

Abstract
Type 1 diabetes (T1D) pathophysiology, while incompletely understood, has in part been attributed to aberrant presentation of self-antigen plus proinflammatory co-stimulation by professional antigen-presenting cells (APCs). Therapies targeting dendritic cells (DCs) offer an avenue to restore antigen-specific tolerance by promoting presentation of self-antigen in an anti-inflammatory or suppressive context. Here, we describe a subcutaneously administered, dual-sized biodegradable microparticle (MP) platform that includes phagocytosable (~1 μm) and non-phagocytosable (~30 μm) MPs to deliver pro-tolerogenic factors both intra- and extracellularly, as well as the T1D-associated autoantigen, insulin, to DCs for amelioration of autoimmunity. This MP platform resulted in increased recruitment of DCs, suppressive skewing of DC phenotype with diminished expression of CD86 and MHC-II, increased regulatory T cell (Treg) frequency, and upregulated expression of the checkpoint inhibitor programmed cell death protein 1 (PD-1) on T cells. When administered concomitantly with anti-CD3 antibody, which provides transient T cell depletion while preserving Treg populations, in 12-week-old non-obese diabetic (NOD) mice, regulatory immune populations persisted out to 20 weeks of age; however, combination anti-CD3 + dual sized MP (dMP) therapy failed to synergistically inhibit diabetes onset.
Keywords: Type 1 diabetes, autoimmunity, antigen specific therapy, tolerance, microparticle, biomaterials, drug delivery
Graphical Abstract
Combination treatment with dual-sized microparticle system plus anti-CD3 fails to synergize in late-stage type 1 diabetes prevention in NOD mice
INTRODUCTION
Type 1 diabetes (T1D) is a pernicious autoimmune disease characterized by aberrant immune activation that results in irrecoverable destruction of insulin producing β-cells in the pancreas [1]. While triggering events that cause T1D onset and disease pathophysiology are still incompletely understood, β-cell loss is marked by a dramatic increase in infiltration of pancreatic islets by pathogenic CD4+ and CD8+ T cells as well as CD11c+ antigen-presenting cells (APCs) [2]. Numerous recent clinical trials, attempting to corral the adaptive immune response by administering agents that induce T cell depletion (e.g., anti-CD3, anti-thymocyte globulin (ATG)) and/or inhibit T cell activation (e.g., CTLA4-Ig (abatacept)), have shown promise providing transient immunomodulation and/or temporary preservation of insulin production in patients with recent-onset T1D but have ultimately failed to provide long term tolerance induction or preservation of β-cell function [3–8]. Alternatively, strategies targeting APCs (i.e., upstream of T cell activation), may prove capable of inducing long-lasting immunological tolerance [9, 10]. Dendritic cells (DCs) are the most prominent APC in directing adaptive immunity [11] with tolerogenic or suppressive DCs representing a subset that are notable in their capacity to limit T cell activation and proliferation [12]. Tolerogenic DCs possess numerous modalities to mediate immune tolerance, including upregulation of inhibitory co-receptors (e.g., PD-L1, ILT, Fas) which can interfere with T cell co-stimulation or induce T cell apoptosis directly, release of anti-inflammatory cytokines (e.g., IL-10) to promote regulatory T cell (Treg) differentiation, and antigen presentation in the absence of co-stimulation which can result in T cell anergy [13, 14].
We previously developed a biodegradable microparticle (MP) system designed to modulate DCs in an antigen-specific, pro-tolerogenic manner [15–17]. This poly (lactic-co-glycolic acid) (PLGA) MP system was composed of two ~1 μm phagocytosable MPs (vitamin D3 (VD3)-loaded MPs and autoantigen-loaded MPs) and two ~30 μm non-phagocytosable MPs loaded with transforming growth factor beta 1 (TGF-β1) and granulocyte-macrophage colony-stimulating factor (GM-CSF). The large, ~30 μm MPs are inherently non-phagocytosable due to their size, as phagocytosis for DCs and macrophages is roughly limited to particles < 10–15 μm in diameter [18–20]. The guiding principle behind the subcutaneously injectable dual-sized MP (dMP) platform is to 1) provide extracellular release of a DC chemokine (GM-CSF) and the pro-tolerogenic cytokine (TGF-β1) to recruit and tolerize DCs, respectively, together with 2) phagocytosis of small MPs encapsulating autoantigen and an additional immunomodulatory factor, VD3, by locally recruited DCs in order to promote presentation of autoantigen in a tolerogenic context to reeducate aberrant autoreactivity. An early iteration of this dMP system established the efficacy of this dMP platform to prevent T1D in 4-week-old non-obese diabetic (NOD) mice when utilizing insulin B9–23 peptide as antigen [16]. However, when this dMP formulation was administered to 8-week-old pre-diabetic NOD mice, which display comparatively advanced β-cell loss and T cell infiltration in pancreatic islets [21, 22], diabetes prevention was not realized [17]. We sought to enhance the applicability of the dMP in 8-week-old NOD mice; the dMP was reformulated to encapsulate whole, denatured insulin (the intent being to ward against epitope spreading, wherein the autoantigen repertoire expands as pancreatic tissue damage releases neoantigens [23]), as well as increased loading of the tolerogenic and chemotactic factors in PLGA MPs. This reengineered dMP formulation improved therapeutic efficacy when administered in 8-week-old NOD mice and reversed hyperglycemia for up to 100 days in recent-onset NOD mice [17]. Prevention using the dMP was associated with an increase in Treg frequency, upregulation of PD-1 on CD4+ and CD8+ T cells, and an increase in suppressive DCs. However, T1D prevention using the reformulated dMP failed to impart protection against T1D when attempted in late-stage pre-diabetic, 12-week-old NOD mice (p = 0.14; Supplemental Figure 1).
In previous studies, we investigated the impact of each individual MP-encapsulated factor in preventing T1D [16, 17]. While only the full, four-factor dMP significantly inhibited T1D incidence compared to unloaded MP control, treatment with TGF-β1 MPs plus insulin MPs resulted in greater T1D protection than any other single or paired MP treatment. Because the previous dMP injection schedules delivered TGF-β1 doses well below toxicity levels [24], we speculated that boosting loading of TGF-β1 in MPs may improve tolerogenicity without compromising safety. Along this line, the previous dMP dosing for GM-CSF was also well below the GM-CSF doses of 1–7 μg, which were reported to recruit dose-dependent numbers of dendritic cells capable of trafficking to lymph nodes [25]. We therefore endeavored to increase factor loading of TGF-β1 and GM-CSF via an updated dMP (udMP) formulation, conceptualized to improve both DC recruitment and suppressive conditioning.
Additionally, we explore combination treatment utilizing a low dose of T cell depleting anti-CD3 plus the previous dMP formulation (lower dose) in 12-week-old NOD mice. A landmark study recently completed in relatives of patients with T1D showed Teplizumab, an Fc receptor–nonbinding anti-CD3 monoclonal antibody (aCD3), delayed diabetes onset in high-risk participants by over two years [26]. Similarly, previous research has shown that low-dose aCD3 provides age-dependent efficacy toward diabetes prevention in NOD mice: aCD3 had no effect when administered in 4- and 8-week-old NOD mice but delayed diabetes onset to roughly 20 weeks of age when administered to 12-week-old NOD mice [27]. This window of euglycemia closed shortly thereafter, however, with precipitous onset of hyperglycemia and final diabetes incidence being comparable to untreated mice by roughly 24 weeks of age. We hypothesized that low-dose aCD3 in combination with dMP treatment would deplete pathogenic T cells providing a favorable immune profile within which repopulating naïve T cells would be amendable to tolerogenic instruction from the dMP-modulated DCs.
RESEARCH DESIGN AND METHODS
Microparticle Fabrication and Characterization
PLGA (MW ~44,000 g/mol; Corbion Purac, Gorinchem, Netherlands) MPs were fabricated by standard oil-in-water single emulsion or water-in-oil-in-water double emulsion methods, as previously described [15]. All factors were separately encapsulated in distinct MPs. Briefly, phagocytosable MPs were loaded with 1-α 25-Dihydroxyvitamin D3 (VD3; Thermo Fisher Scientific, Waltham, MA), denatured human recombinant insulin (Sigma-Aldrich, St. Louis, MO), or ovalbumin (OVA; Sigma-Aldrich) while non-phagocytosable MPs encapsulated TGF-β1 or GM-CSF (Millipore Sigma).
Insulin was rendered inactive by chemical means and heat denaturation. First, insulin was reconstituted in ultrapure water (Barnstead GenPure, Thermo Fisher Scientific) and 1 M HCl added dropwise to promote solubility of insulin in solution (~ pH 2.5). Peptide disulfide bonds were cleaved by adding 10 mM 2-mercaptoethanol (Sigma-Aldrich). This solution was incubated in a water bath at 95°C for five minutes to ensure complete denaturation of insulin. After cooling to room temperature, 0.1 M NaOH was added dropwise until pH reached between 7.0–7.5 without inducing protein aggregation out of solution. This solution was filtered through a low-binding 0.22 μm filter and concentration confirmed by spectrophotometry (NanoDrop ND-1000, Thermo Fisher Scientific) against a standard curve.
Phagocytosable MPs were fabricated by dissolving 500 mg PLGA in methylene chloride at a 5% weight/volume (w/v) ratio. 50 μg VD3 in 1 mL methanol (Thermo Fisher Scientific) was added directly to the methylene chloride/PLGA solution and shaken at 150 rpm for 10 min. This solution was added to 50 mL 5% w/v poly-vinyl alcohol (PVA; MW ~15,000 g/mol; Themo Fisher Scientific) and homogenized at 35,000 rpm for 180 s using a tissue-miser homogenizer (Thermo Fisher Scientific) to form an oil-in-water emulsion. The MP solution was subsequently added to 100 mL 1% PVA and stirred for 4–6 h for solvent evaporation and MP hardening. For water-soluble denatured insulin MPs or denatured OVA MPs (as an irrelevant antigen control), 12.9 mg of protein in 2 mL PBS was added to the 5% methylene chloride/PLGA solution and homogenized at 35,000 rpm for 120 s to form a primary emulsion. This emulsion was added to 50 mL of 5% PVA, homogenized again at 35,000 rpm for 180 s to form the secondary emulsion, and added to 100 mL of stirring 1 % PVA.
Non-phagocytosable MPs encapsulating TGF-β1 or GM-CSF were fabricated by first dissolving 500 mg PLGA in methylene chloride at a 20% w/v ratio. Human TGF-β1 (25 μg for dMP or 75 μg for udMP) was reconstituted in 10 mM HCl and 2 mg/mL bovine serum albumin (BSA) in 250 μL PBS. Recombinant mouse GM-CSF (40 μg for dMP or 320 μg for udMP) was reconstituted in 400 μL PBS. Protein solutions were added to the methylene chloride/PLGA solution and vortexed at the highest setting (~3200 rpm) for 30 s to generate the primary emulsion. This emulsion was added to 5 mL 2.5% PVA, vortexed again at 3200 rpm for 60 s to form the secondary emulsion and added to 100 mL of stirring 1% PVA. Unloaded MPs were generated using methanol as a control for VD3 or using or PBS as a control for insulin, OVA, TGF-β1 and GM-CSF.
After stirring for 4–6 h, phagocytosable and non-phagocytosable MP solutions were centrifuged at 10,000 × g for 10 min to collect MPs and washed three times with ultrapure water (Barnstead GenPure, Thermo Fisher Scientific). MPs were flash-frozen in liquid nitrogen, lyophilized for 24 h, and stored at −20°C until use.
Encapsulation efficiency was assessed by μBCA (Thermo Fisher Scientific). Briefly, a known mass of insulin, OVA, TGF-β1, or GM-CSF MPs was dissolved in a 0.2 M NaOH/5% sodium dodecyl sulfate (SDS) solution. An analogous process with unloaded MPs plus soluble drug was performed as a control. Solution pH was neutralized with a small volume of HCl and protein concentration measured by μBCA. Serial dilutions of the unloaded MP/soluble drug solution was used as a standard curve to determine the encapsulation efficiency for each drug-loaded MP. Vitamin D3 MPs were measured by dissolving 100 mg of MPs into 2 mL methylene chloride and re-precipitating the PLGA with a known volume of methanol. The suspension was centrifuged and the supernatant collected. Following evaporation, residue remaining in the tube was concentrated in DMSO and the solution concentration measured by spectrophotometer.
Experimental Animals
Female NOD/ShiLtj and C57BL/6 mice, ages 6–8 weeks, were purchased from either Jackson Laboratory (Bar Harbour, ME) or University of Florida Animal Care Services (ACS) (Gainesville, FL). All animals were housed in a specific pathogen free-environment in University of Florida ACS facilities, and experiments were performed in accordance with detailed protocols approved by University of Florida Institutional Animal Care and Use Committee (IACUC).
Site of Injection Nodule Characterization
Characterization of recruited leukocytes at the site of injection was carried out via flow cytometry. Eight-week-old, female C57BL/6 mice were injected subcutaneously in the abdominal region using a 20 G needle (BD Biosciences) with 10mg MPs total (1:1:1:1 MP mass ratio) in 0.2 mL PBS. Injection site nodules were excised seven days after injection, enzymatically digested with 2 mg/mL collagenase type XI (Sigma-Aldrich, St. Louis, MO, USA) at 37°C for 30 min, filtered through a 30 μm filter to remove large particulates, and cells stained for flow cytometry.
udMP Treatment in 8-Week-Old NOD Mice
Eight-week-old, female NOD mice were randomized into two treatment groups as follows: (1) blank unloaded MPs (BMP) or (2) udMP (VD3 MPs + TGF-β1 MPs + GM-CSF MPs + insulin MPs) (n = 15/group). Animals were injected subcutaneously in the right upper abdominal region, anatomically proximal to the pancreas using a 20 G needle (BD Biosciences) with the described formulations (i.e., 10 mg MPs total (1:1:1:1 MP mass ratio for the udMP; 1:1 mass ratio of phagocytosable:non-phagocytosable MPs for the BMP) in 0.2 mL PBS) once per week for the first three weeks (8, 9 and 10 weeks of age), and given a booster once monthly thereafter for four months (12, 16, 20, and 24 weeks of age), as previously described [17]. Blood glucose levels were monitored once weekly for 20 weeks, after which remaining non-diabetic mice were euthanized, and diabetes onset was defined as blood glucose levels ≥ 240 mg/dL for two consecutive days.
Anti-CD3 Antibody Plus dMP Treatment in 12-Week-Old NOD Mice
Non-diabetic, twelve-week-old, female NOD mice were randomized into seven treatment groups as follows: (1) no treatment, (2) dMP OVA (VD3 MPs + TGF-β1 MPs + GM-CSF MPs + OVA MPs) + control hamster IgG F(ab)2 fragments, (3) dMP (VD3 MPs + TGF-β1 MPs + GM-CSF MPs + insulin MPs), (4) low-dose aCD3, (5) low-dose aCD3 + dMP, (6) ultra-low-dose aCD3, and (7) ultra-low-dose aCD3 + dMP (n = 16–22/group). Control hamster F(ab)2 fragments and hamster anti-mouse CD3 145–2C11 monoclonal antibody (mAb) F(ab)2 fragments were purchased from BioXCell (West Lebanon, NH). At 12 weeks of age, mice were injected intraperitoneally (i.p.) for 5 consecutive days with either PBS (groups 1 and 3), a control hamster F(ab)2 fragments (5 μg/day; group 2), low-dose aCD3 (5 μg/day; groups 4 and 5), or ultra-low-dose aCD3 (1 μg/day; groups 6 and 7). The low-dose aCD3 regimen is a suboptimal dose shown to reduce potential toxicity and to lower efficacy, enhancing the potential ability to identify synergistic effects with combination therapy [28]. Mice were injected subcutaneously in the right upper abdominal region, anatomically proximal to the pancreas with the described MP formulation every five days beginning at 12 weeks of age on the same day that aCD3 treatment began, for a total of four initial MP injections. Subsequently, mice received three monthly MP booster injections. Blood glucose levels were monitored once weekly to 30 weeks of age, after which remaining non-diabetic mice were euthanized. Diabetes onset was defined as blood glucose levels ≥ 240mg/dL for two consecutive days.
Kinetic Tracking of Circulating Leukocytes
Five to ten mice per treatment group from the aCD3+dMP study were serially bled for flow cytometry analysis to assess the impact of aCD3 and dMP treatment on circulating leukocyte frequency and phenotype. Roughly 100 μL blood was collected in EDTA-coated microtubes (Braintree Scientific, Braintree, MA) by submandibular bleed one week before treatment began (11 weeks of age) and three days following completion of the five-day aCD3 or control injection regimen. Whole blood was lysed with 750 μL ACK lysis buffer (Thermo Fisher Scientific) for 5 min at RT. Subsequently, 250 μL 4x PBS and 10% FBS (Hyclone, GE Healthcare Life Sciences, Marlborough, MA) were added, cells spun down, and supernatant discarded. Washing was repeated once more to fully deplete red blood cells, and leukocytes were stained for flow cytometry.
Midpoint Analyses of Secondary Lymphoid Organs
Animals were randomly enrolled in the seven treatment groups for the aCD3 plus dMP study (n = 4–5/group), as described above, but were not included in survival curves. At 20 weeks of age, prior to the week 20 booster MP injection, animals were euthanized and MP draining lymph nodes (combined inguinal and axillary), pancreatic lymph nodes, and spleen were harvested and analyzed by flow cytometry to interrogate cellular phenotypes as potential indicators of therapeutic mechanisms.
Flow Cytometry
For experiments involving flow cytometry, staining was performed as follows: cells were (1) isolated into a single cell suspension and stained with fixable viability dye (Green or Near-IR; Life Technologies) for 10 min at RT, (2) washed and blocked with anti-CD16/32 (FCγ III/II receptor, clone 2.4G2, Thermo Fisher Scientific) for 15 min on ice, (3) fluorescent conjugated antibodies incubated at a 1:50–1:250 dilution for 30 min on ice, (4) washed and data immediately acquired or fixed with paraformaldehyde for acquisition within 24 hours. If intracellular staining was required, after step (3), cells were washed, fixed/permeabilized with Intracellular Fixation and Permeabilization Buffer Set (eBioscience) according to the manufacturer’s instructions, and intracellular markers stained with fluorescent conjugated antibodies. Data were collected on an LSR II (BD Biosciences) or a Guava Easycyte flow cytometer (Millipore Sigma) and analyzed with FlowJo software (Tree Star, Ashland, OR).
Cells were stained with the following antibodies: anti-B220 (brilliant violet (BV) 785; clone: RA3–6B2), anti-CD11b (BV605; clone: M1/70), anti-CD11c (PE; clone: N418), anti-CD3ε (AF700; clone: 500A2), anti-CD4 (BV605, BV785, AF488; clone: RM4–5), anti-CD44 (PE-Cy7; clone: IM7), CD45 (APC-Cy7; clone: 30-F11), anti-CD62L (PE; clone: MEL-14), anti-CD8a (BV605; clone: 53–6.7), anti-CD80 (APC; clone: 16–10A1), anti-CD86 (PE-Cy7; clone: GL1), anti-Foxp3 (eFluor450; clone: FJK-16s), anti-Helios (AF488; clone: 22F6), anti-ICOS (PerCP-Cy5.5; clone: C398.4A), anti-Foxp3 (eFluor450; clone: FJK-16s), anti-Ly6G (AF700; clone: 1A8), anti-MHC-II (AF488, PE; clones: M5/114.15.2, 39-10-8), anti-PD-1 (APC; clone: 29F.1A12), and anti-PD-L1 (BV421; clone: 10F.9G2). All antibodies were purchased from Biolegend (San Diego, CA).
Statistical Analysis
GraphPad Prism software v7 (La Jolla, CA) was used for all statistical analyses. Unpaired t-test, one-way ANOVA, and Kaplan-Meier estimator were applied, as indicated. Bonferroni’s and Tukey’s post hoc tests were used to account for multiple comparisons, as indicated. Data are presented as mean ± SEM, with P values ≤ 0.05 considered significant.
RESULTS
Microparticle Fabrication and Characterization
A udMP formulation was fabricated to deliver higher doses of the DC chemokine GM-CSF and the pro-tolerogenic factor TGF-β1 in an otherwise identical MP fabrication protocol as we have previously reported [17]. MPs were loaded with an individual agent (GM-CSF, TGF-β1, VD3, or denatured insulin) using single- or double-emulsion solvent evaporation techniques as dictated by the solubility of the encapsulated factors. Large, non-phagocytosable (~30 μm) MPs contained GM-CSF or TGF-β1, and small, phagocytsoable (~1 μm) MPs encapsulated VD3, denatured insulin, or OVA. Distinct MP sizes were achieved by varying the viscosity of the oil phase (PLGA in methylene chloride), the concentration and volume of surfactant, and the agitation rate. MP encapsulation of loaded factors was characterized, and encapsulation efficiencies of GM-CSF and TGF-β1 were 57% and 50%, respectively (Table 1), as compared to 61% and 57% for the previously reported dMP formulation [17]. Due to the increased mass of drug loaded per mg/PLGA, this translated to roughly 7-fold and 2.5-fold increases in GM-CSF and TGF-β1 delivered per injection, respectively. These doses are well-grounded in relevant literature, and represent a reasonable entry point for investigation. For example, at the updated 190 ng TGF-β1 dose per injection, there is no concern of exceeding > 2 μg/day per mouse, which can induce cachexia (severe wasting, weight loss), cause system fibrosis and be fatal [24]. Regarding GM-CSF, doses of 1–7 μg were previously investigated [25], and the updated 900 ng GM-CSF dose per injection falls in line with the 1–3 μg doses found to recruit increased numbers of dendritic cells which were capable of subsequently trafficking to lymph nodes.
Table 1. Characterization of updated dMP (udMP) formulation.
The loading efficiencies of encapsulated agents and mass delivered per 2.5 mg PLGA injection was calculated (n = 3). Data is represented by mean ± SD.
| Encapsulated Agent | Mass Loaded/PLGA (μg/ 100 mg) | Encapsulation Efficiency ± SD (%) | Mass Delivered per Injection ± SD (ng) |
|---|---|---|---|
| Denatured Insulin | 2580 | 65 ± 7 | 41,500 ± 4,400 |
| Vitamin D3 | 10 | 66 ± 3 | 165 ± 10 |
| TGF-β1 | 15 | 50 ± 5 | 190 ± 20 |
| GM-CSF | 64 | 57 ± 6 | 900 ± 100 |
Increased GM-CSF and TGF-β1 Loading in the udMP Enhanced DC Recruitment and Tolerization
We sought to examine whether the udMP boosts DC recruitment and tolerization. Previously, we reported that subcutaneous dMP administration results in a palpable nodule at the injection site composed of significant leukocyte infiltration that resorbed within a month of injection as the PLGA bolus degraded [15]. Here, we isolated nodule-infiltrating cells following unloaded blank MP (BMP), dMP, and udMP injection in C57BL/6 mice and assessed the cellular composition by flow cytometry. Comparing recruited DC frequencies for MPs containing bioactive factors yielded augmented recruitment at the injection site nodule by 57% and 111%, for the dMP and udMP respectively, as compared to BMP control; however, only the udMP realized statistical significance (Figure 1; gating scheme – Supplemental Figure 8). Total numbers of DCs were similarly increased for the udMP nodules, recruiting 30.7 × 104 DCs as compared to 7.1 × 104 and 9.8 × 104 DCs for the BMP and dMP respectively (Supplemental Figure 2). Furthermore, dMP and udMP treatment reduced frequencies of CD86+ and MHC-II+ DCs within the nodule consistent with a tolerogenic or suppressive phenotype, and this effect appeared to be dose dependent (Figure 2; gating scheme – Supplemental Figure 8). Similar trends in CD86 and MHC-II expression were also seen on recruited Ly6G−CD11b+CD11c− monocytes/macrophages isolated from excised nodules (data not shown).
Figure 1. Increased GM-CSF and TGF-β1 loading in the udMP enhanced DC recruitment.
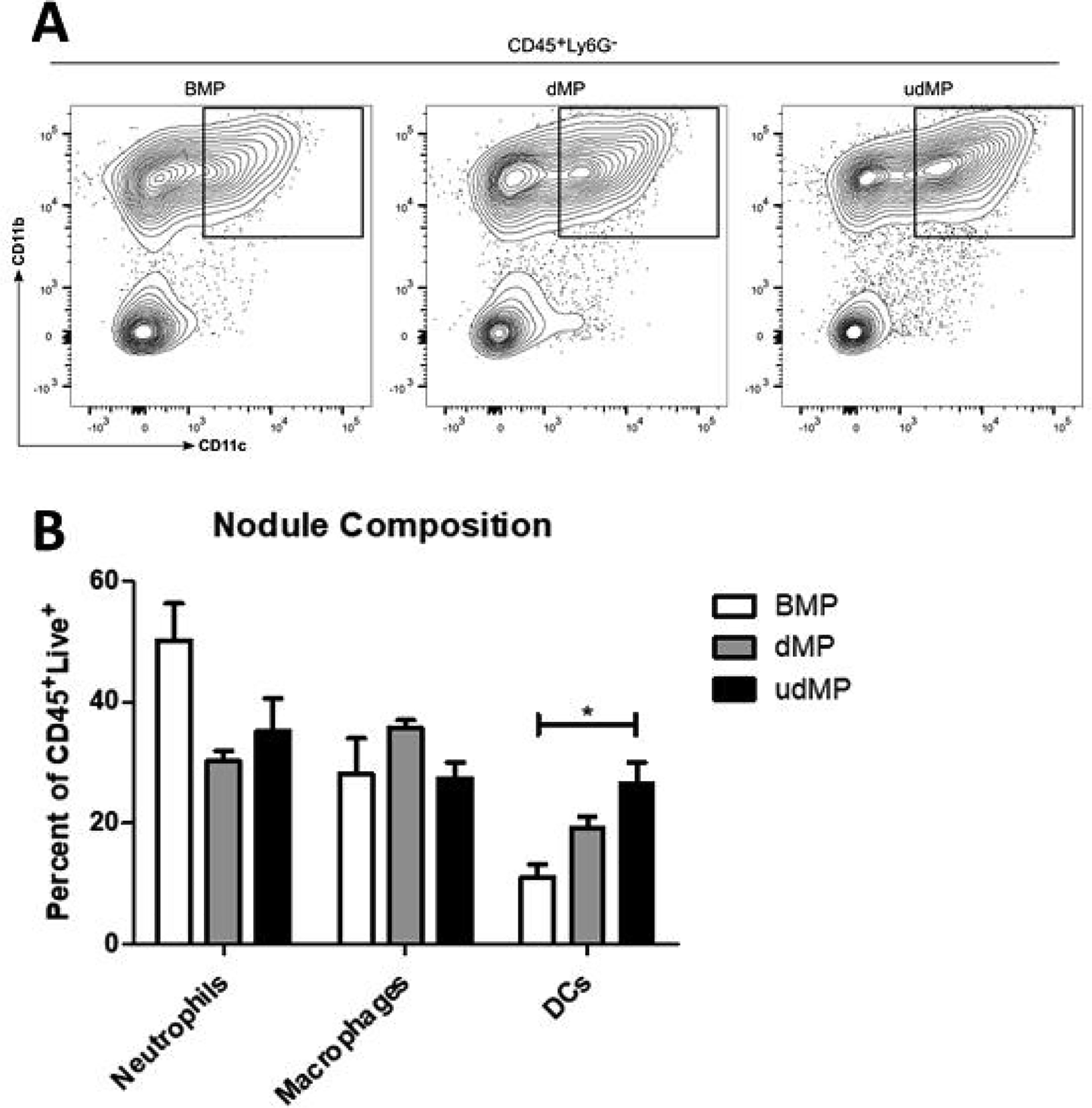
C57BL/6 mice were injected subcutaneously in the abdominal region with either blank PLGA MPs (BMP), the dMP, or the udMP (n = 4–5). (A) Injection site nodules were excised seven days later, enzymatically digested, and leukocytes analyzed by flow cytometry. (B) Leukocyte frequencies (Ly6G+CD11b+ neutrophils; Ly6G−CD11b+CD11c− macrophages; Ly6G−CD11b+CD11c+ DCs) as a as a percentage of CD45+ live cells. The frequency of DCs recruited to the site of MP injection increased in a bioactive factor dose-dependent manner. P-values (* = ≤ 0.05) were obtained by one-way ANOVA with Tukey’s significance test. Data is represented by mean ± SEM. Full gating scheme – see Supplemental Figure 8.
Figure 2. High dose GM-CSF and TGF-β1 loading in the udMP led to reduced costimulatory marker expression on DCs.
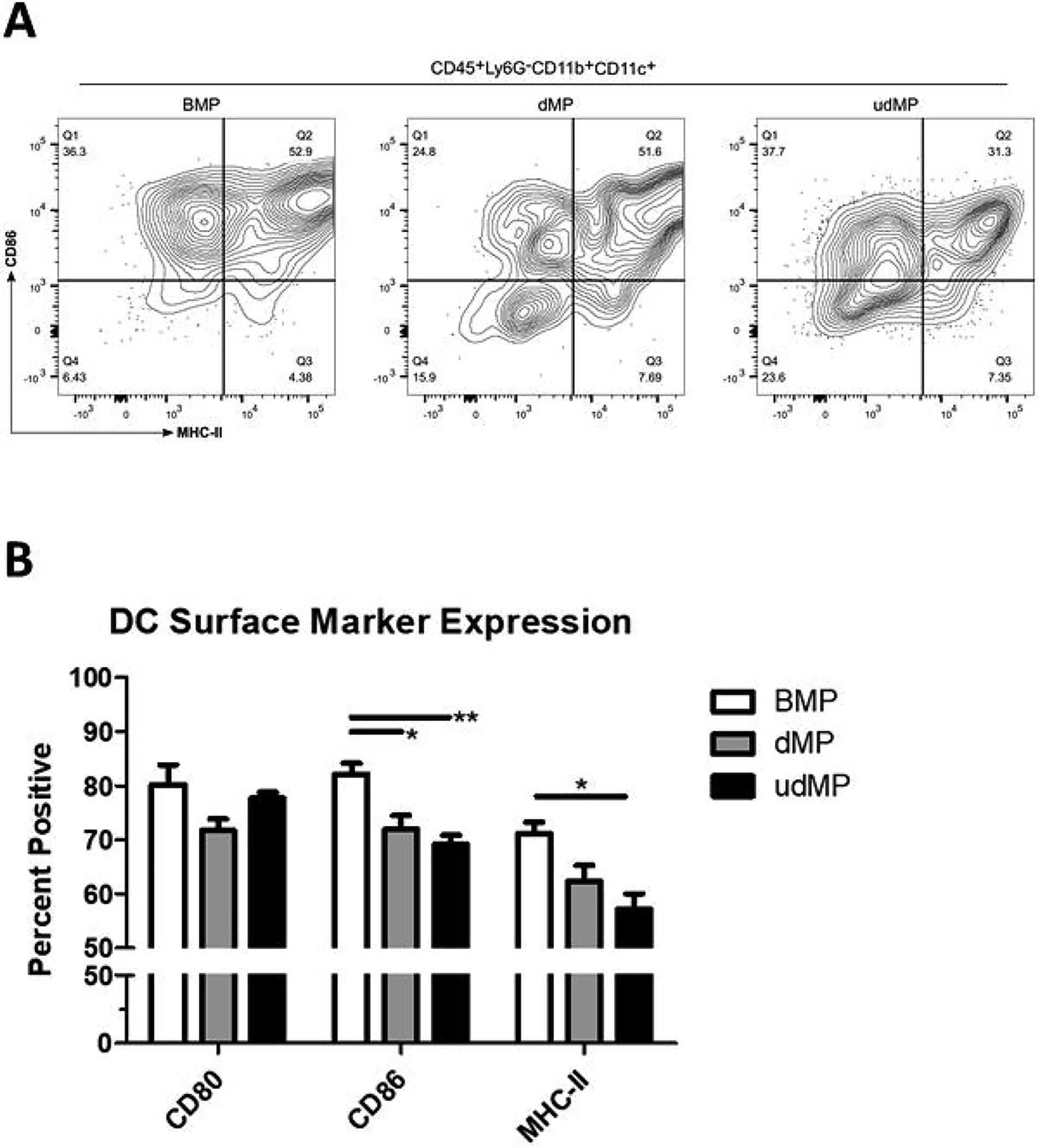
C57BL/6 mice were injected subcutaneously in the abdominal region with either blank PLGA MPs (BMP), the dMP, or the udMP (n = 4–5). (A) Injection site nodules were excised seven days later, enzymatically digested, and leukocytes analyzed by flow cytometry for CD86 and MHC-II expression on DCs recruited to the site of injection. (B) Frequency of surface marker expression (CD80, CD86, and MHC-II) was characterized on DCs to assess the capacity of the udMP to induce a suppressive phenotype. P-values (* = ≤ 0.05, ** = ≤ 0.01) were obtained by one-way ANOVA with Tukey’s significance test. Data is represented by mean ± SEM. Full gating scheme – see Supplemental Figure 8.
Increased GM-CSF and TGF-β1 Loading in the udMP Failed to Improve Diabetes Prevention in 8-week-old NOD Mice
Eight-week-old pre-diabetic female NOD mice were treated with udMP or BMP over 16 weeks to ascertain whether increased doses of GM-CSF and TGF-β1 would improve diabetes prevention compared to historical data on the original dMP formulation (60% prevention at 28 weeks of age) [17]. The injection schedule consisted of subcutaneous udMP injections once per week for the first three weeks (8, 9 and 10 weeks of age) and a booster injection once monthly thereafter for four months (12, 16, 20, and 24 weeks of age), identical to the previous dMP injection schedule which significantly prevented diabetes in 8-week-old NOD mice [17]. In the current study, diabetes incidence in udMP-treated mice was comparable to historical results with the original dMP (i.e., 60% of animals remaining euglycemic at 28 weeks of age); however, with 40% of BMP control treated mice remaining non-diabetic at the end of the study, the difference was not statistically significant (Figure 3).
Figure 3. High dose GM-CSF and TGF-b1 in the udMP do not prevent diabetes in 8-week-old NOD mice compared to blank MPs.
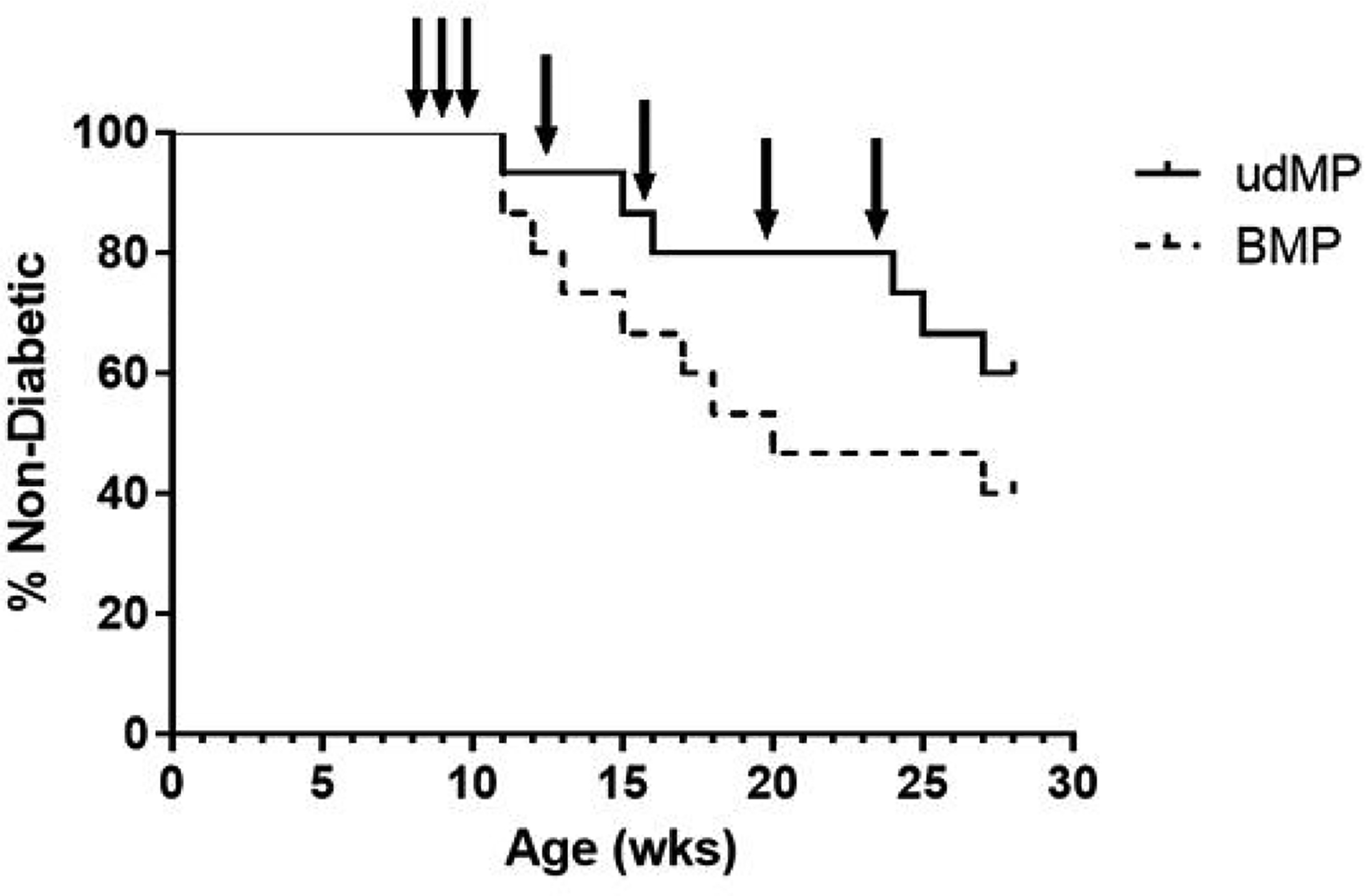
A cohort of 8-week-old NOD mice (n = 15/group) were injected at a subcutaneous site anatomically proximal to the pancreas with the described MP formulations over 16 weeks. Animals received MP injections (arrows) once a week for the first three weeks (8, 9 and 10 weeks of age) and a booster injection once monthly thereafter for four months (12, 16, 20, and 24 weeks of age). Blank MPs were included as a control treatment group. Animals were monitored weekly until week 28, after which remaining non-diabetic mice were euthanized, and mice were considered diabetic when blood glucose levels were ≥ 240 mg/dL on two consecutive days. Survival data is fit using the Kaplan–Meier non-parametric survival analysis model and statistical analysis (p-value = 0.21) performed via log-rank test (Mantel-Cox method).
MP Fabrication and Characterization for Combination Treatment with aCD3
We hypothesized that combination therapy with low-dose aCD3, which has been shown to delay diabetes onset in 12-week-old NOD mice, might provide an opportune euglycemic window with which repopulating T cells may be amenable to dMP-induced tolerogenic instruction. Because the udMP formulation did not provide improved protection against T1D onset (Figure 3), for this late-stage T1D prevention study, we reverted to the original dMP formulation. As before, MPs were loaded with an individual agent (GM-CSF, TGF-β1, VD3, denatured insulin, or denatured OVA (control)) and characterized for encapsulation efficiency (Table 2). The reduced loading dose of factors in non-phagocytosable MPs altered the encapsulation efficiency slightly, increasing the encapsulation efficiency of GM-CSF from 57% to 60% and reducing the encapsulation efficiency of TGF-β1 from 50% to 44%. This translated to roughly 7.5-fold and 3.5-fold lower doses of GM-CSF and TGF-β1 delivered per dMP injection compared to the udMP, respectively. Scanning electron microscopy and drug release kinetics from PLGA MPs were measured previously [17]. Note there is large difference in the reported 41.5 μg mass of insulin delivered vs. the 6.4 μg reported in [17], although no MP fabrication changes were made. This is due to an updated insulin encapsulation efficiency measurement. Previously, encapsulation efficiency for insulin MPs was measured by solvent extraction. However, it has since been determined this method resulted in a substantial amount of encapsulated insulin releasate prone to aggregation/precipitation, unavailable for quantitation in the aqueous phase. The current method is less harsh, and more accurately reflects the encapsulated insulin dose in the dMP formulation. Comparable insulin loading was confirmed by testing preserved sample of the previously reported dMP.
Table 2. Characterization of the dMP formulation.
The loading efficiencies of encapsulated agents and mass delivered per 2.5 mg PLGA injection was calculated (n = 3). Data is represented by mean ± SD.
| Encapsulated Agent | Mass Loaded/PLGA (μg/ 100 mg) | Encapsulation Efficiency ± SD (%) | Mass Delivered per Injection ± SD (ng) |
|---|---|---|---|
| Denatured Insulin | 2580 | 65 ± 7 | 41,500 ± 4,400 |
| Vitamin D3 | 10 | 66 ± 3 | 165 ± 10 |
| TGF-β1 | 5 | 44 ± 12 | 55 ± 15 |
| GM-CSF | 8 | 60 ± 7 | 120 ± 15 |
aCD3 + dMP Treatment Did Not Synergize to Improve T1D Prevention in 12-week-old NOD Mice
Female NOD mice were randomized into seven treatment groups as follows: (1) no treatment, (2) dMP OVA (VD3 MPs + TGF-β1 MPs + GM-CSF MPs + OVA MPs) + control hamster IgG, (3) dMP (VD3 MPs + TGF-β1 MPs + GM-CSF MPs + insulin MPs), (4) low-dose aCD3 (5 μg/day), (5) low-dose aCD3 + dMP, (6) ultra-low-dose aCD3 (1 μg/day), and (7) ultra-low-dose aCD3 + dMP as shown in Figure 4A. Mice were monitored weekly for hyperglycemia beginning at 10 weeks of age, and hyperglycemic animals removed from the study prior to enrollment at 12 weeks of age. Weekly blood glucose checks continued to 30 weeks of age. Although the sample size per group ranged from 17–22, the study was not sufficiently powered to detect a statistically significant difference in T1D incidence across all seven groups as assessed by Kaplan–Meier survival analysis with Bonferroni correction for multiple comparisons (p=0.34; Figure 4B). While 18%,19%, and 33% of NOD mice treated with low-dose aCD3, low-dose aCD3 + dMP, and dMP, respectively, remained euglycemic by 30-weeks-old (p=0.62, 0.51, and 0.39, respectively, versus untreated controls (8%) without Bonferroni correction), 41% of mice treated with ultra-low-dose aCD3 or ultra-low-dose aCD3 + dMP remained non-diabetic at the end of the study (p=0.08 and p=0.03, respectively, as compared to untreated controls without Bonferroni correction). Hence, dMP did not appear to synergize with aCD3 to improve T1D prevention, but these data suggest ultra-low-dose aCD3 might be more effective than low-dose aCD3 toward preventing T1D progression in late-stage pre-diabetic mice. Additionally, dMP treatment alone was ineffective at preventing diabetes onset at this stage in contrast to results demonstrated in 4- and 8-week old NOD mice and in temporarily reversing recent onset T1D [16, 17] Though the combination therapy did not effectively prevent T1D, local and systemic immunomodulation was observed following treatment.
Figure 4. Treatment schedule for aCD3 and dMP combination therapy and diabetes incidence.
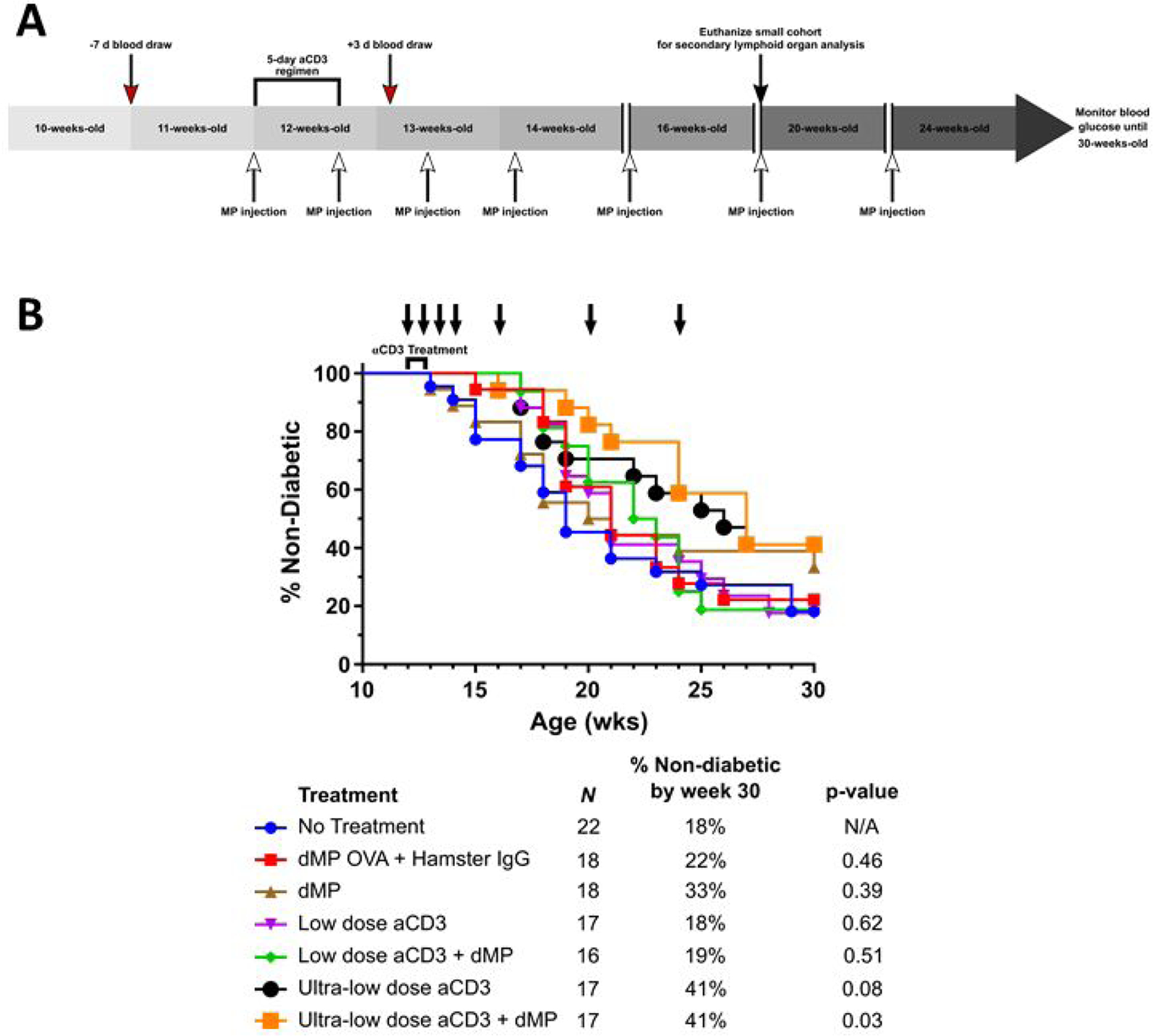
(A) A cohort of female NOD mice were randomized into seven treatment groups. Mice were monitored weekly for hyperglycemia beginning at 10-weeks-old and any diabetic animals (blood glucose levels were ≥ 240 mg/dL on two consecutive days) removed prior to study enrollment. At 11 weeks of age, a week before aCD3 and dMP treatment began, and three days after aCD3 treatment a selection of mice from each group was bled (red arrows) to assess circulating blood leukocyte frequencies and phenotype. At 12 weeks of age, mice began aCD3 treatment and/or received MP injections (white arrows). Animals received either PBS, control IgG F(ab)2 fragments (5 μg/day), low-dose aCD3 (5 μg/day), or ultra-low-dose aCD3 (1 μg/day) i.p. for five consecutive days. Simultaneously, beginning at 12 weeks of age, mice were subcutaneously injected with either PBS, dMP OVA, or the dMP every five days for a total of four initial MP injections. Subsequently mice received three monthly booster dMP injections at 16-, 20-, and 24-weeks-old. A small cohort of animals excluded from Kaplan-Meier survival analysis were euthanized at 20 weeks of age (black arrow) to analyze secondary lymphoid organs. Weekly blood glucose measurements continued up to 30 weeks of age, after which remaining non-diabetic mice were euthanized, and mice were considered diabetic when blood glucose levels were ≥ 240 mg/dL on two consecutive days. (B) dMP treatment failed to synergize with aCD3 to improve diabetes prevention in 12-week-old NOD mice.. Survival data is fit using the Kaplan–Meier non-parametric survival analysis model and statistical analysis performed via log-rank test (Mantel-Cox method). Descriptive characteristics for each treatment group highlight the n per group, percent of mice non-diabetic by the end of study, and unadjusted p-value as compared to the no treatment group. Statistical significance was not realized when accounting for multiple comparisons via Bonferroni correction, as the study was not powered to resolve this large number of groups. However, pairwise comparison between survival curves of mice that received the ultra-low-dose aCD3 + dMP and untreated mice resulted in a p-value < 0.05, suggesting a difference between treatments.
Peripheral Immunomodulation Following aCD3 + dMP Treatment in 12-week-old NOD Mice
One week before treatment began (11 weeks of age) and on day 8 following the initiation of treatment (Figure 4A), mice from each group were bled to assess circulating blood lymphocyte frequency and phenotype via flow cytometry. As expected, pre-diabetic NOD mice from each group displayed similar lymphocyte composition before treatment (Supplemental Figure 3), and following low-dose aCD3, the frequency of circulating CD4+ and CD8+ T cells was dramatically reduced (Figure 5A–C; gating scheme – Supplemental Figure 9) concomitant with a significant increase in the CD4+/CD8+ T cell ratio (Figure 5D), in agreement with previous studies [4, 29, 30]. The ultra-low-dose aCD3 regimen did not significantly reduce circulating T cell frequencies, with or without the addition of the dMP; Figure 5A–C). Interestingly, T cell frequency upon the addition of dMP suggests there may be potential to partially rescue low-dose aCD3 induced CD8+ T cell depletion (Figure 5B), although these values are not statically significant, and dMP inclusion negated the effect on CD4+:CD8+ T cell ratio (Figure 5D).
Figure 5. Circulating T cell frequencies were reduced in low-dose aCD3 treated mice.
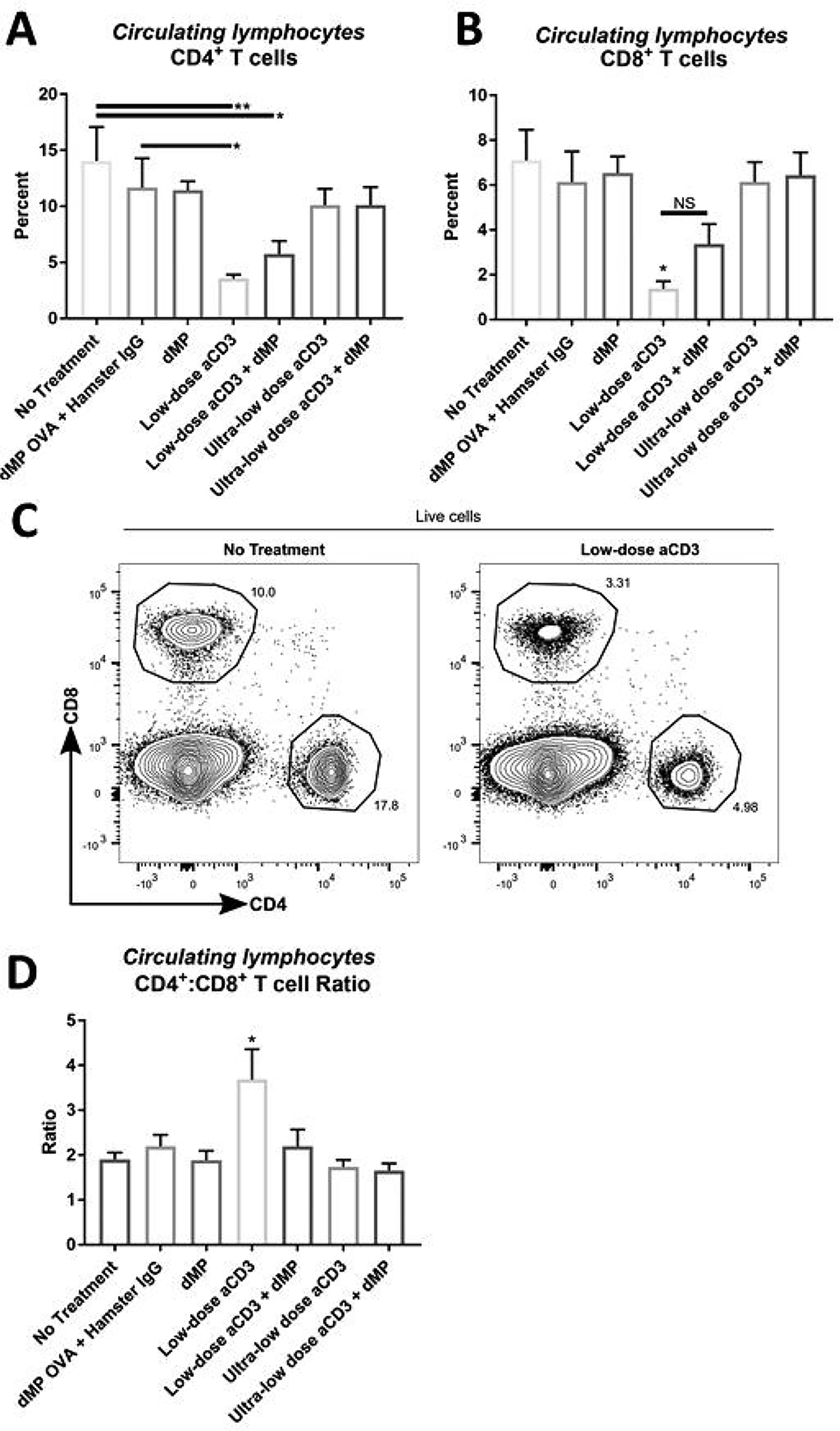
At ~13 weeks of age, three days after completing a five-day aCD3 treatment regimen, a selection of mice from each group was bled to assess circulating blood lymphocyte frequencies (n = 9–10/group). Frequency of (A) CD4+ and (B) CD8+ T cells in blood was determined by flow cytometry. (C) Representative flow analysis. (D) CD4+ to CD8+ T cell ratio. P-values (* ≤ 0.05, ** ≤ 0.01) were determined by one-way ANOVA with Tukey’s significance test. Significance (*) in (B) reflects low-dose aCD3 is significant against every group except low-dose aCD3 + dMP (NS; non-significant). Significance in (D) reflects low-dose aCD3 is significant against every other treatment group. Data is represented by mean ± SEM. Full gating scheme – see Supplemental Figure 9.
While ultra-low-dose aCD3 treatment did not significantly reduce CD4+ or CD8+ T cell frequencies, both low-dose and ultra-low-dose aCD3 imparted a shift in T cell phenotype, significantly reducing the CD44loCD62Lhi naïve / CD44hiCD62Llo memory CD4+ T cell ratio as compared to untreated and OVA dMP control-treated mice (Figure 6; gating scheme – Supplemental Figure 10). Once again, the addition of dMP partially counteracted this effect for both low-dose aCD3 + dMP and ultra-low-dose aCD3 + dMP groups, respectively (Figure 6B).
Figure 6. Anti-CD3 treatment selectively depleted naïve CD4+ T cells.
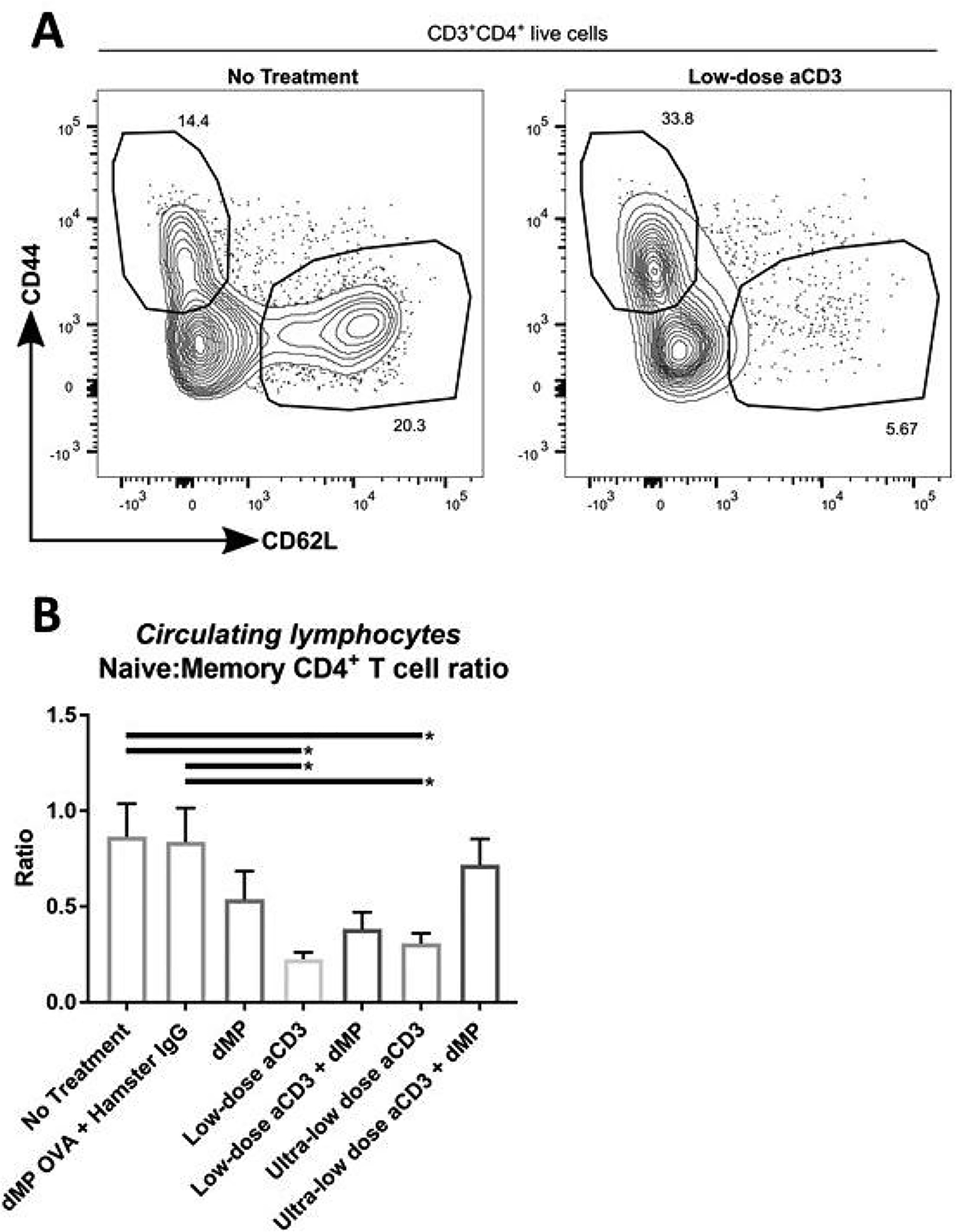
At ~13 weeks of age, three days after completing a five-day aCD3 treatment regimen, a selection of mice from each group was bled to assess circulating blood lymphocyte phenotype (n = 9–10/group). (A) Representative flow analysis is shown. (B) The ratio of naïve (CD62L+CD44−) to memory (CD62L−CD44+) CD4+ T cells in blood. *P-values ≤ 0.05 were determined by one-way ANOVA with Tukey’s multiple comparison test. Data is represented by mean ± SEM. Full gating scheme – see Supplemental Figure 10.
Low-dose aCD3-mediated selective depletion of naïve CD4+ T cells resulted in a subsequent escalation in Foxp3+ Treg frequency in circulation (Figure 7A–C; gating scheme – Supplemental Figure 10). Combination treatment with the dMP slightly mitigated this effect, diminishing the frequency of Tregs to levels that were not significantly different from untreated animals (Figure 7C). These patterns were supported by significantly higher frequencies of CD4+ T cells expressing the immunoregulatory costimulatory molecules, PD-1 and inducible costimulator (ICOS) (Figure 7D–E) in low-dose aCD3 treated animals. However, PD-1 and ICOS expressing cells were not increased in mice that received ultra-low-dose aCD3 or combination therapy with aCD3+dMP (Figure 7D–E).
Figure 7. Non-depleted T cells displayed a regulatory phenotype.
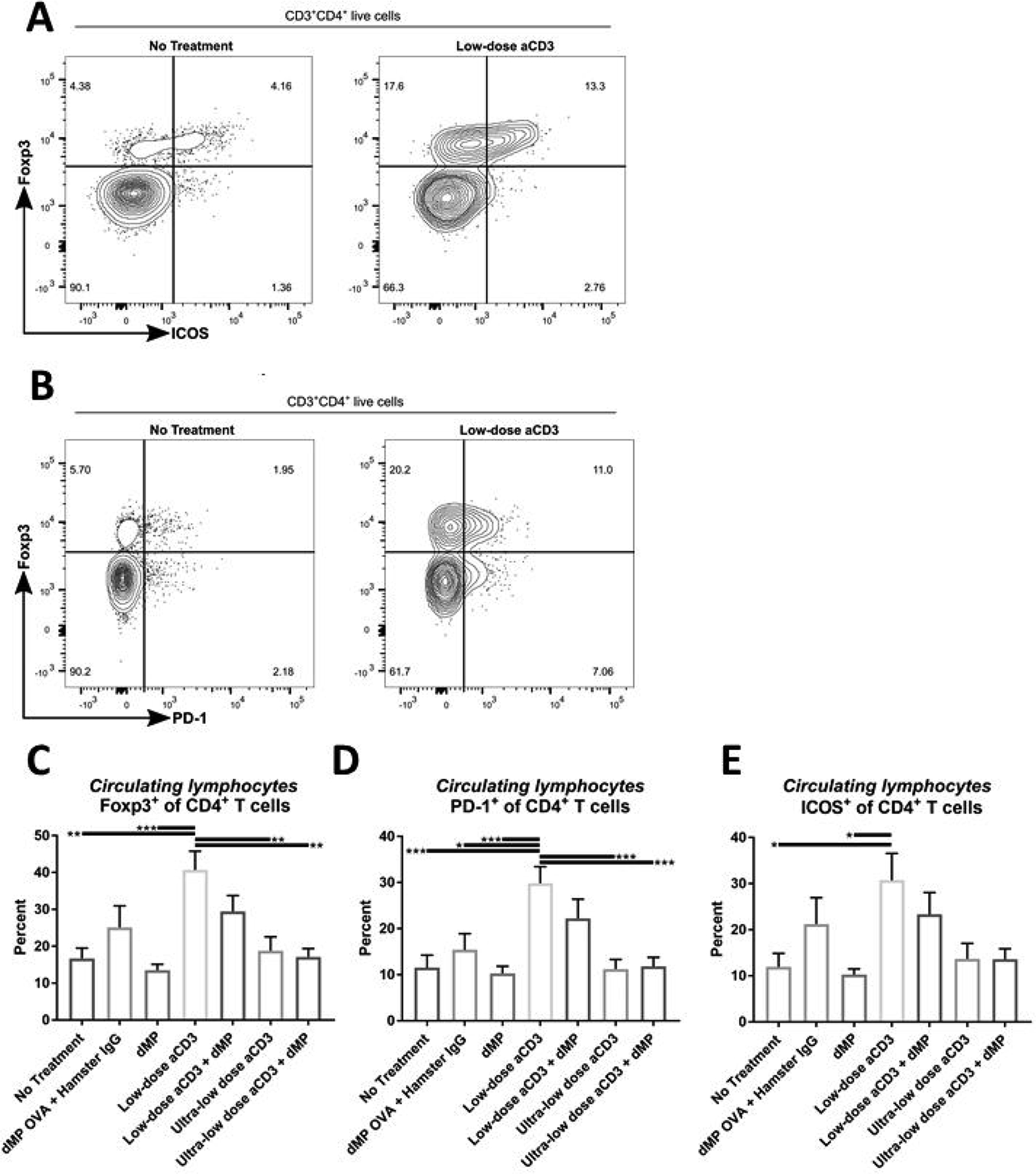
(A-B) At ~13 weeks of age, three days after completing a five-day aCD3 treatment regimen, a selection of mice from each group was bled to assess circulating blood lymphocyte frequency and phenotype by flow cytometry (n = 9–10/group). (C) The frequency of Foxp3+ Tregs as a percentage of total CD4+ T cells in blood. Percentage of CD4+ T cells expressing (D) PD-1 or (E) ICOS+ in blood. P-values (* ≤ 0.05, ** ≤ 0.01, *** ≤ 0.001) were determined by one-way ANOVA with Tukey’s multiple comparisons test. Data is represented by mean ± SEM. Full gating scheme – see Supplemental Figure 10.
In addition to monitoring changes in lymphocyte subpopulations, we characterized changes in innate immune cell frequency and phenotype in circulation (Supplemental Figure 4). Minimal changes were detected following aCD3 or dMP treatment. There was a trend toward increased circulating granulocytes in animals that received dMP (with or without aCD3), but statistically significant differences in granulocyte frequency was only observed with a single pairwise comparison between low-dose aCD3 + dMP and untreated groups (Supplemental Figure 4A). Neither total nor PD-L1+ suppressive DC frequencies were significantly changed across treatment groups (Supplemental Figure 4B–C). Similarly, expression levels of the maturation marker, MHC-II, on DCs were comparable across all treatment groups except for a small but significant increase in MHC-II expression on DCs isolated from ultra-low-dose aCD3 + dMP mice as compared to low-dose aCD3 treated animals (Supplemental Figure 4D).
dMP Treatment Modulated DCs Toward a Suppressive Phenotype and Increased PD- 1 +CD4+ T cells and Foxp3+ Treg Frequency in dMP-Draining Lymph Nodes
A small cohort of NOD mice, which were excluded from Kaplan–Meier survival analysis (Figure 4B), were enrolled in the seven treatment groups and euthanized at 20 weeks of age to examine leukocyte frequency and phenotype in lymphoid organs (Figures 8–11). Flow cytometric analysis revealed that dMP downregulated markers of DC activation in dMP-draining lymph nodes (combined inguinal and axillary lymph nodes; Figure 8; gating scheme – Supplemental Figure 11). While CD80 expression on DCs was relatively consistent across treatment groups (Figure 8B), CD86 (Figure 8C) and MHC-II (Figure 8D) expression was dramatically reduced in all groups that received dMP except for mice that received ultra-low-dose aCD3 + dMP combination therapy. This observation held true irrespective of the antigen encapsulated in the dMP formulation, as dMP containing either OVA or denatured insulin diminished CD86 and MHC-II expression (Figure 8C–D). These changes were not exhibited on DCs in distal lymphoid organs (spleen and pancreatic lymph nodes; data not shown). dMP treated animals also exhibited a greater frequency of CD4+ T cells expressing PD-1 in dMP-draining lymph nodes, again regardless of the encapsulated antigen (Figure 9A–B; gating scheme – Supplemental Figure 12). A similar trend toward increased CD4+ T cells expressing the inhibitory surface marker, ICOS, was also noted across dMP treated mice, albeit not statistically significant (Figure 9C). Finally, Foxp3+CD4+ Treg frequencies were increased in the dMP draining lymph nodes in all treatment groups (including dMP OVA) compared to untreated mice (Figure 10A; gating scheme – Supplemental Figure 12), but Treg frequency was not increased in the pancreatic lymph nodes (Figure 10B) or spleen (Figure 10C). Mice that were already hyperglycemic at this cross-sectional time point exhibited roughly comparable leukocyte frequencies and phenotypes to their euglycemic counterparts (data not shown), consistent with research that suggests longitudinal frequencies of leukocyte populations do not accurately predict diabetes in NOD mice [31].
Figure 8. Administration of the dMP reduced markers of activation on DCs in dMP-draining lymph nodes.
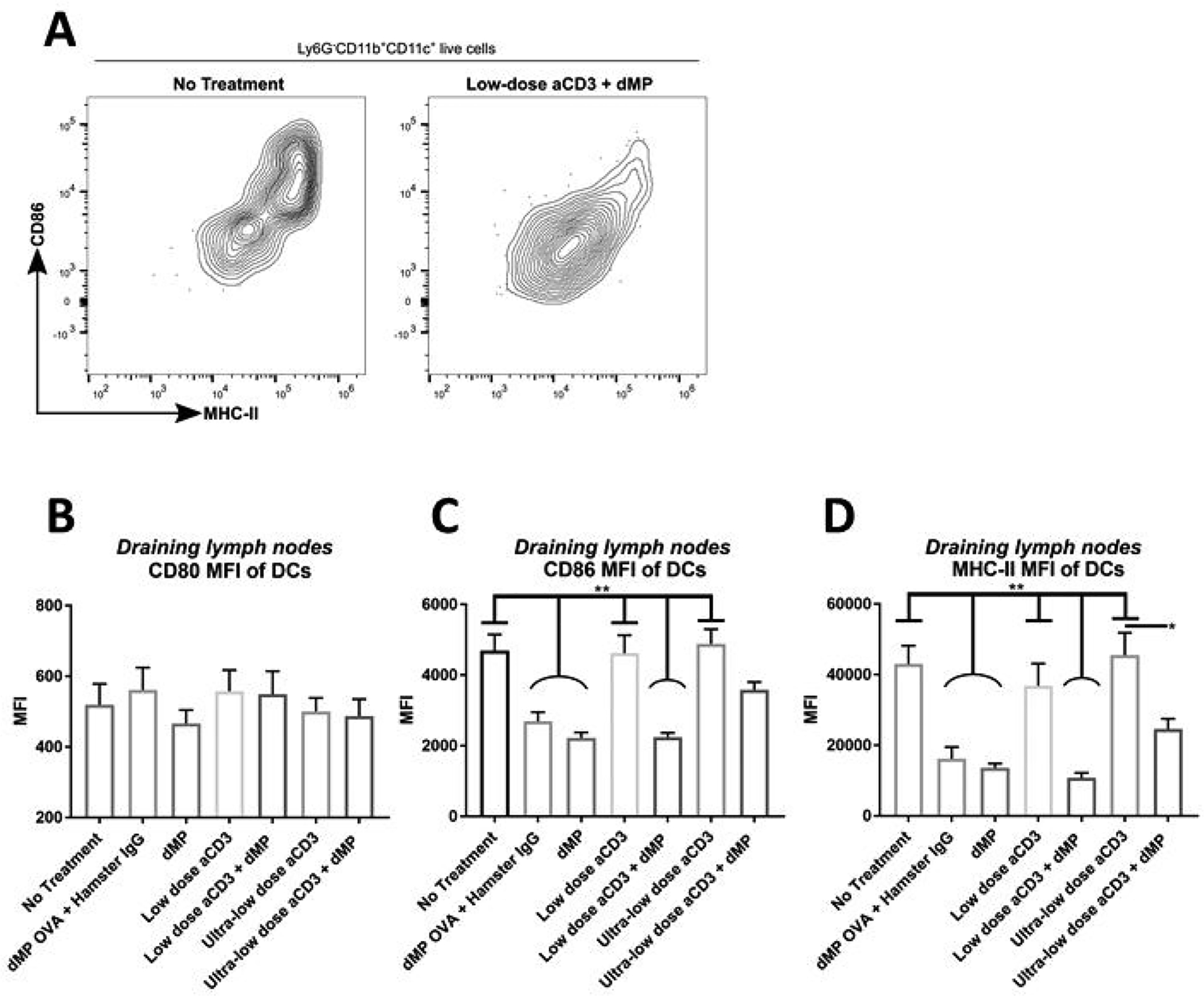
Twelve-week-old pre-diabetic NOD mice received aCD3 and dMP treatment at identical time points as in the prevention study and were euthanized at 20 weeks of age, prior to the sixth dMP injection. As before, MP injections were administered subcutaneously on the right side of the abdomen, proximal to the pancreas. (A) Ipsilateral dMP-draining lymph nodes (combined inguinal and axillary) were excised and stained for flow cytometry (n = 4–5/group). Mean fluorescent intesnsity (MFI) of CD80 (B), CD86 (C), and MHC-II (D) expression was characterized on Ly6G−CD11b+CD11c+ DCs. P-values (* ≤ 0.05, ** ≤ 0.01) were determined by one-way ANOVA with Tukey’s multiple comparison test. Significance in (C-D) is illustrated by pairwise comparisons between groups with flat bars (no treatment, low-dose aCD3, and ultra-low dose aCD3 groups)against each group with curved bars (dMP OVA + control IgG, dMP, and low-dose aCD3 + dMP groups) (nine significant pairwise comparisons total). Data is represented by mean ± SEM. Full gating scheme – see Supplemental Figure 11.
Figure 11. ICOS expression was upregulated on splenic CD4+ T cells in ultra-low-dose aCD3 treated mice.
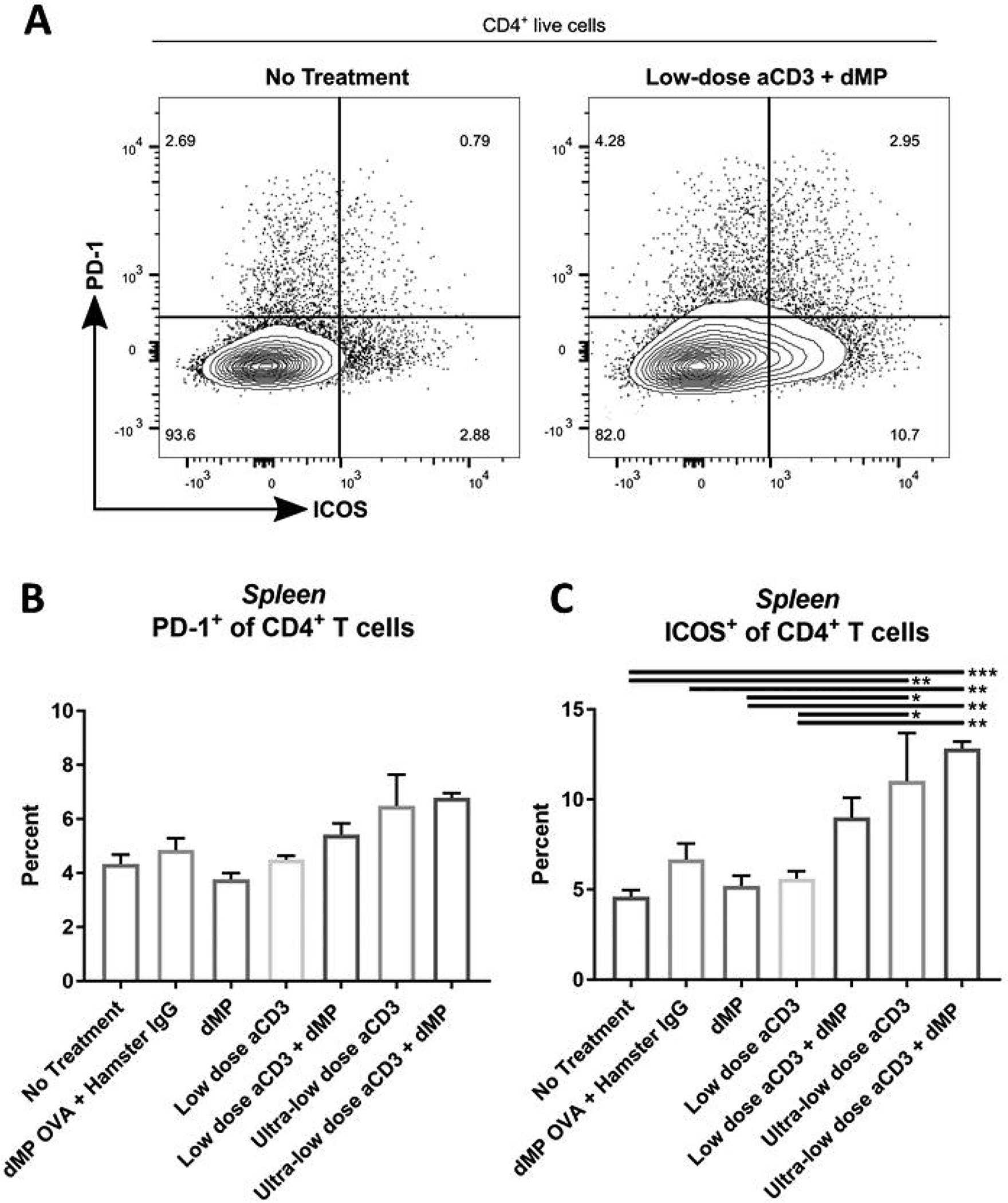
Twelve-week-old pre-diabetic NOD mice received aCD3 and dMP treatment at identical time points as in the prevention study (Figure 4A) and were euthanized at 20 weeks of age, prior to the sixth dMP injection. As before, MP injections were administered subcutaneously on the right side of the abdomen and lymphoid organs excised and (A) stained for flow cytometry (n = 4–5/group). Positive expression of regulatory markers (B) PD-1 and (C) ICOS was characterized on splenic CD4+ T cells. P-values (* ≤ 0.05, ** ≤ 0.01, *** ≤ 0.001) were obtained by one-way ANOVA with Tukey’s significance test. Data is represented by mean ± SEM. Full gating scheme – see Supplemental Figure 12.
Figure 9. Administration of the dMP increased PD-1 on CD4+ T cells in dMP-draining lymph nodes.
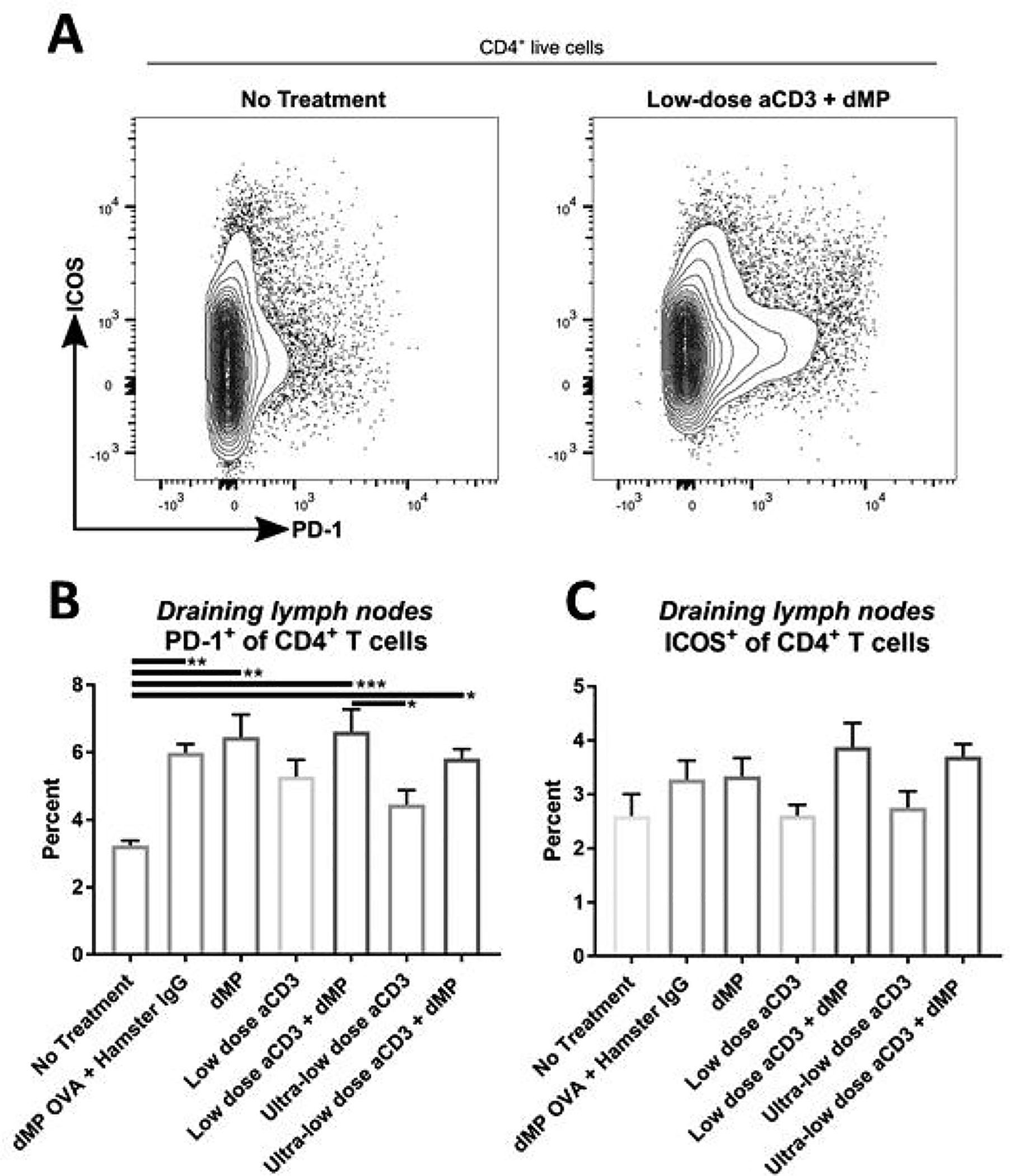
Twelve-week-old pre-diabetic NOD mice received aCD3 and dMP treatment at identical time points as in the prevention study and were euthanized at 20 weeks of age, prior to the sixth dMP injection. As before, MP injections were administered subcutaneously on the right side of the abdomen, proximal to the pancreas. (A) Ipsilateral dMP-draining lymph nodes (combined inguinal and axillary) were excised and stained for flow cytometry (n = 4–5/group). Positive expression of regulatory markers (B) PD-1 and (C) ICOS was characterized on CD4+ T cells. P-values (* ≤ 0.05, ** ≤ 0.01, *** ≤ 0.001) were obtained by one-way ANOVA with Tukey’s multiple comparison test. Data is represented by mean ± SEM. Full gating scheme – see Supplemental Figure 12.
Figure 10. Treg frequency was elevated in all treatment groups compared to untreated mice in dMP-draining lymph nodes, but comparable across groups in pancreatic lymph nodes and spleen.
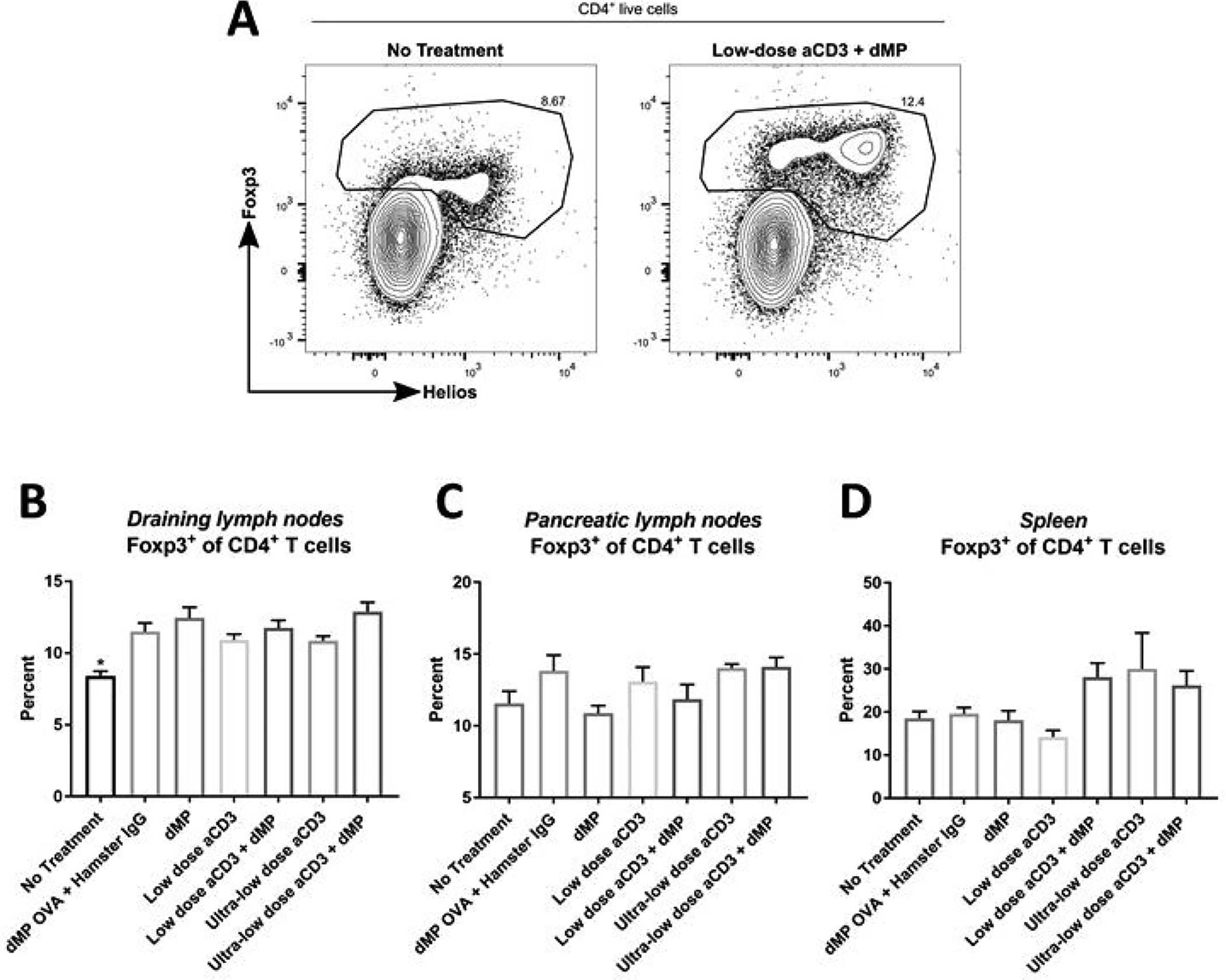
Twelve-week-old pre-diabetic NOD mice received aCD3 and dMP treatment at identical time points as in the prevention study (Figure 4A) and were euthanized at 20 weeks of age, prior to the sixth dMP injection. As before, MP injections were administered subcutaneously on the right side of the abdomen, proximal to the pancreas. (A) Ipsilateral dMP-draining lymph nodes (combined inguinal and axillary), pancreatic lymph nodes, and spleen were excised and stained for flow cytometry (n = 4–5/group). Frequency of Foxp3+ Tregs as a percentage of total CD4+ T cells was characterized in (B) dMP-draining lymph nodes, (C) pancreatic lymph nodes, and (D) spleen. P-values (* P ≤ 0.05) were determined by one-way ANOVA with Tukey’s multiple comparison test. Data is represented by mean ± SEM. Full gating scheme – see Supplemental Figure 12.
Ultra-Low-Dose aCD3 Treatment was Associated with an Increase in Splenic ICOS+CD4+ T Cells, a Reduction in Splenic CD4+ and CD8+ T Cells, and an Increase in DCs and Granulocytes in the Spleen
Cellular composition of lymphoid organs was notably different between low-dose and ultra-low-dose aCD3 groups as well (Supplemental Figure 5). The frequency of CD4+ T cells in pancreatic lymph nodes was slightly reduced in ultra-low-dose aCD3 + dMP treated mice compared to both low-dose aCD3 groups (Supplemental Figure 5A). Differences were also seen when comparing the frequency of CD8+ T cells in pancreatic lymph nodes (Supplemental Figure 5B). Integrating these results together, the ratio of CD4+:CD8+ T cells increased in low-dose aCD3 and low-dose aCD3 + dMP treatment groups (Supplemental Figure 5C).
Splenic T cell frequencies were markedly lower in ultra-low-dose aCD3 treated mice for both CD4+ and CD8+ T cells (Supplemental Figure 5D–F). These changes in cellular composition were reflected in the elevated splenic CD4+:CD8+ T cell ratios observed for low-dose aCD3 and low-dose aCD3 + dMP treatment groups (Supplemental Figure 5F). Treg frequencies in the spleen were not significantly different across treatment groups; however, a trend suggests ultralow-dose aCD3 treatment may have increased Treg frequencies (Figure 10D).While no difference in PD-1 expression was observed on splenic CD4+ T cells across treatment groups at 20 weeks of age (Figure 11B; gating scheme – Supplemental Figure 12), ultra-low-dose aCD3 treated mice displayed increased ICOS expression compared to untreated and even low-dose aCD3 treated mice (Figure 11C). Conversely, low-dose aCD3 treated mice showed similar ICOS expression to untreated animals. Hence, the trend toward improved therapeutic efficacy seen in ultra-low-dose aCD3 and ultra-low-dose aCD3 + dMP treated mice (Figure 4B) may be explained by altered leukocyte frequencies and phenotypes seen in these animals.
T cell phenotype alterations in ultra-low-dose aCD3 and ultra-low-dose aCD3 + dMP treated mice can in part be explained by a corresponding increase in DC and B220−CD11b+CD11c− granulocyte frequencies (Supplemental Figure 6A–B) and phenotypic shifts. The vast majority of B220−CD11b+CD11c− were granulocytes, as indicated by an SSChi phenotype (>70%; similar across treatment groups). Phenotypic analysis also revealed a reduction in markers of maturation on splenic DCs and granulocytes (Supplemental Figure 7). Diminished CD86 (Supplemental Figure 7B) and MHC-II (Supplemental Figure 7C) expression was seen on DCs for mice treated with low-dose aCD3 + dMP and ultra-low dose aCD3, respectively. However, all four groups receiving aCD3 (with or without dMP) exhibited reduced CD86 (Supplemental Figure 7E) and MHC-II (Supplemental Figure 7F) expression on B220−CD11b+CD11c− cells, as compared to treatment groups that did not receive aCD3. Interestingly, low-dose aCD3 + dMP treated mice displayed trends in phenotypic alterations similar to ultra-low-dose aCD3 and ultra-low-dose aCD3 + dMP treatment groups across these flow cytometric analyses.
DISCUSSION
We previously demonstrated efficacy of earlier formulations of this dMP system in preventing T1D in pre-diabetic, 4- and 8-week-old NOD mice, reversing hyperglycemia in recent-onset NOD mice, and in a mouse model of multiple sclerosis upon substitution to myelin-based antigen [15–17]. Furthermore, another group has recently used this dMP platform to promote remission of collagen-induced arthritis in mice [32]. We sought to expand therapeutic efficacy of this biomaterial platform by modifying the dMP formulation to deliver higher doses of a DC chemokine and pro-tolerogenic factor. Initial results investigating the udMP formulation, which incorporated 7x GM-CSF and 2.5x TGF-β1 compared to the earlier iteration of the dMP [16, 17], were promising, with the udMP augmenting DC recruitment. Similarly, in accordance with our previous findings [15], expression of T cell activating makers on DCs, CD86 and MHC-II, were reduced at the site of subcutaneous injection, consistent with hallmarks of a suppressive DC phenotype [12, 13]. However, the udMP formulation, although accomplishing what was intended in early time point in vivo experiments, failed to translate to improved T1D prevention when administered to 8-week-old NOD mice. Specifically, 60% of NOD mice treated with the udMP remained non-diabetic by 28 weeks of age, which is a cohort fraction equivalent to historical outcomes for 8-week-old NOD mice treated with the dMP [17]. Additionally, there is a lack of significance between the udMP and BMP groups. Notably, prevention for the BMP group appears unexpectedly high, and higher (40% vs. 20%) than we previously reported [17]. Larger cohorts in future studies may be able to rectify this seeming difference as well as possibly resolve any potential difference between the udMP and BMP. Nevertheless, regarding the present goal, it is clear the udMP failed to provide a dramatic improvement in prevention. While there are limitations with such comparison to historical data (e.g., disparate cohort and cohort sizes) confounding the ability to draw statistical conclusions between the udMP and dMP formulations, the lack of increased prevention in the udMP cohort, as well as lack of significance between the udMP and BMP groups suggests that the udMP formulation modifications were insufficient to make a substantial improvement in NOD mouse T1D prevention.
The reason for a lack of corresponding improvement in diabetes prevention is unclear, but may be due to several factors. Dendritic cell recruitment to the site of MP injection was much higher upon increased GM-CSF and TGF-β1 doses in the udMP, more than doubling the frequency of DCs at the site of injection compared to blank MPs. However, a concomitant increase in a tolerogenic DC phenotype was less forthcoming. Although statistically significant, DC expression of CD86 and MHC-II was reduced by only 16% and 20%, respectively, compared to DCs isolated from blank MP nodules. The marginal improvement in DC tolerogenicity may not have provided enough of a boost to overcome widespread inflammation typically seen in 8-week-old NOD mice [22, 33]. Alternatively, it is possible the PLGA biomaterial itself may influence a suppressive phenotype, as seen in cultured DCs [34]. Another explanation may be that kinetics of udMP immunomodulation may be too slow, potentially taking weeks to be fully realized, by which time NOD mice can exhibit more pronounced insulitis and an irrecoverable loss of β-cell mass [35]. Evidence to support this premise can be inferred from a comparable PLGA-based GM-CSF delivery system [25]. In this work, a PLGA matrix that delivered controlled release of GM-CSF over ~15 days, comparable to our own system, took ≥ 10 days after subcutaneous administration to detect augmented DC numbers in draining lymph nodes, suggesting DC egression from the injection site occurred only after GM-CSF reservoirs were depleted. If applied to our system, udMP administration in 8-week-old mice might not influence immunity until ~10 weeks of age, at which time blood leukocyte frequencies in NOD mice begin to diverge from non-obese diabetes resistant (NOR) mice, exhibiting increased circulating B cells and granulocytes, and some NOD mice may even exhibit overt hyperglycemia [21, 22]. Despite the disappointing lack of improved therapeutic efficacy of the udMP, there is clearly a large parameter space to investigate here and optimize, potentially leaving opportunity for future success in this general approach.
Limitations regarding therapeutic efficacy of the udMP despite improved secondary outcomes motivated investigation of the dMP in combination with aCD3. Building on previous research that demonstrated low-dose aCD3 provides a euglycemic window when administered to 12-week-old NOD mice [27], we hypothesized that aCD3 treatment in combination with dMP administration would provide a favorable immune profile with which repopulating naïve T cells will be amendable to tolerogenic instruction from the dMP. Here again, results suggested the guiding principles behind the study were well founded, but synergistic effects toward T1D prevention were not realized.
Findings from this study showed that low-dose aCD3 treatment modified the T cell population consistent with results from previous research [4, 27, 29, 36–38]. Specifically, transient depletion of CD4+ and CD8+ T cells was observed in low-dose aCD3 treated mice, with more pronounced depletion of CD8+ T cells. Furthermore, low-dose aCD3 selectively depleted naïve T cells, subsequently increasing the proportion of Tregs as a percentage of total CD4+ T cells. The remaining circulating CD4+ T cell population was also characterized as having additional markers of immunoregulation, exhibiting increased surface expression of PD-1 and ICOS compared to T cells in untreated mice. In contrast, ultra-low-dose aCD3 did not deplete circulating T cell levels, although some impact was noted due to the change in naïve:memory CD4+ T cell ratio.
Similarly, administration of the dMP modulated local immunity toward a suppressive phenotype consistent with previous results [15–17, 32]. Characterization of blood leukocytes revealed the dMP did not alter circulating immune cell frequency or phenotype. However, analysis of secondary lymphoid organs showed that dendritic cells isolated from lymph nodes proximal to the dMP injection site had diminished CD86 and MHC-II expression and PD-1 was upregulated on CD4+ T cells. These findings were demonstrated in dMP-draining lymph nodes, but not seen in distal lymphoid organs including the pancreatic lymph nodes or spleen. The localized response of the dMP has typically been suggested as an advantageous design feature. In contrast to the global immunosuppression employed for autoimmunity [39, 40], which leaves patients at increased risk for opportunistic infections and cancer development [41], the dMP has been shown to prevent T1D and a mouse model of multiple sclerosis without inducing systemic suppression, likely due to the antigen-specificity in the dMP system. In this current model, however, the local immunomodulation did not impart tolerance capable of inhibiting autoimmunity.
One potential explanation for this lack of autoimmune protection is that the dMP does not provide enough pro-tolerogenic support to overcome the progressive pathogenic activity in 12 week old NOD mice. Some evidence to support this includes the observed increased frequency of Tregs in dMP-draining lymph nodes but not in distal lymphoid organs. Previous findings demonstrated dMP treatment increased systemic Treg frequencies at 10, 12, and 14 weeks of age in NOD mice which began treatment at 8-weeks-old [17]. However, results here suggest the changes are either transient, as Treg frequencies were comparable to untreated mice in distal lymphoid organs (spleen and pancreatic lymph nodes) by 20 weeks of age (over eight weeks removed from the initial repeated dMP injection battery) or that 12-week-old NOD mice are significantly more resistant to immunomodulation than their younger counterparts. Alternatively, an explanation for the absence of success may be found by examining the mechanisms of action for aCD3-mediated T1D protection. Although the complete mechanisms of action are still under investigation, some evidence, while requiring further validation, points to an enhanced regulatory population rather than the depletion of autoreactive cells as a driving factor for autoimmune protection [27, 29, 36]. Specifically, researchers demonstrated that insulitis persisted in protected animals that received aCD3 and that these mice were diabetes-resistant even upon the adoptive transfer of diabetogenic splenocytes [27]. Notably, upregulated tolerogenic modalities include an increased Treg frequency and PD-1 on CD4+ T cells [29, 36], a few of the same mechanisms that the dMP also influences. Similarly, work by Perruche et al. demonstrated that aCD3 increased upregulated suppressive pathways in innate immune cells, increasing TGF-β production by macrophages and DCs following ingestion of apoptotic T cells [30]. Tolerance to apoptotic debris in APCs has also been linked to indoleamine 2,3-dioxygenase expression [42], an immunoregulatory enzyme similarly upregulated in response to the dMP in DCs [17], which is capable of programming suppressive DCs [43], again suggesting that the dMP and aCD3 may induce tolerance through overlapping mechanisms. Thus, it is possible that the combination treatment attempted to amplify redundant, potentially saturated pathways, where neither treatment has been shown to provide long-lasting autoimmune protection when administered to 12-week-old NOD mice.
Lastly, findings from this study suggested that ultra-low-dose aCD3 (1 μg/day for five days) may have protective effects in 12-week-old NOD mice. While statistical significance was not realized when including all seven groups in the Kaplan-Meier survival analysis using Bonferroni correction, pairwise comparisons between survival curves of mice that received the ultra-low-dose aCD3 or ultra-low-dose aCD3 + dMP and untreated mice resulted in p-values of 0.08 and 0.03, respectively, suggesting that ultra-low-dose aCD3 may have a protective effect against T1D onset.
A cohort of mice that were euthanized at 20 weeks of age revealed several possible markers of therapeutic mechanism associated with T1D prevention. Surface expression of ICOS was upregulated on splenic CD4+ T cells, as was a trend of increased Treg frequencies and PD-1 in the spleen. While often implicated in T cell activation, ICOS expression is essential for functional stability of Foxp3 in Tregs [44] and inhibition of the ICOS pathway in in NOD neonates or BDC2.5-NOD (BDC2.5) mice exacerbates T1D [45]. Differential immunosuppressive capacity of Foxp3+ Tregs is an emerging topic in immunobiology, for example, metabolic differences between Treg subsets is reportedly critical in maintaining survival and homeostatic suppressive function [46]. While the frequencies of Tregs in NOD mice were not statistically significant different between groups when evaluated at 20 weeks of age, functional differences in Treg activity due to ICOS expression may possibly explain the protective effect exhibited. Additionally, T cell frequencies were noticeably different in secondary lymphoid organs in ultra-low-dose aCD3 mice. These animals displayed diminished CD4+ T cell frequencies in the pancreatic lymph nodes and reduced CD4+ and CD8+ splenic T cell frequencies. Most dramatic, however, was the increase in splenic DC and granulocyte frequencies. While full characterization of innate immunity was not performed, spleens from ultra-low-dose aCD3 treated mice showed that ≥ 20% of leukocytes were B220−CD11b+CD11c−, over 70% of which were SSChi granulocytes. Various reports in recent years have suggested that granulocytes, neutrophils in particular, play a pivotal role in T1D pathogenesis in both mice and humans [47–49]. Notably, T1D patients and those at risk of developing disease exhibit circulating neutropenia, however, display enhanced neutrophil numbers in pancreatic tissue [48]. It may be that sequestration of neutrophils in the spleen rather than pancreatic tissue could limit β-cell destruction. However, many of these results were mirrored in low-dose aCD3 + dMP treated mice, which displayed an equivalent diabetes incidence rate as low-dose aCD3 treated mice, suggesting that these findings may not be a primary mechanism for the therapeutic benefit. Worth mention are the reductions seen in DC and granulocyte maturation markers CD86 and MHC-II. In contrast to the largely transient changes seen in alterations in T cell numbers and preservation of regulatory cells, the lasting suppressive phenotype on DCs persisted more than two months following aCD3 treatment. These changes likely reflect the complex interplay between innate and adaptive immunity, potentially through reciprocal tolerization of DCs via the elevated Treg numbers [50, 51].
These results establish the capacity of a biodegradable MP system to modulate immunity toward an immunosuppressive state in situ. However, the lack of diabetes prevention even with a concomitant increase in DC tolerogenicity underscores limitations surrounding the lack of understanding of fundamental aspects of T1D, as well as the need for improved understanding of timing of administration of such a platform, which appears to depend upon the specific stage of disease. These findings were parlayed into a subsequent approach, where we anticipated that depletion of the T cell population using aCD3 would generate a favorable immune milieu to reprogram with the dMP. Our results suggested the dMP inhibited DC immunogenicity and modestly increased markers of tolerance on T cells. However, the dMP failed to synergize with aCD3 administration to provide improved late stage diabetes prevention. Lack of synergy could be explained in part by the apparently overlapping immunosuppressive mechanisms of aCD3 and the dMP, neither of which demonstrated long-lasting diabetes protection when administered at 12 weeks. Taking these two experiments together suggests that supplementing a complementary mechanism of immune tolerance to the dMP platform may be necessary to overcome autoimmunity at this disease stage. However, more work is needed to fully understand the implications of these results.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the National Institutes of Health (R01 DK091658 and R01 AI133623 to BGK and T32 DK108736 to JMS), the University of Florida Clinical and Translational Science Institute Network Science Pilot award, and the McJunkin Family Charitable Foundation, Inc.
Footnotes
Conflicts of Interest
BGK is a cofounder of OneVax, LLC, a preclinical biotechnology company with interest in commercializing the dual-microparticle technology. The authors declare no other relevant conflicts of interest.
REFERENCES
- [1].Atkinson MA, Eisenbarth GS, Michels AW, Type 1 diabetes, Lancet 383(9911) (2014) 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Gregg BE, Moore PC, Demozay D, Hall BA, Li M, Husain A, Wright AJ, Atkinson MA, Rhodes CJ, Formation of a human beta-cell population within pancreatic islets is set early in life, J. Clin. Endocrinol. Metab 97(9) (2012) 3197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, Herold KC, Lares A, Lee MR, Li K, Liu W, Long SA, Masiello LM, Nguyen V, Putnam AL, Rieck M, Sayre PH, Tang Q, Type 1 diabetes immunotherapy using polyclonal regulatory T cells, Sci. Transl. Med 7(315) (2015) 315ra189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Herold KC, Hagopian W, Auger JA, Poumian-Ruiz E, Taylor L, Donaldson D, Gitelman SE, Harlan DM, Xu D, Zivin RA, Bluestone JA, Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus, N. Engl. J. Med 346(22) (2002) 1692–8. [DOI] [PubMed] [Google Scholar]
- [5].Sherry N, Hagopian W, Ludvigsson J, Jain SM, Wahlen J, Ferry RJ Jr., Bode B, Aronoff S, Holland C, Carlin D, King KL, Wilder RL, Pillemer S, Bonvini E, Johnson S, Stein KE, Koenig S, Herold KC, Daifotis AG, Protege Trial I, Teplizumab for treatment of type 1 diabetes (Protege study): 1-year results from a randomised, placebo-controlled trial, Lancet 378(9790) (2011) 487–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Orban T, Bundy B, Becker DJ, Dimeglio LA, Gitelman SE, Goland R, Gottlieb PA, Greenbaum CJ, Marks JB, Monzavi R, Moran A, Peakman M, Raskin P, Russell WE, Schatz D, Wherrett DK, Wilson DM, Krischer JP, Skyler JS, Type G 1 Diabetes TrialNet Abatacept Study, Costimulation modulation with abatacept in patients with recent-onset type 1 diabetes: follow-up 1 year after cessation of treatment, Diabetes Care 37(4) (2014) 1069–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Haller MJ, Gitelman SE, Gottlieb PA, Michels AW, Perry DJ, Schultz AR, Hulme MA, Shuster JJ, Zou B, Wasserfall CH, Posgai AL, Mathews CE, Brusko TM, Atkinson MA, Schatz DA, Antithymocyte Globulin Plus G-CSF Combination Therapy Leads to Sustained Immunomodulatory and Metabolic Effects in a Subset of Responders With Established Type 1 Diabetes, Diabetes 65(12) (2016) 3765–3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Haller MJ, Gitelman SE, Gottlieb PA, Michels AW, Rosenthal SM, Shuster JJ, Zou B, Brusko TM, Hulme MA, Wasserfall CH, Mathews CE, Atkinson MA, Schatz DA, Anti-thymocyte globulin/G-CSF treatment preserves beta cell function in patients with established type 1 diabetes, J. Clin. Invest 125(1) (2015) 448–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Creusot RJ, Giannoukakis N, Trucco M, Clare-Salzler MJ, Fathman CG, It’s time to bring dendritic cell therapy to type 1 diabetes, Diabetes 63(1) (2014) 20–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Keselowsky BG, Xia CQ, Clare-Salzler M, Multifunctional dendritic cell-targeting polymeric microparticles: engineering new vaccines for type 1 diabetes, Human vaccines 7(1) (2011) 37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Banchereau J, Steinman RM, Dendritic cells and the control of immunity, Nature 392(6673) (1998) 245–252. [DOI] [PubMed] [Google Scholar]
- [12].Steinman RM, Hawiger D, Nussenzweig MC, Tolerogenic dendritic cells, Annu. Rev. Immunol 21 (2003) 685–711. [DOI] [PubMed] [Google Scholar]
- [13].Maldonado RA, von Andrian UH, How tolerogenic dendritic cells induce regulatory T cells, Adv. Immunol 108 (2010) 111–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Appleman LJ, Boussiotis VA, T cell anergy and costimulation, Immunol. Rev 192 (2003) 161–80. [DOI] [PubMed] [Google Scholar]
- [15].Cho JJ, Stewart JM, Drashansky TT, Brusko MA, Zuniga AN, Lorentsen KJ, Keselowsky BG, Avram D, An antigen-specific semi-therapeutic treatment with local delivery of tolerogenic factors through a dual-sized microparticle system blocks experimental autoimmune encephalomyelitis, Biomaterials 143 (2017) 79–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lewis JS, Dolgova NV, Zhang Y, Xia CQ, Wasserfall CH, Atkinson MA, Clare-Salzler MJ, Keselowsky BG, A combination dual-sized microparticle system modulates dendritic cells and prevents type 1 diabetes in prediabetic NOD mice, Clin. Immunol (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lewis JS, Stewart JM, Marshall GP, Carstens MR, Zhang Y, Dolgova NV, Xia C, Brusko TM, Wasserfall CH, Clare-Salzler MJ, Atkinson MA, Keselowsky BG, Dual-Sized Microparticle System for Generating Suppressive Dendritic Cells Prevents and Reverses Type 1 Diabetes in the Nonobese Diabetic Mouse Model, ACS biomaterials science & engineering 5(5) (2019) 2631–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Foged C, Brodin B, Frokjaer S, Sundblad A, Particle size and surface charge affect particle uptake by human dendritic cells in an in vitro model, Int. J. Pharm 298(2) (2005) 315–22. [DOI] [PubMed] [Google Scholar]
- [19].Cannon GJ, Swanson JA, The macrophage capacity for phagocytosis, J. Cell Sci 101 (Pt 4) (1992) 907–13. [DOI] [PubMed] [Google Scholar]
- [20].Zent CS, Elliott MR, Maxed out macs: physiologic cell clearance as a function of macrophage phagocytic capacity, FEBS J 284(7) (2017) 1021–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Miyazaki A, Hanafusa T, Yamada K, Miyagawa J, Fujino-Kurihara H, Nakajima H, Nonaka K, Tarui S, Predominance of T lymphocytes in pancreatic islets and spleen of pre-diabetic non-obese diabetic (NOD) mice: a longitudinal study, Clin. Exp. Immunol 60(3) (1985) 622–30. [PMC free article] [PubMed] [Google Scholar]
- [22].Pearson JA, Wong FS, Wen L, The importance of the Non Obese Diabetic (NOD) mouse model in autoimmune diabetes, J. Autoimmun 66 (2016) 76–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Prasad S, Kohm AP, McMahon JS, Luo X, Miller SD, Pathogenesis of NOD diabetes is initiated by reactivity to the insulin B chain 9–23 epitope and involves functional epitope spreading, J. Autoimmun 39(4) (2012) 347–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zugmaier G, Paik S, Wilding G, Knabbe C, Bano M, Lupu R, Deschauer B, Simpson S, Dickson RB, Lippman M, Transforming growth factor beta 1 induces cachexia and systemic fibrosis without an antitumor effect in nude mice, Cancer Res 51(13) (1991) 3590–4. [PubMed] [Google Scholar]
- [25].Ali OA, Huebsch N, Cao L, Dranoff G, Mooney DJ, Infection-mimicking materials to program dendritic cells in situ, Nature materials 8(2) (2009) 151–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Herold KC, Bundy BN, Long SA, Bluestone JA, DiMeglio LA, Dufort MJ, Gitelman SE, Gottlieb PA, Krischer JP, Linsley PS, Marks JB, Moore W, Moran A, Rodriguez H, Russell WE, Schatz D, Skyler JS, Tsalikian E, Wherrett DK, Ziegler AG, Greenbaum CJ, Type G 1 Diabetes TrialNet Study, An Anti-CD3 Antibody, Teplizumab, in Relatives at Risk for Type 1 Diabetes, N. Engl. J. Med (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chatenoud L, Primo J, Bach JF, CD3 antibody-induced dominant self tolerance in overtly diabetic NOD mice, J. Immunol 158(6) (1997) 2947–54. [PubMed] [Google Scholar]
- [28].Gill RG, Pagni PP, Kupfer T, Wasserfall CH, Deng S, Posgai A, Manenkova Y, Bel Hani A, Straub L, Bernstein P, Atkinson MA, Herold KC, von Herrath M, Staeva T, Ehlers MR, Nepom GT, A Preclinical Consortium Approach for Assessing the Efficacy of Combined Anti-CD3 Plus IL-1 Blockade in Reversing New-Onset Autoimmune Diabetes in NOD Mice, Diabetes 65(5) (2016) 1310–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Penaranda C, Tang Q, Bluestone JA, Anti-CD3 therapy promotes tolerance by selectively depleting pathogenic cells while preserving regulatory T cells, J. Immunol 187(4) (2011) 2015–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Perruche S, Zhang P, Liu Y, Saas P, Bluestone JA, Chen W, CD3-specific antibody-induced immune tolerance involves transforming growth factor-beta from phagocytes digesting apoptotic T cells, Nat. Med 14(5) (2008) 528–35. [DOI] [PubMed] [Google Scholar]
- [31].Telieps T, Kohler M, Treise I, Foertsch K, Adler T, Busch DH, Hrabe de Angelis M, Verschoor A, Adler K, Bonifacio E, Ziegler AG, Longitudinal Frequencies of Blood Leukocyte Subpopulations Differ between NOD and NOR Mice but Do Not Predict Diabetes in NOD Mice, Journal of diabetes research 2016 (2016) 4208156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Allen R, Chizari S, Ma JA, Raychaudhuri S, Lewis JS, Combinatorial, Microparticle-Based Delivery of Immune Modulators Reprograms the Dendritic Cell Phenotype and Promotes Remission of Collagen-Induced Arthritis in Mice, ACS Applied Bio Materials 2(6) (2019) 2388–2404. [DOI] [PubMed] [Google Scholar]
- [33].Anderson MS, Bluestone JA, The NOD mouse: a model of immune dysregulation, Annu. Rev. Immunol 23 (2005) 447–85. [DOI] [PubMed] [Google Scholar]
- [34].Allen RP, Bolandparvaz A, Ma JA, Manickam VA, Lewis JS, Latent, Immunosuppressive Nature of Poly(lactic-co-glycolic acid) Microparticles, ACS biomaterials science & engineering 4(3) (2018) 900–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wang P, Fiaschi-Taesch NM, Vasavada RC, Scott DK, Garcia-Ocana A, Stewart AF, Diabetes mellitus--advances and challenges in human beta-cell proliferation, Nat. Rev. Endocrinol 11(4) (2015) 201–12. [DOI] [PubMed] [Google Scholar]
- [36].Belghith M, Bluestone JA, Barriot S, Megret J, Bach JF, Chatenoud L, TGF-beta-dependent mechanisms mediate restoration of self-tolerance induced by antibodies to CD3 in overt autoimmune diabetes, Nat. Med 9(9) (2003) 1202–8. [DOI] [PubMed] [Google Scholar]
- [37].Chatenoud L, Thervet E, Primo J, Bach JF, Anti-CD3 antibody induces long-term remission of overt autoimmunity in nonobese diabetic mice, Proc. Natl. Acad. Sci. U. S. A 91(1) (1994) 123–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Herold KC, Bluestone JA, Montag AG, Parihar A, Wiegner A, Gress RE, Hirsch R, Prevention of autoimmune diabetes with nonactivating anti-CD3 monoclonal antibody, Diabetes 41(3) (1992) 385–91. [DOI] [PubMed] [Google Scholar]
- [39].Couri CE, Oliveira MC, Stracieri AB, Moraes DA, Pieroni F, Barros GM, Madeira MI, Malmegrim KC, Foss-Freitas MC, Simoes BP, Martinez EZ, Foss MC, Burt RK, Voltarelli JC, C-peptide levels and insulin independence following autologous nonmyeloablative hematopoietic stem cell transplantation in newly diagnosed type 1 diabetes mellitus, JAMA 301(15) (2009) 1573–9. [DOI] [PubMed] [Google Scholar]
- [40].Voltarelli JC, Couri CE, Stracieri AB, Oliveira MC, Moraes DA, Pieroni F, Coutinho M, Malmegrim KC, Foss-Freitas MC, Simoes BP, Foss MC, Squiers E, Burt RK, Autologous nonmyeloablative hematopoietic stem cell transplantation in newly diagnosed type 1 diabetes mellitus, JAMA 297(14) (2007) 1568–76. [DOI] [PubMed] [Google Scholar]
- [41].Caspi RR, Immunotherapy of autoimmunity and cancer: the penalty for success, Nat. Rev. Immunol 8(12) (2008) 970–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ravishankar B, Liu H, Shinde R, Chandler P, Baban B, Tanaka M, Munn DH, Mellor AL, Karlsson MC, McGaha TL, Tolerance to apoptotic cells is regulated by indoleamine 2,3-dioxygenase, Proc. Natl. Acad. Sci. U. S. A 109(10) (2012) 3909–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Bracho-Sanchez E, Hassanzadeh A, Brusko MA, Wallet MA, Keselowsky BG, Dendritic Cells Treated with Exogenous Indoleamine 2,3-Dioxygenase Maintain an Immature Phenotype and Suppress Antigen-specific T cell Proliferation, Journal of immunology and regenerative medicine 5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Landuyt AE, Klocke BJ, Colvin TB, Schoeb TR, Maynard CL, Cutting Edge: ICOS-Deficient Regulatory T Cells Display Normal Induction of Il10 but Readily Downregulate Expression of Foxp3, J. Immunol 202(4) (2019) 1039–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kornete M, Sgouroudis E, Piccirillo CA, ICOS-dependent homeostasis and function of Foxp3+ regulatory T cells in islets of nonobese diabetic mice, J. Immunol 188(3) (2012) 1064–74. [DOI] [PubMed] [Google Scholar]
- [46].Wang H, Franco F, Tsui YC, Xie X, Trefny MP, Zappasodi R, Mohmood SR, Fernandez-Garcia J, Tsai CH, Schulze I, Picard F, Meylan E, Silverstein R, Goldberg I, Fendt SM, Wolchok JD, Merghoub T, Jandus C, Zippelius A, Ho PC, CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors, Nat. Immunol 21(3) (2020) 298–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Diana J, Simoni Y, Furio L, Beaudoin L, Agerberth B, Barrat F, Lehuen A, Crosstalk between neutrophils, B-1a cells and plasmacytoid dendritic cells initiates autoimmune diabetes, Nat. Med 19(1) (2013) 65–73. [DOI] [PubMed] [Google Scholar]
- [48].Valle A, Giamporcaro GM, Scavini M, Stabilini A, Grogan P, Bianconi E, Sebastiani G, Masini M, Maugeri N, Porretti L, Bonfanti R, Meschi F, De Pellegrin M, Lesma A, Rossini S, Piemonti L, Marchetti P, Dotta F, Bosi E, Battaglia M, Reduction of circulating neutrophils precedes and accompanies type 1 diabetes, Diabetes 62(6) (2013) 2072–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Wang Y, Xiao Y, Zhong L, Ye D, Zhang J, Tu Y, Bornstein SR, Zhou Z, Lam KS, Xu A, Increased neutrophil elastase and proteinase 3 and augmented NETosis are closely associated with beta-cell autoimmunity in patients with type 1 diabetes, Diabetes 63(12) (2014) 4239–48. [DOI] [PubMed] [Google Scholar]
- [50].Hoebe K, Janssen E, Beutler B, The interface between innate and adaptive immunity, Nat. Immunol 5(10) (2004) 971–4. [DOI] [PubMed] [Google Scholar]
- [51].Rissoan MC, Soumelis V, Kadowaki N, Grouard G, Briere F, de Waal Malefyt R, Liu YJ, Reciprocal control of T helper cell and dendritic cell differentiation, Science 283(5405) (1999) 1183–6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.