

INTRODUCTION
Bladder cancer is a major cause of cancer-related morbidity and mortality and is characterized by significant molecular and histologic heterogeneity.1 Despite exhibiting a high rate of potentially actionable genomic alterations1 (Figure 1), outside of FGFR3 inhibition, efforts to tailor therapy based on such biomarkers have had limited success. The failure of many initially promising biomarkers to impact the clinical care of bladder cancer patients thus far highlights the importance of thorough biomarker validation prior to implementation in clinical practice. In this review, we will summarize precision medicine efforts to date for patients with bladder cancer and review ongoing work in this area (Table 1). We will also review obstacles to the advancement of precision medicine and potential solutions.
Figure 1.
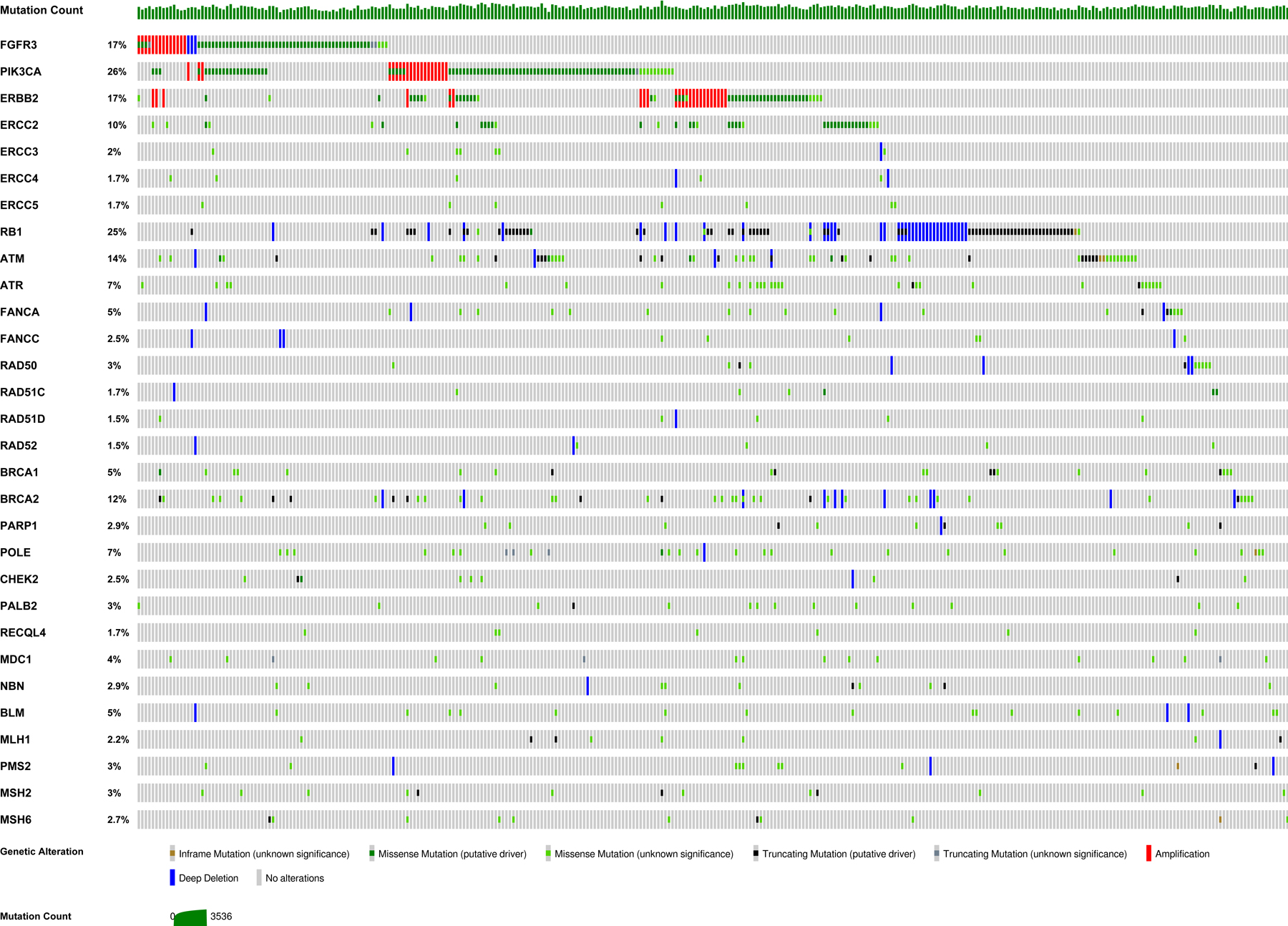
Oncoprint from The Cancer Genome Atlas (TCGA) depicting the distribution of potentially predictive genomic biomarkers in muscle-invasive bladder cancer.1,106,107 Each column depicts the genomic profile of an individual patient. Y-axis labels include the percentage of muscle-invasive bladder cancers in the TCGA with alterations in each gene. TCGA patients without alterations in one or more of the selected genes are not shown.
Table 1.
Summary of notable biomarkers investigated in bladder cancer and associated study findings.
| Biomarker | Studies | Key findings |
|---|---|---|
| p53 | Stadler et al.5 |
|
| FGFR3 alterations | Loriot et al.14 Milowsky et al.10 Pal et al.11 |
|
| FGFR3 mRNA overexpression | Schuler et al.16 Rosenberg et al.17 |
|
| HER2 | Hussain et al.26 Powles et al.29 Wulfing et al.105 Choudhury et al.30 |
|
| EGFR | Petrylak et al.23 Philips et al.24 Pruthi et al.25 |
|
| PIK3CA | Munster et al.39 Seront et al.40 McPherson et al.41 |
|
| Molecular subtypes / gene expression profiling | Choi et al.42 McConkey et al.51 Seiler et al.43 Rosenberg et al.52 Sharma et al.53 Kim et al.54 Flaig et al.59 |
|
| DDR gene alterations | Van Allen et al.65 Plimack et al.69 Miron et al.71 Teo et al.70 Iyer et al.72 Teo et al.75 |
|
| PD-L1 | Powles et al.80 Galsky et al.81 |
|
| Tumor mutational burden | Balar et al.55 Galsky et al.83 |
|
| Microsatellite instability | Iyer et al.87 |
|
Abbreviations: DDR = DNA damage response and repair; EGFR = epidermal growth factor receptor; FGFR = fibroblast growth factor receptor; MSI = microsatellite instability; mUC = metastatic or advanced urothelial carcinoma; adjuvant MVAC = methotrexate, vinblastine, doxorubicin, and cisplatin; PD-L1 = programmed cell death ligand 1; PI3K = phosphoinositide 3-kinase; TMB = tumor mutational burden; UC = urothelial carcinoma.
DISCUSSION
Early efforts: Personalizing treatment based on p53 status
Prior studies showed dysregulation of the tumor suppressor p53 to be a marker of poor prognosis in bladder cancer2 and that p53 inactivation may predict benefit from DNA-damaging chemotherapy.3,4 In the first trial in bladder cancer to assign treatment based on molecular alterations,5 patients with p53 mutant tumors were randomized to adjuvant MVAC (methotrexate, vinblastine, doxorubicin, and cisplatin) versus observation, while p53 wild-type patients were observed. Accrual was stopped after a futility analysis demonstrated no difference in recurrence by p53 status. However, the study suffered from notable limitations, including a low event rate, frequent patient refusal to receive adjuvant chemotherapy, and a high number of patients who did not receive their assigned therapy. The trial also relied on defining p53 expression by immunohistochemistry (IHC). While wildtype p53 is not detectable by IHC due to rapid degradation, mutant p53 is stabilized and detectable in the nucleus.6 However, this method fails to detect p53 mutations that prevent expression of p53 and cannot differentiate between tumors with heterozygous versus homozygous loss of p53 function. These limitations can now be circumvented with genetic sequencing of TP53.5
Personalizing therapy based on FGFR alterations and expression
Deregulated fibroblast growth factor (FGF) signaling, often through gain-of-function alterations of FGF receptor (FGFR) 3, contributes to tumorigenesis in multiple cancers, including 32% of urothelial carcinomas (UC).7 FGFR alterations occur in up to 37% of patients with upper tract UC8 and are especially common in UC of the luminal I subtype.9 FGFR signaling is crucial to tissue homeostasis and angiogenesis.9 Upon binding of extracellular FGF ligands, the FGFR family of transmembrane receptor tyrosine kinases (RTKs), consisting of FGFR1–4, trigger intracellular signaling via multiple pathways including RAS/MAPK/ERK, PI3K/AKT, PLCγ and STAT.9
Early efforts to target FGFR3 using small molecule inhibitors included a phase II trial of dovitinib, a multi-targeted tyrosine kinase inhibitor (TKI), in advanced/metastatic UC (mUC) patients with and without FGFR3 mutations detected using a mass spectrometry assay.10 Dovitinib failed to show significant activity regardless of mutation status.10 Moreover, only 12 FGFR3 mutant patients were enrolled, due in part to some false positive results requiring reclassification as wild-type and because the rate of FGFR3 mutations is 15–20% in invasive disease.10 This study highlights the crucial importance of a reliable assay to detect the biomarker of relevance as well as the need for drugs that effectively inhibit the target of interest.
Other FGFR inhibitors are also being investigated in the context of FGFR alterations. In a population of 67 patients with mUC and FGFR3 alterations who were platinum-refractory, -intolerant, or -ineligible, the FGFR1–4 inhibitor infigratinib demonstrated modest activity, with a response rate of 25.4%, median progression-free survival of 3.75 months, and overall survival of 7.75 months.11 A randomized trial of adjuvant infigratinib versus placebo for invasive UC with specific FGFR3 alterations is now ongoing (PROOF 302; NCT04197986). The FGFR1–3 inhibitor, pemigatinib, has been approved in cholangiocarcinoma with FGFR2 fusions12 and has shown clinical activity in UC with an ORR of 25%.13 Ongoing or planned studies of pemigatinib for FGFR-altered UC include FIGHT-205, a trial of pemigatinib with or without pembrolizumab for mUC (NCT04003610), and PEGASUS, a study of adjuvant pemigatinib for high-risk UC (NCT04294277).
The phase 2 BLC2001 trial tested the pan-FGFR TKI erdafitinib in metastatic FGFR2/3 altered UC and demonstrated a confirmed objective response rate (ORR) of 40%.14 Median progression-free and overall survival were 5.5 and 13.8 months, respectively.14 These results led to FDA-approval of erdafitinib as the first targeted therapy for mUC with a specific genetic alteration.15 A confirmatory phase III study (THOR) is currently open and randomizes patients with mUC and pre-specified FGFR alterations to erdafitinib versus vinflunine, docetaxel or pembrolizumab following receipt of one to two prior lines of therapy (NCT03390504).
FGFR inhibitors are also being explored in patients with FGFR mRNA over-expression. A phase I dose-escalation and dose-expansion study explored the pan-FGFR inhibitor rogaratinib for patients with advanced cancers, including 52 with UC, characterized by FGFR1–3 mRNA overexpression and activating mutations, and demonstrated that rogaratinib had a favorable safety profile and promising anti-tumor activity.16 In the cohort of patients with UC, 24% (n = 11) had an objective response, including one complete response; 49% had stable disease and 14% had progressive disease. All but one of the patients with UC who experienced an objective response were positive for FGFR3 mRNA expression. Of the 11 responders, six had no alterations of the FGFR gene. In the FORT-2 trial of first-line rogaratinib plus atezolizumab for cisplatin-ineligible patients with mUC and FGFR1/3 mRNA overexpression, the ORR in 23 evaluable patients was 39%, with a complete response rate of 13%.17 Notably, most patients in FORT-2 were FGFR wildtype, and 96% had little to no expression of programmed cell death ligand 1 (PD-L1).
Mechanisms of FGFR inhibitor resistance remain incompletely understood. Preclinical studies have suggested the existence of both on-target mechanisms, such as mutations of the tyrosine kinase domain and the ATP binding cleft, and off-target mechanisms, including upregulation of parallel signaling pathways (RAS/PI3K).18 Cell-free DNA (cfDNA) sequencing in patients treated with infigratinib identified ATP binding cleft mutations, or so-called gatekeeper mutations, in four patients who ultimately progressed.11 Further characterization of resistance mechanisms will be necessary to optimize patient selection for FGFR-targeted therapies and to design means of overcoming FGFR inhibitor resistance.
A primary limitation of current FGFR TKIs is the spectrum of toxicity produced by simultaneous inhibition of multiple FGFRs, resulting in hand-foot syndrome, nail toxicities, hyperphosphatemia, gastrointestinal side effects, and central serous retinopathy.12,14,16,19 FGFR isoform-specific inhibitors, e.g. inhibitors specific to FGFR3, may substantially reduce toxicity burden, allowing for increased dosing levels to enhance target inhibition. Also, anti-FGFR3 antibody therapy may represent a less toxic means of signaling inhibition. Vofatamab is a fully human monoclonal antibody to FGFR3 which blocks both wild-type and mutant receptors.20 Preliminary results from the FIERCE-21 study indicated that vofatamab with or without docetaxel in mUC with FGFR3 mutations/fusions was well-tolerated, with only 1 patient discontinuing treatment due to an adverse event.20 However, only 7 of 55 patients had an objective response, suggesting insufficient efficacy.20
HER2-Targeted Therapy in UC
The ErbB family of RTKs consists of EGFR, HER2, ErbB3, and ErbB4.21 Amplifications and somatic alterations of the ErbB family occur in a significant percentage of UC, including EGFR amplification (11%), ERBB2 amplification (7%), ERBB2 mutations (12%), and ERBB3 mutations (10%).1,22 Trials targeting the ErbB family in UC have produced mixed results, including two negative trials for gefitinib in chemotherapy-resistant UC23,24 and one trial of neoadjuvant erlotinib in muscle-invasive bladder cancer (MIBC) that showed pathologic downstaging in 12 of 20 patients.25 A single arm phase II trial of trastuzumab plus chemotherapy for patients with mUC and HER2 positivity—defined as HER2 overexpression by IHC, ERBB2 amplification, or elevated serum HER2—resulted in a response rate of 70% but also grade 1 to 3 cardiotoxicity in 22.7% of patients (7% grade 3 toxicity).26 Notably, the trial’s primary endpoint was the rate of cardiotoxicity, and the trial was not powered to detect differences in ORR or survival by HER2 expression or amplification subgroups. In the phase IIa basket study MyPathway, nine patients with advanced refractory bladder cancer with ERBB2 amplification/overexpression were treated with trastuzumab plus pertuzumab, resulting in one CR ongoing at 15 months, 2 PRs lasting 1 and 6 months, and two patients with stable disease lasting more than 4 months.27 A separate multicenter, phase II trial randomized 61 patients with advanced/metastatic urothelial cancer overexpressing HER2 to gemcitabine plus platinum with trastuzumab versus without trastuzumab and found that the addition of trastuzumab was well-tolerated.28 However, there was no statistically significant difference in progression-free or overall survival.28 A phase III, double-blind, randomized trial of maintenance lapatinib versus placebo after first-line chemotherapy in patients with EGFR or HER2 positive UC failed to show a statistically significant difference in clinical outcomes, including in the subgroup of patients with 3+ expression by IHC.29
Promising findings were reported by a phase II trial of afatinib for platinum-refractory UC.30 The median progression-free survival of patients with ERBB2 and/or ERBB3 alterations (n = 6) treated with afatinib was 6.6 months, a three-fold improvement over historical controls.30,31 One patient with both ERBB3 mutation and ERBB2 amplification never progressed but did discontinue therapy at 10.3 months due to cardiac toxicity.30 Notably, all patients without alterations of either ERBB2 or ERBB3 (n = 15) progressed or died within 3 months, including patients with EGFR amplification and EGFR protein overexpression, perhaps because the EGFR exon 19 and 21 alterations for which aftanitib is approved in non-small cell lung cancer were not found in these patients.32
Notably, a consensus on the optimal method of measuring HER2 positivity in UC is lacking due to the poor correlation between ERBB2 gene amplification and HER2 overexpression in this disease.33 Moreover, studies regarding which method is of greater prognostic relevance have yielded conflicting results.34–36 Though earlier trials of HER-targeted therapy primarily used IHC for patient selection,23,26 the results of the aforementioned afatinib trial suggested that ERBB2 amplification as measured by qPCR or FISH may be superior in UC.30 While 75% of patients in the afatinib study with ERBB2 amplification reached a progression-free survival of at least 3 months, this only occurred in 25% of patients with 2+ or 3+ HER2 overexpression by IHC.30
Promising trials of HER2-directed therapy for mUC continue, including a phase 1b study (NCT03523572) of nivolumab plus the highly active antibody-drug conjugate trastuzumab deruxtecan, which was recently FDA-approved for the treatment of metastatic HER2-positive breast cancer.37,38 Trastuzumab deruxtecan has shown efficacy in HER2-positive breast cancer even after treatment with the earlier HER2-directed antibody-drug conjugate, ado-trastuzumab emtansine, likely due to trastuzumab deruxtecan’s higher cytotoxic payload and its efficacy at lower levels of HER2 expression.37
Personalizing therapy based on alterations in PIK3CA
PIK3CA is altered in approximately 20% of MIBC.1 Early stage trials of PI3K inhibitors in bladder cancer have shown significant treatment-related toxicity but also occasional treatment responses.39–41 Some studies suggest that responses to PI3K inhibition in bladder cancer do not always occur in the context of PI3K pathway alterations.39,40 Recently, a phase II trial testing the pan-isoform class I PI3K inhibitor buparlisib for mUC with PI3K pathway alterations reported modest activity in patients with somatic loss of function of TSC1, achieving a partial response in one such patient and stable disease in three.41 However, trial accrual was halted early after no evaluable patients achieved disease control at 8 weeks. Treatment efficacy was likely impaired by dose-reductions for toxicity in 40% of trial participants. The authors concluded that future trials should employ isoform-selective PI3K inhibitors in genomically selected patients to increase on-target efficacy and minimize off-target toxicity.41
Molecular subtypes based on gene expression profiling
The application of gene expression profiling to UC tumors defined discrete molecular subtypes, broadly categorized as luminal and basal similar to breast cancer subtypes, which are being intensely explored for correlation with response to various therapies.1,22,42–45 While basal tumors display aggressive behavior and expression patterns similar to less-differentiated stem-like or mesenchymal cells, luminal tumors express FOXA1 and GATA3 in similar fashion to luminal-differentiated breast cancer cells and often display superficial papillary growth patterns.42,46–49 Additional neuronal or neuroendocrine-like subtypes of UC are characterized by a gene expression profile consistent with neuronal and neuroendocrine differentiation, often in the absence of neuroendocrine histopathologic features.1,50
Multiple studies have reported that basal subtype tumors derive the greatest benefit from neoadjuvant cisplatin-based chemotherapy, while tumors with a p53-like phenotype, characterized by an activated wild-type p53 gene expression signature, are resistant to neoadjuvant chemotherapy.42,43,51 The sensitivity of basal subtype UC to cisplatin-based chemotherapy may be attributable to an inherently higher proliferation rate given its aggressive natural history.51
Studies investigating the capacity of molecular subtyping to predict response to immunotherapy have generated conflicting results. In the phase II trial IMvigor 210, ORR to the anti-PD-L1 inhibitor atezolizumab was highest in patients with the TCGA luminal cluster II subtype at 34%.52 However, results from CheckMate 275 suggested that patients with basal I subtype benefited most often from the anti-PD-1 inhibitor nivolumab with an ORR of 30%.53 Both trials showed responses to anti-PD-1/PD-L1 therapy across all subtypes and suggested that, compared to the luminal II subtype, tumors of the luminal I subtype respond less often to immunotherapy.52,53 Notably, luminal I tumors are characterized by absence of tumor-infiltrating immune cells, low expression of PD-L1, and enrichment for FGFR3 mutations.52,53
In IMvigor 210, patients with tumors of the neuronal molecular subtype experienced impressive benefit from atezolizumab, with a partial response rate of 75% and complete response rate of 25%, compared to 14% and 9% in the overall trial population, respectively,54,55 even though these tumors lacked features otherwise associated with immunotherapy response, such as high mutational burden and immune-inflammation.54 The response to immunotherapy was especially noteworthy given the poor prognosis associated with the neuroendocrine-like subtype in other contexts.56
A recent publication described a consensus classification consisting of six molecular subtypes of MIBC that will hopefully serve as a basis for prospective validation in future clinical trials.57
The predictive capacity of a different expression-based signature was tested in SWOG S1314 (NCT02177695), a phase II trial in which 237 patients with MIBC were randomized to neoadjuvant dose dense MVAC versus gemcitabine and cisplatin. The investigators tested for associations between pathologic response and the COXEN score, a dichotomized gene expression model which accurately predicted sensitivity to cisplatin-based therapy in a prior cohort of bladder cancer patients.58 Preliminary results from S1314 indicated that the COXEN scores were not significant predictors of response in the individual arms, though the COXEN gemcitabine-cisplatin score did predict for pathologic downstaging in pooled arms.59 No interaction between COXEN score and chemotherapy regimen as a predictor of treatment response was identified.59 Additional analyses focusing on genomic predictors of response are planned from S1314 specimens.
Alterations in DNA Damage Response and Repair (DDR) Genes
While successive randomized trials have demonstrated a survival benefit with neoadjuvant cisplatin-based chemotherapy for MIBC, uptake in community practice has been low given regimen toxicity and inability to predict which patients are most likely to benefit.60–64 Subsequent investigations have attempted to identify biomarkers of chemotherapy response to aid in patient selection. A study of whole-exome sequencing in patients with MIBC treated with neoadjuvant cisplatin-based chemotherapy identified an association between somatic ERCC2 mutations and cisplatin-sensitivity.65 ERCC2, a nucleotide excision repair gene, is mutated in 10–18% of bladder cancers, higher than in any other cancer type.66 ERCC2 helicase domain mutations appear to be especially critical markers of cisplatin sensitivity.67 The association between alteration of ERCC2 and platinum sensitivity was validated in a later study68 and was followed by identification of defects in other DNA repair genes as predictors of response to cisplatin-based therapy.69,70 In a study of patients enrolled in clinical trials of neoadjuvant cisplatin-based chemotherapy, Plimack et al found that alterations in any one of three DNA repair genes—ATM, RB1, and FANCC—predicted pathologic response with 87% sensitivity and 100% specificity, as well as longer overall survival.69,71 In a subsequent analysis of patients with mUC, Teo et al found that the presence of deleterious alterations in various DDR genes was associated with longer progression-free and overall survival on platinum-based therapy (overall survival 23.7 months in DDR mutant patients versus 13 months among DDR wild-type patients).70 Finally, a multicenter phase II trial of neoadjuvant dose-dense gemcitabine plus cisplatin for MIBC found the presence of deleterious DDR gene alterations to have positive predictive value for pathologic downstaging of 89%.72 At a median follow-up of 2 years, no patients with deleterious DDR gene alterations had developed disease recurrence.
Two groups are currently testing a bladder-sparing approach following neoadjuvant cisplatin-based chemotherapy for patients with DDR gene-altered MIBC. In Alliance A031701 (NCT03609216), MIBC patients whose tumors harbor deleterious DDR alterations who experience a clinical complete response or non-invasive residual disease after neoadjuvant chemotherapy are offered bladder sparing surveillance in place of definitive local therapy. The RETAIN trial (NCT02710734) is also investigating bladder preservation for MIBC following neoadjuvant accelerated MVAC in patients with alterations in ATM, RB1, FANCC, or ERCC2 and no clinical evidence of disease following neoadjuvant chemotherapy.73 Other patients in the trial receive bladder-directed therapy in the form of intravesical therapy, chemoradiation, or cystectomy. If proven effective, such biomarker-driven bladder sparing approaches could substantially improve patient quality of life by avoiding the morbidity of cystectomy or chemoradiation.
DDR gene alterations may also predict clinical benefit from immune checkpoint inhibitors.74 In a study of 60 patients with mUC treated with nivolumab or atezolizumab, the presence of deleterious alterations in DDR genes proved superior to mutational load as a predictor of treatment response, overall survival, and progression-free survival.75 Deleterious DDR alterations were associated with a response rate of 80% versus 19% among DDR gene wild-type patients.75 At a median follow-up of 19.6 months, median overall and progression-free survival for patients with deleterious DDR alterations were not reached75 versus 9.3 months and 2.9 months, respectively, in DDR gene wild-type patients.75 The association between DDR alterations and immunotherapy response is thought to be related to an increased frequency of immune-stimulating cancer neoantigens caused by defective DNA damage repair, as supported by an association between DDR alterations and higher mutational load.75 Alterations in DDR genes may also predict immunotherapy response in other cancer types, such as esophagogastric and non-small-cell lung cancer.76,77
Predictors of immune checkpoint response in clinical use: PD-L1, tumor mutational burden (TMB), and microsatellite instability (MSI)
Five anti-PD-1/PD-L1 immune checkpoint inhibitors are now FDA approved for the treatment of mUC in the second-line setting. However, approximately 80% of patients do not respond to these agents, and the ability to predict response is limited.78 As in many solid tumors, the most extensively studied biomarker in this context is expression of PD-L1. While high levels of tumor and immune cell PD-L1 expression are more frequently found in immune checkpoint inhibitor responders, responses have been observed in the absence of PD-L1 expression. Therefore, PD-L1 is not used to guide treatment decisions in the second-line setting.78 A significant barrier to defining the predictive capacity of PD-L1 is the lack of standardization in PD-L1 assessment. This includes variations in PD-L1 assays, thresholds for PDL1 positivity, and inclusion versus exclusion of PD-L1 expression on tumor-infiltrating immune cells.78 For example, while expression of PD-L1 is used to decide between carboplatin-based chemotherapy versus checkpoint blockade with pembrolizumab or atezolizumab in cisplatin-ineligible patients with treatment-naïve mUC, pembrolizumab is only used if the combined positive score is ≥10, integrating PD-L1 expression on both tumor and tumor-infiltrating immune cells as determined by the Dako 22C3 Assay, while atezolizumab requires PD-L1 staining of immune cells covering ≥5% of the tumor area as determined by the Ventana SP142 Assay.79–81
As in other tumor types, high TMB has also been explored extensively as a predictor of immunotherapy response in bladder cancer.78,82 Elevated TMB is thought to correlate with an increased frequency of neo-antigens that may prompt anti-tumor immune recognition and response.78 In IMvigor 210, patients treated with atezolizumab in the highest TMB quartile experienced longer overall survival compared to the rest of the trial cohort.55 Patients treated with nivolumab in Checkmate 275 with tumors in the highest tertile of TMB also experienced a higher ORR of 31.9% compared to patients in the middle tertile (17.4%) and lowest tertile (10.9%).83
Of note, the anti-PD-1 agent pembrolizumab was recently approved by the FDA for all patients with advanced solid tumors with ≥10 mutations per megabase who have progressed on prior treatment and have no satisfactory alternative therapies84 based on results from KEYNOTE-158, which enrolled patients with high microsatellite instability/deficient mismatch repair non-colorectal tumors.84,85 While this approval does not impact the management of mUC, it does apply to patients with advanced bladder cancers with high TMB and non-urothelial histology.
Similar to TMB, MSI or mismatch repair deficiency is also a marker of immunotherapy response.86 The FDA approved pembrolizumab for all solid tumors with MSI that have already progressed on prior therapies without satisfactory alternative treatments options.86 MSI or mismatch repair deficiency occurs in 3–5% of UC, predominantly upper tract UC tumors in patients with Lynch Syndrome or somatic MMR deficiency.75,87,88 A retrospective study identified 13 of 424 UC patients with MSI. Of the 5 who received immune checkpoint blockade for metastatic disease, all achieved near-complete or complete responses, and all were alive at 27 months of follow-up.87
Innovative trial designs and associated limitations
Validation of biomarkers within single cancer types can prove challenging, especially given that such biomarkers are typically found in the minority of tumors, robust preclinical data confirming their biologic relevance and mechanism of action are often limited, and because a standardized method to screen for these biomarkers may not be established. In response to these limitations, innovative trial designs have been developed, including basket trials, umbrella trials, and adaptive platform trials (Figure 2). Collectively, these have been referred to as master protocols.89 Basket trials enroll patients with cancers of various types on the basis of a shared targetable trait—e.g. alteration of a specific gene—to investigate a single targeted therapy. Umbrella trials focus on a single cancer histology or lineage and match patients by theoretically targetable alterations, such as FGFR alteration or HER2 amplification, to one of several rational targeted therapies. Adaptive platform trials feature multiple interventions in a single disease that may enter or exit the platform over time as directed by a decision algorithm (Figure 3). Such adaptive platform trials allow investigators to revise study designs in light of newly generated data in real-time.89 Potential limitations of master protocols include the simultaneous testing of multiple hypotheses leading to reduced statistical power, longer trial timelines that may suffer due to changes in standard of care during the study period, and the increased planning required for trial complexity.89 Examples of master protocols include NCI-MATCH (NCT02465060) and MPACT (NCT01827384), ongoing NCI-funded, histology-agnostic, multicenter basket trials of targeted therapies for patients with advanced cancers.
Figure 2.
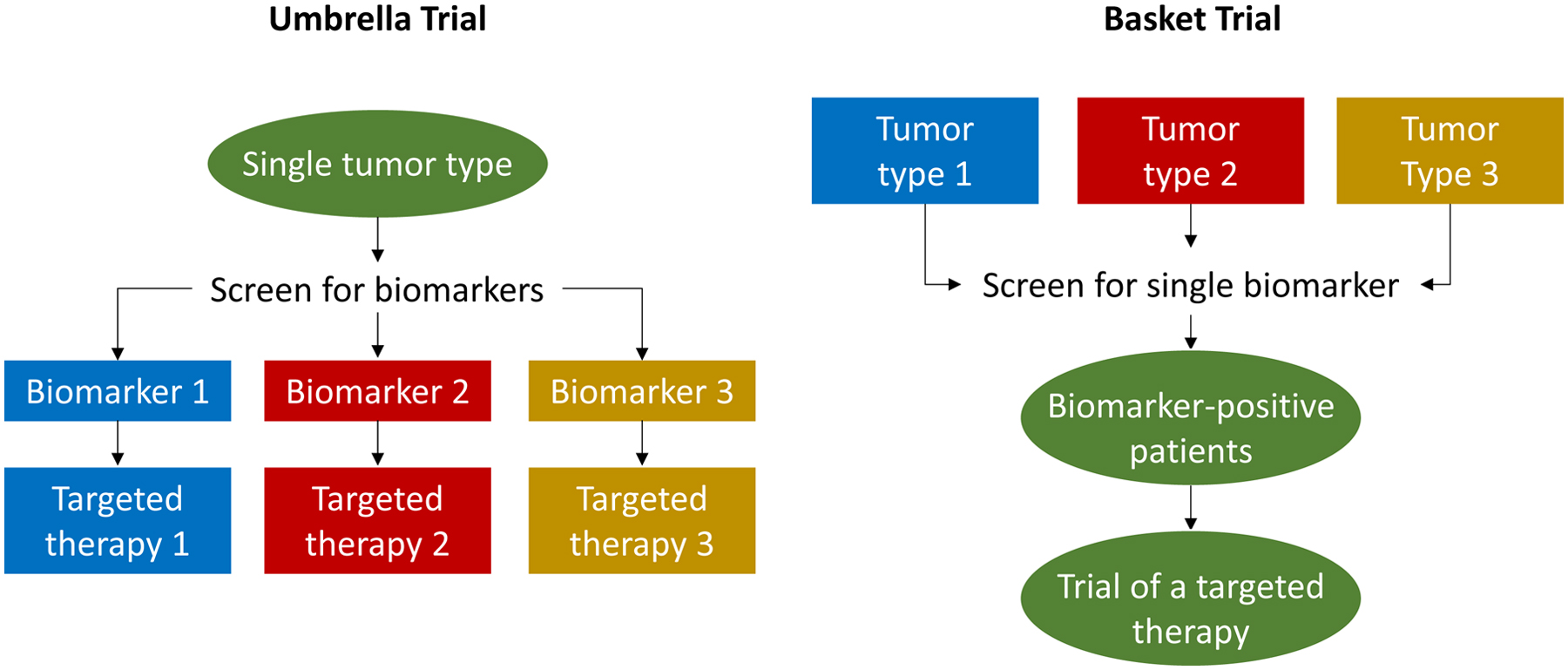
Umbrella trials match patients with a single disease to one of multiple rational targeted therapies based on the presence of informative biomarkers. Basket trials enroll patients with various diseases based on a shared targetable trait to facilitate investigation of a single targeted therapy.
Figure 3.
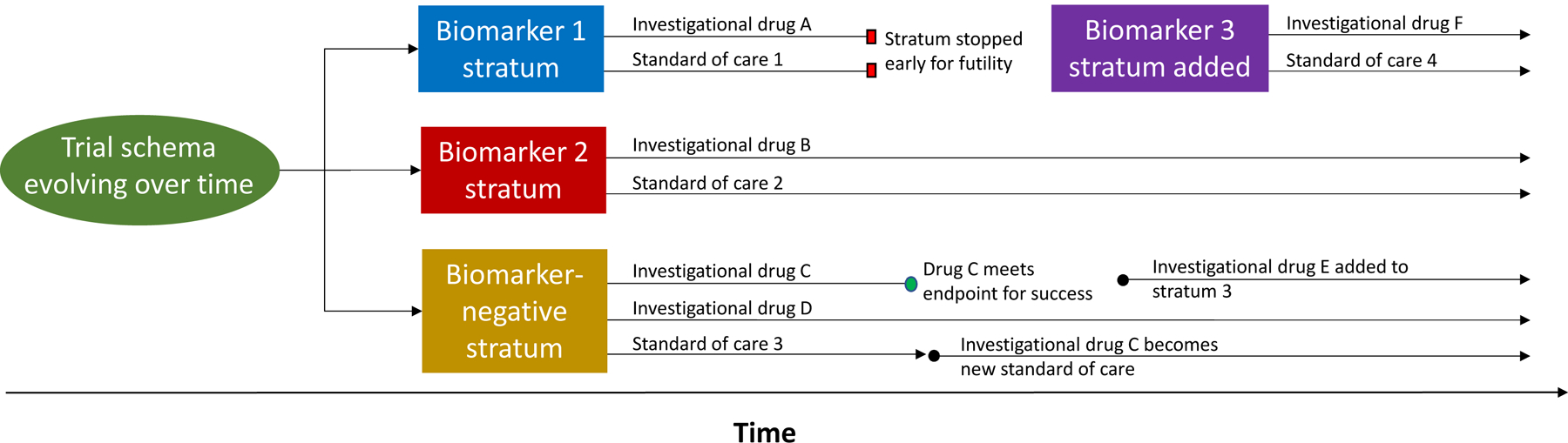
Example schema for an adaptive platform trial. The schema depicts evolution of a platform trial’s design over time. In this example, patients are screened and matched to a trial stratum based on the presence or absence of targetable biomarkers. Each stratum features one or more investigational therapies personalized to patient biomarker status compared to a standard of care. As evidence from the trial accrues, each stratum or arm within a stratum can be individually stopped early for success or futility, while remaining strata and arms may be left open for continued enrollment. New strata (e.g. Biomarker 3 stratum) and treatment arms (e.g. investigational drug E) can be added as the trial proceeds. If a stratum closes early, patients enrolled in that stratum can be enrolled in another (e.g. transition from Biomarker 1 stratum to the Biomarker-negative stratum). The overall trial does not necessarily feature a fixed stop date.
The phase 1b BISCAY study is a multi-drug, biomarker-directed, umbrella trial for patients with muscle-invasive UC that has progressed on prior therapy.90 Treatments investigated in this study include the PD-L1 inhibitor durvalumab alone or in combination with various targeted agents, including the FGFR1–3 inhibitor AZD4547 for patients with FGFR alterations, the PARP inhibitor olaparib for patients with alterations in BRCA1/2, ATM, and homologous recombination repair gene alterations, the mTOR inhibitor vistusertib, the Wee1 inhibitor AZD1775, and the antisense oligonucleotide STAT3 inhibitor AZD9150.90 Patients without targetable biomarkers received durvalumab monotherapy.90 Preliminary results from BISCAY showed that the combinations of durvalumab with targeted therapies were reasonably tolerated and that treatment responses occurred across all study arms, with ORRs ranging from 20% to 35.7%.90 However ORRs in all arms reported to date have failed to meet the prespecified efficacy endpoint of 50%. The trial is ongoing and will have additional arms to report in the future.
The BISCAY trial’s failure to meet its prespecified endpoints despite the matching of multiple targeted therapies to rationally selected biomarkers highlights an important obstacle to precision oncology: differences in drug efficacy are often dependent on tumor lineage. For example, while the PARP inhibitor olaparib is an effective standard agent in the management of prostate and epithelial ovarian cancers with homologous recombination repair deficiency,91,92 the BISCAY trial reported an ORR to olaparib plus durvalumab of 35.7% despite a PD-L1 positivity rate in this cohort of 50%.90 Disappointing findings for PARP inhibition in bladder cancer were also reported in preliminary results of the ATLAS trial, in which rucaparib monotherapy failed to demonstrate significant activity in unselected patients with previously treated mUC.93 Notably, 20.6% of the 66 patients were ultimately determined to have homologous recombination deficiency. While additional trials of PARP inhibition in bladder cancers with somatic DDR alterations are ongoing (NCT03448718, NCT03375307), currently available findings suggest that PARP inhibition may not succeed in bladder cancer even in genomically selected patients.
A possible explanation for the failure of PARP inhibitors and other targeted agents in bladder cancer may lie in the results of a study by Jonsson et al.94 The study characterized BRCA-mediated phenotypes across a variety of cancer lineages and found that BRCA1/2 mutations only conferred sensitivity to PARP inhibition in cancer types with increased heritable risk in BRCA1/2 carriers.94 The authors concluded that BRCA1/2 alterations in non-BRCA associated cancers are often passenger mutations that play little role in tumor pathogenesis, and therefore do not predict response to BRCA-targeted therapies.94 If these findings prove generalizable to biomarkers beyond BRCA1/2, then targeted agents may only succeed in specific tumor types where their respective targets are foundational drivers of oncogenesis. Basket trials are therefore needed to interrogate genomic alterations in the context of multiple tumor lineages simultaneously. For example, while HER2 mutations are prevalent in UC, a basket trial of the HER kinase inhibitor neratinib failed to achieve a partial response in any of 16 patients with ERBB2 mutant UC.95
Overcoming tumor heterogeneity
Inter- and intratumor genomic heterogeneity poses a major barrier to precision medicine in bladder cancer.96 Biopsy specimens from a single tumor site may miss informative alterations present in synchronous metastases or even subclonal alterations within the biopsied tumor.96 Such heterogeneity can lead to proliferation of resistant subclones, limiting the utility of targeted therapies. Application of cfDNA sequencing may complement mutation data extracted from the primary tumor and encompass subclonal alterations missed by single site sampling.97 By sampling tumor DNA from blood and urine, cfDNA sequencing provides a noninvasive means to detect somatic alterations potentially shed by any tumor site in the body.97 Such liquid biopsies also offer a means to non-invasively assess the genomic evolution of tumors over time, which under selective pressures inevitably generates mechanisms of resistance to precision cancer therapies.97 In mUC, cfDNA has already proven useful in studies of FGFR inhibition, wherein plasma cfDNA sequencing detected FGFR3 alterations in 79% of patients with FGFR3 altered tumors.98 Plasma cfDNA from patients on infigratinib also identified the emergence of putative resistance mutations and tracked changes in plasma mutant FGFR3 allele fractions that correlated with changes in tumor volume.11
Multi-omic platforms and computational methods
Efforts to define biomarkers of treatment response in bladder cancer are ongoing. A key example is the multi-omic investigation underway within the completed trial CALGB 90601.99 This phase III trial of gemcitabine and cisplatin plus either bevacizumab or placebo for mUC failed to demonstrate a significant improvement in overall survival with the addition of bevacizumab. However, the large, well-annotated trial cohort of 506 patients provides an opportunity to investigate biomarkers of response to cisplatin based and anti-VEGF therapy. A multi-institutional endeavor is underway to define the predictive capacity of DDR alterations, angiogenesis signatures and expression subtypes, germline single nucleotide polymorphisms, circulating angiokines, and other alterations associated with treatment sensitivity. Ultimately, this collaboration aims to develop composite biomarkers of response for investigation in future studies. While many biomarkers investigated in bladder cancer are enriched in responders, such as DDR alterations in the case of cisplatin-based therapy or PD-L1 in immunotherapy, the positive and negative predictive value of most individual biomarkers is limited. Composite biomarkers may allow prediction of response with precision sufficient for clinical decision making. Historically, many bladder cancer biomarker studies have been limited in size. Use of larger cohorts such as CALGB 90601 are critical to characterize definitive composite biomarkers of response, which may require integration of larger datasets across multi-omic platforms.
Multi-omic analyses are essential to the development of successful precision medicine approaches in bladder cancer, allowing for integration of aberrations at the genetic, trascriptomic, epigenetic, and proteomic levels that may all influence sensitivity to therapy. For example, resistance to the antibody-drug conjugate enfortumab-vedotin may be mediated in some cases by loss of surface expression of nectin-4, enfortumab-vedotin’s cell surface target.100 Therefore, characterization of expression and localization of proteins such as nectin-4 through transcriptomics and proteomics may be necessary to fully understand response and resistance to oncologic therapies. The multi-omic platforms employed by TCGA, PanCancer Atlas, and others offer role models for future endeavors.1,101
Such complex multi-omic analyses will only be feasible with increasingly sophisticated computation methods.102 Machine-learning has already shown promise in precision oncology as an investigatory means of improving central nervous system tumor classification, a historically challenging task fraught with inter-observer variability.103 Such advanced computational methods will allow rapid analysis of vast datasets, as demonstrated by a PanSoftware analysis of 9,423 tumor exomes from the PanCancer Atlas that identified 59 novel likely oncodriver genes.104
SUMMARY
Ongoing efforts to implement precision medicine in bladder cancer have yielded pivotal improvements in the care of patients with mUC through targeting FGFR2/3 alterations with the pan-FGFR inhibitor, erdafitinb. Investigation of PD-L1 expression has also led to improvements in patient selection for first-line immunotherapy for mUC. A variety of other promising biomarkers to further advance precision medicine in bladder cancer remain investigational. Such biomarkers offer the potential for personalization of care in both the neoadjuvant and metastatic settings and include molecular subtypes of UC, DDR gene alterations, and alterations of PIK3CA and genes encoding the ErbB family of RTKs. Barriers to the advancement of precision medicine in bladder cancer remain, but innovative trial designs, cell-free DNA sequencing, multi-omic platforms, and increasingly sophisticated computational methods offer promising solutions for future studies.
CLINICS CARE POINTS.
Alterations of FGFR2/3 are the only genetic feature of metastatic urothelial cancer currently used to select patients for targeted therapy. Erdafitinib is the only targeted therapy specifically approved for FGFR altered urothelial cancer.
Selection of patients with metastatic urothelial carcinoma to receive first-line immunotherapy is informed by expression of PD-L1. Such patients must also be cisplatin-ineligible, but may be carboplatin-eligible if PD-L1 expression is sufficiently high.
Patients with metastatic urothelial carcinoma who are ineligible for any platinum-based chemotherapy may receive immunotherapy in the first-line regardless of PD-L1 expression.
High tumor mutational burden and microsatellite instability may be used to select patients for treatment with immunotherapy in cases of advanced bladder cancer with some non-urothelial histologies in which patients have already progressed on all other satisfactory alternatives. Such cases are rare.
Promising biomarkers may eventually lead to greater personalization of care for patients with bladder cancer, but remain investigational. These include but are not limited to: molecular subtypes of urothelial carcinoma; alterations of DNA damage response and repair genes; FGFR amplifications; alterations/amplifications of genes encoding ErbB receptor tyrosine kinases; and alterations of PIK3CA.
KEY POINTS.
Biomarkers such as alteration of FGFR3 and expression of programmed death ligand 1 have led to advances in precision medicine for bladder cancer
Additional candidate biomarkers to personalize bladder cancer care are under investigation, but significant obstacles must be overcome before they can be implemented in clinical practice
Promising innovations likely to advance precision medicine in bladder cancer include multi-omic approaches, innovative trials designs, cell-free DNA, and machine learning algorithms
SYNOPSIS.
The hallmark of precision medicine involves tailoring treatment to patient and/or tumor-specific biomarkers. Candidate biomarkers in bladder cancer are abundant, but few have been validated in clinical practice. Significant obstacles to precision medicine in bladder cancer include: limited predictive value of candidate biomarkers; lack of standardization in biomarker assessment; heterogeneity in biomarker expression and function; and limited insight into the biologic factors that influence biomarker expression and predictive capacity. This review summarizes key biomarkers explored in bladder cancer and outlines innovative trial designs to approach these obstacles.
DISCLOSURE STATEMENT
All authors receive institutional support from Memorial Sloan Kettering’s NIH/NCI Cancer Center Support Grant P30 CA008748. Dr. Guercio is supported by NIH/NCI award No. T32-CA009207. Drs. Iyer and Rosenberg are supported by MSK’s NCI SPORE in Bladder Cancer (award No. P50 CA221745-01) and a DOD Congressionally Directed Medical Research Program Translational Team Science Award. Dr. Rosenberg is also supported by the NCI Biomarker, Imaging and Quality of Life Studies Funding Program (BIQSFP). Dr. Guercio reports honoraria from Medscape and institutional research funding from Bristol-Myers Squibb, Genentech, Eli Lilly, Pfizer, and Sanofi, outside the submitted work. Dr. Iyer reports grants and personal fees from Mirati Therapeutics, grants from Novartis, grants from DeBioPharm, grants and personal fees from Janssen, outside the submitted work.
Dr. Rosenberg reports honoraria, consulting/advisory role, and expenses from Bristol-Myers Squibb, institutional research funding, honoraria, and consulting/advisory role from AstraZeneca, honoraria from Chugai Pharma, consulting/advisory role for Lilly, consulting/advisory role for Merck, consulting/advisory role and institutional research funding from Astellas, consulting/advisory role, expenses, and institutional research funding from Genentech/Roche, consulting/advisory role for Pfizer, consulting/advisory role and institutional research funding from Seattle Genetics, consulting/advisory role and institutional research funding from Bayer, consulting/advisory role for BioClin Therapeutics, consulting/advisory role and institutional research funding from QED Therapeutics, consulting/advisory role for Pharmacyclics, consulting/advisory role for GlaxoSmithKline, consulting/advisory role for Janssen Oncology, consulting/advisory role for Boehringer Ingelheim, consulting/advisory role for Mirati Therapeutics, outside the submitted work; In addition, Dr. Rosenberg has a patent Predictor of platinum sensitivity owned by Memorial Sloan Kettering Cancer Center.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Robertson AG, Kim J, Al-Ahmadie H, et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell. 2017;171(3):540–556 e525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goebell PJ, Groshen SG, Schmitz-Drager BJ, International Study-Initiative on Bladder C. p53 immunohistochemistry in bladder cancer--a new approach to an old question. Urol Oncol. 2010;28(4):377–388. [DOI] [PubMed] [Google Scholar]
- 3.Ferreira CG, Tolis C, Giaccone G. p53 and chemosensitivity. Ann Oncol. 1999;10(9):1011–1021. [DOI] [PubMed] [Google Scholar]
- 4.Cote RJ, Esrig D, Groshen S, Jones PA, Skinner DG. p53 and treatment of bladder cancer. Nature. 1997;385(6612):123–125. [DOI] [PubMed] [Google Scholar]
- 5.Stadler WM, Lerner SP, Groshen S, et al. Phase III study of molecularly targeted adjuvant therapy in locally advanced urothelial cancer of the bladder based on p53 status. J Clin Oncol. 2011;29(25):3443–3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Esrig D, Spruck CH 3rd, Nichols PW, , et al. p53 nuclear protein accumulation correlates with mutations in the p53 gene, tumor grade, and stage in bladder cancer. Am J Pathol. 1993;143(5):1389–1397. [PMC free article] [PubMed] [Google Scholar]
- 7.Helsten T, Elkin S, Arthur E, Tomson BN, Carter J, Kurzrock R. The FGFR Landscape in Cancer: Analysis of 4,853 Tumors by Next-Generation Sequencing. Clin Cancer Res. 2016;22(1):259–267. [DOI] [PubMed] [Google Scholar]
- 8.Li Q, Bagrodia A, Cha EK, Coleman JA. Prognostic Genetic Signatures in Upper Tract Urothelial Carcinoma. Curr Urol Rep. 2016;17(2):12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haugsten EM, Wiedlocha A, Olsnes S, Wesche J. Roles of fibroblast growth factor receptors in carcinogenesis. Mol Cancer Res. 2010;8(11):1439–1452. [DOI] [PubMed] [Google Scholar]
- 10.Milowsky MI, Dittrich C, Duran I, et al. Phase 2 trial of dovitinib in patients with progressive FGFR3-mutated or FGFR3 wild-type advanced urothelial carcinoma. Eur J Cancer. 2014;50(18):3145–3152. [DOI] [PubMed] [Google Scholar]
- 11.Pal SK, Rosenberg JE, Hoffman-Censits JH, et al. Efficacy of BGJ398, a Fibroblast Growth Factor Receptor 1–3 Inhibitor, in Patients with Previously Treated Advanced Urothelial Carcinoma with FGFR3 Alterations. Cancer Discov. 2018;8(7):812–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abou-Alfa GK, Sahai V, Hollebecque A, et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. Lancet Oncol. 2020;21(5):671–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Necchi A, Pouessel D, Leibowitz-Amit R, et al. Interim results of FIGHT-201, a phase 2, open-label, multicenter study of INCB054828 in patients (pts) with metastatic or surgically unresectable urothelial carcinoma (UC) harboring fibroblast growth factor (FGF)/FGF receptor (FGFR) genetic alterations (GA). Ann Oncol. 2018;29(suppl_8):viii303–viii331. [Google Scholar]
- 14.Loriot Y, Necchi A, Park SH, et al. Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma. N Engl J Med. 2019;381(4):338–348. [DOI] [PubMed] [Google Scholar]
- 15.U.S. Food & Drug Administration. FDA grants accelerated approval to erdafitinib for metastatic urothelial carcinoma. 2019. Available at: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-erdafitinib-metastatic-urothelial-carcinoma:~:text=On%20April%2012%2C%202019%2C%20the,progressed%20during%20or%20following%20platinum%2D. Accessed August 2, 2020.
- 16.Schuler M, Cho BC, Sayehli CM, et al. Rogaratinib in patients with advanced cancers selected by FGFR mRNA expression: a phase 1 dose-escalation and dose-expansion study. Lancet Oncol. 2019;20(10):1454–1466. [DOI] [PubMed] [Google Scholar]
- 17.Rosenberg JE, Gajate P, Morales-Barrera R, et al. Safety and preliminary efficacy of rogaratinib in combination with atezolizumab in a phase Ib/II study (FORT-2) of first-line treatment in cisplatin-ineligible patients (pts) with locally advanced or metastatic urothelial cancer (UC) and FGFR mRNA overexpression. J Clin Oncol. 2020;38(15_suppl):5014–5014. [Google Scholar]
- 18.Babina IS, Turner NC. Advances and challenges in targeting FGFR signalling in cancer. Nat Rev Cancer. 2017;17(5):318–332. [DOI] [PubMed] [Google Scholar]
- 19.Joerger M, Cassier P, Penel N, et al. Rogaratinib treatment of patients with advanced urothelial carcinomas prescreened for tumor FGFR mRNA expression. J Clin Oncol. 2018;36(6_suppl):494–494. [Google Scholar]
- 20.Necchi A, Castellano DE, Mellado B, et al. Fierce-21: Phase II study of vofatmab (B-701), a selective inhibitor of FGFR3, as salvage therapy in metastatic urothelial carcinoma (mUC). J Clin Oncol. 2019;37(7_suppl):409–409. [Google Scholar]
- 21.Mooso BA, Vinall RL, Mudryj M, Yap SA, deVere White RW, Ghosh PM. The role of EGFR family inhibitors in muscle invasive bladder cancer: a review of clinical data and molecular evidence. J Urol. 2015;193(1):19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cancer Genome Atlas Research N. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507(7492):315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petrylak DP, Tangen CM, Van Veldhuizen PJ Jr., et al. Results of the Southwest Oncology Group phase II evaluation (study S0031) of ZD1839 for advanced transitional cell carcinoma of the urothelium. BJU Int. 2010;105(3):317–321. [DOI] [PubMed] [Google Scholar]
- 24.Philips GK, Halabi S, Sanford BL, et al. A phase II trial of cisplatin (C), gemcitabine (G) and gefitinib for advanced urothelial tract carcinoma: results of Cancer and Leukemia Group B (CALGB) 90102. Ann Oncol. 2009;20(6):1074–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pruthi RS, Nielsen M, Heathcote S, et al. A phase II trial of neoadjuvant erlotinib in patients with muscle-invasive bladder cancer undergoing radical cystectomy: clinical and pathological results. BJU Int. 2010;106(3):349–354. [DOI] [PubMed] [Google Scholar]
- 26.Hussain MH, MacVicar GR, Petrylak DP, et al. Trastuzumab, paclitaxel, carboplatin, and gemcitabine in advanced human epidermal growth factor receptor-2/neu-positive urothelial carcinoma: results of a multicenter phase II National Cancer Institute trial. J Clin Oncol. 2007;25(16):2218–2224. [DOI] [PubMed] [Google Scholar]
- 27.Hainsworth JD, Meric-Bernstam F, Swanton C, et al. Targeted Therapy for Advanced Solid Tumors on the Basis of Molecular Profiles: Results From MyPathway, an Open-Label, Phase IIa Multiple Basket Study. J Clin Oncol. 2018;36(6):536–542. [DOI] [PubMed] [Google Scholar]
- 28.Oudard S, Culine S, Vano Y, et al. Multicentre randomised phase II trial of gemcitabine+platinum, with or without trastuzumab, in advanced or metastatic urothelial carcinoma overexpressing Her2. Eur J Cancer. 2015;51(1):45–54. [DOI] [PubMed] [Google Scholar]
- 29.Powles T, Huddart RA, Elliott T, et al. Phase III, Double-Blind, Randomized Trial That Compared Maintenance Lapatinib Versus Placebo After First-Line Chemotherapy in Patients With Human Epidermal Growth Factor Receptor 1/2-Positive Metastatic Bladder Cancer. J Clin Oncol. 2017;35(1):48–55. [DOI] [PubMed] [Google Scholar]
- 30.Choudhury NJ, Campanile A, Antic T, et al. Afatinib Activity in Platinum-Refractory Metastatic Urothelial Carcinoma in Patients With ERBB Alterations. J Clin Oncol. 2016;34(18):2165–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sonpavde G, Pond GR, Fougeray R, et al. Time from prior chemotherapy enhances prognostic risk grouping in the second-line setting of advanced urothelial carcinoma: a retrospective analysis of pooled, prospective phase 2 trials. Eur Urol. 2013;63(4):717–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chaux A, Cohen JS, Schultz L, et al. High epidermal growth factor receptor immunohistochemical expression in urothelial carcinoma of the bladder is not associated with EGFR mutations in exons 19 and 21: a study using formalin-fixed, paraffin-embedded archival tissues. Hum Pathol. 2012;43(10):1590–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kruger S, Weitsch G, Buttner H, et al. Overexpression of c-erbB-2 oncoprotein in muscle-invasive bladder carcinoma: relationship with gene amplification, clinicopathological parameters and prognostic outcome. Int J Oncol. 2002;21(5):981–987. [PubMed] [Google Scholar]
- 34.Fleischmann A, Rotzer D, Seiler R, Studer UE, Thalmann GN. Her2 amplification is significantly more frequent in lymph node metastases from urothelial bladder cancer than in the primary tumours. Eur Urol. 2011;60(2):350–357. [DOI] [PubMed] [Google Scholar]
- 35.Bellmunt J, Werner L, Bamias A, et al. HER2 as a target in invasive urothelial carcinoma. Cancer Med. 2015;4(6):844–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jimenez RE, Hussain M, Bianco FJ Jr., et al. Her-2/neu overexpression in muscle-invasive urothelial carcinoma of the bladder: prognostic significance and comparative analysis in primary and metastatic tumors. Clin Cancer Res. 2001;7(8):2440–2447. [PubMed] [Google Scholar]
- 37.Modi S, Saura C, Yamashita T, et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N Engl J Med. 2020;382(7):610–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.U.S. Food & Drug Administration. FDA approves fam-trastuzumab deruxtecan-nxki for unresectable or metastatic HER2-positive breast cancer. 2019. Available at: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-famtrastuzumab-deruxtecan-nxki-unresectable-or-metastatic-her2-positive-breast-cancer. Accessed August 17, 2020.
- 39.Munster P, van der Noll R, Voest E, et al. PI3K Kinase Inhibitor GSK2126458 (GSK458): Clinical Activity in Select Patient (PT) Populations Defined by Predictive Markers (STUDY P3K112826). Ann Oncol. 2012;23(suppl_9):IX153–IX154. [Google Scholar]
- 40.Seront E, Rottey S, Filleul B, et al. Phase II study of dual phosphoinositol-3-kinase (PI3K) and mammalian target of rapamycin (mTOR) inhibitor BEZ235 in patients with locally advanced or metastatic transitional cell carcinoma. BJU Int. 2016;118(3):408–415. [DOI] [PubMed] [Google Scholar]
- 41.McPherson V, Reardon B, Bhayankara A, et al. A phase 2 trial of buparlisib in patients with platinum-resistant metastatic urothelial carcinoma. Cancer. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Choi W, Porten S, Kim S, et al. Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell. 2014;25(2):152–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Seiler R, Ashab HAD, Erho N, et al. Impact of Molecular Subtypes in Muscle-invasive Bladder Cancer on Predicting Response and Survival after Neoadjuvant Chemotherapy. Eur Urol. 2017;72(4):544–554. [DOI] [PubMed] [Google Scholar]
- 44.Damrauer JS, Hoadley KA, Chism DD, et al. Intrinsic subtypes of high-grade bladder cancer reflect the hallmarks of breast cancer biology. Proc Natl Acad Sci U S A. 2014;111(8):3110–3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sjodahl G, Lauss M, Lovgren K, et al. A molecular taxonomy for urothelial carcinoma. Clin Cancer Res. 2012;18(12):3377–3386. [DOI] [PubMed] [Google Scholar]
- 46.Papafotiou G, Paraskevopoulou V, Vasilaki E, Kanaki Z, Paschalidis N, Klinakis A. KRT14 marks a subpopulation of bladder basal cells with pivotal role in regeneration and tumorigenesis. Nat Commun. 2016;7:11914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dadhania V, Zhang M, Zhang L, et al. Meta-Analysis of the Luminal and Basal Subtypes of Bladder Cancer and the Identification of Signature Immunohistochemical Markers for Clinical Use. EBioMedicine. 2016;12:105–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Volkmer JP, Sahoo D, Chin RK, et al. Three differentiation states risk-stratify bladder cancer into distinct subtypes. Proc Natl Acad Sci U S A. 2012;109(6):2078–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Warrick JI, Walter V, Yamashita H, et al. FOXA1, GATA3 and PPAR Cooperate to Drive Luminal Subtype in Bladder Cancer: A Molecular Analysis of Established Human Cell Lines. Sci Rep. 2016;6:38531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sjodahl G, Eriksson P, Liedberg F, Hoglund M. Molecular classification of urothelial carcinoma: global mRNA classification versus tumour-cell phenotype classification. J Pathol. 2017;242(1):113–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McConkey DJ, Choi W, Shen Y, et al. A Prognostic Gene Expression Signature in the Molecular Classification of Chemotherapy-naive Urothelial Cancer is Predictive of Clinical Outcomes from Neoadjuvant Chemotherapy: A Phase 2 Trial of Dose-dense Methotrexate, Vinblastine, Doxorubicin, and Cisplatin with Bevacizumab in Urothelial Cancer. Eur Urol. 2016;69(5):855–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rosenberg JE, Hoffman-Censits J, Powles T, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. 2016;387(10031):1909–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sharma P, Retz M, Siefker-Radtke A, et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): a multicentre, single-arm, phase 2 trial. Lancet Oncol. 2017;18(3):312–322. [DOI] [PubMed] [Google Scholar]
- 54.Kim J, Kwiatkowski D, McConkey DJ, et al. The Cancer Genome Atlas Expression Subtypes Stratify Response to Checkpoint Inhibition in Advanced Urothelial Cancer and Identify a Subset of Patients with High Survival Probability. Eur Urol. 2019;75(6):961–964. [DOI] [PubMed] [Google Scholar]
- 55.Balar AV, Galsky MD, Rosenberg JE, et al. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: a single-arm, multicentre, phase 2 trial. Lancet. 2017;389(10064):67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Batista da Costa J, Gibb EA, Bivalacqua TJ, et al. Molecular Characterization of Neuroendocrine-like Bladder Cancer. Clin Cancer Res. 2019;25(13):3908–3920. [DOI] [PubMed] [Google Scholar]
- 57.Kamoun A, de Reynies A, Allory Y, et al. A Consensus Molecular Classification of Muscle-invasive Bladder Cancer. Eur Urol. 2020;77(4):420–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kothari S, Gustafson D, Killian K, et al. COXEN prediction of antineoplastic drug sensitivity in bladder cancer patients. J Clin Oncol. 2016;34(2_suppl):365–365. [Google Scholar]
- 59.Flaig TW, Tangen CM, Daneshmand S, et al. SWOG S1314: A randomized phase II study of co-expression extrapolation (COXEN) with neoadjuvant chemotherapy for localized, muscle-invasive bladder cancer. J Clin Oncol. 2019;37(15_suppl):4506–4506. [Google Scholar]
- 60.Sternberg CN, Skoneczna I, Kerst JM, et al. Immediate versus deferred chemotherapy after radical cystectomy in patients with pT3-pT4 or N+ M0 urothelial carcinoma of the bladder (EORTC 30994): an intergroup, open-label, randomised phase 3 trial. Lancet Oncol. 2015;16(1):76–86. [DOI] [PubMed] [Google Scholar]
- 61.Advanced Bladder Cancer Meta-analysis C. Neoadjuvant chemotherapy in invasive bladder cancer: a systematic review and meta-analysis. Lancet. 2003;361(9373):1927–1934. [DOI] [PubMed] [Google Scholar]
- 62.Winquist E, Kirchner TS, Segal R, Chin J, Lukka H, Genitourinary Cancer Disease Site Group CCOPiE-bCPGI. Neoadjuvant chemotherapy for transitional cell carcinoma of the bladder: a systematic review and meta-analysis. J Urol. 2004;171(2 Pt 1):561–569. [DOI] [PubMed] [Google Scholar]
- 63.Advanced Bladder Cancer Meta-analysis C. Neoadjuvant chemotherapy in invasive bladder cancer: update of a systematic review and meta-analysis of individual patient data advanced bladder cancer (ABC) meta-analysis collaboration. Eur Urol. 2005;48(2):202–205; discussion 205–206. [DOI] [PubMed] [Google Scholar]
- 64.Raj GV, Karavadia S, Schlomer B, et al. Contemporary use of perioperative cisplatin-based chemotherapy in patients with muscle-invasive bladder cancer. Cancer. 2011;117(2):276–282. [DOI] [PubMed] [Google Scholar]
- 65.Van Allen EM, Mouw KW, Kim P, et al. Somatic ERCC2 mutations correlate with cisplatin sensitivity in muscle-invasive urothelial carcinoma. Cancer Discov. 2014;4(10):1140–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Abbosh PH, Plimack ER. Molecular and Clinical Insights into the Role and Significance of Mutated DNA Repair Genes in Bladder Cancer. Bladder Cancer. 2018;4(1):9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li Q, Damish AW, Frazier Z, et al. ERCC2 Helicase Domain Mutations Confer Nucleotide Excision Repair Deficiency and Drive Cisplatin Sensitivity in Muscle-Invasive Bladder Cancer. Clin Cancer Res. 2019;25(3):977–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu D, Plimack ER, Hoffman-Censits J, et al. Clinical Validation of Chemotherapy Response Biomarker ERCC2 in Muscle-Invasive Urothelial Bladder Carcinoma. JAMA Oncol. 2016;2(8):1094–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Plimack ER, Dunbrack RL, Brennan TA, et al. Defects in DNA Repair Genes Predict Response to Neoadjuvant Cisplatin-based Chemotherapy in Muscle-invasive Bladder Cancer. Eur Urol. 2015;68(6):959–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Teo MY, Bambury RM, Zabor EC, et al. DNA Damage Response and Repair Gene Alterations Are Associated with Improved Survival in Patients with Platinum-Treated Advanced Urothelial Carcinoma. Clin Cancer Res. 2017;23(14):3610–3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Miron B, Hoffman-Censits JH, Anari F, et al. Defects in DNA Repair Genes Confer Improved Long-term Survival after Cisplatin-based Neoadjuvant Chemotherapy for Muscle-invasive Bladder Cancer. Eur Urol Oncol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Iyer G, Balar AV, Milowsky MI, et al. Multicenter Prospective Phase II Trial of Neoadjuvant Dose-Dense Gemcitabine Plus Cisplatin in Patients With Muscle-Invasive Bladder Cancer. J Clin Oncol. 2018;36(19):1949–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Geynisman DM, Abbosh P, Zibelman MR, et al. A phase II trial of risk-adapted treatment for muscle invasive bladder cancer after neoadjuvant accelerated MVAC. J Clin Oncol. 2018;36(6_suppl):TPS537–TPS537. [Google Scholar]
- 74.Mouw KW, Goldberg MS, Konstantinopoulos PA, D’Andrea AD. DNA Damage and Repair Biomarkers of Immunotherapy Response. Cancer Discov. 2017;7(7):675–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Teo MY, Seier K, Ostrovnaya I, et al. Alterations in DNA Damage Response and Repair Genes as Potential Marker of Clinical Benefit From PD-1/PD-L1 Blockade in Advanced Urothelial Cancers. J Clin Oncol. 2018;36(17):1685–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hsiehchen D, Hsieh A, Samstein RM, et al. DNA Repair Gene Mutations as Predictors of Immune Checkpoint Inhibitor Response beyond Tumor Mutation Burden. Cell Rep Med. 2020;1(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang J, Shih DJH, Lin SY. Role of DNA repair defects in predicting immunotherapy response. Biomark Res. 2020;8:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhu J, Armstrong AJ, Friedlander TW, et al. Biomarkers of immunotherapy in urothelial and renal cell carcinoma: PD-L1, tumor mutational burden, and beyond. J Immunother Cancer. 2018;6(1):4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.U.S. Food and Drug Administration. FDA alerts health care professionals and oncology clinical investigators about an efficacy issue identified in clinical trials for some patients taking Keytruda (pembrolizumab) or Tecentriq (atezolizumab) as monotherapy to treat urothelial cancer with low expression of PD-L1. 2018. Available at: https://www.fda.gov/drugs/drug-safety-and-availability/fda-alerts-health-care-professionals-and-oncology-clinical-investigators-about-efficacy-issue, January 18, 2020.
- 80.Powles T, Loriot Y, Gschwend JE, et al. KEYNOTE-361: phase 3 trial of pembrolizumab ± chemotherapy versus chemotherapy alone in advanced urothelial cancer. European Urology, supplements. 2018;17(2):e1147–e1148. [Google Scholar]
- 81.Galsky MD, Arija JAA, Bamias A, et al. Atezolizumab with or without chemotherapy in metastatic urothelial cancer (IMvigor130): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet. 2020;395(10236):1547–1557. [DOI] [PubMed] [Google Scholar]
- 82.Samstein RM, Lee CH, Shoushtari AN, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. 2019;51(2):202–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Galsky MD, Saci A, Szabo PM, et al. Impact of tumor mutation burden on nivolumab efficacy in second-line urothelial carcinoma patients: Exploratory analysis of the phase II checkmate 275 study. Ann Oncol. 2017;28(suppl_5):296–297. [Google Scholar]
- 84.U.S. Food and Drug Administration. FDA approves pembrolizumab for adults and children with TMB-H solid tumors. Available at: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-pembrolizumab-adults-and-children-tmb-h-solid-tumors. Accessed July 24, 2020.
- 85.Marabelle A, Fakih MG, Lopez J, et al. Association of tumor mutational burden with outcomes in patients with select advanced solid tumors treated with pembrolizumab in KEYNOTE-158. Ann Oncol. 2019;30(suppl_5):v475–v532. [Google Scholar]
- 86.U.S. Food and Drug Administration. FDA approves first cancer treatment for any solid tumor with a specific genetic feature. 2017. Available at: https://www.fda.gov/news-events/press-announcements/fda-approves-first-cancer-treatment-any-solid-tumor-specific-genetic-feature. Accessed August 1, 2020.
- 87.Iyer G, Audenet F, Middha S, et al. Mismatch repair (MMR) detection in urothelial carcinoma (UC) and correlation with immune checkpoint blockade (ICB) response. J Clin Oncol. 2017;35(15_suppl):4511–4511. [Google Scholar]
- 88.Pradere B, Lotan Y, Roupret M. Lynch syndrome in upper tract urothelial carcinoma: significance, screening, and surveillance. Curr Opin Urol. 2017;27(1):48–55. [DOI] [PubMed] [Google Scholar]
- 89.Woodcock J, LaVange LM. Master Protocols to Study Multiple Therapies, Multiple Diseases, or Both. N Engl J Med. 2017;377(1):62–70. [DOI] [PubMed] [Google Scholar]
- 90.Powles TB, Balar A, Gravis G, et al. An adaptive, biomarker directed platform study in metastatic urothelial cancer (BISCAY) with durvalumab in combination with targeted therapies. Ann Oncol. 2019;30(suppl_5):v356–v402. [Google Scholar]
- 91.Tew WP, Lacchetti C, Ellis A, et al. PARP Inhibitors in the Management of Ovarian Cancer: ASCO Guideline. J Clin Oncol. 2020:JCO2001924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.U.S. Food and Drug Administration. FDA approves olaparib for HRR gene-mutated metastatic castration-resistant prostate cancer. 2020. Available at: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-olaparib-hrr-gene-mutated-metastatic-castration-resistant-prostate-cancer. Accessed August 16, 2020.
- 93.Grivas P, Loriot Y, Feyerabend S, et al. Rucaparib for recurrent, locally advanced, or metastatic urothelial carcinoma (mUC): Results from ATLAS, a phase II open-label trial. J Clin Oncol. 2020;38(6_suppl):440–440. [Google Scholar]
- 94.Jonsson P, Bandlamudi C, Cheng ML, et al. Tumour lineage shapes BRCA-mediated phenotypes. Nature. 2019;571(7766):576–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hyman DM, Piha-Paul SA, Won H, et al. HER kinase inhibition in patients with HER2- and HER3-mutant cancers. Nature. 2018;554(7691):189–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Meeks JJ, Al-Ahmadie H, Faltas BM, et al. Genomic heterogeneity in bladder cancer: challenges and possible solutions to improve outcomes. Nat Rev Urol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Corcoran RB, Chabner BA. Application of Cell-free DNA Analysis to Cancer Treatment. N Engl J Med. 2018;379(18):1754–1765. [DOI] [PubMed] [Google Scholar]
- 98.Pal SK, Bajorin D, Dizman N, et al. Infigratinib in upper tract urothelial carcinoma versus urothelial carcinoma of the bladder and its association with comprehensive genomic profiling and/or cell-free DNA results. Cancer. 2020;126(11):2597–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rosenberg JE, Ballman KV, Halabi S, et al. CALGB 90601 (Alliance): Randomized, double-blind, placebo-controlled phase III trial comparing gemcitabine and cisplatin with bevacizumab or placebo in patients with metastatic urothelial carcinoma. J Clin Oncol. 2019;37(15_suppl):4503–4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rosenberg JE, O’Donnell PH, Balar AV, et al. Pivotal Trial of Enfortumab Vedotin in Urothelial Carcinoma After Platinum and Anti-Programmed Death 1/Programmed Death Ligand 1 Therapy. J Clin Oncol. 2019;37(29):2592–2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cancer Genome Atlas Research N, Weinstein JN, Collisson EA, et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013;45(10):1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Azuaje F. Artificial intelligence for precision oncology: beyond patient stratification. NPJ Precis Oncol. 2019;3:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Capper D, Jones DTW, Sill M, et al. DNA methylation-based classification of central nervous system tumours. Nature. 2018;555(7697):469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bailey MH, Tokheim C, Porta-Pardo E, et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell. 2018;173(2):371–385 e318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wulfing C, Machiels JP, Richel DJ, et al. A single-arm, multicenter, open-label phase 2 study of lapatinib as the second-line treatment of patients with locally advanced or metastatic transitional cell carcinoma. Cancer. 2009;115(13):2881–2890. [DOI] [PubMed] [Google Scholar]
- 106.Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]