

1. Introduction
Nonalcoholic steatohepatitis (NASH) cirrhosis is a leading etiology of liver disease worldwide, paralleling the rise in the metabolic syndrome [1]. While nonalcoholic fatty liver disease affects approximately 25% of the global population [2], progression to NASH occurs less frequently and varies amongst individuals due to comorbid conditions and genetic risk factors [3]. Two recent population studies [4,5] implicate a single nucleotide polymorphism (SNP) in the Perilipin-2 (PLIN2) gene, rs35568725, in atherosclerosis and insulin resistance. This SNP codes a Ser251Pro variant and is associated with smaller, more numerous lipid droplets (LD) and reduced lipolysis in macrophages [4]. Germline and hepatic loss of PLIN2 have been shown to abrogate hepatic steatosis in animal models of NASH [6] but investigations of the PLIN2 Ser251Pro variant have not been reported in the context of liver disease.
Our study is the first to investigate rs35568725 in NASH, a disease driven by altered hepatocyte LD metabolism. We determined the prevalence of rs35568725 in a population of adults with and without NASH. To determine if the Pro251 variant of PLIN2 affects LD phenotype, we assessed hepatocyte LDs containing the wild type and Pro251 variants within an in vitro model of hepatic steatogenesis. Our findings suggest rs35568725 conveys risk for NASH in humans and that the mechanism may be due to phenotypic changes in hepatocyte LDs under steatogenic conditions.
2. Materials and methods
The Nebraska Biobank, a biorepository with human genomic DNA and linked deidentified health data was utilized. Subjects gave written informed consent and the study was approved by the University of Nebraska Medical Center Institutional Review Board. All subjects age ≥ 18 with a diagnosis of NASH excluding other etiologies of liver disease (viral, alcohol, autoimmune, etc.) were included (n = 116). Using previous population studies of rs35568725 with minor allele frequencies of 0.06, 0.05 [4], and 0.03 [5], and a 5% margin of error with a 95% confidence interval, a sample size of 67 age- and gender-matched controls lacking NASH and other liver disease was deemed appropriate for this pilot study.
Genomic DNA was extracted from blood with the DNA QIAcube HT kit (Qiagen, Germantown, MD USA). PCR of the PLIN2 region of interest was performed using forward (5′AGGTTAGAGTCCAGGCCTTAT3′) and reverse (5′GAATATGGAGACAGCTCACAGAA3′) primers, yielding a 408 bp product verified by agarose gel. Amplicons were purified using ExoSAP-IT™ PCR Cleanup Reagent (ThermoFisher Scientific, Waltham, MA USA) and sequenced using Applied Biosystems 3730 DNA Analysis Instrument (Life Technologies, Grand Island, NY USA). Two authors (CSF and LLJ) remained blinded to subject status and used SnapGene (GSL Biotech, Chicago, IL USA) to determine genotypes.
2.1. Genetic models and statistical analysis
The Hardy–Weinberg equilibrium, allelic odds ratio (OR) and genotypic OR using dominant and recessive models were calculated as reviewed [7]. To determine differences between groups for all variables, t-test, Mann-Whitney U and Chi-square test were used as indicated. p values < 0.05 were deemed significant. All analyses were performed using GraphPadPrism 8.1.2 (GraphPad Software, La Jolla, CA USA).
2.2. In vitro phenotype of PLIN2 variants
Using site-directed mutagenesis (QuikChange Lightning, Agilent, Santa Clara, CA USA), the Pro251 allele was insetted into the green fluorescent protein (GFP)-PLIN2 plasmid (Addgene #87161). Sequences were verified in forward and reverse directions using primers EGFP-N and SV40pA-R, respectively. Huh7 hepatocytes were reverse transfected using Lipofectamine2000 (ThermoFisher Scientific, Waltham, MA USA) and steatosis was induced for 48 h with 0.8 mM oleic acid (OA) as described [8]. For all experiments with imaging endpoints, cells were fixed for 10 min at room temperature with 4% paraformaldehyde. To visualize endoplasmic reticulum (ER), cells were incubated at 4 °C for 16 h with 1:100 calnexin monoclonal antibody (AF18, Invitrogen, Waltham, MA USA) followed by 1:150 goat anti-mouse Alexa Flour 555 secondary antibody (A-21422 Invitrogen, Waltham, MA USA) for 1 h at 37 °C. Neutral lipid within cells was demarcated with HCS LipidTOX Red Neutral Lipid Stain (ThermoFisher Scientific, Waltham, MA USA) diluted 1:2000 in PBS for 1 h at 37 °C. Images were taken using an epifluorescence microscope (Axio Observer; Carl Zeiss MicroImaging) using the 63× objective and were adjusted with Zen Black software (Carl Zeiss MicroImaging) uniformly to the entire image. For comparative morphometries of GFP-PLIN2 and GFP-PLIN2Pro251 coated LDs, > 10 cells with similar expression levels under all experimental conditions from three independent experiments were photographed using confocal (Zeiss LSM 800 with Airyscan, Carl Zeiss, Obercohen, Germany) and epifluorescent microscopy (Echo Revolve, San Diego, CA USA) and quantified using ImageJ software as described [9]. LD measurements are expressed as mean ± one standard deviation. For quantification of intracellular triglycerides, steatotic cells were trypsinized, stained with propidium iodide (PI) and sorted by flow cytometry (BD Biosciences LSRFortessa, San Jose, CA USA) to select for viable, transfected cells (GFP-positive, PI-negative). Sorted cells were pelleted via centrifugation and lysed with 1% triton x-100 in PBS. Total cellular protein was quantified with the Bradford dye-binding method (BioRad, Hercules, CA USA) and triglyceride was measured with Triglyceride (GPO) Reagent Set (Pointe Scientific, Canton, MI USA). Four independent replicates were performed and data were subjected to the Wilcoxon matched-pairs signed rank test using GraphPadPrism 8.1.2 (GraphPad Software, La Jolla, CA USA).
3. Results
Control and NASH groups were matched for gender and age and discordant for comorbidities associated with NASH as expected (Supplemental Table 1). rs35568725 was in Hardy–Weinberg equilibrium (χ2 3.52, p < 0.05) in our subject population. Of 116 NASH subjects, 81.9% (n = 95) lacked, 15.5% (n = 18) had a single copy, and 2.66% (n = 3) had two copies of rs35568725. Of 67 controls, 92.5% (n = 62) lacked, 7.5% (n = 5) had one copy and none were homozygous for rs35568725 (Fig. 1A). Due to its lower frequency, rs35568725 was deemed to be the minor allele (a) and wild type was deemed the major allele (A). Allelic OR for NASH conveyed by the minor allele was found to be 2.98 (1.12–7.31, p = 0.02) (Fig. 1B). In a dominant inheritance model, the OR for NASH conveyed by the minor allele was 2.74 (1.01–6.92, p = 0.04) (Fig. 1B). Due to lack of minor allele homozygotes within the control cohort, a recessive model of inheritance was indeterminate. Within the NASH cohort, rs35568725 carriers did not have higher rates of common NASH comorbidities including obesity, type 2 diabetes and hyperlipidemia or cirrhosis compared to non-carriers (Supplemental Table 2).
Fig. 1.
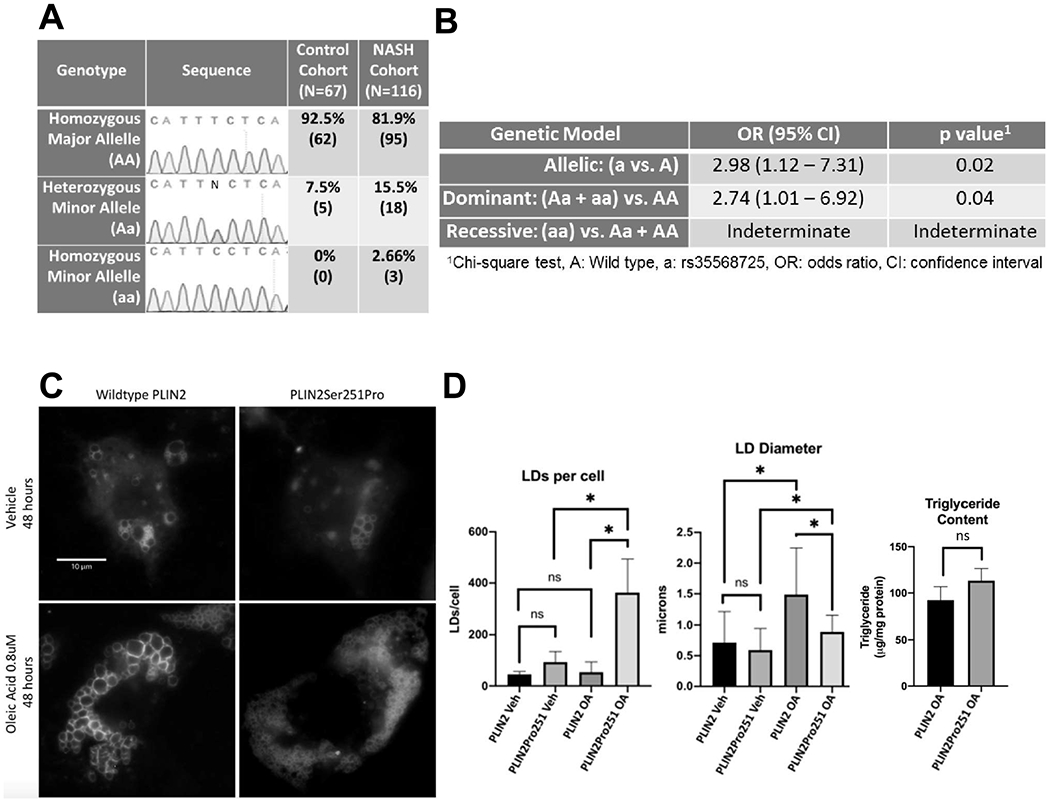
A) Representative electropherograms and distribution of PLIN2 genotypes in study subjects, stratified by control and NASH cohorts. B) Allelic, dominant and recessive inheritance models and odds ratios (OR) for NASH conveyed by the rs35568725, aka the minor allele (a). C) Fluorescence microscopy of representative single Huh7 hepatocytes expressing green fluorescent protein (GFP) tagged-PLIN2 (left) and GFP-PLIN2Pro251 (right) localized to LDs treated with vehicle (top) and 0.8 μm oleic acid for 48 h (bottom), scale bar = 10 μm. D) LDs per cell (left) and LD diameter (middle) quantified from ≥10 cells with similar GFP-PLIN2 and GFP-PLIN2Pro251 expression levels under all experimental conditions from 3 independent experiments (N = 3) and triglyceride levels (right) of steatotic flow cytometry-sorted GFP-positive Huh7 hepatocytes expressing GFP-PLIN2 versus GFP-PLIN2Pro251 normalized to total cellular protein (N = 4). Veh, vehicle treatment; OA, oleic acid 0.8 mM 48 h; ns, not significant; error bar = 1 standard deviation. *p < 0.05.
Within transfected Huh7 human hepatocytes, overexpressed GFP-PLIN2 and GFP-PLIN2Pro251 localized to neutral lipid-containing, cytosolic LDs (Supplemental Fig. 1). Under basal conditions, hepatocytes expressing GFP-PLIN2Pro251 compared to GFP-PLIN2 showed similar diameter LDs (0.59 ± 0.35 μm vs 0.71 ± 0.51 μm, p = 0.09) and number of LDs per cell (93 ± 41.68 vs 45.33 ± 10.69 p = 0.13) (Fig. 1C, D). Under steatogenic conditions however, hepatocytes expressing GFP-PLIN2Pro251 had smaller diameter (0.89 ± 0.27 vs 1.49 ± 0.76 μm, p ≤ 0.0001) and more numerous LDs per cell (362.7 ± 131.2 vs 53.67 ± 40.15, p = 0.0175) than did GFP-PLIN2 expressing hepatocytes (Fig. 1C, D). Cells expressing GFP-PLIN2Pro251 showed a modest, yet insignificant trend toward harboring more triglyceride after treatment with OA than cells expressing GFP-PLIN2 (113.1 ± 14.42 vs 92.34 ± 13.18 μg/mg cellular protein, p = 0.06).
4. Discussion
Within a discovery cohort, we found a SNP within the PLIN2 gene, rs35568725, may convey risk for NASH. In our population the allelic OR for NASH in subjects carrying rs35568725 was ~3 compared to non-carriers. rs35568725 may not portend a more aggressive trajectory of liver disease, as carriers had the same rate of cirrhosis as non-carriers in the NASH group. Limitations of this pilot study are the racial homogeneity of our population and relatively small sample size. No subjects in our control cohort were homozygous for rs35568725 precluding use of additional genotypic models of inheritance (e.g. recessive).
Our in vitro work herein reveals the impact that the PLIN2Pro251 variant protein has on hepatocyte LD phenotype. Under basal conditions, hepatocyte LDs harboring wild type PLIN2 and PLIN2Pro251 are similar in size and abundance. Under 48 h of steatogenic conditions however, hepatocytes expressing PLIN2Pro251 become heavily laden with small neutral lipid-filled LDs, harboring up to ~7 fold more LDs that are approximately half the diameter of LDs circumscribed with wild type PLIN2. LDs in PLIN2Pro251 expressing hepatocytes also appear more homogeneous in phenotype, with less size variability than those in wild type PLIN2 expressing hepatocytes. Most interesting is that this striking increase in LD number does not convey a significant increase in lipid storage capacity (volume) but more so an increase in LD-associated protein docking capacity (surface area). This underscores the importance of the LD as a protein storage depot [10] with a dynamic proteome which signals within and beyond the regulation of lipid storage [11]. The profound increase in tiny, homogeneous LDs within hepatocytes expressing PLIN2Pro251 is reminiscent of hepatocytes with microvesicular steatosis. To determine if this polymorphism conveys risk for this poorly understood type of fatty liver disease [12], we are currently surveying the prevalence of the rs35568725 polymorphism amongst individuals with hepatic microvesicular (as opposed to macrovesicular) steatosis.
As PLIN2 plays regulatory roles in many aspects of LD biology, we are undertaking a comprehensive mechanistic assessment of PLIN2Pro251. Others have shown that PLIN2Pro251 decreases lipolysis in macrophages [4] via a tertiary change in PLIN2, which may decrease hormone sensitive lipase [13] or adipose triglyceride lipase [14] docking to PLIN2 on the LD surface. While we observed a modest trend of increased triglyceride within cells harboring PLIN2Pro251, this tertiary change could affect a number of other protein–protein or protein-organelle interactions on the surface of the LD. Our work demonstrates that the Pro251 mutation does not cause LD sequestration within the ER however it is possible that this variant could impact phosphatidylcholine cycling between the ER and the LD, an event controlling LD size, number and microvesicular steatosis phenotype [15]. Further research is needed to delineate how the Pro251 variant impacts rate of LD biogenesis and scission from the ER [16] as well as LD fusion [17] and autophagy [14] (Supplemental Fig. 2).
In summary, we demonstrate that a coding SNP of PLIN2, rs35568725, may convey risk for NASH in a discovery cohort. When expressed under steatogenic conditions, human hepatocytes harboring the PLIN2Pro251 variant protein become heavily laden with small, homogenous LDs. More work is needed to validate rs35568725 as a risk factor for NASH in broader, more diverse populations and to delineate the mechanism(s) of its effects on LD phenotype.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bbalip.2020.158637.
Supplementary Material
Acknowledgements
We thank Ms. Mary Anne Phillippi and Dr. Cody Wehrkamp (UNMC) for assistance with PCR and cell culture. We acknowledge U54GM115458/NIGMS NIH HHS/United States, the Great Plains IDeA-CTR grant and the Nebraska Research Initiative, which subsidize the Nebraska Biobank repository. We also acknowledge the Mayo Clinic Center for Cell Signaling in Gastroenterology Optical Microscopy Core supported by NIH P30DK084567, the UNMC Advanced Microscopy Core Facility, the UNMC Center for Cellular Signaling CoBRE NIH P30GM106397 and the UNMC DNA Sequencing Core Facility, supported by NE-INBRE P20GM103427-14, CoBRE P30GM110768 and P30CA036727.
Funding sources
LLJ has received support from the UNMC College of Medicine Program of Excellence Physician-Scientist Training Program and the Mayo Foundation.
Footnotes
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, et al. , Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention, Nat. Rev. Gastroenterol. Hepatol 15 (2017) 11. [DOI] [PubMed] [Google Scholar]
- [2].Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M, Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes, Hepatology 64 (1) (2016) 73–84. [DOI] [PubMed] [Google Scholar]
- [3].Dongiovanni P, Anstee QM, Valenti L, Genetic predisposition in NAFLD and NASH: impact on severity of fiver disease and response to treatment, Curr. Pharm. Des 19 (29) (2013) 5219–5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Magne J, Aminoff A, Perman Sundelin J, Mannila MN, Gustafsson P, Hultenby K, et al. , The minor allele of the missense polymorphism Ser251Pro in perifipin 2 (PLIN2) disrupts an alpha-helix, affects lipolysis, and is associated with reduced plasma triglyceride concentration in humans, FASEB J. 27 (8) (2013) 3090–3099. [DOI] [PubMed] [Google Scholar]
- [5].Sentinelfi F, Capoccia D, Incani M, Bertoccini L, Severino A, Pani MG, et al. , The perifipin 2 (PLIN2) gene Ser251Pro missense mutation is associated with reduced insulin secretion and increased insulin sensitivity in Italian obese subjects, Diabetes Metab. Res. Rev 32 (6) (2016) 550–556. [DOI] [PubMed] [Google Scholar]
- [6].Najt CP, Senthivinayagam S, Aljazi MB, Fader KA, Olenic SD, Brock JR, et al. , Liver-specific loss of Perilipin 2 alleviates diet-induced hepatic steatosis, inflammation, and fibrosis, Am. J. Physiol. Gastrointest. Liver Physiol 310 (9) (2016) G726–G738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lewis CM, Genetic association studies: design, analysis and interpretation, Brief. Bioinform 3 (2) (2002) 146–153. [DOI] [PubMed] [Google Scholar]
- [8].Lu S, Mott JL, Harrison-Findik DD, Saturated fatty acids induce post-transcriptional regulation of HAMP mRNA via AU-rich element-binding protein, human antigen R (HuR), J. Biol. Chem 290 (40) (2015) 24178–24189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Jophlin LL, Koutalos Y, Chen C, Shah V, Rockey DC, Hepatic stellate cells retain retinoid-laden lipid droplets after cellular transdifferentiation into activated myofibroblasts, Am. J. Physiol. Gastrointest. Liver Physiol 315 (5) (2018) G713–G721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Welte MA, Gould AP, Lipid droplet functions beyond energy storage, Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1862 (10 Pt B) (2017) 1260–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bersuker K, Olzmann JA, Establishing the lipid droplet proteome: mechanisms of lipid droplet protein targeting and degradation, Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1862 (10 Pt B) (2017) 1166–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Tandra S, Yeh MM, Brunt EM, Vuppalanchi R, Cummings OW, Ünalp-Arida A, et al. , Presence and significance of microvesicular steatosis in nonalcoholic fatty liver disease, J. Hepatol 55 (3) (2011) 654–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wang H, Hu L, Dalen K, Dorward H, Marcinkiewicz A, Russell D, et al. , Activation of hormone-sensitive lipase requires two steps, protein phosphorylation and binding to the PAT-1 domain of lipid droplet coat proteins, J. Biol. Chem 284 (46) (2009) 32116–32125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kaushik S, Cuervo AM, Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis, Nat. Cell Biol 17 (6) (2015) 759–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Nicholls HT, Hornick JL, Cohen DE, Phosphatidylcholine transfer protein/StarD2 promotes microvesicular steatosis and fiver injury in murine experimental steatohepatitis, Am. J. Physiol. Gastrointest Liver Physiol 313 (1) (2017) G50–G61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Henne M, Goodman JM, Hariri H, Spatial compartmentalization of lipid droplet biogenesis, S1388-1981(19)30128-3, Biochim Biophys Acta Mol Cell Biol Lipids. 1865 (1) (2020), 10.1016/j.bbalip.2019.07.008 Epub 2019 Jul 25. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gao G, Chen FJ, Zhou L, Su L, Xu D, Xu L, et al. , Control of lipid droplet fusion and growth by CIDE family proteins, Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1862 (10 Pt B) (2017) 1197–1204. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.