

Summary
Although type 1 innate lymphoid cells (ILC1) have been originally found as liver-resident ILC, their pathophysiological role in the liver remains poorly investigated. Here we demonstrated that carbon tetrachloride (CCl4) injection into mice activated ILC1, but not natural killer (NK) cells, in the liver. Activated ILC1 produced interferon-γ (IFN-γ) and protected mice from CCl4-induced acute liver injury. IFN-γ released from activated ILC1 promoted the survival of hepatocytes through upregulation of Bcl-xL. An activating NK receptor, DNAM-1, was required for the optimal activation and IFN-γ production of liver ILC1. Extracellular adenosine triphosphate accelerated interleukin-12-driven IFN-γ production by liver ILC1. These findings suggest that ILC1 are critical for tissue protection during acute liver injury.
Keywords: ILC1, IFN-γ, acute liver injury, CCl4, DNAM-1, IL-7, ATP, IL-12, hepatocyte, Bcl-xL
Introduction
Innate lymphoid cells (ILC), the most recently discovered group of innate lymphocytes, are characterized by the lack of B cell receptors and T cell receptors, which are expressed on adaptive immune cells; ILC are able to invoke immediate inflammation and innate immune responses in tissues (Artis and Spits, 2015; Spits and Di Santo, 2011). As ILC are largely tissue-resident innate immune cells, they sense proinflammatory cytokines in the context of pathogen-derived and danger signals at local sites of pathogen invasion and tissue damage and promptly mount local immune responses by producing cytokines (Artis and Spits, 2015; Eberl et al., 2015; Spits and Di Santo, 2011; Vivier et al., 2018). The immune regulation triggered by tissue-resident ILC contributes not only to inflammation for host protection against pathogens but also to tissue repair, homeostasis, and metabolism (Artis and Spits, 2015; Colonna, 2018; Ebbo et al., 2017; Eberl et al., 2015; Klose and Artis, 2016; McKenzie et al., 2014; Vivier et al., 2018). ILC are classified into 3 subsets (group 1, group 2, and group 3) by the ability to produce effector cytokines and the requirement for specific transcriptional programs for development (Artis and Spits, 2015; Eberl et al., 2015; Vivier et al., 2018). Group 1 ILC respond to intracellular pathogens such as viruses, intracellular bacteria, and certain parasites. They comprise natural killer (NK) cells and type 1 ILC (ILC1).
ILC1 were initially found in the liver of mice as an atypical subset of NK cells expressing the TNF superfamily member TNF-related apoptosis-inducing ligand (TRAIL) or not expressing an NK cell marker, CD49b (DX5), and were redefined as a subset distinct from NK cells by the differential requirement for transcription factors Nfil3 and Eomes (Daussy et al., 2014; Geiger et al., 2014; Seillet et al., 2014; Takeda et al., 2001). ILC1 and NK cells share many common characteristics; they (i) produce large amounts of IFN-γ, (ii) are functionally dependent on the transcription factor T-bet, (iii) require interleukin-15 (IL-15) for their development and homeostasis, and (iv) have considerably overlapping gene expression signatures and expression profiles of NK cell receptors and cytokine receptors (Colonna, 2018; Ebbo et al., 2017; Jiao et al., 2016; Robinette et al., 2015; Seillet et al., 2016; Spits et al., 2016). The function of NK cell receptors on the surfaces of ILC1 remains largely unknown (Colonna, 2018; Jiao et al., 2016; Seillet et al., 2016; Spits et al., 2016). Despite the above common features, ILC1 and NK cells are developmentally, phenotypically, and functionally distinct subsets. NK cells exert cytotoxicity and circulate in the bloodstream, whereas ILC1 are weakly or non-cytotoxic and are largely tissue resident (Colonna, 2018; Spits et al., 2016; Vivier et al., 2018).
Phenotypic definition of ILC1 is still controversial because of the lack of unique and stable ILC1 markers to clearly distinguish them from NK cells (Colonna, 2018; Jiao et al., 2016; Spits et al., 2016; Vivier et al., 2018). Mouse ILC1 are defined as non-T, non-B, NK1.1+NKp46+CD49a+DX5− lymphocytes with high capacity to produce IFN-γ in the steady state, although these markers can be modulated by ILC1 activation status and environmental factors in the resident tissues; ILC1 are found in the liver, intestines, salivary glands, skin, thymus, lymph nodes, adipose tissues, peritoneal cavity, and uterus (Daussy et al., 2014; Gasteiger et al., 2015; Klose et al., 2014; O’Sullivan et al., 2016; Peng et al., 2013). It is increasingly evident that tissue-resident ILC1 not only play important roles in host defense against pathogens but also elicit prompt inflammation in response to tissue damage in several organs. Intestinal ILC1 function as the first line of host defense against certain intestinal pathogens such as Clostridium difficile, Listeria monocytogenes, Salmonella enterica, and Toxoplasma gondii; they produce IFN-γ and subsequently recruit and activate inflammatory myeloid cell subsets (Abt et al., 2015; Geiger et al., 2014; Klose et al., 2013, 2014; Reynders et al., 2011). Intestinal ILC1 also amplify mucosal inflammation and exacerbate anti-CD40-induced colitis in mice, which is reminiscent of ILC1 accumulation in inflamed intestines in individuals with Crohn’s disease (Bernink et al., 2013; Fuchs et al., 2013). In the kidney, ILC1 mediate ischemia-reperfusion injury in mice (Victorino et al., 2015), suggesting that ILC1 are involved in the pathology of tissue injury. Adipose-resident ILC1 promote a shift toward an inflammatory environment; this shift is mediated by IFN-γ-driven proinflammatory macrophage polarization and cause obesity-related insulin resistance (Boulenouar et al., 2017; O’Sullivan et al., 2016). Yet, although ILC1 in the liver has recently been reported to limit the replication of mouse cytomegalovirus (MCMV) at the early stage of infection in an IFN-γ-dependent manner (Weizman et al., 2017), little is known about their physiological roles in the liver injury, homeostasis, and regeneration, in spite of the fact that ILC1 were originally discovered as a liver-resident ILC subset and reside in the liver as the predominant ILC subset except for NK cells (Daussy et al., 2014; Peng et al., 2013; Sojka et al., 2014; Takeda et al., 2001). In this study, we investigated the role of liver ILC1 in the pathogenesis of drug-induced acute liver injury by using carbon tetrachloride (CCl4).
Results
Liver ILC1 are activated and produce IFN-γ after CCl4 injection
Increasing evidence indicates that the group 2 ILC (ILC2) and group 3 ILC (ILC3) subsets orchestrate immune regulation during the injury, repair, and homeostasis of their resident tissues (Artis and Spits, 2015; Ebbo et al., 2017; Eberl et al., 2015; Klose and Artis, 2016; McKenzie et al., 2014; Vivier et al., 2018), strongly suggesting that these tissue-resident ILC subsets have unique roles as the first responders to threats against the maintenance of their resident tissue homeostasis; however, the pathophysiological role of ILC1 in liver injury remains to be elucidated. We hypothesized that liver ILC1 are involved in the pathogenesis of acute liver injury. To test this hypothesis, we intraperitoneally injected carbon tetrachloride (CCl4) into wild-type (WT) mice to induce acute liver injury (Knockaert et al., 2012). Liver NK cells were identified as either NK1.1+TCRβ−B220−DX5+CD49a−CD200R− or NK1.1+NKp46+DX5+CD49a−CD200R− lymphocytes (Weizman et al., 2017), both before and 18 h after CCl4 injection (Figures 1A and S1A). Under the same condition, liver ILC1 were identified as either NK1.1+TCRβ−B220−DX5−CD49a+CD200R+ or NK1.1+NKp46+DX5−CD49a+CD200R+ lymphocytes and stably expressed CD200R, illustrating that the ILC1 gating strategy based on DX5 and CD49a is not likely to include contamination of liver NK cells in the experimental settings (Figures 1A and S1A). The frequency and the absolute number of NK cells in the liver increased after CCl4 injection, whereas those of ILC1 were not affected (Figure 1B). ILC1 but not NK cells showed upregulation of the activation marker CD69 on their surfaces after CCl4 injection (Figures 1C, 1D, and S1B). In addition, after CCl4 injection, the T cell activation marker CD25 was upregulated on ILC1 but not NK cells (Figures 1E, 1F, and S1C). The expression of CD69 and CD25 on ILC1 peaked on day 1 after CCl4 injection (Figures S1B and S1C). The CD25+ ILC1 in CCl4-injected mice displayed a more activated phenotype than did CD25− ILC1 in the liver of naïve and CCl4-injected mice, as demonstrated by higher expression of CD69 on liver CD25+ ILC1 (Figures 1G and 1H). More importantly, liver CD25+ ILC1 in CCl4-inejcted mice preferentially produced IFN-γ and degranulated ex vivo (Figures 1I–K and S1D). Unlike liver ILC1, NK1.1+TCRβ+DX5− lymphocytes (mainly composed of NKT cells), NK1.1−TCRβ+DX5− lymphocytes (conventional αβ T cells and MAIT cells), and NK1.1−TCRβ−DX5− lymphocytes which include γδ T cells in the liver of CCl4-injected mice did not show obvious IFN-γ production (Figure S1E). WT mice and Rag-1-deficient (Rag1−/−) mice which lack adaptive immune cells showed similar concentrations of alanine aminotransferase (ALT) in the plasma and expression of CD69 and CD25 on ILC1 18 h after CCl4 injection (Figure S2). These results demonstrate that liver ILC1 but not NK cells are activated and produce IFN-γ after CCl4 injection independently of adaptive immune cells, suggesting a unique function of ILC1 during acute liver injury.
Figure 1. Liver ILC1 but not NK cells are activated and produce IFN-γ after CCl4 injection.
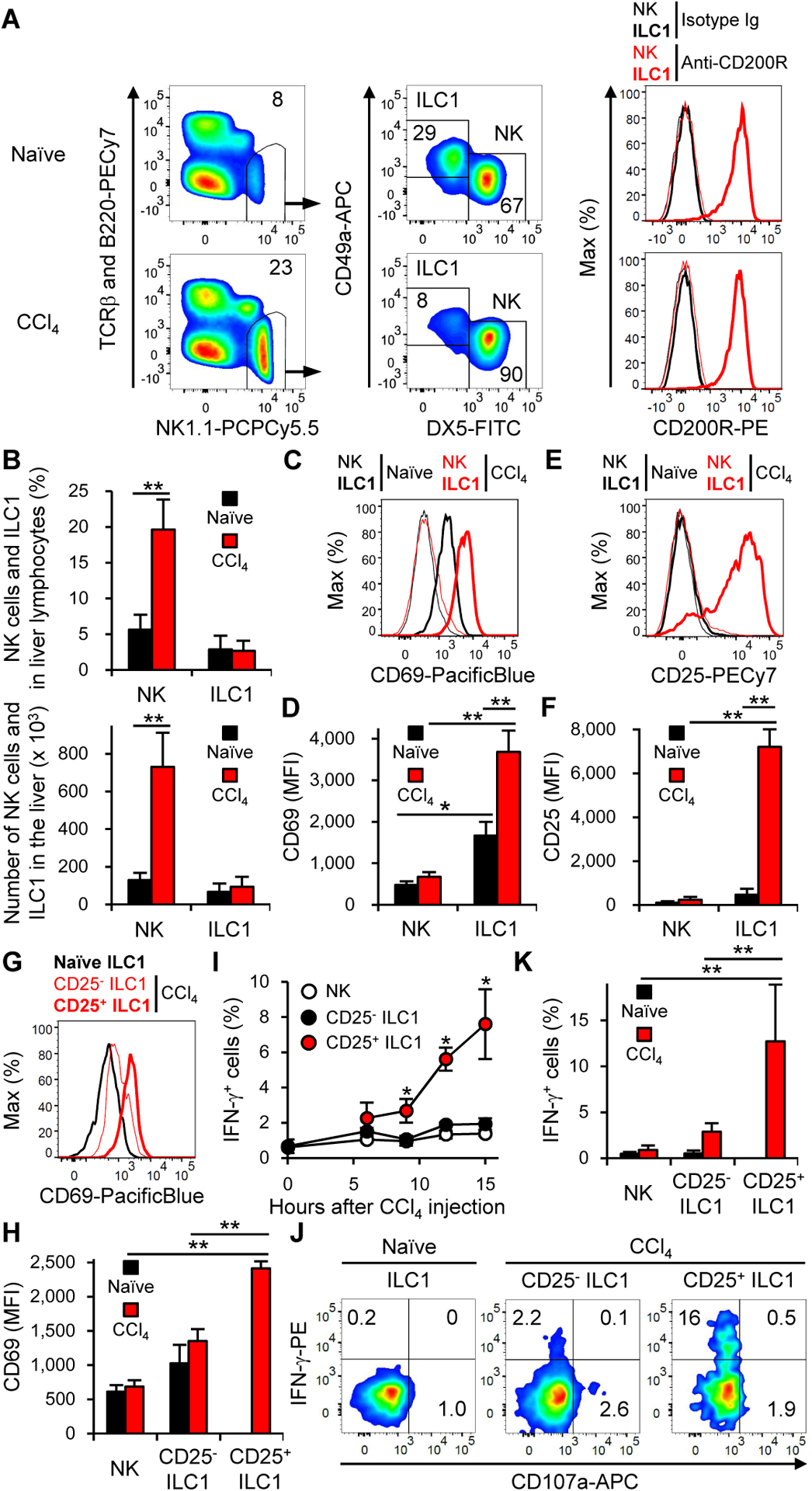
(A) Gating strategy and expression of CD200R for NK cells and ILC1 from the liver of mice before (naïve) and 18 h after CCl4 injection (n = 2–5 for gating strategy and n = 3–4 for CD200R). (B) The percentages and the number of NK cells and ILC1 in the liver before (naïve) and 18 h after CCl4 injection. Data were pooled from 6 experiments (n = 5–14). (C) Expression of CD69 on NK cells and ILC1 in the liver before and 18 h after CCl4 injection (n = 2–5). (D) Mean fluorescence intensity (MFI) of CD69 on NK cells and ILC1 in the liver before and 18 h after CCl4 injection. Data were pooled from 4 experiments (n = 4–14). (E) Expression of CD25 on NK cells and ILC1 in the liver before and 18 h after CCl4 injection (n = 2–5). (F) MFI of CD25 on NK cells and ILC1 in the liver before and 18 h after CCl4 injection. Data were pooled from 2 experiments (n = 4). (G) Expression of CD69 on ILC1 in the liver of mice before CCl4 injection and on CD25− ILC1 and CD25+ ILC1 in the liver 15 h after CCl4 injection (n = 2–5). (H) MFI of CD69 on NK cells and ILC1 in the liver of mice before CCl4 injection and on NK cells, CD25− ILC1, and CD25+ ILC1 in the liver 15 h after CCl4 injection. Data were pooled from 3 experiments (n = 9–13). (I) Kinetics of the percentages of IFN-γ+ cells in NK cells, CD25− ILC1, and CD25+ ILC1 in the liver after CCl4 injection (n = 4). *p<0.05 vs. NK cells and CD25− ILC1. (J) The percentages of IFN-γ+ cells in ILC1 in the liver before CCl4 injection and those in CD25− ILC1 and CD25+ ILC1 in the liver 15 h after CCl4 injection (n = 2–5). (K) The percentages of IFN-γ+ cells in NK cells and ILC1 in the liver before CCl4 injection and those in NK cells, CD25− ILC1, and CD25+ ILC1 in the liver 15 h after CCl4 injection. Data were pooled from 3 experiments (n = 9–13). Data are representative of more than 20 (A, C, and E), 3 (G and J), and 2 (I, and A (CD200R)) independent experiments. **p<0.005. Error bars show s.d. See also Figures S1 and S2.
ILC1-derived IFN-γ suppresses CCl4-induced acute liver injury
Like liver ILC1 in WT mice (Figures 1F and S1C), those cells, but neither NK cells nor non-group 1 innate lymphocytes (NK1.1−NKp46−DX5− lymphocytes), in the liver of Rag1−/− mice upregulated CD25 and increased in number after CCl4 injection (Figures 2A and 2B). To investigate the role of liver ILC1 in the development of CCl4-induced acute liver injury, we depleted activated CD25+ ILC1 in Rag1−/− mice by injecting a monoclonal antibody (mAb) against CD25 (Figure 2B). CCl4 injection increased the IFN-γ concentration in the plasma of mice that had been injected with isotype-matched immunoglobulin (Ig), but not of those injected with anti-CD25 mAb (Figure 2B), consistent with IFN-γ production by CD25+ ILC1 after CCl4 injection (Figures 1I–K). In Rag1−/− mice injected with anti-CD25 mAb, the increase in the plasma ALT after CCl4 injection was higher than that in mice injected with isotype-matched Ig (Figure 2B). These results suggest that activated ILC1-derived IFN-γ protects the liver from CCl4-induced acute injury. To confirm these findings, we depleted either NK cells alone by injecting the optimized dose of anti-asialo GM1 polyclonal antibody (pAb) or both ILC1 and NK cells by injecting a depletion mAb against NK1.1 (Victorino et al., 2015) (Figures S3A and S3B). Depletion of NK cells and ILC1 in the liver prevented an increase in IFN-γ concentrations and elevated ALT in the plasma after CCl4 injection (Figures S3C and S3D). On the contrary, preferential depletion of NK cells elevated plasma concentrations of IFN-γ but not of ALT after CCl4 injection (Figures S3C and S3D). These results indicate that activated ILC1 rather than NK cells in the liver are the major source of IFN-γ after CCl4 injection. To confirm whether liver ILC1, but not NK cells, are required for the amelioration of CCl4-induced acute liver injury, we utilized Hobit-deficient (Zfp683−/−) mice that are severely reduced in liver ILC1 (Mackay et al., 2016), but neither NK cells nor other peripheral ILC1 (Weizman et al., 2017), and NcrcreEomesfl/fl mice that selectively lack NK cells (Gordon et al., 2012; Narni-Mancinelli et al., 2011), but not tissue-resident ILC1 (Weizman et al., 2017). Zfp683−/− mice, but not Ncr1creEomesfl/fl mice, showed increased ALT in the plasma after CCl4 injection (Figures 2C and 2D).
Figure 2. IFN-γ released from liver ILC1 has a protective role in CCl4-induced acute liver injury.
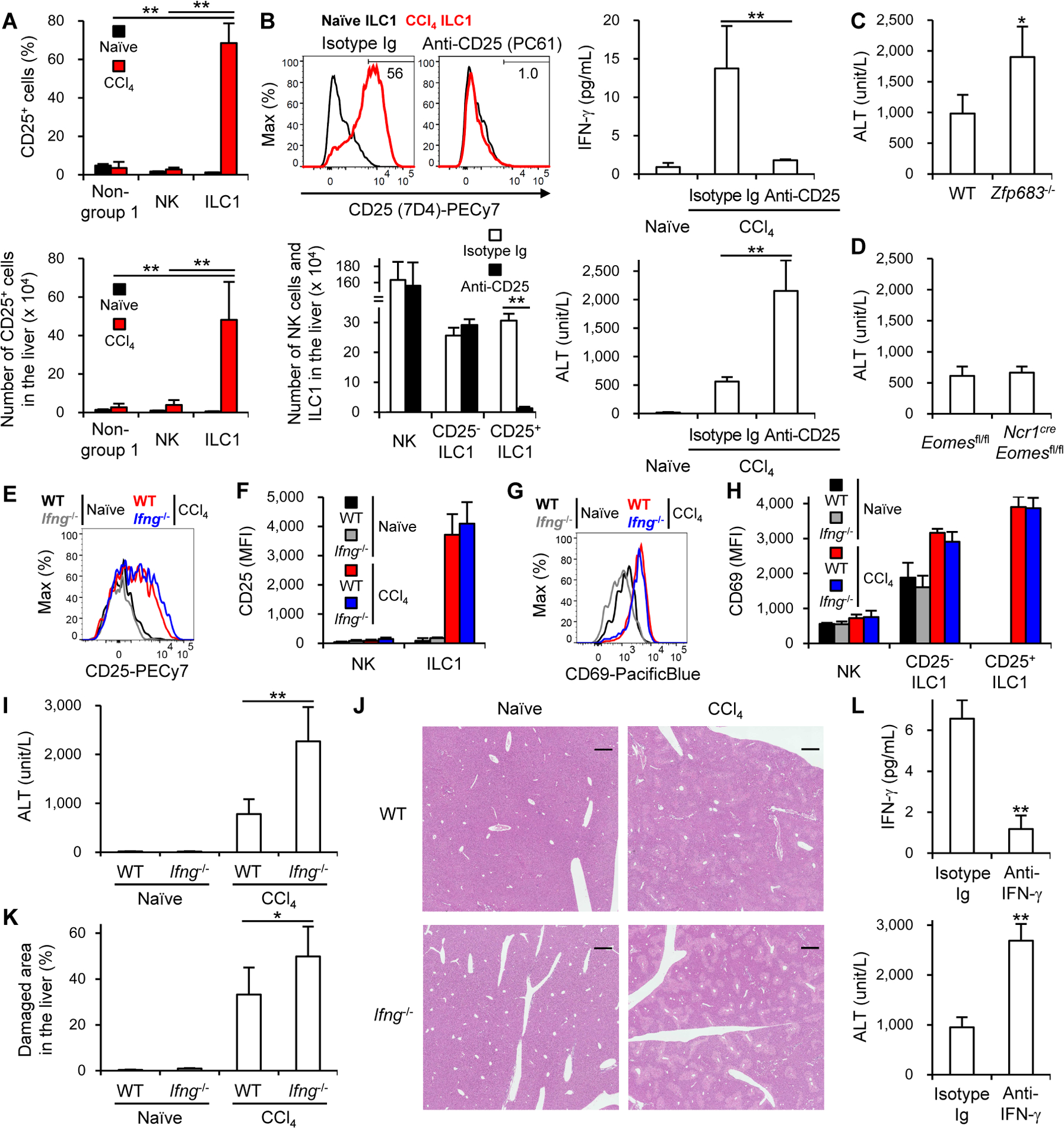
(A) The percentages and number of CD25+ cells in the liver of Rag1−/− mice before (naïve) and 18 h after CCl4 injection. Data were pooled from 2 experiments (n = 4–6). (B) The percentages of CD25+ (7D4+) ILC1 (upper left) (n = 3) and number of each lymphocyte subset in the liver (lower left) and plasma concentrations of IFN-γ (upper right) and ALT (lower right) of Rag1−/− mice that had been injected with a depletion anti-CD25 mAb (PC61) or isotype-matched Ig (Isotype Ig) 18 h after CCl4 injection. Data were pooled from 2 (lower panels) (n = 6) and 3 (upper right) (n = 6–10) experiments. (C) Plasma concentrations of ALT of WT and Zfp683−/− mice 18 h after CCl4 injection. Data were pooled from 2 experiments (n = 4–8). (D) Plasma concentrations of ALT of Eomesfl/fl and Ncr1creEomesfl/fl mice 18 h after CCl4 injection. Data were pooled from 2 experiments (n = 7). (E) Expression of CD25 on ILC1 in the liver of WT and Ifng−/− mice before (naïve) and 18 h after CCl4 injection (n = 2–4). (F) MFI of CD25 on NK cells and ILC1 in the liver of WT and Ifng−/− mice before and 18 h after CCl4 injection. Data were pooled from 2 experiments (n = 3–7). (G) Expression of CD69 on ILC1 in the liver of WT and Ifng−/− mice before and 18 h after CCl4 injection (n = 2–4). (H) MFI of CD69 on NK cells, CD25− ILC1, and CD25+ ILC1 in the liver of WT and Ifng−/− mice before and 18 h after CCl4 injection. Data were pooled from 2 experiments (n = 3–7). (I) Plasma concentrations of ALT before and 18 h after CCl4 injection. Data were pooled from 3 experiments (n = 4–10). (J) Histology of the liver (hematoxylin and eosin staining) before and 18 h after CCl4 injection (n = 2–4). Scale bars represent 500 μm. (K) Quantified damaged areas around the central veins and the portal veins of mice before and 18 h after CCl4 injection. Data were pooled from 2 experiments (n = 3–7). (L) Plasma concentrations of IFN-γ (upper) and ALT (lower) of mice (that had been injected with a neutralizing anti-IFN-γ mAb or isotype Ig 6 h before and 6 h after CCl4 injection) 18 h after CCl4 injection. Data were pooled from 2 experiments (n = 8). Data are representative of 2 independent experiments (B (upper left), E, G, and J). *p<0.05, **p<0.005. Error bars show s.d. See also Figure S3.
We further evaluated the protective effect of IFN-γ secreted from activated ILC1 in CCl4-induced acute liver injury by using IFN-γ-deficient (Ifng−/−) mice. Although liver ILC1 expressed both CD25 and CD69 at similar amounts in WT and Ifng−/− mice before and after CCl4 injection (Figures 2E–H), Ifng−/− mice had significantly higher concentrations of ALT and larger damaged areas around the central veins and the portal triads in the liver after CCl4 injection in comparison with WT mice (Figures 2I–K). Consistent with these results, treatment with a neutralizing mAb against IFN-γ significantly decreased IFN-γ concentrations and increased ALT concentrations after CCl4 injection in comparison with isotype-matched Ig (Figure 2L). Selective depletion of activated CD25+ ILC1, depletion of both ILC1 and NK cells, and neutralization of IFN-γ partially increased concentrations of IL-4 and IL-5 in the plasma after CCl4 injection (Figures S3E–G), suggesting that ILC1 partially but inadequately counterbalance T helper-2 (Th2) cell-associated cytokine-mediated responses by producing IFN-γ in CCl4-induced acute liver injury. Depletion of both ILC1 and NK cells, preferential depletion of NK cells, selective depletion of activated CD25+ ILC1, and neutralization of IFN-γ did not affect the recovery of the acute liver injury on day 3 after CCl4 injection, as compared with vehicle and isotype-matched Ig controls (Figures S3H–J). Zfp683−/− and Ncr1creEomesfl/fl mice showed similar concentrations of ALT at the recovery phase of the acute liver injury on day 3 after CCl4 injection (Figures S3K and S3L). Taken together, these results indicate that IFN-γ derived from activated ILC1 has a protective role in CCl4-induced acute liver injury.
To confirm the protective effect of liver ILC1 in acute liver injury, we also established a low-dose acetaminophen (APAP)-induced mild liver injury in Rag1−/− mice. The number of NK cells in the liver slightly increased after APAP injection, whereas that of ILC1 was not affected (Figure S3M). However, liver ILC1, but neither NK cells nor non-group 1 ILC, upregulated CD25 after APAP injection (Figures S3N and S3O). Liver CD25+ ILC1 in APAP-injected mice displayed a more activated phenotype than did liver CD25− ILC1 and NK cells in naïve and APAP-injected mice, as demonstrated by the higher expression of CD69 (Figure S3P). These liver CD25+ ILC1 in APAP-injected mice preferentially produced IFN-γ ex vivo (Figure S3Q). Depletion of CD25+ ILC1 by using anti-CD25 mAb exacerbated the acute liver damage (Figure S3R). Consistent with findings in CCl4-induced acute liver injury, Ifng−/− mice had significantly higher concentrations of ALT after APAP injection (Figure S3S). These results suggest that activated ILC1-derived IFN-γ protects the liver from not only CCl4-induce acute injury but also APAP-induced acute injury.
DNAM-1 and IL-7 are required for optimal activation of liver ILC1 after CCl4 injection
Although ILC1 express a variety of NK cell receptors (Jiao et al., 2016; Seillet et al., 2016; Spits et al., 2016), it is largely unknown whether signals though these receptors regulate activation of ILC1. The activating NK receptor DNAM-1 (CD226) was expressed more highly on liver ILC1 than on liver NK cells (Figure 3A). A DNAM-1 ligand, CD155, is expressed in the liver (Ravens et al., 2003). We found that the expression of Pvr encoding CD155 was strongly upregulated in the liver after CCl4 injection (Figure 3B), consistent with previous reports that cellular stress such as reactive free radical-induced DNA double strand break, which is the key mechanism of CCl4-mediated hepatotoxicity (Knockaert et al., 2012), increases CD155 expression (Soriani et al., 2014). When WT and DNAM-1-deficient (Cd226−/−) mice were injected with CCl4, liver ILC1 in Cd226−/− mice expressed smaller amounts of CD69 and CD25 than did WT mice (Figures 3C–F). In contrast, the expression of these activation markers on NK cells did not change between WT and Cd226−/− mice after CCl4 injection (Figures 3D and 3F). In line with these findings in vivo, crosslinking DNAM-1 with anti-DNAM-1 mAb increased the expression of CD25 on liver-derived ILC1 but not NK cells in vitro (Figure 3G). These results indicate that DNAM-1 is involved in the optimal activation of liver ILC1 after CCl4 injection.
Figure 3. DNAM-1 and IL-7 are required for optimal activation of liver ILC1 after CCl4 injection.
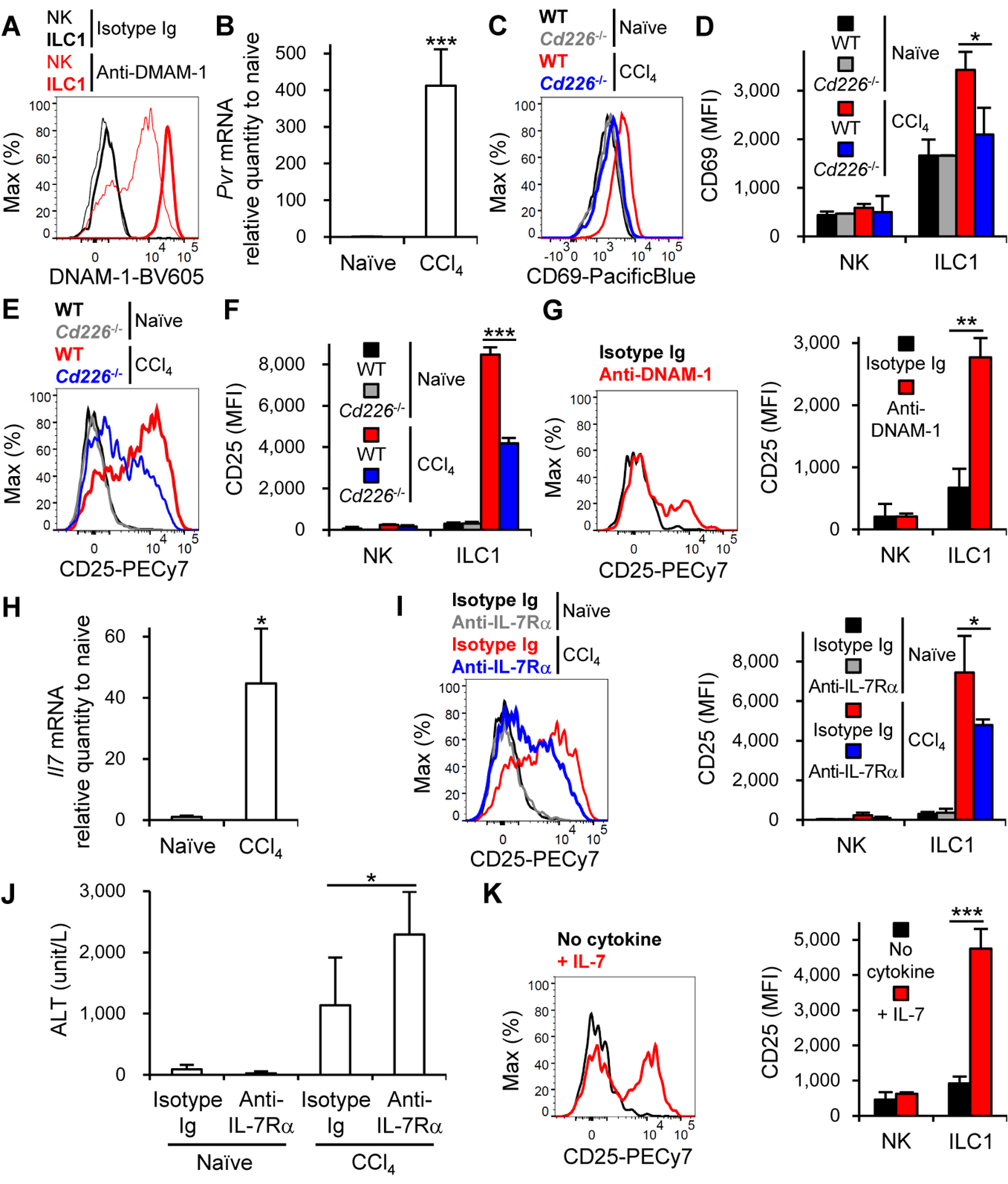
(A) Expression of DNAM-1 on NK cells and ILC1 in the liver (n = 2–5). (B) Expression of Pvr mRNA in the liver of mice before (naïve) and 18 h after CCl4 injection (n = 3–4). (C) Expression of CD69 on ILC1 in the liver of WT and Cd226−/− mice before and 18 h after CCl4 injection. (D) MFI of CD69 on NK cells and ILC1 in the liver of WT and Cd226−/− mice before and 18 h after CCl4 injection (n = 2–5). (E) Expression of CD25 on ILC1 in the liver of WT and Cd226−/− mice before and 18 h after CCl4 injection. (F) MFI of CD25 on NK cells and ILC1 in the liver of WT and Cd226−/− mice before and 18 h after CCl4 injection (n = 2–5). (G) Expression of CD25 on liver ILC1 (left) (n = 2–4) and MFI of CD25 on liver NK cells and liver ILC1 (right) after crosslinking with isotype Ig or anti-DNAM-1 mAb. Data were pooled from 3 experiments (right) (n = 8). (H) Expression of Il7 mRNA in the liver of mice before and 18 h after CCl4 injection (n = 3–4). (I) Expression of CD25 on ILC1 (left) (n = 3) and MFI of CD25 on NK cells and ILC1 (right) in the liver of mice (that had been injected with a neutralizing anti-IL-7Rα mAb or isotype Ig 6 h before and 6 h after CCl4 injection) before and 18 h after CCl4 injection. Data were pooled from 2 experiments (right) (n = 3–6). (J) Plasma concentrations of ALT of mice (that had been injected with a neutralizing anti-IL-7Rα mAb or isotype Ig 6 h before and 6 h after CCl4 injection) before and 18 h after CCl4 injection. Data were pooled from 2 experiments (n = 3–6). (K) Expression of CD25 on liver ILC1 (left) (n = 2) and MFI of CD25 on liver NK cells and liver ILC1 (right) in the absence or presence of IL-7. Data were pooled from 3 experiments (right) (n = 4). Data are representative of 3 (A, G (left), and K (left)), 2 (B, H, and I (left)), and 4 (C-F) independent experiments. *p<0.05, **p<0.01, ***p<0.005. Error bars show s.d.
Because DNAM-1 deficiency did not completely abolish the upregulation of CD25 on liver ILC1 after CCl4 injection, we searched for another factor that increases CD25 expression on ILC1 but not NK cells after CCl4 injection. Unlike NK cells, all ILC subsets, including liver ILC1, express IL-7Rα (Robinette et al., 2015; Sojka et al., 2014; Weizman et al., 2017); IL-7 is dispensable for the development of ILC1 but not ILC2 and ILC3 (Daussy et al., 2014; Klose et al., 2014). IL-7 is produced by hepatocytes and regulates the responses of hepatic lymphocytes (Liang et al., 2012). We examined whether IL-7-IL-7R signaling affects the generation of activated CD25+ ILC1 after CCl4 injection. The amount of Il7 transcripts in the liver increased in response to CCl4 injection (Figure 3H). Blockade of IL-7-IL-7R signaling with a neutralizing mAb against IL-7Rα reduced the upregulation of CD25 on ILC1 after CCl4 injection (Figure 3I). Treatment with a neutralizing mAb against IL-7Rα significantly increased ALT concentrations after CCl4 injection in comparison with isotype-matched Ig (Figure 3J), consistent with exacerbated liver injury in mice depleted of activated CD25+ ILC1 (Figure 2B). IL-7 upregulated the expression of CD25 on ILC1 but not on NK cells in vitro (Figure 3K). These findings indicate that activating signaling through DNAM-1 and IL-7R is required for activation of ILC1 in the liver after CCl4 injection.
DNAM-1 is critical for IFN-γ production by liver ILC1 after CCl4 injection
DNAM-1 promotes IFN-γ production by NK cells (Nabekura et al., 2014; Zhang et al., 2015). After CCl4 injection, the frequencies of CD25+ and CD25− ILC1 subsets producing IFN-γ in the liver were significantly lower in Cd226−/− mice than in WT mice (Figures 4A and 4B). Cd226−/− ILC1 were unable to upregulate Ifng transcripts in response to CCl4 injection (Figure S4A). Consequently, the plasma concentrations of IFN-γ after CCl4 injection were significantly lower in Cd226−/− mice than in WT mice (Figure 4C). Consistent with our findings of low IFN-γ concentrations concomitant with exaggerated liver injury in mice with depleted CD25+ ILC1, Ifng−/− mice, and anti-IFN-γ mAb-treated mice after CCl4 injection (Figures 2B, I, and L), CCl4-induced liver injury was significantly more severe in Cd226−/− mice than in WT mice irrespective of the existence of adaptive immune cells (Figure 4C). However, Rag1−/− and Rag1−/−Cd226−/− mice had similar magnitude of liver injury after CCl4 injection when CD25+ ILC1 were depleted with anti-CD25 mAb (Figure 4D). WT and Cd226−/− mice showed similar concentrations of ALT at the recovery phase of the acute liver injury on day 3 after CCl4 injection (Figure S4B). Crosslinking DNAM-1 with anti-DNAM-1 mAb enhanced IFN-γ production from freshly isolated and cytokine-primed liver ILC1 (Figure 4E). Together, these results demonstrate that DNAM-1 is critical for IFN-γ production from activated CD25+ ILC1 and suppresses acute liver injury after CCl4 injection.
Figure 4. DNAM-1 is critical for IFN-γ production by liver ILC1 after CCl4 injection.

(A) The percentages of IFN-γ+ cells in ILC1, CD25− ILC1, and CD25+ ILC1 in the liver of in WT and Cd226−/− mice before (naïve) and 15 h after CCl4 injection (n = 2–5). (B) The percentages of IFN-γ+ cells in NK cells, CD25− ILC1, and CD25+ ILC1 in the liver of in WT and Cd226−/− mice before and 15 h after CCl4 injection. Data were pooled from 2 experiments (n = 5–9). (C) Plasma concentrations of IFN-γ (left) and ALT (middle) of WT and Cd226−/− mice or Rag1−/− and Rag1−/−Cd226−/− mice (right) before and 18 h after CCl4 injection. Data were pooled from 4 (left) (n = 4–15), 5 (middle) (n = 5–17), and 3 (right) (n = 3–10) experiments. (D) Plasma concentrations of ALT of Rag1−/− and Rag1−/−Cd226−/− mice (that had been injected with a depletion anti-CD25 mAb or isotype Ig) 18 h after CCl4 injection (n = 3). (E) The percentages of IFN-γ+ cells in liver ILC1 (that had been primed with IL-2 and IL-7 in vitro) (left) (n = 2–5) and those in naïve or primed liver NK cells and liver ILC1 (right) (n = 5–7) after crosslinking with anti-DNAM-1 mAb or Isotype Ig. Data were pooled from 2 experiments (right). Data are representative of 2 (A and D) and 3 (E (left)) independent experiments. *p<0.05, **p<0.005. Error bars show s.d. See also Figure S4.
Adenosine triphosphate (ATP) accelerates IL-12-driven IFN-γ production by liver ILC1
We investigated which damage-associated molecular pattern (DAMP) molecule is responsible for IFN-γ production by liver ILC1 during CCl4-induced acute liver injury. Tissue injury, including acute liver injury, results in an immediate ATP release, which regulates the activation and functions of myeloid cells and adaptive immune cells expressing P2 purinergic receptors (Cekic and Linden, 2016; Savio et al., 2018). We found that liver ILC1, but not NK cells, expressed the purinergic receptor P2RX7 (Figure 5A), consistent with the previous reports (Robinette et al., 2015; Sojka et al., 2014). CCl4 injection elevated the concentration of extracellular ATP (Figure 5B). To assess the effect of ATP-P2RX7 signaling on the activation of and IFN-γ production by ILC1, we treated mice with a selective P2RX7 antagonist Brilliant Blue G (BBG) (Donnelly-Roberts and Jarvis, 2007) before CCl4 injection. BBG did not affect the upregulation of CD25 or CD69 on liver ILC1 after CCl4 injection (Figures 5C and 5D). However, CD25+ and CD25− ILC1 from BBG-treated mice showed attenuated IFN-γ production, and these mice had more severe acute liver injury after CCl4 injection (Figures 5E–G). These results indicate that ATP-P2RX7 signaling is required for optimal IFN-γ production by liver ILC1 but not for activation of ILC1 during CCl4-induced acute liver injury.
Figure 5. ATP accelerates IL-12-driven IFN-γ production by liver ILC1.

(A) Expression of P2RX7 on NK cells and ILC1 in the liver (n = 3–5). (B) Plasma concentration of extracellular ATP from the proximal inferior vena cava before (naïve) and 15 h after CCl4 injection. Data were pooled from 2 experiments (n = 6–10). (C) Expression of CD25 on NK cells and ILC1 in the liver of mice (that had been injected or not with BBG) before and 18 h after CCl4 injection (n = 2–5). (D) Expression of CD69 on NK cells, CD25− ILC1, and CD25+ ILC1 in the liver of mice (that had been injected or not with BBG) before and 18 h after CCl4 injection (n = 2–5). (E) The percentages of IFN-γ+ cells in ILC1, CD25− ILC1, and CD25+ ILC1 in the liver of mice (that had been injected or not with BBG) before and 15 h after CCl4 injection (n = 2–5). (F) The percentages of IFN-γ+ cells in NK cells, CD25− ILC1, and CD25+ ILC1 in the liver of mice (that had been injected or not with BBG) before and 15 h after CCl4 injection. Data were pooled from 2 experiments (n = 5–10). (G) Plasma concentration of ALT in mice (that had been injected or not with BBG) before and 18 h after CCl4 injection. Data were pooled from 2 experiments (n = 4–8). (H) Expression of Il12a and Il12b mRNA in the liver before and 18 h after CCl4 injection (n = 3–4). (I) Expression of Il12a and Il12b mRNA in myeloid cell subsets (CD11b− DC, CD11b+ DC, Kupffer cells (KC), and macrophages (Mϕ)) in the liver before and 18 h after CCl4 injection. Data were pooled from 3 experiments (n = 4–5). (J) The percentages of IFN-γ+ cells in liver NK cells and liver ILC1 after stimulation with or without IL-12, ATP or both. Data were pooled from 2 experiments (n = 6). (K) The percentages of IFN-γ+ cells in liver NK cells, liver CD25− ILC1, and liver CD25+ ILC1 (isolated from mice before (naïve) and 18 h after CCl4 injection) after stimulation with or without IL-12 and ATP. Data were pooled from 3 experiments (n = 10–16). Data are representative of 7 (A) and 2 (C, D, E, and H) independent experiments. *p<0.05, **p<0.005. Error bars show s.d. See also Figure S5.
IL-12 strongly drives IFN-γ production by tissue-resident ILC1 (Bernink et al., 2013; Fuchs et al., 2013; Klose et al., 2014; O’Sullivan et al., 2016; Weizman et al., 2017). CCl4 injection increased the amounts of Il12a and Il12b transcripts in the liver (Figure 5H), and these increases were not affected by BBG (Figure S5A). To determine the major cellular source of IL-12 in the liver after CCl4 injection, we sorted myeloid cell subsets in the liver of naïve and CCl4-injected mice and quantified the expression of Il12 transcripts (Figure S5B). In CCl4-injected mice, the Il12b transcript was upregulated in the CD11b− dendritic cell (DC) subset (CD45.2+TCRβ−B220−NK1.1−F4/80−CD11c+I-Ab+CD11b− cells), mainly including conventional DC1 (Figure 5I). Extracellular ATP alone did not induce IFN-γ production by liver ILC1 or NK cells isolated from the liver of naïve mice, but a significantly higher proportion of liver ILC1, but not NK cells, produced IFN-γ in response to IL-12 and ATP (Figure 5J), demonstrating that ATP enhances IL-12-driven IFN-γ production by the liver ILC1. When ILC1 were isolated from the liver of CCl4-injected mice and stimulated with IL-12 and ATP, the majority of CD25+ ILC1 but not CD25− ILC1 or NK cells produce IFN-γ (Figure 5K). Liver ILC1 displayed the greater responsiveness to IL-12 than liver NK cells in vitro in their ability to produce IFN-γ (Figures 5J and 5K), consistent with a prior study (Weizman et al., 2017). In support of these results, we quantified the expression of IL-12 receptors in these cells. Liver ILC1 expressed larger amounts of Il12rb transcripts than did liver NK cells (Figure S5C). These results demonstrate that ATP accelerates IL-12-driven IFN-γ production by ILC1, especially CD25+ ILC1, in CCl4-induced acute liver injury.
IFN-γ contributes to the survival of hepatocytes through upregulation of Bcl-xL after CCl4 injection
IFN-γ suppresses the activation of hepatic stellate cells (HSC), which exacerbate acute and chronic liver injury and fibrosis (Baroni et al., 1996; Grossman et al., 1998). To evaluate the effect of IFN-γ on HSC activation in the case of CCl4-induced liver injury, we used a neutralizing anti-IFN-γ mAb and quantified the expression of the activation markers (Col1a1, Acta2, and Timp1) of HSC (Ghiassi-Nejad et al., 2013). However, neutralization of IFN-γ had little effect on activation of HSC following CCl4 injection (Figure S6A). In addition, it also had no obvious effect on the elevation of Th2 cell-associated cytokines and the activation of HSC after CCl4 injection (Figures S3G and S6A). IFN-γ deficiency did not affect the recruitment of immune cell subsets after CCl4 injection (Figure S6B). Therefore, we directly investigated the effect of IFN-γ on hepatocytes in naïve and CCl4-injected WT and Ifng−/− mice. Hepatocytes expressed IFN-γ receptor (Figure 6A) and Bcl-2 family proteins Bcl-2, Bcl-xL, and Mcl-1 (Figure 6B), which promote hepatocyte survival during acute liver injury (Hikita et al., 2009; Kovalovich et al., 2001). After CCl4 injection, the hepatocytes of WT mice increased amounts of Bcl-2, and more obviously Bcl-xL, but not Mcl-1, whereas the hepatocytes of Ifng−/− mice did not (Figures 6B and 6C). Injection of a neutralizing mAb against IFN-γ, but not isotype-matched Ig, prevented the upregulation of Bcl-2 and Bcl-xL expressions (Figures S6C and S6D). Hepatocytes of Cd226−/− mice, in which liver ILC1 had an impaired ability to produce IFN-γ, also failed to increase the amounts of Bcl-2 and Bcl-xL after CCl4 injection (Figures S6E and S6F). Together, these results suggest that IFN-γ helps the survival of hepatocytes damaged by CCl4 through upregulation of Bcl-2 and Bcl-xL expressions.
Figure 6. IFN-γ contributes to the survival of hepatocytes through upregulation of Bcl-xL.
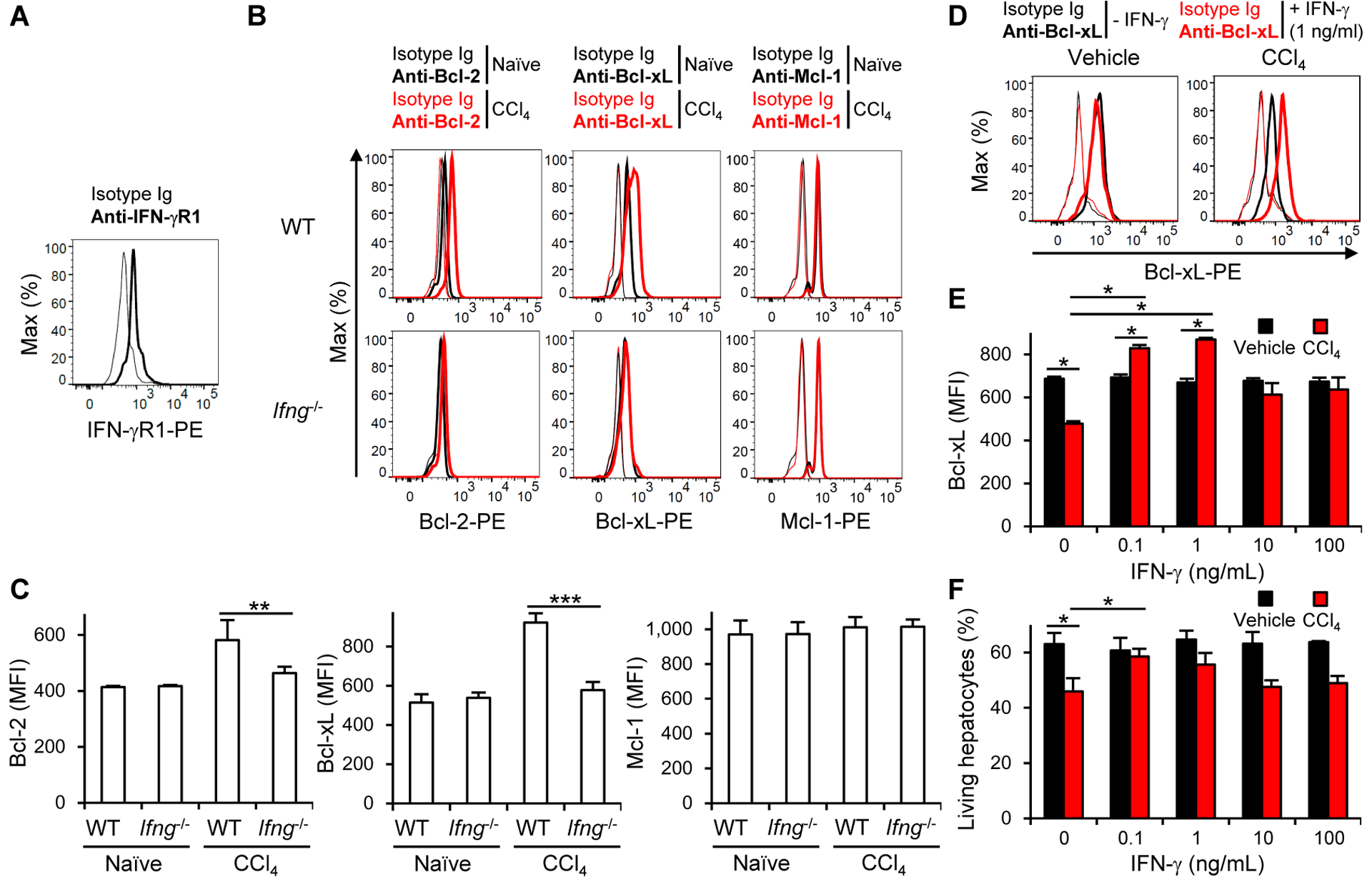
(A) Expression of IFN-γR1 on hepatocytes (n = 2). (B) Expression of intracellular Bcl-2, Bcl-xL, and Mcl-1 in hepatocytes of WT and Ifng−/− mice before (naïve) and 18 h after CCl4 injection (n = 2–3). (C) MFI of intracellular Bcl-2, Bcl-xL, and Mcl-1 in hepatocytes of WT and Ifng−/− mice before and 18 h after CCl4 injection. Data were pooled from 2 experiments (n = 2–6). (D) Expression of Bcl-xL in hepatocytes cultured in the presence or absence of CCl4, IFN-γ, or both (n = 3). (E) MFI of Bcl-xL in hepatocytes cultured in the presence or absence of CCl4, IFN-γ, or both (n = 3). (F) Survival of hepatocytes cultured in the presence or absence of CCl4, IFN-γ, or both (n = 4). The percentages of living hepatocytes are shown. Data are representative of 3 (A, D, E, and F) and 2 (B) independent experiments. *p<0.05. **p<0.01, ***p<0.005. Error bars show s.d. See also Figure S6.
To determine whether IFN-γ directly promotes the survival of CCl4-damaged hepatocytes, we cultured hepatocytes in the presence or absence of IFN-γ, CCl4, or both. In the presence of CCl4 alone, hepatocytes decreased amounts of Bcl-xL (Figures 6D and 6E). However, in the presence of both IFN-γ and CCl4, hepatocytes expressed significantly larger amounts of Bcl-xL than in the presence of CCl4 alone (Figures 6D and 6E). Only low concentrations of IFN-γ (0.1 and 1 ng/mL) restored the expression of Bcl-xL in CCl4-damaged hepatocytes (Figures 6D and 6E) and rescued the survival of CCl4-damaged hepatocytes (Figure 6F). IFN-γ did not affect the expression of Bcl-xL and the survival in the absence of CCl4 (Figures 6D–F). These results demonstrate that IFN-γ contributes to the survival of CCl4-damaged hepatocytes by upregulating Bcl-xL expression.
Liver ILC1-derived IFN-γ is sufficient to ameliorate CCl4-induced acute liver injury
To prove that activated liver ILC1 producing IFN-γ are necessary and sufficient for the amelioration of CCl4-induced acute liver injury, we purified liver ILC1 (NK1.1+NKp46+DX5−CD49a+TRAIL+CD69+ lymphocytes) from Rag1−/− B6 mice and injected them together with or without IL-15 into the portal vein of Rag-2 and IL-2 receptor common γ chain double-deficient (Rag2−/−Il2rg−/−) mice, which lack all lymphocyte subsets, including lLC1 (Song et al., 2010) (Figures 7A and S7A). Despite the low frequency and the small number of donor ILC1 engrafted into the liver of recipient Rag2−/−Il2rg−/− mice on day 1 post-transfer, donor ILC1 proliferated homeostatically, and their frequency and number increased significantly in the liver of recipient mice by day 7 (Figures 7B and 7C). The homeostatic proliferation of donor ILC1 was enhanced by IL-15 (Figures 7B and 7C). Donor ILC1 in the liver of recipient mice displayed the same phenotype as naïve liver ILC1, irrespective of the IL-15 supplementation (Figures 7B and S7A). On day 7 post-transfer, the recipient mice were injected with CCl4 and donor ILC1 in the liver were analyzed. CCl4 injection showed upregulation of CD25 on donor ILC1 in the liver of recipient mice (Figures 7D and 7E), which was observed in the liver of WT mice as well (Figures 1E, 1F, and S1C). Donor-derived CD25+ ILC1 in the liver of CCl4-injected recipient mice expressed larger amounts of CD69 than did donor-derived CD25− ILC1 in the liver of naïve or CCl4-injected mice (Figures 7F and 7G). These donor CD25+ ILC1 produced more IFN-γ ex vivo than did CD25− ILC1 (Figures 7H–J).
Figure 7. Liver ILC1-derived IFN-γ is sufficient to ameliorate CCl4-induced acute liver injury.
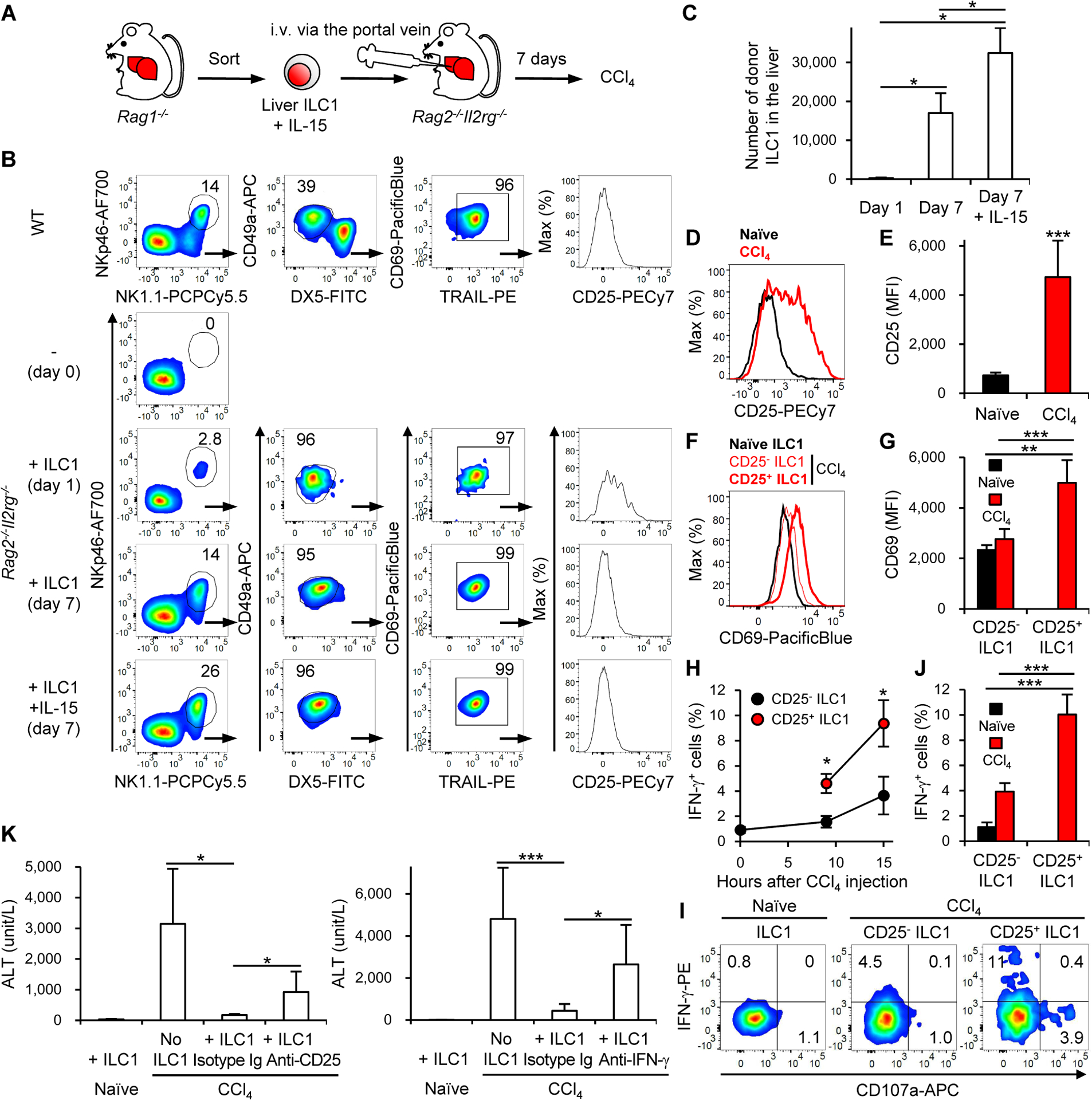
(A) Experimental design of ILC1 transfer into Rag2−/−Il2rg−/− mice. (B) Phenotypical characterization of donor-derived ILC1 in the liver of recipient Rag2−/−Il2rg−/− mice that had been transferred with purified donor ILC1 together with or without IL-15 (n = 2–4). (C) The number of donor-derived ILC1 in the liver of recipient Rag2−/−Il2rg−/− mice that had been transferred with purified donor ILC1 together with or without IL-15. Data were pooled from 4 experiments (n = 4). (D) Expression of CD25 on donor-derived ILC1 in the liver of Rag2−/−Il2rg−/− mice (that had been transferred with purified donor ILC1 together with IL-15) before (naïve) and 18 h after CCl4 injection on day 7 post-transfer (n = 2–3). (E) MFI of CD25 on donor-derived ILC1 in the liver of Rag2−/−Il2rg−/− mice (that had been transferred with purified donor ILC1 together with IL-15) before and 18 h after CCl4 injection on day 7 post-transfer. Data were pooled from 2 experiments (n = 4). (F) Expression of CD69 on donor-derived ILC1, CD25− ILC1, and CD25+ ILC1 in the liver of Rag2−/−Il2rg−/− mice (that had been transferred with purified donor ILC1 together with IL-15) before and 18 h after CCl4 injection on day 7 post-transfer (n = 2–3). (G) MFI of CD69 on donor-derived CD25− ILC1 and CD25+ ILC1 in the liver of Rag2−/−Il2rg−/− mice (that had been transferred with purified donor ILC1 together with IL-15) before and 18 h after CCl4 injection on day 7 post-transfer. Data were pooled from 2 experiments (n = 4). (H) Kinetics of IFN-γ+ cells in donor-derived CD25− ILC1 and CD25+ ILC1 in the liver of Rag2−/−Il2rg−/− mice (that had been transferred with purified donor ILC1 together with IL-15) after CCl4 injection. Data were pooled from 4 experiments (n = 5–15). *p<0.005 vs. CD25− ILC1. (I) The percentages of IFN-γ+ cells in donor-derived ILC1, CD25− ILC1, and CD25+ ILC1 in the liver of Rag2−/−Il2rg−/− mice (that had been transferred with purified donor ILC1 together with IL-15) before and 15 h after CCl4 injection (n = 4–6). (J) The percentages of IFN-γ+ cells in donor-derived CD25− ILC1 and CD25+ ILC1 in the liver of Rag2−/−Il2rg−/− mice (that had been transferred with purified donor ILC1 together with IL-15) before and 15 h after CCl4 injection (n = 4–6). (K) Plasma concentrations of ALT in Rag2−/−Il2rg−/− mice (that had been transferred or not with purified donor ILC1 together with IL-15) before and 18 h after CCl4 injection on day 7 post-transfer, which were also injected with a depletion anti-CD25 mAb (left), a neutralizing anti-IFN-γ mAb (right), or isotype Ig 6 h before and 6 h after CCl4 injection. Data were pooled from 2 (left) (n = 4–5) and 3 (right) (n = 6–7) experiments. Data are representative of 4 (B) and 3 (D, F, I, and J) independent experiments. *p<0.05, **p<0.01, ***p<0.005. Error bars show s.d. See also Figure S7.
Next, we injected a neutralizing mAb against IFN-γ or isotype-matched Ig into the recipient Rag2−/−Il2rg−/− mice that had been engrafted with donor ILC1, and then injected CCl4. Neutralization of IFN-γ had little effect on the activation of donor ILC1 by CCl4, as evidenced by normal upregulation of CD25 and CD69 on donor ILC1 after CCl4 injection (Figures S7B–E). Donor ILC1 transfer significantly ameliorated acute liver injury after CCl4 injection (Figure 7K). This therapeutic effect of donor ILC1 was partially abolished by depletion of CD25+ ILC1 with anti-CD25 mAb and neutralization of IFN-γ (Figure 7K). Ifng−/− donor ILC1 also did not show sufficient therapeutic effect after CCl4 injection (Figure S7F). These results demonstrate that IFN-γ from activated liver ILC1 is necessary and sufficient for the amelioration of CCl4-induced acute liver injury.
Discussion
In the current study, we investigated the role of ILC1 in acute liver injury. Whereas ILC1-derived IFN-γ amplifies inflammation in intestinal injury (Bernink et al., 2013; Fuchs et al., 2013; Peters et al., 2016; Victorino et al., 2015), we found that IFN-γ production by liver ILC1 ameliorated CCl4-induced acute liver injury. IFN-γ is intricately involved in not only the pathogenesis of liver injury but also liver tissue protection (Heymann and Tacke, 2016; Horras et al., 2011). The detrimental role of IFN-γ has been found in mouse models of fulminant liver injury induced by concanavalin A or high doses of APAP (Ishida et al., 2002; Küsters et al., 1996). Yet, the protective effect of IFN-γ was found in mild acute liver injury, such as that induced by CCl4 injection or bile duct ligation (Cheng et al., 2011; Grossman et al., 1998). Our finding is consistent with the protective role of liver ILC1-derived IFN-γ in CCl4-induced and low dose APAP-induced acute liver injury. Fulminant and mild liver injury models differ in the magnitude of elevation of IFN-γ during the progression of acute liver injury. In the present study, IFN-γ concentrations after CCl4 injection increased much less than in fulminant liver injury models, most likely because liver ILC1 but not the other lymphocyte subsets were activated and produced IFN-γ. We showed that low concentrations of IFN-γ promoted survival of damaged hepatocytes in vitro more efficiently than did high concentrations; this is consistent with the magnitude and kinetics of IFN-γ production defining whether the effect of IFN-γ on the pathogenesis of acute liver injury is protective or deleterious. Elucidation of the spatiotemporal regulation of IFN-γ release from liver ILC1 would be key to understanding the mechanism underlying its protective effect in acute liver injury. Activated ILC1 may also produce other-tissue protective molecules because neutralization of IFN-γ and IFN-γ deficiency in donor ILC1 in Rag2−/−Il2rg−/− mice do not completely abolish the therapeutic effect of ILC1 on CCl4-induced acute liver injury.
The varying biological effects of IFN-γ are mediated through IFN-γ receptor-JAK-STAT-1-dependent and -independent signaling pathways and subsequent dynamic regulation of gene expression (Ramana et al., 2002). We found that IFN-γ positively regulates the amounts of Bcl-xL in damaged but not healthy hepatocytes, suggesting that the different modes of action of IFN-γ between healthy and damaged hepatocytes. One possible scenario is that a molecule in damaged hepatocytes interacts with the IFN-γ receptor signaling pathway and modulates the amounts of Bcl-xL by affecting its degradation or post-translational modifications. However, this hypothetical key molecule is still unidentified and further analyses are required to better understand the molecular mechanism underlying this previously unappreciated role of IFN-γ.
Because ILC subsets provide the first line of host defense and elicit local inflammation in their resident tissues, it is likely that DAMP molecules, such as ATP, IL-1, and IL-33, critically affect the effector functions of ILC. For instance, proliferation and Th2 cell cytokine production of ILC2 are triggered by epithelial-derived IL-33, IL-25, and TLSP in the mucosal barrier (Hammad and Lambrecht, 2015). Extracellular ATP is released from damaged tissue, and purinergic ATP receptor signaling regulates the effector functions of both innate and adaptive immune cells (Cekic and Linden, 2016; Di Virgilio et al., 2017). The role of ATP in ILC functions is poorly documented, and no studies have assessed whether extracellular ATP affects the functions of liver ILC1. We found that ATP-P2RX7 signaling is required for optimal IFN-γ production by liver ILC1 after CCl4 injection. P2RX7-deficient mice are highly susceptible to Toxoplasma gondii infection and have impaired IFN-γ responses (Corrêa et al., 2017). These findings support the hypothesis that extracellular ATP at infection sites stimulates ILC1 to produce IFN-γ in a P2RX7-dependent manner, and that ILC1-derived IFN-γ contributes to clearance of the parasite, consistent with an essential role of ILC1 in host protection against the parasite (Klose et al., 2014). We also found that ATP accelerated IL-12-driven IFN-γ production by naïve and activated liver ILC1. Liver ILC1 rapidly produce IFN-γ in response to conventional DC1-derived IL-12 early in MCMV infection (Weizman et al., 2017). In accordance with this study, we demonstrated that liver ILC1, but not NK cells, produced IFN-γ in response to CD11b− DC-derived IL-12 in CCl4-induced acute liver injury. The selective responsiveness of liver ILC1 to IL-12 can be explained partly by the expression of P2RX7 on liver ILC1 but not NK cells and by the rapid elevation of extracellular ATP concentrations in the liver after CCl4 injection and presumably also after MCMV infection. P2RX7 is an ATP-gated cation-selective ion channel that allows the influx of calcium ions (Savio et al., 2018; Di Virgilio et al., 2017), implicating that increased intracellular calcium concentrations might enhance IL-12-driven IFN-γ production in liver ILC1.
Although NK cells accumulated in the liver after CCl4 injection, these cells did not produce appreciable amounts of IFN-γ in vivo following CCl4 injection, whereas liver ILC1 displayed sufficient IFN-γ production. However, both liver NK cells and ILC1 had the ability to produce IFN-γ in response to IL-12 in vitro. The discrepancy in IL-12-driven IFN-γ production by NK cells in vivo and in vitro might be explained by different activation status of and sensation of activating signals for liver NK cells from liver ILC1 after CCl4 injection. Unlike liver ILC1, liver NK cells did not show any signs of activation, as demonstrated by expression of CD69 and CD25, after CCl4 injection partly because of no IL-7Rα and low expression of DNAM-1 as compared with liver ILC1. In addition to lower DNAM-1 expression, liver NK cells did not express P2RX7 in their ability to produce IFN-γ. Furthermore, liver NK cells expressed smaller amounts of Il12rb than liver ILC1 both in naïve state and after CCl4 injection. Therefore, insufficient activation status of and suboptimal activating signals for NK cells may render liver NK cells unresponsive to CCl4-induced acute liver injury in vivo.
ILC1 express a variety of NK cell receptors (Jiao et al., 2016; Seillet et al., 2016; Spits et al., 2016). However, it largely remains to be elucidated whether these receptors regulate the activation and function of ILC1. We revealed that the activating receptor DNAM-1 is required for the optimal activation of and IFN-γ production by ILC1 in CCl4-induced acute liver injury. Activating and inhibitory NK cell receptors, including DNAM-1, regulate activation and the effector functions of NK cells as well as the adaptive immune features of NK cells, i.e. antigen-specific proliferation of effector NK cells and differentiation into memory NK cells (Nabekura and Lanier, 2016a, 2016b; Nabekura et al., 2014; Sun et al., 2009). Proliferation activity and memory differentiation of ILC1 in an antigen-specific and non-specific manner have not yet been examined. Further studies of the roles of NK cell receptors on ILC1 are needed to better understand the regulation of activation, functions, and adaptive immune features of ILC1; such studies might provide important insights into therapeutic interventions which modulate the functions of tissue-resident ILC1 for the prevention of tissue damage and organ failure.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Akira Shibuya (ashibuya@md.tsukuba.ac.jp).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
WT C57BL/6 (B6) mice were purchased from CLEA Japan (Tokyo, Japan). Rag-1-deficient (Rag1−/−) mice (Mombaerts et al., 1992) and IFN-γ-deficient (Ifng−/−) mice (Dalton et al., 1993) on the B6 background and Rag-2 and IL-2 receptor common γ chain double-deficient (Rag2−/−Il2rg−/−) mice (Song et al., 2010) were purchased from Jackson Laboratory (Bar Harbor, Maine, USA). DNAM-1-deficient (Cd226−/−) mice on the B6 background were generated as described previously (Iguchi-Manaka et al., 2008). Rag-1 and DNAM-1 double-deficient (Rag1−/−Cd226−/−) B6 mice were generated by crossing Rag1−/− B6 mice with Cd226−/− B6 mice. Ncr1creEomesfl/fl mice (Gordon et al., 2012; Narni-Mancinelli et al., 2011) and Hobit-deficient (Zfp683−/−) mice (Mackay et al., 2016) on the B6 background were generated as described previously (Weizman et al., 2017). Male and female mice between 6 and 22 weeks of age were used for experiments in a gender-matched manner. All mice were housed and maintained under specific-pathogen-free conditions and all procedures were approved by the University of Tsukuba animal ethics committee (17–313, 18–279, 19–235) and the University of California, Los Angeles (UCLA) Institutional Animal Care and Use Committee (ARC#2017-084-01F) and performed at the laboratory animal resource centers of University of Tsukuba and UCLA in accordance with the guidelines of the institutional animal ethics committees.
CCl4-induced liver injury
Mice on the B6 background were intraperitoneally injected with 10 μl of 10% CCl4 (Sigma-Aldrich, St. Luis, Missouri, USA) in corn oil (Fujifilm Wako Pure Chemical Corporation, Tokyo, Japan) per gram body weight. Rag2−/−Il2rg−/− mice exhibited severe acute liver injury (20,000–30,000 unit/L) 18 h after injection of the same dose of CCl4 as that used for mice on the B6 background (data not shown). Thus, the dose of CCl4 for Rag2−/−Il2rg−/− mice was titrated down to the similar magnitude of acute liver injury to that of mice on the B6 background (1,000–3,000 unit/L). Rag2−/−Il2rg−/− mice were intraperitoneally injected with 10 μl of 0.04% CCl4 in corn oil per gram body weight. Blood was collected 18 h (1 day), and 2 and 3 days after CCl4 injection and plasma samples were prepared by centrifugation at 2500 rpm (500 xg) for 15 min at 4°C. Concentrations of ALT in the plasma were measured by using DRI-CHEM 7000V and GPT/ALT-PIII slides (Fujifilm, Tokyo, Japan).
To deplete specific cell populations and block cytokine signaling, mice were intraperitoneally injected with 200–250 μg depletion mAb against mouse CD25 (clone PC61), a neutralizing mAb against mouse IFN-γ (clone R4–6A2, Bio × Cell, West Lebanon, New Hampshire, U.S.A.), a neutralizing mAb against mouse IL-7Rα (clone A7R34, Bio × Cell), or isotype-matched immunoglobulins (Igs) (Bio × Cell) 6 h before and after the injection of CCl4. Anti-mouse CD25 mAb (clone 7D4) was used for detection of CD25+ cells. Anti-mouse CD25 mAbs (clones PC61 and 7D4) do not compete with the other for binding to CD25 (Moreau et al., 1987).
To deplete both NK cells and ILC1 or NK cells only, mice were intraperitoneally injected with 200 μg depletion mAb against mouse NK1.1 (clone PK136, ATCC, Manassas, Virginia, U.S.A.) or 10 μg anti-asialo GM1 rabbit pAb (Fujifilm Wako Pure Chemical Corporation), respectively, or the same volume of PBS (vehicle control) 36 h before the injection of CCl4. The doses of anti-mouse NK1.1 mAb and anti-asialo GM1 rabbit pAb described above are optimal for depletion of both NK cells (defined as NKp46+DX5+CD49a− lymphocytes) and ILC1 (defined as NKp46+DX5−CD49a+ lymphocytes) and of NK cells but not ILC1 in the liver, respectively (Figures S3A and B).
For selective blockade of the ATP receptor P2RX7, mice were intraperitoneally injected with 50 μg Brilliant Blue G (BBG, Sigma-Aldrich) in PBS per gram body weight 1 day before CCl4 injection.
Low dose APAP-induced liver injury
Mice (female and 8–14 weeks of age for evaluation of the acute liver injury) were fasted for 24 h with free access to water but not bedding and feces. Fifteen mg per ml APAP (Sigma-Aldrich) was freshly prepared in 0.9% isotonic sodium chloride solution (Otsuka Pharmaceutical, Tokyo, Japan) at 37°C. WT and Ifng−/− mice and Rag1−/− mice were intraperitoneally injected with 100 and 150 mg/ kg body weight of APAP, respectively, and had free access to food, water, bedding, and feces after APAP injection. Blood was collected 18 h after APAP injection and plasma samples were prepared by centrifugation at 2500 rpm (500 xg) for 15 min at 4°C. Concentrations of ALT in the plasma were measured by using DRI-CHEM 7000V and GPT/ALT-PIII slides (Fujifilm).
To deplete CD25+ cells, Rag1−/− mice were intraperitoneally injected with 250 μg mAb against mouse CD25 (clone PC61) 6 h before and after the injection of APAP. Anti-mouse CD25 (clone 7D4) was used for staining CD25 after depletion of CD25+ cells by using PC61.
Preparation of liver lymphocytes
The liver of naïve, CCl4-injected, and APAP-injected male or female mice at the indicated hours or days in Figure legends after the injection was perfused with PBS, isolated, and mechanically homogenized by using a glass homogenizer (Sanyo, Tokyo, Japan). Liver homogenates were washed with RPMI-1640 culture medium (Sigma-Aldrich) followed by PBS, resuspended with 40% Percoll PLUS density gradient medium (GE Healthcare, Chicago, Illinois, USA), overlaid on 60% Percoll PLUS medium, and centrifuged at 800 xg for 35–45 min at 20°C. Buffy coats were collected and washed with RPMI-1640 medium followed by PBS supplemented with 5% fetal calf serum (FCS, Thermo Fisher Scientific, Massachusetts, USA), and used as liver lymphocytes. Liver NK cells were identified as either NK1.1+TCRβ−B220−DX5+CD49a−CD200R− or NK1.1+NKp46+DX5+CD49a−CD200R− lymphocytes, liver ILC1 were identified as either NK1.1+TCRβ−B220−DX5−CD49a+CD200R+ or NK1.1+NKp46+DX5−CD49a+CD200R+ lymphocytes. Liver T cells and NKT cells were identified as NK1.1−TCRβ+ and NK1.1+TCRβ+ lymphocytes, respectively. Non-group 1 innate lymphocytes in the liver of Rag1−/− mice were identified as NK1.1−NKp46−DX5− lymphocytes.
To purify NK cells and ILC1, liver lymphocytes from naïve and CCl4-injected male or female mice were stained for cell surface molecules at the indicated hours in Figure legends after the injection, and NK cells (NK1.1+NKp46+CD3ε−DX5+CD49a− lymphocytes), CD25− ILC1 (NK1.1+NKp46+CD3ε−DX5−CD49a+CD25− lymphocytes), and CD25+ ILC1 (NK1.1+NKp46+CD3ε−DX5−CD49a+CD25+ lymphocytes) were sorted by using BD FACSAria III (BD Biosciences, San Jose, California, USA).
Preparation of liver myeloid cell subsets
The liver of naïve and CCl4-injected male or female mice 18 h after the injection was isolated after tissue perfusion with PBS, cut into small pieces in 5 ml Krebs-Ringer modified buffer with 5 mg collagenase type IV (Sigma-Aldrich) and 600 μg deoxyribonuclease I (Worthington Biochemical Corporation, Lakewood, New Jersey, USA) per liver, and incubated at 37°C and for 30 min with rotation at 7 rpm. The cell suspension was vortexed, and cells were collected by being passed through a ϕ100 μm cell strainer (BD Biosciences). The remaining pieces of liver tissue were homogenized in a glass homogenizer, and cells were collected by being passed through a ϕ100 μm cell strainer. These cells were combined, centrifuged, and washed with PBS supplemented with 5% FCS. Cells were stained for cell surface molecules, and myeloid cells were gated on leukocytes with large FSC-SSC values followed by gating on CD45.2+TCRβ−B220−NK1.1− cells, and CD11b− DC (F4/80−CD11c+I-Ab+CD11b−), CD11b+ DC (F4/80−CD11c+I-Ab+CD11b+), Kupffer cells (KC: F4/80+ and highCD11c−CD11b−), and macrophages (Mϕ: F4/80+CD11c−CD11bhigh), and sorted by using BD FACSAria III.
In some experiments, myeloid cell subsets in the liver were gated on leukocytes with large FSC-SSC values followed by gating on CD45.2+TCRβ−NK1.1− cells, and CD11b− DC, CD11b+ DC, KC, Mϕ and monocytes (Mono: F4/80+CD11c−CD11b+ and high), and neutrophils (Neu: F4/80−CD11c−I-Ab−Ly-6G+) were analyzed. B cells in the liver were gated on leukocytes with small FSC-SSC values followed by gating on CD45.2+TCRβ−NK1.1− cells, and identified as F4/80−CD11c−Ly-6G−B220+ cells.
Isolation of hepatocytes
Hepatocytes were isolated as described previously (Mederacke et al., 2015) with some modifications. At 18 h after CCl4 injection, the liver of naïve and CCl4-injected male or female mice was perfused at 42°C with 20 ml Ca2+ chelator buffer (8 g NaCl, 0.4 g KCl, 0.1 g NaH2PO4-2H2O, 0.12 g Na2HPO4, 0.35 g NaHCO3, 0.9 g glucose, 2.38 g HEPES, 0.38 g EGTA per liter at pH 7.4) followed by 20 ml collagenase liver perfusion buffer (10 mg collagenase type IV, 8 g NaCl, 0.4 g KCl, 0.735 g CaCl2-2H2O, 0.078 g NaH2PO4-2H2O, 0.151 g Na2HPO4-12H2O, 0.35 g NaHCO3, 0.9 g glucose, 2.38 g HEPES per liter at pH7.4) by using a 25G needle (Terumo Corporation, Tokyo, Japan) and a Pharmacia Biotech Pump P-1 (GE Healthcare) at a flow rate of 20 (100 mL/ h) via the portal vein. The liver was transferred into a 10 cm dish with 10 ml PBS supplemented with 10% FCS and incubated for 10 min at 42°C. The liver was shaken gently, and pinched repeatedly with forceps to release hepatocytes, and the cell suspension was collected by being passed a ϕ100 μm cell strainer. The remaining liver tissues were ground on the ϕ100 μm cell strainer by using a plunger of 10 ml syringe, resuspended in PBS supplemented with 10% FCS, and collected by being passed through the ϕ100 μm cell strainer. Both cell suspensions were combined, washed twice by centrifugation at 50 xg for 1 min, resuspended with 4 ml PBS, gently mixed with 5 ml 90% Percoll PLUS medium, and centrifuged at 100 xg for 10 min at 20°C. The cell pellet was washed by centrifugation at 70 g for 5 min with PBS supplemented with 10% FCS, gently resuspended with cold PBS supplemented with 10% FCS, and used as hepatocytes. Hepatocytes were analyzed ex vivo by flow cytometry or cultured in vitro.
Ex vivo culture of NK cells and ILC1
NK cells and ILC1 were enriched from the liver of naïve, CCl4-injected, and APAP-injected male mice 15 h or the indicated hours in Figure legends after the injection, and one million or the smaller number of cells were cultured in 96-well cell culture plates for 5 h at 5% CO2 and 37°C in the dark in RPMI-1640 culture medium supplemented with 10% FCS, 50 μM 2-mercaptoethenol, 2 mM L-glutamine, 100 U penicillin, 0.1 mg/mL streptomycin (Sigma-Aldrich), 10 mM HEPES, 1 mM sodium pyruvate, and 100 μM MEM non-essential amino acids (Thermo Fisher Scientific) in the presence of BD GolgiStop (BD Biosciences) and APC-conjugated anti-mouse CD107a mAb (clone 1D4B, BioLegend, San Diego, California, USA). The cells were then collected, washed, stained for surface molecules and intracellular IFN-γ, and analyzed by flow cytometry. Degranulation of NK cells and ILC1 was determined by the frequencies of CD107a+ cells.
In vitro culture of NK cells and ILC1
NK cells and ILC1 were enriched from the liver of naïve male or female mice, and one million or the smaller number of cells were cultured in 96-well cell culture plates for 24 h at 5% CO2 and 37°C in RPMI-1640 culture medium supplemented with 10% FCS, 50 μM 2-mercaptoethenol, 2 mM L-glutamine, 100 U penicillin, 0.1 mg/mL streptomycin, 10 mM HEPES, 1 mM sodium pyruvate, and 100 μM MEM non-essential amino acids in the presence or absence of 25 ng/mL recombinant mouse (rm) IL-7 (Bio-Techne Corporation, Minneapolis, Minnesota, USA).
NK cells and ILC1 were enriched from the liver of naïve male or female mice and were stimulated by crosslinking DNAM-1 as described previously (Nabekura and Lanier, 2016b) with some modifications. Wells of 96 -well cell culture plates were coated with 1 mg/mL 1,2-dioleoyl-3-trimethylammoniumpropane (DOTAP; Cayman Chemical, Ann Arbor, Michigan, USA) in 50% ethanol for 10 min at room temperature and washed with PBS. Ten microgram purified anti-DNAM-1 mAb (TX42.1) (Iguchi-Manaka et al., 2008) or isotype-matched Ig in 50 mM carbonate buffer (pH 9.6) was added to DOTAP-coated wells, and the wells were incubated for 24 h at 4°C; they were then washed with RPMI-1640 culture medium supplemented with 10% FCS and blocked with the same culture medium for 10 min at room temperature. NK cells and ILC1 were enriched from the liver of naïve male or female mice and one million or the smaller number of cells were cultured in mAb-coated wells of 96-well cell culture plates for 24 h at 5% CO2 and 37°C in RPMI-1640 culture medium supplemented with 10% FCS, 50 μM 2-mercaptoethenol, 2 mM L-glutamine, 100 U penicillin, 0.1 mg/mL streptomycin, 10 mM HEPES, 1 mM sodium pyruvate, and 100 μM MEM non-essential amino acids.
The cells were then collected, washed, stained for surface molecules, and analyzed by flow cytometry.
Ex vivo stimulation of NK cells and ILC1
To prepare cytokine-primed NK cells and ILC1, NK cells and ILC1 were enriched from the liver of naïve male or female mice and cultured in 96-well cell culture plates for 24 h at 5% CO2 and 37°C in RPMI-1640 culture medium supplemented with 10% FCS, 50 μM 2-mercaptoethenol, 2 mM L-glutamine, 100 U penicillin, 0.1 mg/mL streptomycin, 10 mM HEPES, 1 mM sodium pyruvate, and 100 μM MEM non-essential amino acids in the presence or absence of 25 ng/mL rm IL-7 and 50 ng/mL rm IL-2 (BD Biosciences). For stimulation by crosslinking DNAM-1, one million or the smaller number of freshly isolated naïve or cytokine-primed cells were resuspended in RPMI-1640 culture medium supplemented with 10% FCS, 50 μM 2-mercaptoethenol, 2 mM L-glutamine, 100 U penicillin, 0.1 mg/mL streptomycin, 10 mM HEPES, 1 mM sodium pyruvate, and 100 μM MEM non-essential amino acids, and cultured for 5 h at 5% CO2 and 37°C in the dark in the presence of BD GolgiStop in the wells of 96-well cell culture plates coated with anti-DNAM-1 mAb or isotype-matched Ig, as described previously (Nabekura and Lanier, 2016b).
For stimulation with IL-12, ATP, or both, NK cells and ILC1 were enriched from the liver of naïve and CCl4-injected male or female mice 18 h after the injection and resuspended in RPMI-1640 culture medium supplemented with 10% FCS, 50 μM 2-mercaptoethenol, 2 mM L-glutamine, 100 U penicillin, 0.1 mg/mL streptomycin, 10 mM HEPES, 1 mM sodium pyruvate, and 100 μM MEM non-essential amino acids; one million or the smaller number of NK cells and ILC1 from the liver of naïve male or female mice were cultured in 96-well cell culture plates for 5 h at 5% CO2 and 37°C in the dark in the presence of BD GolgiStop and stimulated with 10 ng/mL rm IL-12 (BD Biosciences), 500 nM ATP (Sigma-Aldrich), or both. Alternatively, one million or the smaller number of NK cells and ILC1 from the liver of naïve and CCl4-injected male or female mice were cultured as above and stimulated with 25 ng/mL rm IL-12 and 500 nM ATP.
After stimulation, cells were collected, washed, stained for surface molecules and intracellular IFN-γ, and analyzed by flow cytometry.
Primary culture of hepatocytes
One hundred thousand hepatocytes were cultured at 37°C and 10% CO2 in DMEM/F12 culture medium (Sigma-Aldrich) supplemented with 10% FCS, 50 μM 2-mercaptoethenol, 4 mM L-glutamine, 100 U penicillin, 0.1 mg/mL streptomycin, 20 mM HEPES, 100 nM dexamethasone, 0.2 mM L-ascorbic acid 2-phosphate sesquimagnesium salt hydrate, and 2 μg/mL insulin from bovine pancreas (Sigma-Aldrich) in the wells of 24-well cell culture plates coated with 5 μg/cm2 mouse natural collagen type IV (Corning, Corning, New York, U.S.A.). Three hours after plating, to remove floating dead hepatocytes, the culture medium was replaced with fresh medium supplemented with 2 mM CCl4 or the same volume of DMSO (vehicle control), and adherent hepatocytes were cultured in the presence or absence of 0.1–100 ng/mL rm IFN-γ (BioLegend) for 24 h at 37°C and 10% CO2. Hepatocytes were washed twice with room temperature PBS supplemented with 1 mM EDTA, and treated with 0.25% trypsin-EDTA (Sigma-Aldrich) for 5 min at 37°C and 10% CO2, DMEM culture medium (Sigma-Aldrich) supplemented with 10% FCS was then added, and cells collected by gentle pipetting, and washed with PBS supplemented with 10% FCS. Hepatocytes were analyzed by flow cytometry.
METHOD DETAILS
Flow cytometry
Fc receptors (CD16 and CD32) were blocked with mAb clone 2.4G2 (Tonbo Biosciences, San Diego, California, USA) before surface and intracellular staining with the indicated fluorochrome-conjugated mAbs or isotype-matched control antibodies (Thermo Fisher Scientific, Tonbo Biosciences, BD Biosciences, and BioLegend). mAbs used were FITC-conjugated anti-mouse CD49b (clone DX5), FITC-conjugated anti-mouse I-Ab (clone AF6–120.1), PE-conjugated anti-mouse CD4 (clone RM4–5), PE-conjugated anti-mouse CD8α (clone 53–6.7), PE-conjugated anti-mouse CD11b (clone M1/70), PE-conjugated anti-mouse CD200R (clone OX-110), PE-conjugated anti-mouse P2RX7 (clone 1F11), PE-conjugated anti-mouse TRAIL (clone N2B2), PE-conjugated streptavidin, PerCPCy5.5-conjugated anti-mouse B220 (clone RA3–6B2), PerCPCy5.5-conjugated anti-mouse NK1.1 (clone PK136), PerCPCy5.5-conjugated anti-mouse NKp46 (clone 29A1.4), APC-conjugated anti-mouse CD11b (clone HL3), APC-conjugated anti-mouse CD49a (clone HMα1), APC-conjugated anti-mouse NK1.1 (clone PK136), Alexa Fluor (AF) 700-conjugated anti-mouse Ly-6G (clone 1A8), AF700-conjugated anti-mouse NKp46 (clone 29A1.4), PECy7-conjugated anti-mouse B220 (clone RA3–6B2), PECy7-conjugated anti-mouse CD3ε (clone 145–2C11), PECy7-conjugated anti-mouse CD25 (clone PC61), PECy7-conjugated anti-mouse F4/80 (clone BM8), PECy7-conjugated anti-mouse TCRβ (clone H57–597), Pacific Blue-conjugated anti-mouse CD3ε (clone 145–2C11), Pacific Blue-conjugated anti-mouse CD69 (clone H1.2F3), Brilliant Violet (BV) 421-conjugated anti-mouse CD45.2 (clone 104), biotinylated anti-mouse B220 (clone RA3–6B2), biotinylated anti-mouse CD25 (clone 7D4), biotinylated anti-mouse DNAM-1 (clone TX42.1), biotinylated anti-mouse IFN-γR1 (clone GR20), biotinylated anti-mouse NK1.1 (clone PK136), biotinylated anti-mouse TCRβ (clone H57–597), and Brilliant Violet 605-conjugated streptavidin.
For staining of intracellular IFN-γ, liver NK cells and ILC1, cells were fixed and permeabilized with BD Cytofix/Cytoperm solution (BD Biosciences), washed with Intracellular Staining Perm Wash Buffer (BioLegend), and stained with PE-conjugated anti-mouse IFN-γ mAb (clone XMG1.2, BioLegend).
For staining of intracellular Bcl-2 family proteins, hepatocytes were fixed, permeabilized, and washed with eBioscience Foxp3 / Transcription Factor Staining Buffer Set (Thermo Fisher Scientific), and stained with PE-conjugated anti-mouse Bcl-2 mAb (clone BCL/10C4, BioLegend), PE-conjugated anti-mouse Bcl-xL mAb (clone 7B2.5, Abcam, Cambridge, United Kingdom), or PE-conjugated anti-mouse Mcl-1 (clone D2W9E, Cell Signaling Technology, Massachusetts, U.S.A.). To exclude white blood cells, hepatocytes were defined as cells with very large FSC (forward scatter) and SSC (side scatter) parameters. Hepatic stellate cells were excluded as vitamin A- and retinol-positive cells by using a violet laser and the Pacific Blue detection channel (450/50 nm filter) of BD LSRFortessa (BD Biosciences). Cells were stained with Zombie NIR Fixable Viability Kit (BioLegend) and living hepatocytes were defined as Zombie NIR-negative cells.
In all experiments, doublet cells were excluded by FSC-A and FSC-H gating followed by SSC-A and SSC-W gating, and cells with bright autofluorescence including dead cells were excluded by using AmCyan (525/50 nm filter) as a dump channel of BD LSRFortessa and BD FACSAria III. In some experiments, cells were stained with Zombie NIR Fixable Viability Kit and living cells were defined as Zombie NIR-negative cells.
Samples were run on BD LSRFortessa, BD FACSAria III, or BD FACS Calibur (BD Biosciences) and the data were analyzed with FlowJo software (FlowJo, Ashland, Oregon, U.S.A.).
Measurement of cytokines in the plasma
Blood was collected 18 h after CCl4 injection and plasma samples were prepared by centrifugation at 2500 rpm (500 xg) for 15 min at 4°C. Concentrations of IFN-γ, IL-4, and IL-5 in the plasma were measured with BD Cytometric Bead Array Mouse Flex Sets for mouse IFN-γ, mouse IL-4, and mouse IL-5 (BD Biosciences).
Quantitative reverse transcription (RT)-PCR
The liver of naïve and CCl4-injected mice 18 h after the injection was isolated after tissue perfusion with PBS, 5 mm3 pieces of the liver were excised, and homogenized in Isogen reagent (Nippon Gene, Tokyo, Japan) by using frosted slide glasses (Matsunami Glass Industry, Osaka, Japan).
In some experiments, NK cells, CD25− ILC1, and CD25+ ILC1 purified from the liver of naïve and CCl4-injected mice 12 h after the injection, or CD11b− DC, CD11b+ DC, KC, and Mϕ purified from the liver of naïve and CCl4-injected mice 18 h after the injection were resuspended with TRIzol reagent (Thermo Fisher Scientific).
Total RNA was isolated using the Isogen or TRIzol and first-strand DNA was synthesized by using a High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). Quantitative RT-PCR was performed on an ABI 7500 Fast real-time PCR system with ABI Power SYBR Green PCR Master Mix (both from Thermo Fisher Scientific) and 50–250 nM primers. The primers were as follows: Acta2 forward, 5’-GTCCCAGACATCAGGGAGTAA-3’; Acta2 reverse, 5’-TCGGATACTTCAGCGTCAGGA-3’; Actb forward, 5’-ACTGTCGAGTCGCGTCCA-3’; Actb reverse, 5’-GCAGCGATATCGTCATCCAT-3’; Col1a1 forward, 5’-GCTCCTCTTAGGGGCCACT-3’; Col1a1 reverse, 5’-CCACGTCTCACCATTGGGG-3’; Ifng forward, 5’-ACAGCAAGGCGAAAAAGGATG-3’; Ifng reverse, 5’-TGGTGGACCACTCGGATGA-3’; Il7 forward, 5’-TTCCTCCACTGATCCTTGTTCT-3’; Il7 reverse, 5’-AGCAGCTTCCTTTGTATCATCAC-3’; Il12a forward, 5’-CCCTTGCCCTCCTAAACCAC-3’; Il12a reverse, 5’-AAGGAACCCTTAGAGTGCTTACT-3’; Il12b forward, 5’-GGAGACCCTGCCCATTGAACT-3’; Il12b reverse, 5’-CAACGTTGCATCCTAGGATCG-3’; Il12rb1 forward, 5’-ATGGCTGCTGCGTTGAGAA-3’; Il12rb1 reverse, 5’-AGCACTCATAGTCTGTCTTGGA-3’; Il12rb2 forward, 5’-AGAGAATGCTCATTGGCACTTC-3’; Il12rb2 reverse, 5’-AACTGGGATAATGTGAACAGCC-3’; Pvr forward, 5’-CAACTGGTATGTTGGCCTCA-3’; Pvr reverse, 5’-ATTGGTGACTTCGCACACAA-3’; Timp1 forward, 5’-GCAACTCGGACCTGGTCATAA-3’; and Timp1 reverse, 5’-CGGCCCGTGATGAGAAACT-3’. Relative quantities and copy numbers of gene transcripts were normalized to those of Actb transcripts.
Histology
The liver of naïve and CCl4-injected mice was fixed with formalin and embedded in paraffin 18 h after the injection. Fixed histological sections were stained with hematoxylin and eosin. Images were acquired under a BZ-X710 microscope (Keyence, Osaka, Japan), and damaged areas around the central veins and the portal triads in one lobe of the liver were quantified with BZ-X analyzer (Keyence).
Measurement of extracellular ATP in the plasma
Blood was collected from the proximal inferior vena cava (after which the hepatic veins join) of naïve and CCl4-injected mice 15 h after the injection. Plasma samples were prepared by centrifugation at 2500 rpm (500 xg) for 15 min at 4°C. Concentrations of ATP were measured by a bioluminescence-based assay using a Tissue ATP assay kit (Fujifilm Wako Pure Chemical) and a Turner BioSystems Luminometer (Promega, Madison, Wisconsin, USA).
Adoptive transfer of liver ILC1
Lymphocytes were enriched from the liver of WT, Ifng−/−, or Rag1−/− B6 male or female mice by Percoll gradient centrifugation and stained for surface molecules, and ILC1 were gated on NK1.1+NKp46+DX5−CD49a+TRAIL+CD69+ lymphocytes and sorted by using BD FACSAria III. Thirty thousand purified donor liver ILC1 were mixed with or without 2 μg rm IL-15 (BioLegend) and adoptively transferred into recipient Rag2−/−Il2rg−/− mice via the portal vein. On days 1 and 7 post-transfer, donor ILC1 were enriched from the liver of the recipient mice and analyzed by flow cytometry.
Recipient mice that received donor liver ILC1 and IL-15 were injected with or without CCl4 on day 7 post-transfer. In some experiments, they were intraperitoneally injected with 200–250 μg depletion mAb against mouse CD25, neutralizing mAb against mouse IFN-γ, or an isotype-matched Ig 6 h before and 6 h after the injection of CCl4. Donor ILC1 were enriched from the liver of naïve and CCl4-injected recipient mice 18 h after the injection and the expression of cell surface molecules, including activation markers, on donor ILC1 were analyzed by flow cytometry. To evaluate IFN-γ production by donor ILC1, they were enriched from the liver of naïve and CCl4-injected recipient mice 9 h and 15 h after the injection and cultured in 96-well cell culture plates for 5 h at 5% CO2 and 37°C in the dark in RPMI-1640 culture medium supplemented with 10% FCS, 50 μM 2-mercaptoethenol, 2 mM L-glutamine, 100 U penicillin, 0.1 mg/mL streptomycin, 10 mM HEPES, 1 mM sodium pyruvate, 100 μM MEM non-essential amino acids, BD GolgiStop, and APC-conjugated anti-mouse CD107a mAb. Cells were collected, washed, stained for surface molecules and intracellular IFN-γ, and analyzed by flow cytometry. Degranulation of ILC1 was determined by the frequency of CD107a+ cells.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical methods
The Student’s t-test, the Mann-Whitney U test, and one-way Analysis of variance (ANOVA) were used to compare the data by using GraphPad Prism software (GraphPad Software, San Diego, California, U.S.A.). p<0.05 was considered statistically significant. Each data point represents the average of samples. Error bars show s.d.
Supplementary Material
Acknowledgments
We are grateful to Tsuyoshi Kiniwa (RIKEN, Japan) and Yun-Wen Zheng (University of Tsukuba, Japan) for helpful comments on the isolation and the culture of hepatocytes, respectively. We thank Eric Vivier, Steven L. Reiner, and Klaas P. J. M. van Gisbergen for mice. We also thank Yumi Yamashita-Kanemaru and Fumie Abe (University of Tsukuba, Japan) for veterinary procedures. This work was supported by JSPS KAKENHI Grant Number JP16H06387 and 18H05022 (A.S.), the Naito Foundation, the Kato Memorial Bioscience Foundation, Life Science Foundation of Japan, the Uehara Memorial Foundation, and the Kanae Foundation for the Promotion of Medical Science (T.N.), and the National Institutes of Health (AI145997 and P30 DK063491 to T.E.O.).
Footnotes
Declaration of Interests
The authors declare no competing interests.
References
- Abt MC, Lewis BB, Caballero S, Xiong H, Carter RA, Sušac B, Ling L, Leiner I, and Pamer EG (2015). Innate Immune Defenses Mediated by Two ILC Subsets Are Critical for Protection against Acute Clostridium difficile Infection. Cell Host Microbe 18, 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artis D, and Spits H (2015). The biology of innate lymphoid cells. Nature 517, 293–301. [DOI] [PubMed] [Google Scholar]
- Baroni GS, D’Ambrosio L, Curto P, Casini A, Mancini R, Jezequel AM, and Benedetti A (1996). Interferon gamma decreases hepatic stellate cell activation and extracellular matrix deposition in rat liver fibrosis. Hepatology 23, 1189–1199. [DOI] [PubMed] [Google Scholar]
- Bernink JH, Peters CP, Munneke M, te Velde AA, Meijer SL, Weijer K, Hreggvidsdottir HS, Heinsbroek SE, Legrand N, Buskens CJ, et al. (2013). Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat Immunol 14, 221–229. [DOI] [PubMed] [Google Scholar]
- Boulenouar S, Michelet X, Duquette D, Alvarez D, Hogan AE, Dold C, O’Connor D, Stutte S, Tavakkoli A, Winters D, et al. (2017). Adipose Type One Innate Lymphoid Cells Regulate Macrophage Homeostasis through Targeted Cytotoxicity. Immunity 46, 273–286. [DOI] [PubMed] [Google Scholar]
- Cekic C, and Linden J (2016). Purinergic regulation of the immune system. Nat Rev Immunol 16, 177–192. [DOI] [PubMed] [Google Scholar]
- Cheng CW, Duwaerts CC, Rooijen N, Wintermeyer P, Mott S, and Gregory SH (2011). NK cells suppress experimental cholestatic liver injury by an interleukin-6-mediated, Kupffer cell-dependent mechanism. J Hepatol 54, 746–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colonna M (2018). Innate Lymphoid Cells: Diversity, Plasticity, and Unique Functions in Immunity. Immunity 48, 1104–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrêa G, Almeida Lindenberg C, Moreira-Souza AC, Savio LE, Takiya CM, Marques-da-Silva C, Vommaro RC, and Coutinho-Silva R (2017). Inflammatory early events associated to the role of P2X7 receptor in acute murine toxoplasmosis. Immunobiology 222, 676–683. [DOI] [PubMed] [Google Scholar]
- Dalton DK, Pitts-Meek S, Keshav S, Figari IS, Bradley A, and Stewart TA (1993). Multiple defects of immune cell function in mice with disrupted interferon-gamma genes. Science (80-. ). 259, 1739–1742. [DOI] [PubMed] [Google Scholar]
- Daussy C, Faure F, Mayol K, Viel S, Gasteiger G, Charrier E, Bienvenu J, Henry T, Debien E, Hasan UA, et al. (2014). T-bet and Eomes instruct the development of two distinct natural killer cell lineages in the liver and in the bone marrow. J Exp Med 211, 563–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly-Roberts DL, and Jarvis MF (2007). Discovery of P2X7 receptor-selective antagonists offers new insights into P2X7 receptor function and indicates a role in chronic pain states. Br J Pharmacol 151, 571–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebbo M, Crinier A, Vély F, and Vivier E (2017). Innate lymphoid cells: major players in inflammatory diseases. Nat Rev Immunol 17, 665–678. [DOI] [PubMed] [Google Scholar]
- Eberl G, Colonna M, Di Santo JP, and McKenzie AN (2015). Innate lymphoid cells. Innate lymphoid cells: a new paradigm in immunology. Science (80-. ). 348, aaa6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, Newberry RD, Cella M, and Colonna M (2013). Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-γ-producing cells. Immunity 38, 769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger G, Fan X, Dikiy S, Lee SY, and Rudensky AY (2015). Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science (80-. ). 350, 981–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger TL, Abt MC, Gasteiger G, Firth MA, O’Connor MH, Geary CD, O’Sullivan TE, van den Brink MR, Pamer EG, Hanash AM, et al. (2014). Nfil3 is crucial for development of innate lymphoid cells and host protection against intestinal pathogens. J Exp Med 211, 1723–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiassi-Nejad Z, Hernandez-Gea V, Woodrell C, Lang UE, Dumic K, Kwong A, and Friedman SL (2013). Reduced hepatic stellate cell expression of Kruppel-like factor 6 tumor suppressor isoforms amplifies fibrosis during acute and chronic rodent liver injury. Hepatology 57, 786–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon SM, Chaix J, Rupp LJ, Wu J, Madera S, Sun JC, Lindsten T, and Reiner SL (2012). The Transcription Factors T-bet and Eomes Control Key Checkpoints of Natural Killer Cell Maturation. Immunity 36, 55–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman HJ, White D, Grossman VL, and Bhathal PS (1998). Effect of interferon gamma on intrahepatic haemodynamics of the cirrhotic rat liver. J Gastroenterol Hepatol 13, 1058–1060. [DOI] [PubMed] [Google Scholar]
- Hammad H, and Lambrecht BN (2015). Barrier Epithelial Cells and the Control of Type 2 Immunity. Immunity 43, 29–40. [DOI] [PubMed] [Google Scholar]
- Heymann F, and Tacke F (2016). Immunology in the liver--from homeostasis to disease. Nat Rev Gastroenterol Hepatol 13, 88–110. [DOI] [PubMed] [Google Scholar]
- Hikita H, Takehara T, Shimizu S, Kodama T, Li W, Miyagi T, Hosui A, Ishida H, Ohkawa K, Kanto T, et al. (2009). Mcl-1 and Bcl-xL cooperatively maintain integrity of hepatocytes in developing and adult murine liver. Hepatology 50, 1217–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horras CJ, Lamb CL, and Mitchell KA (2011). Regulation of hepatocyte fate by interferon-γ. Cytokine Growth Factor Rev 22, 35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iguchi-Manaka A, Kai H, Yamashita Y, Shibata K, Tahara-Hanaoka S, Honda S, Yasui T, Kikutani H, Shibuya K, and Shibuya A (2008). Accelerated tumor growth in mice deficient in DNAM-1 receptor. J Exp Med 205, 2959–2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida Y, Kondo T, Ohshima T, Fujiwara H, Iwakura Y, and Mukaida N (2002). A pivotal involvement of IFN-gamma in the pathogenesis of acetaminophen-induced acute liver injury. FASEB J 16, 1227–1236. [DOI] [PubMed] [Google Scholar]
- Jiao Y, Huntington ND, Belz GT, and Seillet C (2016). Type 1 Innate Lymphoid Cell Biology: Lessons Learnt from Natural Killer Cells. Front Immunol 7, 426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose CS, and Artis D (2016). Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat Immunol 17, 765–774. [DOI] [PubMed] [Google Scholar]
- Klose CS, Kiss EA, Schwierzeck V, Ebert K, Hoyler T, d’Hargues Y, Göppert N, Croxford AL, Waisman A, Tanriver Y, et al. (2013). A T-bet gradient controls the fate and function of CCR6-RORγt+ innate lymphoid cells. Nature 494, 261–265. [DOI] [PubMed] [Google Scholar]
- Klose CSN, Flach M, Möhle L, Rogell L, Hoyler T, Ebert K, Fabiunke C, Pfeifer D, Sexl V, Fonseca-Pereira D, et al. (2014). Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell 157, 340–356. [DOI] [PubMed] [Google Scholar]
- Knockaert L, Berson A, Ribault C, Prost PE, Fautrel A, Pajaud J, Lepage S, Lucas-Clerc C, Bégué JM, Fromenty B, et al. (2012). Carbon tetrachloride-mediated lipid peroxidation induces early mitochondrial alterations in mouse liver. Lab Invest 92, 396–410. [DOI] [PubMed] [Google Scholar]
- Kovalovich K, Li W, DeAngelis R, Greenbaum LE, Ciliberto G, and Taub R (2001). Interleukin-6 protects against Fas-mediated death by establishing a critical level of anti-apoptotic hepatic proteins FLIP, Bcl-2, and Bcl-xL. J Biol Chem 276, 26605–26613. [DOI] [PubMed] [Google Scholar]
- Küsters S, Gantner F, Künstle G, and Tiegs G (1996). Interferon gamma plays a critical role in T cell-dependent liver injury in mice initiated by concanavalin A. Gastroenterology 111, 462–471. [DOI] [PubMed] [Google Scholar]
- Liang B, Hara T, Wagatsuma K, Zhang J, Maki K, Miyachi H, Kitano S, Yabe-Nishimura C, Tani-Ichi S, and Ikuta K (2012). Role of hepatocyte-derived IL-7 in maintenance of intrahepatic NKT cells and T cells and development of B cells in fetal liver. J Immunol 189, 4444–4450. [DOI] [PubMed] [Google Scholar]
- Mackay LK, Zaid A, Freestone D, Braun A, Wynne-Jones E, Pellicci DG, Godfrey DI, Gebhardt T, Carbone FR, Minnich M, et al. (2016). Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science (80-. ). 352, 459–463. [DOI] [PubMed] [Google Scholar]
- McKenzie ANJ, Spits H, and Eberl G (2014). Innate lymphoid cells in inflammation and immunity. Immunity 41, 366–374. [DOI] [PubMed] [Google Scholar]
- Mederacke I, Dapito DH, Affò S, Uchinami H, and Schwabe RF (2015). High-yield and high-purity isolation of hepatic stellate cells from normal and fibrotic mouse livers. Nat Protoc 10, 305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, and Papaioannou VE (1992). RAG-1-deficient mice have no mature B and T lymphocytes. Cell 68, 869–877. [DOI] [PubMed] [Google Scholar]
- Moreau J‐L, Nabholz M, Diamantstein T, Malek T, Shevach E, and Thèze J (1987). Monoclonal antibodies identify three epitope clusters on the mouse p55 subunit of the interleukin 2 receptor: relationship to the interleukin 2‐binding site. Eur. J. Immunol 17, 929–935. [DOI] [PubMed] [Google Scholar]
- Nabekura T, and Lanier LL (2016a). Activating Receptors for Self-MHC Class I Enhance Effector Functions and Memory Differentiation of NK Cells during Mouse Cytomegalovirus Infection. Immunity 45, 74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabekura T, and Lanier LL (2016b). Tracking the fate of antigen-specific versus cytokine-activated natural killer cells after cytomegalovirus infection. J Exp Med 213, 2745–2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabekura T, Kanaya M, Shibuya A, Fu G, Gascoigne NR, and Lanier LL (2014). Costimulatory molecule DNAM-1 is essential for optimal differentiation of memory natural killer cells during mouse cytomegalovirus infection. Immunity 40, 225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narni-Mancinelli E, Chaix J, Fenis A, Kerdiles YM, Yessaad N, Reynders A, Gregoire C, Luche H, Ugolini S, Tomasello E, et al. (2011). Fate mapping analysis of lymphoid cells expressing the NKp46 cell surface receptor. Proc Natl Acad Sci U S A 108, 18324–18329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan TE, Rapp M, Fan X, Weizman OE, Bhardwaj P, Adams NM, Walzer T, Dannenberg AJ, and Sun JC (2016). Adipose-Resident Group 1 Innate Lymphoid Cells Promote Obesity-Associated Insulin Resistance. Immunity 45, 428–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng H, Jiang X, Chen Y, Sojka DK, Wei H, Gao X, Sun R, Yokoyama WM, and Tian Z (2013). Liver-resident NK cells confer adaptive immunity in skin-contact inflammation. J Clin Invest 123, 1444–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters CP, Mjösberg JM, Bernink JH, and Spits H (2016). Innate lymphoid cells in inflammatory bowel diseases. Immunol Lett 172, 124–131. [DOI] [PubMed] [Google Scholar]
- Ramana CV, Gil MP, Schreiber RD, and Stark GR (2002). Stat1-dependent and -independent pathways in IFN-gamma-dependent signaling. Trends Immunol 23, 96–101. [DOI] [PubMed] [Google Scholar]
- Ravens I, Seth S, Förster R, and Bernhardt G (2003). Characterization and identification of Tage4 as the murine orthologue of human poliovirus receptor/CD155. Biochem Biophys Res Commun 312, 1364–1371. [DOI] [PubMed] [Google Scholar]
- Reynders A, Yessaad N, Vu Manh TP, Dalod M, Fenis A, Aubry C, Nikitas G, Escalière B, Renauld JC, Dussurget O, et al. (2011). Identity, regulation and in vivo function of gut NKp46+RORγt+ and NKp46+RORγt- lymphoid cells. EMBO J 30, 2934–2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinette ML, Fuchs A, Cortez VS, Lee JS, Wang Y, Durum SK, Gilfillan S, Colonna M, and Consortium IG (2015). Transcriptional programs define molecular characteristics of innate lymphoid cell classes and subsets. Nat Immunol 16, 306–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savio LEB, de Andrade Mello P, da Silva CG, and Coutinho-Silva R (2018). The P2X7 Receptor in Inflammatory Diseases: Angel or Demon? Front Pharmacol 9, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seillet C, Huntington ND, Gangatirkar P, Axelsson E, Minnich M, Brady HJ, Busslinger M, Smyth MJ, Belz GT, and Carotta S (2014). Differential requirement for Nfil3 during NK cell development. J Immunol 192, 2667–2676. [DOI] [PubMed] [Google Scholar]
- Seillet C, Belz GT, and Huntington ND (2016). Development, Homeostasis, and Heterogeneity of NK Cells and ILC1. Curr Top Microbiol Immunol 395, 37–61. [DOI] [PubMed] [Google Scholar]
- Sojka DK, Plougastel-Douglas B, Yang L, Pak-Wittel MA, Artyomov MN, Ivanova Y, Zhong C, Chase JM, Rothman PB, Yu J, et al. (2014). Tissue-resident natural killer (NK) cells are cell lineages distinct from thymic and conventional splenic NK cells. Elife 3, e01659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Willinger T, Rongvaux A, Eynon EE, Stevens S, Manz MG, Flavell RA, and Galán JE (2010). A mouse model for the human pathogen Salmonella typhi. Cell Host Microbe 8, 369–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriani A, Iannitto ML, Ricci B, Fionda C, Malgarini G, Morrone S, Peruzzi G, Ricciardi MR, Petrucci MT, Cippitelli M, et al. (2014). Reactive oxygen species- and DNA damage response-dependent NK cell activating ligand upregulation occurs at transcriptional levels and requires the transcriptional factor E2F1. J Immunol 193, 950–960. [DOI] [PubMed] [Google Scholar]
- Spits H, and Di Santo JP (2011). The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nat Immunol 12, 21–27. [DOI] [PubMed] [Google Scholar]
- Spits H, Bernink JH, and Lanier L (2016). NK cells and type 1 innate lymphoid cells: partners in host defense. Nat Immunol 17, 758–764. [DOI] [PubMed] [Google Scholar]
- Sun JC, Beilke JN, and Lanier LL (2009). Adaptive immune features of natural killer cells. Nature 457, 557–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Hayakawa Y, Smyth MJ, Kayagaki N, Yamaguchi N, Kakuta S, Iwakura Y, Yagita H, and Okumura K (2001). Involvement of tumor necrosis factor-related apoptosis-inducing ligand in surveillance of tumor metastasis by liver natural killer cells. Nat Med 7, 94–100. [DOI] [PubMed] [Google Scholar]
- Victorino F, Sojka DK, Brodsky KS, McNamee EN, Masterson JC, Homann D, Yokoyama WM, Eltzschig HK, and Clambey ET (2015). Tissue-Resident NK Cells Mediate Ischemic Kidney Injury and Are Not Depleted by Anti-Asialo-GM1 Antibody. J Immunol 195, 4973–4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Virgilio F, Dal Ben D, Sarti AC, Giuliani AL, and Falzoni S (2017). The P2X7 Receptor in Infection and Inflammation. Immunity 47, 15–31. [DOI] [PubMed] [Google Scholar]
- Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie ANJ, Mebius RE, et al. (2018). Innate Lymphoid Cells: 10 Years On. Cell 174, 1054–1066. [DOI] [PubMed] [Google Scholar]
- Weizman OE, Adams NM, Schuster IS, Krishna C, Pritykin Y, Lau C, Degli-Esposti MA, Leslie CS, Sun JC, and O’Sullivan TE (2017). ILC1 Confer Early Host Protection at Initial Sites of Viral Infection. Cell 171, 795–808.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Wu N, Lu Y, Davidson D, Colonna M, and Veillette A (2015). DNAM-1 controls NK cell activation via an ITT-like motif. J Exp Med 212, 2165–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.