiss ST. Body fat distribution, blood pressure, and hypertension. A prospective cohort study of men in the normative aging study. Ann Epidemiol. 1990;1:33–48. [DOI] [PubMed] [Google Scholar]
- 9. Kissebah AH, Vydelingum N, Murray R, et al. Relation of body fat distribution to metabolic complications of obesity. J Clin Endocrinol Metab. 1982;54:254–260. [DOI] [PubMed] [Google Scholar]
- 10. Krotkiewski M, Bjorntorp P, Sjostrom L, Smith U. Impact of obesity on metabolism in men and women. Importance of regional adipose tissue distribution. J Clin Invest. 1983;72:1150–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kalkhoff RK, Hartz AH, Rupley D, et al. Relationship of body fat distribution to blood pressure, carbohydrate tolerance, and plasma lipids in healthy obese women. J Lab Clin Med. 1983;102:621–627. [PubMed] [Google Scholar]
- 12. Ferrannini E, Buzzigoli G, Bonadonna R, et al. Insulin resistance in essential hypertension. N Engl J Med. 1987;317:350–357. [DOI] [PubMed] [Google Scholar]
- 13. Modan M, Halkin H, Almog S, et al. Hyperinsulinemia. A link between hypertension obesity and glucose intolerance. J Clin Invest. 1985;75:809–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Landsberg L. The metabolic syndrome: diabetes, obesity and hypertension. In: Birkenager WH, Robertson JIS, Zanchetti A, eds. Handbook of Hypertension. Hypertension in the Twentieth Century: Concepts and Achievements. Vol 22. Elsevier: Amsterdam; 2004:245–261. [Google Scholar]
- 15. Landsberg L. Insulin‐mediated sympathetic stimulation: role in the pathogenesis of obesity‐related hypertension (or, how insulin affects blood pressure, and why). J Hypertens. 2001;19(3 Pt 2):523–528. [DOI] [PubMed] [Google Scholar]
- 16. Rowe JW, Young JB, Minaker KL, et al. Effect of insulin and glucose infusions on sympathetic nervous system activity in normal man. Diabetes. 1981;30:219–225. [DOI] [PubMed] [Google Scholar]
- 17. Hausberg M, Mark AL, Hoffman RP, et al. Dissociation of sympathoexcitatory and vasodilator actions of modestly elevated plasma insulin levels. J Hypertens. 1995;13:1015–1021. [DOI] [PubMed] [Google Scholar]
- 18. Troisi RJ, Weiss ST, Parker DR, et al. Relation of obesity and diet to sympathetic nervous system activity. Hypertension. 1991;17:669–677. [DOI] [PubMed] [Google Scholar]
- 19. Grassi G, Seravalle G, Cattaneo BM, et al. Sympathetic activation in obese normotensive subjects. Hypertension. 1995;25:560–563. [DOI] [PubMed] [Google Scholar]
- 20. Scherrer U, Randin D, Tappy L, et al. Body fat and sympathetic nerve activity in healthy subjects. Circulation. 1994;89:2634–2640. [DOI] [PubMed] [Google Scholar]
- 21. Grassi G, Seravalle G, Colombo M, et al. Body weight reduction, sympathetic nerve traffic, and arterial baroreflex in obese normotensive humans. Circulation. 1998;97:2037–2042. [DOI] [PubMed] [Google Scholar]
- 22. DeFronzo RA. Insulin and renal sodium handling: clinical implications. Int J Obes (Lond). 1981;5(suppl 1):93–104. [PubMed] [Google Scholar]
- 23. Kennedy A, Gettys TW, Watson P, et al. The metabolic significance of leptin in humans: gender‐based differences in relationship to adiposity, insulin sensitivity, and energy expenditure. J Clin Endocrinol Metab. 1997;82:1293–1300. [DOI] [PubMed] [Google Scholar]
- 24. Tang‐Christensen M, Havel PJ, Jacobs RR, et al. Central administration of leptin inhibits food intake and activates the sympathetic nervous system in rhesus macaques. J Clin Endocrinol Metab. 1999;84:711–717. [DOI] [PubMed] [Google Scholar]
- 25. Haynes WG, Morgan DA, Walsh SA, et al. Receptor‐mediated regional sympathetic nerve activation by leptin. J Clin Invest. 1997;100:270–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mantzoros CS. The role of leptin in human obesity and disease: a review of current evidence. Ann Intern Med. 1999;130:671–680. [DOI] [PubMed] [Google Scholar]
- 27. Shek EW, Brands MW, Hall JE. Chronic leptin infusion increases arterial pressure. Hypertension. 1998;31:409–414. [DOI] [PubMed] [Google Scholar]
- 28. Dunbar JC, Hu Y, Lu H. Intracerebroventricular leptin increases lumbar and renal sympathetic nerve activity and blood pressure in normal rats. Diabetes. 1997;46:2040–2043. [DOI] [PubMed] [Google Scholar]
- 29. Agata J, Masuda A, Takada M, et al. High plasma immunoreactive leptin level in essential hypertension. Am J Hypertens. 1997;10(10 Pt 1):1171–1174. [DOI] [PubMed] [Google Scholar]
- 30. Kazumi T, Kawaguchi A, Katoh J, et al. Fasting insulin and leptin serum levels are associated with systolic blood pressure independent of percentage body fat and body mass index. J Hypertens. 1999;17:1451–1455. [DOI] [PubMed] [Google Scholar]
- 31. Landsberg L. Diet, obesity and hypertension: an hypothesis involving insulin, the sympathetic nervous system, and adaptive thermogenesis. Q J Med. 1986;61:1081–1090. [PubMed] [Google Scholar]
- 32. Engeli S, Bohnke J, Gorzelniak K, et al. Weight loss and the renin‐angiotensin‐aldosterone system. Hypertension. 2005;45:356–362. [DOI] [PubMed] [Google Scholar]
- 33. Goodfriend TL. Obesity, sleep apnea, aldosterone, and hypertension. Curr Hypertens Rep. 2008;10:222–226. [DOI] [PubMed] [Google Scholar]
- 34. Sarzani R, Salvi F, Dessi‐Fulgheri P, Rappelli A. Renin‐angiotensin system, natriuretic peptides, obesity, metabolic syndrome, and hypertension: an integrated view in humans. J Hypertens. 2008;26:831–843. [DOI] [PubMed] [Google Scholar]
- 35. Bomback AS, Klemmer PJ. Interaction of aldosterone and extracellular volume in the pathogenesis of obesity‐associated kidney disease: a narrative review. Am J Nephrol. 2009;30:140–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bogaert YE, Linas S. The role of obesity in the pathogenesis of hypertension. Nat Clin Pract Nephrol. 2009;5:101–111. [DOI] [PubMed] [Google Scholar]
- 37. Ahmed SB, Fisher ND, Stevanovic R, Hollenberg NK. Body mass index and angiotensin‐dependent control of the renal circulation in healthy humans. Hypertension. 2005;46:1316–1320. [DOI] [PubMed] [Google Scholar]
- 38. Hall JE, Guyton AC, Coleman TG, et al. Regulation of arterial pressure: role of pressure natriuresis and diuresis. Fed Proc. 1986;45:2897–2903. [PubMed] [Google Scholar]
- 39. Rocchini AP, Key J, Bondie D, et al. The effect of weight loss on the sensitivity of blood pressure to sodium in obese adolescents. N Engl J Med. 1989;321:580–585. [DOI] [PubMed] [Google Scholar]
- 40. Adamczak M, Wiecek A, Funahashi T, et al. Decreased plasma adiponectin concentration in patients with essential hypertension. Am J Hypertens. 2003;16:72–75. [DOI] [PubMed] [Google Scholar]
- 41. Van Cauter SK. Sleep as a mediator of the relationship between socioeconomic status and health: a hypothesis. Annals N Y Acad Sci. 1999;896:254–261. [DOI] [PubMed] [Google Scholar]
- 42. Narkiewicz K, Somers VK. Sympathetic nerve activity in obstructive sleep apnoea. Acta Physiol Scand. 2003;177:385–390. [DOI] [PubMed] [Google Scholar]
- 43. Wolk R, Shamsuzzaman AS, Somers VK. Obesity, sleep apnea, and hypertension. Hypertension. 2003;42:1067–1074. [DOI] [PubMed] [Google Scholar]
- 44. Spiegel K, Leproult R, L’Hermite‐Baleriaux M, et al. Leptin levels are dependent on sleep duration: relationships with sympathovagal balance, carbohydrate regulation, cortisol, and thyrotropin. J Clin Endocrinol Metab. 2004;89:5762–5771. [DOI] [PubMed] [Google Scholar]
- 45. Barker DJ. Fetal programming of coronary heart disease. Trends Endocrinol Metab. 2002;13:364–368. [DOI] [PubMed] [Google Scholar]
- 46. Silverman BL, Landsberg L, Metzger BE. Fetal hyperinsulinism in offspring of diabetic mothers. Association with the subsequent development of childhood obesity. Ann N Y Acad Sci. 1993;699:36–45. [DOI] [PubMed] [Google Scholar]
- 47. Hausberg M, Barenbrock M, Kosch M. Elevated sympathetic nerve activity: the link between low birth size and adult‐onset metabolic syndrome? J Hypertens. 2004;22:1087–1089. [DOI] [PubMed] [Google Scholar]
- 48. Caballero AE. Endothelial dysfunction in obesity and insulin resistance: a road to diabetes and heart disease. Obes Res. 2003;11:1278–1289. [DOI] [PubMed] [Google Scholar]
- 49. Barrett EJ, Eggleston EM, Inyard AC, et al. The vascular actions of insulin control its delivery to muscle and regulate the rate‐limiting step in skeletal muscle insulin action. Diabetologia. 2009;52:752–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gillespie C, Kuklina EV, Briss PA, et al. Vital signs: prevalence, treatment, and control of hypertension: United States, 1999–2002 and 2005–2008. MMWR Morb Mortal Wkly Rep. 2011;60:103–108. [PubMed] [Google Scholar]
- 51. Roger VL, Go AS, Lloyd‐Jones DM, et al. Heart disease and stroke statistics – 2011 update: a report from the American Heart Association. Circulation. 2011;123:e18–e209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang Y, Wang QJ. The prevalence of prehypertension and hypertension among US adults according to the new Joint National Committee guidelines. Arch Intern Med. 2004;164:2126–2134. [DOI] [PubMed] [Google Scholar]
- 53. Wilson PWF, D’Agostino RB, Sullivan L, et al. Overweight and obesity as determinants of cardiovascular risk: the Framingham experience. Arch Intern Med. 2002;162:1867–1872. [DOI] [PubMed] [Google Scholar]
- 54. Burt VL, Whelton P, Roccella EJ, et al. Prevalence of hypertension in the US adult population: results from the Third National Health and Nutrition Examination Survey, 1988–1991. Hypertension. 1995;25:305–313. [DOI] [PubMed] [Google Scholar]
- 55. Franklin SS, Gustin W, Wong ND, et al. Hemodynamic patterns of age‐related changes in blood pressure. The Framingham Heart Study. Circulation. 1997;96:308–315. [DOI] [PubMed] [Google Scholar]
- 56. Lloyd‐Jones DM, Liu K, Colangelo LA, et al. Consistently stable or decreased body mass index in young adulthood and longitudinal changes in metabolic syndrome components: the Coronary Artery Risk Development in Young Adults Study. Circulation. 2007;115:1004–1011. [DOI] [PubMed] [Google Scholar]
- 57. Mamun AA, Lawlor DA, O’Callaghan MJ, et al. Effect of body mass index changes between ages 5 and 14 on blood pressure at age 14: findings from a birth cohort study. Hypertension. 2005;45:1083–1087. [DOI] [PubMed] [Google Scholar]
- 58. Collaboration PS. Age‐specific relevance of usual blood pressure to vascular mortality: a meta‐analysis of individual data for one million adults in 61 prospective studies. Lancet. 2002;360:1903–1913. [DOI] [PubMed] [Google Scholar]
- 59. Neaton JD, Kuller L, Stamler J, Wentworth DN. Impact of systolic and diastolic blood pressure on cardiovascular mortality. In: Laragh JH, Brenner BM, eds. Hypertension: Pathophysiology, Diagnosis, and Management, 2nd ed. New York, NY: Raven Press; 1995:127–144. [Google Scholar]
- 60. Stamler J, Stamler R, Neaton JD. Blood pressure, systolic and diastolic, and cardiovascular risks. US population data. Arch Intern Med. 1993;153:598–615. [DOI] [PubMed] [Google Scholar]
- 61. Lloyd‐Jones DM, Evans JC, Levy D. Hypertension in adults across the age spectrum: current outcomes and control in the community. JAMA. 2005;294:466–472. [DOI] [PubMed] [Google Scholar]
- 62. Lloyd‐Jones DM, Evans JC, Larson MG, et al. Cross‐classification of JNC VI blood pressure stages and risk groups in the Framingham Heart Study. Arch Intern Med. 1999;159:2206–2212. [DOI] [PubMed] [Google Scholar]
- 63. D’Agostino RB, Grundy SM, Sullivan LM, et al. Validation of the Framingham coronary heart disease prediction scores: results of a multiple ethnic groups investigation. JAMA. 2001;286:180–187. [DOI] [PubMed] [Google Scholar]
- 64. Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham Study. Stroke. 1991;22:8:983‐8. [DOI] [PubMed] [Google Scholar]
- 65. Seshadri S, Beiser A, Kelly‐Hayes M, et al. The lifetime risk of stroke: estimates from the Framingham Study. Stroke. 2006;37:345–350. [DOI] [PubMed] [Google Scholar]
- 66. Wolf P, D’Agostino R, Belanger A, Kannel W. Probability of stroke: a risk profile from the Framingham Study. Stroke. 1991;22:312–318. [DOI] [PubMed] [Google Scholar]
- 67. Lloyd‐Jones DM, Larson MG, Leip EP, et al. Lifetime risk for developing congestive heart failure: the Framingham Heart Study. Circulation. 2002;106:3068–3072. [DOI] [PubMed] [Google Scholar]
- 68. Levy D, Larson MG, Vasan RS, et al. The progression from hypertension to congestive heart failure. JAMA. 1996;275:1557–1562. [PubMed] [Google Scholar]
- 69. Hsu C‐Y, McCulloch CE, Darbinian J, et al. Elevated blood pressure and risk of end‐stage renal disease in subjects without baseline kidney disease. Arch Intern Med. 2005;165:923–928. [DOI] [PubMed] [Google Scholar]
- 70. Hsu CY, McCulloch CE, Iribarren C, et al. Body mass index and risk for end‐stage renal disease. Ann Intern Med. 2006;144:21–28. [DOI] [PubMed] [Google Scholar]
- 71. Stamler J, Dyer AR, Shekelle RB, et al. Relationship of baseline major risk factors to coronary and all‐cause mortality, and to longevity: findings from long‐term follow‐up of Chicago cohorts. Cardiology. 1993;82:191–222. [DOI] [PubMed] [Google Scholar]
- 72. Sever PS, Dahlof B, Poulter NR, et al. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower‐than‐average cholesterol concentrations, in the Anglo‐Scandinavian Cardiac Outcomes Trial – Lipid Lowering Arm (ASCOT‐LLA): a multicentre randomised controlled trial. Lancet. 2003;361:1149–1158. [DOI] [PubMed] [Google Scholar]
- 73. Hansson L, Zanchetti A, Carruthers SG, et al. Effects of intensive blood‐pressure lowering and low‐dose aspirin in patients with hypertension: principal results of the Hypertension Optimal Treatment (HOT) randomised trial. Lancet. 1998;351:1755–1762. [DOI] [PubMed] [Google Scholar]
- 74. Haffner SM, Lehto S, Ronnemaa T, et al. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med. 1998;339:229–234. [DOI] [PubMed] [Google Scholar]
- 75. Juutilainen A, Lehto S, Ronnemaa T, et al. Type 2 diabetes as a “coronary heart disease equivalent”: an 18‐year prospective population‐based study in Finnish subjects. Diabetes Care. 2005;28:2901–2907. [DOI] [PubMed] [Google Scholar]
- 76. Hansen BC, Bray GA. The Metabolic Syndrome Epidemiology, Clinical Treatment, and Underlying Mechanisms. Totowa, NJ: Humana Press; 2008. [Google Scholar]
- 77. Reaven GM. Metabolic Syndrome To Be or Not to Be? In: Hansen BC, Bray GA, eds. The Metabolic Syndrome Epidemiology, Clinical Treatment, and Underlying Mechanisms. Totowa, NJ: Humana Press; 2008:11–36. [Google Scholar]
- 78. Ervin R. Prevalence of metabolic syndrome among adults 20 years of age and over, by sex, age, race and ethnicity, and body mass index: United States, 2003–2006. Natl Health Stat Reports. 2009;13:1–8. [PubMed] [Google Scholar]
- 79. Grundy SM, Brewer HB Jr, Cleeman JI, et al. Definition of metabolic syndrome: report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition. Circulation. 2004;109:433–438. [DOI] [PubMed] [Google Scholar]
- 80. Sanchez‐Lozada LG, Tapia E, Jimenez A, et al. Fructose‐induced metabolic syndrome is associated with glomerular hypertension and renal microvascular damage in rats. Am J Physiol Renal Physiol. 2007;292:F423–F429. [DOI] [PubMed] [Google Scholar]
- 81. Lee JE, Kim YG, Choi YH, et al. Serum uric acid is associated with microalbuminuria in prehypertension. Hypertension. 2006;47:962–967. [DOI] [PubMed] [Google Scholar]
- 82. Johnson RJ, Segal MS, Sautin Y, et al. Potential role of sugar (fructose) in the epidemic of hypertension, obesity and the metabolic syndrome, diabetes, kidney disease, and cardiovascular disease. Am J Clin Nutr. 2007;86:899–906. [DOI] [PubMed] [Google Scholar]
- 83. Choi J. Sugar‐sweetened soft drinks, diet soft drinks, and serum uric acid level: the Third National Health and Nutrition Examination Survey. Arthritis Rheum. 2008;59:109–116. [DOI] [PubMed] [Google Scholar]
- 84. Spiegel K, Leproult R, Van Cauter E. Impact of sleep debt on metabolic and endocrine function. Lancet. 1999;354:1435–1439. [DOI] [PubMed] [Google Scholar]
- 85. Bass J, Turek FW. Sleepless in America: a pathway to obesity and the metabolic syndrome? Arch Intern Med. 2005;165:15–16. [DOI] [PubMed] [Google Scholar]
- 86. Turek FW, Joshu C, Kohsaka A, et al. Obesity and metabolic syndrome in circadian Clock mutant mice. Science. 2005;308:1043–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Nedeltcheva AV, Kilkus JM, Imperial J, et al. Insufficient sleep undermines dietary efforts to reduce adiposity. Ann Intern Med. 2010;153:435–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. l’Allemand‐Jander D. Clinical diagnosis of metabolic and cardiovascular risks in overweight children: early development of chronic diseases in the obese child. Int J Obes (Lond). 2010;34(suppl 2):S32–S36. [DOI] [PubMed] [Google Scholar]
- 89. Srinivasan SR, Myers L, Berenson GS. Changes in metabolic syndrome variables since childhood in prehypertensive and hypertensive subjects: the Bogalusa Heart Study. Hypertension. 2006;48:33–39. [DOI] [PubMed] [Google Scholar]
- 90. Juonala M, Magnussen CG, Berenson GS, et al. Childhood adiposity, adult adiposity, and cardiovascular risk factors. N Engl J Med. 2011;365:1876–1885. [DOI] [PubMed] [Google Scholar]
- 91. Craigie AM, Lake AA, Kelly SA, et al. Tracking of obesity‐related behaviours from childhood to adulthood: a systematic review. Maturitas. 2011;70:266–284. [DOI] [PubMed] [Google Scholar]
- 92. Eriksson J, Forsen T, Tuomilehto J, et al. Fetal and childhood growth and hypertension in adult life. Hypertension. 2000;36:790–794. [DOI] [PubMed] [Google Scholar]
- 93. Huang RC, Mori TA, Burke V, et al. Synergy between adiposity, insulin resistance, metabolic risk factors, and inflammation in adolescents. Diabetes Care. 2009;32:695–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Huang RC, Burke V, Newnham JP, et al. Perinatal and childhood origins of cardiovascular disease. Int J Obes (Lond). 2007;31:236–244. [DOI] [PubMed] [Google Scholar]
- 95. Chivers PT, Hands B, Parker H, et al. Longitudinal modelling of body mass index from birth to 14 years. Obes Facts. 2000;2:302–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Waters E, de Silva‐Sanigorski A, Hall BJ, et al. Interventions for preventing obesity in children. Cochrane Database Syst Rev. 2011;12:CD001871. [DOI] [PubMed] [Google Scholar]
- 97. Vandongen R, Jenner DA, Thompson C, et al. A controlled evaluation of a fitness and nutrition intervention program on cardiovascular health in 10‐ to 12‐year‐old children. Prev Med. 1995;24:9–22. [DOI] [PubMed] [Google Scholar]
- 98. Burke V, Milligan RA, Thompson C, et al. A controlled trial of health promotion programs in 11‐year‐olds using physical activity “enrichment” for higher risk children. J Pediatr. 1998;132:840–848. [DOI] [PubMed] [Google Scholar]
- 99. Milligan RA, Thompson C, Vandongen R, et al. Clustering of cardiovascular risk factors in Australian adolescents: association with dietary excesses and deficiencies. J Cardiovasc Risk. 1995;2:515–523. [PubMed] [Google Scholar]
- 100. Taylor BJ, Heath AL, Galland BC, et al. Prevention of Overweight in Infancy (POI.nz) study: a randomised controlled trial of sleep, food and activity interventions for preventing overweight from birth. BMC Public Health. 2011;11:942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Berenson GS. Cardiovascular health promotion for children: a model for a Parish (County)‐wide program (implementation and preliminary results). Prev Cardiol. 2010;13:23–28. [DOI] [PubMed] [Google Scholar]
- 102. Sobko T, Svensson SV, Ek A, et al. A randomised controlled trial for overweight and obese parents to prevent childhood obesity‐Early STOPP (Stockholm Obesity Prevention Program). BMC Public Health. 2011;11:336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Christie D, Hudson L, Mathiot A, et al. Assessing the efficacy of the healthy eating and lifestyle programme (HELP) compared with enhanced standard care of the obese adolescent in the community: study protocol for a randomized controlled trial. Trials. 2011;12:242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Borys JM, Le Bodo Y, Jebb SA, et al. EPODE approach for childhood obesity prevention: methods, progress and international development. Obes Rev. 2011;13:299–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Mozaffarian D, Hao T, Rimm EB, et al. Changes in diet and lifestyle and long‐term weight gain in women and men. N Engl J Med. 2011;364:2392–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Wang Y, Beydoun MA. The obesity epidemic in the United States – gender, age, socioeconomic, racial/ethnic, and geographic characteristics: a systematic review and meta‐regression analysis. Epidemiol Rev. 2007;29:6–28. [DOI] [PubMed] [Google Scholar]
- 107. Popkin BM, Adair LS, Ng SW. Global nutrition transition and the pandemic of obesity in developing countries. Nutr Rev. 2011;70:3–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Zhai F, Wang H, Wang Z, et al. Closing the energy gap to prevent weight gain in China. Obes Rev. 2008;9(suppl 1):107–112. [DOI] [PubMed] [Google Scholar]
- 109. Akers JD, Estabrooks PA, Davy BM. Translational research: bridging the gap between long‐term weight loss maintenance research and practice. J Am Diet Assoc. 2010;110:1511–1522, 22 e1‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Anderson SE, Whitaker RC. Prevalence of obesity among US preschool children in different racial and ethnic groups. Arch Pediatr Adolesc Med. 2009;163:344–348. [DOI] [PubMed] [Google Scholar]
- 111. Neter JE, Stam BE, Kok FJ, et al. Influence of weight reduction on blood pressure: a meta‐analysis of randomized controlled trials. Hypertension. 2003;42:878–884. [DOI] [PubMed] [Google Scholar]
- 112. Aucott L, Poobalan A, Smith WC, et al. Effects of weight loss in overweight/obese individuals and long‐term hypertension outcomes: a systematic review. Hypertension. 2005;45:1035–1041. [DOI] [PubMed] [Google Scholar]
- 113. Aucott L, Rothnie H, McIntyre L, et al. Long‐term weight loss from lifestyle intervention benefits blood pressure?: a systematic review. Hypertension. 2009;54:756–762. [DOI] [PubMed] [Google Scholar]
- 114. Siebenhofer A, Jeitler K, Berghold A, et al. Long‐term effects of weight‐reducing diets in hypertensive patients. Cochrane Database Syst Rev. 2011;9:CD008274. [DOI] [PubMed] [Google Scholar]
- 115. Bray GA. Lifestyle and pharmacological approaches to weight loss: efficacy and safety. J Clin Endocrinol Metab. 2008;93(11 suppl 1):S81–S88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Makris A, Foster GD. Dietary approaches to the treatment of obesity. Psychiatr Clin North Am. 2011;34:813–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Avenell A, Broom J, Brown TJ, et al. Systematic review of the long‐term effects and economic consequences of treatments for obesity and implications for health improvement. Health Technol Assess. 2004;8:iii–iv, 1–182. [DOI] [PubMed] [Google Scholar]
- 118. Nordmann AJ, Nordmann A, Briel M, et al. Effects of low‐carbohydrate vs low‐fat diets on weight loss and cardiovascular risk factors: a meta‐analysis of randomized controlled trials. Arch Intern Med. 2006;166:285–293. [DOI] [PubMed] [Google Scholar]
- 119. Appel LJ. ASH position paper: dietary approaches to lower blood pressure. J Am Soc Hypertens. 2009;3:321–331. [DOI] [PubMed] [Google Scholar]
- 120. Appel LJ, Moore TJ, Obarzanek E, et al. A clinical trial of the effects of dietary patterns on blood pressure. DASH Collaborative Research Group. N Engl J Med. 1997;336:1117–1124. [DOI] [PubMed] [Google Scholar]
- 121. Joint National Committee on Prevention E, and Treatment of High Blood Pressure . The Seventh Report of the Joint National Committee on Prevention, Evaluation, and Treatment of High Blood Pressure. Bethesda, MD: US Department of Health and Human Services; 2004. NIH publication 04‐5230. [Google Scholar]
- 122. Kastorini CM, Milionis HJ, Esposito K, et al. The effect of Mediterranean diet on metabolic syndrome and its components: a meta‐analysis of 50 studies and 534,906 individuals. J Am Coll Cardiol. 2011;57:1299–1313. [DOI] [PubMed] [Google Scholar]
- 123. Beilin LJ, Burke V. Vegetarian diet components, protein and blood pressure: which nutrients are important? Clin Exp Pharmacol Physiol. 1995;22:195–198. [DOI] [PubMed] [Google Scholar]
- 124. Miller ER 3rd, Erlinger TP, Young DR, et al. Results of the Diet, Exercise, and Weight Loss Intervention Trial (DEW‐IT). Hypertension. 2002;40:612–618. [DOI] [PubMed] [Google Scholar]
- 125. Elmer PJ, Obarzanek E, Vollmer WM, et al. Effects of comprehensive lifestyle modification on diet, weight, physical fitness, and blood pressure control: 18‐month results of a randomized trial. Ann Intern Med. 2006;144:485–495. [DOI] [PubMed] [Google Scholar]
- 126. Jehn ML, Patt MR, Appel LJ, Miller ER 3rd. One year follow‐up of overweight and obese hypertensive adults following intensive lifestyle therapy. J Hum Nutr Diet. 2006;19:349–354. [DOI] [PubMed] [Google Scholar]
- 127. Lien LF, Brown AJ, Ard JD, et al. Effects of PREMIER lifestyle modifications on participants with and without the metabolic syndrome. Hypertension. 2007;50:609–616. [DOI] [PubMed] [Google Scholar]
- 128. Blumenthal JA, Babyak MA, Hinderliter A, et al. Effects of the DASH diet alone and in combination with exercise and weight loss on blood pressure and cardiovascular biomarkers in men and women with high blood pressure: the ENCORE study. Arch Intern Med. 2010;170:126–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Gao SK, Fitzpatrick AL, Psaty B, et al. Suboptimal nutritional intake for hypertension control in 4 ethnic groups. Arch Intern Med. 2009;169:702–707. [DOI] [PubMed] [Google Scholar]
- 130. Burke V, Beilin LJ, Cutt HE, et al. Effects of a lifestyle programme on ambulatory blood pressure and drug dosage in treated hypertensive patients: a randomized controlled trial. J Hypertens. 2005;23:1241–1249. [DOI] [PubMed] [Google Scholar]
- 131. Sacks FM, Svetkey LP, Vollmer WM, et al. Effects on blood pressure of reduced dietary sodium and the Dietary Approaches to Stop Hypertension (DASH) diet. DASH‐Sodium Collaborative Research Group. N Engl J Med. 2001;344:3–10. [DOI] [PubMed] [Google Scholar]
- 132. Fujita T. Mineralocorticoid receptors, salt‐sensitive hypertension, and metabolic syndrome. Hypertension. 2010;55:813–818. [DOI] [PubMed] [Google Scholar]
- 133. Cook NR, Cutler JA, Obarzanek E, et al. Long term effects of dietary sodium reduction on cardiovascular disease outcomes: observational follow‐up of the trials of hypertension prevention (TOHP). BMJ. 2007;334:885–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. The Hypertension Prevention Trial: three‐year effects of dietary changes on blood pressure. Hypertension Prevention Trial Research Group. Arch Intern Med. 1990;150:153–162. [PubMed] [Google Scholar]
- 135. Whelton PK, Kumanyika SK, Cook NR, et al. Efficacy of nonpharmacologic interventions in adults with high‐normal blood pressure: results from phase 1 of the Trials of Hypertension Prevention. Trials of Hypertension Prevention Collaborative Research Group. Am J Clin Nutr. 1997;2(suppl):652S–660S. [DOI] [PubMed] [Google Scholar]
- 136. Stevens VJ, Obarzanek E, Cook NR, et al. Long‐term weight loss and changes in blood pressure: results of the Trials of Hypertension Prevention, phase II. Ann Intern Med. 2001;134:1–11. [DOI] [PubMed] [Google Scholar]
- 137. Kumanyika SK, Cook NR, Cutler JA, et al. Sodium reduction for hypertension prevention in overweight adults: further results from the Trials of Hypertension Prevention Phase II. J Hum Hypertens. 2005;19:33–45. [DOI] [PubMed] [Google Scholar]
- 138. Appel LJ, Espeland MA, Easter L, et al. Effects of reduced sodium intake on hypertension control in older individuals: results from the Trial of Nonpharmacologic Interventions in the Elderly (TONE). Arch Intern Med. 2001;161:685–693. [DOI] [PubMed] [Google Scholar]
- 139. Thorogood A, Mottillo S, Shimony A, et al. Isolated aerobic exercise and weight loss: a systematic review and meta‐analysis of randomized controlled trials. Am J Med. 2011;124:747–755. [DOI] [PubMed] [Google Scholar]
- 140. Douketis JD, Macie C, Thabane L, Williamson DF. Systematic review of long‐term weight loss studies in obese adults: clinical significance and applicability to clinical practice. Int J Obes (Lond). 2005;29:1153–1167. [DOI] [PubMed] [Google Scholar]
- 141. Fagard RH. Exercise is good for your blood pressure: effects of endurance training and resistance training. Clin Exp Pharmacol Physiol. 2006;33:853–856. [DOI] [PubMed] [Google Scholar]
- 142. Cox KL, Puddey IB, Morton AR, et al. Exercise and weight control in sedentary overweight men: effects on clinic and ambulatory blood pressure. J Hypertens. 1996;14:779–790. [DOI] [PubMed] [Google Scholar]
- 143. Cornelissen VA, Fagard RH, Coeckelberghs E, Vanhees L. Impact of resistance training on blood pressure and other cardiovascular risk factors: a meta‐analysis of randomized, controlled trials. Hypertension. 2011;58:950–958. [DOI] [PubMed] [Google Scholar]
- 144. Shaw K, Gennat H, O’Rourke P, Del Mar C. Exercise for overweight or obesity. Cochrane Database Syst Rev. 2006;CD003817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Fogelholm M, Kukkonen‐Harjula K. Does physical activity prevent weight gain – a systematic review. Obes Rev. 2000;1:95–111. [DOI] [PubMed] [Google Scholar]
- 146. Jakicic JM. The effect of physical activity on body weight. Obesity (Silver Spring). 2009;17(suppl 3):S34–S38. [DOI] [PubMed] [Google Scholar]
- 147. Proper KI, Singh AS, van Mechelen W, Chinapaw MJ. Sedentary behaviors and health outcomes among adults: a systematic review of prospective studies. Am J Prev Med. 2011;40:174–182. [DOI] [PubMed] [Google Scholar]
- 148. Puddey IB, Beilin LJ. Alcohol is bad for blood pressure. Clin Exp Pharmacol Physiol. 2006;33:847–852. [DOI] [PubMed] [Google Scholar]
- 149. Costanzo S, Di Castelnuovo A, Donati MB, et al. Wine, beer or spirit drinking in relation to fatal and non‐fatal cardiovascular events: a meta‐analysis. Eur J Epidemiol. 2011;26:833–850. [DOI] [PubMed] [Google Scholar]
- 150. Sayon‐Orea C, Martinez‐Gonzalez MA, Bes‐Rastrollo M. Alcohol consumption and body weight: a systematic review. Nutr Rev. 2011;69:419–431. [DOI] [PubMed] [Google Scholar]
- 151. Puddey IB, Parker M, Beilin LJ, et al. Effects of alcohol and caloric restrictions on blood pressure and serum lipids in overweight men. Hypertension. 1992;20:533–541. [DOI] [PubMed] [Google Scholar]
- 152. Canoy D, Wareham N, Luben R, et al. Cigarette smoking and fat distribution in 21,828 British men and women: a population‐based study. Obes Res. 2005;13:1466–1475. [DOI] [PubMed] [Google Scholar]
- 153. Rhee MY, Na SH, Kim YK, et al. Acute effects of cigarette smoking on arterial stiffness and blood pressure in male smokers with hypertension. Am J Hypertens. 2007;20:637–641. [DOI] [PubMed] [Google Scholar]
- 154. Simpson SA, Shaw C, McNamara R. What is the most effective way to maintain weight loss in adults? BMJ. 2011;343:d8042. [DOI] [PubMed] [Google Scholar]
- 155. Van Dorsten B, Lindley EM. Cognitive and behavioral approaches in the treatment of obesity. Med Clin North Am. 2011;95:971–988. [DOI] [PubMed] [Google Scholar]
- 156. Greaves CJ, Sheppard KE, Abraham C, et al. Systematic review of reviews of intervention components associated with increased effectiveness in dietary and physical activity interventions. BMC Public Health. 2011;11:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Svetkey LP, Stevens VJ, Brantley PJ, et al. Comparison of strategies for sustaining weight loss: the weight loss maintenance randomized controlled trial. JAMA. 2008;299:1139–1148. [DOI] [PubMed] [Google Scholar]
- 158. Funk KL, Stevens VJ, Appel LJ, et al. Associations of internet website use with weight change in a long‐term weight loss maintenance program. J Med Internet Res. 2010;12:e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Tuomilehto J, Lindstrom J, Eriksson JG, et al. Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. N Engl J Med. 2001;344:1343–1350. [DOI] [PubMed] [Google Scholar]
- 160. Knowler WC, Barrett‐Connor E, Fowler SE, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346:393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. McCullough ML, Patel AV, Kushi LH, et al. Following cancer prevention guidelines reduces risk of cancer, cardiovascular disease, and all‐cause mortality. Cancer Epidemiol Biomark Prev. 2011;20:1089–1097. [DOI] [PubMed] [Google Scholar]
- 162. Ketola E, Sipila R, Makela M. Effectiveness of individual lifestyle interventions in reducing cardiovascular disease and risk factors. Ann Med. 2000;32:239–251. [DOI] [PubMed] [Google Scholar]
- 163. Angermayr L, Melchart D, Linde K. Multifactorial lifestyle interventions in the primary and secondary prevention of cardiovascular disease and type 2 diabetes mellitus – a systematic review of randomized controlled trials. Ann Behav Med. 2010;40:49–64. [DOI] [PubMed] [Google Scholar]
- 164. Ebrahim S, Taylor F, Ward K, et al. Multiple risk factor interventions for primary prevention of coronary heart disease. Cochrane Database Syst Rev. 2011;CD001561. [DOI] [PubMed] [Google Scholar]
- 165. Mancia G, De Backer G, Dominiczak A, et al. Guidelines for the management of arterial hypertension: the Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and or the European Society of Cardiology (ESC). Eur Heart J. 2007;28:1462–1536. [DOI] [PubMed] [Google Scholar]
- 166. Moore LL, Visioni AJ, Qureshi MM, et al. Weight loss in overweight adults and the long‐term risk of hypertension: the Framingham study. Arch Intern Med. 2005;165:1298–1303. [DOI] [PubMed] [Google Scholar]
- 167. Allcock DM, Sowers JR. Best strategies for hypertension management in type 2 diabetes and obesity. Curr Diab Rep. 2010;10:139–144. [DOI] [PubMed] [Google Scholar]
- 168. Jandeleit‐Dahm KA, Tikellis C, Reid CM, et al. Why blockade of the renin‐angiotensin system reduces the incidence of new‐onset diabetes. J Hypertens. 2005;23:463–473. [DOI] [PubMed] [Google Scholar]
- 169. Sharma AM, Engeli S. The role of renin‐angiotensin system blockade in the management of hypertension associated with the cardiometabolic syndrome. J Cardiometab Syndr. 2006;1:29–35. [DOI] [PubMed] [Google Scholar]
- 170. Lindholm LH, Ibsen H, Borch‐Johnsen K, et al. Risk of new‐onset diabetes in the Losartan Intervention For Endpoint reduction in hypertension study. J Hypertens. 2002;20:1879–1886. [DOI] [PubMed] [Google Scholar]
- 171. Gupta AK, Dahlof B, Dobson J, et al. Determinants of new‐onset diabetes among 19,257 hypertensive patients randomized in the Anglo‐Scandinavian Cardiac Outcomes Trial – Blood Pressure Lowering Arm and the relative influence of antihypertensive medication. Diabetes Care. 2008;31:982–988. [DOI] [PubMed] [Google Scholar]
- 172. Lardizabal JA, Deedwania PC. The role of renin‐angiotensin agents in altering the natural history of type 2 diabetes mellitus. Curr Cardiol Rep. 2010;12:464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Wing LM, Reid CM, Ryan P, et al. A comparison of outcomes with angiotensin‐converting – enzyme inhibitors and diuretics for hypertension in the elderly. N Engl J Med. 2003;348:583–592. [DOI] [PubMed] [Google Scholar]
- 174. Redon J, Cifkova R, Laurent S, et al. The metabolic syndrome in hypertension: European Society of Hypertension position statement. J Hypertens. 2008;26:1891–1900. [DOI] [PubMed] [Google Scholar]
- 175. Kjeldsen SE, Julius S, Mancia G, et al. Effects of valsartan compared to amlodipine on preventing type 2 diabetes in high‐risk hypertensive patients: the VALUE trial. J Hypertens. 2006;24:1405–1412. [DOI] [PubMed] [Google Scholar]
- 176. Grimm C, Koberlein J, Wiosna W, et al. New‐onset diabetes and antihypertensive treatment. GMS Health Technol Assess. 2010;6:Doc03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. National High Blood Pressure Education Program . The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Bethesda, MD: National Heart, Lung and Blood Institute (US); 2004;Report No.: 04‐5230. [PubMed] [Google Scholar]
- 178. Lee P, Kengne AP, Greenfield JR, et al. Metabolic sequelae of beta‐blocker therapy: weighing in on the obesity epidemic? Int J Obes (Lond). 2011;35:1395–1403. [DOI] [PubMed] [Google Scholar]
- 179. Messerli FH, Bell DS, Fonseca V, et al. Body weight changes with beta‐blocker use: results from GEMINI. Am J Med. 2007;120:610–615. [DOI] [PubMed] [Google Scholar]
- 180. Sharma AM, Pischon T, Hardt S, et al. Hypothesis: beta‐adrenergic receptor blockers and weight gain: a systematic analysis. Hypertension. 2001;37:250–254. [DOI] [PubMed] [Google Scholar]
- 181. Manrique C, Whaley‐Connell A, Sowers JR. Nebivolol in obese and non‐obese hypertensive patients. J Clin Hypertens (Greenwich). 2009;11:309–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. NHLBI Obesity Education Initiative Working Group . The Practical Guide: Identification, Evaluation and Treatment of Overweight and Obesity in Adults. Bethesda, MD: NIH Publication 00‐4084; 2000. [Google Scholar]
- 183. Lawrence CB, Turnbull AV, Rothwell NJ. Hypothalamic control of feeding. Curr Opin Neurobiol. 1999;9:778–783. [DOI] [PubMed] [Google Scholar]
- 184. Garfield AS, Lam DD, Marston OJ, et al. Role of central melanocortin pathways in energy homeostasis. Trends Endocrinol Metab. 2009;20:203–215. [DOI] [PubMed] [Google Scholar]
- 185. van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, et al. Cold‐activated brown adipose tissue in healthy men. N Engl J Med. 2009;360:1500–1508. [DOI] [PubMed] [Google Scholar]
- 186. Bostrom P, Wu J, Jedrychowski MP, et al. A PGC1‐alpha‐dependent myokine that drives brown‐fat‐like development of white fat and thermogenesis. Nature. 2012;481:463–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Bordicchia M, Liu D, Amri EZ, et al. Cardiac natriuretic peptides act via p38 MAPK to induce the brown fat thermogenic program in mouse and human adipocytes. J Clin Invest. 2012;122:1022–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Torgerson JS, Hauptman J, Boldrin MN, Sjostrom L. XENical in the prevention of diabetes in obese subjects (XENDOS) study: a randomized study of orlistat as an adjunct to lifestyle changes for the prevention of type 2 diabetes in obese patients. Diabetes Care. 2004;27:155–161. [DOI] [PubMed] [Google Scholar]
- 189. Genentech USA, Inc . Xenical (Orlistat) Package Insert. South San Francisco, CA: Genentech USA, Inc; 2010. [Google Scholar]
- 190. Over‐the‐counter weight loss with orlistat? Drug Ther Bull. 2009;47:125–127. [DOI] [PubMed] [Google Scholar]
- 191. Hauptman J, Lucas C, Boldrin MN, et al. Orlistat in the long‐term treatment of obesity in primary care settings. Arch Fam Med. 2000;9:160–167. [DOI] [PubMed] [Google Scholar]
- 192. Rossner S, Sjostrom L, Noack R, et al. Weight loss, weight maintenance, and improved cardiovascular risk factors after 2 years treatment with orlistat for obesity. European Orlistat Obesity Study Group. Obes Res. 2000;8:49–61. [DOI] [PubMed] [Google Scholar]
- 193. Kaplan LM. Pharmacological therapies for obesity. Gastroenterol Clin North Am. 2005;34:91–104. [DOI] [PubMed] [Google Scholar]
- 194. Li Z, Maglione M, Tu W, et al. Meta‐analysis: pharmacologic treatment of obesity. Ann Intern Med. 2005;142:532–546. [DOI] [PubMed] [Google Scholar]
- 195. Bray GA, Ryan DH. Drug treatment of the overweight patient. Gastroenterology. 2007;132:2239–2252. [DOI] [PubMed] [Google Scholar]
- 196. Haddock CK, Poston WS, Dill PL, et al. Pharmacotherapy for obesity: a quantitative analysis of four decades of published randomized clinical trials. Int J Obes Relat Metab Disord. 2002;26:262–273. [DOI] [PubMed] [Google Scholar]
- 197. Hendricks EJ, Greenway FL, Westman EC, Gupta AK. Blood pressure and heart rate effects, weight loss and maintenance during long‐term phentermine pharmacotherapy for obesity. Obesity (Silver Spring). 2011;19:2351–2360. [DOI] [PubMed] [Google Scholar]
- 198. Bristol‐Myers Squibb Company , Glucophage (metformin) Package Insert. Uxbridge, UK: Bristol‐Myers Squibb Company; 2009. [Google Scholar]
- 199. DeFronzo RA, Goodman AM. Efficacy of metformin in patients with non‐insulin‐dependent diabetes mellitus. The Multicenter Metformin Study Group. N Engl J Med. 1995;333:541–549. [DOI] [PubMed] [Google Scholar]
- 200. Robinson AC, Burke J, Robinson S, et al. The effects of metformin on glycemic control and serum lipids in insulin‐treated NIDDM patients with suboptimal metabolic control. Diabetes Care. 1998;21:701–705. [DOI] [PubMed] [Google Scholar]
- 201. Amylin Pharmaceuticals and Eli Lilly & Company . Byetta Package Insert. Cincinnati, OH: Amylin Pharmaceuticals and Eli Lilly & Company; 2010. [Google Scholar]
- 202. Moretto TJ, Milton DR, Ridge TD, et al. Efficacy and tolerability of exenatide monotherapy over 24 weeks in antidiabetic drug‐naive patients with type 2 diabetes: a randomized, double‐blind, placebo‐controlled, parallel‐group study. Clin Ther. 2008;30:1448–1460. [DOI] [PubMed] [Google Scholar]
- 203. DeFronzo RA, Ratner RE, Han J, et al. Effects of exenatide (exendin‐4) on glycemic control and weight over 30 weeks in metformin‐treated patients with type 2 diabetes. Diabetes Care. 2005;28:1092–1100. [DOI] [PubMed] [Google Scholar]
- 204. Buse JB, Henry RR, Han J, et al. Effects of exenatide (exendin‐4) on glycemic control over 30 weeks in sulfonylurea‐treated patients with type 2 diabetes. Diabetes Care. 2004;27:2628–2635. [DOI] [PubMed] [Google Scholar]
- 205. Kendall DM, Riddle MC, Rosenstock J, et al. Effects of exenatide (exendin‐4) on glycemic control over 30 weeks in patients with type 2 diabetes treated with metformin and a sulfonylurea. Diabetes Care. 2005;28:1083–1091. [DOI] [PubMed] [Google Scholar]
- 206. Blonde L, Klein EJ, Han J, et al. Interim analysis of the effects of exenatide treatment on A1C, weight and cardiovascular risk factors over 82 weeks in 314 overweight patients with type 2 diabetes. Diabetes Obes Metab. 2006;8:436–447. [DOI] [PubMed] [Google Scholar]
- 207. Buse JB, Klonoff DC, Nielsen LL, et al. Metabolic effects of two years of exenatide treatment on diabetes, obesity, and hepatic biomarkers in patients with type 2 diabetes: an interim analysis of data from the open‐label, uncontrolled extension of three double‐blind, placebo‐controlled trials. Clin Ther. 2007;29:139–153. [DOI] [PubMed] [Google Scholar]
- 208. Horton ES, Silberman C, Davis KL, Berria R. Weight loss, glycemic control, and changes in cardiovascular biomarkers in patients with type 2 diabetes receiving incretin therapies or insulin in a large cohort database. Diabetes Care. 2010;33:1759–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Amylin Pharmacueticals . Symlin (pramlintide) Package Insert. 812003‐CC. Cincinnati, OH: Amylin Pharmacueticals; 2008. [Google Scholar]
- 210. Ratner RE, Dickey R, Fineman M, et al. Amylin replacement with pramlintide as an adjunct to insulin therapy improves long‐term glycaemic and weight control in Type 1 diabetes mellitus: a 1‐year, randomized controlled trial. Diabet Med. 2004;21:1204–1212. [DOI] [PubMed] [Google Scholar]
- 211. Younk LM, Mikeladze M, Davis SN. Pramlintide and the treatment of diabetes: a review of the data since its introduction. Expert Opin Pharmacother. 2011;12:1439–1451. [DOI] [PubMed] [Google Scholar]
- 212. Hollander PA, Levy P, Fineman MS, et al. Pramlintide as an adjunct to insulin therapy improves long‐term glycemic and weight control in patients with type 2 diabetes: a 1‐year randomized controlled trial. Diabetes Care. 2003;26:784–790. [DOI] [PubMed] [Google Scholar]
- 213. Aronne L, Fujioka K, Aroda V, et al. Progressive reduction in body weight after treatment with the amylin analog pramlintide in obese subjects: a phase 2, randomized, placebo‐controlled, dose‐escalation study. J Clin Endocrinol Metab. 2007;92:2977–2983. [DOI] [PubMed] [Google Scholar]
- 214. Aronne LJ, Isoldi KK, Roarke DT. The Johns Hopkins University Textbook of Dyslipidemia Chapter 24: Therapeutic Options for Modifying Obesity and Cardiometabolic Risk Factors. 288.
- 215. Smith SR, Aronne LJ, Burns CM, et al. Sustained weight loss following 12‐month pramlintide treatment as an adjunct to lifestyle intervention in obesity. Diabetes Care. 2008;31:1816–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Gadde KM, Allison DB, Ryan DH, et al. Effects of low‐dose, controlled‐release, phentermine plus topiramate combination on weight and associated comorbidities in overweight and obese adults (CONQUER): a randomised, placebo‐controlled, phase 3 trial. Lancet. 2011;377:1341–1352. [DOI] [PubMed] [Google Scholar]
- 217. Greenway FL, Fujioka K, Plodkowski RA, et al. Effect of naltrexone plus bupropion on weight loss in overweight and obese adults (COR‐I): a multicentre, randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet. 2010;376:595–605. [DOI] [PubMed] [Google Scholar]
- 218. Rubino F, R’Bibo SL, del Genio F, et al. Metabolic surgery: the role of the gastrointestinal tract in diabetes mellitus. Nat Rev Endocrinol. 2010;6:102–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Buchwald H, Avidor Y, Braunwald E, et al. Bariatric surgery: a systematic review and meta‐analysis. JAMA. 2004;292:1724–1737. [DOI] [PubMed] [Google Scholar]
- 220. Sjostrom L, Lindroos AK, Peltonen M, et al. Lifestyle, diabetes, and cardiovascular risk factors 10 years after bariatric surgery. N Engl J Med. 2004;351:2683–2693. [DOI] [PubMed] [Google Scholar]
- 221. Delling L, Karason K, Olbers T, et al. Feasibility of bariatric surgery as a strategy for secondary prevention in cardiovascular disease: a report from the Swedish obese subjects trial. J Obes. 2010;2010:102341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Saxe JS. Promoting healthy lifestyles and decreasing childhood obesity: increasing physician effectiveness through advocacy. Ann Fam Med. 2011;9:546–548. [DOI] [PMC free article] [PubMed] [Google Scholar]