

Abstract
OBJECTIVE:
12-LOX (12-lipoxygenase) produces a number of bioactive lipids including 12(S)-HETE that are involved in inflammation and platelet reactivity. The GPR31 (G-protein–coupled receptor 31) is the proposed receptor of 12(S)-HETE; however, it is not known whether the 12(S)-HETE-GPR31 signaling axis serves to enhance or inhibit platelet activity.
APPROACH AND RESULTS:
Using pepducin technology and biochemical approaches, we provide evidence that 12(S)-HETE-GPR31 signals through Gi to enhance PAR (protease-activated receptor)-4–mediated platelet activation and arterial thrombosis using both human platelets and mouse carotid artery injury models. 12(S)-HETE suppressed AC (adenylyl cyclase) activity through GPR31 and resulted in Rap1 (Ras-related protein 1) and p38 activation and low but detectable calcium flux but did not induce platelet aggregation. A GPR31 third intracellular (i3) loop–derived pepducin, GPR310 (G-protein–coupled receptor 310), significantly inhibited platelet aggregation in response to thrombin, collagen, and PAR4 agonist, AYPGKF, in human and mouse platelets but relative sparing of PAR1 agonist SFLLRN in human platelets. GPR310 treatment gave a highly significant 80% protection (P=0.0018) against ferric chloride–induced carotid artery injury in mice by extending occlusion time, without any effect on tail bleeding. PAR4-mediated dense granule secretion and calcium flux were both attenuated by GPR310. Consistent with these results, GPR310 inhibited 12(S)-HETE–mediated and PAR4-mediated Rap1-GTP and RASA3 translocation to the plasma membrane and attenuated PAR4-Akt and ERK activation. GPR310 caused a right shift in thrombin-mediated human platelet aggregation, comparable to the effects of inhibition of the Gi-coupled P2Y12 receptor. Co-immunoprecipitation studies revealed that GPR31 and PAR4 form a heterodimeric complex in recombinant systems.
CONCLUSIONS:
The 12-LOX product 12(S)-HETE stimulates GPR31-Gi–signaling pathways, which enhance thrombin-PAR4 platelet activation and arterial thrombosis in human platelets and mouse models. Suppression of this bioactive lipid pathway, as exemplified by a GPR31 pepducin antagonist, may provide beneficial protective effects against platelet aggregation and arterial thrombosis with minimal effect on hemostasis.
Keywords: blood platelets, calcium, lipids, platelet activation, thrombosis
Graphical Abstract
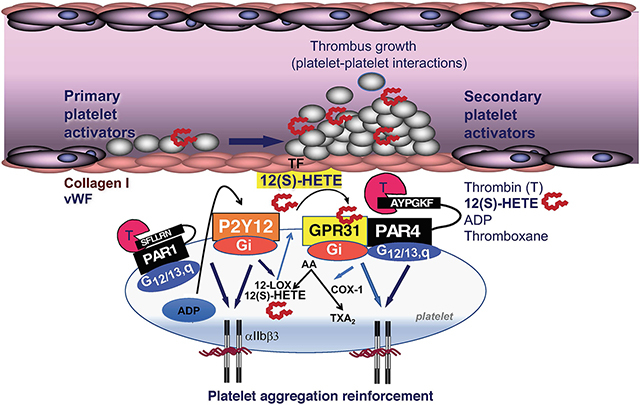
Platelets play an essential role in the pathogenesis of coronary artery disease with coronary thrombosis and occlusion being the ultimate pathogenic mechanism leading to myocardial ischemia and infarction.1 Thrombin is the most potent agonist of platelets, causing the fastest increase in platelet shape change and integrin engagement, thereby playing a critical role in the development of thrombosis under conditions of arterial shear stress.2,3 Thrombin activates platelets by cleaving the cell surface PARs (protease-activated receptors) PAR1 and PAR4, both of which play a critical role in platelet aggregation and arterial thrombus formation in humans.4 As chronic inhibition of the high-affinity PAR1 receptor with the long-acting oral agent, vorapaxar,5 has led to a significant increase in bleeding,6,7 recent interest in targeting the PAR4 thrombin receptor has been enhanced.8,9 Despite being a lower affinity receptor for thrombin,10,11 PAR4 plays a major role in the stability of platelet-platelet aggregates and arterial thrombosis in both humans and rodents.4,12,13
PAR1 and PAR4 are members of the GPCR (G-protein–coupled receptor) superfamily, which are important in regulating normal cardiovascular functions and comprise the largest group of pharmacologically active receptors in vascular cells and circulating platelets.14 Cardiovascular GPCRs respond to a diversity of water- and lipid-soluble agonists in both a paracrine and autocrine manner.15,16 Thrombin activation of PAR1 and PAR4 triggers G12/13 and Gq signaling, which leads to rapid platelet shape change and biphasic calcium flux.10,17 The P2Y12 ADP receptor, the target of clopidogrel and other thienopyridines, acts solely through Gi, which does not cause platelet aggregation but instead serves to strengthen the avidity of integrin αIIbβ3-fibrinogen interactions and amplify signaling induced by other primary receptors such as PAR1 and PAR4.17
The most widely used antithrombotic agent, namely aspirin, prevents COX (cyclooxygenase) 1/2–dependent production of TXA2 thromboxane from arachidonic acid. In a less known pathway, arachidonic acid generated from cytosolic phospholipase A2 is also available for oxidation by LO (lipoxygenase) enzymes.18 LOs are a family of nonheme iron dioxygenases that catalyze the oxidation of polyunsaturated fatty acids to conjugated hydroperoxidases.19 12-LOX (12(S)-LO) is highly expressed in platelets20 and is known to potentiate the activation of signaling pathways via PAR4 and the ITAM (immunoreceptor tyrosine activation motif)-containing receptor complex, GPVI-FcγRIIa.21 Inhibition of 12-LOX in platelets has been shown to attenuate thrombin-mediated calcium flux and platelet aggregation and FcγRIIa-mediated platelet activation.22,23 The oxidation of arachidonic acid by 12-LOX results in the production of a number of bioactive lipids including the metabolite 12(S)-HETE.24,25 Guo et al24 identified the lipid receptor GPR31 (G-protein–coupled receptor 31)—an orphan class A GPCR—as a 12-HETE receptor that was involved in regulatory inflammatory signals in prostate PC3 cells, but it is unknown whether GPR31 mediates 12(S)-HETE signaling in platelets. Here, we show that GPR31 is a Gi-coupled receptor expressed in platelets, which signals through 12(S)-HETE to potentiate platelet aggregation, calcium flux, and dense granule release through the PAR4 thrombin receptor. We discovered that GPR31 and PAR4 form a stable heterodimer complex and provide evidence that GPR31/PAR4 functions in partnership to modulate platelet activation. A new GPR31 cell-penetrating pepducin, GPR310 (G-protein–coupled receptor 310), was used to delineate the role of GPR31 in thrombin-mediated platelet activation both in human platelets and in mouse models of arterial thrombosis and hemostasis.
MATERIALS AND METHODS
The authors declare that all supporting data are available within the article and its Data Supplement.
Human Platelet RNA Extraction, cDNA Generation, and Quantitative Reverse Transcriptase Polymerase Chain Reaction (Real-Time RT-PCR)
Gel-purified human platelets were prepared as described previously,26 and the total RNA was extracted using RNeasy Mini kit (Qiagen; catalog No. 74104) according to the manufacturer’s instructions. cDNA preparation was performed using MMLV RT (Moloney murine leukemia virus reverse transcriptase) enzyme (Invitrogen; catalog No. 28025–013). Gene-specific primers are GPR31-F 5′-ATCATCTGGCAGGAAGCACT-3′; GPR31-R 5′-AGGGCAGAAAGCACAGAGCA −3′; PAR1-F 5′-GTCTATCATTCGATGTCTTAGC-3′; PAR1-R 5′-TAGACGTACCTCTGGCACTC- 3′; PAR4-F 5′-AACCTCTATGGTGCCTACGTGC-3′; PAR4-R 5′-CCAAGCCCAGCTAATTTTTG-3′; β-actin-F: 5′-GGCTCTTCCAGCCTTCCTTCCT-3′; β-actin-R 5′-CACAGAGTACTTGCGCTCAGGAGG-3′. Actin was used as control. Semiquantitative and quantitative analysis was performed.
Cre-Luciferase Activation Assay
Chinese hamster ovary (CHO) cells were cotransfected (10×10 cm plates) with 8 μg each of Cre (cAMP-responsive element)-luciferase or GPR31 plasmid DNA and 40 μL of lipofectamine (ThermoFisher Scientific). Transfected cells were plated in 96-well plates and treated with 10-μM forskolin and ±300 nmol/L 12(S)-HETE for 6 hours after which cells were lysed and luciferase activity was measured (relative light units).
Rap 1 and RASA3 Activation Assay
GPR310 is a GPR31 i3 loop–derived 21 amino acid pepducin10 that is conjugated to palmitate lipid to form an N-palmitoylated peptide, which has a molecular formula of C125H225N41O30 (MW 2782.4 Da). GPR310 was synthesized by fluorenylmethoxy-carbonyl protecting group solid-phase chemistry and purified to >99% purity by reverse phase high-performace liquid chromatography as described previously.27 Gel-purified platelets were stimulated with up to 160 μM AYPGKF for 5 minutes, 15 minutes, 30 minutes, and 1 hour at 37 °C and 0 time (vehicle control). Samples were pretreated with 30 μM AZD1283, plus 160 μM AYPGKF for 5 minutes, 15 minutes, 30 minutes, and 1 hour at 37 °C and 0 time (vehicle control). Platelet lysates were prepared using extraction lysate buffer (25 mmol/L tris(hydroxymethyl)aminomethane [Tris])-HCl at pH 7.5, 150 mmol/L NaCl, 5 mmol/L MgCl2, 1% Nonidet P-40, 1 mmol/L dithiothreitol, 5% glycerol, 1 g/mL aprotinin, 1 g/mL leupeptin, and 1 mmol/L phenylmethylsulphonyl fluoride on ice for 10 minutes; cells were centrifuged at 14 000 g for 10 minutes at 4 °C. Aliquots of the lysate were used to detect total Rap1 (Ras-related protein 1) levels. The remaining supernatants were incubated with 50 μL of Ral GDS-RBD (GPS Rap-binding domain) agarose slurry (EMD Millipore, MA) for 60 minutes at 4 °C. Proteins were separated by 15% SDS-PAGE and transferred to nitrocellulose membranes (BioRad, Hercules, CA). Rap1 levels were detected with rabbit polyclonal antibodies (EMD Millipore; catalog No. 07–916) followed by HRP (horseradish peroxidase)-conjugated goat anti-rabbit antibodies (Zymed, South San Francisco, CA). For RASA3 (Ras P21 protein activator 3), platelet lysates were ultracentrifuged at 100K for 2 hours at 4 °C and membrane protein quantified using the Bradford assay, equally loaded and resolved by SDS-PAGE and Western blot and Rasa3 levels detected with Rasa3 Ab (Proteintech; No. 27835–1-AP).
Platelet Aggregation Studies
In accordance with the informed consent procedures approved by the Tufts Medical Center Institutional Review Board, venous whole blood was drawn from healthy volunteers with an 18-gauge needle into a 30-mL syringe containing 3 mL of 4% sodium citrate (0.4% vol/vol final). Platelets were isolated by gel filtration chromatography of platelet-rich plasma (PRP) with the use of Sepharose 2B (Pharmacia, Piscataway, NJ) in modified PIPES (piperazine-N,N’-bis(2-ethanesulfonic acid) buffer.12 Platelet aggregation was measured with the use of a Chrono-log 560VS/490 aggregometer (Chrono-Log Corporation, Havertown, PA) after induction with the agonists AGPYK, SFLLRN, thrombin, collagen, or vehicle with and without pretreatment with GPR310 pepducin, 12-LOX inhibitor ML355 (Selleckchem), or P2Y12 inhibitor AZD1283 (BioVision, Milpitas, CA). Reactions were conducted in final volumes of 250 μL at 37 °C, stirring at 900 rpm. For dual granule release and aggregation studies, ATP release was recorded using a Model 700 Aggregometer followed by the addition of luciferase substrate/luciferase mixture (12.5 μL; Chrono-Log) and calibrated using ATP standard control.
Fluorescence-Activated Cell Sorting Analysis
Gel-purified platelets were fixed with 4% paraformaldehyde in PBS for 30 minutes at room temperature (RT) washed, and pellet resuspended in 0.5% Triton X-100/ PBS for 30 minutes at RT. After washing (PBS), platelets are incubated with primary antibody for GPR31, PAR1 (ATAP22), and PAR4 (GYP-18) polyclonal ab (GYPGQVSANDSDTLELPC) for 45 minutes at RT. After washing with PBS, cells are incubated with fluorescein isothiocyanate secondary antibody for 45 minutes at RT. Mean fluorescence was quantified on a BD Canto II flow cytometer.
Immunoprecipitation of PAR4 and GPR31
CHO cells (10×10 cm plates) were cotransfected with 8 μg each of T7-PAR4 and GPR31 plasmid DNA and 40 μL of lipofectamine (ThermoFisher Scientific). After 48 hours of transfection, immunoprecipitation buffer containing 0.11% (vol/vol) Triton X-100, 50 mmol/L Tris pH 7.4, 100 mmol/L NaCl, 5 mmol/L EDTA, 50 mmol/L NaF, and 1 mmol/L phenylmethylsulphonyl fluoride was used to prepare the cell lysate (750 μL per plate). The sample was sonicated, clarified by centrifugation, and the supernatant transferred to a 2-mL Eppendorf tube. Following Bradford assay to assess protein concentration, 3 mg of protein was taken from each immunoprecipitation buffer and incubated for 24 hours with 50 μL of T7 agarose beads (Novagen, Madison, WI) or with GPR31-Ab (40 μg)-protein A/G PLUS-Agarose beads overnight at 4 °C in a final volume of 2 mL. The agarose beads were washed 2× with cold immunoprecipitation buffer and once with 0.1% SDS in immunoprecipitation buffer. For immunoprecipitation without PNGase F, samples were incubated with 1× SDS loading dye at 42 °C for 45 minutes to elute protein. For immunoprecipitation with PNGase F, 5 μL (500 U) PNGase F enzyme (Biolabs, Beverly, MA) was added to 35 μL of distilled water, 5 μL 10× deglycosylation buffer (0.5 M sodium phosphate), and 5 μL 10% NP40, for a total volume of 50 μL and incubated at 37 °C overnight. The sample was incubated with 10 μL of 6× SDS loading dye at 42 °C for 45 minutes to elute the protein. The sample was resolved by SDS-PAGE and Western blot using the GPR31-Ab (Santa Cruz Biotechnology, CA) and PAR4-Ab.10,28 Immunoprecipitation of human platelet PAR4 was performed by preparing the membrane fraction by ultracentrifuge. Membrane was treated with 100 nmol/L thrombin for 15 minutes and used in pull-down assay with the GPR31 antibody coupled to Cyanogen bromide–activated Sepharose beads (Amersham, Uppsala, Sweden) with a procedure similar to that described above. The platelet lysates and GPR31-Ab beads were incubated for overnight at 4 °C.
Calcium Studies
Following gel purification, platelets were incubated with 2.5 μM fura-2/AM (molecular probes) for 30 minutes at 37 °C. Platelets were centrifuged at 800 rpm for 5 minutes without brake. Pellets were resuspended in PIPES-buffered saline (25 mmol/L PIPES, 137 mmol/L NaCl, 4 mmol/L KCl, and 0.1% glucose, pH 6.7). Calcium flux was performed using a LS508 spectrometer, and fluorescent emission was recorded at 510 nm and dual excitation at 340 and 380 nm as described.4
Clot Retraction Assay
PRP was adjusted to a platelet count of 150 000 platelets/mL in borosilicate 12×75 mm glass tubes (VWR International). Per tube, 200 μL of PRP without EDTA added was combined with 800 μL of PBS and 10 μL of red blood cells. Calcium chloride was added for a final concentration of 0.5 mmol/L tube. Following treatment with GPR310 pepducin or AZD1283 (10 and 30 μM), clot formation and retraction was initiated with 3 nmol/L thrombin (Haematologic Technologies, Essex Junction, VT) and 10 nmol/L thrombin at RT. Digital photographs were taken of the clots at 0 minutes, 5 minutes, 10 minutes, 15 minutes, 30 minutes, 1 hour, 2 hours, and 4 hours.
Mouse Carotid Artery Injury Model
All procedures and manipulations of animals were approved by the Institutional Animal Care and Use Committee of the Tufts Medical Center in accordance with the US Public Health Service Policy on the Humane Care and Use of Animals and the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Six- to 8-week-old male CD-1 mice were purchased from Charles River Laboratories (Wilmington, MA). Male mice were used because previous data showed that there were no sex-related differences in injury. Ferric chloride (FeCl3) injury of the carotid artery was performed as described previously.10,29 Briefly, occlusion of the carotid artery was measured by recording blood flow with a Transonic Animal Research Flowmeter, model T106/T206 series. A 0.5 V, V-Series flow probe was placed on the carotid artery isolated by dissection and adjusted to ensure baseline flow between 0.3 and 0.9 mL/s. 10% FeCl3 was applied to filter paper (5×10 mm) and the filter paper placed around the carotid artery caudal to the probe. After 5 minutes, the filter paper was removed, and the flow was measured. Initial and final data points were collected per second, and the time to achieve a stable occlusion was calculated. An occlusion was determined to be stable when the flow was reduced to 0 mL/s for at least 30 seconds.
Assessment of Tail Bleeding Time
The tail bleeding time assay was performed as described previously.4 Four hours after subcutaneous administration of GPR310 (5 and 10 mg/kg) or vehicle, mice were anesthetized and placed on a homeothermic blanket (37 °C) before the tail was transected 5 mm from the tip using a sterile scalpel. After transection, the tail was immediately immersed in saline (37 °C), and the time to bleeding cessation was measured. Bleeding stoppage was not considered complete until bleeding had ceased for 1 minute.
Pharmacokinetics of Subcutaneously Injected Pepducins in Mouse Plasma
CD-1 mice were injected subcutaneously with 10 mg/kg of GPR310 in 10% dimethyl sulfoxide, and blood was collected at various time points from the vena cava into 5 μL 1000 U heparin. Pepducins were extracted and injected onto a liquid chromatography–mass spectrometry (NovaBioAssays LLC).
Immunoblot Detection of p38, AKT, and ERK Phosphorylation in Human Platelets
Gel-purified platelets were incubated with 10 nmol/L 12(S)-HETE in the presence of 1 and 3 μM GPR310 or with 30 μM SFLLRN in a platelet aggregometer at 37 °C for various times. Platelets were solubilized in SDS-PAGE sample buffer.12 In other experiments, gel-purified platelets were incubated with 3 nmol/L thrombin in the presence or absence of 1 μM GPR310. Equal amounts of protein were resolved by 10% SDS-PAGE gel for Western analysis. The primary antibodies used were human phospho-p38 (Thr180/Tyr182; Cell Signaling; No. 4631), total p38 (Cell Signaling; No. 9212), phospho-Akt (Ser473; Cell Signaling; No. 4058S), total-Akt (No. 9272), phospho-Erk (Thr202/Tyr204; Cell Signaling; No. 4370), total-Erk (No. 9102S), and actin (Sigma Aldrich; No. A1978).
Statistical Analysis
Unless otherwise indicated, all results are presented as mean±SE.
For 2-group comparisons of parametric data, a 2-tailed distribution unequal variance Student t test was performed. For multiple-group comparisons, 2-way ANOVA tests were performed followed by Bonferroni post test analysis. Data analysis was performed using GraphPad PRISM (version 5.0a). Kaplan-Meier survival curves were analyzed using log-rank (Mantel-Cox) test.
Normality and variance were not tested. Significance was accepted at P<0.05.
RESULTS
GPR31 Couples to Gi and Is Expressed in Human Platelets
12(S)-HETE was originally found to bind with high affinity to GPR31 in recombinant systems and PC3 prostate cancer cells.30 Therefore, we set out to determine whether GPR31 is a downstream target of the 12-LOX metabolite 12-HETE in platelets. To validate that the GPR31 receptor was expressed in human platelets, we isolated platelet mRNA and performed transcriptional analysis for Gpr31, Par1, and Par4. Gpr31 receptor mRNA was highly expressed in human platelets, similar to Par1 and Par4 expression (Figure 1A). To validate these data, we obtained discarded platelets (on day 5) collected by plateletpheresis from our hospital qualified for human use. These platelets have <1:1×106 ratio of leukocyte contamination in the preparation:platelets. Similar polymerase chain reaction–based detection of transcript was observed as previously shown in Figure 1A using gel-purified human platelets (Figure IA in the Data Supplement). Therefore, there is an extremely low probability that the presence of the GPR31 transcript is due to leukocyte contamination. In addition, we also confirmed GPR31, PAR1, and PAR4 platelet membrane expression with FACS (fluorescence-activated cell sorting) analysis of human platelets as shown in Figure IA and IB in the Data Supplement.
Figure 1. Characterization of GPR31 (G-protein–coupled receptor 31) receptor antagonist in platelets and recombinant systems.
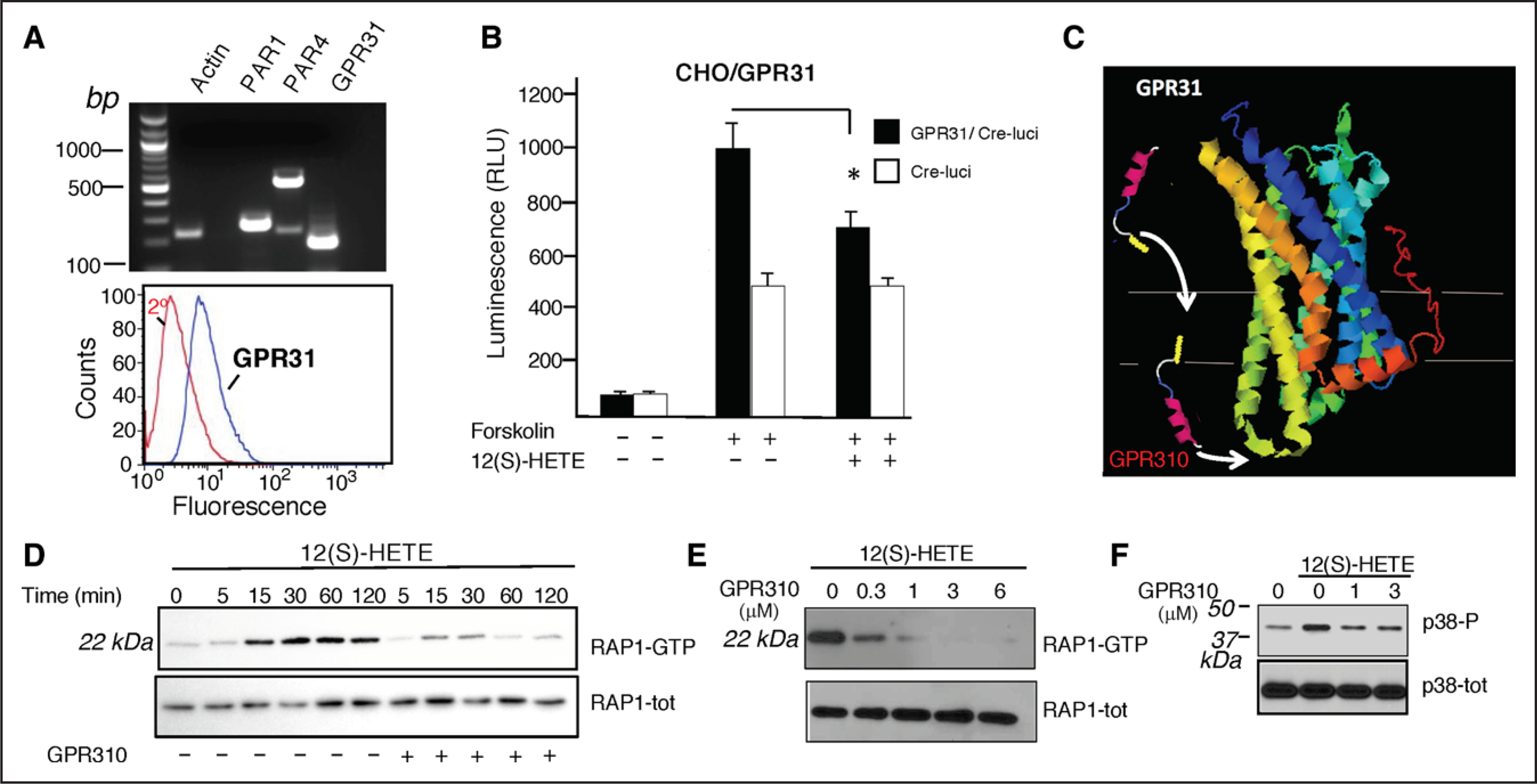
A, Top, Expression of mRNA encoding PAR (protease-activated receptor)-1, PAR4, and GPR31 in human platelets. Human platelets were gel purified, and total RNA was reverse transcribed using an oligo(dT), and cDNA was amplified with polymerase chain reaction (PCR) using specific sets of primers for PAR1, PAR4, GPR31, and actin described in the Methods section. RT-PCR products were run on a 1% agarose gel. A, Bottom, Gel-purified platelets were analyzed for GPR31 expression by flow cytometry using GPR31 specific ab. B, Chinese hamster ovary (CHO)-K1 cells expressing GPR31 and Cre (cAMP-responsive element) reporter stimulated with 10 μM forskolin in the presence or absence of 300 nmol/L 12(S)-HETE. C, Predicted GPCR (G-protein–coupled receptor) structural model of GPR31 and model of GPR310 (G-protein–coupled receptor 310) pepducin using GPCR-I-TASSER58 (CCR5 [C-C chemokine receptor type 5] PBID ID: 4MBS). D, Time course of Rap1 (Ras-related protein 1) activation in human platelets stimulated with 100 nmol/L 12(S)-HETE in the absence (−) or presence (+) of 1 μM GPR310. The lower panel shows Rap1 as loading control. E, Dose response of human platelets pretreated with GPR310 as indicated and activated with 100 nmol/L 12(S)-HETE. The lower panel shows Rap1 as loading control. F, Immunoblot showing 100 nmol/L 12(S)-HETE rapidly (15 min) activates p38 MAPK (mitogen-activated protein kinase) phosphorylation in human platelets. *P<0.05, unpaired 2-tailed t test.
Early studies26 showed that 12(S)-HETE can stimulate enhanced GTPγS (guanosine-5’-triphosphate γS) binding to GPR31-transfected cells but the coupled G proteins were not identified. CHO-K1 cells expressing GPR31 and a cAMP-responsive element–luciferase reporter showed 10-fold induction after stimulation with forskolin that activates AC (adenylyl cyclase) to produce cAMP. Addition of 12(S)-HETE resulted in a significant inhibition of AC activity in GPR31-transfected cells that was not observed in Cre-luci controls (Figure 1B), demonstrating a GPR31-Gi–mediated receptor response.
GPR310 Is an Antagonist of 12(S)-HETE Activity in Human Platelets
To develop an inhibitor against GPR31 using pepducin technology,26 we first created a model of GPR31 using the GPCR-Iterative Threading Assembly Refinement27,31,32 program that predicts protein structure based on homology modeling and threading to the known GPCR structural database. Iterative Threading Assembly Refinement modeled the 3-dimensional topological homology of GPR31 to the x-ray structure of the CCR5 (C-C chemokine receptor type 5) chemokine receptor33,34 (Figure 1C). A series of intracellular loop 1 (i1) to intacellular loop 3 (i3) pepducins were synthesized and screened in the GPR31-CHO cell Gi assay, culminating in the identification of the i3 loop–derived GPR310—a synthetic 21mer peptide that is conjugated to palmitate to form an N-palmitoylated lipopeptide. Next, we examined the effect of 12(S)-HETE on small GTPase Rap1 activation in human platelets—a key regulator of integrin αIIbβ3 outside-in signaling.34 12(S)-HETE strongly activated the Rap1-GTP signaling pathway with a robust signal detected at 15 to 120 minutes (Figure 1D). The GPR310 pepducin was a potent inhibitor of 12(S)-HETE–mediated Rap1-GTP activation with an IC50 (a half maximal inhibitory concentration) of 0.3 μM (Figure 1E). Lastly, 12(S)-HETE rapidly (15 minutes) activated p38 MAPK (mitogen-activated protein kinase) phosphorylation in human platelets, which are completely attenuated with 1 to 3 μM GPR310 (Figure 1F).
Specificity of GPR310 for GPR31 was tested against GPCRs including PAR1, PAR2, ADP receptor, IL (interleukin)-8 receptor CXCR1/2 (C-X-C chemokine receptor type 1 and type 2), and CXCR4 (C-X-C chemokine receptor 4) receptor. GPR310 (6 μM) did not inhibit by calcium flux assays in human neutrophils using 50 nmol/L IL-8 (CXCR1/2), 100 nmol/L SDF1α (CXCR4), or ADP receptors (20 μM ADP; Figure IIA in the Data Supplement). There was minimal inhibition of PAR1 using HEK293 (human embryonic kidney 293) recombinant system by calcium flux assay with PAR1 agonist peptide (100 μM SFLLRN) and no detected inhibition of recombinant PAR2 with PAR2 agonist peptide (100 μM SLIGRL) with GPR310 (6 μM; Figure IIB in the Data Supplement). Together, these data provide compelling evidence for GPR310 specificity against several diverse GPCRs.
GPR310 Blocks PAR4 and Collagen-Mediated Human Platelet Activation
The 2 thrombin-activated receptors on human platelets, the Gq-coupled high-affinity PAR1 and the low-affinity PAR4 result in activation of αIIbβ3, platelet aggregation, and arterial thrombosis, which are sensitive to inhibition of the P2Y12-Gi–coupled receptor.10,13,35,36 Our recent work29 suggested an additional P2Y12-independent signaling mechanism that contributes to the PAR4-mediated platelet activation. We hypothesized that Gi-coupled GPR31 may mediate an important alternative 12(S)-HETE autocrine mechanism in addition to the well-characterized Gi-coupled P2Y12 ADP receptor. Unlike ADP, which uses P2Y1-Gq coupling in addition to P2Y12-Gi to stimulate aggregation, 12(S)-HETE at a range of concentrations failed to effectively activate platelet aggregation in PRP human platelets (Figure 2A). We then tested the effect of the GPR31 blockade on PAR4-mediated platelet aggregation using the AYPGKF agonist. We showed that 3 to 10 μmol/L GPR310 significantly inhibited (P<0.01–0.05) the majority of PAR4-mediated platelet aggregation in human platelets (Figure 2B and 2D), similar to inhibition using the 12-LOX inhibitor, 30 μmol/L ML355.37 We then tested the effect of GPR31 blockade on PAR1-mediated platelet aggregation. As shown in Figure 2C, there was significant inhibition of 3 μM SFLLRN with 10 μmol/L GPR310 but no significant effect on 10 μmol/L SFLLRN. Consistent with the concentration-dependent effect of GPR310, ML355 (12-LOX inhibitor) also showed considerable inhibition with 3 μmol/L SFLLRN but not 10 μmol/L SFLLRN indicative of a potential role for the 12-LOX-GPR31 pathway with low amounts of PAR1 activation. Higher concentration of SFLLRN (10 and 30 μmol/L) overcomes both GPR31 and 12-LOX inhibition. Overall, GPR31 primarily inhibits PAR4 response with relative sparing of PAR1. This is especially prominent upon examining ATP release from dense granules (Figure 2D). We conducted more detailed studies with ATP (dense granule) release and an AYPGKF (EC50 [a half maximal effective concentration], ≈30–90 μmol/L) and SFLLRN (EC50, ≈3 μmol/L) dose response. Consistent with our previous results, there is a greater dependence of PAR4 on GPR31-mediated dense granule release as compared with PAR1. Collagen-mediated platelet aggregation was almost completely inhibited by GPR310 (P<0.001) to a similar extent as 12-LOX inhibitor (Figure 2F).
Figure 2. Role of GPR31 (G-protein–coupled receptor 31) in thrombin-mediated human platelet aggregation.

A, Effect of 5, 10, and 20 μmol/L 12(S)-HETE or 5 μM ADP (in red) on platelet aggregation using human platelet-rich plasma. B, Human platelets were preincubated for 2 min with 0, 3, and 10 μmol/L GPR310 (G-protein–coupled receptor 310) and 30 μmol/L ML355 before the addition of 160 μmol/L AYPGKF, and aggregation was assessed at 15 min. C, Gel-purified human platelets stimulated with 3, 10, and 30 μmol/L SFLLRN in the presence of 3 and 10 μmol/L GPR310 or 30 μmol/L ML355 as indicated. D, Platelet for ATP secretion was measured in gel-purified human platelets stimulated with indicated concentrations of AYPGKF and SFLLRN in the presence of 3 μmol/L GPR310 and vehicle. E, Platelet aggregation stimulated with 10 μg/mL collagen in the presence or absence of 3 μmol/L GPR310 or 30 μmol/L ML355. F, Platelets were stimulated with 10 nmol/L thrombin (T) in the presence of 3 μmol/L GPR310, 30 μmol/L 12-LOX (12-lipoxygenase) inhibitor (12-LOX-Inh; ML355), and 10 μmol/L P2Y12 ADP receptor Inh (AZD1283) as indicated. ANOVA followed by Bonferonni correction; *P<0.05, **P<0.01, ***P<0.001, unpaired 2-tailed t test.
To compare the relative contributions of GPR31, 12-LOX, and P2Y12 to platelet aggregation, thrombin-induced platelet aggregation was evaluated in the presence of GPR310, ML355, and the P2Y12 inhibitor AZD1283,38 respectively. Platelets were preincubated for 2 minutes with each inhibitor before thrombin activation. GPR310 markedly right shifted the thrombin aggregation curve by 5-fold, ML355 by 3-fold, and AZD1283 by 7-fold (Figure 2G). GPR310 and ML355 in combination with AZD1283 both further right shifted the thrombin activation curve by 19- and 10-fold, respectively, demonstrating synergy between P2Y12 and GPR31 on thrombin-platelet activation. We have addressed the synergism or additivity between GPR31 and P2Y12 inhibition for PAR1 and PAR4 using aggregation. As shown in Figure III in the Data Supplement, it appears that the PAR1 response is more dependent on P2Y12, whereas the PAR4 response is more dependent on GPR31—both Gi-coupled receptors. However, there is some synergy (superadditivity) between both receptors, GPR31 and P2Y12, upon AYPGKF stimulation, whereas PAR1 effects appear additive. Further studies are warranted to understand the complexity of this interesting and potentially important synergy between PARs, GPR31, and P2Y12.
GPR31 and PAR4 Form a Stable Heteromeric Complex
Because GPR31 provides assistance to PAR4-mediated platelet activation, we postulated that GPR31 and PAR4 may form a heterodimer on the platelet surface, as shown previously for PAR1-PAR4.39 To provide direct evidence that GPR31 and PAR4 can form stable heterodimers or heteromers, we performed co-immunoprecipitation experiments using CHO cells that were transiently transfected with T7-tagged PAR4 and GPR31. In human platelets and recombinant systems, PAR4 exists as both an N-linked Asn56 low glycosylated species that migrate at 32 to 38 kDa, with nonglycosylated PAR4 detected at 29 kDa and highly glycosylated species (monomer and homodimer) at >50 kDa as described previously.4 PAR4 strongly associated with GPR31 when GPR31-Ab was used in a pull-down assay for immunoprecipitation (Figure 3A). Conversely, GPR31 strongly associated with T7-PAR4 when T7-Ab was used in the pull-down (Figure 3B). Unlike PAR4, PAR1 did not form a stable complex with GPR31 (Figure 3C).
Figure 3. GPR31 (G-protein–coupled receptor 31) and PAR (protease-activated receptor)-4 form a heterodimer in Chinese hamster ovary (CHO)-K1 recombinant system.
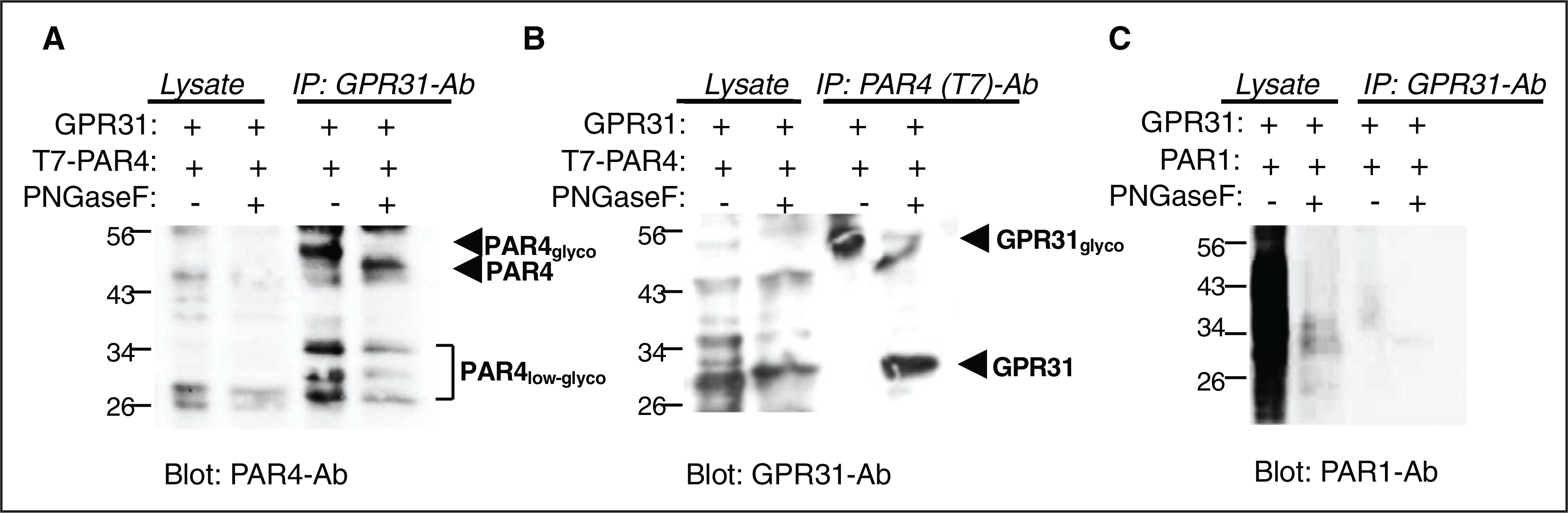
A and B, Co-immunoprecipitation (IP) experiments using T7-tagged PAR4 and GPR31 transiently transfected in CHO cells. Lysates from T7-PAR4/GPR31 cotransfected cells, co-IP with GPR31 ab, or T7 (PAR4) ab. C, Co-IP experiments using PAR1 and GPR31 transiently transfected in CHO cells were used as control. Western blot analysis performed using PAR4-ab, GPR31-ab, and ATAP2 PAR1-ab as indicated. For deglycosylated IP, washed beads were treated with PNFGase F (endoglycocidase removes N-linked oligosaccharides) enzyme overnight before eluting and running for Western blot analysis.
To determine whether GPR31/PAR4 stable interaction could be detected under physiological conditions, we isolated platelet membranes. We coupled GPR31-Ab to beads and conducted pull-down assays from human platelet membranes activated with 100 nmol/L thrombin and probed for PAR4 using the GYP-Ab as described in the Methods section. We have reported previously that the PAR4 GYP-Ab did not strongly stain uncleaved PAR4 from resting platelets; however, thrombin treatment of platelets produced a newly reactive glycosylated PAR4 monomer band migrating at 32 to 38 kDa and dimer at ≈95 kDa that was not present in untreated platelets.4 Treatment with PNGase F yielded a homogenous band migrating at 29 kDa, consistent with nearly complete removal of N-linked sugars from platelet PAR4. Using a similar approach, we coupled GPR31-Ab to beads and conducted pull-down assays from human platelet membranes activated with 100 nmol/L thrombin and probed for PAR4 using the GYP-Ab as described in the Methods section. As shown below in Figure IV in the Data Supplement, we were able to detect the expected pattern of bands that correspond to the different PAR4 species in human platelets thereby confirming that GPR31 and PAR4 form a heterodimer/heteromer in the relevant cell type under normal expression levels.
GPR31 Contributes to Thrombin-Platelet Clot Retraction
Thrombin-induced platelet clot retraction requires platelet integrin αIIbβ3-fibrin/fibrinogen engagement and actin-dependent contraction. PRP was preincubated with GPR310 pepducin, AZD1283 P2Y12 inhibitor, or vehicle and activated by 3 nmol/L thrombin. Clot retraction was evident by 15 minutes, and by 2 hours, there was extensive and complete clot retraction (P<0.0001) induced by thrombin in the absence of inhibitors (Figure 4A and 4B). Both the GPR310- and AZD1283-treated PRP had significantly (P<0.0001) longer clot retraction times compared with vehicle from 15 minutes to the 4-hour time point. Although GPR310 treatment was visually more protective (Figure 4A), it did not reach statistical significance compared with AZD1283. Similar inhibition of clot retraction was observed for GPR310 and AZD1283 using higher amounts of thrombin (10 nmol/L; Figure V in the Data Supplement). These data indicate that blockade of GPR31 may afford similar protective effects as P2Y12 inhibition in thrombin-induced platelet activity on a longer time scale.
Figure 4. Effect of inhibition of GPR31 (G-protein–coupled receptor 31) on thrombin initiated clot retraction.
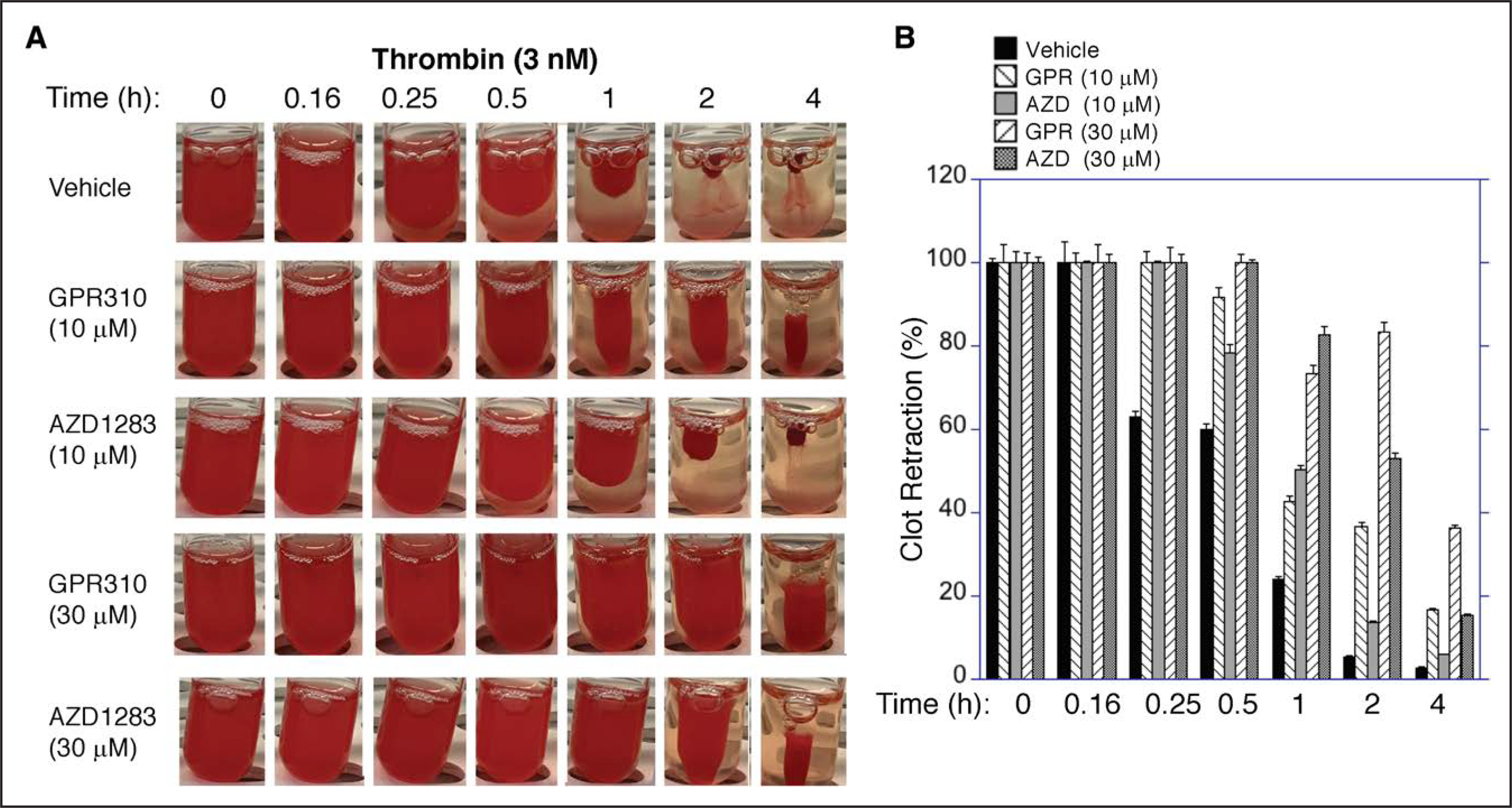
A, Platelet-rich plasma was isolated from whole blood and anticoagulated with 4% sodium citrate. Clot retraction was done essentially as described previously.40 Clot retraction was done with samples pretreated with 10 or 30 μM GPR310 (G-protein–coupled receptor 310) or 10 or 30 μM AZD1283 or vehicle, and clot formation was initiated with 3 nmol/L thrombin. Samples were incubated at room temperature over time as indicated and then photographed with a digital camera. B, Two-dimensional retraction clots in A were measured using National Institutes of Health ImageJ and expressed as percent retraction (mean±SD). Statistical significance was determined using ANOVA followed by Bonferonni correction; ****P<0.0001.
Effect of Inhibition of GPR31 on PAR4 Platelet Activation and Carotid Artery Thrombosis and Hemostasis in Mice
Human and mouse share 71% overall homology in GPR31 receptor sequence and the intracellular loops, which couple to G proteins, share higher 84% identity. To investigate the role of GPR31 in mouse platelet aggregation and dense granule secretion, we examined the effect of the GPR310 inhibitor on platelet aggregation and ATP release induced by PAR4 agonist peptide AYPGKF using mouse PRP. As shown in Figure 5A and 5B, both aggregation and ATP release from mouse platelets were inhibited by spiked GPR310. Next, the therapeutic potential of GPR31 blockade was evaluated in the FeCl3-induced arterial injury model4 in mice. Carotid artery blood flow was completely occluded in all vehicle-treated mice within 18 to 26 minutes by application of FeCl3. GPR310 administration (10 mg/kg, sc) afforded striking protection (P<0.0018) against FeCl3-induced carotid artery injury in mice with complete patency sustained in 80% of the GPR310 cohort, versus 0% in the vehicle cohort (Figure 5C).
Figure 5. GPR310 (G-protein–coupled receptor 310) protects mice from arterial thrombosis.
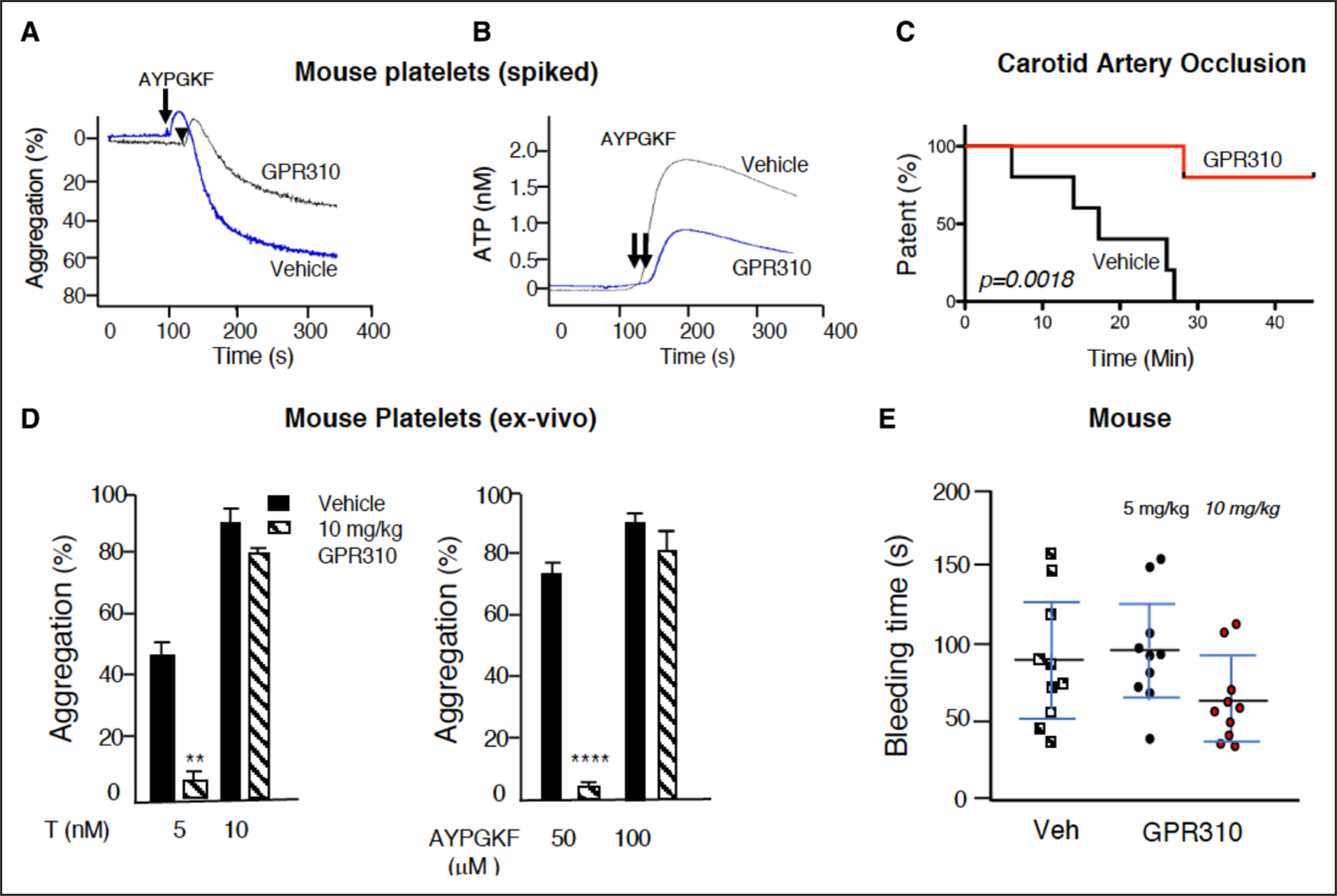
A and B, Aggregation and ATP secretion was measured in mouse platelet-rich plasma stimulated with 100 μM AYPGKF in the presence of 30 μM GPR310, and ATP release was measured, and aggregation was simultaneously measured. C, Thrombosis was induced with 10% ferric chloride (FeCl3) application to the carotid artery during which time flow was monitored using Doppler probe. The time to occlusion was determined and plotted for mice treated with 10 mg/kg GPR310 or vehicle (Veh; n=5) delivered subcutaneously 4 h before FeCl3 injury. Carotid artery occlusion in mice plotted as Kaplan-Meier curves showed significant (P=0.0018, log-rank Mantel-Cox test) protection against FeCl3-induced carotid artery injury measured over 45 min. D, Ex vivo analysis of mouse platelets collected at 4 h after injection with 10 mg/kg GPR310 or Veh (n=5) stimulated with 50 and 100 μM AYPGKF and 5 and 10 nmol/L thrombin as indicated. Student t test; **P<0.01, ****P<0.0001. E, Mouse tail bleeding was measured 4 h after GPR310 or vehicle sc injection (n=10) as indicated.
To determine the systemic pharmacodynamic effect of GPR310 on ex vivo platelet function, mice were injected sc with 10 mg/kg of GPR310 or vehicle. Blood was collected 4 hours later for evaluation of PAR4-dependent (mice lack PAR1 on platelets) ex vivo platelet pharmacodynamic effects. Platelets from GPR310-treated animals were 88% to 95% protected against 5 nmol/L thrombin and 50 μmol/L PAR4 AYPGKF-mediated platelet aggregation as compared with vehicle-treated mice (Figure 5D). The antiplatelet effects of 10 mg/kg GPR310 were reversed by high concentrations of AYPGKF (100 μmol/L) or thrombin (10 nmol/L) agonist. There was no difference between the tail bleeding times of animals 4 hours after treatment with GPR310 (5 and 10 mg/kg) versus vehicle control (Figure 5E).
We measured the plasma levels of GPR310 (10 mg/kg) as shown in Figure VI in the Data Supplement. Pharmacokinetics of plasma levels of GPR310 were determined at 5, 15, and 30 minutes and 1, 4, 5, 14, and 24 hours after subcutaneous injection of GPR310 (10 mg/kg) using liquid chromatography–mass spectrometry to determine peak pepducin drug levels and rate of terminal elimination. As shown in Figure VI in the Data Supplement, the peak plasma level of GPR310 reached 4.3 μmol/L at 1 hour with sustained high levels between 15 minutes and 6 hours (1.6–4.3 μmol/L) followed by elimination overnight to reach 10 nmol/L at the 14-hour time point. At the 4-hour experimental time point when hemostasis and arterial thrombosis experiments were conducted, the plasma levels of GPR310 were 3.5 μmol/L. Therefore, GPR310 has an excellent pharmacokinetic profile with high systemic bioavailability that may be attractive for further therapeutic development.
GPR310 Blocks Thrombin Receptor–Mediated Calcium Flux, AKT, and ERK Activation in Human Platelets
Early studies suggested that 12(S)-HETE triggered the formation of diacylglycerol and D-myo-inositol 1,4,5-triphosphate in melanoma cells through a Giβγ pathway41; however, 12(S)-HETE did not induce calcium mobilization in HUVECS (human umbilical vein endothelial cells).42 We found that 2(S)-HETE could mobilize low but detectable calcium transients in CHO/GPR31 cells (Figure 6A). Calcium flux induced by 12(S)-HETE was negligible in human platelets (Figure 6B). Despite 12(S)-HETE not directly generating a calcium response, addition of 3 μM GPR310 inhibitor suppressed the AYPGKF-mediated Ca2+ response by 80% (Figure 6C), suggesting that the PAR4-mediated calcium response is dependent upon potentiation by GPR31.
Figure 6. GPR31 (G-protein–coupled receptor 31) regulates PAR (protease-activated receptor)-4–mediated platelet activation resulting in orchestrated control of Rap1-RASA3–mediated αIIbβ3 activation.
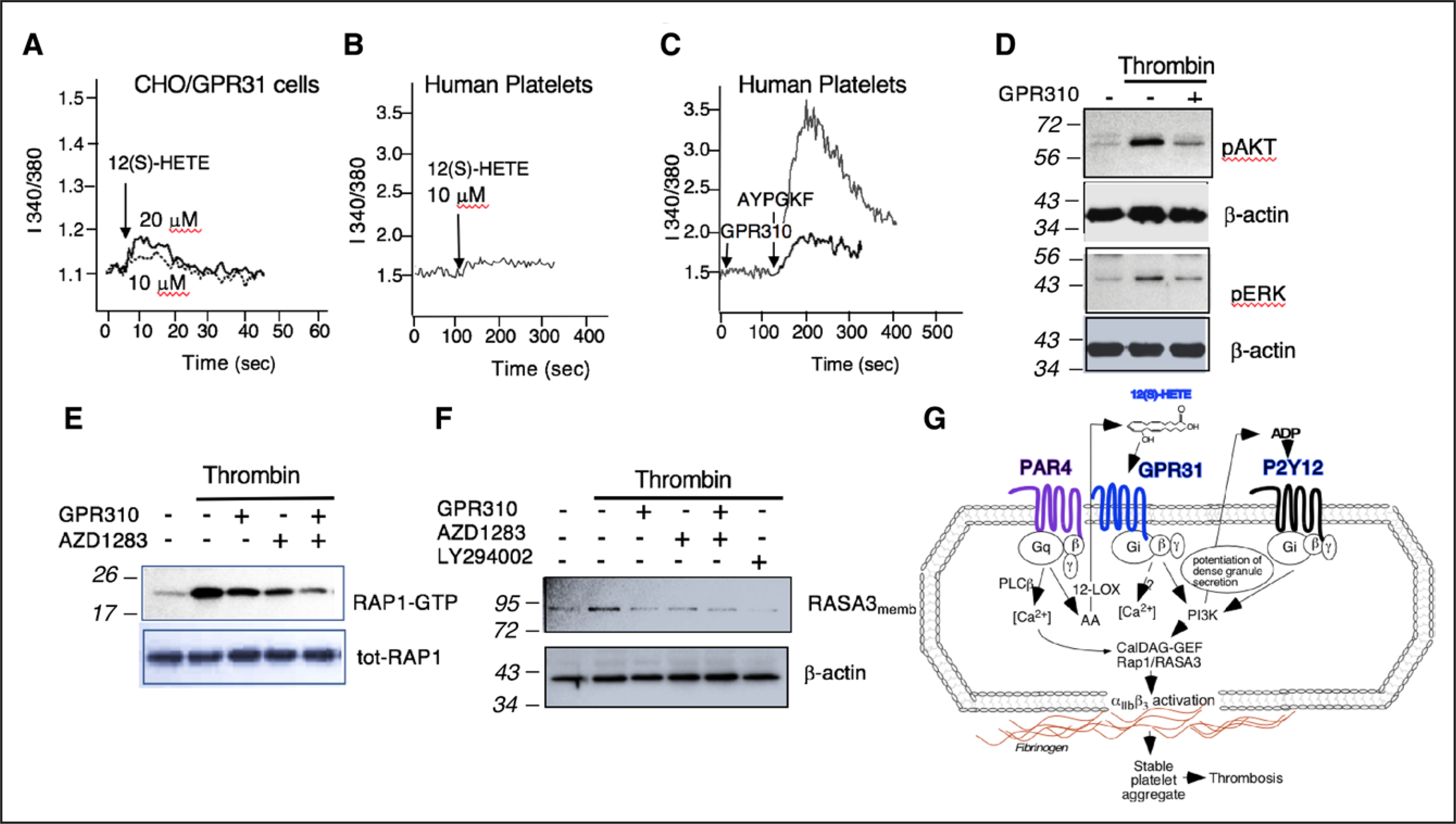
A, Fura-2–labeled transiently transfected GPR31/Chinese hamster ovary (CHO) cells were stimulated with 10 and 20 μM 12(S)-HETE, and Ca2+ flux was measured over time. B, Fura-2–labeled human platelets were stimulated with 10 μM 12(S)-HETE, and Ca2+ flux was measured over time. C, GPR310 (G-protein–coupled receptor 310) blocks PAR4-mediated calcium flux on human platelets. Platelets were preincubated with 3 μM GPR310 or vehicle before stimulation with 160 μM AYPGKF. D, Immunoblot showing 3 nmol/L thrombin activation of pAKT or pERK (phospho extracellular signal-regulated kinase) phosphorylation after 15 min in human platelets pretreated with 1 μM GPR310 as indicated. β-actin is used as loading control. E, Gel-purified platelets were treated with GPR310 (3 μM), AZD1283 (10 μM), or dual inhibition before thrombin stimulation (3 nmol/L) for 15 min, and Rap1 activation was measured. The bottom panels represent total Rap1 as loading control. F, Human platelets were treated with GPR310 (1 μM), AZD1283 (10 μM), dual inhibition, or LY294002 (10 μM), before thrombin stimulation with 3 nmol/L thrombin for 5 min. After membrane preparation, samples were immunoblotted with RASA3 ab. β-actin is used as loading control. G, Schematic of the PAR4, GPR31, and P2Y12 signaling in human platelet. Molecular weight markers (kDa). 12-LOX indicates 12-lipoxygenase; GTP, guanosine triphosphate; pAkt, protein kinase B; pERK, phospho extracellular signal-regulated kinase; PLCβ, phospholipase C beta; Rap1, Ras-related protein 1; and RASA3, Ras P21 protein activator 3.
In previous studies,26,43 it was suggested that ADP-P2Y12-Gi and not thrombin-Gq or G12/13 signaling pathway activates Akt and ERK1/2 by demonstrating that thrombin-mediated activation of Akt and ERK1/2 could be abrogated through blockade of P2Y12. We found that stimulation of platelets with thrombin results in phosphorylation (Thr308/Ser473) of Akt, which was attenuated with GPR310 (Figure 6D). Likewise, GPR310 inhibited thrombin-mediated ERK activation (Figure 6D), consistent with GPR31-Gi potentiating both the thrombin-pAKT and pERK (phospho extracellular signal-regulated kinase) pathways.
Effect of GPR31 Inhibition in PAR4-Mediated Rap1 and RASA3 Activation
Given that blockade of GPR31 had significant inhibitory effects on thrombin and PAR4-dependent activation of essentially all tested signaling pathways in platelets, we compared the effects of blocking the 2 individual Gi-autocrine pathways, 12(S)-HETE-GPR31 and ADP-P2Y12, in thrombin activation of the critical common downstream Rap1 GTPase, which controls the αIIbβ3 integrin on state.44 As shown in Figure 6E, thrombin induced strong Rap1 activation, which was partially suppressed by either GPR31 or P2Y12 receptor inhibitors. The combination of both GPR310 and AZD1283 gave nearly complete inhibition of thrombin activation of Rap1 in an additive manner (Figure 6E). To determine the contribution of GPR31 to PAR4-mediated Rap1, platelets were stimulated with the PAR4-activating peptide AYPGKF. The robust Rap1-GTP signal peaked at 5 minutes post-AYPGKF activation, which was significantly attenuated by GRP310 with the peak of Rap1-GTP shifting to 15 minutes, albeit at a greatly reduced intensity (Figure VII in the Data Supplement).
Lastly, we compared the effects of GPR31 versus P2Y12 blockade on thrombin activation of the dual Rap and Ras GAP (GTPase-activating protein), RASA3. Stimulation of platelets with 3 nmol/L thrombin in the presence of GPR310, AZD1283, or the both inhibitors together resulted in complete suppression of RASA3 appearance in plasma membrane at 15 minutes (Figure 6F). Blockade of PI3K (phosphatidylinositol 3-kinase) with LY294002 gave similar complete blockade of thrombin-induced RASA3 membrane localization. Therefore, both Gi-coupled platelet receptors GPR31 and P2Y12 were contributing to platelet RASA3 membrane localization via a downstream PI3K-mediated response as depicted in the mechanism of Figure 6G.
DISCUSSION
The major bioactive lipid in platelets produced by 12-LOX, namely 12(S)-HETE, has been shown to have both prothrombotic and antithrombotic effects, akin to the dual effects seen with prostaglandins and thromboxanes produced by COX from the common arachidonic acid precursor.45 Due to a lack of inhibitors and undefined G-protein coupling profile to the proposed 12(S)-HETE receptor GPR31,18 it has been difficult to determine what role, in any, GPR31 plays in critical cardiovascular and other inflammatory functions. Here, we present a study of the role of GPR31 in hemostasis and thrombosis. We used cell-penetrating pepducin technology26 to produce an inhibitor of GPR31, which was used to help uncover the functions of GPR31 in human platelets and in mouse models.
We found that GPR31 couples to Gi and is expressed on human platelets, analogous to the ADP P2Y12 Gi-coupled receptor—the target of clopidogrel and other thienopyridines. Unlike blockade of the thromboxane receptor with aspirin, which gave slight effects in the mouse carotid artery injury model,46 we demonstrated that inhibition of the GPR31 receptor with the GPR310 pepducin gave nearly complete protection against FeCl3-induced carotid artery occlusion in mice. The GPR31 agonist 12(S)-HETE did not induce platelet aggregation by itself; instead, it served to potentiate platelet aggregation through PAR4 and collagen, relatively sparing PAR1. However, the complexity of this interesting and potentially important synergy between PARs, GPR31, and P2Y12 needs to be further investigated. Despite preventing carotid artery thrombosis, GPR31 blockade with GPR310 had no discernable effect on hemostasis in mice. However, thrombosis and bleeding in mice have 1 major limitation in that they differ in platelet PAR1 expression as compared with humans. Therefore, further studies using either guinea pigs or cynomolgus monkeys would be beneficial. Together, these data support GPR31 as a promising new target in terms of efficacy and safety for the prevention and treatment of coronary arterial diseases and thrombotic stroke.
Autocrine pathways dependent on dense granule secretion in platelets have been a central focus of numerous studies because of their importance in regulating a second wave of signaling and calcium flux amplification from metabolites such as ADP46 and epinephrine that are important regulators of irreversible platelet aggregation through Gi-signaling pathways.10 A recent report suggests that platelet PAR4 may perform a unique function compared with PAR1 in release of dense granules.13 Our data show that PAR4-mediated dense granule secretion is strongly dependent on autocrine 12(S)-HETE-GPR31 pathways in both human and mouse platelets. Despite 12(S)-HETE producing a minimal calcium response itself, GPR31 was found to greatly amplify PAR4-dependent calcium mobilization consistent with the previous reports of prolonged PAR4 signaling48 being dependent on both calcium flux and dense granule release.10
Co-immunoprecipitation studies revealed that GPR31 and PAR4 form a heterodimeric (or heteromeric) complex, as previously shown for PAR1-PAR449 and P2Y12-PAR4.4 The dependence of PAR4 on GPR31 for activation of Rap1, RASA3, Akt, ERK, dense granule release, platelet aggregation, and clot retraction is highly reminiscent of the autocrine ADP-P2Y12-Gi signaling effects on PAR4.43,44,50 Rapid and sustained Rap1-GTP translocation to the platelet plasma membrane, which was strongly activated by 12(S)-HETE, is necessary for talin recruitment and inside-out signaling of integrin αIIbβ3 in the locked-in state.45 Rap1 is a highly abundant small GTPase and member of the RAS family expressed in endothelial cells, leukocytes, and platelets.35 Both GEFs (guanine nucleotide exchange factors) and GAPs tightly regulate Rap1 by GTP loading and subsequent GTP hydrolysis.51–53 CalDAG-GEFI is a Rap-GEF expressed in platelets. Upon platelet stimulation, CalDAG-GEFI is necessary for rapid, calcium-dependent activation of Rap1 and integrin αIIbβ3 activation.54 Loss of RASA3 function—the most abundant Rap-GAP expressed in platelets55—is associated with severe thrombocytopenia in mice, confirming the importance of RASA3 as a key regulator of Rap1 in platelet biology.56 Therefore, both Rap1 and RASA3 signaling induced by thrombin are critical for the formation of a stable platelet plug and clot retraction,56 and we found that both were blocked by GPR31 and P2Y12 inhibitors, consistent with a common Gi pathway as proposed in Figure 6G.
In conclusion, we identify GPR31 as a novel target in human platelets that impacts both PAR4- and collagen-mediated signaling, however, relatively sparing PAR1. The GPR310 pepducin inhibitor was highly effective in inhibiting occlusive arterial thrombosis without detectable effects on hemostasis in animal models, suggesting consideration of 12(S)-HETE-GPR31 as a new antithrombotic and antistroke target.
Supplementary Material
Highlights.
GPR31 (G-protein–coupled receptor 31)—a G-protein–coupled receptor for the 12(S)-HETE bioactive lipid—has not previously been shown to play a role in platelet function or thrombosis.
We found that GPR31 was expressed on human platelets and formed a heterodimeric complex with the PAR (protease-activated receptor)-4 thrombin receptor.
A pepducin-based inhibitor, GPR310 (G-protein–coupled receptor 310), was used to delineate the role of human and mouse GPR31 in platelet function and arterial thrombosis.
GPR310 suppressed platelet aggregation and ATP release to PAR4 agonists in human and mouse platelets and significantly reduced arterial thrombosis without affecting hemostasis in mice.
These data indicate that GPR31 is a promising new target for suppression of platelet activation and arterial thrombosis.
Acknowledgments
We thank Dr Emily Michael for providing help with the cAMP assay.
Sources of Funding
This work was funded, in part, by grants from the National Institutes of Health by R41HL134517, R01HL136485, and P50 HL110789 (L. Covic and A. Kuliopulos).
Nonstandard Abbreviations and Acronyms
- 12-LOX
12-lypoxygenase
- AC
adenylyl cyclase
- CHO
Chinese hamster ovary
- COX
cyclooxygenase
- Cre
cAMP-responsive element
- FeCl3
ferric chloride
- GAP
GTPase-activating protein
- GPCR
G-protein–coupled receptor
- GPR31
G-protein–coupled receptor 31
- GPR310
GPR31 pepducin inhibitor
- HRP
horseradish peroxidase
- IL
interleukin
- LO
lipoxygenase
- PAR
protease-activated receptor
- PRP
platelet-rich plasma
- Tris
tris(hydroxymethyl)aminomethane
Footnotes
A. Kuliopulos and L. Covic report serving as Founders of Oasis Pharmaceuticals. The other authors report no conflicts.
Disclosures
The Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/ATVBAHA.120.315154.
Contributor Information
Layla Van Doren, Division of Hematology/Oncology, Center for Hemostasis and Thrombosis Research, Tufts Medical Center, Boston, MA.
Nga Nguyen, Division of Hematology/Oncology, Center for Hemostasis and Thrombosis Research, Tufts Medical Center, Boston, MA.
Christopher Garzia, Division of Hematology/Oncology, Center for Hemostasis and Thrombosis Research, Tufts Medical Center, Boston, MA.
Elizabeth K. Fletcher, Division of Hematology/Oncology, Center for Hemostasis and Thrombosis Research, Tufts Medical Center, Boston, MA
Ryan Stevenson, Division of Hematology/Oncology, Center for Hemostasis and Thrombosis Research, Tufts Medical Center, Boston, MA.
Athan Kuliopulos, Division of Hematology/Oncology, Center for Hemostasis and Thrombosis Research, Tufts Medical Center, Boston, MA; Department of Medicine, Tufts University School of Medicine, Boston, MA; Department of Biochemistry, Tufts University School of Medicine, Boston, MA.
Lidija Covic, Division of Hematology/Oncology, Center for Hemostasis and Thrombosis Research, Tufts Medical Center, Boston, MA; Department of Medicine, Tufts University School of Medicine, Boston, MA; Department of Biochemistry, Tufts University School of Medicine, Boston, MA.
REFERENCES
- 1.van der Meijden PEJ, Heemskerk JWM. Platelet biology and functions: new concepts and clinical perspectives. Nat Rev Cardiol. 2019;16:166–179. doi: 10.1038/s41569-018-0110-0 [DOI] [PubMed] [Google Scholar]
- 2.Leger AJ, Covic L, Kuliopulos A. Protease-activated receptors in cardiovascular diseases. Circulation. 2006;114:1070–1077. doi: 10.1161/CIRCULATIONAHA.105.574830 [DOI] [PubMed] [Google Scholar]
- 3.Koukos G, Sevigny L, Zhang P, Covic L, Kuliopulos A. Serine and metalloprotease signaling through PAR1 in arterial thrombosis and vascular injury. IUBMB Life. 2011;63:412–418. doi: 10.1002/iub.465 [DOI] [PubMed] [Google Scholar]
- 4.Leger AJ, Jacques SL, Badar J, Kaneider NC, Derian CK, Andrade-Gordon P, Covic L, Kuliopulos A. Blocking the protease-activated receptor 1–4 heterodimer in platelet-mediated thrombosis. Circulation. 2006;113:1244–1254. doi: 10.1161/CIRCULATIONAHA.105.587758 [DOI] [PubMed] [Google Scholar]
- 5.Bliden K, Chaudhary R, Kuliopulos A, Tran H, Taheri H, Tehrani B, Rosenblatt A, Navarese E, Tantry US, Gurbel PA. Effects of vorapaxar on clot characteristics, coagulation, inflammation, and platelet and endothelial function in patients treated with mono- and dual-antiplatelet therapy. J Thromb Haemost. 2020;18:23–35. doi: 10.1111/jth.14616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tricoci P, Huang Z, Held C, Moliterno DJ, Armstrong PW, Van de Werf F, White HD, Aylward PE, Wallentin L, Chen E, et al. ; TRACER Investigators. Thrombin-receptor antagonist vorapaxar in acute coronary syndromes. N Engl J Med. 2012;366:20–33. doi: 10.1056/NEJMoa1109719 [DOI] [PubMed] [Google Scholar]
- 7.Morrow DA, Braunwald E, Bonaca MP, Ameriso SF, Dalby AJ, Fish MP, Fox KA, Lipka LJ, Liu X, Nicolau JC, et al. ; TRA 2P–TIMI 50 Steering Committee and Investigators. Vorapaxar in the secondary prevention of atherothrombotic events. N Engl J Med. 2012;366:1404–1413. doi: 10.1056/NEJMoa1200933 [DOI] [PubMed] [Google Scholar]
- 8.Wilson SJ, Ismat FA, Wang Z, Cerra M, Narayan H, Raftis J, Gray TJ, Connell S, Garonzik S, Ma X, et al. PAR4 (protease-activated receptor 4) antagonism with BMS-986120 inhibits human ex vivo thrombus formation. Arterioscler Thromb Vasc Biol. 2018;38:448–456. doi: 10.1161/ATVBAHA.117.310104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li S, Tarlac V, Hamilton JR. Using PAR4 inhibition as an anti-thrombotic approach: why, how, and when? Int J Mol Sci. 2019;20:5629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Covic L, Gresser AL, Kuliopulos A. Biphasic kinetics of activation and signaling for PAR1 and PAR4 thrombin receptors in platelets. Biochemistry. 2000;39:5458–5467. doi: 10.1021/bi9927078 [DOI] [PubMed] [Google Scholar]
- 11.Jacques SL, Kuliopulos A. Protease-activated receptor-4 uses dual prolines and an anionic retention motif for thrombin recognition and cleavage. Biochem J. 2003;376:733–740. doi: 10.1042/BJ20030954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Covic L, Misra M, Badar J, Singh C, Kuliopulos A. Pepducin-based intervention of thrombin-receptor signaling and systemic platelet activation. Nat Med. 2002;8:1161–1165. doi: 10.1038/nm760 [DOI] [PubMed] [Google Scholar]
- 13.Covic L, Singh C, Smith H, Kuliopulos A. Role of the PAR4 thrombin receptor in stabilizing platelet-platelet aggregates as revealed by a patient with Hermansky-Pudlak syndrome. Thromb Haemost. 2002;87:722–727. [PubMed] [Google Scholar]
- 14.Hauser AS, Attwood MM, Rask-Andersen M, Schiöth HB, Gloriam DE. Trends in GPCR drug discovery: new agents, targets and indications. Nat Rev Drug Discov. 2017;16:829–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Husted AS, Trauelsen M, Rudenko O, Hjorth SA, Schwartz TW. GPCR-mediated signaling of metabolites. Cell Metab. 2017;25:777–796. doi: 10.1016/j.cmet.2017.03.008 [DOI] [PubMed] [Google Scholar]
- 16.Ibrahim P, Wifling D, Clark T. Universal activation index for class A GPCRs. J Chem Inf Model. 2019;59:3938–3945. doi: 10.1021/acs.jcim.9b00604 [DOI] [PubMed] [Google Scholar]
- 17.Gurbel PA, Kuliopulos A, Tantry US. G-protein-coupled receptors signaling pathways in new antiplatelet drug development. Arterioscler Thromb Vasc Biol. 2015;35:500–512. doi: 10.1161/ATVBAHA.114.303412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yeung J, Hawley M, Holinstat M. The expansive role of oxylipins on platelet biology. J Mol Med (Berl). 2017;95:575–588. doi: 10.1007/s00109-017-1542-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tersey SA, Bolanis E, Holman TR, Maloney DJ, Nadler JL, Mirmira RG. Minireview: 12-lipoxygenase and islet β-cell dysfunction in diabetes. Mol Endocrinol. 2015;29:791–800. doi: 10.1210/me.2015-1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yeung J, Holinstat M. 12-lipoxygenase: a potential target for novel antiplatelet therapeutics. Cardiovasc Hematol Agents Med Chem. 2011;9:154–164. doi: 10.2174/187152511797037619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yeung J, Apopa PL, Vesci J, Stolla M, Rai G, Simeonov A, Jadhav A, Fernandez-Perez P, Maloney DJ, Boutaud O, et al. 12-lipoxygenase activity plays an important role in PAR4 and GPVI-mediated platelet reactivity. Thromb Haemost. 2013;110:569–581. doi: 10.1160/TH13-01-0014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yeung J, Tourdot BE, Fernandez-Perez P, Vesci J, Ren J, Smyrniotis CJ, Luci DK, Jadhav A, Simeonov A, Maloney DJ, et al. Platelet 12-LOX is essential for FcγRIIa-mediated platelet activation. Blood. 2014;124:2271–2279. doi: 10.1182/blood-2014-05-575878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coffey MJ, Jarvis GE, Gibbins JM, Coles B, Barrett NE, Wylie OR, O’Donnell VB. Platelet 12-lipoxygenase activation via glycoprotein VI: involvement of multiple signaling pathways in agonist control of H(P)ETE synthesis. Circ Res. 2004;94:1598–1605. doi: 10.1161/01.RES.0000132281.78948.65 [DOI] [PubMed] [Google Scholar]
- 24.Morgan LT, Thomas CP, Kühn H, O’Donnell VB. Thrombin-activated human platelets acutely generate oxidized docosahexaenoic-acid-containing phospholipids via 12-lipoxygenase. Biochem J. 2010;431:141–148. [DOI] [PubMed] [Google Scholar]
- 25.Katoh A, Ikeda H, Murohara T, Haramaki N, Ito H, Imaizumi T. Platelet-derived 12-hydroxyeicosatetraenoic acid plays an important role in mediating canine coronary thrombosis by regulating platelet glycoprotein IIb/IIIa activation. Circulation. 1998;98:2891–2898. doi: 10.1161/01.cir.98.25.2891 [DOI] [PubMed] [Google Scholar]
- 26.Guo Y, Zhang W, Giroux C, Cai Y, Ekambaram P, Dilly AK, Hsu A, Zhou S, Maddipati KR, Liu J, et al. Identification of the orphan G protein-coupled receptor GPR31 as a receptor for 12-(S)-hydroxyeicosatetraenoic acid. J Biol Chem. 2011;286:33832–33840. doi: 10.1074/jbc.M110.216564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Covic L, Gresser AL, Talavera J, Swift S, Kuliopulos A. Activation and inhibition of G protein-coupled receptors by cell-penetrating membrane-tethered peptides. Proc Natl Acad Sci USA. 2002;99:643–648. doi: 10.1073/pnas.022460899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuliopulos A, Covic L, Seeley SK, Sheridan PJ, Helin J, Costello CE. Plasmin desensitization of the PAR1 thrombin receptor: kinetics, sites of truncation, and implications for thrombolytic therapy. Biochemistry. 1999;38:4572–4585. doi: 10.1021/bi9824792 [DOI] [PubMed] [Google Scholar]
- 29.Zhang P, Gruber A, Kasuda S, Kimmelstiel C, O’Callaghan K, Cox DH, Bohm A, Baleja JD, Covic L, Kuliopulos A. Suppression of arterial thrombosis without affecting hemostatic parameters with a cell-penetrating PAR1 pepducin. Circulation. 2012;126:83–91. doi: 10.1161/CIRCULATIONAHA.112.091918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuliopulos A, Mohanlal R, Covic L. Effect of selective inhibition of the p38 MAP kinase pathway on platelet aggregation. Thromb Haemost. 2004;92:1387–1393. doi: 10.1160/TH04-03-0187 [DOI] [PubMed] [Google Scholar]
- 31.Zhang Y I-TASSER: fully automated protein structure prediction in CASP8. Proteins. 2009;77(suppl 9):100–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang J, Zhang Y. I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res. 2015;43:W174–W181. doi: 10.1093/nar/gkv342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roy A, Yang J, Zhang Y. COFACTOR: an accurate comparative algorithm for structure-based protein function annotation. Nucleic Acids Res. 2012;40:W471–W477. doi: 10.1093/nar/gks372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tan Q, Zhu Y, Li J, Chen Z, Han GW, Kufareva I, Li T, Ma L, Fenalti G, Li J, et al. Structure of the CCR5 chemokine receptor-HIV entry inhibitor maraviroc complex. Science. 2013;341:1387–1390. doi: 10.1126/science.1241475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stefanini L, Paul DS, Robledo RF, Chan ER, Getz TM, Campbell RA, Kechele DO, Casari C, Piatt R, Caron KM, et al. RASA3 is a critical inhibitor of RAP1-dependent platelet activation. J Clin Invest. 2015;125:1419–1432. doi: 10.1172/JCI77993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kimmelstiel C, Badar J, Covic L, Waxman S, Weintraub A, Jacques S, Kuliopulos A. Pharmacodynamics and pharmacokinetics of the platelet GPIIb/IIIa inhibitor tirofiban in patients undergoing percutaneous coronary intervention: implications for adjustment of tirofiban and clopidogrel dosage. Thromb Res. 2005;116:55–66. doi: 10.1016/j.thromres.2004.11.011 [DOI] [PubMed] [Google Scholar]
- 37.Kimmelstiel C, Stevenson R, Nguyen N, Van Doren L, Zhang P, Perkins J, Kapur NK, Weintraub A, Castaneda V, Kuliopulos A, et al. Enhanced potency of prasugrel on protease-activated receptors following bivalirudin treatment for PCI as compared to clopidogrel. Thromb Res. 2019;177:59–69. doi: 10.1016/j.thromres.2019.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Adili R, Tourdot BE, Mast K, Yeung J, Freedman JC, Green A, Luci DK, Jadhav A, Simeonov A, Maloney DJ, et al. First selective 12-LOX inhibitor, ML355, impairs thrombus formation and vessel occlusion in vivo with minimal effects on hemostasis. Arterioscler Thromb Vasc Biol. 2017;37:1828–1839. doi: 10.1161/ATVBAHA.117.309868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang J, Zhang K, Gao ZG, Paoletta S, Zhang D, Han GW, Li T, Ma L, Zhang W, Müller CE, et al. Agonist-bound structure of the human P2Y12 receptor. Nature. 2014;509:119–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trivedi V, Boire A, Tchernychev B, Kaneider NC, Leger AJ, O’Callaghan K, Covic L, Kuliopulos A. Platelet matrix metalloprotease-1 mediates thrombogenesis by activating PAR1 at a cryptic ligand site. Cell. 2009;137:332–343. doi: 10.1016/j.cell.2009.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lockyer S, Kambayashi J. Demonstration of flow and platelet dependency in a ferric chloride-induced model of thrombosis. J Cardiovasc Pharmacol. 1999;33:718–725. doi: 10.1097/00005344-199905000-00007 [DOI] [PubMed] [Google Scholar]
- 42.Liu B, Khan WA, Hannun YA, Timar J, Taylor JD, Lundy S, Butovich I, Honn KV. 12(S)-hydroxyeicosatetraenoic acid and 13(S)-hydroxyoctadecadienoic acid regulation of protein kinase C-alpha in melanoma cells: role of receptor-mediated hydrolysis of inositol phospholipids. Proc Natl Acad Sci USA. 1995;92:9323–9327. doi: 10.1073/pnas.92.20.9323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim S, Jin J, Kunapuli SP. Akt activation in platelets depends on Gi signaling pathways. J Biol Chem. 2004;279:4186–4195. doi: 10.1074/jbc.M306162200 [DOI] [PubMed] [Google Scholar]
- 44.Shankar H, Garcia A, Prabhakar J, Kim S, Kunapuli SP. P2Y12 receptor-mediated potentiation of thrombin-induced thromboxane A2 generation in platelets occurs through regulation of Erk1/2 activation. J Thromb Haemost. 2006;4:638–647. doi: 10.1111/j.1538-7836.2006.01789.x [DOI] [PubMed] [Google Scholar]
- 45.Stefanini L, Bergmeier W. RAP1-GTPase signaling and platelet function. J Mol Med (Berl). 2016;94:13–19. doi: 10.1007/s00109-015-1346-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O’Callaghan K, Kuliopulos A, Covic L. Turning receptors on and off with intracellular pepducins: new insights into G-protein-coupled receptor drug development. J Biol Chem. 2012;287:12787–12796. doi: 10.1074/jbc.R112.355461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang X, Cheng Q, Xu L, Feuerstein GZ, Hsu MY, Smith PL, Seiffert DA, Schumacher WA, Ogletree ML, Gailani D. Effects of factor IX or factor XI deficiency on ferric chloride-induced carotid artery occlusion in mice. J Thromb Haemost. 2005;3:695–702. doi: 10.1111/j.1538-7836.2005.01236.x [DOI] [PubMed] [Google Scholar]
- 48.Rigg RA, Healy LD, Chu TT, Ngo ATP, Mitrugno A, Zilberman-Rudenko J, Aslan JE, Hinds MT, Vecchiarelli LD, Morgan TK, et al. Protease-activated receptor 4 activity promotes platelet granule release and platelet-leukocyte interactions. Platelets. 2019;30:126–135. doi: 10.1080/09537104.2017.1406076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bilodeau ML, Hamm HE. Regulation of protease-activated receptor (PAR) 1 and PAR4 signaling in human platelets by compartmentalized cyclic nucleotide actions. J Pharmacol Exp Ther. 2007;322:778–788. doi: 10.1124/jpet.107.121830 [DOI] [PubMed] [Google Scholar]
- 50.Khan A, Li D, Ibrahim S, Smyth E, Woulfe DS. The physical association of the P2Y12 receptor with PAR4 regulates arrestin-mediated Akt activation. Mol Pharmacol. 2014;86:1–11. doi: 10.1124/mol.114.091595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frische EW, Zwartkruis FJ. Rap1, a mercenary among the Ras-like GTPases. Dev Biol. 2010;340:1–9. doi: 10.1016/j.ydbio.2009.12.043 [DOI] [PubMed] [Google Scholar]
- 52.Franke B, Akkerman JW, Bos JL. Rapid Ca2+-mediated activation of Rap1 in human platelets. EMBO J. 1997;16:252–259. doi: 10.1093/emboj/16.2.252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cifuni SM, Wagner DD, Bergmeier W. CalDAG-GEFI and protein kinase C represent alternative pathways leading to activation of integrin alphaIIbbeta3 in platelets. Blood. 2008;112:1696–1703. doi: 10.1182/blood-2008-02-139733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stefanini L, Roden RC, Bergmeier W. CalDAG-GEFI is at the nexus of calcium-dependent platelet activation. Blood. 2009;114:2506–2514. doi: 10.1182/blood-2009-04-218768 [DOI] [PubMed] [Google Scholar]
- 55.Stefanini L, Bergmeier W. CalDAG-GEFI and platelet activation. Platelets. 2010;21:239–243. doi: 10.3109/09537101003639931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Blanc L, Ciciotte SL, Gwynn B, Hildick-Smith GJ, Pierce EL, Soltis KA, Cooney JD, Paw BH, Peters LL. Critical function for the Ras-GTPase activating protein RASA3 in vertebrate erythropoiesis and megakaryopoiesis. Proc Natl Acad Sci USA. 2012;109:12099–12104. doi: 10.1073/pnas.1204948109 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.