

Abstract
Objective
To prospectively determine the value of optical coherence tomography (OCT) as a surrogate outcome measure for the progression of myelopathy in males with adrenoleukodystrophy.
Methods
Retinal nerve fiber layer (RNFL) and ganglion cell layer (GCL) thickness were measured at baseline, 1‐ and 2‐year follow‐up in patients and age‐matched controls. We assessed the severity of myelopathy with clinical parameters: Expanded Disability Status Scale (EDSS), Severity Scoring system for Progressive Myelopathy (SSPROM), and timed up‐and‐go. Linear mixed model analysis was used to compare changes in retinal layer thickness of patients to controls. In addition, we correlated changes in retinal layer thickness with changes in clinical parameters.
Results
Longitudinal data were available for 28 patients and 29 controls. Peripapillary RNFL (pRNFL) thickness decreased significantly in patients compared to controls (−1.75µm, p = 0.001), whereas change in macular GCL and RNFL was not different between groups. Analysis of the symptomatic subgroup showed that, apart from a similar decrease in pRNFL thickness, GCL thickness decreased significantly (−0.55 µm, p = 0.014). There were moderately strong correlations between changes in retinal layer thickness and changes in clinical parameters of severity of myelopathy.
Interpretation
This prospective study demonstrates the potential of OCT‐measured retinal neurodegeneration as a surrogate outcome measure for the progression of myelopathy in adrenoleukodystrophy. As differences were small, our findings need to be confirmed with longer follow‐up and/or in a larger patient sample.
Introduction
Progressive myelopathy is the main cause of disability in adrenoleukodystrophy (ALD), affecting virtually all male and over 80% of female patients. 1 , 2 ALD is a neurometabolic disorder caused by mutations in the ABCD1‐gene on the X‐chromosome. 3 , 4 ABCD1‐deficiency results in accumulation of very long‐chain fatty acids (VLCFA) in plasma and tissues. 5 , 6 The myelopathy of ALD is characterized pathologically by axonal degeneration of the corticospinal tracts and dorsal columns. 7 VLCFA‐induced oxidative stress, mitochondrial dysfunction, and endoplasmic reticulum stress have been implicated in the pathophysiology of this axonal degeneration. 2 , 6 Clinically, it manifests as a slowly progressive gait disorder due to spastic paraparesis and sensory ataxia. 8 Treatment of myelopathy in ALD is currently supportive only, but disease‐modifying therapies are being developed. Evaluation of these therapies in clinical trials using “traditional” clinical outcomes would require large numbers of patients and long follow‐up, due to the high intra‐ and interrater variability of these clinical outcomes and the slow disease progression. 9 Therefore, there is a need for more sensitive and reproducible outcome measures.
Using optical coherence tomography (OCT), our group recently showed that the retinal nerve fiber layer (RNFL) is thinner in ALD patients with myelopathy compared to healthy controls. Moreover, this neuroretinal thinning correlated with disease severity. 10 The more recent study of Bianchi‐Marzoli et al. showed similar results, which were not significant probably due to the smaller number of patients with myelopathy in this cohort. 11 These studies suggest that neurodegeneration of the spinal cord in ALD is reflected in the retina, a concept that has already been illustrated in a number of other neurodegenerative diseases. 12 , 13 , 14 As OCT is a fast, noninvasive and reproducible technique, retinal neurodegeneration may be valuable as a new surrogate outcome measure in ALD. 14
In this longitudinal study, we further evaluated the potential of OCT‐measured retinal neurodegeneration as a surrogate outcome measure for myelopathy in ALD. We investigated whether retinal neurodegeneration is progressive over 2‐year follow‐up and if it corresponds with clinical parameters of disease progression. In previous studies, we showed that male ALD patients have small, but statistically significant progression of myelopathy on clinical parameters during 2‐year follow‐up. 9 In female patients, however, disease progression is much slower, with only minor progression over a period of 8 years. 15 Therefore, this longitudinal study was limited to the male subgroup of this cohort.
Methods
Baseline data of this cohort were previously published. 10 We used the same methodology (with minor adjustments) as applied in that study, which is summarized below.
Study design and participants
This prospective cohort study was performed at the Amsterdam UMC between June 2015 and July 2019, as part of a large natural history study (the Dutch ALD cohort). For this particular study, we included male patients over 16 years of age with a confirmed diagnosis of ALD (very long‐chain fatty acid (VLCFA) and genetic analysis). Patients with cerebral ALD were excluded, apart from one patient with arrested cerebral ALD with a very low and stable lesion load. Other exclusion criteria were a history of neurodegenerative (other than ALD) or ophthalmological disease, diabetes mellitus, and comorbidity interfering with the assessment of myelopathy. Patients underwent neurological assessment, ophthalmological examination and OCT imaging on the same day. The ophthalmological examination was performed by an experienced staff member and included visual acuity measurement (ETDRS‐card with Sloan letters), measurement of intraocular pressure with air‐puff tonometry, slit‐lamp biomicroscopy, and fundus photography. Eyes with reduced visual acuity (>0.1 LogMar), high refractive errors (>6 diopter), high intra‐ocular pressure (>21 mmHg), substantial media opacities, and optic nerve disease or retinal disease as defined in the OSCAR‐IB criteria were excluded. 16 Sex‐ and age‐matched controls without a history of diabetes, neurological or ophthalmological disease and a normal visual acuity (≤0.1 Logmar) were recruited via public advertisement. Both patients and controls were examined with regular 1‐year intervals (baseline, year 1, and year 2). The local Institutional Review Board approved the study protocol (METC 2014_302) and all participants provided written informed consent.
Neurological assessment
Assessment of myelopathy in this cohort has been previously described. 9 In short, patients underwent a detailed neurological history and examination. They were scored as symptomatic if both signs and symptoms of myelopathy were present. Clinical outcome measures used to quantify myelopathy were the Expanded Disability Status Scale (EDSS), Severity Scoring system for Progressive Myelopathy (SSPROM), and timed up‐and‐go. The EDSS measures neurological disability ranging from 0 (no disability) to 10 (death). 17 SSPROM measures the severity of myelopathy ranging from 0 to 100, with lower scores indicating a higher degree of impairment. 18 , 19 The timed up‐and‐go is used to assess walking function by recording the time that the patient needs to get up from an armchair, walk 3 meters, turn around, walk back, and sit down again. 20 , 21
Imaging protocol and image analysis
OCT‐imaging was performed by four OCT‐operators under dimmed‐light conditions on two identical Heidelberg Spectralis OCT‐scanners (Heidelberg Engineering GmbH, Germany). Images of both the macula and optic nerve (peripapillary scan) were obtained. Scans with poor quality or retinal disease as defined in the OSCAR‐IB criteria were excluded. 16 The macula was scanned in the horizontal direction in an area of 6 × 6 mm (20 degrees) with 49 b‐scans; each b‐scan was the average of 15 scans. The optic nerve head was scanned with a 3.5 mm diameter circle centered on the optic disc, containing 768 × 496 pixels. Macular and peripapillary scans were segmented using Heidelbergs built‐in segmentation algorithm (version 1.910.0). Macular RNFL and GCL thickness were calculated for the total ETDRS grid‐surface. 22 Peripapillary RNFL (pRNFL) thickness was calculated for both the total peripapillary ring and each of the four quadrants (temporal, superior, nasal, and inferior). Figure 1 shows an example of a peripapillary scan of a healthy control (upper panel, A‐C) and a symptomatic patient (lower panel, D‐F).
Figure 1.
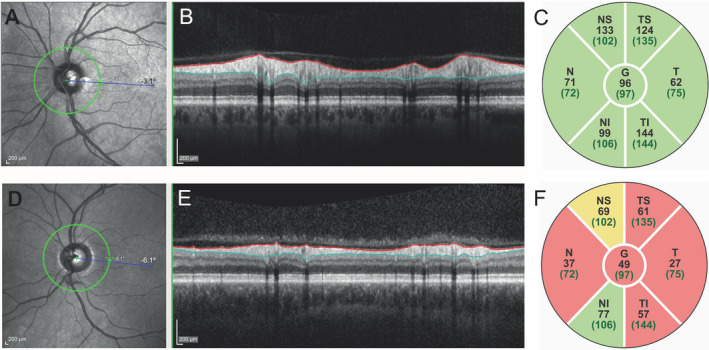
Peripapillary RNFL thickness of a healthy control and a patient. The pRNFL is shown of a healthy control (upper panel, A‐C) and a symptomatic patient with an EDSS of 7.0 (D‐F). A and D: optic nerve scans with the peripapillary ring (green) where the pRNFL thickness is measured. B and E: cross section of the retina at the peripapillary ring; the pRNFL is marked by the colored lines and is much thinner in the patient (E) than in the healthy control (B). C and F: pRNFL thickness (µm) per segment of the peripapillary ring. Green segments indicating normal pRNFL thickness, orange moderately reduced and red more severely reduced pRNFL thickness. Numbers between brackets are reference values. EDSS, expanded disability status scale; I, inferior quadrant; N, nasal quadrant; OCT, optical coherence tomography; pRNFL, peripapillary retinal nerve fiber layer; S, superior quadrant; T, temporal quadrant.
We allowed for the inclusion of one eye if the other eye was not eligible for inclusion. If both eyes were eligible, the mean layer thickness of both eyes was used for analysis.
Statistical analysis
Clinical characteristics at baseline were summarized using descriptive statistics. We used linear mixed model analyses for repeated measures to assess changes in retinal layer thickness. Linear mixed model analyses were chosen to fully use the available data, including cases with missing data. First, a mixed model was built with group (patient versus control), time and their two‐way interaction (group * time) and a random intercept on subject level. With this model, retinal layer thickness at each time point and the change during follow‐up for each group were estimated. The group * time interaction was the effect of interest, as it demonstrates whether the change in retinal layer thickness over time of patients differs from controls. Second, we repeated this analysis including only symptomatic patients, because in our previous study statistically significant change on the clinical parameters was limited to the symptomatic subgroup. In an exploratory analysis, we also evaluated change in retinal layer thickness for the asymptomatic subgroup; because of the small sample size we did not perform statistical analyses on this subgroup. In accordance with our previous study, 9 we compared clinical parameters at baseline and at 2‐year follow‐up using a paired t‐test for normally distributed data or Wilcoxon signed rank test for non‐normally distributed data; mixed model analysis was not used for these analyses because of the large proportion of non‐normally distributed data. We evaluated the association between changes in retinal layer thickness and clinical parameters with Pearson’s correlation (in case of normal distribution) or Spearman’s rank order correlation (non‐normal distribution). Finally, similar to our clinical study in this cohort, 9 we calculated the number of patients that would be needed for a placebo‐controlled trial using retinal layer thickness as a surrogate outcome measure—assuming a 1:1 ratio of active substance versus placebo, a 50% decrease in progression rate, and 80% power. 23
Statistical analyses were conducted with IBM SPSS statistics version 24 (IBM Inc.). For all tests, significance level was set at 0.05. Because of the exploratory character of this study, we did not correct for multiple comparisons.
Results
Participant characteristics
Figure 2 provides an overview of subject enrollment and exclusions. Of 70 subjects screened, 62 were included: 29 patients and 33 controls. Because we continued to include new patients during the course of the study, data for year 2 were not available for all patients. In contrast, some participants in the control group did not show up at year 1 (but did at year 2), causing the number of controls to increase from year 1 to 2. Longitudinal data (either baseline and year 1, baseline and year 2, or all time points) were available for 28 patients and 29 controls. One of both eyes was excluded in two patients (one due to amblyopia, one due to low quality of the OCT) and one control (due to amblyopia). There were no patients with progressive white matter lesions on MRI during follow‐up; the one patient with arrested cerebral ALD had a small asymptomatic lesion in the splenium of the corpus callosum, which has been stable for years. The median age of patients (42.0, range 16‐68) and controls (41.0, range 21‐65) was not statistically significantly different (p = 0.78), nor was mean follow‐up time (23.3 ± 1.5 vs. 23.9 ± 0.9, p = 0.08).
Figure 2.
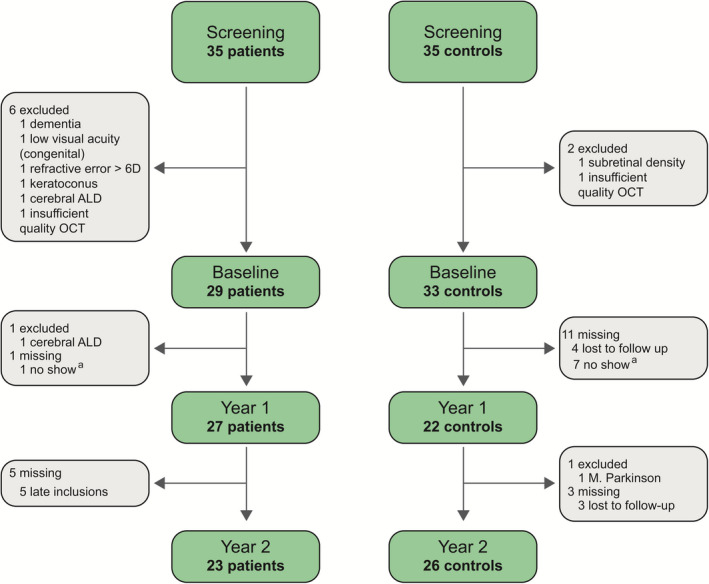
Subject enrollment and exclusions. Flow diagram of subject enrollment, exclusions, and missing data for both the patients (left) and controls (right). a these subjects did not attend their study visit at year 1, but did at year 2. ALD, adrenoleukodystrophy; OCT, optical coherence tomography.
Neurological characteristics of this cohort are described in detail elsewhere. 9 In summary, 20/28 (71%) patients were symptomatic, having both signs and symptoms of myelopathy. One patient (age 23) converted from asymptomatic to symptomatic during follow‐up. Baseline scores on the clinical parameters of severity of myelopathy (EDSS, SSPROM, and timed up‐and‐go) can be found in Table 1; these scores indicate a moderate average level of disability.
Table 1.
Change in retinal layer thickness during follow‐up for patients and controls.
|
Baseline (P = 28, C = 29) |
Year 1 (P = 27, C = 22) |
Year 2 (P = 23, C = 26) |
Change (95% CI) | p‐value | ||
|---|---|---|---|---|---|---|
| pRNFL (total) a , µm | patient | 88.84 (2.10) | 87.45 (2.10) | 87.08 (2.11) | −1.75 (−2.58 to −0.93) | 0.001 |
| control | 92.19 (2.07) | 92.63 (2.07) | 92.32 (2.07) | 0.13 (−0.98 to 1.25) | ||
| Superior | patient | 107.30 (2.93) | 107.88 (2.93) | 107.68 (2.94) | 0.37 (−0.79 to 1.53) | 0.990 |
| control | 116.52 (2.88) | 117.13 (2.89) | 117.00 (2.88) | 0.48 (−1.36 to 1.80) | ||
| Nasal | patient | 69.98 (2.38) | 66.00 (2.38) | 65.12 (2.41) | −.87 (−6.59 to −3.14) | <0.001 |
| control | 66.26 (2.34) | 67.25 (2.37) | 65.80 (2.35) | −0.46 (−2.80 to 1.86) | ||
| Inferior | patient | 114.64 (3.24) | 113.08 (3.24) | 112.66 (3.25) | −1.98 (−3.09 to −0.88) | 0.018 |
| control | 115.14 (3.18) | 115.45 (3.19) | 114.95 (3.19) | −0.19 (−1.69 to 1.31) | ||
| Temporal | patient | 63.41 (2.48) | 62.85 (2.48) | 62.83 (2.49) | −0.58 (−1.59 to 0.42) | 0.165 |
| control | 70.85 (2.43) | 70.66 (2.44) | 71.56 (2.44) | 0.72 (−0.52 to 2.08) | ||
| RNFL, µm | patient | 31.68 (0.69) | 32.28 (0.69) | 31.80 (0.69) | 0.12 (−0.29 to 0.54) | 0.140 |
| control | 34.21 (0.68) | 33.97 (0.68) | 34.09 (0.68) | −0.12 (−0.70 to 0.45) | ||
| GCL, µm | patient | 37.21 (0.70) | 37.14 (0.70) | 36.85 (0.70) | −0.36 (−0.60 to −0.11) | 0.119 |
| control | 37.81 (0.68) | 37.61 (0.69) | 37.69 (0.68) | −0.11 (−0.45 to 0.22) | ||
| EDSS | 3.5 (0‐7.0) | 3.5 (0‐7.5) | 4.0 (0‐7.5) | 0.44 (0.09 to 0.78) | 0.014 | |
| SSPROM | 85.25 (65‐100) | 83.00 (59‐100) | 81.50 (45.5‐100) | −2.80 (−5.11 to −0.50) | 0.018 | |
| Timed up‐and‐go, s | 6.66 (2.57‐13.54) | 7.39 (2.97‐15.25) | 8.81 (2.77‐14.49) | 0.60 (−11 to 1.31) | 0.107 |
Retinal layer thickness and changes during follow‐up are the estimated values from the linear mixed‐effects models and summarized as means (standard errors). pRNFL (total) and GCL thickness are also presented in Figure 3 (green line for total patient group, blue lines for controls). P‐values represent the significance level of the interaction term “group * time” and signify whether the change in retinal layer thickness over time of patients differs from controls. For patients, change on clinical parameters is also reported. Values are summarized as median (range), change is reported as mean paired change between baseline and year 2 with corresponding p‐value (Wilcoxon signed rank test).
EDSS, expanded disability status scale; GCL, ganglion cell layer; pRNFL, peripapillary retinal nerve fiber layer; RNFL, retinal nerve fiber layer; SSPROM, severity scoring system for progressive myelopathy.
Mean thickness of the total peripapillary ring followed by each of the four quadrants.
Retinal layer thinning
Table 1 and Figure 3 show the changes in retinal layer thickness during follow‐up for patients and controls. For patients, the change in clinical parameters is also reported. pRNFL thickness (total ring, nasal, and inferior quadrant) decreased significantly during follow‐up in the patient group compared to the control group, whereas macular RNFL and GCL thickness did not. Of the clinical parameters, the EDSS increased and SSPROM decreased significantly over time, indicating an increase in disability between baseline and follow‐up. The increase on the timed up‐and‐go was not statistically significant.
Figure 3.
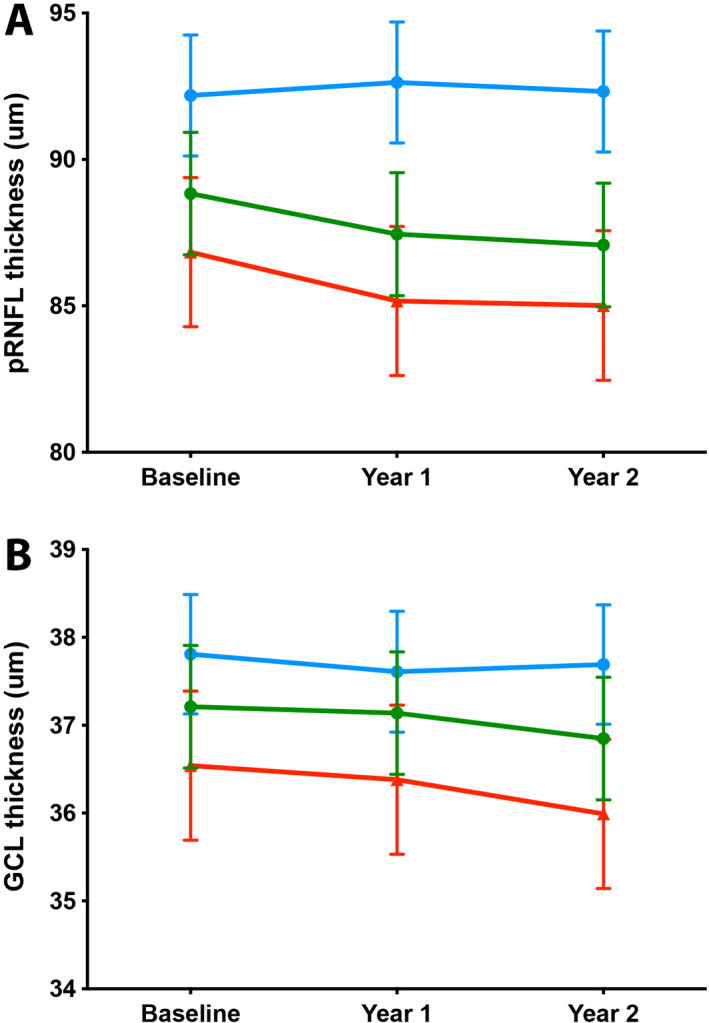
Change in retinal layer thickness over time. Mean pRNFL thickness (A) and GCL thickness (B) in controls (blue), the total patient group (green), and the symptomatic subgroup (red). Values are the estimates from the linear mixed model analysis, bars represent standard errors. Baseline patient n = 28, control n = 29; Year 1 patient n = 27, control n = 22; Year 2 patient n = 23, control n = 26. pRNFL, peripapillary retinal nerve fiber layer; GCL, ganglion cell layer.
When looking at symptomatic patients only (Table 2), the differences in pRNFL thickness were slightly larger than for the total patient group. Also, in contrast to the total patient group, the macular GCL showed significant thinning compared to the control group (−0.55µm, p = 0.014). Absolute changes in clinical parameters were also slightly larger, although due to the smaller sample size significance levels of these changes were slightly weaker than for the total patient group. Changes in retinal layer thickness for the one patient with arrested cerebral ALD (and myelopathy with EDSS 3.5) were not very different from the rest of the symptomatic group (total pRNFL = 83.44 µm on both baseline and 2‐year follow‐up; GCL thickness 38.84 µm on baseline and 38.69 on follow‐up).
Table 2.
Change in retinal layer thickness during follow‐up for symptomatic patients and controls.
|
Baseline (P = 20, C = 29) |
Year 1 (P = 20, C = 22) |
Year 2 (P = 17, C = 26) |
Change (95% CI) | p‐value | ||
|---|---|---|---|---|---|---|
| pRNFL (total) a , µm | patient | 86.83 (2.55) | 85.17 (2.55) | 85.01 (2.56) | −1.82 (−2.68 to −0.97) | <0.001 |
| control | 92.19 (2.07) | 92.63 (2.07) | 92.32 (2.07) | 0.13 (−0.98 to 1.25) | ||
| Superior | patient | 103.88 (3.48) | 104.13 (3.48) | 104.19 (3.49) | 0.32 (−0.86 to 1.50) | 0.874 |
| control | 116.52 (2.88) | 117.13 (2.89) | 117.00 (2.88) | 0.48 (−1.36 to 1.80) | ||
| Nasal | patient | 70.80 (2.93) | 66.63 (2.93) | 65.63 (2.97) | −5.17 (−7.27 to −3.06) | <0.001 |
| control | 66.26 (2.34) | 67.25 (2.37) | 65.80 (2.35) | −0.46 (−2.80 to 1.86) | ||
| Inferior | patient | 112.83 (3.35) | 110.93 (1.04) | 110.82 (4.05) | −2.00 (−3.07 to −0.94) | 0.002 |
| control | 115.14 (3.18) | 115.45 (3.19) | 114.95 (3.19) | −0.19 (−1.69 to 1.31) | ||
| Temporal | patient | 59.83 (2.69) | 59.00 (2.69) | 59.32 (2.70) | −0.51 (−1.67 to −0.66) | 0.254 |
| control | 70.85 (2.43) | 70.66 (2.44) | 71.56 (2.44) | 0.72 (−0.52 to 2.08) | ||
| RNFL, µm | patient | 30.71 (0.79) | 31.43 (0.79) | 30.91 (0.79) | 0.19 (−0.31 to 0.70) | 0.140 |
| control | 34.21 (0.68) | 33.97 (0.68) | 34.09 (0.68) | −0.12 (−0.70 to 0.45) | ||
| GCL, µm | patient | 36.54 (0.85) | 36.38 (0.85) | 35.99 (0.85) | −0.55 (−0.81 to −0.29) | 0.014 |
| control | 37.81 (0.68) | 37.61 (0.69) | 37.69 (0.68) | −0.11 (−0.45 to 0.22) | ||
| EDSS | 4.0 (0‐7.0) | 5.0 (1.5‐7.5) | 6.0 (2.0‐7.5) | 0.47 (0.01 to 0.93) | 0.045 | |
| SSPROM | 80.25 ± 9.94 | 79.65 ± 9.11 | 75.27 ± 11.78 | −3.71 (−6.76 to −0.65) | 0.021 | |
| Timed up‐and‐go, s | 8.45 ± 2.84 | 8.68 ± 2.98 | 9.45 ± 2.99 | 0.96 (−0.04 to 1.95) | 0.058 |
Retinal layer thickness and changes during follow‐up are the estimated values from the linear mixed‐effects models and summarized as means (standard errors). pRNFL (total) and GCL thickness are also presented in Figure 3 (red lines for symptomatic patients, blue lines for controls). P‐values represent the significance level of the interaction term “group * time” and signify whether change in retinal layer thickness over time of patients differs from controls. For patients, change on clinical parameters is also reported. Values are summarized as mean ± SD or median (range) depending on the distribution of the data; change is reported as mean paired change between baseline and year 2 with corresponding p‐value (paired t‐test/Wilcoxon signed rank test).
EDSS, Expanded Disability Status Scale; GCL, ganglion cell layer; pRNFL, peripapillary retinal nerve fiber layer; RNFL, retinal nerve fiber layer; SSPROM, Severity Scoring system for Progressive Myelopathy.
Mean thickness of the total peripapillary ring followed by each of the four quadrants.
Exploratory analysis of the asymptomatic subgroup (Table 3) showed a similar decrease in pRNFL thickness (total ring, superior, and inferior quadrant) as in the total patient group. Because of the small sample size of this subgroup, we did not perform statistical tests to compare this change to the control group.
Table 3.
Change in retinal layer thickness during follow‐up for asymptomatic patients.
|
Baseline (N = 8) |
Year 1 (N = 7) |
Year 2 (N = 6) |
Change | |
|---|---|---|---|---|
| pRNFL (total) a , µm | 93.84 (2.55) | 93.26 (2.58) | 92.39 (2.61) | −1.61 |
| Superior | 115.88 (4.06) | 116.43 (4.10) | 116.31 (4.15) | 0.43 |
| Nasal | 67.94 (3.32) | 64.45 (3.35) | 63.83 (3.81) | −4.10 |
| Inferior | 119.19 (3.16) | 118.59 (3.21) | 117.22 (3.28) | −1.96 |
| Temporal | 72.38 (5.52) | 72.58 (5.53) | 71.59 (5.55) | −0.79 |
| RNFL, µm | 34.09 (1.29) | 34.42 (1.30) | 34.04 (1.30) | −0.04 |
| GCL, µm | 38.88 (0.78) | 39.03 (0.78) | 39.05 (0.79) | 0.17 |
| EDSS | 1.0 (0‐3.0) | 1.0 (0‐3.0) | 1.0 (0‐3.5) | 0.33 |
| SSPROM | 100 (98.0‐100) | 100 (99.0‐100) | 100 (95.5‐100) | −0.25 |
| Timed up‐and‐go, s | 3.69 ± 0.69 | 3.53 ± 0.42 | 3.52 ± 0.50 | −0.18 |
Retinal layer thickness and changes during follow‐up are the estimated values from the linear mixed‐effects models and summarized as means (standard errors). Change on clinical parameters is also reported; values are summarized as mean ± SD or median (range) depending on the distribution of the data; change is reported as mean paired change between baseline and year 2 with corresponding P‐value (paired t‐test/Wilcoxon signed rank test).
EDSS, expanded disability status scale; GCL, ganglion cell layer; pRNFL, peripapillary retinal nerve fiber layer; RNFL, retinal nerve fiber layer; SSPROM, severity scoring system for progressive myelopathy.
Mean thickness of the total peripapillary ring followed by each of the four quadrants.
Correlation between change on OCT and clinical parameters
There was a moderately strong correlation between change on the EDSS and thinning of the nasal quadrant of the pRNFL (Spearman’s rho = 0.51, p = 0.02) and change on the timed up‐and‐go and thinning of the pRNFL total ring (Spearman’s rho = 0.51, p = 0.04). Correlations between change on timed up‐and‐go and thinning of the nasal quadrant of the pRNFL (Spearman’s rho = 0.46, p = 0.06) and GCL (Spearman’s rho = 0.44, p = 0.06) were on the border of statistical significance. No other significant correlations were found.
Sample size calculation
The number of patients needed per treatment arm for a placebo‐controlled trial of 2 years using retinal layer thickness as a surrogate outcome measure (assuming a 50% reduction of disease progression and 80% power) would be 95 for the pRNFL total ring, 95 for the pRNFL inferior quadrant and 93 for the pRNFL nasal quadrant.
Discussion
As potential treatments emerge, finding a sensitive surrogate outcome measure for myelopathy in ALD is essential. In this prospective cohort study, we provide evidence that retinal neurodegeneration on OCT may serve as a surrogate outcome measure for the progression of myelopathy in men with ALD. Previously, in a cross‐sectional analysis, we showed that ALD patients have a thinner neuroretina compared to healthy controls. Moreover, neuroretinal layer thickness correlated with the severity of myelopathy. 10 We now provide evidence that neuroretinal layer thinning in ALD patients is progressive over 2‐year follow‐up and that this thinning differs significantly from a healthy control group. In addition, we found moderately strong correlations between changes in retinal layer thickness and changes in severity of myelopathy, suggesting that the retinal neurodegeneration has a clinical equivalent.
Our findings support the hypothesis that neurodegeneration of the spinal cord is reflected in the retina, and are in line with several other studies that have showed neuroretinal thinning in neurodegenerative and neuro‐inflammatory disorders, including a recent paper describing OCT in patients with ALD. 11 , 12 , 13 , 14 However, similar to those studies, we cannot prove that the same pathological process (a dying‐back axonopathy) is occurring in both the spinal cord and retina. Although it seems unlikely that there is a second, unrelated cause for the retinal neurodegeneration in patients with ALD, only a pathological study in which both spinal cord and retinal nerve fibers are examined could definitively resolve this issue.
The most substantial decrease in retinal layer thickness was found for the peripapillary RNFL (Tables 1 and 2, Fig. 3). The RNFL contains the axons of neurons projecting from the retina to the thalamus. These axons converge at the optic disk (or optic papilla) to form the optic nerve; therefore the RNFL is thickest at this peripapillary ring. 24 It follows that axonal degeneration, the pathological hallmark of ALD that is presumed to occur simultaneously both in the spinal cord and retina, is best measured at this point. 7 Indeed, our cross‐sectional study already showed that the absolute differences in retinal layer thickness were largest for the pRNFL, but due to the substantial spread of pRNFL thickness between subjects these differences were not statistically significant. 10 Because of the high reproducibility of pRNFL measurements within the same subject over time, within‐subject changes can be detected in this longitudinal analysis.
In our cross‐sectional study, we found significant between‐group differences in the pRNFL temporal and superior quadrant, 10 whereas in this study the pRNFL total ring, nasal quadrant, and inferior quadrant showed significant thinning over time. A possible explanation for this difference is that some retinal layers or regions may degenerate slower (or do not degenerate any further) in advanced compared to the early stages of the disease. This so‐called ceiling effect is also present for other outcome measures for myelopathy in ALD (such as the EDSS and timed walking activities). Conversely, GCL thickness did not decrease significantly in the total patient group, whereas it did in the symptomatic subgroup (Table 2). The GCL contains the cell bodies of the neurons that form the RNFL. One would expect that, as axonal degeneration progresses, the cell bodies of the neurons would also eventually be lost, resulting in atrophy of the GCL. This could, however, be a feature of advanced disease and therefore be restricted to (more severely) affected patients (a so‐called floor effect). Indeed, explorative analysis showed a correlation between disease severity and decrease in GCL thickness (correlation coefficient for SSPROM 0.45, p = 0.03; EDSS −0.41, p = 0.05), supporting this hypothesis. Therefore, GCL thinning could be valuable as a marker of disease progression in advanced disease.
Ideally, a surrogate outcome measure for myelopathy in ALD is also able to detect changes in a presymptomatic state, allowing interventions to be implemented and evaluated before disability appears. In our cohort, exploratory analysis in the asymptomatic group showed a similar decrease in pRNFL thickness as the total patient group. Because of the small sample size, we did not statistically test whether this change was significant. Although confirmation in a larger sample is needed, it does suggest that retinal layer thickness on OCT may be valuable even in a presymptomatic state.
Interestingly, retinal layer thinning seemed more pronounced in the first compared to the second year of follow‐up (Fig. 3). Although progression of myelopathy in an individual patient may not be linear, one would expect this to average in the total patient group. It might just be variability that would not be remarkable over a longer period, but longer term studies are needed to clarify this.
In addition to retinal layer thinning in patients compared to controls, we found correlations between retinal thinning and disease progression on clinical parameters. These correlations were moderately strong (correlation coefficients between 0.44‐0.51), but not present for all clinical parameters. There are some possible explanations for this variation. First, the clinical parameters of myelopathy are neither very sensitive nor specific. They cannot measure all aspects of disability due to myelopathy, but are at the same time influenced by factors other than myelopathy (for example, the timed up‐and‐go is influenced by patient motivation or can be reduced after strenuous exercise). In addition, both inter‐ and intra‐observer variability of clinical assessments are substantial. 25 , 26 , 27 Second, anatomical changes do not always directly lead to functional changes. It is likely that a certain threshold of axonal damage has to be reached before symptoms appear, limiting the correlation for presymptomatic patients. Finally, the myelopathy of ALD is slowly progressive, with disability accumulating over years or decades. Two years of follow‐up is a relatively short period in this regard, therefore it is not surprising that the differences we found were small. Long‐term follow‐up, which is ongoing in this cohort, will hopefully confirm the correlation between neurodegeneration on OCT and clinical parameters.
Although the changes in retinal layer thickness were small, the sample size calculation indicates that OCT is more sensitive than “traditional” clinical outcome measures—EDSS, SSPROM, and timed activities. In a previous study using these clinical outcomes, we calculated that 219‐314 patients would be needed per treatment arm for a placebo‐controlled trial assuming a 50% reduction of disease progression, depending on the outcome measure chosen. 9 Using the OCT‐measured pRNFL as a surrogate outcome measure, this number would be 93‐95 per treatment arm depending on the region chosen, a reduction of >50% compared to the clinical outcome measures.
Strengths of this study are the structured ophthalmological and neurological assessments occurring on the same day and with regular intervals, and the use of an age‐matched control group. A limitation is the relatively large number of exclusions in the patient group (Fig. 2). Although we used the conventional exclusion criteria as defined in the OSCAR‐IB criteria, 16 this led to exclusion of 6/35 (17.1%) patients. Such an exclusion rate could be problematic if retinal neurodegeneration on OCT is to be used as an outcome measure in clinical trials.
In conclusion, in this longitudinal study, we demonstrate the potential of retinal neurodegeneration measured by OCT as a surrogate outcome measure for myelopathy in ALD. OCT has several promising advantages—disease progression can be measured over a relatively short follow‐up period in symptomatic and possibly also presymptomatic patients, and there is a correlation with clinical parameters. In addition, OCT is fast, noninvasive, and reproducible (provided that subjects are followed on the same scanner). The main disadvantages are the high exclusion rate due to (largely opthalmological) comorbidity. As differences were small, our findings need to be confirmed in studies with longer follow‐up and/or a larger cohort.
Conflict of Interest
The authors declare the following potential conflicts of interest: IH has received unrestricted research grants from Vertex. ME has received unrestricted research grants from Vertex, Swanbio Therapeutics, Bluebird Bio, and Minoryx Therapeutics. No other potential conflicts of interest apply.
Funding Information
This study was funded by the Netherlands Organization for Scientific Research (VENI grant: 016.156.033 to M.E. and VIDI grant: 016.196.310 to M.E.).
Funding Statement
This work was funded by Nederlandse Organisatie voor Wetenschappelijk Onderzoek grants 016.156.033 and 016.196.310.
References
- 1. Engelen M, Barbier M, Dijkstra IM, et al. X‐linked adrenoleukodystrophy in women: a cross‐sectional cohort study. Brain 2014;137(Pt 3):693–706. [DOI] [PubMed] [Google Scholar]
- 2. Kemp S, Huffnagel IC, Linthorst GE, et al. Adrenoleukodystrophy ‐ neuroendocrine pathogenesis and redefinition of natural history. Nat Rev Endocrinol 2016;12(10):606–615. [DOI] [PubMed] [Google Scholar]
- 3. Engelen M, Kemp S, de Visser M, et al. X‐linked adrenoleukodystrophy (X‐ALD): clinical presentation and guidelines for diagnosis, follow‐up and management. Orphanet J Rare Dis 2012;7:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mosser J, Douar AM, Sarde CO, et al. Putative X‐linked adrenoleukodystrophy gene shares unexpected homology with ABC transporters. Nature 1993;361(6414):726–730. [DOI] [PubMed] [Google Scholar]
- 5. Singh I, Moser AE, Moser HW, Kishimoto Y. Adrenoleukodystrophy: impaired oxidation of very long chain fatty acids in white blood cells, cultured skin fibroblasts, and amniocytes. Pediatr Res 1984;18(3):286–290. [DOI] [PubMed] [Google Scholar]
- 6. Berger J, Forss‐Petter S, Eichler FS. Pathophysiology of X‐linked adrenoleukodystrophy. Biochimie 2014;98(100):135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Powers JM, DeCiero DP, Ito M, et al. Adrenomyeloneuropathy: a neuropathologic review featuring its noninflammatory myelopathy. J Neuropathol Exp Neurol 2000;59(2):89–102. [DOI] [PubMed] [Google Scholar]
- 8. Moser HW, Moser AB, Naidu S, Bergin A. Clinical aspects of adrenoleukodystrophy and adrenomyeloneuropathy. Dev Neurosci 1991;13(4–5):254–261. [DOI] [PubMed] [Google Scholar]
- 9. Huffnagel IC, van Ballegoij WJC, van Geel BM, et al. Progression of myelopathy in males with adrenoleukodystrophy: towards clinical trial readiness. Brain 2019;142(2):334–343. [DOI] [PubMed] [Google Scholar]
- 10. van Ballegoij WJC, Kuijpers SC, Huffnagel IC, et al. Optical coherence tomography shows neuroretinal thinning in myelopathy of adrenoleukodystrophy. J Neurol 2020;267(3):679–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bianchi‐Marzoli S, Fenu S, Melzi L, et al. Optical coherence tomography in adult adrenoleukodystrophy: a cross‐sectional and longitudinal study. Neurol Sci 2021;42(1):235–241. [DOI] [PubMed] [Google Scholar]
- 12. Mutlu U, Colijn JM, Ikram MA, et al. Association of retinal neurodegeneration on optical coherence tomography with dementia: a population‐based study. JAMA Neurol 2018;75(10):1256–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Petzold A, Balcer LJ, Calabresi PA, et al. Retinal layer segmentation in multiple sclerosis: a systematic review and meta‐analysis. Lancet Neurol 2017;16(10):797–812. [DOI] [PubMed] [Google Scholar]
- 14. Maldonado RS, Mettu P, El‐Dairi M, Bhatti MT. The application of optical coherence tomography in neurologic diseases. Neurol Clin Pract 2015;5(5):460–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huffnagel IC, Dijkgraaf MGW, Janssens GE, et al. Disease progression in women with X‐linked adrenoleukodystrophy is slow. Orphanet J Rare Dis 2019;14(1):30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tewarie P, Balk L, Costello F, et al. The OSCAR‐IB consensus criteria for retinal OCT quality assessment. PLoS One 2012;7(4):e34823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology 1983;33(11):1444–1452. [DOI] [PubMed] [Google Scholar]
- 18. Castilhos RM, Blank D, Netto CB, et al. Severity score system for progressive myelopathy: development and validation of a new clinical scale. Braz J Med Biol Res 2012;45(7):565–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. D'Souza M, Yaldizli O, John R, et al. Neurostatus e‐Scoring improves consistency of Expanded Disability Status Scale assessments: a proof of concept study. Mult Scler 2017;23(4):597–603. [DOI] [PubMed] [Google Scholar]
- 20. Podsiadlo D, Richardson S. The timed "Up & Go": a test of basic functional mobility for frail elderly persons. J Am Geriatr Soc 1991;39(2):142–148. [DOI] [PubMed] [Google Scholar]
- 21. van Hedel HJ, Wirz M, Dietz V. Assessing walking ability in subjects with spinal cord injury: validity and reliability of 3 walking tests. Arch Phys Med Rehabil 2005;86(2):190–196. [DOI] [PubMed] [Google Scholar]
- 22. Chew EY, Klein ML, Ferris FL 3rd, et al. Association of elevated serum lipid levels with retinal hard exudate in diabetic retinopathy. Early Treatment Diabetic Retinopathy Study (ETDRS) Report 22. Arch Ophthalmol. 1996;114(9):1079–1084. [DOI] [PubMed] [Google Scholar]
- 23. Rosner B. Fundamentals of biostatistics. Boston: Brooks/Cole, Cengage Learning, 2011. [Google Scholar]
- 24. De Moraes CG. Anatomy of the visual pathways. J Glaucoma 2013;22:S2–S7. [DOI] [PubMed] [Google Scholar]
- 25. Noseworthy JH, Vandervoort MK, Wong CJ, Ebers GC. Interrater variability with the expanded disability status scale (EDSS) and functional systems (FS) in a multiple sclerosis clinical trial. The Canadian Cooperation MS Study Group. Neurology 1990;40(6):971–975. [DOI] [PubMed] [Google Scholar]
- 26. Amato M, Fratiglioni L, Groppi C, et al. Interrater reliability in assessing functional systems and disability on the kurtzke scale in multiple sclerosis. Arch Neurol 1988;45(7):746–748. [DOI] [PubMed] [Google Scholar]
- 27. Meyer‐Moock S, Feng Y‐S, Maeurer M, et al. Systematic literature review and validity evaluation of the expanded disability status scale (EDSS) and the multiple sclerosis functional composite (MSFC) in patients with multiple sclerosis. BMC Neurol 2014;14:58. [DOI] [PMC free article] [PubMed] [Google Scholar]